
Aminotriazine derived N-doped mesoporous carbon with a tunable nitrogen content and their improved oxygen reduction reaction performance†
Jefrin M.
Davidraj
a,
C. I.
Sathish
 *a,
Premkumar
Selvarajan
b,
Mohammed
Fawaz
a,
Vibin
Perumalsamy
a,
Xiaojiang
Yu
c,
Mark B. H.
Breese
c,
Jiabao
Yi
*a and
Ajayan
Vinu
*a
*a,
Premkumar
Selvarajan
b,
Mohammed
Fawaz
a,
Vibin
Perumalsamy
a,
Xiaojiang
Yu
c,
Mark B. H.
Breese
c,
Jiabao
Yi
*a and
Ajayan
Vinu
*a
aGlobal Innovative Centre for Advanced Nanomaterials (GICAN), School of Engineering, The University of Newcastle, Callaghan, New South Wales 2308, Australia. E-mail: sathish.ci@newcastle.edu.au; jiabao.yi@newcastle.edu.au; ajayan.vinu@newcastle.edu.au
bDepartment of Physics, School of Advanced Sciences, Vellore Institute of Technology, Vellore, India
cSingapore Synchrotron Light Source, National University of Singapore, 119260, Singapore
First published on 29th July 2024
Abstract
The electrocatalytic activity of carbon materials is highly dependent on the controlled modulation of their composition and porosity. Herein, mesoporous N-doped carbon with different amounts of nitrogen was synthesized through a unique strategy of using a high nitrogen containing CN precursor, 3-amino 1,2,4 triazine (3-ATZ) which is generally used for the preparation of carbon nitrides, integrated with the combination of a templating method and high temperature treatment. The nitrogen content and the graphitisation of the prepared materials were finely tuned with the simple adjustment of the carbonisation temperature (800–1100 °C). The optimised sample as an electrocatalyst for oxygen reduction reaction (ORR) exhibited an onset potential of 0.87 V vs. RHE with a current density of 5.1 mA cm−2 and a high kinetic current density (Jk) of 33.1 mA cm−2 at 0.55 V vs. RHE. The characterisation results of the prepared materials indicated that pyridinic and graphitic nitrogen in the carbon framework promoted ORR activity with improved four-electron selectivity and excellent methanol tolerance and stability. DFT calculations demonstrated that the structural and planar defects in the N-doped carbon regulated the surface electronic properties of the electrocatalyst, leading to a reduction in the energy barrier for the ORR activity. This strategy has the potential to unlock a platform for designing a series of catalysts for electrochemical applications.
1. Introduction
The surge in the development of renewable energy storage and conversion technologies is driven by the urgent need to address the environmental crisis caused by over-reliance on fossil fuel-based energy generation. With ongoing developments, proton exchange membrane fuel cells (PEMFC) emerge as promising green energy alternatives because of their impressive efficiency in converting different fuels into electric energy, offering a clean and sustainable way to mitigate the global energy crisis. However, there are still limiting factors that put hurdle in the path of their performance and commercialisation, including the sluggish nature of the ORR.1–7 To overcome these limitations, many noble metal-based nanoparticles (Pt, Ir and Ru) and their alloys have been widely used as catalysts for ORR. However, CO poisoning, limited stability, scarcity, and the high cost of noble metals restrict their wide industrial applications. Owing to these reasons, there has been significant interest recently in developing highly efficient and low-cost electrocatalysts for ORR.8 In recent years, heteroatom-doped carbon materials have been broadly studied,2,9–13 but those containing nitrogen substitutions, particularly, have gained significant attention as ORR catalysts owing to their excellent electron transfer kinetics, long-term stability, scalability and low cost.4,8,14–17 Among them, graphitic carbon nitride (g-C3N4) with a high nitrogen content is a promising candidate for boosting the ORR activity as the N functionalities in g-C3N4 can induce a more positive charge density on adjacent carbon atoms due to their different atomic sizes and higher electronegativity than carbon.2,15 This effect is beneficial for weakening the O–O bond, promoting oxygen molecule chemisorption and the overall ORR catalytic process.17–19 However, the lack of electronic conductivity and the poor specific surface area of g-C3N4 are the major drawbacks which limit its performance in the ORR, highlighting an area for further exploration.20–24Previous studies focused on introducing micropores and mesopores into the carbon nitride structure with a high nitrogen content to improve its specific surface area and pore volume, contributing to enhanced active sites and ion transport.25–28 Although a family of high nitrogen-containing mesoporous carbon nitrides (MCNs) like C3N5, C3N6 and C3N7 with unique chemical structures and a high specific surface area has been successfully prepared by varying the nature of the carbon nitride precursors,2,25,26,29–31 the poor conductivity of these materials is the major issue as it significantly affects the efficiency of the catalysts in the ORR. Therefore, developing high-performance ORR catalysts with a high number of active sites and high electronic conductivity for ORR remains a topic of interest. It is surmised that catalysts with the properties of both CN and graphitic carbon can offer high conductivity but at the same time change the local density state of bonded carbon atoms with nitrogen to increase the charge density on the adjacent carbon atoms. This simple manipulation is believed to be a key factor in enhancing the ORR performance as it may increase the number of active sites and Lewis basic sites.2,32,33
In this work, we developed a mesoporous nitrogen-doped porous carbon through a hard templating approach using SBA-15 and 3-amino 1,2,4 triazine (3-ATZ) as a dual carbon and nitrogen precursor, which is generally used for the preparation of carbon nitride materials, by treating at different high carbonization temperatures. The prepared materials exhibit a mesoporous structure with structural and planar defects favouring the formation of pyridinic, pyrrolic and graphitic N at the edge sites which contribute more π electrons to the π system. The prepared catalyst displayed a high specific surface area of 759.6 m2 g−1 and a high pore volume of 1.61 cm3 g−1. The nitrogen content in the mesoporous carbon can be tuned with the simple adjustment of the carbonisation temperature which in turn controls the number of active sites and the conductivity of the materials. The optimised N-doped and ordered mesoporous carbon sample treated at high carbonisation temperature showed superior electrocatalytic performance for ORR with a half-wave potential (E1/2) of 0.78 V, excellent stability and resistance towards methanol. With the help of DFT calculations, we predicted that the pyridinic and pyrrolic N contents at the edge sites, formed after treating at high carbonisation temperature, are crucial in promoting and enhancing the ORR activity in ordered mesoporous N-doped carbon.
2. Experimental method
2.1. Synthesis of mesoporous N-doped carbon from 3-ATZ using SBA-15 as a template
Mesoporous N-doped carbons were synthesized using SBA-15 as a sacrificial template and 3-ATZ as a C and N precursor.34 Initially, the SBA-15 template was prepared using pluronic P123 surfactant via a soft templating method at 150 °C through a procedure described in the earlier reports.34–37 For the preparation of mesoporous N-doped carbon, 1 g of the SBA-15 template was mixed and ground thoroughly with 4 g of 3-ATZ using a mortar and pestle. A solution containing 0.64 g of concentrated HCl in 5 ml water was added to the ground mixture. This solution helps in filling the pores of the silica template with 3-ATZ using a pore-filling method. The resulting sample was then placed in a hot-air oven and heated at 100 °C for 6 h, followed by heating at 160 °C for another 6 h. After the above steps, the composite was carbonized at various temperatures (800, 900, 1000, and 1100 °C) with a heating rate of 5 °C min−1 for 5 h. The carbonization process was carried out in a tubular furnace under a nitrogen atmosphere. Once the carbonization was completed, the silica template was removed by washing with hydrofluoric acid (HF), and then the sample was extensively washed with ethanol to eliminate any remaining HF. The samples were then dried in a 100 °C oven for overnight and the final product was labelled as NMC-AT-800, NMC-AT-900, NMC-AT-1000 and NMC-AT-1100 based on the carbonization temperature at 800, 900, 1000, and 1100 °C respectively. The high carbonisation temperature was used to improve the crystallinity and further to tune the N content in the graphitic carbon framework of NMC-AT materials. The color of the final NMC-AT materials was black.2.2. Characterization
The structure of NMC-AT samples was studied using a powder X-ray diffractometer Empyrean from PANalytical instruments. The sample was ground into fine powder and loaded into the sample holder and irradiated using Cu-Kα radiation (λ = 1.5405 A) which operates at a voltage and current of 40 kV and 40 mA, respectively. XRD patterns were collected in the region 2θ = 10°–80° using a step size of 0.007. The d-spacing was calculated using the Braggs law λ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ), where λ is the wavelength of the X-ray beam, d is the distance and θ is the diffraction angle. N2 adsorption–desorption isotherms were obtained at liquid nitrogen temperature (−196 °C) using a Micromeritics ASAP 2420 analyser. All the samples were degassed at 200 °C for 12 h prior to the measurements. The surface area was obtained using the Brunauer–Emmett–Teller (BET) method. The total pore volume was taken from the adsorption point at a relative pressure (P/P0) of 0.99. The surface morphology of the sample was investigated using scanning electron microscopy (SEM) from JEOL with the model JSM-7900F. FTIR analysis was performed using a Frontier MIR/NIR spectrometer (PerkinElmer) using the standard KBr method. Raman spectra were recorded on a Renishaw confocal spectrometer with 633 nm laser excitation. X-ray photoelectron spectroscopy (XPS) analysis was carried out using the multiprobe photoelectron spectrometer from Omicron Nanotechnology. Peak fitting and atomic concentrations were carried out using the CasaXPS software. Near-edge X-ray absorption fine structure (NEXAFS) was obtained on the Soft X-ray Spectroscopy Beamline at the Singapore synchrotron facility in Singapore.
