
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-membrane pH-decoupling aqueous batteries using proton-coupled electrochemistry for pH recovery†
Dawei
Xi
 a,
Zheng
Yang
a,
Abdulrahman M.
Alfaraidi
a,
Yan
Jing
b,
Roy G.
Gordon
ab and
Michael J.
Aziz
*a
a,
Zheng
Yang
a,
Abdulrahman M.
Alfaraidi
a,
Yan
Jing
b,
Roy G.
Gordon
ab and
Michael J.
Aziz
*a
aJohn A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA, USA. E-mail: maziz@harvard.edu
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA, USA
First published on 8th July 2024
Abstract
pH-decoupling in aqueous redox flow batteries (ARFBs) represents a promising strategy for enhancing cell voltage and expanding the repertoire of redox pair combinations. Effective management of acid–base crossover and the implementation of cost-effective pH recovery methods are pivotal for long-term stability of pH-decoupling ARFBs. We introduce a pH-decoupling design integrated into a conventional single-membrane ARFB architecture. This approach reduces the area specific resistance while suppressing acid–base crossover to an acceptable level. We explore various electrolyte pairs, ranging from anions to cations, acids to bases, always dissolved to electrodepositing, showing the flexibility afforded by this design in selecting electrolyte compositions. Furthermore, we demonstrate the utility of proton-coupled electrochemical reactions as proton pumps, facilitating in situ or ex situ pH recovery within pH-decoupling batteries. Our findings potentially offer benefits including improved energy efficiency, increased areal power output, and decreased capital costs, thereby advancing the prospects for scalable and sustainable energy storage solutions.
Broader contextAqueous redox flow batteries (ARFBs) stand out as a leading solution for economical and scalable storage of intermittent renewable energy derived from sources like wind and solar. For long time storage (>8 h), these batteries offer a unique advantage by decoupling energy storage from power generation, providing a level of design versatility and scalability that traditional rechargeable batteries can hardly match. Traditionally, ARFBs maintain a uniform pH in both negolyte and posolyte to ensure consistent and stable operation over the long term. However, introducing disparate pH levels within a single cell, especially using acid for posolyte and base for negolyte, would broaden the choice of high voltage candidate redox couples. This approach also facilitates the pairing of redox couples that necessitate distinct pH environments, thereby enhancing the versatility and efficacy of the system. Our research demonstrated that different pH can be used in a conventional ARFB architecture, while the acid–base crossover is not a fatal drawback. We also showed that proton-coupled electrochemistry can be used to maintain the pH difference across the cell, paving the way toward the commercialization of pH-decoupling batteries. |
Driven by decarbonization, the rapid expansion of renewable energy sources like wind and solar incentivizes the development of robust energy storage systems to regulate their fluctuating power output.1 Aqueous redox flow batteries (ARFBs) emerge as a viable option, offering safety, long discharge duration at rated power, and the potential for both longevity and affordability.2 Through the transition from solid electrodes to liquid flowable electrolytes, ARFBs facilitate the independent optimization of energy capacity and power output, thereby augmenting system design versatility.3 Advancements in redox-active materials are also bringing us closer to realizing the practical feasibility of grid-scale storage.4–8
