
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of a crystalline Na4Fe3(PO4)2(P2O7) cathode material for sodium-ion batteries†
Yaprak
Subaşı
 *a,
Laura
Altenschmidt
a,
Fredrik
Lindgren
a,
Tore
Ericsson
b,
Lennart
Häggström
b,
Cheuk-Wai
Tai
c,
Haidong
Liu
*a and
Reza
Younesi
a
*a,
Laura
Altenschmidt
a,
Fredrik
Lindgren
a,
Tore
Ericsson
b,
Lennart
Häggström
b,
Cheuk-Wai
Tai
c,
Haidong
Liu
*a and
Reza
Younesi
a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Uppsala, 75121, Sweden. E-mail: yaprak.subasi@kemi.uu.se; haidong.liu@kemi.uu.se
bDepartment of Physics and Astronomy, Uppsala University, Uppsala, 75121, Sweden
cDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, 10691, Sweden
First published on 5th August 2024
Abstract
Na4Fe3(PO4)2(P2O7) (NFPP) as a promising cathode material for sodium-ion batteries possesses excellent structural stability, minimal volume change, low cost, and non-toxicity. However, its practical application is hindered by the formation of impurity phases and its intrinsically low electronic conductivity. Herein, crystalline high purity carbon-coated NFPP (NFPP/CC) is synthesized by performing a green and scalable combustion method to enhance its overall electrochemical performance. The effects of pre-treatment and the calcination atmosphere on the structure and purity of NFPP are systematically investigated for a variety of synthesis parameters. The electrochemical performance of NFPP cathodes is evaluated in both half-cells with the sodium metal anode and full-cells with the hard-carbon anode via galvanostatic charge–discharge cycling measurements. The “combustion” synthesized NFPP/CC cathode delivers a reversible discharge capacity of ∼102 mA h g−1 at 0.1C in an operating voltage window of 1.8–3.8 V (vs. Na/Na+) retaining 99.7% of its initial capacity over 100 cycles. Furthermore, it demonstrates enhanced rate capability in comparison to the NFPP/CC cathode synthesized via the conventional calcination route. This study sheds light on using the combustion method as a facile and effective strategy to simultaneously mitigate the formation of impurity phases, reduce the carbon content, enhance the quality of carbon coating, improve the homogeneity of nanoparticles and pores within the structure, and enhance the electronic conductivity and physical stability of NFPP cathodes, paving the way for their practical application in high-performance sodium-ion batteries.
1. Introduction
Cost-effective large-scale energy storage systems (ESSs) have been a critical issue over the past few years to fully implement the use of solar, wind, geothermal, and tidal energy.1–3 Lithium-ion batteries (LIBs) have rapidly entered different sectors including EESs, however, there is an increase in demand for abundant and low-cost alternative batteries due to the limited reserves and high costs of lithium compounds. Sodium-ion batteries (SIBs) have received great attention because of their potential to provide a cost-effective alternative.4,5 The current focus is to increase the energy density and lifetime of SIBs to make them fully competitive with LIBs. Therefore, different cathode materials including layered transition metal oxides,6,7 Prussian blue analogues (PBAs),8,9 and polyanionic compounds10,11 have been heavily researched and developed. Polyanionic compounds have been suggested as promising cathode materials with regard to their high working voltage, structural stability, thermal stability, and small volume change upon cycling.12 Among the polyanionic compounds, mixed phosphates, Na4M3(PO4)2P2O7 (M = Co, Ni, Mn, Mg, Fe) show fast Na+ diffusion ascribed to their open framework along the b direction which would enable fast charge and discharge.13,14 Iron-based phosphates have attracted significant interest due to their natural abundance, potentially low cost, environmental friendliness, and stability, therefore different formulations such as NaFePO4, Na3Fe2(PO4)3, Na2FePO4F, Na3.64Fe2.18(P2O7)2, and Na3Fe2(PO4)(P2O7) have been investigated.15–19 Among them, NASICON-type mixed Na4Fe3(PO4)2(P2O7) (NFPP) combines the benefits of both phosphate and pyrophosphate characteristics, offering relatively high average working potential (∼3.1 V vs. Na+/Na), favorable theoretical capacity (129 mA h g−1), and low volume change (<4%) due to its open framework composed of [Fe3P2O13] layers interconnected by (P2O7)4− groups. This architectural configuration ensures not only high structural and thermal stabilities but also the presence of 3D ion channels, thus lowering the activation barriers for Na+ transport.20,21 However, the synthesis of NFPP is prone to the formation of impurities such as maricite NaFePO4 and Na2FeP2O7, which can arise from the synthesis temperature and adopted reactants, potentially restricting the electrochemical performance of NFPP. Wu et al. reported that NFPP calcined at 400 °C showed poor rate capability and huge capacity decay at high rates because of its lower crystallinity and the presence of impurities compared to the one calcined at 500 °C.22 Additionally, the inherent electronically insulating characteristics of the (PO4)3− group in NFPP may result in reduced electronic conductivity and slow ion diffusion, which hinders its practical application in SIBs.23 Facing these challenges, various strategies have been developed to enhance conductivity such as nanosizing, carbon coating, and metal ion doping.24–26 The 3D holey graphene decorated NFPP cathode was produced by ball-milling of the starting chemicals, sodium phytate and ferrous oxalate. This material achieved a reversible capacity of 118 mA h g−1 at 0.2C in the voltage range 1.5–4.2 V.25 In another study, Cao et al. prepared NFPP in the nanosphere form and combined it with surface-modified carbon nanotubes as a high performance future large-scale cathode material.24 Tao et al. concurrently carried out manganese ion doping and surface carbon coating strategies to improve the electronic conductivity and structural stability of the NFPP cathode. The as-fabricated Na4Fe2.9Mn0.1(PO4)2P2O7@C delivered an initial capacity of 119.6 mA h g−1 at 0.1C showing a capacity retention of 97.4% over 100 cycles at 1C.26NFPP can be synthesized by various methods like solid-state,27 sol–gel,20 spray-drying15 and combustion techniques.28 Among them, spray-drying and combustion approaches can be considered practical techniques to obtain high quality products for large scale production and commercialization.13,26,29 Kim et al. synthesized NFPP for the first time by ball-milling stoichiometric amounts of Na4P2O7, Fe2C2O4·2H2O, NH4H2PO4 and pyromellitic acid followed by calcination under an inert atmosphere, which effectively increased NFPP's conductivity. It was reported that 3Na+ could be reversibly extracted with a volume change of less than 4% and a notable reversible capacity of 113.5 mA h g−1 at 0.1C.17,23 However, the conventional solid-state synthesis demands specialized equipment often being time- and energy-intensive. Conversely, several studies have shown that methods involving homogeneous atomic-level mixing, such as sol–gel and solution combustion approaches, can achieve higher purity, smaller particle size with narrow distribution range, and more homogeneous dispersion of nanoparticles within the structure, thanks to the controlled synthesis conditions.30–32 Wu et al. performed a sol–gel method for the first time by using iron metal powder as a precursor and citric acid serving both as a chelating agent and carbon source. The NFPP/CC calcined at 500 °C delivered a first discharge capacity of 110 mA h g−1 at 0.05C with 89% of capacity retention after 300 cycles, making a significant improvement in NFPP synthesis.22 Very recently, Gezović et al. performed a citric acid-assisted sol–gel process, using both phosphate and pyrophosphate reactants with pH adjustment in order to reach the theoretical capacity based on the extraction of 3Na+ ions at a high current of 1 A g−1.33 Li et al. fabricated Na4Fe3−xMnx(PO4)2P2O7/rGO microspheres through the spray-drying method by using iron nitrate as the precursor and graphene as the conductive source. The replacement of Fe2+ with Mn2+ was related to their similar ionic size resulting in the maintained crystal structure of NFPP. The enhanced electrochemical performance even at low temperature (−20 °C) was attributed to the reduced Na+ diffusion barriers and a narrow energy bandgap in the lattice. The Na4Fe2.7Mn0.3(PO4)2P2O7/rGO showed an initial capacity of 131.5 mA h g−1 at 0.1C retaining 97.2% of its initial value after 2000 cycles at 10C achieved by combining the synergistic effects of lattice doping and graphene modification.34 For the feasible flash-combustion approach, Lin et al. used the iron nitrate precursor for the synthesis of coral-like Na3.12Fe2.44(P2O7)2 by performing a brief calcination (3 min) under an inert atmosphere.32 Senthilkumar et al. extended the application of NFPP by synthesizing NFPP thin films through a solution combustion method, followed by annealing and pulsed laser deposition tailored for Na-ion microbatteries.13 Contrary to the sol–gel method, the combustion process may result in the formation of a narrower pore size distribution, more uniform, smaller particle size with a lower degree of agglomeration, and higher quality of carbon coating owing to the elimination of the pre-calcination step whereas the carbon-content of polyanionic cathodes prepared by sol–gel can be above 10 wt% that may be responsible for low energy density.35,36
To the best of our knowledge there is no report focusing on the effects of synthesis parameters on the purity and electrochemical performance of NFPP with the combination of sol–gel and combustion approaches. Despite the valuable previous studies on various methods to synthesize NFPP, the potential of the combustion method and the critical effect of the calcination atmosphere have not been fully investigated. This study aims to bridge this knowledge gap by comprehensively comparing the “combustion” and “calcination” approaches, elucidating the impact of the synthesis conditions on the structural, morphological, and electrochemical properties of NFPP. Through a combination of advanced characterization techniques, including X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Mössbauer spectroscopy, scanning electron microscopy (SEM), and transmission electron microscopy (TEM), this work provides a deeper understanding of the structureproperty relationships in NFPP cathodes. Furthermore, the electrochemical performance of NFPP is extensively investigated in both half-cells with the Na metal anode and full-cells with the hard-carbon anode via galvanostatic charge–discharge cycling measurements, providing valuable insights into the practical viability of this material for applications in SIBs. By addressing the fundamental challenges associated with NFPP synthesis and optimization, this work paves the way for the development of high-performance, low-cost, and environmentally benign cathode materials for advanced SIBs.