sin(θ), where λ is the wavelength of the X-ray beam, d is the distance and θ is the diffraction angle. N2 adsorption–desorption isotherms were obtained at liquid nitrogen temperature (−196 °C) using a Micromeritics ASAP 2420 analyser. All the samples were degassed at 200 °C for 12 h prior to the measurements. The surface area was obtained using the Brunauer–Emmett–Teller (BET) method. The total pore volume was taken from the adsorption point at a relative pressure (P/P0) of 0.99. The surface morphology of the sample was investigated using scanning electron microscopy (SEM) from JEOL with the model JSM-7900F. FTIR analysis was performed using a Frontier MIR/NIR spectrometer (PerkinElmer) using the standard KBr method. Raman spectra were recorded on a Renishaw confocal spectrometer with 633 nm laser excitation. X-ray photoelectron spectroscopy (XPS) analysis was carried out using the multiprobe photoelectron spectrometer from Omicron Nanotechnology. Peak fitting and atomic concentrations were carried out using the CasaXPS software. Near-edge X-ray absorption fine structure (NEXAFS) was obtained on the Soft X-ray Spectroscopy Beamline at the Singapore synchrotron facility in Singapore.
2.3. Electrochemical ORR measurement
Electrochemical ORR studies were carried out in a three-electrode system at room temperature using a Pine electrochemical workstation coupled with a galvanostat/potentiostat. A glassy carbon electrode (GCE) of 5 mm diameter was used as a working electrode, which was well-cleaned before electrochemical experiments. The sample ink was prepared in a 2 ml vial, where 5 mg of the sample, 50 μL of 5 wt% Nafion solution and 950 μL isopropyl alcohol were sonicated for an hour to obtain a well-homogenized suspension. About 10 μL of the suspension was added dropwise onto the GCE and dried at room temperature. In a basic O2-saturated aqueous 0.1 M KOH electrolyte environment, a platinum wire was immersed as the counter electrode and Ag/AgCl as the reference electrode, and RDE measurements were carried out. The number of electrons transferred per oxygen molecule was calculated using the Koutecky–Levich equation as given below:where, j is the measured current density; jk and jd are the kinetic and diffusion-limited current density, respectively; n is the overall electron number transferred per oxygen molecule during the ORR; F represents the Faraday constant (96
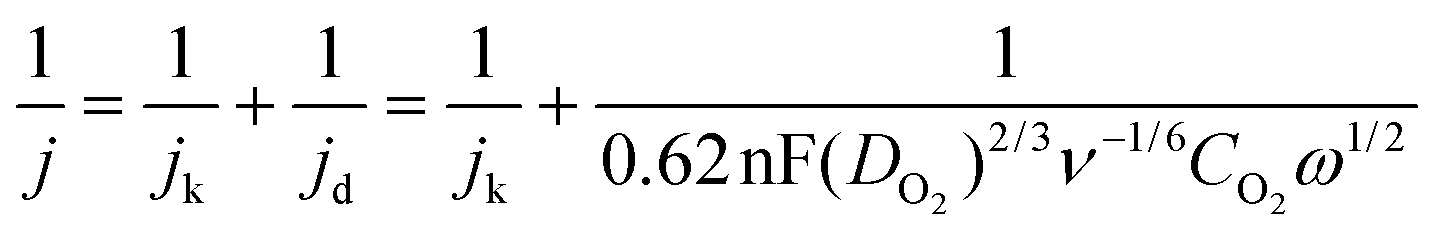 485 C mol−1); DO2 is the diffusion coefficient of O2 in the electrolyte (1.9 × 10−5 cm2 s−1 in 0.1 M KOH); ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1); CO2 is the bulk concentration of O2 in the electrolyte (1.2 × 10−6 mol cm−3 in 0.1 M KOH); ω is the rotating rate of the electrode (rad s−1).
485 C mol−1); DO2 is the diffusion coefficient of O2 in the electrolyte (1.9 × 10−5 cm2 s−1 in 0.1 M KOH); ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1); CO2 is the bulk concentration of O2 in the electrolyte (1.2 × 10−6 mol cm−3 in 0.1 M KOH); ω is the rotating rate of the electrode (rad s−1).
2.4. Computational methods
All the first-principles calculations were performed with the Vienna ab initio Simulation Package (VASP) based on density functional theory.38,39 The projector augmented wave (PAW) method was considered for core electrons, and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was used to represent valence electrons.40 For all the calculations, an energy cutoff of 520 eV was used. A vacuum space of 20 Å along the z direction was considered for all calculations to avoid interactions between the different nearest neighbouring layers. Bader charge analysis was carried out to understand the charge-transfer interactions.41The adsorption energy was calculated using the formula,
| EAds = ENMC-AT+O2 − (ENMC-AT + EO2) |
3. Results and discussion
The synthesis of NMC-AT samples is schematically represented in Fig. 1. From the key synthesis steps schematized, the confinement of 3-amino 1,2,4-triazine into the sacrificial mould of SBA-15 restricted the movement of the precursor molecules, and the thermal treatment promoted the formation of interconnected carbon and nitrogen pore networks. Powder X-ray diffraction was used to investigate the crystal structure of the NMC-AT samples prepared at different carbonisation temperatures. As shown in Fig. 2a, all the samples showed broad diffraction peaks at the 2θ angle of ∼25.8 to 26.07° and ∼44.3° corresponding to the (002) and (101) plane of graphitic carbon. The two tiny peaks at 17.1 and 55.3° in NMC-AT-1100 correspond to the impurities at the time of measurement.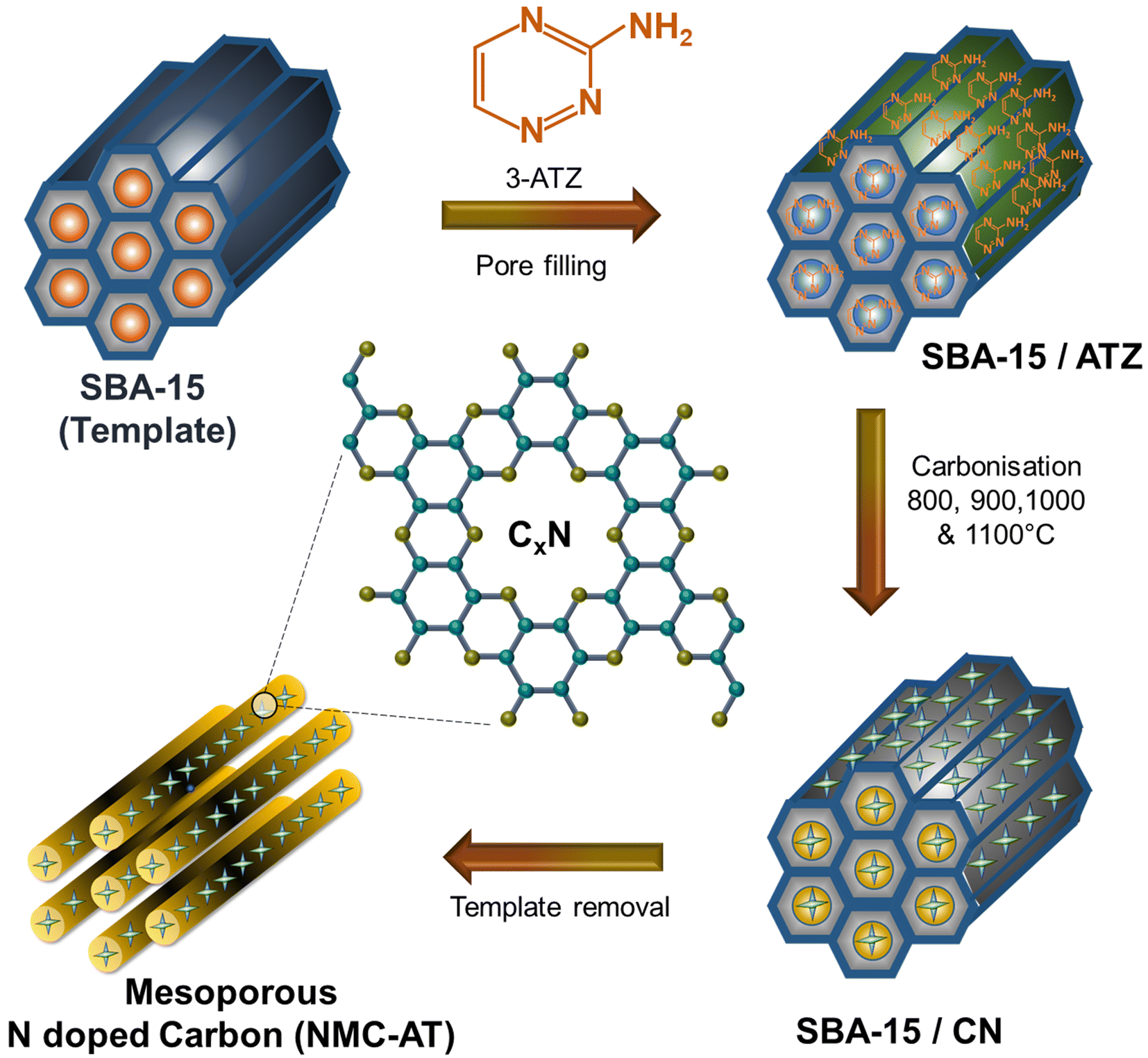 | ||
| Fig. 1 Schematic representation of the synthesis of NMC-AT samples. | ||
 | ||
| Fig. 2 (a) Powder X-ray diffraction patterns and (b) nitrogen adsorption–desorption isotherms of NMC-AT samples. (c) SEM image and (d) TEM image of the NMC-AT-1000 sample. | ||