In the conventional design of ARFBs, a single ion exchange membrane is utilized to separate the negolyte and posolyte.9 For example, an anion-exchange membrane (AEM) can block cationic redox-active species, serving to prevent self-discharge while simultaneously allowing the migration of non-reactive anions to maintain charge balance in the electrolytes. Likewise, a cation-exchange membrane (CEM) is effective for cells that use anionic redox-active species. In most cases, maintaining the same time-averaged value of the pH on both sides the cell is pursued to suppress proton or hydroxide crossover.8
A higher voltage can increase the practicability of ARFBs by raising energy and power density, thereby lowering capital costs.10 The emerging strategy of pH-decoupling ARFBs, employing negolytes and posolytes with distinct pH values, is drawing attention, due to its potential to achieve higher voltages.11 Tailored ion-selective and bipolar membranes have been used in the development of single-membrane pH-decoupling ARFBs.12–14 Moreover, the use of multi-membrane cell architectures has enabled higher flexibility in the selection of negolyte and posolyte active species.15–17 However, pH-decoupling systems are susceptible to proton/hydroxide crossover caused by the pH difference, which compromises longevity and overall efficiency.18 Recently, the acid/base crossover rate in pH-decoupling systems was systematically studied and ex situ pH recovery using bipolar membrane sub-cells was realized, making the pH-decoupling system practical for efficient, long-term operation.19
Unfortunately, most pH-decoupling systems have high area specific resistance, which dramatically lowers the benefits derived from pH-decoupling. Also, the multi-chamber multi-membrane cell architecture or the utilization of a bipolar membrane increases the capital cost and the operational complexity. Herein, we report the decoupling of the pH of the posolyte and negolyte in a single-membrane ARFB of conventional architecture and the investigation of its performance. By employing a variety of redox couples, we studied the cycling patterns and power generation potential inherent in this design, utilizing either an AEM or a CEM. Additionally, we estimated the crossover rate of acid and base, which is a vital factor for ensuring the sustained operation of pH-decoupled flow systems over extended periods. To recover the crossover of acid and base, we utilized a proton pump that runs proton-coupled electron transfer (PCET) reactions inside or outside the original cell. The proton pump can use water splitting, the oxygen reduction reaction (ORR) or the hydrogen oxidation reaction (HOR) as side-reactions to recover the pH, provided that the reductive and oxidative side-reactions are balanced. With such a design and suitable proton pumps, different types of electrolytes – even electrodepositing batteries (aqueous batteries involving electrodeposition on one or both sides)20,21 – can potentially leverage a pH-decoupling architecture to exhibit stable operation at higher voltage.
