
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTruxinates and truxillates: building blocks for architecturally complex polymers and advanced materials
Sara El-Arid†
 a,
Jason M. Lenihan†
a,
Aaron B. Beeler
*a and
Mark W. Grinstaff
*ab
a,
Jason M. Lenihan†
a,
Aaron B. Beeler
*a and
Mark W. Grinstaff
*ab
aDepartment of Chemistry, Boston University, Boston, Massachusetts 02215, USA. E-mail: mgrin@bu.edu; beelera@bu.edu
bDepartment of Biomedical Engineering, Boston University, Boston, Massachusetts 02215, USA
First published on 18th September 2024
Abstract
Significant advancements in the syntheses of cyclobutane containing small molecules and polymers are described in the last 15 years. Small molecule cyclobutanes are under investigation for their diverse pharmacological activities, while polymers with cyclobutane backbones are emerging as novel mechanophores, stress-responsive materials, and sustainable plastics. Within these chemistries, [2 + 2] photocycloadditions to yield truxinates and truxillates are highly efficient offering a versatile strategy to access complex scaffolds. This article provides a comprehensive review on the synthetic methodologies, properties, and applications of polymer truxinates and truxillates, providing the background necessary to understand current developments and envision future applications. Additionally, we highlight the links between the development, discoveries, and synthetic methodologies of small molecules and cyclobutane polymers. We emphasize structure property relationships and discuss methods to control composition and structure for desired applications. We begin with a discussion of synthetic techniques for small molecule and polymer cyclobutanes followed by their greater applications, including pharmacological and material properties with examples including sustainable plastics and stimuli-responsive systems.
 Sara El-Arid | Sara El-Arid received her B.S. in Biochemistry from UC Santa Barbara where she conducted undergraduate research under the guidance of Professor Read de Alaniz in organic and polymer synthesis for the development of photothermal actuators. She is currently a Chemistry Ph.D. candidate at Boston University under the mentorship of Professor Mark W. Grinstaff. Her thesis focuses on scalable organic and polymer syntheses for the development of novel functional materials. |
 Jason M. Lenihan | Jason M. Lenihan obtained his B.S. in Biochemistry from Quinnipiac University, where he conducted undergraduate research in biochemical and biophysical sciences under the guidance of Professor Alex Hodges. He recently completed his Ph.D. in chemistry under the supervision of Professor Aaron Beeler, focusing on the development of novel methodologies involving [2 + 2] photocycloadditions to access truxinate scaffolds. He is currently a Medicinal Chemist at Eikon Therapeutics. |
 Aaron B. Beeler | Aaron Beeler received his B.Sc. in biology from Belmont University in 1998. In 2002 he earned a Ph.D. in medicinal chemistry at the University of Mississippi. He then joined the group of John Porco, Jr. as a postdoctoral fellow eventually moving into the Center for Chemical Methodology and Library Development at Boston University, now the Center for Molecular Discovery (BU-CMD). He served as the Associate Director until 2012 and then joined the Department of Chemistry at Boston University as an Assistant Professor. Aaron's multidisciplinary educational and research background has translated to the work in his lab covering medicinal chemistry, reaction methods, photochemistry, and flow chemistry. |
 Mark W. Grinstaff | Mark W. Grinstaff is the William Fairfield Warren Distinguished Professor, and a Professor of Biomedical Engineering, Chemistry, Materials Science and Engineering, and Medicine at Boston University. He is also the Director of BU's Nanotechnology Innovation Center and the NIH T32 Biomaterials Program. Information about his research interests is found at https://www.grinstaff.org. |
1. Introduction
Truxinate and truxillate natural products represent a captivating class of natural products possessing intricate molecular architectures. Central to their design is a cyclobutane core decorated with two aryl substituents and two carbonyl moieties, forming an intricate and three-dimensional framework (Fig. 1A). Truxillic and Truxinic acids are the products from the hydrolysis of the ester moieties on truxinates and truxillates. Interest in this class of small molecules continues to grow as we uncover their pharmacological properties to include anti-cancer, anti-diabetic, and neuroprotective1–6 as well as recognize their utility as building blocks for sustainable polyesters, photo-responsive materials, and stress-responsive materials. Synthetically, truxinic acids and their counterparts, truxillic acids, form from the dimerization of cinnamic acid units, resulting in two distinct modes of assembly: “head-to-head” for truxinic acids and “head-to-tail” for truxillic acid derivatives. Consequently, the resulting truxinic/truxillic scaffolds display up to four stereocenters, yielding 11 diastereomers with distinct spatial arrangements of their aryl and carbonyl groups. While every homo dimeric truxillic acid scaffold is achiral, the majority of truxinate scaffolds are inherently chiral. The β- and δ-diastereomers (Fig. 1B) predominate in nature, being found in both homo- and hetero-dimeric forms across a multitude of truxinate natural products.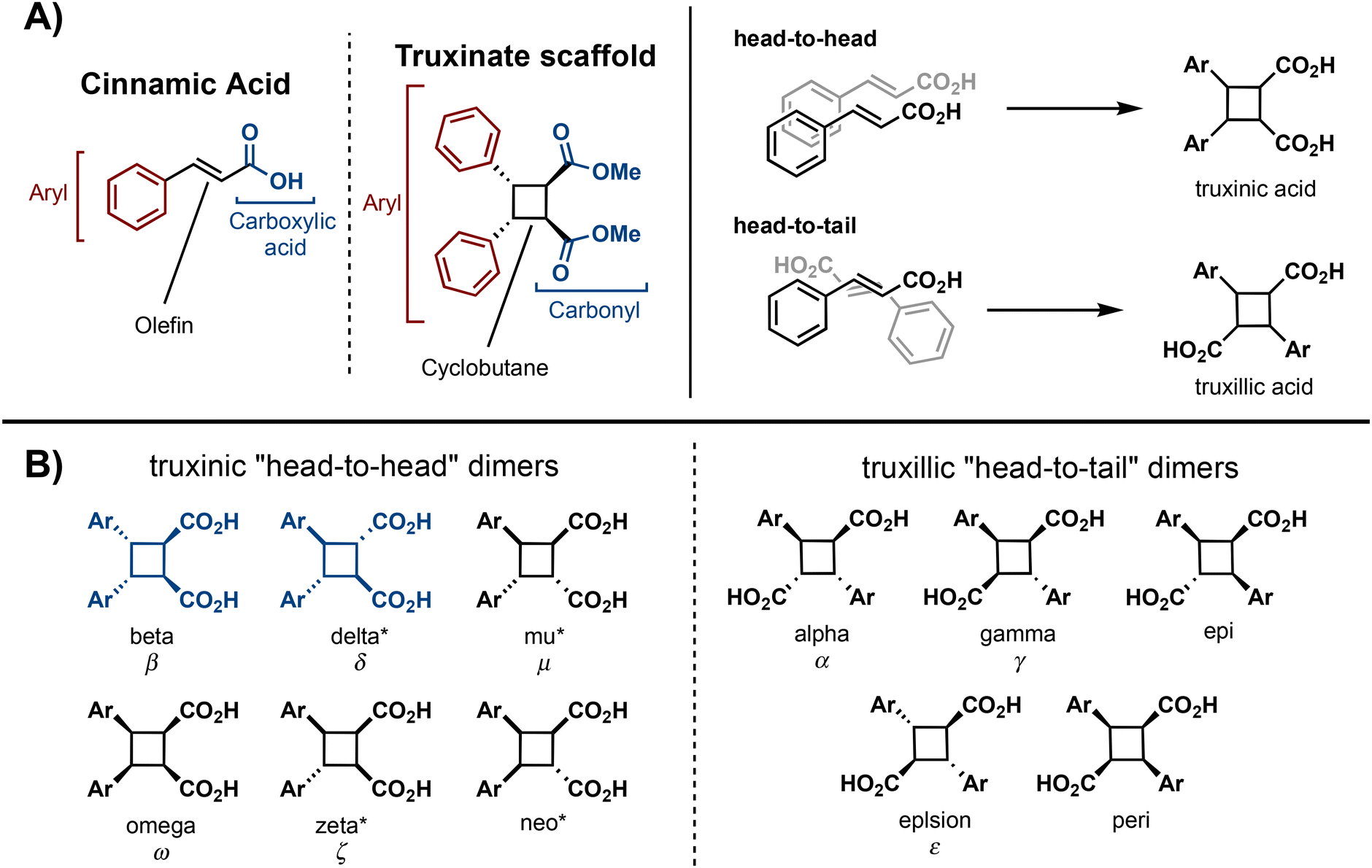 | ||
| Fig. 1 (A) General structure of cinnamic acid and truxinates and general [2 + 2] photocycloadditions resulting in truxinic acids or truxillic acids. (B) All possible cinnamic acid dimer isomers. *Indicates inherent chirality. | ||
While the first reports of [2 + 2] photocycloadditions of cinnamic acids were reported in the 1960s by Schmidt and coworkers, efforts to isolate truxinic and truxillic acids from plants did not occur until 30 years later. In 1990, Balza and Towers7 isolated and characterized truxinic and truxillic acid homodimers and heterodimers from the cellular matrix of Cynadon dactylon grass, these dimers comprise p-coumaric acid (Fig. 2, 1.1) and ferulic acid (1.2) units. Overall, 14 distinct compounds were isolated with six being truxillic acids and the remaining eight being truxinic acids. Their presence within the cell wall is thought to confer cellular protection via a contribution to the crosslinking of wall polysaccharides. Studies on the dimerization of phenolic acids 1.1 and 1.2, by Morrison and colleagues8 reveal the occurrence of both head-to-tail and head-to-head dimeric species, with the former being the predominant product, as determined by advanced NMR spectroscopy and GC-MS analyses (Fig. 2). Mechanistically, the dimerization may transpire via a step-wise radical-mediated processes or through a concerted photocycloaddition induced by UV irradiation. These findings align with earlier investigations on the solid-state dimerization of cinnamic acid,9 which will be expanded upon in subsequent sections, providing additional insights into synthetic requirements and challenges.
 | ||
| Fig. 2 (A) Solid state dimerization of p-coumaric acid. (B) Solid state dimerization of ferulic acid. (C) Solid state cross dimerization of p-coumaric and ferulic acid. | ||
Truxinate and truxillate natural products are of significant interest in the scientific community due to their broad pharmacological properties. Predating the discovery of truxillic and truxinic acids, the discovery of truxinate natural products dates back to the 1960s and 1970s,10 with initial isolation from Erythroxylum coca. Within this class of compounds is a notable subclass, the truxilines, which are comprised of a bare truxinic/truxillic core adorned with two tropane esters.11,12 Within the truxiline family, the α- and β-truxiline isomers are the most prevalent members. While these isomers do not exhibit desirable biological effects, they are notorious for their potent cardiotoxicity.13 Following this seminal discovery, numerous truxinate and truxillate small molecule natural products have been identified with various pharmacological properties which we will highlight, and the reader is referred to several excellent reviews by Bach et al.,14 Huang et al.,15 and Blanco-Ania et al.16 for more comprehensive discussions of small-molecule cyclobutanes.
The first description of a cyclobutane polymer was in 1958, when Koelsch and coworkers reported the photopolymerization and photodimerization of 2,5-distyrylpyrazine (DSP) when irradiated with UV light (Fig. 3).17 Subsequently, reports of cinnamic acid based cyclobutane polymers began to emerge in 1964 when Hirshfeld and Schmidt postulated that appropriate spatial positioning of the olefins in the crystal will facilitate a topochemical photopolymerization.18 Ultimately this hypothesis was validated in 1966, when Schmidt and coworkers reported the photodimerization and photopolymerization of a triene dicarboxylic acid system highlighting the importance of crystal packing for photochemical reactions in solid-state.19 An alternative polymerization strategy emerged in 1966 when Stueben successfully synthesized a library of cyclobutane polyesters and polyamides via a condensation polymerization of 1,2 and 1,3 substituted cyclobutanes subsequently broadening the synthetic pathways to access cyclobutane polymers.20 Accessing ester-based cyclobutane polymers remained quite challenging at this time until Hasegawa and Nakanishi reported in 1970 the synthesis of highly crystalline linear polyesters.21 While numerous reports of cyclobutane polymers were emerging at this time, all the polymers are highly crystalline due to the topochemical polymerization mechanism. Mediating this issue, Rees et al., published a detailed account of the synthesis of numerous amorphous cyclobutane polymers.22 While these polymers are not of the truxinate/truxillate variety, this study laid the foundation to later synthesize structurally complex truxinate and truxillate based cyclobutane polymers via a photopolymerization strategy.
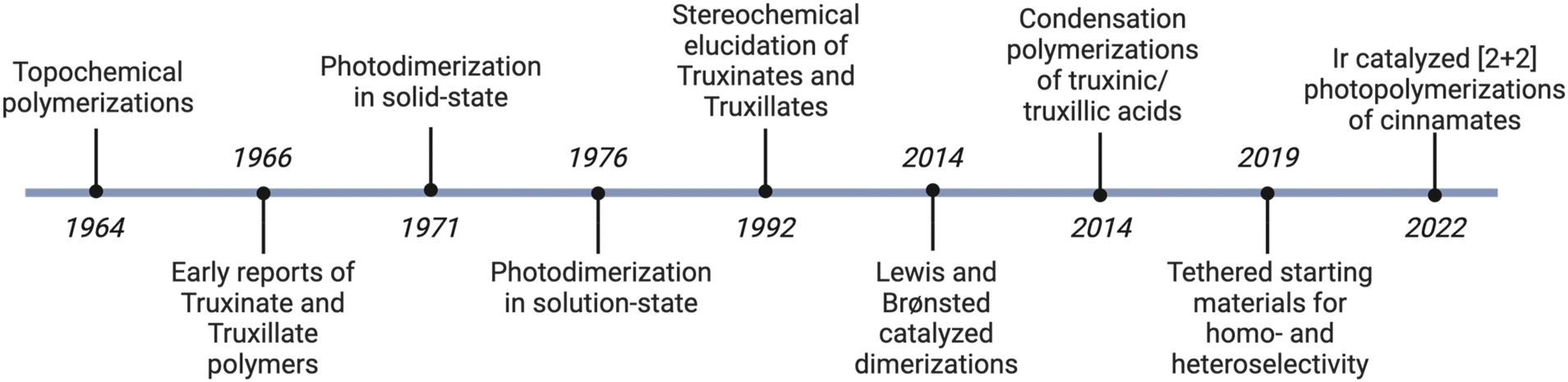 | ||
| Fig. 3 Advancements in truxinate and truxillate syntheses. | ||
More recently, synthetic efforts are primarily focused on optimizing [2 + 2] photopolymerizations both in the solid state and solution state to synthesize truxinate and truxillate based polymers. The resulting polymers show promise as biomaterials, sustainable bioplastics, and photoactive materials. Of these biobased materials, many begin with biocompatible cinnamic acid starting materials. More specifically, hydroxycinnamic acids such as ferulic acid and caffeic acid are used. Strategies to tune the properties of these materials center around modifying the substitution of the aromatic moiety and introducing backbone flexibility or rigidity.
