
Concentration quenching behavior of Stokes and upconversion luminescence for Pr3+-doped Y3Al5O12†
Yuuki
Kitagawa
 *a,
Hitomi
Nakamura
a and
Kenji
Shinozaki
ab
*a,
Hitomi
Nakamura
a and
Kenji
Shinozaki
ab
aNanomaterials Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan. E-mail: kitagawa.yuuki@aist.go.jp
bGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
First published on 15th October 2024
Abstract
Pr3+ ions exhibit Stokes and upconversion luminescence attributed to the 5d–4f interconfigurational and 4f–4f intraconfigurational transitions, whose intensity is sensitive to the Pr3+ concentration because of the interaction between neighboring Pr3+ ions. In this study, the Y3Al5O12 (YAG) ceramics doped with high Pr3+ concentrations (1–20%) were synthesized using the polymerized complex method, and their Stokes and upconversion luminescence properties were characterized. While the Stokes 5d–4f luminescence bands in the ultraviolet (UV) region were insensitive to the Pr3+ concentrations below 5%, the Stokes 4f–4f luminescence in the visible to near-infrared region was notably quenched because of the cross-relaxation of the 3P0 and 1D2 states. The YAG:Pr3+ samples exhibited UV upconversion luminescence in the UV region of 300–400 nm under high-power 455 nm blue laser diode illumination. The upconversion photoluminescence excitation spectra revealed two possible processes for Pr3+: 5d–4f upconversion luminescence, a spin-allowed two-photon absorption process via the intermediate 3PJ′ (J′ = 0, 1, and 2) states, and a multiphonon-relaxation-assisted process via the intermediate 1D2 state. The characterization of the Pr3+ concentration dependence revealed that the UV upconversion intensity was significantly affected by the cross-relaxation rates of the intermediate 3P0 and 1D2 states. In this study, it was demonstrated that compounds with low Pr3+ concentrations or long Pr3+–Pr3+ distances that lower the cross-relaxation rates are advantageous for the development of visible-to-UV upconverting materials activated with Pr3+.
1. Introduction
Pr3+ with a 4f2 electronic configuration, which is one of the lanthanoids, can take 13 2S+1LJ multiplet energy states, resulting in rich energy-level structures. The 4f–4f intraconfigurational transition exhibits numerous sharp luminescence lines in the blue-to-near-infrared (NIR) spectrum. In particular, the 1D2 → 3H4 transition causes sharp red luminescence lines at ∼610 nm, and is used as red (persistent) phosphors for LED applications, optical memories, anticounterfeiting ink, or in vivo imaging.1–6 In addition, because Pr3+ has a relatively low 4f15d1 excited state as well as Ce3+, broadband luminescence attributed to the 5d → 4f allowed interconfigurational transition is observed in some Pr3+-doped compounds. The energy gap between the ground 4f2 (3H4) and degenerated 4f15d1 states in the free Pr3+ ion is ∼61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 (= 7.63 eV).7,8 Due to the nephelauxetic effect by the coordinating anions and the crystal field effect, the lowest 4f15d1 excited level downshifts below the highest 1S0 level at ∼47000 cm−1 (= 5.82 eV),9,10 resulting in the ultraviolet (UV) Pr3+: 5d → 4f emission in fluoride and oxide host compounds.11–14 In particular, the Pr3+ ions in the strong crystal field, such as Pr3+-doped garnets, show UV-blue luminescence at 300–450 nm.15–17 In contrast to Ce3+, Pr3+ has rich 4f energy levels, located approximately half way between the ground 4f and excited 5d states, resulting in high energy 5d → 4f UV luminescence under blue or red laser excitation owing to the two-photon absorption via the intermediate 3P0 or 1D2 states.16,18–21 For the upconversion process, there are two major mechanisms:22 (1) the classical multistep excitation due to the excited state absorption (ESA), which is the photon absorption process from the intermediate excited state populated by one or more photons absorption to an even higher excited state; (2) the energy transfer upconversion (ETU), in which the neighboring Pr3+ ions with the populated excited states interact with each other, resulting in the higher excited state of one Pr3+ ion with two-photon energy by the other Pr3+ losing the energy and relaxing to the lower energy state (sometimes the ground state). In the case of Pr3+-doped compounds, both mechanisms have been reported based on spectroscopic analyses.16,19,23,24 Nevertheless, in either mechanism, the existence of intermediate 4f excited states and their lifetimes are essential parameters for determining the upconversion efficiency of Pr3+ 5d–4f luminescence. Broadband upconversion UV luminescence using a visible light source with a high power density can be applied to new devices, such as solar-blind markers,25,26 photocatalysis,27,28 and light disinfection or UV-bactericidal devices.29,30
500 cm−1 (= 7.63 eV).7,8 Due to the nephelauxetic effect by the coordinating anions and the crystal field effect, the lowest 4f15d1 excited level downshifts below the highest 1S0 level at ∼47000 cm−1 (= 5.82 eV),9,10 resulting in the ultraviolet (UV) Pr3+: 5d → 4f emission in fluoride and oxide host compounds.11–14 In particular, the Pr3+ ions in the strong crystal field, such as Pr3+-doped garnets, show UV-blue luminescence at 300–450 nm.15–17 In contrast to Ce3+, Pr3+ has rich 4f energy levels, located approximately half way between the ground 4f and excited 5d states, resulting in high energy 5d → 4f UV luminescence under blue or red laser excitation owing to the two-photon absorption via the intermediate 3P0 or 1D2 states.16,18–21 For the upconversion process, there are two major mechanisms:22 (1) the classical multistep excitation due to the excited state absorption (ESA), which is the photon absorption process from the intermediate excited state populated by one or more photons absorption to an even higher excited state; (2) the energy transfer upconversion (ETU), in which the neighboring Pr3+ ions with the populated excited states interact with each other, resulting in the higher excited state of one Pr3+ ion with two-photon energy by the other Pr3+ losing the energy and relaxing to the lower energy state (sometimes the ground state). In the case of Pr3+-doped compounds, both mechanisms have been reported based on spectroscopic analyses.16,19,23,24 Nevertheless, in either mechanism, the existence of intermediate 4f excited states and their lifetimes are essential parameters for determining the upconversion efficiency of Pr3+ 5d–4f luminescence. Broadband upconversion UV luminescence using a visible light source with a high power density can be applied to new devices, such as solar-blind markers,25,26 photocatalysis,27,28 and light disinfection or UV-bactericidal devices.29,30
The rich energy levels of Pr3+ can cause severe concentration quenching due to the interaction between adjacent or neighboring Pr3+ ions.31–33 With increasing Pr3+ concentration, the absorption coefficient of the 4f–4f transition and the population of the intermediate 3P0 and 1D2 states for the two-photon absorption increase; however, the quantum efficiency for Pr3+ luminescence decreases because of the cross-relaxation process in the Pr3+–Pr3+ pairs.33–35 Therefore, the Pr3+ concentration is a critical parameter that determines both the Stokes and upconversion luminescence properties, including the quantum efficiency, luminescence intensity, and radiative rate. In relation to Pr3+ upconversion luminescence, the high Pr3+ concentration causes populated intermediate 3P0 and 1D2 states and non-negligible cross-relaxation probabilities, which compete for the upconversion efficiency. The optimum Pr3+ concentration for upconversion UV luminescence depends on the structural and compositional features of the host compounds; for example, the maximum upconversion luminescence intensity was obtained at 1% for LiY9(SiO4)6O2:Pr3+,24 4% for X2-Y2SiO5:Pr3+,36 and 5% for Li2CaSiO4:Pr3+.37
In this study, Pr3+-doped Y3Al5O12 garnet (YAG) ceramics with high Pr3+ concentrations were prepared using a solution-based process. The luminescence properties of the YAG:Pr ceramics have been well examined, but the doping concentrations of Pr3+ ions in the previous studies were just 2% or below.38–41 Previously, we reported the structural and luminescence properties of YAG ceramics doped with super-high concentrations of Ce3+, up to 21% in the Y3+ sites.42 As the concentration of Ce3+ increased, the peak of the 5d–4f luminescence band shifted from 538 to 606 nm, which is possibly because of the dynamic effect of the vibrational mode arising from symmetry modulation at the dodecahedral coordination around the Ce3+ ions.43 It is interesting to observe whether a similar red shift in the 5d–4f luminescence bands is observed in a series of the Pr3+-doped YAG phosphors. We investigated the effect of high Pr3+ concentrations (1–20%) on the upconversion and Stokes luminescence properties regarding the electronic dynamics in the excited states.