2. Experimental section
2.1 Synthesis of Na4Fe3(PO4)2(P2O7)
NFPP was synthesized by a modified combustion process using Fe(NO3)3·9H2O (Sigma-Aldrich, ≥99.99%), ascorbic acid (C6H8O6) (Sigma-Aldrich, ≥99), NaH2PO4·2H2O (Sigma-Aldrich, ≥98) and citric acid (C6H8O7) (Sigma-Aldrich, ≥99.5) as precursors. First, 0.045 mol Fe(NO3)3·9H2O (oxidant) as the Fe3+ precursor was dissolved in 150 mL distilled water at room temperature and was converted into the Fe2+ state by the addition of a pinch of ascorbic acid (C6H8O6) resulting in a color change from brown to colorless jade-green.37 After that, 0.06 mol NaH2PO4·2H2O was added into solution and stirred until dissolution. Finally, 0.034 mol citric acid (fuel) was added to the mixture to trigger the exothermic reaction. The final reaction and the calculation for the required minimum citric acid can be written as follows by using the combustion index:35,38| 4NaH2PO4·2H2O + 3Fe3+(NO3)3·9H2O + 9/4C6H8O7 → Na4Fe2+3(PO4)2(P2O7)/CC + NOx↑ + CO2↑ + H2O↑ | (1) |
The resulting solution was heated using an oil bath at 80 °C until a gel formed. After the evaporation of excess water, the precursor powder was dried in a vacuum oven at 80 °C overnight. The dried metal-citro-nitrate complex gel was then pressed into cylindrical pellets, which hereinafter may be referred to as the “sol–gel precursor”. A first set of sol–gel precursor pellets were fired in a tube furnace at 300 °C for 10 min under an air atmosphere to initiate the combustion which resulted in the evolution of NOx gases. For comparative tests, a second set of sol–gel precursor pellets were calcined at 300 °C for 5 h under an Ar/H2 gas (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 vol%) flow with a heating rate of 3 °C min−1 to eliminate organic components. Finally, the amorphous intermediate products obtained after combustion and calcination, respectively, were ground in an agate mortar and pelletized again using a disc-shaped mold (Φ = 13 mm). The pellets of intermediate “combustion” and “calcination” were further annealed at 500 °C for 10 h under flowing Ar/H2 gas (95:5 vol%) as a reducing atmosphere with a heating rate of 3 °C min−1. During calcination at high temperature, carbonaceous sources are thermally decomposed to form an in situ amorphous carbon coating (CC) layer on the NFPP surface. After naturally cooling down to room temperature, the nanosized NFPP/CC product was obtained. The final samples are hereafter denoted as “NFPP calcination” in the case of the calcination pretreatment or “NFPP combustion” for the combustion pretreatment as shown in Fig. 1a.
:5 vol%) flow with a heating rate of 3 °C min−1 to eliminate organic components. Finally, the amorphous intermediate products obtained after combustion and calcination, respectively, were ground in an agate mortar and pelletized again using a disc-shaped mold (Φ = 13 mm). The pellets of intermediate “combustion” and “calcination” were further annealed at 500 °C for 10 h under flowing Ar/H2 gas (95:5 vol%) as a reducing atmosphere with a heating rate of 3 °C min−1. During calcination at high temperature, carbonaceous sources are thermally decomposed to form an in situ amorphous carbon coating (CC) layer on the NFPP surface. After naturally cooling down to room temperature, the nanosized NFPP/CC product was obtained. The final samples are hereafter denoted as “NFPP calcination” in the case of the calcination pretreatment or “NFPP combustion” for the combustion pretreatment as shown in Fig. 1a.
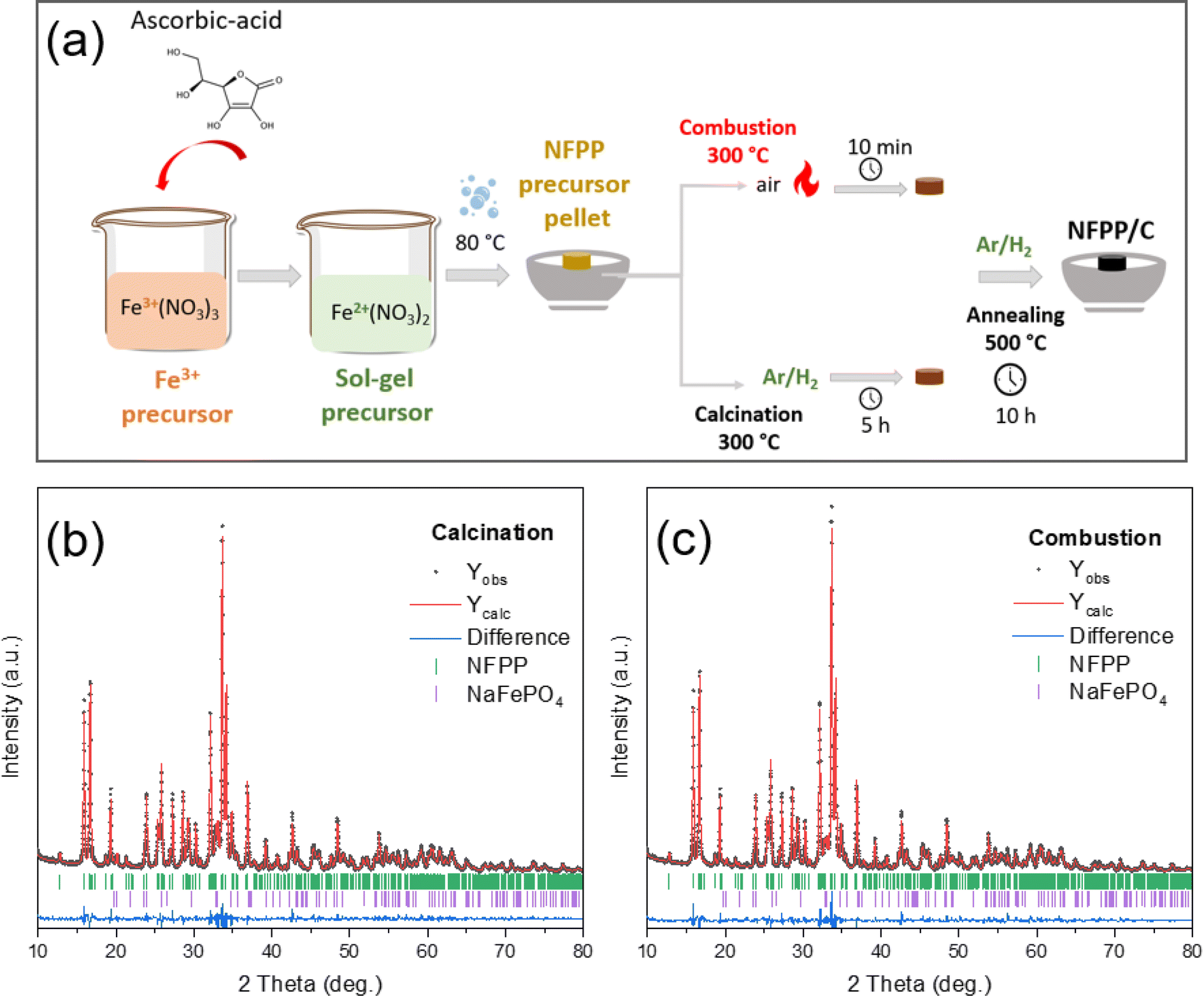 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of NFPP by “calcination” and “combustion”, (b and c) Rietveld refinement of the XRD pattern of the NFPP “combustion” and NFPP “calcination”. Vertical bars represent the Bragg positions for NFPP and maricite NaFePO4. | ||
2.2 Material characterization
Powder X-ray powder diffraction (PXPD) was used to determine the crystal structure and purity of the “combustion” samples and “calcination” samples, respectively, using a Bruker D8 Twin–Twin diffractometer equipped with a CuKα (λ = 1.5406 Å) source operated at 40 kV and 40 mA over the range of 5–70 °(2θ) with a scanning step of 0.01° s−1. The refinement was performed using the Rietveld method39 with the TOPAS software.40 The 57Fe Mössbauer spectra were recorded in transmission mode using a 57Co Rh source at constant acceleration at room temperature. Mössbauer samples were prepared in an argon filled glovebox with a concentration of about 20 mg cm−2. Calibration spectra were recorded from a natural α-Fe metal foil held at 295 K. The resulting spectra were analyzed using a least squares Mössbauer fitting program. Two different pristine materials were examined, one synthesized by “calcination” and one by “combustion” as mentioned in Section 2.1. The electrochemically de-sodiated “NFPP combustion” sample was also studied. Thermogravimetric analysis (TGA) was carried out to determine the thermal stability and carbon content of as-synthesized products by using a TA instruments analyzer Q500 by loading 3–5 mg sample in a platinum pan in the TGA furnace under flowing air (25 mL min−1) in the range of 25 °C to 700 °C with a heating rate of 10 °C min−1. A field-emission scanning electron microscope (FE-SEM) equipped with an energy-dispersive X-ray (EDX) detector was used to investigate the morphology, particle size and elemental distribution of the samples using a Zeiss SEM 1550 with an acceleration voltage of 5.0 kV. As-prepared powders were mounted on an aluminum stub sample holder using carbon tape. For transmission electron microscopy (TEM) measurements, the samples were crushed and then directly dispersed on Cu TEM grids with holey carbon supporting films without any solvent. A double aberration-corrected Thermo Fisher Themis Z equipped with a Super-X EDS detector was used for STEM imaging and EDS experiments. The electron microscope was operated at 300 kV and aberrations in the condenser system lenses were corrected up to fifth order. The convergence angle of 21 mrad and probe current of 120 pA were applied for imaging and EDS analysis. To evaluate