Herein, we describe the various synthetic techniques to access truxillate and truxinate polymers and advanced materials. We begin with a discussion on the various synthetic methodologies to access cyclobutane core structures present in both small molecules and polymers. We discuss [2 + 2] photocycloadditions via direct excitation or in the presence of a photosensitizer, the role of preorganization in regio- and stereo-selective solid-state syntheses, and improvement in enantioselectivity when performing solution-state reactions, as well as describe non-photocatalytic methods. The methodologies are applicable to both small molecule and polymer syntheses. Finally, we discuss small molecule truxinate and truxillate natural products briefly, to give some context, and provide a comprehensive account of the applications of such polymeric materials ranging from biomass derived plastics to functional materials.
2. [2 + 2] photocycloadditions
[2 + 2] photocycloadditions are the most common reaction for the synthesis of cyclobutane products. Initiation of [2 + 2] photocycloaddition occurs from either direct excitation of the starting material, or the use of an appropriate photosensitizer which facilitates the reaction. [2 + 2] photocycloadditions are further differentiated by their reaction medium—solid state or solution state. While traditional [2 + 2] photocycloadditions offer limited selectivity compared to competing synthetic methods such as C–H activation,23 synthetic methods are being developed to improve regio-, diastereo-, chemo-, and enantioselectivity. In general, [2 + 2] photocycloadditions require: (1) excitation from the ground state (S0) to the first excited singlet state (S1); (2) intersystem crossing (ISC) to the lowest-lying triplet state (T1); (3) propagation to the 1,4-diradical intermediate; and, (4) ISC and radical–radical recombination.14,24 Competing photophysical pathways of fluorescence and internal conversion (IC), resulting in E → Z isomerization, plaque energy dissipation in the excited S1 state. To prevent these unfavorable side reactions, achieving rapid ISC promotes transition to the ππ* T1 and subsequent diradical formation. These transformations occur via direct excitation (Fig. 4) or photosensitization which will be described in further detail below.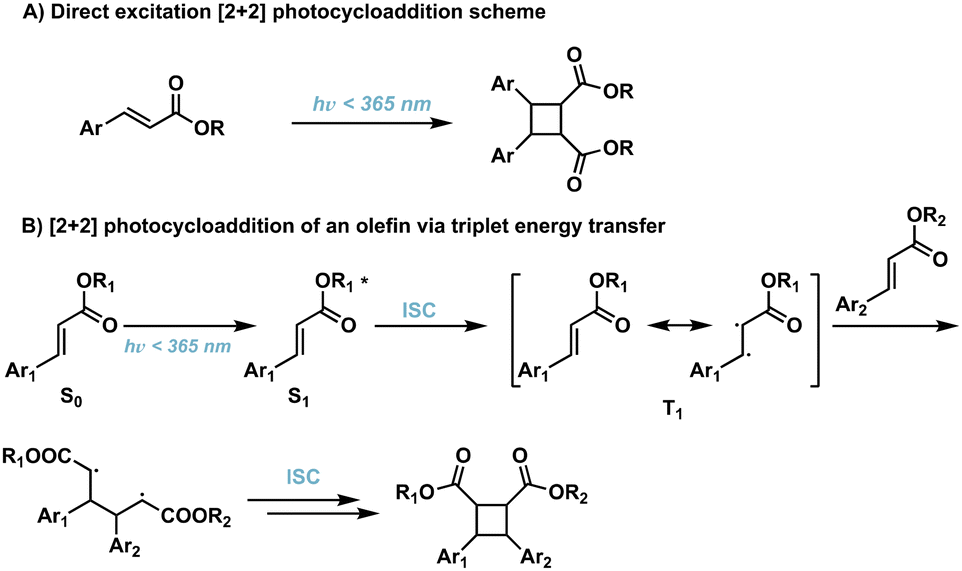 | ||
| Fig. 4 (A) Direct excitation of cinnamic acids to corresponding cyclobutanes; (B) triplet energy transfer pathway resulting in [2 + 2] photocycloadditions of olefins. | ||
2.1. Direct excitation
Direct excitation, which does not require a photosensitizer, is a preferred and facile route to the synthesis of truxinate/truxillate cyclobutanes and truxinic/truxillic acids. α,β-unsaturated carboxylic acid derivatives, such as cinnamic acids, are directly excited to the S1 state (Fig. 4B), and thus, cinnamic acids and cinnamic esters are commonly utilized substrates for direct excitation applications.25,26 However, direct excitation of cinnamic acid derivatives necessitates the use of high-energy light (λmax < 370 nm) to promote the photoreaction. Consequently, there is increased safety risk as well as risk of unfavorable side reactions such as the Fries rearrangement, E/Z isomerization, and material decomposition when using such light. Despite these limitations, numerous direct excitation methods have been developed and are currently in use.In an effort to induce stereochemical control without a photocatalyst, suspension and solid-state [2 + 2] photocycloadditions are utilized. As such, reactions proceed via minimum atomic and molecular movements achieving high homo-selectivity along with total regio- and stereoselectivity. In these instances, olefin–olefin arrangement exhibits less conformational flexibility reducing the amount of stereo- and regioisomers. Within the crystal lattice, the crystal packing of neighboring molecules drives the reactivity rather than intrinsic reactivity of the olefin, thus regioselectivity depends upon orientation of neighboring groups relative to the olefins.27 In a study with two structurally similar cinnamic acids, irradiation affords only the homo anti α-truxillic adducts indicating a preference for homodimerization via an intermolecular [2 + 2] photocycloaddition (Fig. 5A). Access to α-truxillic adducts requires olefin separation between 3.6–4.1 Å in one dimensional monomer stacks. In this packing arrangement, olefin overlap of centrosymmetrically related molecules occurs between adjacent crystal stacks resulting in inversion dimers.28 Even upon increasing to a three or six cinnamic acid system, complete homoselectivity remains (Fig. 5B).29 A subsequent study by Chu et al. implements a similar strategy to target β-truxinic acids by melting cinnamic acid starting materials prior to suspending in solvent to isolate the crystalline β-form of trans-cinnamic acid. β-Type stacking requires olefin distances between 3.9–4.1 Å and face-to-face overlap of neighboring molecules such that olefins are translationally equivalent resulting in mirror dimers.28 Subsequent irradiation of the suspension results in complete conversion to the β homodimer.30 Other landmark studies include those involving direct excitation of both electron rich and nitro cinnamates. Endo et al. describe the direct excitation of electron poor nitrocinnamates in solution resulting in head-to-head dimers,31 while Vantaggi et al. report the photochemical dimerization of methyl methoxy cinnamates in high stereoselectivity.32
 | ||
| Fig. 5 (A) Two cinnamic acid system resulting in complete homodimerization to respective α-truxillates; (B) three cinnamic acid system resulting in complete homodimerization to respective α-truxillates. | ||
Solid-state [2 + 2] photocycloadditions are not restricted to small molecules, and topochemical photopolymerizations are also possible, but are more synthetically challenging and often produce polymers of high crystallinity.22 In these single crystal to single crystal (SCSC) [2 + 2] photopolymerizations, the crystal structure of the monomers dictates the topochemistry of the polymerization allowing for detailed stereo-, regio- and enantioselective control.33 Utilizing metal coordination and hydrogen bonding enables further stereochemical control. Biradha et al. utilize Ag to Ag interactions to induce stereoselectivity in the synthesis of pseudo-truxinate amide dimers and macrocycles.34 In the solid state, the Ag to Ag interactions exhibit linear coordination geometry and position the diolefinic monomers into a reactive conformation which ensures [2 + 2] photocycloadditions to yield macrocycles (Fig. 6A). In a subsequent study by Biradha et al., hydrogen bonding between the diolefin monomers creates two dimensional layers and allows for [2 + 2] photopolymerizations. Irradiating crystals of similar bisamide olefin monomers facilitates SCSC photopolymerizations yielding truxillate and pseudo-truxillate amide polymers with excellent tensile strength (Fig. 6B).35 A subsequent study by Biradha et al., describes a topochemical photopolymerization of coordination polymers from angular dienes coordinated with five metal ions: Co(II), Cu(II), Cd(II), Ni(II) and Zn(II) resulting in highly crystalline monomers. Irradiation of the coordinated crystals with UV light affords amorphous head-to-tail cyclobutane polymers. While the crystal structures of the polymers are difficult to deduce, the crystal structures of the monomers are utilized to extrapolate crystal geometries of the polymers. From these crystal structures, all synthesized polymers exhibit plausible 3D topologies.36 In addition to solid-state [2 + 2] photopolymerizations, photo crosslinking between polymer chains is possible as reported by Carlotti et al. Cinnamoyl groups present within a polyamide backbone photocrosslink upon irradiation with UV (<300 nm) light forming interchain truxinate/truxillate cyclobutanes.37
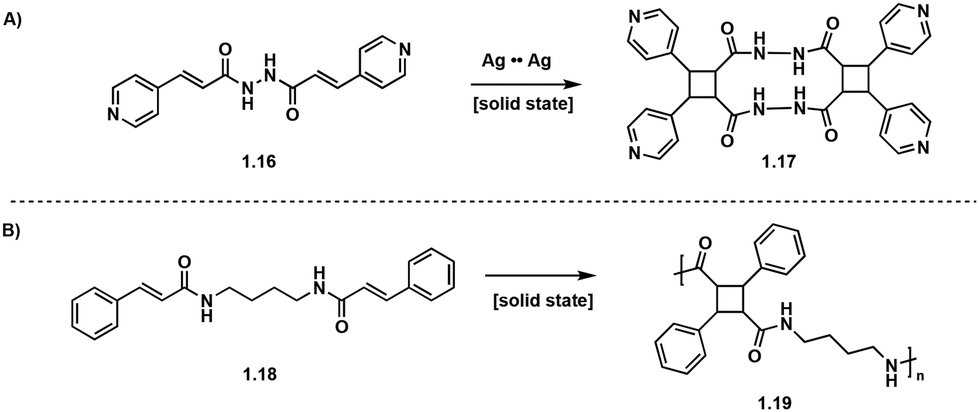 | ||
| Fig. 6 (A) Ag to Ag interactions selectively form tricyclic pseudo-truxillate; (B) SCSC polymerization affording pseudo-truxillate polymers. | ||
In many instances, direct excitation is performed in solid-state to mitigate off-target reactivity, resulting from high energy UV light irradation in solution state syntheses. To mediate this concern, Liao et al. report donor–acceptor diolefinic monomers with electron-donating alkoxy substituted aryl groups that participate in solution-state [2 + 2] photopolymerizations.38 The incorporation of an electron-donating group (EDG) results in red-shifted absorbance spectra of the monomers, allowing direct excitation to the excited state via visible light (400 nm). The subsequent pseudo-truxinate polymers are of molecular weights ranging from 8.1 kDa to 85.9 kDa and exhibit excellent processability and availability for post-polymerization modifications via thiol–ene click reactions (Fig. 7).