2. Experimental procedure
2.1. Synthesis
A series of the Pr3+-doped garnet samples with a composition of (Y1−x/100Prx/100)3Al5O12 (x = 1, 2.5, 5, 10, and 20) were synthesized using a polymerized complex reaction (PCR) method.42,44 Hereafter, the Y3Al5O12 samples with the Pr3+ concentrations of x% are denoted as YAG:Prx (i.e., the 1% Pr3+-doped sample was represented to be “YAG:Pr1”). The starting chemicals of Y(NO3)3·6H2O (99.9%, Kishida Chemical), Al(NO3)3·9H2O (98+%, FUJIFILM Wako), Pr(NO3)3·6H2O (99.9%, Sigma-Aldrich), citric acid monohydrate (CA, 99.5%, Kishida Chemicaand), and ethylene glycol (EG, 99.5%, Kishida Chemical) were used. The metal nitrates were dissolved in 15 mL of distilled water, and CA (1.5 g) and EG (5 mL) were added. The mixed solution was heated at 90 °C for 20 h in an oil bath for gelation. The obtained pale light green transparent gel was heated at 650 °C for three hours and then at 750 °C for three hours in the air atmosphere to allow the combustion of organic substances. The obtained pale light green amorphous precursors were sintered at 1040 °C for six hours in the air atmosphere.2.2. Characterization
The crystalline phase of the prepared YAG:Pr3+ samples was examined through X-ray diffraction (XRD) with a diffractometer (Ultima IV, Rigaku). As a reference, the simulated diffraction pattern of the cubic Y3Al5O12 structure45 was obtained using VESTA.46 The Rietveld refinements for the measured XRD patterns were performed using the RIETAN-FP program.47 Absorption spectra were measured with a UV-visible/NIR spectrophotometer (UH4150, Hitachi High-Tech) equipped with an integrating sphere coated by BaSO4, using the diffuse reflection method. The photoluminescence (PL) and photoluminescence excitation (PLE) spectra were measured using a spectrofluorometer (Fluorolog-3, Horiba). The nanosecond-order luminescence decay measurements at low temperatures (T = 4–300 K) were performed with a Fluorolog-3 spectrofluorometer system using the time-correlated single-photon counting method under excitation with a 300 nm nano-second LED (NanoLED, Horiba). The sample temperature was controlled using a cryostat equipped with a closed-cycle He gas cryogenic refrigerator (Pascal-L101Q-4, Pascal Co., Ltd). The μs-order luminescence decay measurements were performed with a setup consisting of a 455 nm fiber-coupled laser diode (LD) (LSR455SD-4W-FC, CivilLaser), 490 nm and 610 nm bandpass filters with optical density ODabs > 5 and full-width half-maximum bandwidth of 10 nm (FBH490-10 and FBH610-10, Thorlabs), a photomultiplier tube detector (R10699, Hamamatsu Photonics), and an oscilloscope (WaveSurfer 4104HD, Teledyne-LeCroy). A function generator (33120A, Hewlett Packard) generated the pulse signal of the blue LD. The upconversion-PL (UC-PL) and Stokes PL spectra were obtained using a multichannel CCD spectrometer (QEPro, Ocean Optics) connected to an optical fiber under blue LD excitation. The high-power LD excitation light was cut using a UV short-pass (260–390 nm) filter (SWX DUV filter, Asahi Spectra), which has a transparency >10% in the near-infrared region over 750 nm. A tunable supercontinuum laser (SuperK CHROMATUNE, NKT Photonics) was used as the excitation source for the upconversion-PLE (UC-PLE) measurements, whose output optical power was maintained at 1.25 mW in the range 420–680 nm. The UC-PL and Stokes PL intensities obtained at each wavelength were corrected using a calibrated tungsten halogen light source (HL-2000, Ocean Optics) over 350 nm and a calibrated deuterium lamp (SL3, StellarNet) below 350 nm.3. Results and discussion
3.1. Structural analysis
Fig. 1(a) shows the XRD patterns of the YAG:Pr3+ samples with various Pr3+ concentrations of 1, 2.5, 5, 10, and 20%, which were synthesized using the PCR method. All the samples exhibited the single-phase diffraction patterns of the cubic Y3Al5O12 (space group Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d, ICSD No. 41144) without any impurity phases, regardless of the high Pr3+ concentration of 20%. Fig. 1(b) shows the enlarged XRD patterns in the 2θ = 39.4–44° range, in which the observed two peaks are indexed to the 125 and 044 reflections, respectively. With an increasing Pr3+ concentration, all the diffraction peaks linearly shifted to the low-angle side, indicating that the cubic lattice of the YAG:Pr3+ samples expanded. Because the ionic radius of Pr3+ in the eight-fold coordination is larger (1.126 Å) than that of Y3+ (1.019 Å),48 this expansion suggested that the Pr3+ ions are incorporated into the YO8 dodecahedral sites in the YAG lattice. The lattice parameters of the YAG:Pr3+ samples were refined through the Rietveld method, resulting in the lattice constants a of 12.0107(3) Å for YAG:Pr1, 12.0280(4) Å for YAG:Pr2.5, 12.0503(3) Å for YAG:Pr5, 12.0490(2) Å for YAG:Pr10, and 12.0888(4) Å for YAG:Pr20. The other results of the Rietveld refinement are summarized in Fig. S1 and Table S1 (ESI†). The rate of change in the lattice parameter a with Pr3+ concentrations (4.11 × 10−3 Å per Pr3+%) was consistent with those in our previous studies about YAG ceramics with Ce3+ concentrations of 0.6–21% (4.24 × 10−3 Å per Ce3+%).42 Although the YAG:Pr10 sample exhibited a relatively small value, the following results related to the PL intensity suggest that the amount of Pr3+ ions in the YAG:Pr10 sample is between those of the YAG:Pr5 and YAG:Pr20 samples.
d, ICSD No. 41144) without any impurity phases, regardless of the high Pr3+ concentration of 20%. Fig. 1(b) shows the enlarged XRD patterns in the 2θ = 39.4–44° range, in which the observed two peaks are indexed to the 125 and 044 reflections, respectively. With an increasing Pr3+ concentration, all the diffraction peaks linearly shifted to the low-angle side, indicating that the cubic lattice of the YAG:Pr3+ samples expanded. Because the ionic radius of Pr3+ in the eight-fold coordination is larger (1.126 Å) than that of Y3+ (1.019 Å),48 this expansion suggested that the Pr3+ ions are incorporated into the YO8 dodecahedral sites in the YAG lattice. The lattice parameters of the YAG:Pr3+ samples were refined through the Rietveld method, resulting in the lattice constants a of 12.0107(3) Å for YAG:Pr1, 12.0280(4) Å for YAG:Pr2.5, 12.0503(3) Å for YAG:Pr5, 12.0490(2) Å for YAG:Pr10, and 12.0888(4) Å for YAG:Pr20. The other results of the Rietveld refinement are summarized in Fig. S1 and Table S1 (ESI†). The rate of change in the lattice parameter a with Pr3+ concentrations (4.11 × 10−3 Å per Pr3+%) was consistent with those in our previous studies about YAG ceramics with Ce3+ concentrations of 0.6–21% (4.24 × 10−3 Å per Ce3+%).42 Although the YAG:Pr10 sample exhibited a relatively small value, the following results related to the PL intensity suggest that the amount of Pr3+ ions in the YAG:Pr10 sample is between those of the YAG:Pr5 and YAG:Pr20 samples.
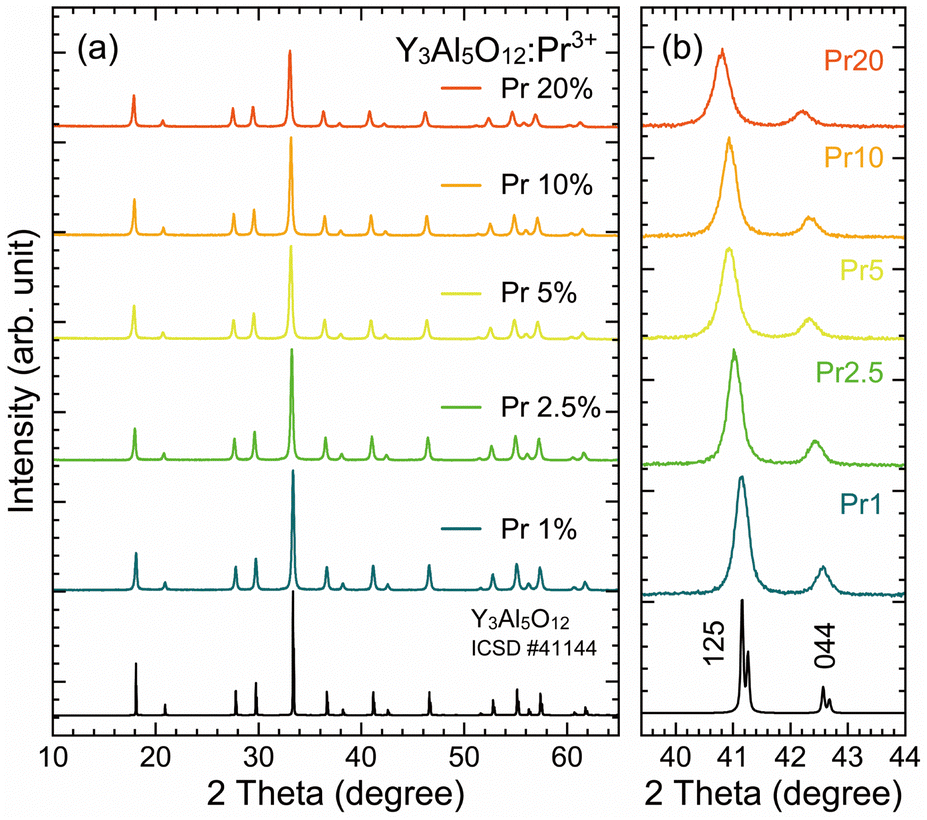 | ||
| Fig. 1 (a) XRD patterns of the prepared YAG:Pr3+ samples with various Pr3+ concentrations of 1–20%, shown with the reference pattern of Y3Al5O12 (ICSD no. 41144). (b) Enlarged XRD patterns in the range of 2θ = 39.4–44°. | ||