The XRD pattern of these samples showed a small shift of the (002) peak towards a lower angle as the carbonisation temperature increased from 800 to 1100 °C. The corresponding interlayer distance calculated from the XRD patterns of the NMC-AT samples increased with increasing the carbonisation temperature, i.e. 0.341 nm (NMC-AT-800), 0.342 nm (NMC-AT-900), 0.344 nm (NMC-AT-1000), and 0.345 nm (NMC-AT-1100), respectively. This shift could be due to the fact that the high carbonisation temperature helps to improve the polymerization and condensation reaction of the 3-ATZ precursor within the nanochannels of the template which leads to improved graphitization of the carbon structure inside the pores of the final samples. The combined effect of the thermal expansion and potential changes in the carbon structure at high temperatures could have led to a higher d-spacing value.42 Noticeably, the samples did not show any lower angle peaks, indicating the absence of a long-range periodic arrangement of the mesoporous structure.43
N2 adsorption–desorption analysis was carried out to understand the surface properties of the NMC-AT samples. The isotherms of NMC-AT samples shown in Fig. 2b display a type IVa isotherm with an H3 hysteresis loop of the IUPAC classification. The feature of broad capillary condensation noticed at the higher relative pressure regions indicates that NMC-AT samples contained a mesoporous structure with disordered pores and broad pore size distribution. The textural properties of the NMC-AT samples are summarized in Table 1. The sample NMC-AT-900 displayed the highest specific surface area of 759.6 m2 g−1 among all the samples, while NMC-AT-800 exhibited a surface area of 634.1 m2 g−1. When the calcination temperature increased above 900 °C, a significant reduction in the specific surface area was observed. The specific surface area was reduced from 682.9 and 589.4 m2 g−1 when the carbonization temperature was increased from 1000 to 1100 °C. The reduction in the specific surface area for NMC-AT-1000 and NMC-AT-1100 could be due to the defects and collapsed structure in the carbon framework that create large pores, adversely affecting the adsorption capacity of the samples. The pore size distribution analysis using the Barrett–Joyner–Halenda (BJH) method indicates that NMC-AT samples have mesopores with a moderate pore size of ∼3.68 to 3.83 nm (Fig. S1†). Among the samples studied, NMC-AT-1000 exhibited the highest specific pore volume (1.61 cm3 g−1) and micropore surface area (90.3 m2 g−1). However, the micropore area (50.2 m2 g−1) and the pore volume (1.24 cm3 g−1) reduced quite significantly when the carbonization temperature was raised to 1100 °C. The change in the specific surface area and the pore volume could be linked to a change in the microporosity and the final crystallinity of the materials at different carbonisation temperatures.
| Sample name | SABET (m2 g−1) | Total pore volume (cm3 g−1) | t-Plot micropore area (m2 g−1) | Micropore volume (cm3 g−1) | Pore diameter (nm) | CHNS (at%) | XPS (at%) |
|---|---|---|---|---|---|---|---|
| C/H/N | C/N/O | ||||||
| NMC-AT-800 | 634.1 | 1.40 | 46.4 | 0.023 | 3.68 | 5.4/1.9/2.36 | 67.4/30.3/2.3 |
| NMC-AT-900 | 759.6 | 1.51 | 43.9 | 0.026 | 3.70 | 6.59/1.4/1.38 | 81.1/17.5/1.4 |
| NMC-AT-1000 | 682.9 | 1.61 | 90.3 | 0.039 | 3.83 | 7.14/1.14/0.92 | 85.9/12.3/1.8 |
| NMC-AT-1100 | 589.4 | 1.24 | 50.2 | 0.024 | 3.73 | 7.76/0.74/0.42 | 92.4/5.7/1.9 |
The surface morphology and the mesoporous structure of the NMC-AT samples were analyzed using SEM and TEM. As shown in Fig. S2,† all the NMC-AT samples displayed a porous structure. At the annealing temperature of 800 and 900 °C, a rod-like morphology was observed (Fig. S2a–d†). When the temperature increased to 1000 and 1100 °C, the morphology was changed from rod to sheet-like structures (Fig. S2e–h†). On the other hand, TEM analysis indicates that all the samples exhibited a sheet-like structure in high resolution, as shown in Fig. 2c, d and Fig. S3.† This change in morphology rather than a mesoporous ordered structure could be due to the collapse of the mesoporous channels and the temperature-assisted graphitization of carbon at elevated temperatures. The inset in Fig. 2d clearly shows both meso and micropores in the sample, as shown by the arrow, which may be due to the evaporation of nitrogen or carbon elements induced pores.
Raman spectroscopy analysis was carried out on the NMC-AT samples to identify the structural defects and N doping. As shown in Fig. 3a, Raman spectra showed two distinct peaks at around 1340 and 1570 cm−1, corresponding to the D and G bands, respectively. The D band is associated with the disorder-induced structural defects on the graphitic plane, while the G band relates to the E2g vibration mode present in the sp2 carbon domains.44–46 The D and G bands of NMC-AT-1100 displayed a shift in the peaks to 1357 and 1583 cm−1, respectively. The peak intensity ratio ID/IG indicates the amount of structural defects in the material and exposure of the edge plane. From the intensity ratio calculated, the NMC-AT-800 sample showed the highest ID/IG ratio of 1.08 and decreased further in NMC-AT-900 (1.06), NMC-AT-1000 (1.05), and NMC-AT-1100 (0.98). The incorporation of more nitrogen atoms into the graphite layers created structural defects and exposed the edge planes, resulting in high ID/IG ratios in NMC-AT-800. Likewise, the decrease in ID/IG ratios to 0.98 in NMC-AT-1100 indicates that the sp2 domains are partially restored, and the graphitic degree is improved.