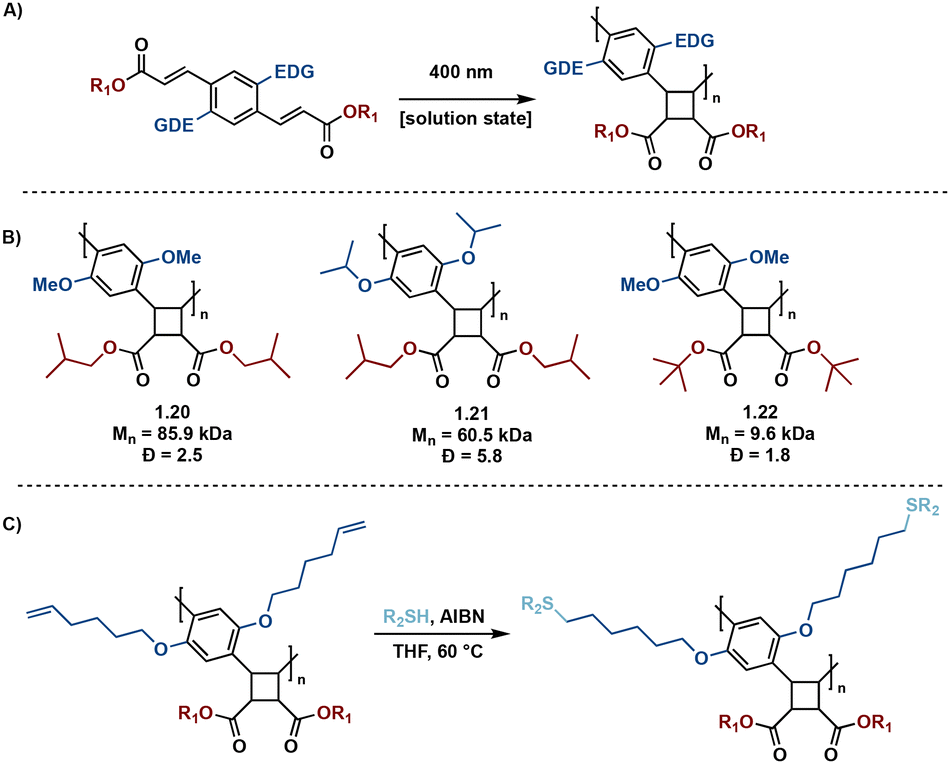 | ||
| Fig. 7 (A) General photopolymerization schematic; (B) selected examples of polymers synthesized; (C) general post-polymerization thiol–ene click reaction. | ||
While intermolecular [2 + 2] photocycloadditions typically favor homodimerization over heterodimerization, tethered starting materials facilitate both intramolecular homodimerization and heterodimerization, contingent on the symmetry or asymmetry of starting materials. For example, in a study by Türkmen et al., tethering of two equivalent or non-equivalent cinnamic acids 4 Å apart via a 1,8-dihydroxynaphthalene linker affords clean conversion to the β homodimer in the case of equivalent cinnamic acids and the β heterodimer in the case of non-equivalent cinnamic acids (Fig. 8A–C).39 Tethering is also utilized in truxinate/truxillate polymer synthesis. Biradha et al. highlight the use of direct excitation of tethered dicarboxylates to synthesize amide containing cyclobutane polymers, a subset of truxinate/truxillate polymers (Fig. 8D).36
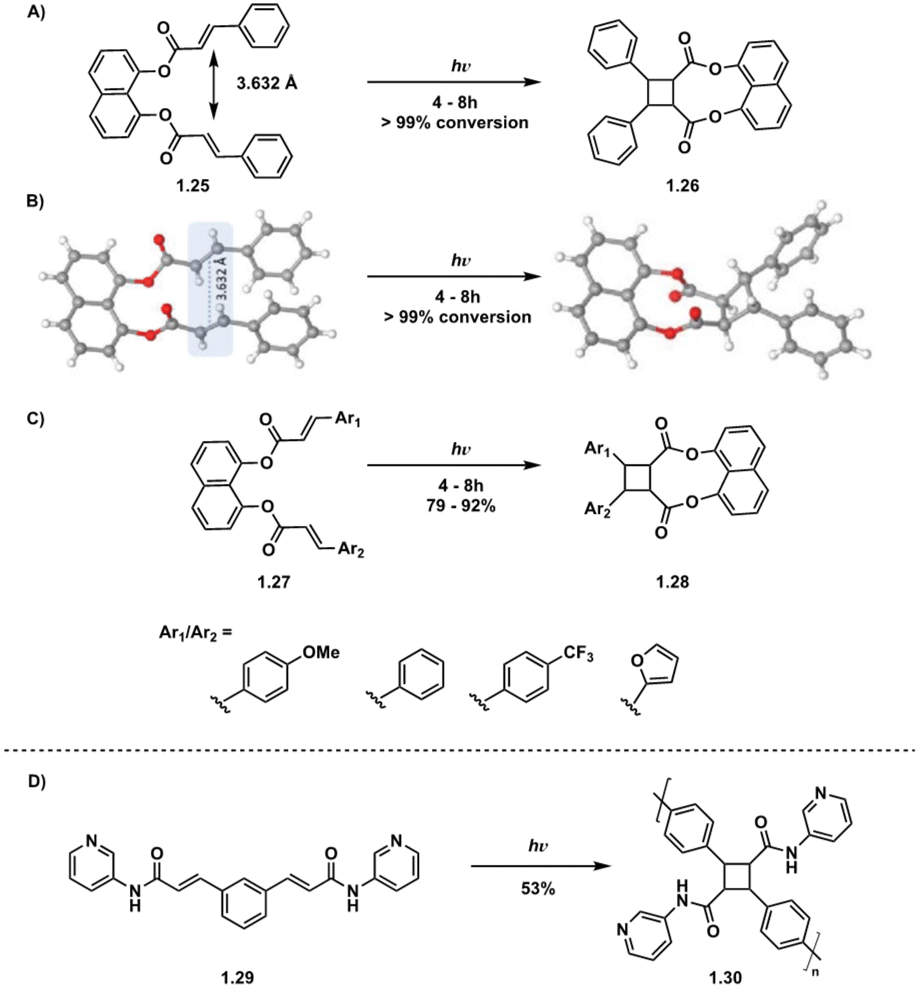 | ||
| Fig. 8 (A) Homodimerization of tethered diesters; (B) crystal structures of 1.25 and 1.26 depicting olefin distances of 3.632 Å. Adapted from ref. 39 with permission from American Chemical Society, Copyright © 2021; (C) heterodimerization of tethered diesters of varying aryl groups; (D) polymerization of tethered diamide. | ||
In effort to introduce enantioselectivity, [2 + 2] photocycloadditions are being performed in the presence of chiral Brønsted and Lewis acids. Lewis acid catalyzed [2 + 2] cycloadditions are among the most successful acid-catalyzed [2 + 2] photocycloaddition reactions.40–45 These syntheses primarily employ chromophore activation, a method defined by Bach.46 In the chromophore activation strategy, the acid complexed substrate absorbs more strongly at lower wavelengths compared to the non-complexed substrate thus allowing for selective singlet excitation of the complexed substrate.46,47 A noteworthy example employs a chiral Brønsted acid chromophore in the synthesis of truxinate natural products: Isatiscycloneolignan A, Barbarumamide C, nigramide R and piperarborenine D.48 This methodology allows access to both dimeric and pseudodimeric truxinates and exhibits excellent diastereoselectivity for the δ and β isomers respectively. Additionally, this approach affords the natural products in high enantioselectivity, overcoming a challenge in previously reported syntheses of truxinate natural products (Fig. 9).
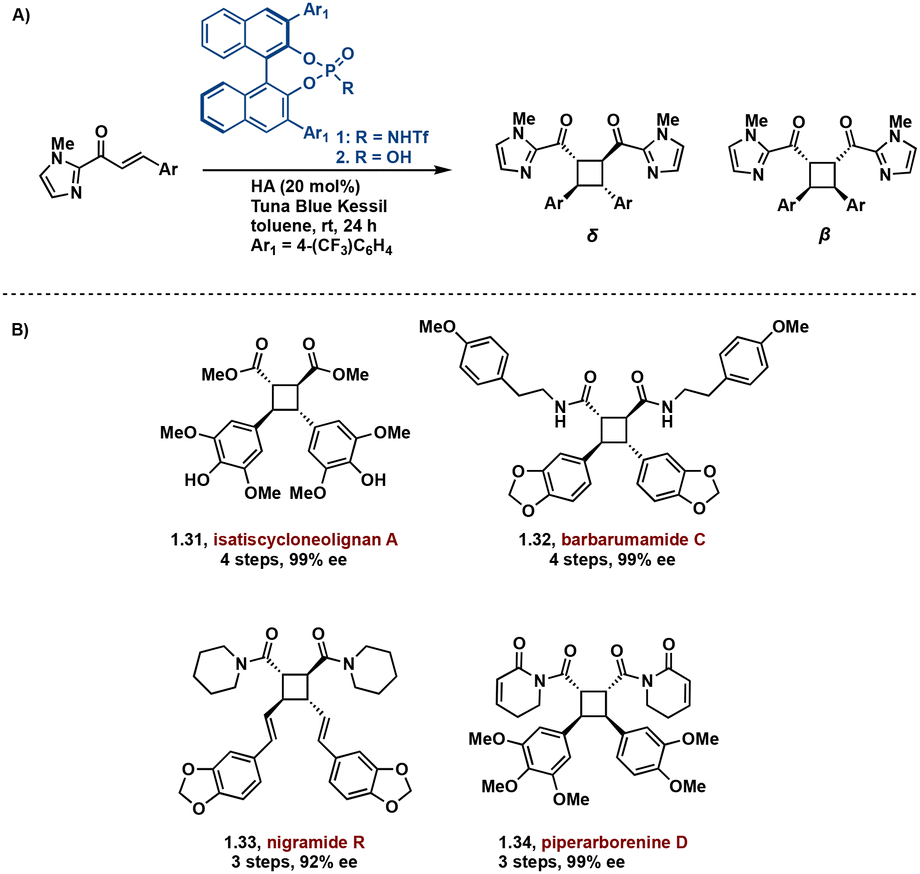 | ||
| Fig. 9 (A) General chiral acid mediated enantioselective [2 + 2] photocycloaddition; (B) synthesized natural products. | ||
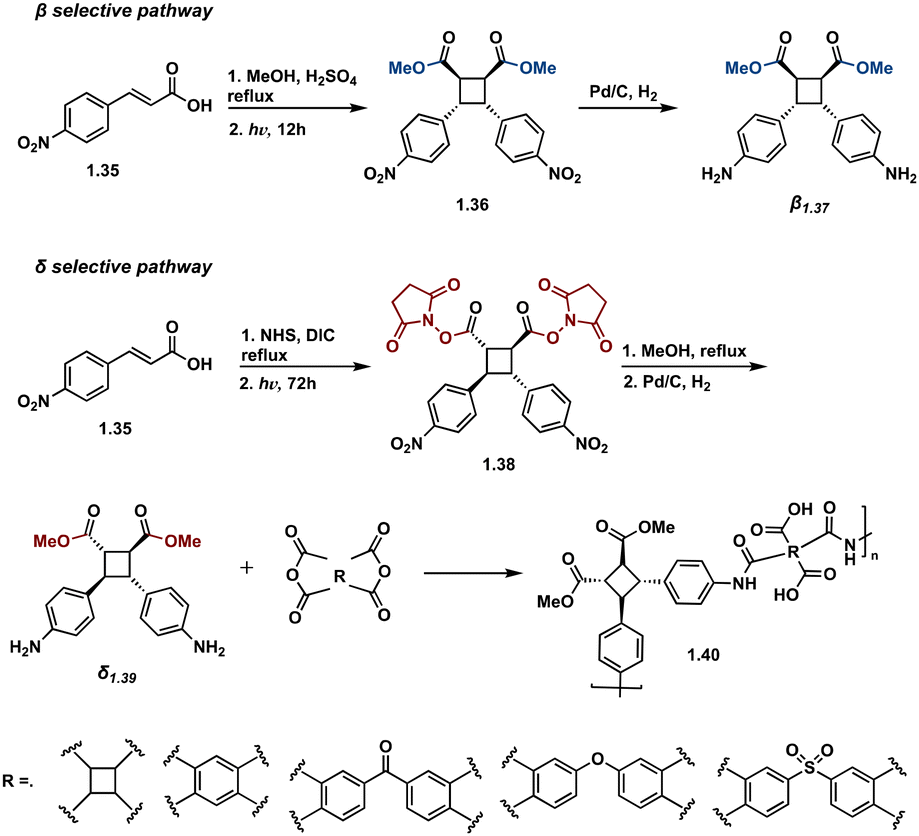 | ||
| Fig. 10 Depiction of both β-selective and δ-selective pathways. Less sterically incumbered methoxy esters selectively form β isomers and sterically hindered NHS esters selectively form δ-isomers. Condensation polymerization of δ-NHS cyclobutane affords δ-polyamides. | ||
2.2. Photosensitized reactions
In many cases direct excitation is not desirable due to high-energy, low wavelength light, that may be required. Alternatively, a triplet sensitizer can be used.52–56 In doing so, greater wavelength tunability is possible. In photosensitized reactions, irradiation of the triplet sensitizer facilitates excitation from the ground state (S0) to the excited singlet state (S1) followed by ISC to the lowest lying triplet state of the sensitizer. Subsequently, Dexter energy transfer occurs,57 transferring energy from the sensitizer to an olefin, in turn facilitating [2 + 2] photocycloadditions (Fig. 11). Successful sensitization requires the triplet energy of the triplet sensitizer to be greater than the triplet energy of the olefin as well close spatial arrangement of the photosensitizer and olefin.58 Desirable triplet sensitizers possess low lying excited singlet states (S1) and high lying triplet states (T1), as well as efficient ISC and long triplet state lifetimes.14 Common triplet sensitizers in use for [2 + 2] photocycloadditions include thioxanthone derivatives,59 benzophenone derivatives60 and iridium-based catalysts.61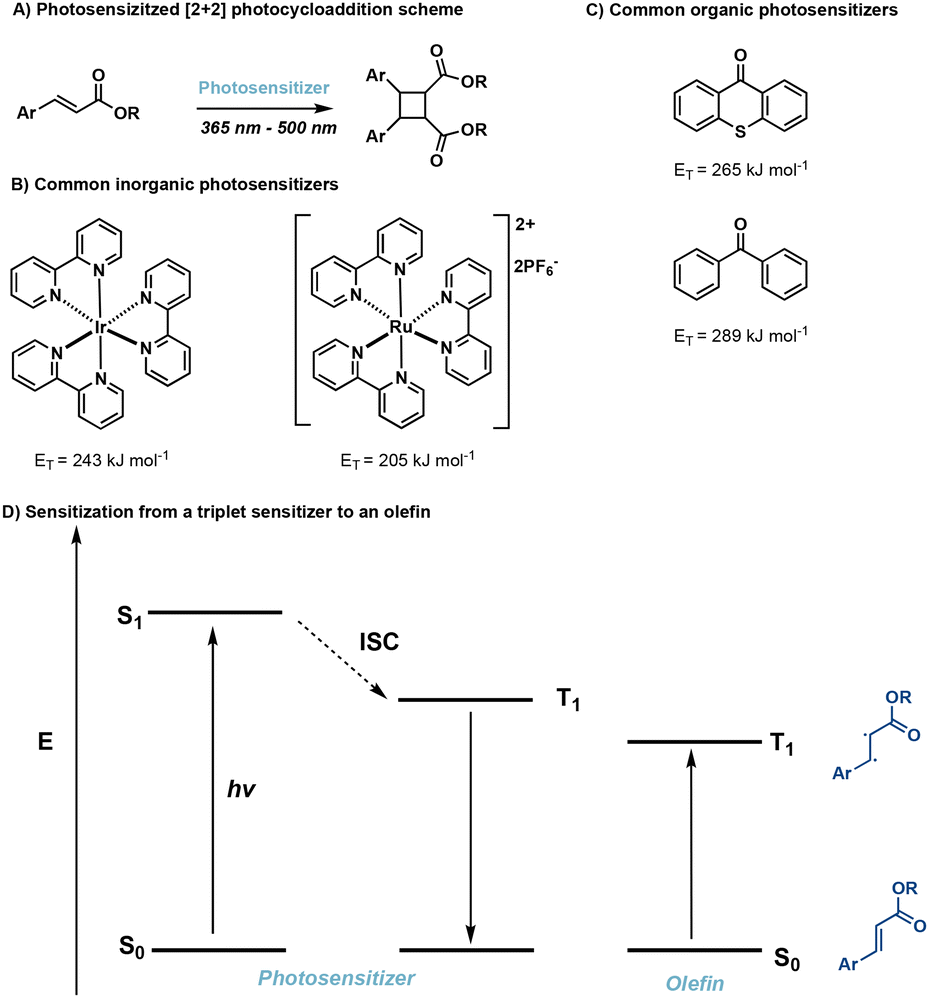 | ||
| Fig. 11 (A) General photosensitized reaction scheme; (B) common inorganic photosensitizers and corresponding triplet energies; (C) common organic photosensitizers and corresponding triplet energies; (D) energy diagram of triplet sensitization pathway. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β diastereoselectivity of up to 49:1 in intermolecular [2 + 2] photocycloadditions using Ir[dF(CF3)ppy]2(dtb-bpy)(PF6) in parallel with a chiral auxiliary compared to previously reported diastereoselectivity values. This method also produces high enantiopurity (>78% ee) of δ truxinates.63
:β diastereoselectivity of up to 49:1 in intermolecular [2 + 2] photocycloadditions using Ir[dF(CF3)ppy]2(dtb-bpy)(PF6) in parallel with a chiral auxiliary compared to previously reported diastereoselectivity values. This method also produces high enantiopurity (>78% ee) of δ truxinates.63
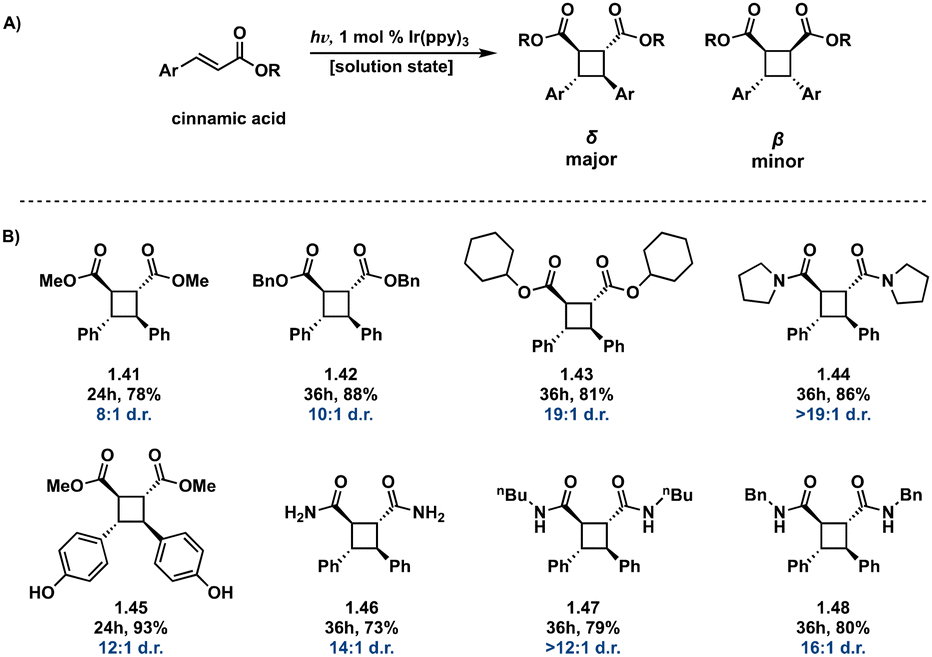 | ||
| Fig. 12 (A) General Ir(ppy)3 photosensitized [2 + 2] of cinnamic acids; (B) selected examples from substrate scope indicating δ:β diastereoselectivity. | ||
Transition metal catalysts also afford improved synthetic control in [2 + 2] photopolymerizations. Solution state [2 + 2] photopolymerizations are challenging due to the lack of monomer pre-assembly and short-lived triplet lifetimes. Overcoming this hurdle, Liao et al. describe energy-transfer catalysis with Ir(ppy)3 for the visible light mediated [2 + 2] photopolymerization of biscinnamate monomers. Additionally, use of visible light eliminates background E/Z isomerization occurring with high energy light. The resulting truxinate polymers range between 14.7 kDa–61.3 kDa and are solely δ-isomers. The polymers exhibit excellent processability and solubility—a notable improvement to previously reported truxinate/truxillate polymers synthesized via direct excitation methodologies (Fig. 13).64
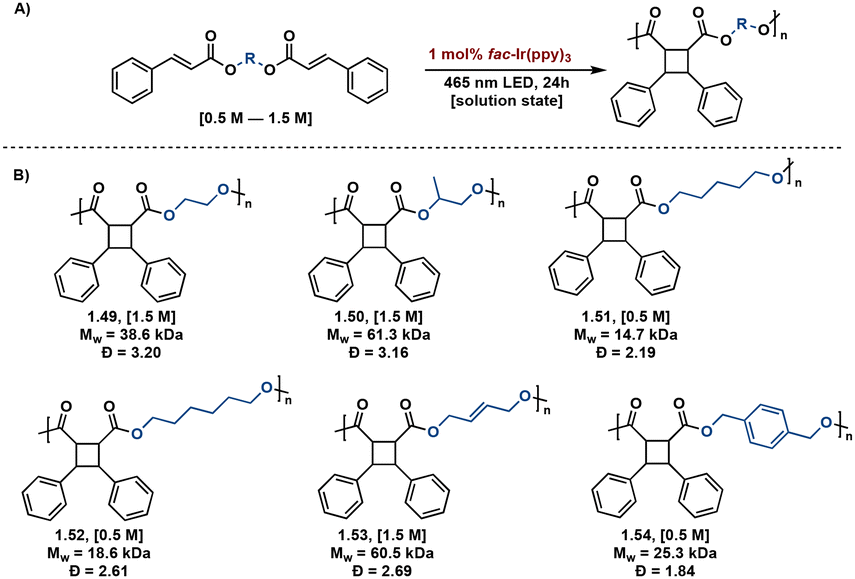 | ||
| Fig. 13 (A) General Ir(ppy)3 catalyzed [2 + 2] photopolymerization with variable diol linker; (B) substrate scope of synthesized polymers with accompanying Mw and Đ values. | ||