the particle size, porosity and elemental distribution of samples, high-angle annular dark-field scanning transmission microscopy (STEM-HAADF) and –BF images were acquired using a Fischione ADF detector and Thermo Fisher BF detector, respectively. The collection angle of HAADF- and BF-STEM images was 63–200 mrad and below 35 mrad, respectively. Imaging and EDS acquisition and analysis were performed using a Thermo Fisher Velox. The atomic ratios of Na, Fe, and P were determined using inductively-coupled plasma-atomic emission spectroscopy (ICP-AES Varian Vista MPX) and carbon contents were determined by using an elemental analyzer (FLASH EA 1112 CHNS-O). X-ray photoelectron spectroscopy (XPS) analysis was performed using a Kratos Ultra DLD (AXIS) instrument with a monochromatic aluminum Kα (1486.6 eV) X-ray source. All peaks were calibrated based on the hydrocarbon species C 1s peak at 284.8 eV. The spectra were analyzed using CasaXPS software. The specific surface area and the pore size distribution of samples were determined using a surface area porosity analyzer (BET, Micromeritics ASAP 2020).2.3 Electrochemical measurements
The electrochemical performance of NFPP samples was investigated in pouch cells assembled in an Ar-filled glovebox in which O2 and H2O levels were below 1 ppm. Electrodes were fabricated by mixing the active material with conductive carbon black (C-ENERGY Super C65, Imerys) and the polyvinylidene fluoride (PVDF) (Kynar HSV 900) binder in an 80:10:10 weight ratio using N-methyl-2-pyrrolidone (NMP) (Sigma-Aldrich®) as the solvent. The resultant slurry was cast onto carbon-coated aluminum foil on a bar coater with a thickness of 400 μm using the doctor blade method. The electrodes were dried in a vacuum oven at 80 °C overnight and further punched into 20 mm diameter discs with an active material loading of 4–5 mg cm−2. The punched electrodes were then dried in a Büchi oven under vacuum at 120 °C overnight inside a glovebox. Pouch cells were assembled in an argon filled glove-box using glass fiber membranes (Whatman®, 26 mm diameter, 240 μm thick) as separators, 22 mm of Na disks punched from metallic Na cubes (Sigma-Aldrich®) serving as the counter and reference electrodes, and 200–250 μL of 1 M NaPF6 (Stella, 99%) in diethylene glycol dimethyl ether (diglyme) (Sigma-Aldrich®, ≥99.99%) as the electrolyte. For full-cell fabrication, hard carbon anodes were prepared by mixing hard carbon (HC) (KURANODE™, Kuraray, Japan) with carboxymethyl cellulose (CMC) (Imerys) binder in ∼6 mL deionized water per gram in a 95:5 ratio by weight. The slurry was coated on carbon-coated aluminum foil using a 200 μm gap applicator rod. The as-fabricated hard carbon anodes were punched into 22 mm diameter discs and dried at 170 °C overnight in a glove box. In full-cells, 200–250 μL of 1 M NaPF6 (Stella, 99%) in propylene carbonate (PC) (Sigma-Aldrich®, ≥99.99%) with 5 wt% fluoroethylene carbonate (FEC) was used as the electrolyte.
The galvanostatic charge–discharge cycling tests of half-cells were carried out using a Neware battery tester BTS4000 in a potential window of 1.8–3.8 V vs. Na/Na+ at 25 °C after 12 h rest to ensure proper wetting of electrodes. The full cells were cycled in the voltage range of 1.0–3.8 V. The areal mass loadings of NFPP and HC are 4.3 mg cm2− and 1.4 mg cm2−, respectively. The negative/positive (N/P) ratio of full cells was 1.1:1. The carbon content within the NFPP/CC was excluded to calculate the specific capacities for each cell. The cyclic voltammetry (CV) measurements were conducted by using a Bio-Logic® MPG2 potentiostat.
3. Results and discussion
3.1 Structure and morphology
NFPP/CC particles were prepared through a sol–gel method combined with either a “calcination” or a modified combustion step referred to as “combustion”, followed by an annealing step. The optimum annealing temperature was determined to be 500 °C by TGA analysis of the sol–gel precursor (Fig. S1a†) under an inert atmosphere. Dehydration of the amorphous intermediate product occurs in the first zone until 180 °C. The primary weight loss in the range of 180–300 °C is attributed to the decomposition of citric acid to form a thin carbon layer on the surface of NFPP. After calcination at 300 °C, the second major loss is observed due to the phase transition of the intermediate complex. An exothermic peak starts to appear after 570 °C indicating the decomposition of NFPP into the thermodynamically stable maricite NaFePO4 impurity.41–44 Therefore, for the first trial, the sol–gel precursor was subjected to 10 min of combustion in a tube furnace at 550 °C in air. However, the XRD pattern (see Fig. S2†) discloses that, after the annealing at 550 °C in air, the obtained orange-reddish powder showed the highly crystalline structure of monoclinic γ-Na3Fe2(PO4)3 with the Fe2O3 impurity. Similar behavior was observed in previous studies where TGA of the as prepared NFPP/CC was performed between room temperature and 800 °C under air flow to determine the carbon content by using the following equations.45–47| 12Na4Fe3(PO4)P2O7 + 9O2 → 16Na3Fe2(PO4)3 + 2Fe2O3 | (2) |
| C + O2 → CO2 | (3) |
These results indicated that preheating or firing at 300 °C may be necessary to form the amorphous carbon coating layer on the NFPP surface by the decomposition of citric acid and subsequent high temperature calcination under a reducing atmosphere should be performed for the transformation of (PO4)3− into (P2O7)4−.48
Scanning electron microscopy (SEM) was performed to evaluate the particle morphology of the samples and the effect of the atmosphere for different synthesis routes. In Fig. S3a,† it can be seen that after only 10 min of calcination, nanosized sodium ferric phosphate particles with some agglomerates below 1 μm were obtained. EDX mapping indicates the uniform distribution of Na, Fe, P and O within the structure (Fig. S3b†). Thus, the formation of crystalline Na3Fe2(PO4)3 nanoparticles by performing a short calcination in air can be adapted to transform into a pyrophosphate phase under a reducing atmosphere by a limited annealing time as well.