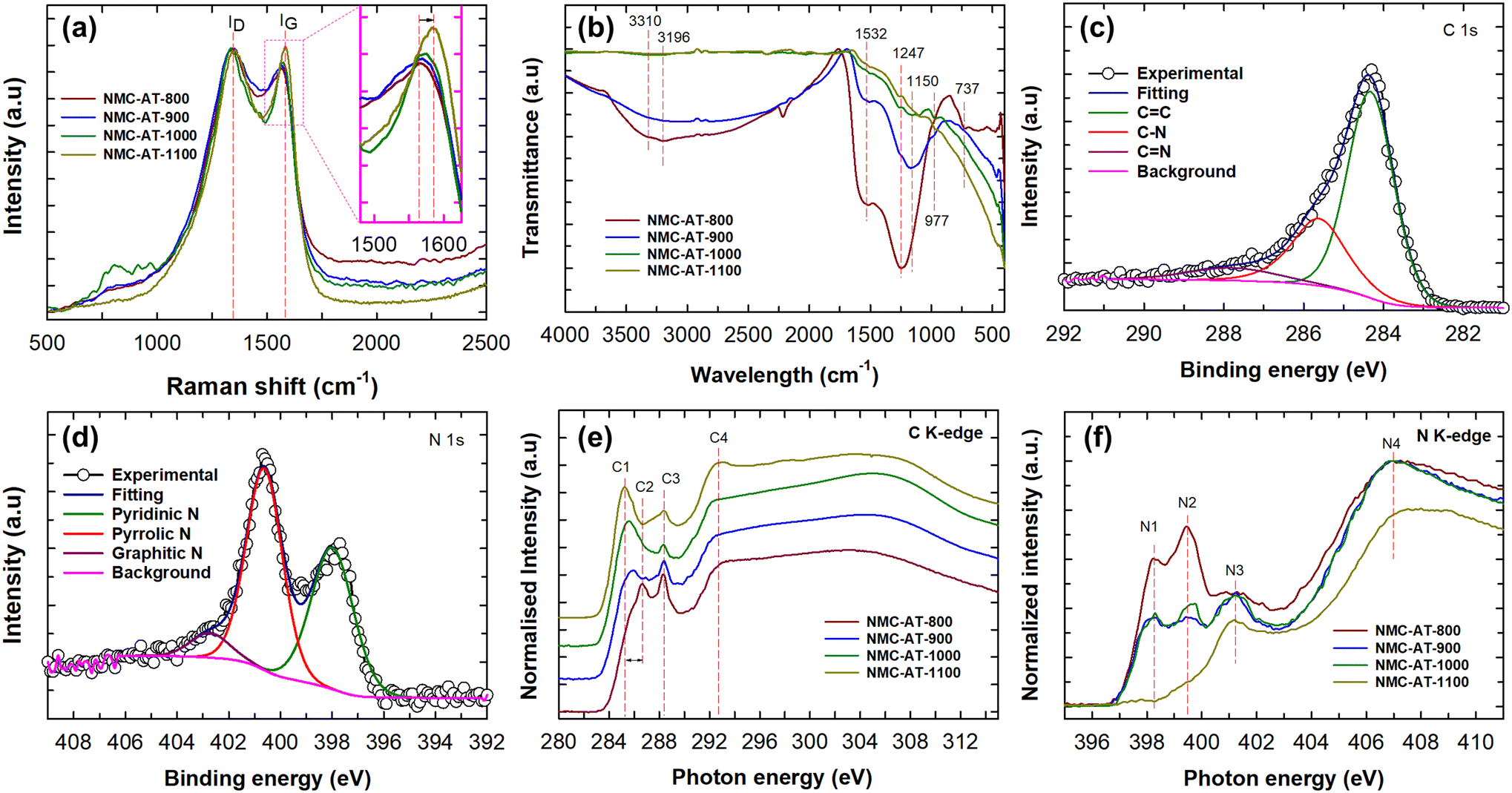 | ||
| Fig. 3 (a) Raman spectra and (b) FTIR of NMC-AT samples. (c) C 1s spectra and (d) N 1s spectra of NMC-AT-1000. (e) C K-edge and (f) N K-edge spectra of NMC-AT samples. | ||
The elemental composition of NMC-AT samples was measured using a CHN analyser, and the corresponding results are summarized in Table 1. The increase in carbon and the decrease in nitrogen content directly correlate with the increase in heat-treatment temperature wherein the nitrogen is partially volatilized through gradual heating. The sample NMC-AT-800 displayed a high nitrogen content of 30.3 wt%, which should be defined as one kind of carbon nitride due to the high concentration of N. With the increasing heating temperature, the concentration of nitrogen gradually reduced to 5.7 wt% in NMC-AT-1100, signalling the formation of NMC-AT. This result is in good agreement with the Raman ID/IG ratios, indicating that the nitrogen content can be fine-tuned to create defects and expose edge planes for catalytic applications through a simple heat treatment process.
The chemical structure and the functional groups of NMC-AT were determined using FTIR spectroscopy. The FTIR spectra of NMC-AT samples are shown in Fig. 3b. The broadband noticed from 2800 to 3600 cm−1 corresponds to the symmetric and antisymmetric stretching vibrations of primary and secondary amines. The peak at 1150, 1247 and 1532 cm−1 corresponds to the sp2 C–N stretching vibrations of the heterocyclics, and the peak at 737 cm−1 corresponds to the aromatic C–C.25,45 As can be seen from the figure, the peak at 1247 cm−1 related to the C–N binding and the intensity of the band gradually decrease with increasing carbonization temperature, which confirms the decrease in N moieties at high temperatures. The intense shoulder peaks at 977 and 1532 cm−1 confirm the pyridinic N nature in the graphene lattice.47 The carbon and nitrogen bonding configuration and the elemental composition of NMC-AT samples were further investigated by X-ray photoelectron spectroscopy (XPS). As shown in Fig. S4,† the survey scan spectra of NMC-AT samples exhibited peaks corresponding to the C 1s, N 1s, and O 1s. From the spectra, we can notice that the N 1s peak intensity diminishes as the carbonization temperature increases and the O 1s spectra should have arisen due to the oxidation of the surface at high temperature. The XPS elemental analysis results are also summarized in Table 1, where the NMC-AT-800 sample shows the highest nitrogen content (30.3 At%) and is consistent with the CHN analysis. The results indicate that at a relatively low heating temperature, the sample may form carbon nitride. When the carbonisation temperature was increased, the transformation of carbon nitride to N-doped mesoporous carbon samples occurred.
The high-resolution C 1s spectra of NMC-AT samples, as shown in Fig. 3c and S5a–c,† are deconvoluted to three different peaks which are centred approximately at 284.4, 285.6, and 287.9 eV, respectively. The main peak in the C 1s spectra at 284.4 eV is assigned to the graphitic sp2 carbon, and the peaks at 285.6 and 287.9 eV are ascribed to nitrogen bonded to sp2-C and sp3-C, respectively. The peak at 284.4 eV shifts to high binding energy in NMC-AT-800 (284.8 eV) but the peak is shifted to lower binding energy for NMC-AT-900 (284.4 eV), NMC-AT-1000 (284.32 eV) and NMC-AT-1100 (284.28 eV) as the carbonisation temperature is increased. A detailed summary of XPS deconvolutions is given in Table S1.† From the table, it can be observed that the peak area of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C increases (41.03 to 71.68%), and a gradual decrease of C–N (50.06 to 18.84%) peak area in NMC-AT-800 to NMC-AT-1100 is noticed. This indicates that the carbon atoms partially replaced the nitrogen atoms in the graphitic domains as the carbonisation temperature is increased. Likewise, the high-resolution N 1s spectra (Fig. 3d and S5d–f†) are deconvoluted to three different peaks at 398.2, 400.4, and 402.7 eV. The peak positioned at 398.2 eV is assigned to the pyridinic nitrogen, which is heterogeneously bonded to two adjacent carbon atoms, whereas the peak at 400.4 eV is assigned to the pyrrolic-nitrogen, while the peak at 402.7 eV is assigned to the graphitic nitrogen.47,48 The N 1s spectra and Table S1† indicate a decrease in the pyridinic-N peak ratio and a simultaneous increase in pyrrolic-N from NMC-AT-800 to NMC-AT-1100 samples, suggesting a shift in the nitrogen bonding environment within the material. The peak intensity ratio ID/IG from Raman analysis also supports this observation, further confirming that pyridinic nitrogen causes structural defects and exposed planar defects.49 Moreover, from the XPS deconvolutions, it can be observed that the peak intensity attributed to the graphitic N significantly increases to a high value of 31.19% in NMC-AT-1100, suggesting that with the increase in carbonisation temperature, the graphitic N content in the graphite layers increases quite significantly.
C increases (41.03 to 71.68%), and a gradual decrease of C–N (50.06 to 18.84%) peak area in NMC-AT-800 to NMC-AT-1100 is noticed. This indicates that the carbon atoms partially replaced the nitrogen atoms in the graphitic domains as the carbonisation temperature is increased. Likewise, the high-resolution N 1s spectra (Fig. 3d and S5d–f†) are deconvoluted to three different peaks at 398.2, 400.4, and 402.7 eV. The peak positioned at 398.2 eV is assigned to the pyridinic nitrogen, which is heterogeneously bonded to two adjacent carbon atoms, whereas the peak at 400.4 eV is assigned to the pyrrolic-nitrogen, while the peak at 402.7 eV is assigned to the graphitic nitrogen.47,48 The N 1s spectra and Table S1† indicate a decrease in the pyridinic-N peak ratio and a simultaneous increase in pyrrolic-N from NMC-AT-800 to NMC-AT-1100 samples, suggesting a shift in the nitrogen bonding environment within the material. The peak intensity ratio ID/IG from Raman analysis also supports this observation, further confirming that pyridinic nitrogen causes structural defects and exposed planar defects.49 Moreover, from the XPS deconvolutions, it can be observed that the peak intensity attributed to the graphitic N significantly increases to a high value of 31.19% in NMC-AT-1100, suggesting that with the increase in carbonisation temperature, the graphitic N content in the graphite layers increases quite significantly.
Near-edge X-ray absorption fine spectroscopy (NEXAFS) was conducted to understand the electronic structure and the chemical bonding environment in the NMC-AT samples. Fig. 3e displays the C K-edge spectra of the NMC-AT samples which show four peaks at 285.2 (C1), 286.6 (C2), 288.3 (C3) and 292.6 eV (C4), respectively. The C1 and C4 peaks are assigned to the electron transition from 1s to π* CC and σ* C–C transition states in graphitic structures, respectively. The C2 peak is assigned to the sp2 hybridized carbon with nitrogen heterocycles CN/C–N, whereas the C3 peak is attributed to π* C–N–C or oxygen-containing functional groups of π* CO.25,46 Interestingly, we can note that the C2 peak in NMC-AT-800 is gradually suppressed with the concomitant increase in the intensity of the C1 peak as the carbonisation temperature is increased. The N K-edge spectra show four different peaks N1–N4, as shown in Fig. 3f. The peaks N1 and N2 at 398.3 and 399.4 eV are attributed to the π* C–N–C excitation and the CN–C, respectively. The N3 peak at 401.4 eV is assigned to the π* N-3C excitation, whereas N4 at 407.1 eV is ascribed to the π* transition similar to pyridine.45 It is evident from the N K-edge spectra that the peak intensity of N1 and N2 peaks in NMC-AT-800 gradually decreases with the increased annealing temperature, indicating the depletion of nitrogen. These results are consistent with FTIR, Raman, CHNS and XPS observations, which suggest that the amounts of nitrogen bonding configurations differ by varying the temperature, indicating the effective tuning of the nitrogen content in carbon using the technique in this work.