In many cases, transition-metal catalysts achieve sufficient diastereo-control, however few enantioselective photocatalytic methods exist. Of the available methods, many utilize an individual transition-metal catalyst or a dual catalysis approach to reduce racemic occurrence. In an effort to increase enantiomeric excess (ee) and reduce racemic background cycloadditions resulting from irradiation of an unbound substrate, many developed methods necessitate high catalyst loading. However, despite high catalyst loading, a degree of racemic background remains. To mediate racemic background, Yoon and coworkers report a unique dual catalysis approach, coined triplet activation, in which use of a stereocontrolling Lewis or Brønsted acid and a transition metal triplet sensitizer in parallel promotes enantionselectivity.65–69 In triplet activation, coordination of the substrate to either a Lewis or Brønsted acid lowers the triplet energy of the substrate facilitating selective activation by an appropriate triplet sensitizer, in turn inducing enantio- and diastereoselectivity of truxinate-like cyclobutanes. Yoon et al. report activation of chalcones by Lewis acid, Sc(OTf)3, and sensitization by Ru(bpy)3(PF6)2. Irradiation with blue LEDs affords pseudo-truxinates in high ee (>85%) as well as synthetic access to norlignane cyclobutane natural products.67 Expanding on this methodology, Yoon et al. utilizes cinnamic esters to directly access cyclobutane carboxylates. Coordination of the cinnamic esters with the chiral Lewis acid, oxazaborolidine, and subsequent irradiation with blue LEDs in presence of [Ir(ppy)2(dtbbpy)](PF6) affords promising ee (89%). Modification of the aryl moiety on the oxazaborolidine Lewis acid to a more electron deficient aryl group improves ee from 89% to 98% (Fig. 14).68 Brønsted acids exhibit similar mechanistic pathways to Lewis acids via triplet activation. A subsequent study by Yoon et al. utilizes a chiral Brønsted acid in an asymmetric [2 + 2] photocycloaddition yielding trans–cis stereochemistry and both diastero- and enantioselectivity in an additional screen of α,β-unsaturated carbonyls.70
 | ||
| Fig. 14 (A) General dual catalysis approach with lewis acid and Ir catalyst; (B) selected substrates with diastereoselectivity and enantioselectivity. | ||
As racemic background remains a challenge in attaining high regio- and diastero-selectivity, host–guest cage confined photocatalysis systems are being leveraged to afford additional selectivity in intermolecular [2 + 2] cycloadditions. Metal–organic containers (MOCs) provide selectivity within [2 + 2] photocycloadditions via confinement driven pre-orientation of the reactive olefins. Su et al. employs a Ru(II)-incorporating metal–organic container (MOC-16) to synthesize homo- and heterocoupled head-to-head truxinates. Specifically, the [Pd6(RuL)8]28+ MOC comprises 8 metalloid ligands, in a truncated octahedron orientation, and facilitates regio- and diastereoselective intermolecular [2 + 2] photocycloadditions of chalcones and cinnamates. para-Functionalized cinnamate starting materials of varying electron densities afford excellent diastereoselectivity upon visible light excitation. Diastereoselectivity decreases in ortho and meta substituted cinnamates due to increasing steric hindrance within the nanospace.71
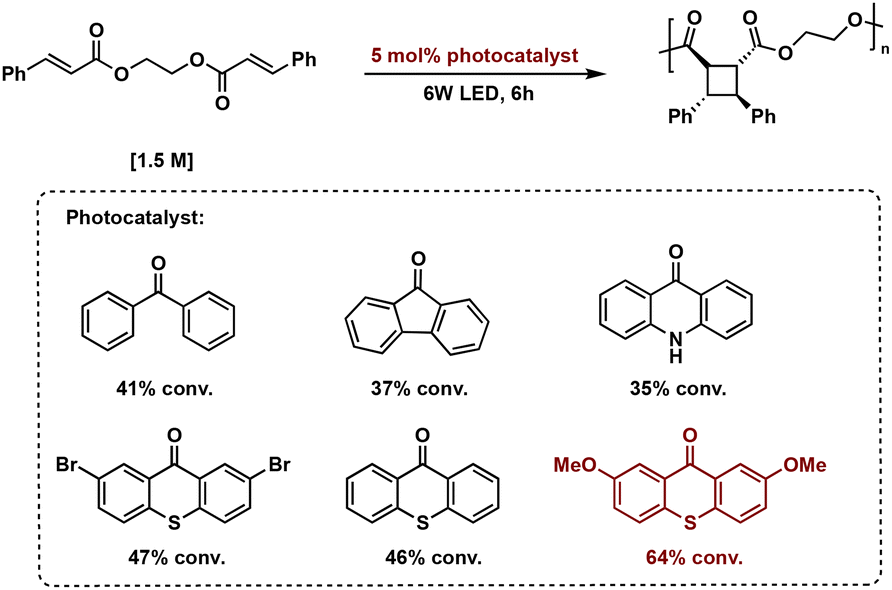 | ||
| Fig. 15 General schematic of organic photosensitized [2 + 2] photopolymerization and photocatalyst screening with percent conversions. The optimal photocatalyst 2,2′-MeOTX is shown in red. | ||
2.3. Continuous flow methodologies
While solid-state photocycloadditions are common and offer advantages such as increased stereo-, diastereo-, and enantioselectivity, scalability and efficiency remain limitations. As such, solution-state methodologies are being investigated. A primary advantage of solution-state methodology is the option of continuous flow technologies to improve the efficiency, scalability, and transformation yield.78 Photochemical batch reactions particularly suffer from poor efficiency on large scale due to inconsistent irradiation of the reaction mixture. In contrast, continuous flow offers consistent irradiation of reaction mixtures due to the increased surface area to volume ratio, shorter path length, and improved temperature control, in turn increasing overall efficiency. In a 2015 report, Beeler et al. utilize a xenon(Hg) light source and cone reactor that allows for consistent irradiation with temperature control even at prolonged irradiation times. Utilizing methyl cinnamate and a hydrogen-bonding bis(thiourea) catalyst with an 8 hour residence time, the percent conversion increases from 25% in batch to 76% in continuous flow. Additionally, diastereoselectivity improves from 2:1 δ:β in batch to 3:1 δ:β in continuous flow.79 In a more recent study, Benaglia et al. employ a continuous flow reactor equipped with blue LEDs in an effort to improve efficiency in the enantioselective syntheses of truxinate cyclobutanes from N-cinnamoyl-oxazolidinones. While enantioselectivity and yield of δ-methyl truxinate remain equal in batch and continuous flow, continuous flow provides a 6-fold productivity increase. Additionally, the space–time–yield (STY) value, a value indicating the product yield per unit volume of catalysts and per unit time, is 73 times higher than that of batch.80
As natural product syntheses often suffer from poor overall yield due to extensive step count and poor yielding individual steps, continuous flow is being investigated as an approach to improve scalability and yield in the synthesis of truxinate and truxillate natural products. Beeler et al. demonstrate the utility of continuous flow in the β-selective total synthesis of piperarborenines C–E. In this report, synthesis of tethered starting materials as [2 + 2] precursors in the construction of truxinate cores facilitates β-selectivity. Incorporating the use of continuous flow in the [2 + 2] photocycloaddition step improves efficiency from batch by reducing irradiation time from 4–12 hours to a 15 minute residence time as well as introduces scalability. Following hydrolysis and subsequent conversion to the corresponding amide affords piperarborenines C–E in overall yields (43%, 68% and 42% respectively), a significant improvement to previously reported syntheses (Fig. 16).81 Notably of the previous total syntheses, Baran et al. employ the triplet sensitization of a non-symmetrical cinnamic acid anhydride by benzophenone in the synthesis and reassignment of piperarborenine D. This total synthetic effort affords milligram quantities of the natural product with an overall yield of 4% and thus lacks scalability.82
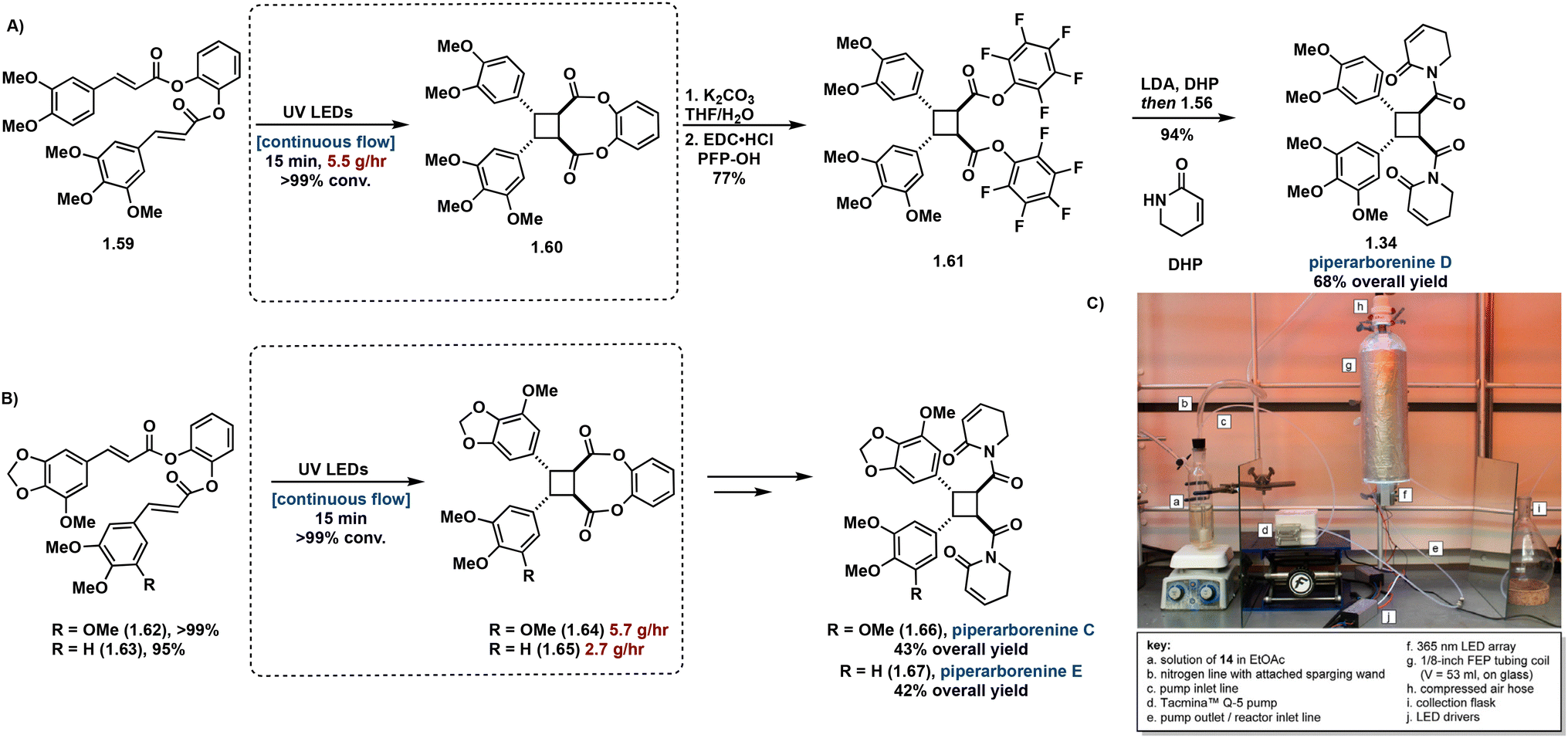 | ||
| Fig. 16 (A) Synthetic route to piperarborenine D with continuous flow step boxed; (B) synthetic routes to piperarborenines C–E with continuous flow step boxed; (C) continuous flow setup. Adapted from ref. 81 with permission from American Chemical Society, Copyright © 2022. | ||
Continuous flow is also applicable to polymer syntheses. Photochemical polymerizations, in particular step-growth polymerizations, often suffer from broad dispersities and uncontrolled molecular weight growth due to poor light irradiation and insufficient mixing. Continuous flow allows for improved molecular weight and dispersity control due to the increased surface area to volume ratio facilitating even irradiation and improved mixing during the polymerization. Grinstaff et al. report an improved method to prepare well-defined and structurally complex truxinate polymers from a library of 42 diesters with varying electron densities and olefin separation via a thioxanthone mediated [2 + 2] photopolymerization. Use of a continuous flow reactor and 365 nm LEDs, affords polymers of greater molecular weight and lower dispersity than compared to batch reactions. At the longest polymerization time of 36 hours, molecular weight increases to 181.3 kDa in continuous flow, a 2.8× increase in molecular weight from batch experiments. Additionally, the dispersity decreases to 1.68 in continuous flow from 3.35 in batch. Scalability also improves with polymer production of 6.5 g per day compared to 200 mg in batch.83