The crystal structure and the purity of NFPP synthesized either by “calcination” or “combustion” were investigated by XRD (see Fig. S4†). Both Ar and Ar/H2 atmospheres lead to the formation of the orthorhombic NFPP crystal structure.27 In addition to the NFPP main phase, maricite (NaFePO4) and Na2FeP2O7 impurities were detected for the NFPP synthesized under an Ar flow as shown in Fig. S4a.† However, using an Ar/H2 (95:5 vol%) reducing atmosphere results in the elimination of Na2FeP2O7 and a significant decrease of maricite (NaFePO4) impurities (Fig. S4a†). This is also reflected in cyclic voltammetry showing that the use of Ar/H2 instead of Ar leads to the elimination of a small peak around 2.51 V assigned to the Na2FeP2O7 impurity (see Fig. S10 and its related discussion in the ESI†). Therefore, the results indicate that the atmosphere plays a crucial role in the reduction of Fe3+ if calcination is used as the method of synthesis.47
Fig. S4b† compares the effect of the intermediate step, combustion vs. calcination, when using an Ar/H2 atmosphere during the 2nd thermal treatment. The diffraction patterns of “NFPP calcination” and “NFPP combustion” show that in both cases, NFPP is formed as the main phase. However, maricite was detected as an impurity regardless of the synthesis conditions.
The quantity of the maricite impurity in the NFPP material obtained by “combustion” and “calcination” was determined by a structural analysis using the Rietveld method (Fig. 1b and c). In both cases, the main NFPP phase can be indexed with the Pn21a space group with lattice parameters and the atomic positions (Tables S1 and S2†) in agreement with the ones reported in the literature.17,27 The four Na positions were refined, but only the Na2 position resulted in a slightly lower occupancy as also previously observed which is due to the Na2 being the first sodium to be extracted from the NFPP material.49 This suggests Na deficiencies in the as-prepared material before the final annealing. From the refinement, the amount of electrochemically inactive NaFePO4 was determined to be ≈5 wt% in the “combustion” sample and about 9 wt% in the “calcination” one. This suggests that using a combustion pre-treatment assists in decreasing the quantity of the undesired NaFePO4 phase.
The chemical composition and the surface electronic valence of iron were evaluated by XPS. The full spectra in Fig. S8a† confirm the presence of Na, Fe, P, O, and C in both compounds. The binding energy of the main peaks in Fe 2p spectra at around 724.2 and 710.5 eV suggests the Fe2+ oxidation state which is consistent with previous reports.29,50 However, XPS reveals only the surface composition of samples, and therefore the possible presence of Fe3+ as an impurity in the bulk could not be detected through XPS.
To further investigate the synthesized material, Mössbauer spectroscopy was carried out to determine the oxidation state and coordination of Fe within the NFPP compound, which has three different Fe2+ sites (Fe1, Fe2, and Fe3).51,52
The two Mössbauer spectra of the pristine materials are almost identical (see Fig. 2 for the combustion sample and Fig. S9† for the calcination sample) and they show mainly a broad two-line pattern. The de-sodiated sample mainly displays a narrow doublet with some extra lines having larger velocities (Fig. 2). Fitted results are presented in Table S3† and the two pristine samples differ only in the amount of the pattern FeW (high-spin Fe3+). The pristine “combustion” sample contains a Fe3+ component having 7% of the total absorption spectrum, while the corresponding Fe3+ intensity for the “calcination” sample is 10%; these components are further discussed later in this section. Otherwise, the results from the fittings were within the margin of error for the two pristine samples. The Mössbauer spectra for the pristine samples in this work are comparable to the ones presented by Kim et al. where another synthesis procedure was utilized17 indicating the similar structural properties achieved using the herein investigated synthesis routes.
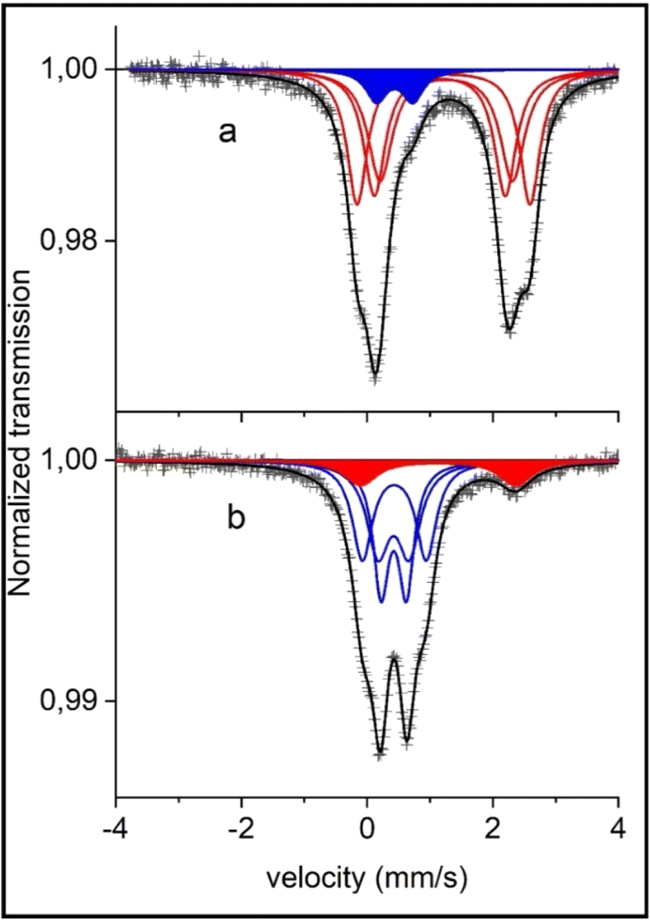 | ||
| Fig. 2 Room temperature 57Fe Mössbauer spectra of (a) combustion synthesized pristine sample, nominally Na4Fe3(PO4)2(P2O7) and (b) its de-sodiated sample NaFe3(PO4)2(P2O7). The red and blue marked resonance lines represent high spin Fe2+ and Fe3+ valences. The existence of minor components of Fe3+ in the pristine sample and of Fe2+ in the de-sodiated sample is discussed in the text. | ||
Fittings of the Fe2+ components in the pristine samples were carried out with the same spectral area for patterns X, Y and Z. These patterns may represent the three different equally occupied sites in the pristine Na4Fe3(PO4)2(P2O7) compound.23 The assignment of the patterns X, Y and Z to the three crystallographic sites Fe(1), Fe(2) and Fe(3) is challenging due to the non-resolved character of the spectra. With the present information of hyperfine parameters, we have to refrain from any such assignments. A neutron diffraction study and a first-principles calculation by Kim et al.23 indicated the same Biso factor for all sites, justifying the assumption of equal resonance areas in the Mössbauer spectra.
Another challenge affecting the fitting results and assignments is the possibility of the previously discussed impurity phase maricite (NaFePO4). The Mössbauer spectrum of pure NaFePO4 at room temperature, studied by Kosova et al.,53 provided a center shift (CS) = 1.20 mm s−1 and an electric quadrupole splitting (QS) = 2.19 mm s−1. These parameters would give a component which overlaps with the present absorption spectrum of Na4Fe3(PO4)2(P2O7) (CS = ∼1.2 and QS = ∼2.1–2.8). During de-sodiation the main peaks of the Na4Fe3(PO4)2(P2O7) that would overlap with NaFePO4 peaks are decreased. If there was a considerable amount of NaFePO4 then these peaks would become more visible (as NaFePO4 is inactive) and there is about 10% Fe2+ remaining in the de-sodiated sample. However, the Fe2+ Mössbauer parameters in the de-sodiated spectra (CS = 1.16, QS = 2.42) and especially the QS indicate that this is more likely the Fe atom in the pristine sample that has not been electrochemically active in the de-sodiation and thereby remains in the Fe2+ state. This is also later supported by electrochemical results as discussed later.
For the pristine samples, 7% Fe3+ in the “combustion” and 10% Fe3+ in the “calcination” synthesized NFPP are observed. The Mössbauer parameters of the Fe3+ in the de-sodiated samples are in good agreement with the fitted peaks for the Fe3+ component in the spectra of the pristine samples. Therefore, it is reasonable to consider these 7 and 10% not as an impurity, but rather as Na deficiencies of the idealized material. Hence the starting material would be Na3.79(6)Fe3(PO4)2(P2O7) for the 7% Fe3+ in the pristine “combustion” sample.