The electrochemical ORR activity of NMC-AT materials was investigated in 0.1 M KOH solution saturated with O2. A linear sweep voltammetry (LSV) measurement with a rotating disk electrode (RDE) was employed to isolate and study the fundamental electrochemical process happening at the electrode interface. The LSV curves for the NMC-AT and Pt/C samples were analyzed at a scanning rate of 5 & 10 mV s−1 and a rotating speed of 1600 rpm as shown in Fig. S6† and Fig. 4a. Noticeably, the sample NMC-AT-1000 displays a high activity towards the ORR in an alkaline electrolyte with an onset potential (Eonset) of 0.87 V (vs. RHE), a half-wave potential (E1/2) of 0.78 V (vs. RHE) and a current density (JL) of 5.1 mAcm−2. Other samples exhibit Eonset, E1/2 and JL in the decreasing order of the NMC-AT-900 (Eonset = 0.83 V vs. RHE, E1/2 = 0.73 V and JL = 4.61 mAcm−2), NMC-AT-1100 (Eonset = 0.82 V vs. RHE, E1/2 = 0.72 V and JL = 4.08 mAcm−2), and NMC-AT-800 (0.78 V vs. RHE, E1/2 = 0.67 V and JL = 4.56 mAcm−2), respectively. The higher specific surface area (682.9 m2 g−1) and higher pore volume (1.61 cm3 g−1) of NMC-AT-1000 compared to other samples influence the limiting current density. In addition, the presence of higher pyridinic and graphitic N in NMC-AT-1000 helps to achieve the highest onset potential. Interestingly, the sample NMC-AT-800, with a high amount of nitrogen, displays a 50 mV negative shift in the onset potential and 100 mV in the half-wave potential. Likewise, the NMC-AT-1100 sample shows a better Eonset, E1/2, but the decline noticed in the reduction current may be due to the high amount of graphitic N and low pyridinic and pyrrolic N compared to other samples.50 The ORR results demonstrate that tailoring the nitrogen content and the annealing temperature plays a key role in the performance of NMC-AT samples in the ORR. Although the NMC-AT-1000 sample shows the highest ORR activity as a metal-free electrocatalyst among the NMC-AT and other samples (Table S2†), the onset, half-wave potentials, and current density are slightly smaller than those of Pt/C (0.93 V vs. RHE, E1/2 = 0.81 V and JL = 5.34 mA cm−2).
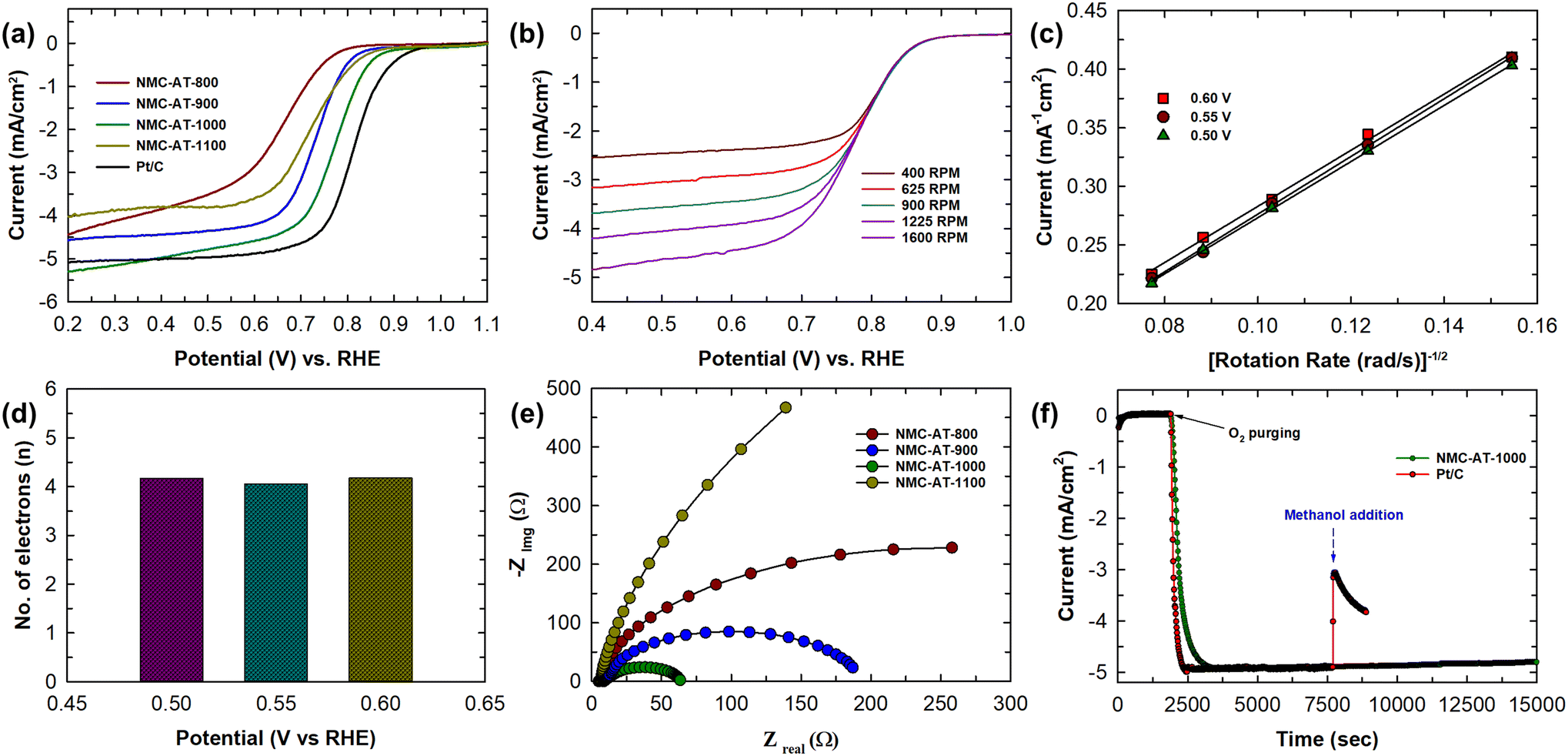 | ||
| Fig. 4 (a) LSV curves obtained at 10 mV s−1 and 1600 RPM. (b) LSV curves obtained at 5 mV s−1 and different speeds. (c) K–L plot derived from LSV curves. (d) Number of electrons calculated at different potentials. (e) EIS spectra of NMC-AT samples. (f) Methanol tolerance of the NMC-AT-1000 sample and Pt/C. | ||
LSV curves of NMC-AT-1000 were obtained at a scanning rate of 5 mV s−1 and various rotating speeds (400–1600 rpm) to clarify the reaction kinetics. As shown in Fig. 4, the current density is enhanced with the increase in the rotation rate, which is attributed to the facilitation of electrolyte diffusion. The potential dependent rate constant of the ORR was analyzed using the Koutecky–Levich equation for a more quantitative analysis. The K–L plot shown in Fig. 4c displays good linearity, exhibiting a first-order reaction kinetics. From the slopes of the K–L plots, it is observed that the sample NMC-AT-1000 exhibits a direct 4e− ORR process. Fig. 4d displays the number of electrons calculated at reaction potentials of 0.5, 0.55 and 0.6 V, which indicates that all the electron transfer numbers are close to 4. Moreover, the kinetic current density (Jk) calculated for the sample NMC-AT-1000 from the K–L plot at 0.55 V vs. RHE is 33.1 mA cm−2, which is higher than that of Pt/C.51 In addition, the Tafel slopes of all the samples were analyzed to understand the kinetics and the plot is displayed in Fig. S7.† The results show that NMC-AT-1000 has a Tafel slope of 82 mV dec−1 which is the lowest among samples like NMC-AT-900 (104 mV dec−1), NMC-AT-800 (109 mV dec−1) and NMC-AT-1100 (109.8 mV dec−1). The lower Tafel slope suggests that the ORR in NMC-AT-1000 proceeds in a faster kinetic way.