Decoupling the pH in a single membrane conventional ARFB architecture has been reported,22–25 yet the long-term acid–base crossover in the cell has not been considered. An AEM type single-membrane pH-decoupling ARFB with cationic redox-active candidates is illustrated in Fig. 1a. To maintain non-hydroxide anions as main charge carriers, the hydroxide concentration in the negolyte must remain dilute. Yet for a highly selective AEM, the cell is workable with an acidic posolyte of low pH. Acid–base crossover is caused by proton crossover and hydroxide exchange through the AEM. Similarly, the posolyte in a CEM type pH-decoupled ARFB with anionic redox-active species requires a dilute proton/hydronium concentration (Fig. 1b), but the cell is workable with a high-pH negolyte. The fluxes for acid–base crossover are depicted at the bottom of the diagram and described in Note S1 (ESI†). The influence of acid–base crossover is decoupled from the electrolyte volume (total capacity). Researchers can find suitable conditions for specific redox active molecules if acid–base crossover remains sufficiently low to avoid significant impacts on energy efficiency and acid–base regeneration is provided. Detailed structures and properties of listed redox molecules are in Table S1 (ESI†).
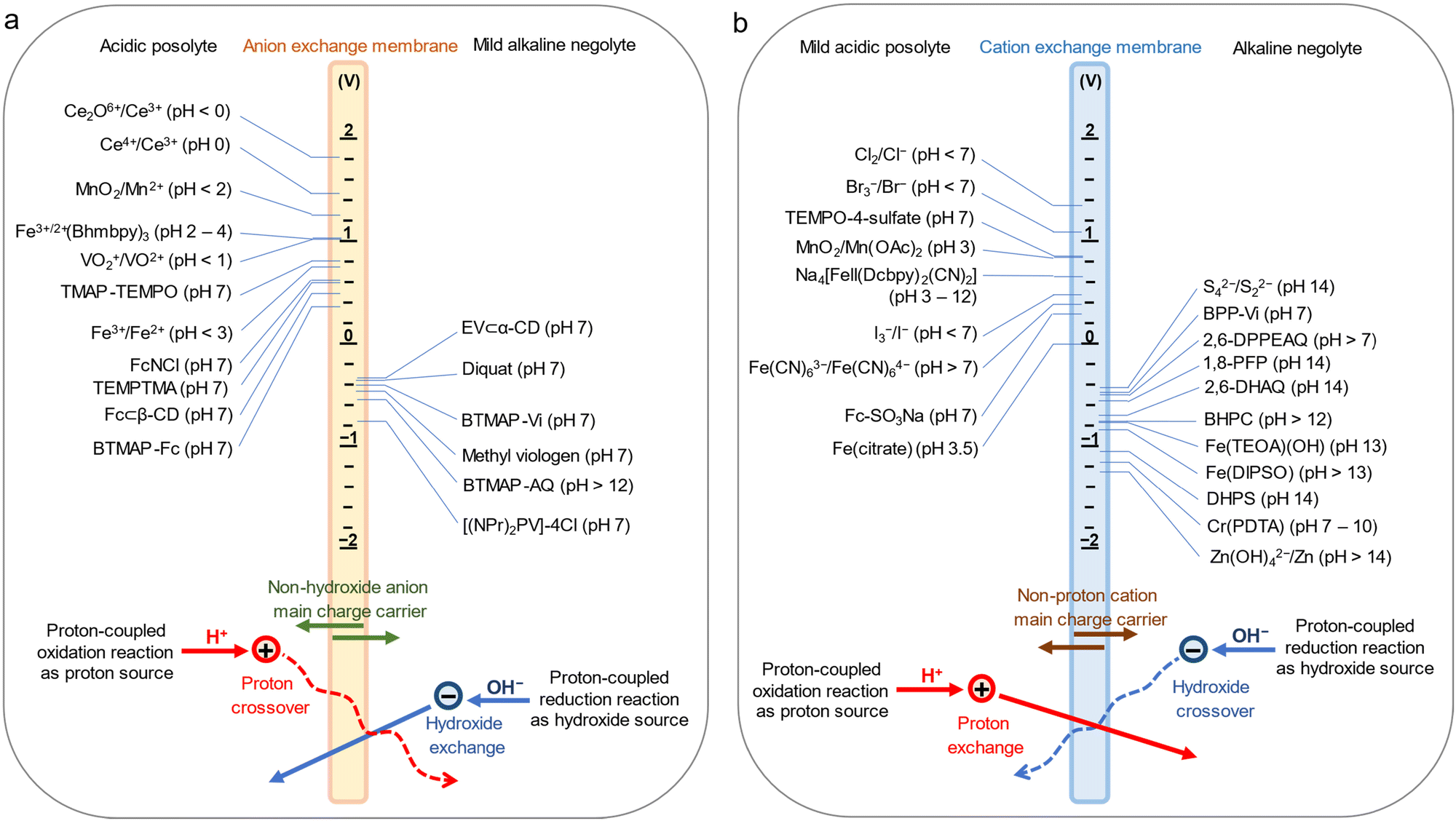 | ||
| Fig. 1 Single-membrane pH-decoupling ARFBs. Schematic of (a) an AEM type and (b) a CEM type single-membrane pH-decoupling ARFB with potential redox candidates with voltage vs. standard hydrogen electrode (SHE) identified. Main charge carriers and possible acid–base crossover processes are labeled at the bottom. | ||
To demonstrate these principles, we paired 5 mL of 0.1 M tris(4,4′-bis(hydroxymethyl)-2,2′-bipyridine) iron dichloride (Fe(Bhmbpy)3) as capacity limiting side with 10 mL 0.1 M 1,4-bis[3-(trimethylammonio)propyl]-anthraquinone (BTMAPAQ) in an AEM type pH-decoupling ARFB (Fig. 2a).26,27 The redox potential of Fe(Bhmbpy)3 is about 0.98 V vs. SHE, and the potential of BTMAPAQ at pH 12 is about −0.55 V vs. SHE.28 The posolyte was buffered by the ligand, operating within a pH range of approximately 2–4. The redox reaction of BTMAPAQ is proton-coupled when the electrolyte pH is below 12, transitioning to a non-proton-coupled reaction at higher pH. Thus, we set the initial pH of the BTMAPAQ at 12. To separate the cell, we employed Selemion DSV-N as the AEM. All cell characterization procedures were conducted within a nitrogen glovebox. We used standard constant-current followed by constant–voltage (CCCV) protocol at 40 mA cm−2 with voltage cut-off values of 1.75 V and 0.4 V, held until the current density dropped below 5 mA cm−2 and 1 mA cm−2 during charge and discharge, respectively. Analysis revealed the acid crossover flux to be lower than 1 nmol s−1 cm−2 (0.1 mA cm−2), with the base exhibiting an approximate flux of 4 nmol s−1 cm−2 (Note S1, ESI†). The cell capacity, Coulombic efficiency and pH of posolyte and negolyte during cell cycling are shown in Fig. 2b and c. The cell accessed >90% of its theoretical capacity and exhibited an average Coulombic efficiency of 98.5%, with Fe(Bhmbpy)3 as the capacity limiting side. Because of the side-reaction of ligand oxidation in the posolyte,26 the Coulombic efficiency of the system is limited by the posolyte side, leading to a drift from initial charge balance toward reduction of both sides. To offset this imbalance during cell cycling, we introduced oxygen into BTMAPAQ when the cell was nearly discharged and the pH of negolyte became low or the cell was out of balance. Each time the negolyte was exposed to oxygen, the cell went back into balance and the pH difference between posolyte and negolyte increased.