The atomic ratio of Na, Fe and P in NFPP synthesized with “calcination” and “combustion” was calculated through ICP where Na/Fe/P = 4.2:2.7:4 and 3.8:3.3:4, respectively. These results are consistent with Mössbauer and refinement findings that Na is deficient in the combustion synthesized sample whereas higher Na content of the calcination product may be related to the higher intensity of the maricite impurity. From CHNS analysis, carbon contents for “calcination” and “combustion” samples were found to be 9% and 5%, respectively. The lower carbon content of the “combustion” synthesized product can be attributed to the firing step under an air atmosphere. The content of carbon in the NFPP compounds synthesized by the sol–gel method reported in the literature can be as high as 8–16 wt% (Table S4†). However, it is crucial to control the carbon content to ensure appropriate electronic conductivity by forming a thin and dense carbon layer. High carbon contents are well known to result in a significant loss in energy density and, thereby not suitable for practical applications.36 Additionally, TGA of the NFPP final products synthesized by both methods was done under an air atmosphere to estimate the carbon contents by using the law of conservation of mass as shown in Fig. S1b.† The mass loss between 300 °C and 570 °C can be related to the amorphous carbon.15,25 The carbon content results for both methods are close to the results obtained by CHNS analysis.
Scanning electron microscopy (SEM) was performed to evaluate the effects of the calcination atmosphere and different synthesis methods on the morphology of NFPP. From Fig. S5,† it can be speculated that the choice of argon gas during calcination leads to incomplete conversion of Fe3+ to Fe2+ with the formation of Fe3+ precipitates on the surface of NFPP particles. These results are in line with Mössbauer findings (Fig. 2).
The NFPP synthesized by “calcination” crystallizes as highly agglomerated micron-sized particles with nanoporous architecture on the surface (Fig. 3a and b). Transmission electron microscopy (TEM) was used to evaluate the particle size and morphology. High-resolution transmission electron microscopy (HRTEM) was used to determine the thickness of the carbon coating and crystallinity. As shown in Fig. 3d, it can be seen that primary nanoparticles below 200 nm are not homogeneously distributed within the bulk structure. The corresponding energy dispersive X-ray spectroscopy (EDS) maps show that the carbon is distributed on the surface of secondary particles non-uniformly. Fig. 4c and S15† suggest that carbon particles or agglomerates are deposited on the particle surface rather than forming a uniform carbon coating. The particle growth and agglomeration of particles could be related to the low quality of the carbon coating (Fig. 3e). However, the combustion method leads to the formation of more uniform and smaller particle size and better nanoporous structure with a lower degree of agglomeration, as well as more uniform distribution of Na, Fe, P, O, and C, as shown in Fig. 4. Additionally, the development of a thin, dense, and uniform carbon cloth takes place due to the control of carbon content that may lead to improvement of the electrical conductivity. The size of the primary nanoparticles is below 100 nm with numerous mesopores and macropores with a size of 80–200 nm, as shown in Fig. S6 and S7.† The specific surface area of NFPP synthesized by “calcination” and “combustion” was determined by Brunauer–Emmet–Teller (BET) theory (Fig S16a and b†). The specific surface areas of “calcination” and “combustion” NFPP were found to be 34.67 and 35.82 m2 g−1, respectively. The Barrett–Joyner–Halenda (BJH) desorption average pore diameter of “calcination” and “combustion” samples are 14.94 and 14.71 nm, respectively. A larger surface area and smaller particle size can significantly facilitate the contact between the electrode and electrolytes shortening the diffusion pathway for Na+, thereby enhancing the electrochemical performance.54
 | ||
| Fig. 3 (a and b) SEM images of NFPP synthesized by “calcination”. (c and d) HAADF- and BF-STEM images and (e) HAADF-STEM image and corresponding EDS mapping results of the “calcination” sample. | ||
 | ||
| Fig. 4 (a and b) SEM images of NFPP synthesized by “combustion”. (c and d) HAADF- and BF-STEM images and (e) HAADF-STEM image and corresponding EDS mapping results of the “combustion” sample. | ||
3.2 Electrochemical performance
The electrochemical performances of NFPP cathodes synthesized by “calcination” and “combustion” were determined by galvanostatic charge–discharge tests in half-cells in the voltage range of 1.8–3.8 V at a current rate of 0.1C, see Fig. 5.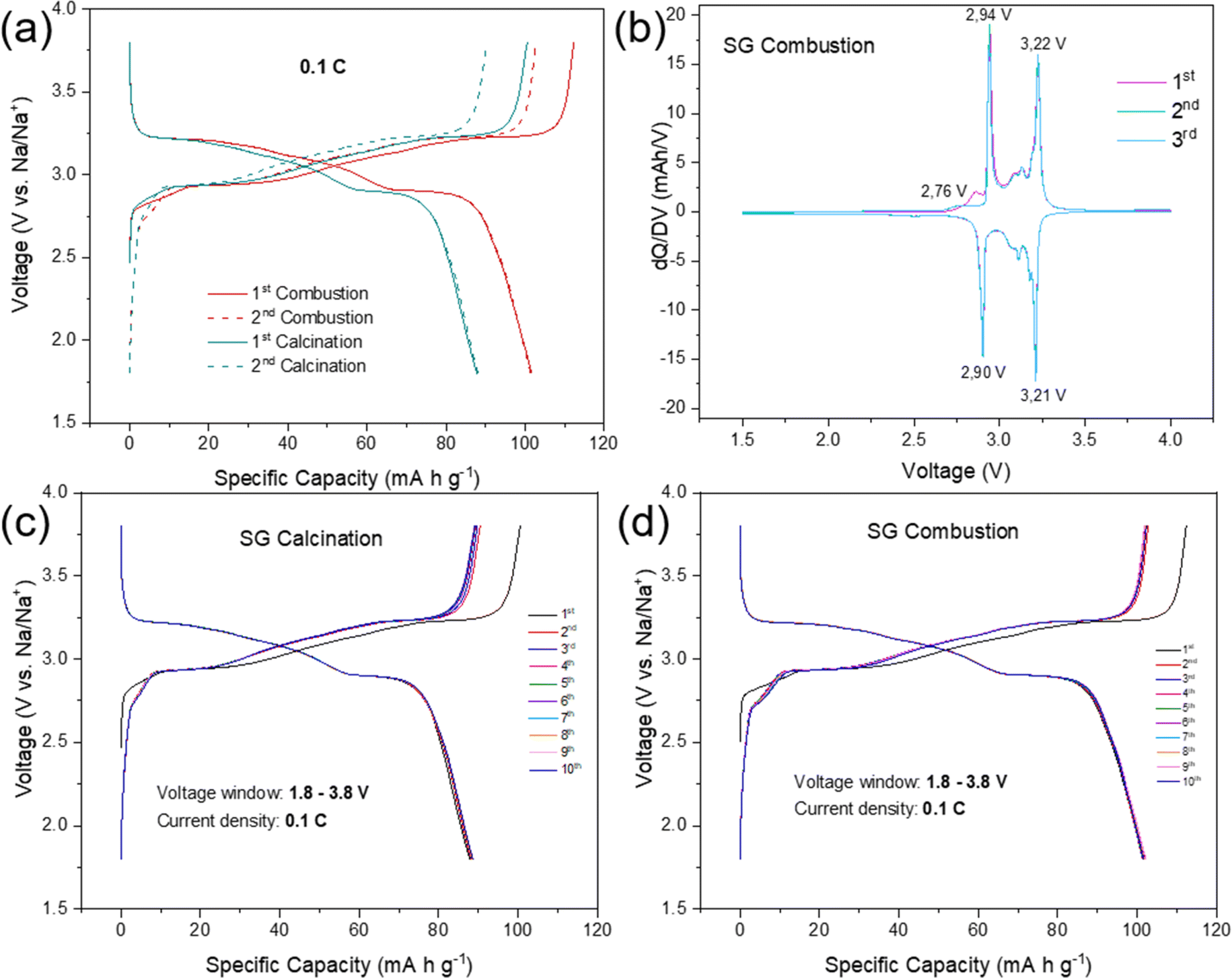 | ||
| Fig. 5 (a) The 1st and 2nd galvanostatic charge–discharge profiles in half-cells of “calcination” and “combustion” samples, (b) corresponding differential capacity vs. voltage (dQ/dV) plots for 3 cycles of the “combustion” sample, and (c) first ten charge–discharge curves of the “calcination” and (d) “combustion”. | ||
NFPP synthesized via “combustion” delivers a reversible capacity of 102 mA h g−1 showing more stable cycling performance whereas, the value remains around 88 mA h g−1 for the “calcination” NFPP cathode due to higher amount of the electrochemically inactive maricite impurity and amorphous carbon residues. The 1st cycle coulombic efficiency (CE) values for the “calcination” and “combustion” synthesized NFPPs were equal to 87% and 91%, respectively (Fig. S12†). After 100 cycles, the cell with NFPP “calcination” retains 98.7% of its initial capacity while the combustion process results in a slightly higher capacity retention of 99.7% as shown in Fig. 6a.