3. Applications
3.1. Brief overview of truxinate and truxillate natural products
Truxinate and truxillate natural products exhibit a diverse array of biological activities encompassing anti-cancer, anti-diabetic, neuroprotective and analgesic properties. The majority of truxinic acid natural products emerge from the dimerization of phenol-rich cinnamic acids, including p-coumaric acid, caffeic acid, and ferulic acid. Among these, the dimerization of p-coumaric acid yields one of the most prevalent phenol-containing truxinate dimers. The most notable among the caffeic acid-based truxinate natural products is undeniably Eucommicin A. Initially isolated from Eucommia ulmoides in 2016,84 this natural product selectively inhibits cancer stem cells, with an IC50 value of 55 μM, compared to the IC50 value of 6900 μM of its monomer, chlorogenic acid (Fig. 17). The exploration of these caffeic acid-derived truxinate natural products holds promise in identifying novel therapeutic agents, with Eucommicin A particularly showcasing its potential to target cancer stem cells, a crucial aspect in the development of effective cancer therapies.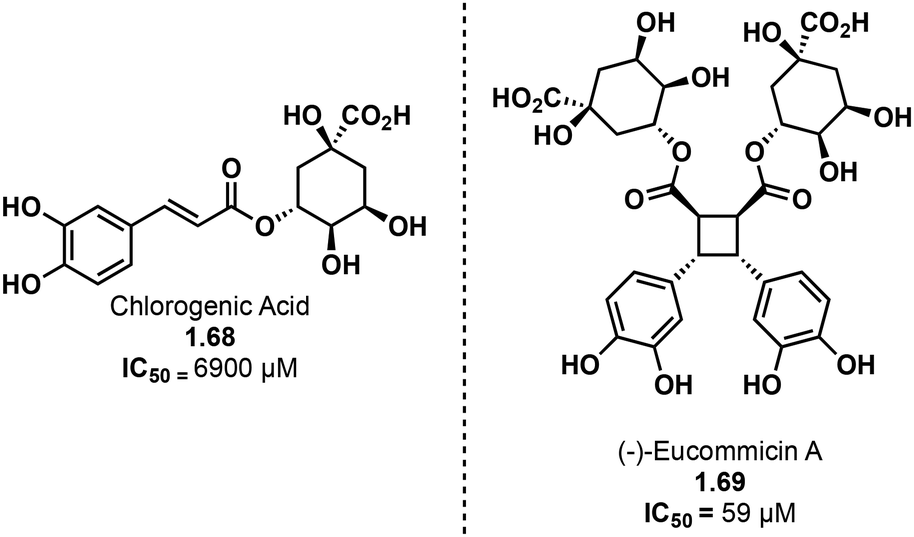 | ||
| Fig. 17 Structure of chlorogenic acid and (−)-eucommicin A. | ||
A noteworthy group of truxinate natural products is the piperarborenines (Fig. 18), which were originally discovered in the plant Piper arboescans, native to Southeast Asia. Piperarborenines A and B (1.70 and 1.71), as well as the piplartine dimer A (1.72), were isolated from the stem of P. arboescans.85 Subsequent isolation efforts in 2005 provided piperarborenines C–E (1.66, 1.34, and 1.67).86 Comparative studies with piperarborenines A–E, piplartine dimer A, and piplartine (commonly known as piperlongumine) against established cancer therapeutics cisplatin and paclitaxel reveal the potential of these natural products as anti-cancer leads.86 While all the isolated compounds demonstrate cytotoxicity against HT-29, P-388, and A549 cancer cell lines in in vitro assays, piperarborenines C–E exhibit impressive IC50 values in the sub micromolar range (0.02–0.3 μM). It is hypothesized that piperarborenines exert their cytotoxic effects through covalent inhibition, engaging with thiols in cells, thereby depleting glutathione or inhibiting proteins due to covalent modification of cystine residues. This mode of action closely resembles that of the structurally related compound piperlongumine. The activity exhibited by piperarborenines C–E warrants further investigation to elucidate the breadth of their cytotoxicity and selectivity. The chemistry and bioactivity of small molecule truxinates and truxillates is rapidly advancing.
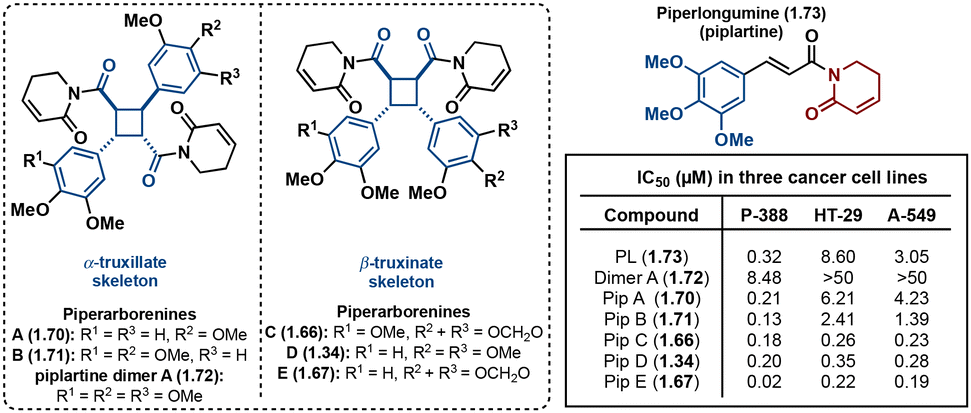 | ||
| Fig. 18 Structure of piperarborenines A–E, and piperlongumine with corresponding cytotoxicity data. | ||
3.2. Polymer advanced materials
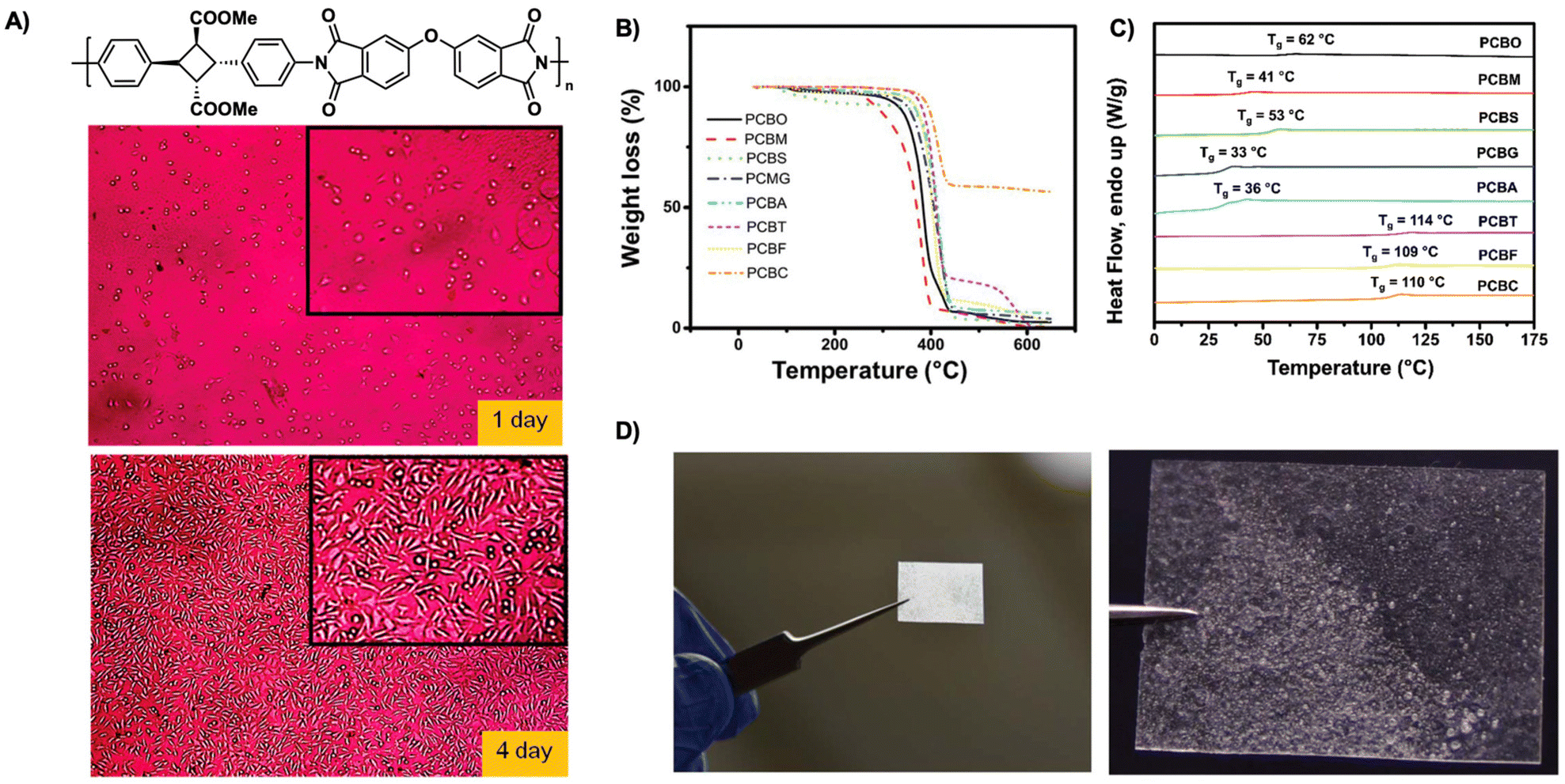 | ||
| Fig. 19 (A) Representative photos of L929 fibroblast cells adhered onto biocompatible polyimide films after incubation of 1 and 4 days. Reproduced from ref. 50 with permission from American Chemical Society, Copyright © 2014; (B) TGA spectra of cyclobutane polyesters from 30 to 600 °C with heating rate of 20 °C min−1 under N2; (C) DSC traces of cyclobutane polyesters; (D) polymer cast of cyclobutane polyester succinate, PCBS showing processability. (B–D) are adapted from ref. 94 with permission from Royal Society of Chemistry, Copyright © 2020. | ||
Sustainable plastics often suffer from low glass transition temperatures (Tg) and poor mechanical performance limiting their use in industries requiring high temperature resistance. An ideal material possesses excellent temperature resistance, high glass transition temperature (Tg), and high mechanical strength. In contrast to many currently available sustainable materials, biobased polyimides provide excellent biocompatibility, degradation, and high material performance. Kaneko et al. describe high-performance biocompatible polyimide films derived from α-truxillates. The polyimides exhibit moderate crystallinity of 30% and good resistance to thermal decomposition with 10% mass loss between 390 °C–425 °C. The polyimide films exhibit high Tg (240–275 °C) with a dependence on backbone structure. Furthermore, the polyimide films demonstrate excellent tensile strength with values of 48–98 MPa, tensile moduli values of 4.2–13.4 GPa and gross mechanical failure at strains of 1.7–4.6%. Additionally, as these polyimides are fully bioderived, the films show excellent cell compatibility with good adherence and growth of L929 fibroblast cells over a course of 1–4 days (Fig. 19A).50 Similarly, polyimide copolymer films possess high thermal resistance and improved toughness compared to previous polyimide films. Previous films are limited in ductility often breaking at low strain, thus hindering their use in industrial sectors. Polyimide copolymer films synthesized from α-truxillates and copolymerized with dianhydrides, show improvements in ductility and toughness resisting breakage even upon folding in half. These films exhibit good heat resistance, Youngs moduli (E) values ranging from 2.9–4.4 GPa, tensile strengths ranging from 28–113 MPa, and strain at break ranging from 0.9–9.4%. As the strain energy is comparable to that of Kapton, these films demonstrate their utility and promise as alternatives.95
Biradha et al. report tunable plastic films of truxillate polyamide polymers. These films resulting from SCSC polymerization and subsequent dissolution into various acids, assemble into two-dimensional hydrogen bonding layers possessing excellent shock-absorption. Modification of the phenyl group to a pyridyl group results in wrinkle-free plastic films that return to their original shape after deformation. Modification of the counterion from formic acid to HCl results in disparate tensile strengths of 15.99 and 7.11 MPa respectively, indicating tunability of the films. Such tunability and potential photoluminescence of the films shows promise for applications as light-emitting diodes.35 A similar film by Kaneko et al. displays unique bending angles in β and δ truxinate imides. Previous reports indicate α-polyimides are brittle and difficult to manipulate films owing to their straight and rigid structures regardless of imide backbone modifications. In contrast, δ-polyimides experience more bond rotation and bending, allowing for increased flexibility and tunability with modifications. The δ-polyimides exhibit high thermal stability with 10% mass loss at 415 °C and high flexibility with 10.2% elongation at break. Solubility of the δ-polyimides increases in organic solvents such as DMF and DMSO in comparison to previous reports of α-polyimides which exhibit limited solubility. As such, film preparation of δ-polyimides is easier.51