3.2. Absorption properties
Fig. 2(a) shows the absorption spectra of the YAG:Pr3+ samples measured using the diffuse reflection method. The measured diffuse reflectance R values were converted to the Kubelka–Munk function (F(R) = (1 − R)2/2R), which is proportional to the absorption coefficients. All the samples exhibited sharp lines in the visible region over 440 nm and a broad band below 320 nm, whose absorption intensity was proportional to the Pr3+ concentration. The sharp lines are attributed to the Pr3+: 4f–4f transition from ground 3H4 to excited 3PJ (J = 0, 1, and 2) and 1I6 (λ = 440–490 nm) or 1D2 (λ = 580–620 nm) states. The broad absorption band in the UV region is attributed to the 5d1 ← 4f (3H4) parity allowed transition. The additional absorption band attributed to the 5d2 ← 4f transition can appear below 250 nm,41,49 but could not be observed due to the limitation of the experimental setup. The YAG:Pr1 sample exhibits a weak 5d ← 4f absorption band because of the relatively low Pr3+ concentration. Although the spectral shapes were not significantly different regardless of the Pr3+ concentrations, the 5d ← 4f absorption band was slightly blue-shifted from 296.8 nm for the YAG:Pr2.5 sample to 291.4 nm for the YAG:Pr20 sample.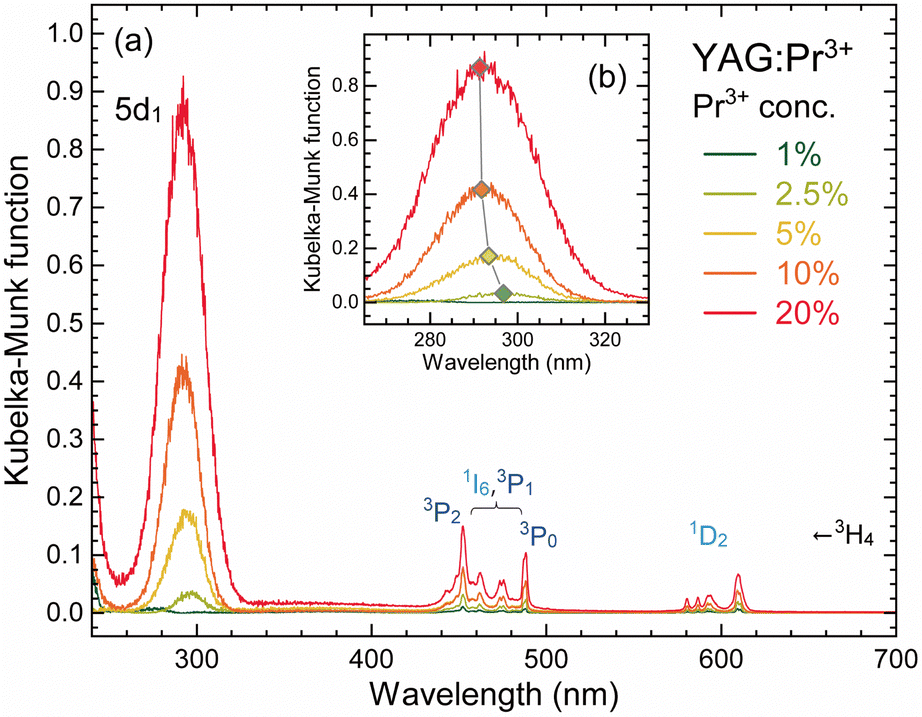 | ||
| Fig. 2 (a) Absorption spectra of the YAG:Pr3+ samples (Pr3+: 1–20%). The y-axes were converted to the Kubelka–Munk functions, which are proportional to the absorption coefficients. (b) Enlarged absorption spectra in the range of 260–330 nm. | ||
3.3. Stokes photoluminescence properties
Fig. 3(a) shows the PL spectra of the YAG:Pr3+ samples with various Pr3+ concentrations, which were measured at room temperature under the 5d ← 4f excitation (λex = 280 nm). All YAG:Pr3+ samples exhibited similar spectral features except for the PL intensities, and their PL spectra could be divided into two regions: over and below 460 nm. Over 460 nm, numerous sharp lines were observed over a wide range of up to 760 nm, which were attributed to the Pr3+:4f–4f transition from the 3P0 and 1D2 excited states. The assignments of each 4f–4f transition are shown in Fig. 3(a). Despite the excitation through the 5d1 ← 4f transition, the 3P0 and 1D2 states were fed through intersystem crossing via the 4f15d1 state.2,33,41 Because the maximum phonon energy of YAG is ∼850 cm−1,50–52 which can bridge the energy gap between the 3P0 and 1D2 levels (∼3750 cm−1) with four or five phonons emissions, the 1D2 state is fed via the multiphonon relaxation (MPR) from the 3P0 level,33 resulting in the relatively intense PL emission attributed to the 1D2 → 3H4 transition for the YAG:Pr1 sample with the lowest Pr3+ concentration. Although the PL intensity of the radiative transitions from both 3P0 and 1D2 decreased with an increasing of the Pr3+ concentration above 1%, their quenching behaviors with the Pr3+ concentration increase were different. The integrated PL intensities of the 3P0 → 3H4 transition (λem = 470–520 nm) and 1D2 → 3H4 transition (λem = 607–614 nm) are plotted as a function of the Pr3+ concentrations in Fig. 3(b), in which the PL intensities were normalized by that of the YAG:Pr1 sample. The nonradiative relaxation pathway for the concentration quenching of Pr3+ ions is the phonon-assisted cross-relaxation process,34,35 which involves energy transfer between neighboring Pr3+ pairs. Because the energy gaps related to the cross-relaxation process are not completely resonant,53 excess or insufficient energy is balanced by phonon emission. Possible cross-relaxation processes for the 3P0 and 1D2 excited states are summarized in Table 1. Because each 2S+1LJ energy level is split into 2J + 1 sublevels due to the crystal field effect (i.e., Stark splitting), the energy gaps between the 2S+1LJ multiplets involved in cross-relaxation cannot be expressed as a single value. The energy gaps listed in Table 1 were calculated by averaging all the energy gaps between the Stark sublevels of the initial and terminal states. The value of ΔE indicates the energy difference between the downward and upward transitions in the relevant cross-relaxation process. For the 3P0 state, the dominant cross-relaxation process can be [3P0, 3H4] → [1D2, 3H6] or [3P0, 3H4] → [3H6, 1D2] because the [3P0, 3H4] → [1G4, 1G4] process hardly occurs owing to the relatively large ΔE of 833 cm−1. Although both processes feed the 1D2 state, the PL intensity attributed to the 1D2 → 3H4 transition decreased significantly with increasing Pr3+ concentration. The cross-relaxation process for the 1D2 state is the near-resonant [1D2, 3H4] → [1G4, 3H6] and [1D2, 3H4] → [3F4, 1G4] processes, which are more efficient than the 3P0 cross-relaxation process because their ΔE values are relatively small (103 cm−1).33,34 In addition, the [1D2, 3H4] → [1G4, 3H6] cross-relaxation can be principal among all cross-relaxation processes because both downward and upward transitions are spin-allowed, resulting in the severe concentration quenching in the 1D2 → 3H4 luminescence, as shown in Fig. 3(b). The relative PL intensity of the 1D2 → 3H4 transition was higher than that of the 3P0 → 3H4 transition only for the YAG:Pr1 sample.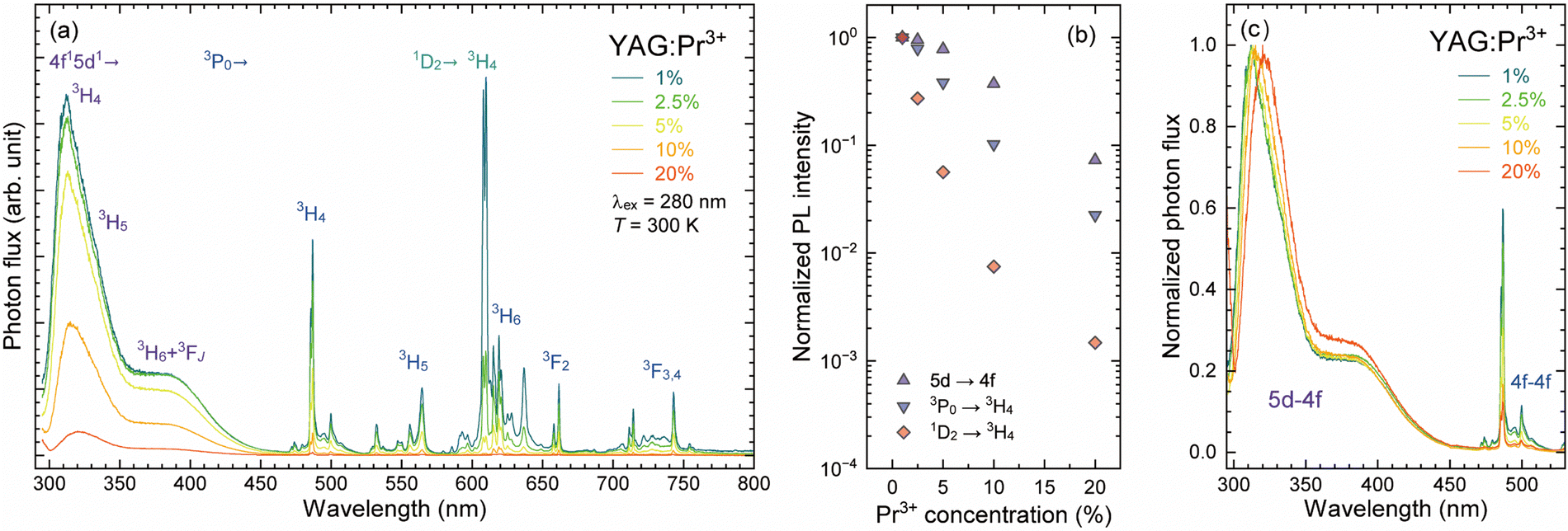 | ||
| Fig. 3 (a) PL spectra of the YAG:Pr3+ samples with different Pr3+ concentrations (1–20%), measured at room temperature. The samples were excited via the 5d–4f (λex = 280 nm) transition. (b) Variation of integrated PL intensity of the YAG:Pr3+ samples as a function of Pr3+ concentrations (1–20%), which was normalized by the intensity of the YAG:Pr1 sample. (c) Normalized PL spectra by the peak intensity of the 5d → 4f (3H4) luminescence band in 295–530 nm. | ||
| Downward (DW) transition | Upward (UP) transition | Energy gapa [cm−1] | ||||
|---|---|---|---|---|---|---|
| Initial state | Terminal state | Initial state | Terminal state | DWb | UPb | ΔE |
| a Energy gaps were estimated from the experimental values for YAG:Pr3+ reported in ref. 53, in which Stark splitting in D2 symmetry was considered. b The average energy gaps for the downward (DW) and upward (UP) transitions of the cross-relaxation process were calculated by considering the transition energies between all Stark sublevels. | ||||||
| 3P0 | 1D2 | 3H4 | 3H6 | 3755 | 4210 | 455 |
| 3P0 | 1G4 | 3H4 | 1G4 | 10483 |
9650 | 833 |
| 3P0 | 3H6 | 3H4 | 1D2 | 15923 |
16378 |
455 |
| 1D2 | 1G4 | 3H4 | 3F4 | 6728 | 6831 | 103 |
| 1D2 | 3F4 | 3H4 | 1G4 | 9547 | 9650 | 103 |