Electrochemical impedance spectroscopy (EIS) is further conducted to compare the reaction kinetics of different NMC-AT samples. It can be observed from Fig. 4e that the Nyquist plot of NMC-AT-1000 converges to a semicircle at lower potentials due to the decrease in the interfacial charge transfer resistance. Also, the lowest diameter of the semicircle indicates a lower polarization resistance in the electrochemical process compared to other samples. A similar trend was observed in the Bode plots as shown in Fig. S8.† NMC-AT-1000 exhibits a more favourable phase angle (<−20°) in the low frequency region, demonstrating the faster ion diffusion. The high specific surface area and porosity of NMC-AT-1000 enhanced the reactant adsorption, consequently reducing the activation energy and promoting a more efficient reaction. Furthermore, the durability test was carried out to understand the stability and tolerance of the electrocatalyst. The methanol tolerance of NMC-AT-1000 was determined by adding 3 M methanol into the 0.1 M KOH electrolyte. As shown in Fig. 4f, no significant change was noticed in the current density of the NMC-AT-1000 sample before and after the addition of methanol at 0.50 vs. RHE. In contrast, the current density of the Pt/C catalyst exhibits a sharp decrease with the addition of CH3OH. From these results, it can be concluded that NMC-AT-1000 emerges as a promising candidate for methanol fuel cells with stability exceeding 15000 seconds, especially considering the inadequate performance of commercial Pt/C. The presence of a high specific surface area, a large pore volume and the structural defects caused by doping nitrogen favoured a better performance in NMC-AT-1000. In addition, the high catalytic activity of NMC-AT-1000 towards the ORR indicates that the presence of optimum pyridinic and pyrrolic N as active sites together with high crystallinity and a larger micropore area and pore volume plays a key role in reducing oxygen through a 4e− pathway.
DFT calculations were further used to investigate the ORR mechanism and the synergistic effects of NMC-AT samples. The feasible geometrical structure of NMC-AT was modelled by considering a 7 × 7 supercell of graphene. In graphene, two carbon atoms were removed to resemble the monolayer of the carbon material and five carbon atoms were replaced by nitrogen atoms to represent the NMC-AT samples. Based on the experimental results obtained from FTIR, XPS and NEXAFS, the model chooses two pyrrolic-N, two pyridinic-N and one graphitic-N atom in the NMC-AT samples. The structure for NMC-AT was modelled and optimized without imposing any constraints, and the finalized structure is visualized in Fig. 5a. The adsorption of O2 is considered one of the important characteristics of ORR activity. Hence, the most feasible O2 adsorption sites and their adsorption energy on NMC-AT were calculated. Utilizing Bader charge analysis, four higher positive charge sites in the NMC-AT have been identified and predicted. The schematic representation of the carbon atoms with positive charge sites in the optimized NMC-AT structure is displayed in Fig. 5b. The O2 molecule has been allowed to interact with the positive sites of the NMC-AT, as shown in Fig. 5c. There are four possibilities for O2 adsorption. Model 1 is the O2 molecule attached to the carbon adjacent to the pyridinic N, Model 2 is the O2 molecule attached to the carbon ring structure, Model-3 is the O2 molecule attached to the C1 site of pyrrolic N, and Model-4 is the O2 attached to the C4 site of pyrrolic N. The calculated adsorption energies of Model-1, Model-2, Model-3 and Model-4 are −1.8, −1.56, −1.4 and −0.96 eV, respectively. A higher O2 adsorption energy was observed in Model I, indicating that the O2 molecule is most likely to adsorb onto the second most positively charged site of the NMC-AT sample. Moreover, the calculated adsorption energy of all the positive charge sites in NMC-AT is negative, demonstrating that carbon atoms around the nitrogen atoms act as active sites for O2 adsorption. The carbon atoms have higher adsorption energy, which promotes O2 adsorption and, hence, the ORR efficiency. The experimental findings of the ORR proved that the NMC-AT samples are undergoing a 4e− pathway oxygen reduction. The DFT calculations reveal that the framework of NMC-AT paves the way for efficient ORR activity.
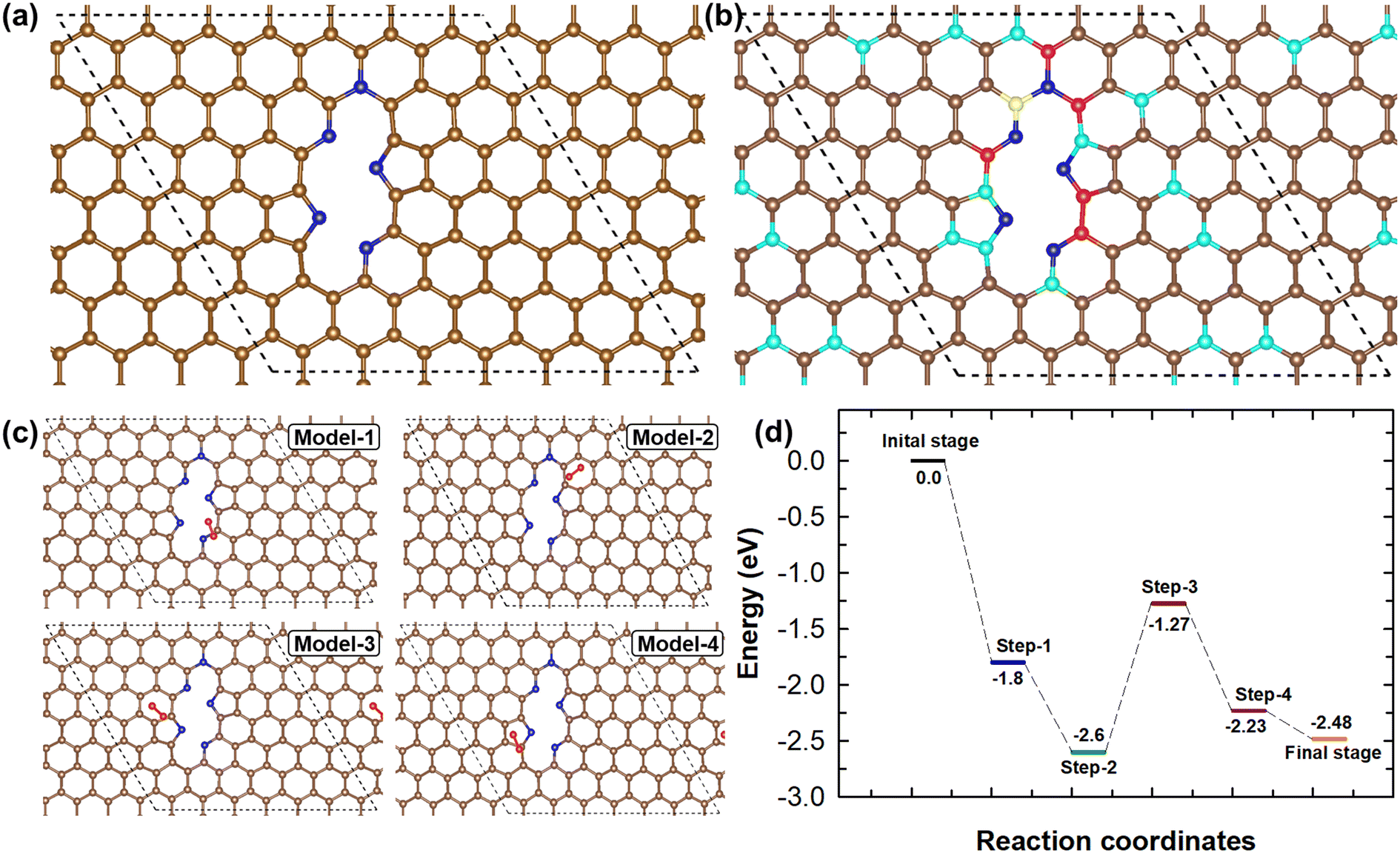 | ||
| Fig. 5 (a) Optimized structure of NMC-AT. (b) Representation of carbon atoms with higher positive charge in NMC-AT samples (yellow: 0.879e, red: 0.4 to 0.5e, green: 0.3 to 0.39e, pink: 0.2 to 0.29e, cyan: 0.1 to 0.19e). (c) Possible models of O2 molecule adsorption orientation on NMC-AT. (d) Electrochemical ORR 4e− pathway energy profile of NMC-AT. | ||
The 4e− ORR pathway and possible intermediate formation during the ORR of electrocatalytic NMC-AT samples were investigated using DFT calculations. The typical 4e− ORR pathway has been considered for the calculation.
| *+O2 → O2* (Step I) |
| O2* + H → OOH* (Step II) |
| OOH* + H → OH* + OH* (Step III) |
| OH* + OH* + H → OH* + H2O* (Step IV) |
| OH* + H2O* + H → 2H2O (Step V) |
Here, * indicates the active catalyst NMC-AT and the intermediate species with * indicates that the species are adsorbed on the NMC-AT. The schematic ORR pathway and the energy profile of the NMC-AT are shown in Fig. S9† and Fig. 5d. In Step 1, the O2 molecule is adsorbed on the electrocatalytic NMC-AT surface with the adsorption energy of −1.8 eV. The adsorption energy indicates that this step is most feasible, and the O2 is readily adsorbed on the NMC-AT surface. After the adsorption, the bond length between oxygen atoms in the O2 molecule increases from 1.16 Å to 1.23 Å, demonstrating the interaction between the O2 molecule and NMC-AT and weakening the O2 bond order. Step 2 represents the O2 hydrogenation, and in this process, the bond length between the O2 molecules increases from 1.23 Å to 1.36 Å and the relative energy is calculated as −2.6 eV. The observed results indicate that the O2 hydrogenation process is more feasible. Step 3 denotes the hydrogenation process, where OOH and H split into two OH radicals and adsorb on the NMC-AT surface. This process is an uphill process as compared to Step 1 and Step 2. The negative energy indicates that the process is feasible; however, the process slows down the overall ORR activity and acts as a rate-determining step. In Step 4, an H2O molecule is formed by the hydrogenation process and desorbed from the NMC-AT surface. During the final hydrogenation process (Step 5), two H2O molecules are formed and desorbed from the NMC-AT surface. The energy profile indicates that both Step 4 and Step 5 are feasible; however, they have slightly higher uphill nature than Step 2. The DFT calculations revealed that the ORR 4e− pathway of the electrocatalytic NMC-AT is feasible, and the formation of two OH radicals acts as a rate-determining step for the overall ORR activity.