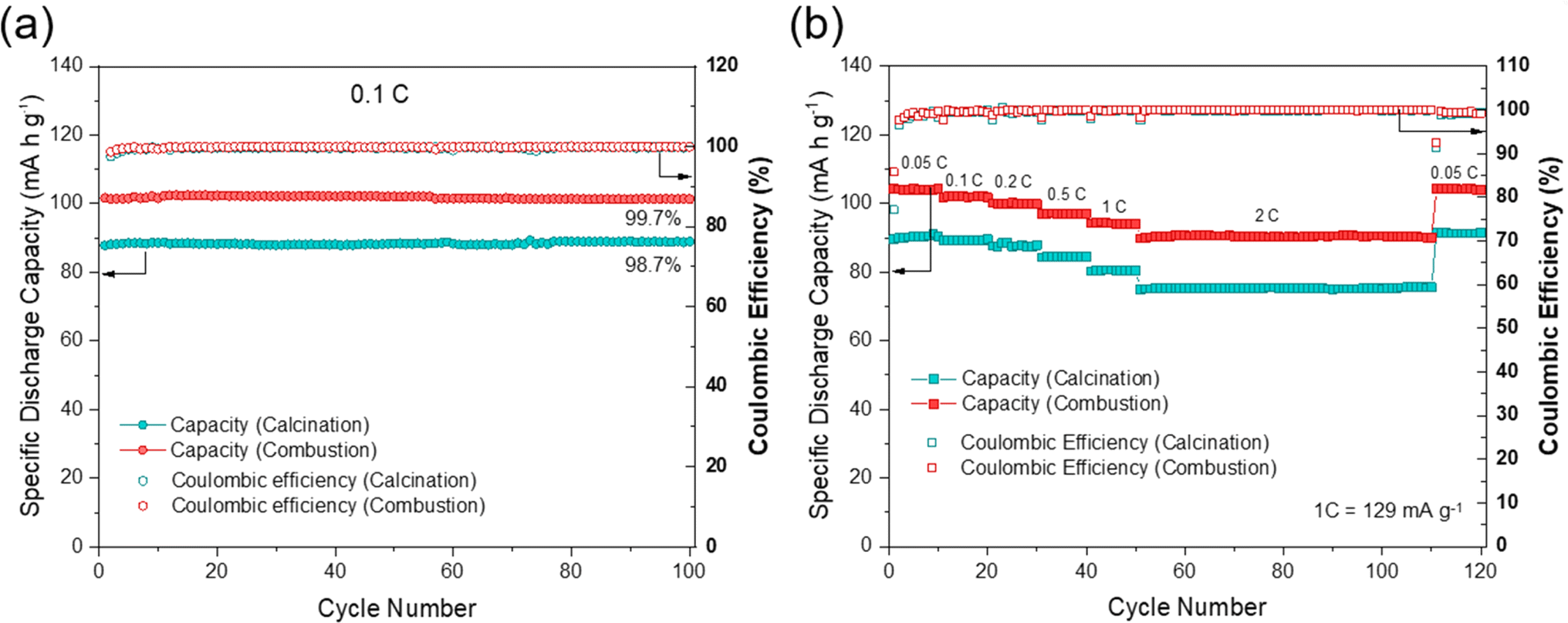 | ||
| Fig. 6 (a) Capacity retention at 0.1C and corresponding coulombic efficiencies for 100 cycles, (b) rate capability at various current rates from 0.05C to 2C of NFPP produced by “calcination” and “combustion” methods in half-cells. | ||
Fig. 6b shows the rate capability tests at different current rates. The “calcination”, provided discharge capacities of 91, 89, 87, 84, 80, and 75 mA h g−1, whereas the combustion sample delivered higher capacities of about 104, 102, 100, 97, 94, to 90 mA h g−1 at 0.05, 0.1, 0.2, 0.5, 1 and 2C rates, respectively. When the rate was changed back to C/20 at 111th cycle, the capacity increased to 104 mA h g−1 and 91 mA h g−1 for the combustion and calcination samples, respectively. The lower electrochemical performance of the “calcination” sample is likely due to the presence of irregular pores and large agglomerates which impede ionic diffusion (Fig. 3a and b).
The electrochemical activity of the material can also be monitored by comparing the Mössbauer spectra for the pristine “combustion” sample to the de-sodiated one. As can be seen in Fig. 2b and Table S3†, most of the Fe2+ are oxidized to Fe3+ during Na-extraction. After de-sodiation there is 10(2)% Fe2+ remaining in this sample. If this is a part of the starting material that was not possible to de-sodiate, it would mean that the sodium content is not 1 but rather 1.30(6) in this sample. Going from 3.79(6) Na (due to a Na-deficiency as explained earlier) to 1.30(6) means that 2.49(12) Na atoms have been extracted out of the theoretical maximum 3, meaning that 83(4)% (2.49/3 = 0.83) of the theoretical capacity has been extracted in the first de-sodiation. If this is now compared to the electrochemical de-sodiation results in the first cycle in Fig. 5a, where it is possible to extract ∼110 mA h g−1 from this sample. This corresponds to 85% of the theoretical capacity (110/129 = 0.85) and this is in good agreement with the 83(4)% suggested by the Mössbauer analysis and these results support the interpretation of the minor peaks in the Mössbauer spectra originating from the deviation of the Na content as compared to the idealized sample.
To evaluate the structure change, the ex situ XRD analysis of the pristine electrode, after the first charge and after the first discharge is carried out (Fig. S13†). It can be seen that the diffraction peak at 45.4° indexed to (424) planes of the pristine NFPP electrode shifts to a smaller angle during charging and returns to its original state after the first discharge indicating the structural stability and high reversibility of NFPP as a promising cathode material.
To further evaluate the practical application of high purity NFPP for SIBs, hard carbon (HC) was used as the anode to assemble full cells. The first five cycles of the HC anode in the voltage range of 0.001–2.5 V at a current density of 30 mA g−1 are shown in Fig. S14.† It delivers a reversible capacity of 281 mA h g−1 with an initial CE of 92%. As given in Fig. 7, the first three formation cycles of the NFPP//HC full-cell were performed at 0.05C followed by 40 cycles at 0.1C in a voltage window of 1.0–3.8 V. NFPP/HC full-cells were cycled with three different electrolytes to investigate the optimum cycling stability. The full-cell using 1 M NaPF6 in EC:DEC with 5 wt% FEC is the worst performing one showing a rapid capacity decay during the formation cycles (Fig. 7b). As shown in Fig. 7a, the cell using 1 M NaPF6 diglyme electrolyte exhibits its first discharge capacity around 80 mA h g−1 at 0.05C and then the C-rate increase to 0.1C; this value fades to 70 mA h g−1 after 10 cycles. Nevertheless, its long-term cycling stability in 1 M NaPF6 diglyme electrolyte was not as good as in half cell configuration. Therefore, cycling tests were performed by using 1 M NaPF6 in PC with the addition of 5 wt% FEC electrolyte as given in Fig. 7c and d. The first discharge capacity during formation cycles was around 80 mA h g−1 whereas 74 mA h g−1 of reversible discharge capacity is delivered at 0.1C with a capacity retention of 90% and a CE above 99.4% during all cycles (Fig. 7d). The average discharge voltage of the NFPP//HC full cell is 2.98 V with a maximum energy density of 219 mW h kg−1. It should also be noted the pre-sodiation of HC was not carried out to decrease the irreversible capacity loss.