 | ||
| Fig. 2 Operation of an AEM pH-decoupling ARFB. (a) Chemistry and properties of the cell. (b) Discharge capacity, charge capacity and Coulombic efficiency of the cell during CCCV cycling under nitrogen. Cell can run out of balance due to the self-discharging of the posolyte. Oxygen was introduced into the cell about day 0.5, 1.5, 2.5 and 3.5 to restore the balance of the posolyte and negolyte. (c) pH of the cell during CCCV cycling showed in b. The pH difference of the posolyte and the negolyte increased after oxygen was introduced. (d) Proton-coupled side reactions in the cell that do not contribute to the cell capacity but can maintain the pH difference. (e) Discharge capacity, charge capacity and Coulombic efficiency of the cell during CCCV cycling in air. (f) Negolyte pH and posolyte pH of the cell during CCCV cycling showed in e. BTMAPAQ constantly reacted with oxygen in the air, without showing obvious pH fluctuations. | ||
The reactions contributing to the recovery of pH are listed in Fig. 2d. These side reactions do not contribute to capacity, but they cause Coulombic efficiency loss. Introducing oxygen into the negolyte containing reduced anthraquinones leads to the oxidation of anthraquinones, generating hydroxide or peroxide.29 The net reactions are the ORR through 2e or 4e pathways.30 During the oxygen exposure periods, the posolyte pH also slightly decreased; the proton source might be the oxidation of peroxide anions generated in the negolyte and crossing the AEM (Fig. S1, ESI†). Additionally, despite being an undesirable side reaction and contributing to the fading observed in Fig. 2b, the oxidation of ligands can act as a proton source (Note S2, ESI†). With future development of a more stable ligand, applying a high positive voltage to the posolyte to drive the oxygen evolution reaction (OER) can possibly be an ideal proton source. By operating the cell outside of the glovebox, aside from the main redox reactions, when all the oxidation reactions happening in the posolyte balanced all the reduction reactions in the negolyte, states of charge (SOC) of both sides of the cell remained balanced (Fig. 2e). Because these minor reactions are all proton-coupled electrochemistry, acting as proton pumps, a relatively stable pH was maintained (Fig. 2f).
We then conducted a galvanostatic and polarization analysis of the cell. During galvanostatic tests, the potential was held at the end of discharge in order to recover Fe(Bhmbpy)3 from dimerization (Fig. S2, ESI†).26 When the current density was increased from 40 to 80 mA cm−2, the Coulombic efficiency of the cell maintained about 98%. Capacity utilization of the cell dropped from 90% to 65%, accompanied by a reduction in round-trip energy efficiency from 68% to 40% (Fig. S3, ESI†). Polarization curves and galvanic power densities are shown in Fig. S4 (ESI†). The cell reached a peak power density of 140 mW cm−2 at 80% SOC. Fig. S5 (ESI†) shows the OCV of the cell is about 1.5 V. The high-frequency area-specific resistance (ASR) of the battery averaged 1.45 Ω cm2 while the average polarization ASR across all SOCs was 2.9 Ω cm2. These values indicate that the ohmic resistance of the cell is low, owing to the single membrane setup, but the charge transfer resistance of the redox molecules, possibly augmented with a mass transport overpotential, also contributed 50% to cell's overall resistance.