Below 460 nm, broad luminescence bands attributed to the 4f15d1 → 4f2 allowed transition were observed, which ranged from 300 nm to 460 nm because of the multiple terminal 4f states of 3H4, 3H5, 3H6, 3F3, and 3F4. As well as the 4f–4f transition, the PL intensity of the 5d → 4f transition decreased with increasing Pr3+ concentration over 1%. The variation of PL intensity is also plotted in Fig. 3(b), indicating that the 5d → 4f transition was less sensitive towards the concentration quenching than the 4f–4f transition. In the YAG:Pr10 sample, the intensity of the 3P0 → 3H4 and 1D2 → 3H4 transitions decreased to less than 10% and 1% of their initial intensity (the YAG:Pr1 sample), yet that of the 5d → 4f transition only decreased to ∼40% of its initial intensity. Despite the 5d ← 4f absorption coefficient increasing from 1 to 2.5%, as shown in Fig. 2, the PL intensities of the YAG:Pr1 and YAG:Pr2.5 samples were equivalent, indicating that the concentration quenching occurred over 1% of Pr3+ doping accompanied by a given nonradiative transition. The normalized PL spectra with the peak intensity of the 5d → 4f (3H4) transition are shown in Fig. 3(c). With the increase in Pr3+ concentration, the 5d → 4f luminescence bands appeared to be red-shifted, and the relative intensity of the 5d → 3H6 or 3F3,4 transition was enhanced. Because it is unlikely that the transition probability of the specific terminal states will increase significantly, this can be due to the self-absorption by the strong 5d ← 4f (3H4) transition with high Pr3+ concentrations.
Fig. 4 shows the PLE spectra of the YAG:Pr samples monitoring the 3P0 → 3F2 emission at 661 nm. Two types of characteristic excitation bands were observed in the PLE spectra, as well as in the PL spectra, which were attributed to the 4f–4f transition over 430 nm and the 5d–4f transition below 310 nm. In 430–500 nm, the numerous excitation bands attributed to the 3PJ′ ← 3H4 (J′ = 0, 1, 2) and 1I6 ← 3H4 transitions overlapped, leading to difficulty in identifying the peaks. All these excitation bands decay monotonously with the Pr3+ concentration. Whereas the 3P0 state has a higher energy (20561 cm−1) than the 1D2 state (16437–17271 cm−1), the 1D2 ← 3H4 transition can pump the 3P0 state via the ETU process between the Pr3+–Pr3+ pair; [1D2, 1D2] → [3P2, 1G4].54,55 Despite the energy mismatch between the 3P2–1D2 and 1D2–1G4 energy gaps, the upconverted 3P0 emission was observed in previous reports of YAG:Pr3+ with low Pr3+ concentrations.55,56 Over 10% of Pr3+ concentration, the 1D2 ← 3H4 excitation bands disappeared because the probability of the competing cross-relaxation process for the 1D2 state exceeded the ETU probability. Below 310 nm, the broad 5d1 ← 4f (3H4) excitation bands were observed. As observed in the absorption spectra, the peaks of the excitation band were slightly blue-shifted with increasing Pr3+ concentration, possibly because of the weaker crystal field of Pr3+-doped YAG compared to that of undoped YAG.
 | ||
| Fig. 4 PLE spectra of the YAG:Pr3+ samples at room temperature with the monitored wavelength of λem = 661 nm (3F2 → 3H4 transition). | ||
The PL spectra of the YAG:Pr3+ manifested that the Pr3+: 5d–4f and 4f–4f luminescence was significantly affected by the concentration quenching over 1% of Pr3+ doping. However, the spectral intensities do not directly reflect the luminescence efficiency, because the variation in the absorption coefficient with increasing Pr3+ concentration is ignored. The influence of the Pr3+ concentration on the luminescence efficiency was investigated using time-resolved spectroscopy. The 4f orbitals of lanthanoid ions are insensitive to crossover quenching between the potential curves due to the small electron–phonon coupling. The dominant quenching route for Pr3+: 4f–4f luminescence is a temperature-dependent energy transfer (ET) process, such as the MPR and cross-relaxation processes.31,57 Because the rates of these temperature-dependent quenching processes do not seem to correlate with the Pr3+ concentration, the 4f luminescence lifetimes of the YAG:Pr3+ samples were characterized at room temperature, and the degrees of increase in the nonradiative rate related to concentration quenching among the samples were compared. Note that the luminescence lifetimes at room temperature of the 3P0 → 3H4 and 1D2 → 3H4 transitions decrease from those at 4 K.31,51
Fig. 5(a) and (b) show the luminescence decay curves of the Pr3+: 3P0 → 3H4 luminescence (λem = 490 nm) and 1D2 → 3H4 luminescence (λem = 610 nm) of the YAG:Pr3+ samples at room temperature, respectively. The decay curve of the 3P0 luminescence of the YAG:Pr1 sample is represented by a single-exponential function. As the Pr3+ concentration increased, the shape of the decay curves changed to a non-exponential shape, indicating the existence of an ET cross-relaxation process for the 3P0 state. If a given ET process is non-negligible, the decay curves for the Pr3+: 4f–4f luminescence are sometimes fitted with the Inokuti–Hirayama function, which does not consider the diffusion and energy migration processes.38,39,55,58 The aim of this study is not to elucidate which interaction is dominant in the ET process, but to investigate how the 4f–4f luminescence intensity decreases with increasing Pr3+ concentration. The 4f–4f decay curves were fitted using the following two-component exponential function:
 | (1) |
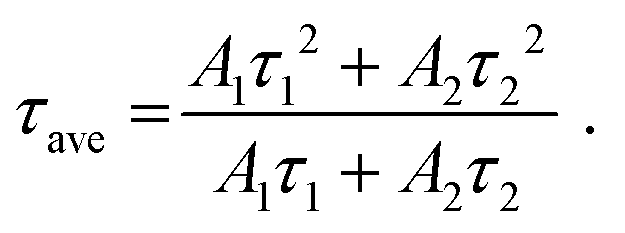 | (2) |
 | ||
| Fig. 5 Luminescence decay curves of the (a) 3P0 → 3H4 (λem = 490 nm) and (b) 1D2 → 3H4 (λem = 610 nm) transitions, measured at room temperature. | ||
The estimated luminescence lifetimes of the 3P0 states of the YAG:Pr3+ samples are summarized in Table 2. The τave for the YAG:Pr1 sample is 7.77 μs, consistent with the 3P0 lifetimes reported in the previous studies.16,51,59,60 For the 1% Pr3+ doping, the influence of the concentration quenching was not significant. With increasing Pr3+ concentrations, the 3P0 lifetime gradually decreased.
| Pr3+ concentration [%] | 1 | 2.5 | 5 | 10 | 20 |
|---|---|---|---|---|---|
| 3P0 | 7.77 | 5.96 | 4.21 | 2.66 | 0.943 |
| 1D2 | 129 | 46.8 | 15.6 | 3.74 | 0.409 |
In contrast, the shape of the decay curve for the 1D2 luminescence in the YAG:Pr1 sample was non-exponential, derived from the significant cross-relaxation process in the 1D2 state between the neighboring Pr3+ ions. The YAG:Pr1 sample also exhibits a slow rise, which overlaps with the decay of the laser pulse because of the feeding process from the 3P0 states through the MPR process. With increasing Pr3+ concentration, the shape of the decay curves was distorted and the decay time was shortened. The 1D2 lifetimes, estimated using eqn (1) and (2) are summarized in Table 2. The 1D2 luminescence lifetime τave of the YAG:Pr1 sample, in which the cross-relaxation rate was smallest among the YAG:Pr3+ samples, was estimated to be 129 μs, which also agreed with the reported lifetimes.16,51,59,60 Nevertheless, the 1D2 state exhibited a relatively long lifetime. Considering that the intermediate states of Pr3+ upconversion luminescence are the 3P0 and 1D2 states, the long lifetime of the 1D2 state is expected to increase its population, resulting in a high probability of two-photon absorption by ESA or ETU. With increasing Pr3+ concentration, the 1D2 luminescence lifetime drastically decreases, and above 5% Pr3+ doping, the 1D2 and 3P0 lifetimes remain almost the same. Time-resolved spectroscopy indicated that the 1D2 state is susceptible to concentration quenching due to its high cross-relaxation probability.