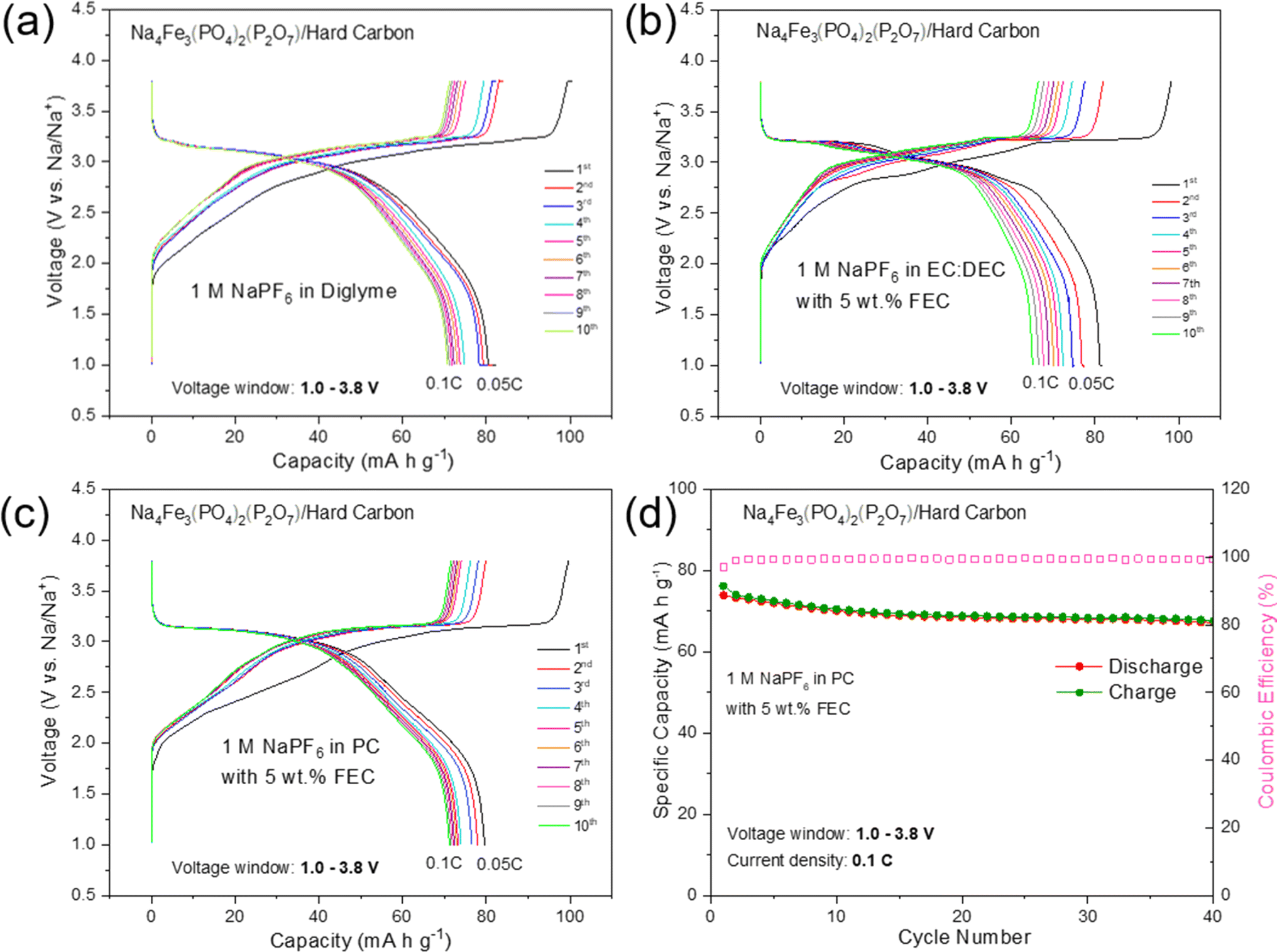 | ||
| Fig. 7 The charge/discharge profiles of NFPP//HC full-cells from 1.0 to 3.8 V at 0.1C cycled with (a) 1 M NaPF6 in diglyme electrolyte (b) 1 M NaPF6 in EC:DEC with 5 wt% FEC electrolyte, and (c) 1 M NaPF6 in PC with 5 wt% FEC electrolyte. (d) Cycling performance of the NFPP//HC full-cell using 1 M NaPF6 in PC with 5 wt% FEC electrolyte. | ||
The electrochemical performance of NFPP fabricated by “calcination” and “combustion” methods in this study was compared with state-of-the-art results reported in the literature, as summarized in Table S4.† The NFPP cathode synthesized via the “combustion” method exhibited a reversible discharge capacity of approximately 102 mA h g−1 at 0.1C, with an impressive capacity retention of 99.7% over 100 cycles. This performance surpasses that of NFPP cathodes modified with some specific carbon coatings, graphene, or nanotubes, which aim to improve electronic conductivity but may compromise the intrinsic properties and energy density of the material.11,15,24,25 The enhanced electrochemical performance of the “combustion” synthesized NFPP reported in this study can be attributed to the effective elimination of impurities and more homogeneous distribution of nanoparticles and pores within the structure. These improvements were realized through the combustion process in combination with the use of a reducing atmosphere during calcination. By addressing the intrinsic limitations of NFPP without obviously sacrificing its energy density, this study demonstrates a promising approach for the development of high-performance cathode materials for SIBs, paving the way for future developments in sustainable energy storage solutions.
4. Conclusions
High-purity NFPP was successfully synthesized by a novel “combustion” method followed by calcination under a reducing atmosphere. This approach effectively minimized the formation of maricite NaFePO4 and Na2FeP2O7 impurities, as confirmed by Rietveld refinement of XRD data. The trace amount of the maricite impurity was reduced to below 5 wt%, which is crucial for enhancing electrochemical performance. Mössbauer spectroscopy revealed that the use of the combustion method, instead of pre-calcination at 300 °C, resulted in the complete transformation of Fe3+ to Fe2+ in NFPP. SEM analysis further confirmed the nanoporous architecture of more uniform particles with the crystallite size below 100 nm.The galvanostatic charge–discharge cycling measurements in the voltage range of 1.8–3.8 V at different C-rates were carried out to evaluate the electrochemical performance of NFPP samples synthesized by the “combustion” and “calcination” methods. NFPP fabricated with the “calcination” method showed a first CE of 87%, while the CE increased up to 91% after performing combustion. The “combustion” synthesized NFPP delivered the highest reversible capacity of around 102 mA h g−1 at 0.1C with an impressive capacity retention of 99.7% after 100 cycles and outstanding rate capability, despite its lower carbon content compared with the “calcination” synthesized NFPP. Additionally, the ex situ XRD results also revealed the structural stability and high reversibility of the “combustion” synthesized NFPP, substantiating its superior cycling stability.
The development of this facile and scalable synthesis method for the environmentally friendly, low-cost, stable and high-purity NFPP cathode material presents a significant advancement in the field of SIBs. This study not only provides a deeper understanding of the structure–property relationships in NFPP cathodes but also offers valuable insights into the design and optimization of high-performance cathode materials for SIBs.
Data availability
Raw data for this article, including XRD, Mössbauer, FESEM, HRTEM, XPS, BET, etc., are available from the corresponding author Yaprak Subasi (E-mail: yaprak.subasi@kemi.uu.se). The data supporting this article have been included as part of the ESI.†Author contributions
Reza Younesi and Haidong Liu supervised the project and revised the manuscript. Yaprak Subasi fabricated and characterized the samples, performed electrochemical measurements, analyzed the data and wrote the manuscript. Laura Altenschmidt carried out the Rietveld refinement of XRD patterns of the samples and revised the manuscript. Fredrik Lindgren, Tore Ericsson, and Lennart Häggström performed Mössbauer spectroscopy and analyzed the data. Cheuk-Wai Tai conducted TEM measurements of the samples. All authors participated in the revision of the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the financial support from VINNOVA, the Swedish Innovation Agency, via project 2021-03735, from ÅForsk Foundation via grant no. 20-675, and from the Swedish Energy Agency (P2020-90112 and P2022-00055) and via StandUp for Energy. This work was performed, in part, at the Myfab-LIMS at Uppsala University and Electron Microscopy Centre, supported by the Department of Materials and Environmental Chemistry and Faculty of Science at Stockholm University, Sweden. Swedish Research Council and Swedish Foundation for Strategic Research are acknowledged for access to ARTEMI, the Swedish National Infrastructure in Advanced Electron Microscopy (2021-00171 and RIF21-0026) are acknowledged for allowing access to the electron microscopy facilities, respectively.References
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- K. M. Tan, T. S. Babu, V. K. Ramachandaramurthy, P. Kasinathan, S. G. Solanki and S. K. Raveendran, J. Energy Storage, 2021, 39, 102591 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- W. Yang, Q. Liu, Y. Zhao, D. Mu, G. Tan, H. Gao, L. Li, R. Chen and F. Wu, Small Methods, 2022, 6, 2200555 CrossRef CAS PubMed.
- N. Tapia-Ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain and Y. Chen, J. Phys.: Energy, 2021, 3, 031503 CAS.
- Y. Xiao, N. M. Abbasi, Y.-F. Zhu, S. Li, S.-J. Tan, W. Ling, L. Peng, T. Yang, L. Wang, X.-D. Guo, Y.-X. Yin, H. Zhang and Y.-G. Guo, Adv. Funct. Mater., 2020, 30, 2001334 CrossRef CAS.
- L. A. Ma, F. Massel, A. J. Naylor, L.-C. Duda and R. Younesi, Commun. Chem., 2019, 2, 125 CrossRef.
- W. R. Brant, R. Mogensen, S. Colbin, D. O. Ojwang, S. Schmid, L. Häggström, T. Ericsson, A. Jaworski, A. J. Pell and R. Younesi, Chem. Mater., 2019, 31, 7203–7211 CrossRef CAS.