4. Conclusion
In summary, we demonstrated the synthesis of a mesoporous nitrogen-doped porous carbon material with a tunable nitrogen content using a high nitrogen-containing precursor, amino-triazine, by simply modifying the annealing temperature and its application in the oxygen reduction reaction. From XPS and NEXAFS analysis, it was found that the optimum doping of pyridinic and pyrrolic N into the mesoporous nitrogen-doped porous carbon material can be achieved at 1000 °C (NMC-AT-1000). Among the samples prepared, NMC-AT-1000 exhibited improved ORR activity with an onset potential of 0.87 V vs. RHE, a half-wave potential of 0.78 V vs. RHE and a high diffusion-limiting current density (JL) of 5.1 mA cm−2. We also demonstrated that the nitrogen-doped carbon species promote ORR activity and improved four-electron selectivity. The high specific surface area, large pore volume and high doping concentration of nitrogen helped to increase the number of active sites required for improving the ORR activity. The DFT calculations suggested that the high ORR activity in NMC-AT-1000 is attributed to its structural and planar defects, which modulate the surface electronic properties of the electrocatalyst, leading to a reduction in the energy barrier for the ORR. The present work highlights the importance of using a triazine-based precursor modulated mesoporous N-doped porous carbon as an efficient catalyst for ORR and a potential replacement for expensive Pt-based catalysts, making it suitable for metal–air batteries and fuel cells.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Y. acknowledges the financial support of ARC discovery project, DP220103045. X. Y. and M. B. would like to acknowledge the Singapore Synchrotron Light Source (SSLS) for providing the facilities necessary for conducting the research. The Laboratory of SSLS is a National Research Infrastructure under the National Research Foundation Singapore.References
- D. H. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts, Science, 2016, 351(6271), 361–365 CrossRef CAS PubMed.
- I. Y. Kim, S. Kim, X. Jin, S. Premkumar, G. Chandra, N. S. Lee, G. P. Mane, S. J. Hwang, S. Umapathy and A. Vinu, Ordered Mesoporous C3N5 with a Combined Triazole and Triazine Framework and Its Graphene Hybrids for the Oxygen Reduction Reaction (ORR), Angew. Chem., Int. Ed., 2018, 57(52), 17135–17140 CrossRef CAS PubMed.
- Q. L. Wei, X. Tong, G. X. Zhang, J. L. Qiao, Q. J. Gong and S. H. Sun, Nitrogen-Doped Carbon Nanotube and Graphene Materials for Oxygen Reduction Reactions, Catalysts, 2015, 5(3), 1574–1602 CrossRef CAS.
- C. C. Wang, W. D. Wu, Y. Wang, D. Xu and F. Yan, Nitrogen doped carbon materials derived from Gentiana scabra Bunge as high-performance catalysts for the oxygen reduction reaction, New J. Chem., 2017, 41(15), 7392–7399 RSC.
- S. H. Wang, L. Liu, S. M. Wang and Z. B. Han, MOF-templated nitrogen-doped porous carbon materials as efficient electrocatalysts for oxygen reduction reactions, Inorg. Chem. Front., 2017, 4(7), 1231–1237 RSC.
- F. Liu, Y. Hu, Z. Qu, X. Ma, Z. Li, R. Zhu, Y. Yan, B. Wen, Q. Ma, M. Liu, S. Zhao, Z. Fan, J. Zeng, M. Liu, Z. Jin and Z. Lin, Rapid production of kilogram-scale graphene nanoribbons with tunable interlayer spacing for an array of renewable energy, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(26), e2303262120 CrossRef PubMed.
- H. Liu, F. Liu, Z. Qu, J. Chen, H. Liu, Y. Tan, J. Guo, Y. Yan, S. Zhao, X. Zhao, X. Nie, X. Ma, Z. Pei and M. Liu, High sulfur loading and shuttle inhibition of advanced sulfur cathode enabled by graphene network skin and N, P, F-doped mesoporous carbon interfaces for ultra-stable lithium sulfur battery, Nano Res. Energy, 2023, 2, e9120049 CrossRef.
- L. Zhang and Z. Xia, Mechanisms of Oxygen Reduction Reaction on Nitrogen-Doped Graphene for Fuel Cells, J. Phys. Chem. C, 2011, 115(22), 11170–11176 CrossRef CAS.
- I. Y. Kim, S. Kim, S. Premkumar, J. H. Yang, S. Umapathy and A. Vinu, Thermodynamically Stable Mesoporous C3N7 and C3N6 with Ordered Structure and Their Excellent Performance for Oxygen Reduction Reaction, Small, 2020, 16(12), 1903572 CrossRef CAS PubMed.
- K. S. Lakhi, D. H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J. H. Choy and A. Vinu, Mesoporous carbon nitrides: synthesis, functionalization, and applications, Chem. Soc. Rev., 2017, 46(1), 72–101 RSC.
- M. Bello, S. M. J. Zaidi, A. Al-Ahmed, S. Basu, D. H. Park, K. S. Lakhi and A. Vinu, Pt–Ru nanoparticles functionalized mesoporous carbon nitride with tunable pore diameters for DMFC applications, Microporous Mesoporous Mater., 2017, 252, 50–58 CrossRef CAS.
- Y. Yan, B. Wen, M. Liu, H. Lei, J. Yang, S. He, Z. Qu, W. Xia, H. Li and J. Zeng, Orienting Electron Fillings in d Orbitals of Cobalt Single Atoms for Effective Zinc–Air Battery at a Subzero Temperature, Adv. Funct. Mater., 2024, 2316100 CrossRef CAS.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S.-M. Hao, X. Cui, Z. Li and Z. Lin, Robust wrinkled MoS2/N-C bifunctional electrocatalysts interfaced with single Fe atoms for wearable zinc-air batteries, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(40), e2110036118 CrossRef CAS PubMed.
- Z. Fang, W. Zhao, T. Shen, D. Qiu, Y. Lv, X. Hou and Y. Hou, Spin-Modulated Oxygen Electrocatalysis, Precis. Chem., 2023, 1(7), 395–417 CrossRef CAS.
- P. Selvarajan, M. Fawaz, C. I. Sathish, M. Li, D. Chu, X. Yu, M. B. H. Breesec, J. Yi and A. Vinu, Activated Graphene Nanoplatelets Decorated with Carbon Nitrides for Efficient Electrocatalytic Oxygen Reduction Reaction, Adv. Energy Sustainability Res., 2021, 2(12), 2100104 CrossRef CAS.
- A. Wutscher, T. Eckhard, D. Hiltrop, K. Lotz, W. Schuhmann, C. Andronescu and M. Muhler, Nitrogen-Doped Metal-Free Carbon Materials Derived from Cellulose as Electrocatalysts for the Oxygen Reduction Reaction, ChemElectroChem, 2019, 6(2), 514–521 CrossRef.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, Observation of Active Sites for Oxygen Reduction Reaction on Nitrogen-Doped Multilayer Graphene, ACS Nano, 2014, 8(7), 6856–6862 CrossRef CAS PubMed.
- L. Mu, R. Wang and C. Tang, Tofu-derived nitrogen-doped mesoporous carbon materials as metal-free catalyst for oxygen reduction reaction, Biomass Convers. Biorefin., 2019, 9, 401–409 CrossRef CAS.
- N. Panangattu Dharmarajan, M. Fawaz, C. Sathish, S. N. Talapaneni, K. Ramadass, A. M. Sadanandan, X. M. C. Ta, M. Huš, V. Perumalsamy, A. Tricoli, B. Likozar, C.-H. Jeon, J.-H. Yang and A. Vinu, Insights into Atomic Level π-Electron Modulations in Supramolecular Carbon Nitride Nanoarchitectonics for Sustainable Green Hydrogen Production, Adv. Energy Mater., 2024, 2400686 CrossRef CAS.
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Triazoles: A New Class of Precursors for the Synthesis of Negatively Charged Carbon Nitride Derivatives, Chem. Mater., 2015, 27(15), 5170–5179 CrossRef CAS.
- M. Fawaz, R. Bahadur, N. Panangattu Dharmarajan, J.-H. Yang, C. I. Sathish, A. M. Sadanandan, V. Perumalsamy, G. Singh, X. Guan, P. Kumar and A. Vinu, Emerging trends of carbon nitrides and their hybrids for photo-/electro-chemical energy applications, Carbon, 2023, 214, 118345 CrossRef CAS.