In contrast, because the 5d excited states are easily affected by the electron–phonon coupling, the activation energy for the thermal quenching of the 5d–4f transition depends on the chemical compositions of the host compounds and the local environments around the luminescence center. Because the reported quenching temperature of YAG:Pr3+, at which the PL intensity or lifetime is half of the initial value at low temperatures, is 320–340 K,40,41 it is difficult to separate the temperature- and concentration-dependent effects on the luminescence decay curves obtained at room temperature. Thus, the luminescence decay curves at low temperatures (T = 4–300 K) were characterized for samples with different Pr3+ concentrations, and the concentration quenching properties of the 5d excited states in YAG:Pr3+ were investigated.
Fig. 6(a) shows the luminescence decay curves for the YAG:Pr3+ samples, which were measured at 4 K. At Pr3+ concentrations of 5% or less, the shape of the decay curves can be expressed with a single-exponential function. In contrast, in the YAG:Pr3+ samples with Pr3+ concentrations over 10%, the shape varied into a non-exponential shape, indicating the existence of a given ET process. A possible ET process is the interaction between adjacent Pr3+ ions, which leads to severe concentration quenching of the 5d–4f luminescence. The average luminescence lifetimes at low temperatures were estimated by fitting the measured decay curves to the two-component exponential functions. Although fitting with a single-exponential function is ideal, the deviation from the single-exponential function is only minor in most cases.61 The initial rising part of the decay curves was analyzed with the convolution of the instrument response function. The decay curves with the fitting results are shown in Fig. S2 (ESI†). The lifetimes obtained for the YAG:Pr3+ samples are plotted as a function of temperature. In the YAG host, the 5d excited states of Pr3+ are relaxed via the thermal activation crossover process because they are significantly affected by the electron–phonon coupling.62 Therefore, these plots were fitted with the single barrier quenching model described below:41
 | (3) |
 | ||
| Fig. 6 (a) Luminescence decay curves of the 5d → 4f transition (λex = 300 nm, λem = 330 nm) for the YAG:Pr3+ samples at 4 K. (b) Temperature dependence of the 5d → 4f luminescence lifetimes for the YAG:Pr3+ samples, estimated by the exponential function in eqn (1) and (2). The depicted curves represent the fitting results with the single barrier quenching model in eqn (3). (c) Configurational coordinate diagram of Pr3+ 4f2 and 4f15d1 states for YAG:Pr3+. | ||
3.4. Upconversion properties
According to the results of the PL measurements, the YAG:Pr1 sample had the highest quantum efficiency for the Pr3+: 4f–4f and 5d–4f transitions among the prepared samples. Before characterizing the concentration dependence, the upconversion luminescence properties of the YAG:Pr1 sample were examined. Fig. 7(a) shows the UC-PL spectra of the YAG:Pr1 sample under various pump power densities using a 455 nm blue LD. By illuminating excitation light with pump power densities of 20–70 W cm−2, the broad 5d → 4f luminescence bands were observed below 400 nm. Note that the tail of the 5d → 4f luminescence bands over 400 nm was absent because of the optical filter used for cutting the excitation light. The 4f–4f luminescence bands were observed in the range over 720 nm, which are assigned to the 3P0 → 3F3,4 (720–760 nm), 1D2 → 3H6 (800–880 nm), and 3P0 → 1G4 (880–950 nm) transitions. Fig. 7(b) shows the variation in the UC-PL and Stokes PL intensities with the laser optical power. The UC-PL intensity is proportional to the nth power of the pumping power, in which n is approximately equal to the number of pump photons required in the upper emitting levels.22,63 Compared to the Stokes PL intensity with slopes of almost unity, the UC-PL intensity under 20–30 W cm−2 excitation illumination had a slope of 1.54, indicating that the UV luminescence below 400 nm was caused through the two-photons excitation pathways. Over 30 W cm−2, the plots for the upconversion intensity deviated from the linear fitting line depicted in Fig. 7(b). There are two possible reasons for this deviation: the saturation of the populated 5d excited states and thermal quenching owing to the relatively high surface temperature under the illumination of high-power LD excitation.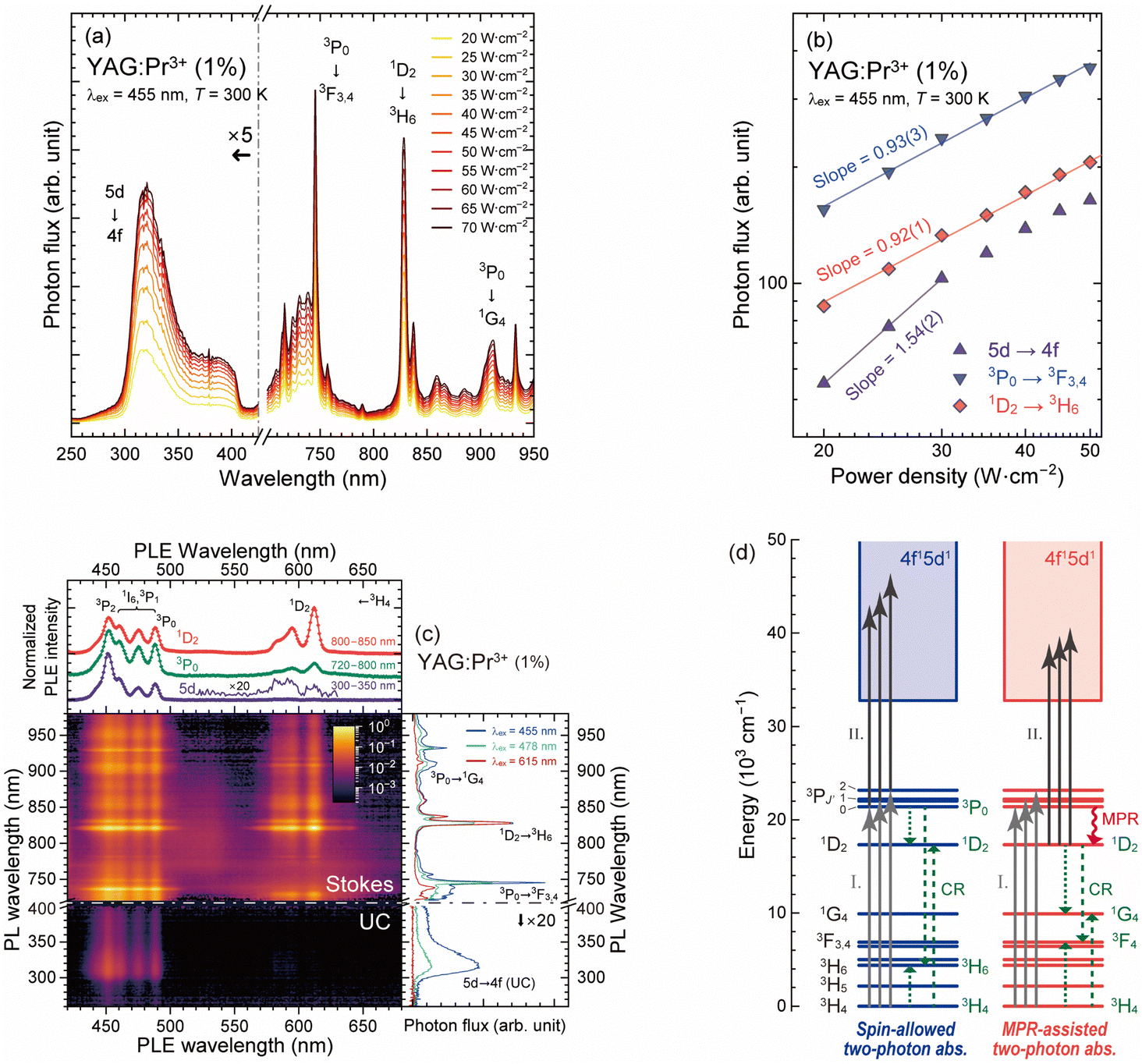 | ||
| Fig. 7 (a) UC-PL (250–420 nm) and Stokes PL (720–950 nm) spectra of the YAG:Pr1 sample under various LD pump power densities of 20–70 W cm−2. (b) log–log scale of UC-PL (5d → 4f) and Stokes PL (3P0 → 3F3,4 and 1D2 → 3H6) intensities with various pumping power densities. (c) Contour plot of UC- and Stokes PL and PLE spectra of the YAG:Pr1 sample at room temperature. The UC- and Stokes PL spectra are displayed in the PL wavelength below 400 and over 720 nm, respectively. The right and top panels show the UC- and Stokes PL and PLE spectra at particular excitation and emission wavelengths. (d) Energy level diagrams illustrating the two-photon absorption processes via the intermediate 3PJ′ and 1D2 states. I and II represent the first and second photon absorption processes, and the cross-relaxation and multiphonon relaxation are abbreviated to CR and MPR, respectively. | ||
Fig. 7(c) shows the PL–PLE contour plot measured using a visible-light-blocking filter (400–720 nm cut). The excitation wavelength was varied from 420 to 680 nm in an increment of 1 nm. In the PL spectra, luminescence bands appeared in the UV (250–400 nm) and NIR (720–950 nm) regions and were attributed to the upconversion 5d–4f and Stokes 4f–4f transitions, respectively. The contour plot illustrates the difference in the excitation characteristics of the upconversion and Stokes luminescence of the Pr3+ ions in the YAG host. The right panel shows the UC- and Stokes PL spectra pumped through the different excitation pathways; the 3P2 ← 3H4 (455 nm), 3P1 ← 3H4 or 1I6 ← 3H4 (478 nm), and 1D2 ← 3H4 (615 nm) transitions. The top panel shows the excitation spectra for the upconversion and Stokes luminescence attributed to the 5d → 4f (280–400 nm), 3P0 → 3F3,4 (720–800 nm), and 1D2 → 3H6 (800–850 nm) transitions. In the Stokes PL spectra, 4f–4f luminescence is observed, the properties of which are similar to those shown in Fig. 3 and 4. When the 1D2 state was directly pumped through the 1D2 ← 3H4 transition, the relative PL intensity of the 1D2 → 3H6 transition peaking at 830 nm was significantly enhanced.