- W. Wang, Y. Gang, J. Peng, Z. Hu, Z. Yan, W. Lai, Y. Zhu, D. Appadoo, M. Ye, Y. Cao, Q.-F. Gu, H.-K. Liu, S.-X. Dou and S.-L. Chou, Adv. Funct. Mater., 2022, 32, 2111727 CrossRef CAS.
- X. Wang, S. Roy, Q. Shi, Y. Li, Y. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9, 1938–1969 RSC.
- Y. Cao, C. Yang, Y. Liu, X. Xia, D. Zhao, Y. Cao, H. Yang, J. Zhang, J. Lu and Y. Xia, ACS Energy Lett., 2020, 5, 3788–3796 CrossRef CAS.
- P. Barpanda, L. Lander, S.-I. Nishimura and A. Yamada, Adv. Energy Mater., 2018, 8, 1703055 CrossRef.
- B. Senthilkumar, A. Rambabu, C. Murugesan, S. B. Krupanidhi and P. Barpanda, ACS Omega, 2020, 5, 7219–7224 CrossRef CAS PubMed.
- F. Sanz, C. Parada, J. M. Rojo and C. Ruíz-Valero, Chem. Mater., 2001, 13, 1334–1340 CrossRef CAS.
- T. Yuan, Y. Wang, J. Zhang, X. Pu, X. Ai, Z. Chen, H. Yang and Y. Cao, Nano Energy, 2019, 56, 160–168 CrossRef CAS.
- Y. Zhu, Y. Xu, Y. Liu, C. Luo and C. Wang, Nanoscale, 2013, 5, 780–787 RSC.
- H. Kim, I. Park, D.-H. Seo, S. Lee, S.-W. Kim, W. J. Kwon, Y.-U. Park, C. S. Kim, S. Jeon and K. Kang, J. Am. Chem. Soc., 2012, 134, 10369–10372 CrossRef CAS PubMed.
- X. Ma, X. Wu and P. Shen, ACS Appl. Energy Mater., 2018, 1, 6268–6278 CrossRef CAS.
- X. Wang, H. Li, W. Zhang, X. Ge, L. He, L. Zhang, S. Li, N. Wen, J. Guo, Y. Lai, S. Li and Z. Zhang, J. Mater. Chem. A, 2023, 11, 6978–6985 RSC.
- J. Gao, Y. Mei, L. Ni, H. Wang, B. Song, W. Deng, G. Zou, H. Hou and X. Ji, Inorg. Chem., 2023, 62, 9099–9110 CrossRef CAS PubMed.
- X. Ma, Z. Pan, X. Wu and P. K. Shen, Chem. Eng. J., 2019, 365, 132–141 CrossRef CAS.
- X. Wu, G. Zhong and Y. Yang, J. Power Sources, 2016, 327, 666–674 CrossRef CAS.
- H. Kim, I. Park, S. Lee, H. Kim, K.-Y. Park, Y.-U. Park, H. Kim, J. Kim, H.-D. Lim, W.-S. Yoon and K. Kang, Chem. Mater., 2013, 25, 3614–3622 CrossRef CAS.
- Y. Cao, X. Xia, Y. Liu, N. Wang, J. Zhang, D. Zhao and Y. Xia, J. Power Sources, 2020, 461, 228130 CrossRef CAS.
- X. Li, Y. Meng and D. Xiao, Chem.–Eur. J., 2023, 29, e202203381 CrossRef CAS PubMed.
- Q. Tao, H. Ding, X. Tang, K. Zhang, J. Teng, H. Zhao and J. Li, Energy Fuels, 2023, 37, 6230–6239 CrossRef CAS.
- A. J. Fernández-Ropero, M. Zarrabeitia, M. Reynaud, T. Rojo and M. Casas-Cabanas, J. Phys. Chem. C, 2018, 122, 133–142 CrossRef.
- B. Senthilkumar, C. Murugesan, K. Sada and P. Barpanda, J. Power Sources, 2020, 480, 228794 CrossRef CAS.
- H. Wang, Z. Pan, H. Zhang, C. Dong, Y. Ding, Y. Cao and Z. Chen, Small Methods, 2021, 5, 2100372 CrossRef CAS PubMed.
- Y. Li, Z. Zhou, X. P. Gao and J. Yan, J. Power Sources, 2006, 160, 633–637 CrossRef CAS.
- R. Gond, S. S. Meena, S. Yusuf, V. Shukla, N. K. Jena, R. Ahuja, S. Okada and P. Barpanda, Inorg. Chem., 2017, 56, 5918–5929 CrossRef CAS PubMed.
- B. Lin, S. Zhang and C. Deng, J. Mater. Chem. A, 2016, 4, 2550–2559 RSC.
- A. Gezović, M. Milović, D. Bajuk-Bogdanović, V. Grudić, R. Dominko, S. Mentus and M. J. Vujković, Electrochim. Acta, 2024, 476, 143718 CrossRef.
- X. Li, Y. Zhang, B. Zhang, K. Qin, H. Liu and Z.-F. Ma, J. Power Sources, 2022, 521, 230922 CrossRef CAS.
- P. Barpanda, T. Ye, S.-C. Chung, Y. Yamada, S.-I. Nishimura and A. Yamada, J. Mater. Chem., 2012, 22, 13455–13459 RSC.
- J. Zeng, D. Guan, W. Wang, X. Tan, Y. Cao, Z. Peng, G. Hu and K. Du, ACS Appl. Energy Mater., 2023, 6, 4238–4248 CrossRef CAS.
- R. Gond, S. S. Meena, S. M. Yusuf, V. Shukla, N. K. Jena, R. Ahuja, S. Okada and P. Barpanda, Inorg. Chem., 2017, 56, 5918–5929 CrossRef CAS PubMed.
- P. Barpanda, Y. Yamashita, Y. Yamada and A. Yamada, J. Electrochem. Soc., 2013, 160, A3095 CrossRef CAS.
- H. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- J. N. Bridson, S. E. Quinlan and P. R. Tremaine, Chem. Mater., 1998, 10, 763–768 CrossRef CAS.
- J. Gao, Y. Tian, Y. Mei, L. Ni, H. Wang, H. Liu, W. Deng, G. Zou, H. Hou and X. Ji, Chem. Eng. J., 2023, 458, 141385 CrossRef CAS.
- N. Jiang, X. Wang, H. Zhou, Y. Wang, S. Sun, C. Yang and Y. Liu, Small, 2024, 2308681 CrossRef CAS PubMed.
- H. Dai, Y. Xu, Y. Wang, F. Cheng, Q. Wang, C. Fang, J. Han and P. K. Chu, ACS Appl. Mater. Interfaces, 2024, 16, 7070–7079 CrossRef CAS PubMed.
- W. Ren, M. Qin, Y. Zhou, H. Zhou, J. Zhu, J. Pan, J. Zhou, X. Cao and S. Liang, Energy Storage Mater., 2023, 54, 776–783 CrossRef.
- J. Gao, H. Chen, Y. Mei, L. Ni, H. Wang, J. Huang, N. Hong, B. Song, Y. Tian, W. Deng, G. Zou, H. Hou and X. Ji, Nano Energy, 2023, 115, 108747 CrossRef CAS.
- T. Yuan, Y. Wang, J. Zhang, X. Pu, X. Ai, Z. Chen, H. Yang and Y. Cao, Nano Energy, 2019, 56, 160–168 CrossRef CAS.
- R. Zhan, Y. Zhang, H. Chen, Q. Xu, Q. Ma, W. Gao, T. Yang, J. Jiang, S. Bao and M. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 5107–5113 CrossRef CAS PubMed.
- H. Moriwake, A. Kuwabara, C. A. Fisher, M. Nose, H. Nakayama, S. Nakanishi, H. Iba and Y. Ikuhara, J. Power Sources, 2016, 326, 220–225 CrossRef CAS.
- X. Wang, Z. Feng, J. Huang, W. Deng, X. Li, H. Zhang and Z. Wen, Carbon, 2018, 127, 149–157 CrossRef CAS.
- A. S. Andersson, B. Kalska, L. Häggström and J. O. Thomas, Solid State Ionics, 2000, 130, 41–52 CrossRef CAS.
- N. V. Kosova and V. A. Belotserkovsky, Electrochim. Acta, 2018, 278, 182–195 CrossRef CAS.
- N. Kosova, V. Podugolnikov, E. Devyatkina and A. Slobodyuk, Mater. Res. Bull., 2014, 60, 849–857 CrossRef CAS.
- M. Liu, M. Li, B. Zhang, H. Li, J. Liang, X. Hu, H. Liu and Z.-F. Ma, ACS Sustain. Chem. Eng., 2023, 11, 18102–18111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03554b |
| This journal is © The Royal Society of Chemistry 2024 |