Leveraging the toughness of α-polyimide materials, α-polyimides form super tough plastic nanomembranes and show promise as effective coating materials. A polycondensation of two diamino-α-truxillic acids affords α-polyamides with high strain-energy densities of 231 MJ m−3. Incorporation of aliphatic chains in the backbone increases elongation at break and increase the toughness as aliphatic content increases. The highest performing polymer with toughness of 231 MJ m−3, exhibits toughness greater than spider silk. Subsequent spin-coating of the polyamides onto glass slides affords nanomembranes of approximately 200 nm thickness. Moreover, these nanomembranes are self-standing despite their thinness and as a result are applicable as electronic displays and organic memory devices.96
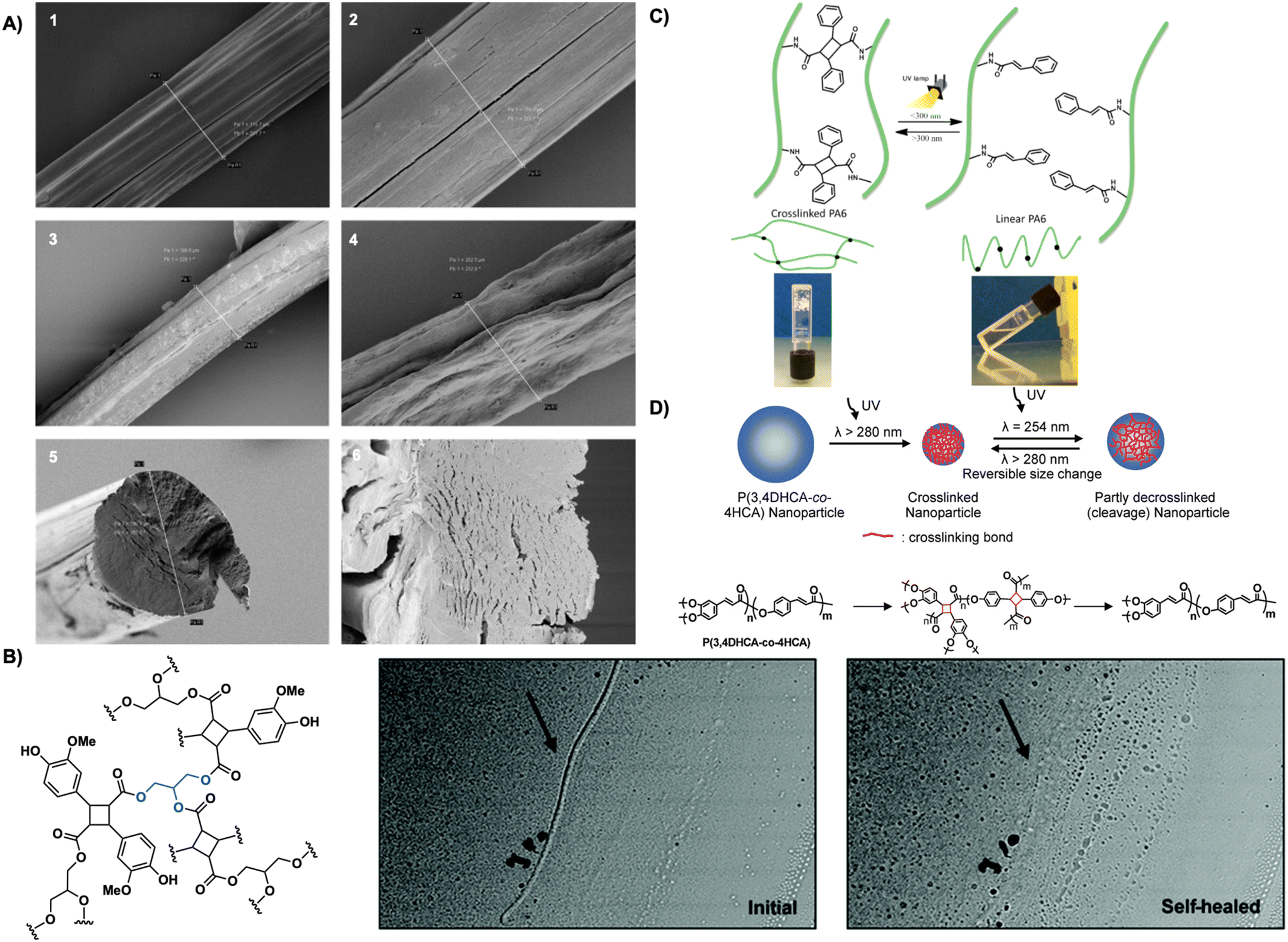 | ||
| Fig. 20 (A) SEM images of hyaluronic acid fibers 1. Drawn, uncrosslinked fibers. 2. Undrawn, uncrosslinked fibers 3. Drawn, crosslinked fibers after 1 h in PBS (pH = 7.4) 4. Undrawn, crosslinked fibers after 1 h in H2O. 5. Cross-section of undrawn, uncrosslinked fiber. 6. Cross-section of undrawn, crosslinked fiber after 1 h in PBS (pH = 7.4). Reproduced from ref. 100 with permission from Elsevier, Copyright © 2015; (B) optical microscope images of crosslinked polymer coated glass slides before and after photo self-healing with UV light. Adapted from ref. 103 with permission from Royal Society of Chemistry, Copyright © 2021; (C) crosslinked gels forming cyclobutane links between polymer chains and linear non-crosslinked chains decorated with cinnamoyl chains. Reproduced from ref. 37 with permission from American Chemical Society, Copyright © 2014; (D) schematic representation of size effect of nanoparticles before and after photo-crosslinking resulting in truxillate nanoparticles. Reproduced from ref. 104 with permission from American Chemical Society, Copyright © 2008. | ||
Cyclobutane crosslinked systems are also stimuli responsive. Stimuli responsive responses including crack healing and photo-reversible dimerization are highly sought after due to their applicability across numerous sectors including energy, consumer goods, and medicine.101 Cyclobutanes are often the target for such materials due to their constrained structure and ability to undergo retro [2 + 2] photocycloadditions readily revealing them as appealing building blocks in stimuli responsive materials development. Carlotti et al. report the photo- and thermo-dimerization of aliphatic polyamides bearing photoactive cinnamoyl pendants forming truxillate crosslinks between polymer chains. Increasing crosslinking percentage of the polyamides induces gelation and increases thermal stability from 290 °C to 390 °C. Subsequent irradiation of the crosslinked gels with 220–280 nm light de-crosslinks the gels, forming a solution. Re-irradiation at 325–380 nm or heating at 140 °C affords re-crosslinked materials (Fig. 20C).37 Avérous et al. employ a similar strategy in the reversible photo-crosslinking of segmented thermoplastic polyurethane with caffeic acid, a biocompatible cinnamic acid. Incorporation of a caffeic acid-based chain-extender into the main chain, rather than as a pendant, enhances thermal properties. Photo-crosslinking with UV light occurs readily and is complete in 2 hours yielding crosslinked thermoplastic polyurethane. Crosslinking increases the plateau modulus and increases flow even at high temperatures of 250 °C. Subsequent mechanical testing reveals an increase in Young's modulus (8.2 MPa after irradiation vs. 6.5 MPa before irradiation) and a 50% reduction in elongation at break thereby decreasing tensile strength. Exposure to high energy 254 nm UVC light promotes decrosslinking, however only 30% crosslinking is accessible. Additionally, the crosslinked thermoplastic polyurethanes display moderate shape-memory behavior with strain fixity of 36% at 20% deformation.102 Similarly, Saito et al. leverage the photo-reversibility of truxillates in a 3-arm photo-crosslinkable system. The 3-arm monomer forms from glycerol and lignin-based vanillin, both heavily present in nature and are inherently biocompatible. Irradiation with 365 nm UV light affords a truxillate crosslinked system that exhibits photo-reversibility upon subsequent irradiation with 254 nm UV light. A self-healing study on the polymer reveals reparation of damage or cracking to the crosslinked system by irradiation with 254 nm light subsequently reducing crack depth by 80% (Fig. 20B).103
Truxinate and truxillate polymers are also incorporated in photo-responsive nanoparticle formulations. Cinnamate copolymers from 3,4-dihydroxycinnamic acid and 4-hydroxy cinnamic acid readily self-organize into nanoparticles in DMF/TFA solutions. Crosslinking of the nanoparticles by irradiation with UV light over 30 minutes decreases particle diameter by 50%. Subsequent irradiation at λ = 254 nm facilitates decrosslinking in 30 s fully recovering the original diameter. Repeated irradiation cycles facilitate a crosslinking and de-crosslinking cycle. Monodispersity maintains through each cycle, however each irradiation event increases the coefficient of variation (Fig. 20D).104
4. Conclusion and future perspectives
Cyclobutanes are ubiquitous in nature as core structures of numerous small molecule natural products and are present in at least 39 (pre)clinical drug candidates being investigated as pain therapeutics and chemotherapeutics among others.16 Their polymer counterparts are less studied but central to the development of new functional materials. The most prevalent method to access cyclobutane materials is the [2 + 2] photocycloaddition due to increased functional group tolerance and direct access to the cyclobutane core. [2 + 2] photocycloadditions occur by both direct excitation and photocatalytic strategies with the latter affording more stereochemical control. Additionally, both solid-state and solution-state methodologies give access to complex structures. Solid-state photocycloadditions are inherently regio-, stereo- and enantio-selective due to preorganization of monomer units. Topochemical polymerizations, a subclass of solid-state [2 + 2] photocycloadditions, are enabling the development of functional materials to target specific material properties that hinge on stereochemistry. While stereochemical control is more difficult in solution-state, novel developments in enantioselective catalysts expand the applicability and use of solution-state methodologies.As monomers containing olefins available for [2 + 2] photocycloadditions are required, cinnamic acids represent a broad class of conjugated molecules that afford direct access to complex truxinate and truxillate scaffolds. Due to the modifiable aryl groups within cinnamic acids, access to highly functionalized materials with targeted material properties is readily achievable. As a result, the construction of structure–property relationships will elucidate key molecular features responsible for a specific functionality of interest to those working in sustainability, biomedical, and nanotechnology fields among others.