Interestingly, the shape of the UC-PLE spectrum differed from that of the Stokes PLE spectra in terms of the peak intensities for each excitation transition. As shown in the contour plot, intense upconversion luminescence bands were observed under excitation light illumination below 500 nm, indicating that the excitation bands related to the upconversion process via the 1D2 state were faint in the range 580–630 nm. According to the absorption spectra shown in Fig. 2, the 5d excitation bands located below 320 nm (>31250 cm−1) indicate that the two-photon absorption process via the 1D2 level at 16437–17271 cm−1 is likely to occur. The UC-PLE spectrum in the top panel was obtained by integrating the UC-PL spectra in the 280–400 nm range. In contrast to the Stokes PLE spectra with the peak of the 1D2 ← 3H4 excitation band at 615 nm, the 1D2 excitation bands in the UC-PLE spectra were barely observed at ∼590 nm. Gayen et al. reported the UC-PLE spectra of YAG:Pr3+ (0.4%) using a dye laser that has a broadband emission at 555–615 nm, in which the upconversion excitation bands related to the intermediate 1D2 state were observed at 581, 587, 593, and 610 nm, and the relative intensity of the excitation bands below 600 nm was higher than that at 610 nm.18 This observation is consistent with the result shown in Fig. 7(c). This suggests that two-photon absorption via the intermediate 1D2 state occurs only at even higher Stark sublevels with a low transition probability. Below 500 nm, the spectral shapes of the UC-PLE (purple line) and Stokes PLE (green and red lines) spectra were inconsistent, and the relative UC-PLE intensity at 460–490 nm was low. This difference indicates that two-photon absorption can occur not only at the 3P0 level but also at the 3P1, 3P2, and 1I6 levels. At 450–490 nm (the energy gap of this range is just ∼2000 cm−1), the 3P0, 3P1, 3P2, and 1I6 states have 1, 3, 5, and 13 Stark sublevels, respectively.53 Because of these dense energy levels, the higher 3P1, 3P2, and 1I6 states are populated by the thermal energy at room temperature, following Boltzmann statistics. Therefore, two-photon absorption in these intermediate states can occur. The weak excitation bands at 460–490 nm were attributed to the excitation from the 3H4 to 3P0, 3P1, and 1I6 states. As the 1I6 ← 3H4 excitation bands extended at 460–490 nm because of the 13 separated Stark sublevels in the dodecahedral sites with D2 symmetry, the decrease in the PLE intensity at 460–490 nm was caused by the absence of the widespread 1I6 bands. The above results show that the upconversion excitation intensities of the 1D2 and 1I6 intermediate states were significantly lower than those of the triplet intermediate states. Considering Hund's rules, the spin multiplicity of the 4f15d1 terminal states for the upconversion process is triplets, implying that the transition probability of two-photon absorption via the intermediate singlet (1D2 and 1I6) states is low due to the spin-forbidden characteristics.
Another upconversion route can be considered; after the MPR from the 3P0 to the 1D2 states following the 3PJ′ ← 3H4 (J′ = 0, 1, and 2) excitation, the 5d excited states are pumped through the ESA or ETU process via the intermediate 1D2 state under 450–500 nm excitation illumination with high power density. Cates and Kim reported that polychromatic excitation by violet (at 447 nm) and yellow (at 589 nm) light can enhance the upconversion UV luminescence of Y2SiO5:Pr3+ by populating the intermediate 1D2 state.20 In the case of the YAG:Pr3+ sample in this study, because the MPR rate between the 3P0 and 1D2 states is relatively high, the upconversion process under blue-cyan light (450–500 nm) illumination via the intermediate 1D2 state populated through the MPR process occurs, even though the upconversion efficiency is relatively low because of the spin-forbidden character. This MPR-assisted upconversion process did not affect the spectral shapes of the UC- and Stokes PLE spectra. Therefore, the above results suggest that the two-photon absorption processes via multiple intermediate 3PJ′ states and the MPR-assisted upconversion process via the intermediate 1D2 state compete in the YAG:Pr3+ samples, as illustrated in Fig. 7(d).
Fig. 8(a) shows the Pr3+ concentration dependence of the UC and Stokes PL spectra of the YAG:Pr3+ samples under 455 nm blue LD excitation with a pump power density of 20 W cm−2. The Stokes PL spectra in the NIR region exhibited the same concentration dependence as the PL spectra in the visible range shown in Fig. 3(a); compared to the 3P0 luminescence, the 1D2 luminescence significantly declined with increasing Pr3+ concentration because of the efficient cross-relaxation process of [1D2, 3H4] → [1G4, 3H6]. The variations in Stokes PL intensity are plotted in Fig. 8(b), in which the degree of the decrease in the integrated PL intensities for the 3P0 → 3F3,4 and 1D2 → 3H6 luminescence was consistent with that in Fig. 3(b), despite a small deviation due to the slight overlap of the 3P0 and 1D2 luminescence bands in the area integration.
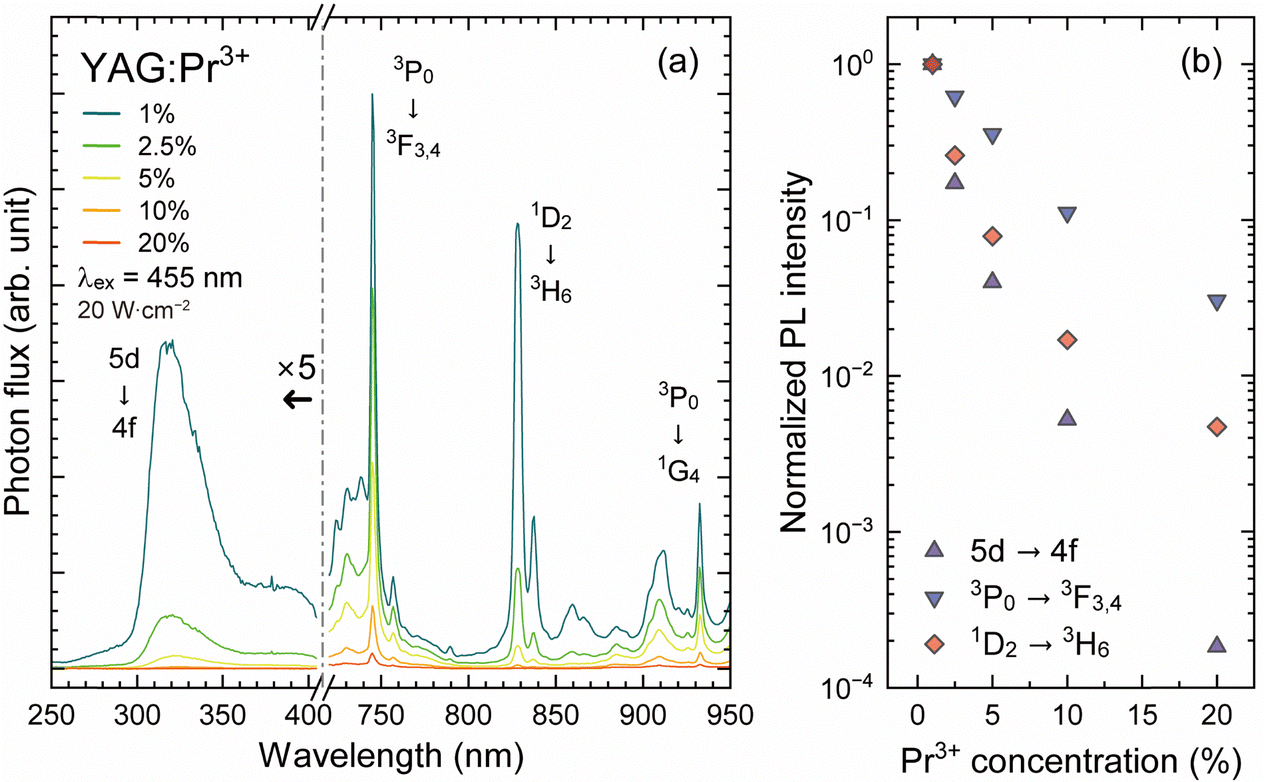 | ||
| Fig. 8 (a) Upconversion and Stokes PL spectra of the YAG:Pr3+ samples at room temperature under 455 nm LD excitation with a pump power density of 20 W cm−2. (b) The variation in the 5d → 4f upconversion-PL and 4f–4f Stokes PL (3P0 → 3F3,4 and 1D2 → 3H6) intensities, plotted as a function of Pr3+ concentration. | ||
In contrast to the Stokes luminescence property, the upconversion luminescence intensity below 400 nm severely decreased with increasing Pr3+ concentration, whereas the Stokes 5d–4f luminescence intensities of the YAG:Pr1 and YAG:Pr2.5 samples were comparable; the upconversion 5d–4f luminescence intensity of the YAG:Pr2.5 sample was at most 20% of that of the YAG:Pr1 sample. The UC-PL intensity of the YAG:Pr10 sample was decayed to less than 1% of the initial UC-PL intensity of the YAG:Pr1 sample. This significant concentration quenching of the upconversion UV luminescence cannot be explained by the concentration quenching of the 5d–4f luminescence and thermal quenching at room temperature. One possible reason for this is the rise in the surface temperature of the YAG:Pr samples due to the LD high-power excitation. As shown in Fig. 6(b), thermal quenching of the 5d–4f luminescence at 300 K is non-negligible, indicating that the 5d excited states, especially for samples with high Pr3+ concentrations, are easily affected by a slight temperature increase. However, because the temperature increase can only be a few degrees under the LD illumination with the power density of 20 W cm−2, it is unlikely that the decline in the UC-PL intensity would be so significant as to alter its order as shown in Fig. 8(b). Another possible reason for the severe concentration quenching of the Pr3+: 5d–4f upconversion luminescence is the increase in the cross-relaxation rate of the intermediate 3P0 and 1D2 states. Because the probability of electronic