- S. Yang, X. Feng, X. Wang and K. Mullen, Graphene-based carbon nitride nanosheets as efficient metal-free electrocatalysts for oxygen reduction reactions, Angew. Chem., Int. Ed., 2011, 50(23), 5339–5343 CrossRef CAS PubMed.
- X. Chu, C. I. Sathish, J.-H. Yang, X. Guan, X. Zhang, L. Qiao, K. Domen, S. Wang, A. Vinu and J. Yi, Strategies for Improving the Photocatalytic Hydrogen Evolution Reaction of Carbon Nitride-Based Catalysts, Small, 2023, 19(41), 2302875 CrossRef CAS PubMed.
- X. Chu, C. I. Sathish, M. Li, J.-H. Yang, W. Li, D.-C. Qi, D. Chu, A. Vinu and J. Yi, Anti-Stoke effect induced enhanced photocatalytic hydrogen production, Battery Energy, 2023, 2(2), 20220041 CrossRef CAS.
- C. Sathish, S. Premkumar, X. Chu, X. Yu, M. B. H. Breese, M. Al-Abri, A. a. H. Al-Muhtaseb, A. Karakoti, J. Yi and A. Vinu, Microporous Carbon Nitride (C3N5.4) with Tetrazine based Molecular Structure for Efficient Adsorption of CO2 and Water, Angew. Chem., Int. Ed., 2021, 60(39), 21242–21249 CrossRef CAS PubMed.
- G. P. Mane, S. N. Talapaneni, K. S. Lakhi, H. Ilbeygi, U. Ravon, K. Al-Bahily, T. Mori, D. H. Park and A. Vinu, Highly Ordered Nitrogen-Rich Mesoporous Carbon Nitrides and Their Superior Performance for Sensing and Photocatalytic Hydrogen Generation, Angew. Chem., Int. Ed., 2017, 56(29), 8481–8485 CrossRef CAS PubMed.
- P. Kumar, G. Singh, X. Guan, J. Lee, R. Bahadur, K. Ramadass, P. Kumar, M. G. Kibria, D. Vidyasagar, J. Yi and A. Vinu, Multifunctional carbon nitride nanoarchitectures for catalysis, Chem. Soc. Rev., 2023, 52(21), 7602–7664 RSC.
- Z. Li, Y. Yan, M. Liu, Z. Qu, Y. Yue, T. Mao, S. Zhao, M. Liu and Z. Lin, Robust ring-opening reaction via asymmetrically coordinated Fe single atoms scaffolded by spoke-like mesoporous carbon nanospheres, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(14), e2218261120 CrossRef CAS PubMed.
- S. Kim, G. Singh, C. Sathish, P. Panigrahi, R. Daiyan, X. Lu, Y. Sugi, I. Y. Kim and A. Vinu, Tailoring the Pore Size, Basicity, and Binding Energy of Mesoporous C3N5 for CO2 Capture and Conversion, Chem. – Asian J., 2021, 16(23), 3999–4005 CrossRef CAS PubMed.
- X. Guan, M. Fawaz, R. Sarkar, C.-H. Lin, Z. Li, Z. Lei, P. D. Nithinraj, P. Kumar, X. Zhang, J.-H. Yang, L. Hu, T. Wu, S. Chakraborty, J. Yi and A. Vinu, S-doped C3N5 derived from thiadiazole for efficient photocatalytic hydrogen evolution, J. Mater. Chem. A, 2023, 11(24), 12837–12845 RSC.
- D. H. Park, K. S. Lakhi, K. Ramadass, M. K. Kim, S. N. Talapaneni, S. Joseph, U. Ravon, K. Al-Bahily and A. Vinu, Energy Efficient Synthesis of Ordered Mesoporous Carbon Nitrides with a High Nitrogen Content and Enhanced CO Capture Capacity, Chem. – Eur. J., 2017, 23(45), 10753–10757 CrossRef CAS PubMed.
- W. J. Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Nitrogen-doped carbon nanotubes and graphene composite structures for energy and catalytic applications, Chem. Commun., 2014, 50(52), 6818–6830 RSC.
- D. Schwarz, Y. Noda, J. Klouda, K. Schwarzova-Peckova, J. Tarabek, J. Rybacek, J. Janousek, F. Simon, M. V. Opanasenko, J. Cejka, A. Acharjya, J. Schmidt, S. Selve, V. Reiter-Scherer, N. Severin, J. P. Rabe, P. Ecorchard, J. He, M. Polozij, P. Nachtigall and M. J. Bojdys, Twinned Growth of Metal-Free, Triazine-Based Photocatalyst Films as Mixed-Dimensional (2D/3D) van der Waals Heterostructures, Adv. Mater., 2017, 29(40), 1–9 CrossRef PubMed.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Direct Syntheses of Ordered SBA-15 Mesoporous Silica Containing Sulfonic Acid Groups, Chem. Mater., 2000, 12(8), 2448–2459 CrossRef CAS.
- M. Miyahara, A. Vinu, K. Z. Hossain, T. Nakanishi and K. Ariga, Adsorption study of heme proteins on SBA-15 mesoporous silica with pore-filling models, Thin Solid Films, 2006, 499(1–2), 13–18 CrossRef CAS.
- A. Vinu, B. M. Devassy, S. B. Halligudi, W. Böhlmann and M. Hartmann, Highly active and selective AlSBA-15 catalysts for the vapor phase-butylation of phenol, Appl. Catal., A, 2005, 281(1–2), 207–213 CrossRef CAS.
- T. Joseph, S. S. Deshpande, S. B. Halligudi, A. Vinu, S. Ernst and M. Hartmann, Hydrogenation of olefins over hydrido chlorocarbonyl tris-(triphenylphosphine) ruthenium(II) complex immobilized on functionalized MCM-41 and SBA-15, J. Mol. Catal. A: Chem., 2003, 206(1–2), 13–21 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- K. Akikubo, T. Kurahashi, S. Kawaguchi and M. Tachibana, Thermal expansion measurements of nano-graphite using high-temperature X-ray diffraction, Carbon, 2020, 169, 307–311 CrossRef CAS.
- D.-H. Park, K. S. Lakhi, K. Ramadass, M.-K. Kim, S. N. Talapaneni, S. Joseph, U. Ravon, K. Al-Bahily and A. Vinu, Energy Efficient Synthesis of Ordered Mesoporous Carbon Nitrides with a High Nitrogen Content and Enhanced CO2 Capture Capacity, Chem. – Eur. J., 2017, 23(45), 10753–10757 CrossRef CAS PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, Catalyst-Free Synthesis of Nitrogen-Doped Graphene via Thermal Annealing Graphite Oxide with Melamine and Its Excellent Electrocatalysis, ACS Nano, 2011, 5(6), 4350–4358 CrossRef CAS PubMed.
- C. Sathish, G. Kothandam, P. Selvarajan, Z. Lei, J. Lee, J. Qu, A. a. H. Al-Muhtaseb, X. Yu, M. B. H. Breese, R. Zheng, J. Yi and A. Vinu, Ordered Mesoporous Boron Carbon Nitrides with Tunable Mesopore Nanoarchitectonics for Energy Storage and CO2 Adsorption Properties, Adv. Sci., 2022, 9(16), 2105603 CrossRef CAS PubMed.
- P. Selvarajan, M. Fawaz, C. Sathish, M. Li, D. Chu, X. Yu, M. B. H. Breesec, J. Yi and A. Vinu, Activated Graphene Nanoplatelets Decorated with Carbon Nitrides for Efficient Electrocatalytic Oxygen Reduction Reaction, Adv. Energy Sustainability Res., 2021, 2(12), 2100104 CrossRef CAS.
- P. Lazar, R. Mach and M. Otyepka, Spectroscopic Fingerprints of Graphitic, Pyrrolic, Pyridinic, and Chemisorbed Nitrogen in N-Doped Graphene, J. Phys. Chem. C, 2019, 123(16), 10695–10702 CrossRef CAS.
- I. Matanovic, K. Artyushkova, M. B. Strand, M. J. Dzara, S. Pylypenko and P. Atanassov, Core Level Shifts of Hydrogenated Pyridinic and Pyrrolic Nitrogen in the Nitrogen-Containing Graphene-Based Electrocatalysts: In-Plane vs. Edge Defects, J. Phys. Chem. C, 2016, 120(51), 29225–29232 CrossRef CAS.
- D. C. Higgins, D. Meza and Z. Chen, Nitrogen-Doped Carbon Nanotubes as Platinum Catalyst Supports for Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells, J. Phys. Chem. C, 2010, 114(50), 21982–21988 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications, ACS Catal., 2012, 2(5), 781–794 CrossRef CAS.
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, Highly Active Nitrogen-Doped Carbon Nanotubes for Oxygen Reduction Reaction in Fuel Cell Applications, J. Phys. Chem. C, 2009, 113(49), 21008–21013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02425g |
| This journal is © The Royal Society of Chemistry 2024 |