The intent of this article is to provide the background necessary to understand current synthetic methods and applications of cyclobutane materials as well as envision future developments and applications. Selectivity in cyclobutane polymer materials remains a challenge and methods to achieve high stereo-, regio- and enantio-selectivity as well as increased functionalization of cinnamic acids are needed. Methods to induce selectivity in small molecule cyclobutanes such as the use of chiral acids, have not been applied to polymer syntheses and offer a starting point to obtain increased stereochemical control of cyclobutane materials. A primary challenge with [2 + 2] photocycloadditions of small molecules is the scalability, selectiveness, and functionalization. Continuous flow strategies are improving scalability, reducing irradiation time, and improving selectivity.112,113 However, continuous flow is under-utilized in truxinate and truxillate polymer synthesis, with only one example to date. A common challenge of [2 + 2] photopolymerizations is the limited molecular weight and dispersity control due to the step growth mechanism. Very few methods exist to control these parameters, especially in solution-state.114,115 Additionally, access to functionalized materials such as those containing imides and amides is limited by currently available methods. Typically, condensation polymerizations employ pre-formed small molecule truxinates and truxillates as monomers, thus two steps are required to afford the desired polymer. Therefore, other techniques to afford polyimides and polyamides directly from [2 + 2] photopolymerizations require exploration, as these polymers show promise as sustainable plastics as we move toward a green society. Further study is also needed on the biodegradation of truxinate and truxillate polymers as they possess readily hydrolysable backbones. Development of high Tg materials with high tensile strength is essential to providing alternatives to commonly utilized plastics such as polyethylene, PET, and poly(vinyl chloride) which slowly degrade in the environment.114–118 Finally, the further exploration of these polymers as stimuli-responsive systems and nanomaterials will be exciting and likely lead to new applications in several fields.
Highly controlled, reproducible, and scalable polymerization methods are key to wide-spread use and to successfully transition these materials into industrial manufacturing processes. In summary the significant advancements to date will empower the next cycle of cyclobutane material development, and we encourage all to continue to explore these polymers given their broad applicability.
Author contributions
S. E. and J. M. L. performed the literature search and background analysis. S. E., J. M. L., A. B. B. and M. W. G. wrote the manuscript. All authors have given approval to the final version of the manuscript. No AI was used in the writing of this manuscript, just revision after revision between the authors.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported in part by Boston University, the William Fairfield Warren Professorship, and the NIH (R01HL159644). We thank Stephanie Laporte for contributions to Fig. 4 and 11.References
- X. Yuan, L. Men, Y. Liu, Y. Qiu, C. He and W. Huang, Truxillic and truxinic acid derivatives: configuration, source, and bioactivities of natural cyclobutane dimers, J. Holistic Integr. Pharm., 2020, 1, 48–69 CrossRef.
- Y.-M. Chi, M. Nakamura, X.-Y. Zhao, T. Yoshizawa, W.-M. Yan, F. Hashimoto, J. Kinjo, T. Nohara and S. Sakurada, Antinociceptive Activities of α-Truxillic Acid and β-Truxinic Acid Derivatives, Biol. Pharm. Bull., 2006, 29, 580–584 CrossRef CAS PubMed.
- Y.-M. Chi, M. Nakamura, T. Yoshizawa, X.-Y. Zhao, W.-M. Yan, F. Hashimoto, J. Kinjo, T. Nohara and S. Sakurada, Anti-inflammatory Activities of α-Truxillic Acid Derivatives and Their Monomer Components, Biol. Pharm. Bull., 2005, 28, 1776–1778 CrossRef CAS PubMed.
- T. Morikawa, K. Ninomiya, J. Akaki, N. Kakihara, H. Kuramoto, Y. Matsumoto, T. Hayakawa, O. Muraoka, L.-B. Wang, L.-J. Wu, S. Nakamura, M. Yoshikawa and H. Matsuda, Dipeptidyl peptidase-IV inhibitory activity of dimeric dihydrochalcone glycosides from flowers of Helichrysum arenarium, J. Nat. Med., 2015, 69, 494–506 CrossRef CAS PubMed.
- M. Carmignani, A. R. Volpe, F. Delle Monache, B. Botta, R. Espinal, S. C. De Bonnevaux, C. De Luca, M. Botta, F. Corelli, A. Tafi, G. Ripanti and G. Delle Monache, Novel Hypotensive Agents from Verbesina ca racasana. 6. Synthesis and Pharmacology of Caracasandiamide, J. Med. Chem., 1999, 42, 3116–3125 CrossRef CAS PubMed.
- I. S. Aljančić, I. Vučković, M. Jadranin, M. Pešić, I. Đorđević, A. Podolski-Renić, S. Stojković, N. Menković, V. E. Vajs and S. M. Milosavljević, Two structurally distinct chalcone dimers from Helichrysum zivojinii and their activities in cancer cell lines, Phytochemistry, 2014, 98, 190–196 CrossRef PubMed.
- R. D. Hartley, W. Herbert Morrison III, F. Balza and G. H. N. Towers, Substituted Truxillic and Truxinic Acids in Cell Walls of Cynodondactylon, Phytochemistry, 1990, 29, 3699–3703 CrossRef CAS.
- R. D. Hartley, D. S. Himmelsbach and W. H. Morrison, Synthesis of Substituted Truxillic Acids from P-Coumaric and Ferulic Acid: Simulation of Photodimerization in Plant Cell Walls, J. Agric. Food Chem., 1992, 40, 768–771 CrossRef.
- G. M. J. Schmidt, Photodimerization in the solid state, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- Anon, Coca leaves from ceylon and federated states, London, 10th edn, 1912 Search PubMed.
- J. M. Moore, J. F. Casale, R. F. X. Klein, D. A. Cooper and J. Lydon, Determination and in-depth chromatographic analyses of alkaloids in South American and greenhouse-cultivated coca leaves, J. Chromatogr. A, 1994, 659, 163–175 CrossRef CAS PubMed.
- I. S. Lurie, J. M. Moore, T. C. Kram and D. A. Cooper, Isolation, identification and separation of isomeric truxillines in illicit cocaine, J. Chromatogr. A, 1990, 504, 391–401 CrossRef CAS.
- V. M. Dembitsky, Bioactive cyclobutane-containing alkaloids, J. Nat. Med., 2008, 62, 1–33 CAS.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- P. Yang, Q. Jia, S. Song and X. Huang, [2 + 2]-Cycloaddition-derived cyclobutane natural products: structural diversity, sources, bioactivities, and biomimetic syntheses, Nat. Prod. Rep., 2023, 40, 1094–1129 RSC.
- M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, Cyclobutanes in Small–Molecule Drug Candidates, ChemMedChem, 2022, 17, 1–22 CrossRef PubMed.
- C. F. Koelsch and W. H. Gumprecht, Some Diazine-N-oxides 1, J. Org. Chem., 1958, 23, 1603–1606 CrossRef CAS.
- F. L. Hirshfeld and G. M. J. Schmidt, Topochemical control of solid–state polymerization, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2181–2190 CrossRef.
- M. Lahav and G. M. J. Schmidt, The photochemistry of crystalline dimethyl all-trans-hexatriene-1,6-dicarboxylate, Tetrahedron Lett., 1966, 7, 2957–2962 CrossRef.
- K. C. Stueben, Cyclobutane polymers from acrylonitrile dimer, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 829–846 CrossRef CAS.
- F. Nakanishi and M. Hasegawa, Four-Center Type Photopolymerization in the Solid State. IV. Polymerization of a,a’-Dicyano-p- benzenediacrylic Acid and Its Derivatives*, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 2151–2160 CrossRef.
- H. Nakanishi, W. Jones, J.-M. Thomas, M. Hasegawa and W.-L. Rees, Topochemically Controlled Solid-State Polymerization, Proc. R. Soc. London, Ser. A, 1980, 369, 307–325 CAS.
- W. R. Gutekunst and P. S. Baran, Applications of C–H Functionalization Logic to Cyclobutane Synthesis, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS PubMed.
- D. I. Schuster, G. Lem and N. A. Kaprinidis, New insights into an old mechanism: [2 + 2] photocycloaddition of enones to alkenes, Chem. Rev., 1993, 93, 3–22 CrossRef CAS.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, 384. Topochemistry. Part II. The photochemistry of trans-cinnamic acids, J. Chem. Soc., 1964, 2000–2013 RSC.
- M. Bertmer, R. C. Nieuwendaal, A. B. Barnes and S. E. Hayes, Solid-State Photodimerization Kinetics of α- trans- Cinnamic Acid to α-Truxillic Acid Studied via Solid-State NMR, J. Phys. Chem. B, 2006, 110, 6270–6273 CrossRef CAS PubMed.
- V. Ramamurthy and K. Venkatesan, Photochemical reactions of organic crystals, Chem. Rev., 1987, 87, 433–481 CrossRef CAS.
- K. Biradha and R. Santra, Crystal engineering of topochemical solid state reactions, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- T. B. Nguyen and A. Al-Mourabit, Remarkably high homoselectivity in [2 + 2] photodimerization of trans-cinnamic acids in multicomponent systems, Photochem. Photobiol. Sci., 2016, 15, 1115–1119 CrossRef CAS PubMed.
- H. Amjaour, Z. Wang, M. Mabin, J. Puttkammer, S. Busch and Q. R. Chu, Scalable preparation and property investigation of a cis -cyclobutane-1,2-dicarboxylic acid from β- trans -cinnamic acid, Chem. Commun., 2019, 55, 214–217 RSC.
- T. Ishigami, T. Murata and T. Endo, The Solution Photodimerization of (E)- p -Nitrocinnamates, Bull. Chem. Soc. Jpn., 1976, 49, 3578–3583 CrossRef CAS.
- M. D'Auria and A. Vantaggi, Photochemical dimerization of methoxy substituted cinnamic acid methyl esters, Tetrahedron, 1992, 48, 2523–2528 CrossRef.
- F. Danusso, P. Ferruti, A. Moro, G. Tieghi and M. Zocchi, Structure and solid state photopolymerization of pentaerythritol tetracinnamate, Polymer, 1977, 18, 161–163 CrossRef CAS.
- R. Santra, K. Banerjee and K. Biradha, Weak Ag⋯Ag and Ag⋯π interactions in templating regioselective single and double [2 + 2] reactions of N,N′-bis(3-(4-pyridyl)acryloyl)–hydrazine: synthesis of an unprecedented tricyclohexadecane ring system, Chem. Commun., 2011, 47, 10740 RSC.
- M. Garai, R. Santra and K. Biradha, Tunable Plastic Films of a Crystalline Polymer by Single–Crystal–to–Single–Crystal Photopolymerization of a Diene: Self–Templating and Shock–Absorbing Two–Dimensional Hydrogen–Bonding Layers, Angew. Chem., Int. Ed., 2013, 52, 5548–5551 CrossRef CAS PubMed.
- R. Mandal and K. Biradha, Organic Polymers of an Angular Diene via Solid State [2 + 2] Polymerization: Coordination Polymers with Dicarboxylates as Templates, Cryst. Growth Des., 2019, 19, 3445–3452 CrossRef CAS.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Reversible Cross-Linking of Aliphatic Polyamides Bearing Thermo- and Photoresponsive Cinnamoyl Moieties, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- Y. Jiang, H. Zhu, J. Chen, Q. Ma and S. Liao, Linear Cyclobutane-Containing Polymer Synthesis via [2 + 2] Photopolymerization in an Unconfined Environment under Visible Light, ACS Macro Lett., 2022, 11, 1336–1342 CrossRef CAS PubMed.
- B. B. Yagci, Y. Zorlu and Y. E. Türkmen, Template-Directed Photochemical Homodimerization and Heterodimerization Reactions of Cinnamic Acids, J. Org. Chem., 2021, 86, 13118–13128 CrossRef CAS PubMed.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2 + 2] Photocycloadditions, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed.
- C. M. Rasik and M. K. Brown, Lewis Acid-Promoted Ketene–Alkene [2 + 2] Cycloadditions, J. Am. Chem. Soc., 2013, 135, 1673–1676 CrossRef CAS PubMed.
- H. Knölker, G. Baum and R. Graf, Lewis Acid Promoted [2 + 2] Cycloaddition of Allylsilanes and Unsaturated Esters: A Novel Method for Cyclobutane Construction, Angew. Chem., Int. Ed. Engl., 1994, 33, 1612–1615 CrossRef.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed.
- F. D. Lewis, S. L. Quillen, P. D. Hale and J. D. Oxman, Lewis acid catalysis of photochemical reactions. 7. Photodimerization and cross-cycloaddition of cinnamic esters, J. Am. Chem. Soc., 1988, 110, 1261–1267 CrossRef CAS.
- T. P. Yoon, Photochemical Stereocontrol Using Tandem Photoredox–Chiral Lewis Acid Catalysis, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- C. Brenninger, J. D. Jolliffe and T. Bach, Chromophore Activation of α,β–Unsaturated Carbonyl Compounds and Its Application to Enantioselective Photochemical Reactions, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS PubMed.
- R. Brimioulle and T. Bach, Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2 + 2] Photocycloaddition Reactions, Science, 2013, 342, 840–843 CrossRef CAS PubMed.
- M. J. Genzink, M. D. Rossler, H. Recendiz and T. P. Yoon, A General Strategy for the Synthesis of Truxinate Natural Products Enabled by Enantioselective [2 + 2] Photocycloadditions, J. Am. Chem. Soc., 2023, 145, 19182–19188 CrossRef CAS PubMed.
- K. Takada, Synthesis of biobased functional materials using photoactive cinnamate derivatives, Polym. J., 2023, 55, 1023–1033 CrossRef CAS.