transition is correlated with the population of the initial state, nonradiative cross-relaxation depleting the 3P0 and 1D2 states causes a lower two-photon absorption probability. The effect of cross-relaxation of the 3P0 and 1D2 states on the upconversion luminescence was assumed from the products of the 5d–4f Stokes PL intensity and the 3P0 or 1D2 Stokes PL intensity shown in Fig. 3(b), plotted as a function of the Pr3+ concentration in Fig. S4 (ESI†) with the 5d–4f upconversion intensity. The experimental values of the decrease in the upconversion intensity were smaller than the simulated values of the products, (Stokes 3P0 or 1D2) × (Stokes 5d–4f), which considered cross-relaxation in the intermediate 3P0 and 1D2 states. In the actual process, the 3P0 and 1D2 cross-relaxations compete in response to their populations, resulting in overestimated simulation values. The experimental and simulated upconversion intensities of the YAG:Pr3+ samples were of the same order. Therefore, the Pr3+: 5d–4f upconversion luminescence intensity decreased because of the significant cross-relaxation of the intermediate 3P0 and 1D2 states with increasing Pr3+ concentrations.
The results in this study suggest the two guidelines for designing the Pr3+-doped phosphors exhibiting the highly efficient upconversion UV luminescence: (1) controlling the concentration and thermal quenching of the broadband UV luminescence along with the 5d → 4f allowed transition and (2) suppressing the cross-relaxation from the intermediate 3PJ′ and 1D2 states. While a relatively low Pr3+ concentration is a perspective for both, it is possible to design a UV upconverter activated with Pr3+ ions, which has a low cross-relaxation probability, maintaining a relatively high Pr3+ concentration. The YAG:Pr3+ samples in this study exhibited severe concentration quenching with 1% Pr3+ doping during the blue-to-UV upconversion luminescence. The dodecahedral Y3+ sites in the cubic YAG structure share edges, leading to Y3+–Y3+ atomic distances of 3.68 Å. In contrast, Li2CaSiO4:Pr3+,37 reported by Schröder et al., exhibited the broadband upconversion luminescence of Pr3+ at 250–320 nm, and its UC-PL intensity did not drop until the Pr3+ concentration reached 5%. In the tetragonal Li2CaSiO4 structure, where the Pr3+ ions substitute at the Ca2+ sites, the Ca2+–Ca2+ atomic distances of 4.82 and 5.05 Å are relatively long due to the corner-sharing.64 Therefore, it is suggested that host compounds with large atomic distances, such as corner-sharing Pr3+ substituted sites, are advantageous as Pr3+-doped phosphors with high visible-to-UV upconversion efficiency.
4. Conclusion
To investigate how the characteristics of the upconversion and Stokes emission of the Pr3+-doped luminescent materials are affected by Pr3+ concentration, the Y3Al5O12 (YAG) ceramics doped with Pr3+ ions up to 20% were synthesized using the polymerized complex method, and their luminescence properties were characterized in a spectroscopic manner. In the Stokes photoluminescence spectra, the peak 5d–4f luminescence bands located at ∼310–320 nm are red-shifted due to self-absorption with increasing Pr3+ concentration. The 5d–4f excitation bands were blue-shifted with increasing Pr3+ concentration because the Pr3+–O2− bond lengths in the YAG:Pr3+ samples increased due to the doped Pr3+ ions, which have a larger ionic radius than Y3+. For the 4f–4f transition, the 3P0 and 1D2 luminescence exhibited severe concentration quenching because of the phonon-assisted cross-relaxation process. The cross-relaxation rate of the 1D2 state is larger than that of the 3P0 state because the energy gaps related to the 1D2 cross-relaxation are more resonant and the [1D2, 3H4] → [1G4, 3H6] cross-relaxation is spin-allowed. In the characterization of the upconversion properties, the contour plots revealed that the 5d–4f upconversion luminescence of YAG:Pr3+ occurred efficiently under the illumination of excitation light only below 500 nm. The Pr3+: 5d–4f upconversion luminescence occurs through the excited-state absorption and energy transfer upconversion processes via the intermediate 3PJ′ (J′ = 0, 1, and 2) and 1D2 states. The experimental results indicated that while the upconversion via the 3PJ′ states has a high transition probability due to the spin-allowed feature, the upconversion via the 1D2 state is assisted by multiphonon relaxation from the 3P0 state despite being spin-forbidden. The concentration dependence of the Pr3+: 5d–4f upconversion luminescence was more significant than that of the 5d–4f Stokes luminescence because of the cross-relaxation of the intermediate 3P0 and 1D2 states. The results of this study suggest that phosphors with lower Pr3+ concentrations or larger distances between crystallographic sites that can accommodate Pr3+ ions are advantageous for suppressing the cross-relaxation rate of the intermediate 3P0 and 1D2 states, resulting in highly efficient Pr3+ visible-to-UV upconversion luminescence.Author contributions
Y. K. conceived the idea of the study. H. N. prepared the materials. Y. K. investigated the structural and optical properties and drafted the original manuscript. K. S. reviewed the manuscript. All authors reviewed the manuscript draft and revised it critically on intellectual content. All authors approved the final version of the manuscript to be submitted.Data availability
The data that support the findings of this study are available from the corresponding author, Y. Kitagawa, upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was financially supported by JSPS KAKENHI (grant numbers 23K23056 and 23K13551).References
- Y. Sato, J. Odahara, R. Yanamoto, S. Noda, T. Hasegawa, S. Yin, J. Jia and M. Kakihana, Chem. Mater., 2024, 36, 313–323, DOI:10.1021/acs.chemmater.3c02119.
- Q. Du, J. Ueda, R. Zheng and S. Tanabe, Adv. Opt. Mater., 2023, 11, 2202612, DOI:10.1002/adom.202202612.
- X. Wang and Y. Mao, J. Mater. Chem. C, 2022, 10, 3626–3646, 10.1039/d2tc00208f.
- S. Tian, P. Feng, S. Ding, Y. Wang and Y. Wang, J. Alloys Compd., 2022, 899, 163325, DOI:10.1016/j.jallcom.2021.163325.
- B. Wang, H. Lin, J. Xu, H. Chen, Z. Lin, F. Huang and Y. Wang, Inorg. Chem., 2015, 54, 11299–11306, DOI:10.1021/acs.inorgchem.5b01894.
- A. Lecointre, A. Bessière, A. J. J. Bos, P. Dorenbos, B. Viana and S. Jacquart, J. Phys. Chem. C, 2011, 115, 4217–4227, DOI:10.1021/jp108038v.
- H. M. Crosswhite, G. H. Dieke and W. J. Carter, J. Chem. Phys., 1965, 43, 2047–2054, DOI:10.1063/1.1697073.
- P. Dorenbos, J. Lumin., 2000, 91, 155–176, DOI:10.1016/s0022-2313(00)00229-5.
- E. van der Kolk, P. Dorenbos, A. P. Vink, R. C. Perego, C. W. E. van Eijk and A. R. Lakshmanan, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 195129, DOI:10.1103/physrevb.64.195129.
- P. Dorenbos, ECS J. Solid State Sci. Technol., 2013, 2, R3001–R3011, DOI:10.1149/2.001302jss.
- D. Wang, S. Huang, F. You, S. Qi, Y. Fu, G. Zhang, J. Xu and Y. Huang, J. Lumin., 2007, 122–123, 450–452, DOI:10.1016/j.jlumin.2006.01.203.
- A. Yoshikawa, K. Kamada, M. Nikl, K. Aoki, H. Sato, J. Pejchal and T. Fukuda, J. Cryst. Growth, 2005, 285, 445–449, DOI:10.1016/j.jcrysgro.2005.08.052.
- M. Broxtermann, D. den Engelsen, G. R. Fern, P. Harris, T. G. Ireland, T. Jüstel and J. Silver, ECS J. Solid State Sci. Technol., 2017, 6, R47–R52, DOI:10.1149/2.0051704jss.
- A. M. Srivastava, A. A. Setlur, H. A. Comanzo, W. W. Beers, U. Happek and P. Schmidt, Opt. Mater., 2011, 33, 292–298, DOI:10.1016/j.optmat.2010.08.026.
- A. Katelnikovas, H. Bettentrup, D. Dutczak, A. Kareiva and T. Jüstel, J. Lumin., 2011, 131, 2754–2761, DOI:10.1016/j.jlumin.2011.06.012.
- F. Schröder and T. Jüstel, Opt. Mater.: X, 2021, 12, 100117, DOI:10.1016/j.omx.2021.100117.
- Q. Du, J. Ueda and S. Tanabe, J. Mater. Chem. C, 2023, 11, 16225–16233, 10.1039/d3tc03619g.
- S. K. Gayen, B. Q. Xie and Y. M. Cheung, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 20–28, DOI:10.1103/physrevb.45.20.
- C. Hu, C. Sun, J. Li, Z. Li, H. Zhang and Z. Jiang, Chem. Phys., 2006, 325, 563–566, DOI:10.1016/j.chemphys.2006.01.037.
- E. L. Cates and J.-H. Kim, Opt. Mater., 2013, 35, 2347–2351, DOI:10.1016/j.optmat.2013.06.030.
- J. Ganem, W. M. Dennis and W. M. Yen, J. Lumin., 1992, 54, 79–87, DOI:10.1016/0022-2313(92)90250-D.
- F. Auzel, Chem. Rev., 2004, 104, 139–173, DOI:10.1021/cr020357g.
- E. L. Cates and F. Li, RSC Adv., 2016, 6, 22791–22796, 10.1039/c6ra01121g.
- N. Rebrova, P. Zdeb and P. J. Dereń, J. Phys. Chem. C, 2024, 128, 9090–9098, DOI:10.1021/acs.jpcc.3c08163.
- X. Wang, Y. Chen, F. Liu and Z. Pan, Nat. Commun., 2020, 11, 2040, DOI:10.1038/s41467-020-16015-z.
- A. Wang, Y. Liu, X.-J. Wang and F. Liu, Opt. Lett., 2020, 45, 2720–2723, DOI:10.1364/OL.393770.
- X. Yan, D. Dai, K. Ma, S. Zuo, W. Liu, X. Li and C. Yao, Front. Mater. Sci., 2020, 14, 43–51, DOI:10.1007/s11706-020-0488-6.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195, DOI:10.1021/acs.chemrev.1c00034.
- E. L. Cates, M. Cho and J.-H. Kim, Environ. Sci. Technol., 2011, 45, 3680–3686, DOI:10.1021/es200196c.
- T. Fukui, T. Niikura, T. Oda, Y. Kumabe, H. Ohashi, M. Sasaki, T. Igarashi, M. Kunisada, N. Yamano, K. Oe, T. Matsumoto, T. Matsushita, S. Hayashi, C. Nishigori and R. Kuroda, PLoS One, 2020, 15, e0235948, DOI:10.1371/journal.pone.0235948.
- M. J. Weber, J. Chem. Phys., 1968, 48, 4774–4780, DOI:10.1063/1.1668061.
- R. C. Naik, N. P. Karanjikar and M. A. N. Razvi, J. Lumin., 1992, 54, 139–144, DOI:10.1016/0022-2313(92)90001-p.
- C. de Mello Donegá, A. Meijerink and G. Blasse, J. Phys. Chem. Solids, 1995, 56, 673–685, DOI:10.1016/0022-3697(94)00183-9.
- H. Dornauf and J. Heber, J. Lumin., 1980, 22, 1–16, DOI:10.1016/0022-2313(80)90040-x.
- R. Naccache, F. Vetrone, A. Speghini, M. Bettinelli and J. A. Capobianco, J. Phys. Chem. C, 2008, 112, 7750–7756, DOI:10.1021/jp711494d.
- F. Lai, X. Xu, J. Shen, Y. Wang, Y. Yan, Y. Nie, W. You, D. Wu, L. Han and Z. Xiao, Silicon, 2022, 15, 1913–1923, DOI:10.1007/s12633-022-02148-x.
- F. Schröder, S. Fischer and T. Jüstel, Aust. J. Chem., 2022, 75, 760–771, DOI:10.1071/ch21311.
- X. Wu, W. M. Dennis and W. M. Yen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 6589–6595, DOI:10.1103/physrevb.50.6589.
- E. Cavalli, L. Esposito, M. Bettinelli, A. Speghini, K. V. Ivanovskikh, R. B. Hughes-Currie and M. de Jong, Mater. Res. Express, 2014, 1, 045903, DOI:10.1088/2053-1591/1/4/045903.
- K. V. Ivanovskikh, J. M. Ogiegło, A. Zych, C. R. Ronda and A. Meijerink, ECS J. Solid State Sci. Technol., 2013, 2, R3148–R3152, DOI:10.1149/2.011302jss.
- J. Ueda, A. Meijerink, P. Dorenbos, A. J. J. Bos and S. Tanabe, Phys. Rev. B, 2017, 95, 014303, DOI:10.1103/physrevb.95.014303.
- H. Nakamura, K. Shinozaki, T. Okumura, K. Nomura and T. Akai, RSC Adv., 2020, 10, 12535–12546, 10.1039/d0ra01381a.
- H. Nakamura, Z. Liu, K. Shinozaki, K. Nomura, T. Akai and K. Kadono, Ceram. Int., 2024, 50, 28498–28504, DOI:10.1016/j.ceramint.2024.05.158.
- M. Kakihana, J. Ceram. Soc. Jpn., 2009, 117, 857–862, DOI:10.2109/jcersj2.117.857.
- K. S. Bagdasarov, N. B. Bolotina and V. I. Kalinin, Kristallografiya, 1991, 36, 715–728 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/s0021889811038970.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20, DOI:10.4028/www.scientific.net/ssp.130.15.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767, DOI:10.1107/S0567739476001551.
- P. Pues, M. Laube, S. Fischer, F. Schröder, S. Schwung, D. Rytz, T. Fiehler, U. Wittrock and T. Jüstel, J. Lumin., 2021, 234, 117987, DOI:10.1016/j.jlumin.2021.117987.
- J. P. Hurrell, S. P. S. Porto, I. F. Chang, S. S. Mitra and R. P. Bauman, Phys. Rev., 1968, 173, 851–856, DOI:10.1103/physrev.173.851.
- G. Özen, O. Forte and B. Di Bartolo, J. Appl. Phys., 2005, 97, 013510, DOI:10.1063/1.1823577.
- M. Poulos, S. Giaremis, J. Kioseoglou, J. Arvanitidis, D. Christofilos, S. Ves, M. P. Hehlen, N. L. Allan, C. E. Mohn and K. Papagelis, J. Phys. Chem. Solids, 2022, 162, 110512, DOI:10.1016/j.jpcs.2021.110512.
- J. B. Gruber, M. E. Hills, R. M. MacFarlane, C. A. Morrison and G. A. Turner, Chem. Phys., 1989, 134, 241–257, DOI:10.1016/0301-0104(89)87159-9.
- S. T. Lai, S. Huang and W. M. Yen, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 2349–2361, DOI:10.1103/physrevb.26.2349.
- M. Malinowski, P. Szczepanski, W. Wolinski, R. Wolski and Z. Frukacz, J. Phys.: Condens. Matter, 1993, 5, 6469–6482, DOI:10.1088/0953-8984/5/35/012.
- G. Özen, O. Forte, B. Di Bartolo and J. M. Collins, J. Appl. Phys., 2007, 102, 023110, DOI:10.1063/1.2753685.
- W. J. Schipper, M. F. Hoogendorp and G. Blasse, J. Alloys Compd., 1993, 202, 283–287, DOI:10.1016/0925-8388(93)90550-7.
- B. Savoini, J. E. Muñoz Santiuste and R. González, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 5856–5865, DOI:10.1103/physrevb.56.5856.
- M. Malinowski, W. Woliński, R. Wolski and W. Strçek, J. Lumin., 1991, 48–49, 235–238, DOI:10.1016/0022-2313(91)90112-9.
- O. L. Malta, E. Antic-Fidancev, M. Lemaitre-Blaise, J. Dexpert-Ghys and B. Piriou, Chem. Phys. Lett., 1986, 129, 557–561, DOI:10.1016/0009-2614(86)80400-6.
- P. Bolek, J. Zeler, C. D. S. Brites, J. Trojan-Piegza, L. D. Carlos and E. Zych, Chem. Eng. J., 2021, 421, 129764, DOI:10.1016/j.cej.2021.129764.
- M. J. Weber, Solid State Commun., 1973, 12, 741–744, DOI:10.1016/0038-1098(73)90326-8.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337, DOI:10.1103/PhysRevB.61.3337.
- J. A. Gard and A. R. West, J. Solid State Chem., 1973, 7, 422–427, DOI:10.1016/0022-4596(73)90171-0.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03386h |
| This journal is © The Royal Society of Chemistry 2024 |