- P. Suvannasara, S. Tateyama, A. Miyasato, K. Matsumura, T. Shimoda, T. Ito, Y. Yamagata, T. Fujita, N. Takaya and T. Kaneko, Biobased Polyimides from 4-Aminocinnamic Acid Photodimer, Macromolecules, 2014, 47, 1586–1593 CrossRef CAS.
- T. Noda, T. Iwasaki, K. Takada and T. Kaneko, Soluble Biobased Polyimides from Diaminotruxinic Acid with Unique Bending Angles, Macromolecules, 2021, 54, 10271–10278 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Mechanistic Studies in Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. L. Dexter, A Theory of Sensitized Luminescence in Solids, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- F. H. Quina and G. T. M. Silva, The photophysics of photosensitization: A brief overview, J. Photochem. Photobiol., 2021, 7, 100042 CrossRef.
- N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Thioxanthone: a powerful photocatalyst for organic reactions, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC.
- C. Prentice, A. E. Martin, J. Morrison, A. D. Smith and E. Zysman-Colman, Benzophenone as a cheap and effective photosensitizer for the photocatalytic synthesis of dimethyl cubane-1,4-dicarboxylate, Org. Biomol. Chem., 2023, 21, 3307–3310 RSC.
- H. Jung, M. Hong, M. Marchini, M. Villa, P. S. Steinlandt, X. Huang, M. Hemming, E. Meggers, P. Ceroni, J. Park and M.-H. Baik, Understanding the mechanism of direct visible-light-activated [2 + 2] cycloadditions mediated by Rh and Ir photocatalysts: combined computational and spectroscopic studies, Chem. Sci., 2021, 12, 9673–9681 RSC.
- T. Lei, C. Zhou, M. Huang, L. Zhao, B. Yang, C. Ye, H. Xiao, Q. Meng, V. Ramamurthy, C. Tung and L. Wu, General and Efficient Intermolecular [2 + 2] Photodimerization of Chalcones and Cinnamic Acid Derivatives in Solution through Visible–Light Catalysis, Angew. Chem., Int. Ed., 2017, 56, 15407–15410 CrossRef CAS PubMed.
- F. Medici, A. Puglisi, S. Rossi, L. Raimondi and M. Benaglia, Stereoselective [2 + 2] photodimerization: a viable strategy for the synthesis of enantiopure cyclobutane derivatives, Org. Biomol. Chem., 2023, 21, 2899–2904 RSC.
- Y. Jiang, Q. Ma, X. Zhang, J. Li and S. Liao, Solution [2 + 2] photopolymerization of biomass-derived nonrigid biscinnamate monomers enabled by energy transfer catalysis, Polym. Chem., 2022, 13, 2538–2544 RSC.
- E. M. Sherbrook, H. Jung, D. Cho, M.-H. Baik and T. P. Yoon, Brønsted acid catalysis of photosensitized cycloadditions, Chem. Sci., 2020, 11, 856–861 RSC.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed.
- Z. D. Miller, B. J. Lee and T. P. Yoon, Enantioselective Crossed Photocycloadditions of Styrenic Olefins by Lewis Acid Catalyzed Triplet Sensitization, Angew. Chem., 2017, 129, 12053–12057 CrossRef.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, Enantioselective [2 + 2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- E. M. Sherbrook, M. J. Genzink, B. Park, I. A. Guzei, M.-H. Baik and T. P. Yoon, Chiral Brønsted acid-controlled intermolecular asymmetric [2 + 2] photocycloadditions, Nat. Commun., 2021, 12, 5735 CrossRef CAS PubMed.
- J.-S. Wang, K. Wu, C. Yin, K. Li, Y. Huang, J. Ruan, X. Feng, P. Hu and C.-Y. Su, Cage-confined photocatalysis for wide-scope unusually selective [2 + 2] cycloaddition through visible-light triplet sensitization, Nat. Commun., 2020, 11, 4675 CrossRef CAS PubMed.
- A. Bossion, K. V. Heifferon, L. Meabe, N. Zivic, D. Taton, J. L. Hedrick, T. E. Long and H. Sardon, Opportunities for organocatalysis in polymer synthesis via step-growth methods, Prog. Polym. Sci., 2019, 90, 164–210 CrossRef CAS.
- C.-L. Sun and Z.-J. Shi, Transition-Metal-Free Coupling Reactions, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed.
- M. Sicignano, R. I. Rodríguez and J. Alemán, Recent Visible Light and Metal Free Strategies in [2 + 2] and [4 + 2] Photocycloadditions, Eur. J. Org. Chem., 2021, 2021, 3303–3321 CrossRef CAS PubMed.
- L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, Rational Design of Triplet Sensitizers for the Transfer of Excited State Photochemistry from UV to Visible, J. Am. Chem. Soc., 2020, 142, 14947–14956 CrossRef CAS PubMed.
- Y. Jiang, H. Zhu, J. Chen and S. Liao, Organocatalytic [2 + 2] Photopolymerization under Visible Light: Accessing Sustainable Polymers from Cinnamic Acids, Macromol. Rapid Commun., 2022, 44, 2200702 CrossRef PubMed.
- F. Pecho, Y. Sempere, J. Gramüller, F. M. Hörmann, R. M. Gschwind and T. Bach, Enantioselective [2 + 2] Photocycloaddition via Iminium Ions: Catalysis by a Sensitizing Chiral Brønsted Acid, J. Am. Chem. Soc., 2021, 143, 9350–9354 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- R. Telmesani, S. H. Park, T. Lynch-Colameta and A. B. Beeler, [2 + 2] Photocycloaddition of Cinnamates in Flow and Development of a Thiourea Catalyst, Angew. Chem., Int. Ed., 2015, 54, 11521–11525 CrossRef CAS PubMed.
- F. Medici, A. Puglisi, S. Rossi, L. Raimondi and M. Benaglia, Stereoselective [2 + 2] photodimerization: a viable strategy for the synthesis of enantiopure cyclobutane derivatives, Org. Biomol. Chem., 2023, 21, 2899–2904 RSC.
- J. M. Lenihan, M. J. Mailloux and A. B. Beeler, Multigram Scale Synthesis of Piperarborenines C-E, Org. Process Res. Dev., 2022, 26, 1812–1819 CrossRef CAS.
- W. R. Gutekunst and P. S. Baran, Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed.
- S. El-Arid, J. Lenihan, A. Jacobsen, A. Beeler and M. Grinstaff, Accessing Cyclobutane Polymers: Overcoming Synthetic Challenges via Efficient Continuous Flow [2 + 2] Photopolymerization, ACS Macro Lett., 2024, 13, 607–613 CrossRef CAS PubMed.
- A. Fujiwara, M. Nishi, S. Yoshida, M. Hasegawa, C. Yasuma, A. Ryo and Y. Suzuki, Eucommicin A, a β-truxinate lignan from Eucommia ulmoides, is a selective inhibitor of cancer stem cells, Phytochemistry, 2016, 122, 139–145 CrossRef CAS PubMed.
- F. P. Lee, Y. C. Chen, J. J. Chen, I. L. Tsai and I. S. Chen, Cyclobutanoid Amides from Piper arborescens, Helv. Chim. Acta, 2004, 87, 463–468 CrossRef CAS.
- I. L. Tsai, F. P. Lee, C. C. Wu, C. Y. Duh, T. Ishikawa, J. J. Chen, Y. C. Chen, H. Seki and I. S. Chen, New cytotoxic cyclobutanoid amides, a new furanoid lignan and anti-platelet aggregation constituents from Piper arborescens, Planta Med., 2005, 71, 535–542 CrossRef CAS PubMed.
- D. G. Bucknall, Plastics as a materials system in a circular economy, Philos. Trans. R. Soc., A, 2020, 378, 20190268 CrossRef CAS PubMed.
- E. MacArthur, Beyond plastic waste, Science, 2017, 358, 843–843 CrossRef PubMed.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- A. Pellis, M. Malinconico, A. Guarneri and L. Gardossi, Renewable polymers and plastics: Performance beyond the green, New Biotechnol., 2021, 60, 146–158 CrossRef CAS PubMed.
- H. Karan, C. Funk, M. Grabert, M. Oey and B. Hankamer, Green Bioplastics as Part of a Circular Bioeconomy, Trends Plant Sci., 2019, 24, 237–249 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Chemically recyclable polymers: a circular economy approach to sustainability, Green Chem., 2017, 19, 3692–3706 RSC.
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- R. K. Shahni, M. Mabin, Z. Wang, M. Shaik, A. Ugrinov and Q. R. Chu, Synthesis and characterization of BPA-free polyesters by incorporating a semi-rigid cyclobutanediol monomer, Polym. Chem., 2020, 11, 6081–6090 RSC.
- H. Shin, S. Wang, S. Tateyama, D. Kaneko and T. Kaneko, Preparation of a Ductile Biopolyimide Film by Copolymerization, Ind. Eng. Chem. Res., 2016, 55, 8761–8766 CrossRef CAS.
- Y. Funahashi, Y. Yoshinaka, K. Takada and T. Kaneko, Self-Standing Nanomembranes of Super-Tough Plastics, Langmuir, 2022, 38, 5128–5134 CrossRef CAS PubMed.
- P. Gupta, S. R. Trenor, T. E. Long and G. L. Wilkes, In Situ Photo-Cross-Linking of Cinnamate Functionalized Poly(methyl methacrylate- co, -2-hydroxyethyl acrylate) Fibers during Electrospinning, Macromolecules, 2004, 37, 9211–9218 CrossRef CAS.
- S. Alven and B. A. Aderibigbe, Hyaluronic Acid-Based Scaffolds as Potential Bioactive Wound Dressings, Polymers, 2021, 13, 2102 CrossRef CAS PubMed.
- D. Rachmiel, I. Anconina, S. Rudnick-Glick, M. Halperin-Sternfeld, L. Adler-Abramovich and A. Sitt, Hyaluronic Acid and a Short Peptide Improve the Performance of a PCL Electrospun Fibrous Scaffold Designed for Bone Tissue Engineering Applications, Int. J. Mol. Sci., 2021, 22, 2425 CrossRef CAS PubMed.
- T. Bobula, J. Běťák, R. Buffa, M. Moravcová, P. Klein, O. Židek, V. Chadimová, R. Pospíšil and V. Velebný, Solid-state photocrosslinking of hyaluronan microfibres, Carbohydr. Polym., 2015, 125, 153–160 CrossRef CAS PubMed.
- G. Kaur, P. Johnston and K. Saito, Photo-reversible dimerisation reactions and their applications in polymeric systems, Polym. Chem., 2014, 5, 2171–2186 RSC.
- A. Duval and L. Avérous, From thermoplastic polyurethane to covalent adaptable network via reversible photo-crosslinking of a biobased chain extender synthesized from caffeic acid, Polym. Chem., 2023, 14, 2685–2696 RSC.
- P. Sinha Roy, M. M. Mention, M. A. P. Turner, F. Brunissen, V. G. Stavros, G. Garnier, F. Allais and K. Saito, Bio-based photo-reversible self-healing polymer designed from lignin, Green Chem., 2021, 23, 10050–10061 RSC.
- D. Shi, M. Matsusaki, T. Kaneko and M. Akashi, Photo-Cross-Linking and Cleavage Induced Reversible Size Change of Bio-Based Nanoparticles, Macromolecules, 2008, 41, 8167–8172 CrossRef CAS.
- E. M. Nofen, N. Zimmer, A. Dasgupta, R. Gunckel, B. Koo, A. Chattopadhyay and L. L. Dai, Stress-sensing thermoset polymer networks via grafted cinnamoyl/cyclobutane mechanophore units in epoxy, Polym. Chem., 2016, 7, 7249–7259 RSC.
- K. Hema, A. Ravi, C. Raju, J. R. Pathan, R. Rai and K. M. Sureshan, Topochemical polymerizations for the solid-state synthesis of organic polymers, Chem. Soc. Rev., 2021, 50, 4062–4099 RSC.
- J. W. Colson and W. R. Dichtel, Rationally synthesized two-dimensional polymers, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- Z. Wang, K. Randazzo, X. Hou, J. Simpson, J. Struppe, A. Ugrinov, B. Kastern, E. Wysocki and Q. R. Chu, Stereoregular Two-Dimensional Polymers Constructed by Topochemical Polymerization, Macromolecules, 2015, 48, 2894–2900 CrossRef CAS.
- W. Wei and H. Xiong, Liquid-Crystalline Polymers: Molecular Engineering, Hierarchical Structures, and Applications, Langmuir, 2022, 38, 11514–11520 CrossRef CAS PubMed.
- T. Kato, J. Uchida, T. Ichikawa and B. Soberats, Functional liquid-crystalline polymers and supramolecular liquid crystals, Polym. J., 2018, 50, 149–166 CrossRef CAS.
- H. Kihara and N. Tamaoki, A Main–Chain Liquid–Crystalline Oligomer Prepared by in situ Photopolymerization of an LC Monomer Having Cinnamate Moieties, Macromol. Rapid Commun., 2006, 27, 829–834 CrossRef CAS.
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Polymer Synthesis in Continuous Flow Reactors, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, Polymerizations in Continuous Flow: Recent Advances in the Synthesis of Diverse Polymeric Materials, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- X. Wu, P. Liu, X. Zhao, J. Wang, M. Teng and S. Gao, Critical effect of biodegradation on long-term microplastic weathering in sediment environments: A systematic review, J. Hazard. Mater., 2022, 437, 129287 CrossRef CAS PubMed.
- P. K. Roy, M. Hakkarainen, I. K. Varma and A.-C. Albertsson, Degradable Polyethylene: Fantasy or Reality, Environ. Sci. Technol., 2011, 45, 4217–4227 CrossRef CAS PubMed.
- S. Ghatge, Y. Yang, J.-H. Ahn and H.-G. Hur, Biodegradation of polyethylene: a brief review, Appl. Biol. Chem., 2020, 63, 27 CrossRef CAS.
- Y. Tokiwa, B. Calabia, C. Ugwu and S. Aiba, Biodegradability of Plastics, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS PubMed.
Footnote |
| † Denotes equal authorship. |
| This journal is © The Royal Society of Chemistry 2024 |