In a demonstration of a pH-decoupling ARFB with a CEM design, bromide/bromine was utilized in the posolyte and an anionic anthraquinone was utilized in the negolyte. We chose (((9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(oxy))bis(propane-3,1-diyl))bis(phosphonic acid) (2,6-DPPEAQ) for the negolyte due to its high solubility at pH ≳ 9.31 The initial pH was set to 12 for 0.25 M DPPEAQ. Poly acrylic acid (PAA, MW = 2000) was added to 1 M NaBr in the posolyte to stabilize the posolyte pH at approximately 4. Nafion 212 was used as the CEM without pre-treatment. Galvanostatic tests of the cell with a cutoff voltage of 2 V and 0.7 V are shown in Fig. 3a. During galvanostatic tests, the Coulombic efficiency of the cell maintained about 95% to 98%. Capacity utilization of the cell dropped from 92% to 65%, and the round-trip energy efficiency dropped from 77% to 55% as the current density was increased from 20 to 80 mA cm−2 (Fig. S6, ESI†). From polarization tests, the OCV of the cell was approximately 1.6 V, with a high frequency ASR of 1.6 Ω cm2 and a polarization ASR of 4.6 Ω cm2 (Fig. S7, ESI†). The cell did not exhibit signs of mass transport limitation under 300 mA cm−2 during discharge, due to a higher concentration of the redox active molecules than in the Fe(Bhmbpy)3/BTMAPAQ cell. The cell reached a peak power density of 190 mW cm−2 at 70% SOC (Fig. S8, ESI†). During cell cycling in air, we used a CCCV protocol at 40 mA cm−2 with voltage cut-off values of 2.0 V and 0.7 V, held until the current density dropped below 10 mA cm−2 and 5 mA cm−2 during charge and discharge, respectively. During cell cycling, the charge and discharge capacity (Fig. 3b) remained stable, with a Coulombic efficiency about 99%. The pH of the cell was relatively stable, due to low acid/base crossover (estimated as lower than 1 nmol s−1 cm−2), and proton-coupled side reactions (Fig. 3c). Mediated ORR served as a hydroxide source on the anthraquinone side,29 while on the bromide side, the oxidation of carbon and PAA buffer by bromine, although unfavorable side reactions, provided protons and maintained cell balance.32 With the future development of a more stable electrode and buffer chemistry for bromide posolyte in a practical long-term cell cycling, water oxidation can serve as an ideal proton pump to balance hydroxide crossover (Note S2, ESI†).
 | ||
| Fig. 3 Operation of a CEM pH-decoupling ARFB. (a) Galvanostatic charge–discharge voltage-capacity profile for the DPPEAQ-NaBr cell. Capacity is reported in Ampere-hours per liter of negolyte. (b) Charge capacity, discharge capacity and Coulombic efficiency of the cell during CCCV cycling under nitrogen. (c) Negolyte and posolyte pH during CCCV cycling shown in (b). | ||
In pH-decoupling ARFBs where both the posolyte and negolyte redox couples are soluble and flowable, a pH-recovery system employing a bipolar membrane (BPM) for water dissociation can be implemented.19,33 However, for pH-decoupling electrodepositing batteries in which one state of the redox couple exists in solid form, it becomes challenging to introduce them into a pH-recovery BPM sub-cell for discharge and subsequent water dissociation. Instead, in situ pH recovery can be done inside the main cell. Several pH-decoupling electrodepositing batteries have been reported.34,35 For example, alkaline Zn – halogen cells have reported high voltage, high power and high energy density.20,24 Alkaline Zn-Br ARFB can exhibit an OCV of 2.3 V.36 The Zn(OH)42−/Zn couple, composing the negolyte, exhibits a low potential of −1.22 V vs. SHE at pH 14, whereas halogen/halide redox couples, being highly positive, necessitate operation at pH levels below 7 to avoid disproportionation. Additionally, MnO2/Mn2+ has also been demonstrated in pH-decoupling cells,21,35 showing promising cell cycling and energy efficiency. These works, however, did not address the management of acid–base crossover. Proton-coupled electrochemistry, as demonstrated with proton pumps, could also be employed in such systems to ensure pH stability. As a demonstration, we assembled a pH-decoupling Zn–Mn aqueous battery that uses water splitting to generate acid and base. For the posolyte, we used an acetic acid (HOAc)/sodium acetate (NaOAc) buffer to support MnO2/Mn(OAc)2, whose redox reaction is mediated by low concentration iodine.21 For the negolyte, we used Na2Zn(OH)4/Zn, known for its high voltage and cost-effectiveness but prone to dendrite formation (Fig. 4a).18 1 M Mn(OAc)2 in 3 M HOAc, with 1 M NaOAc and 0.05 M KI as posolyte, and 1 M Na2Zn(OH)4 with 2 M NaOH as negolyte were pumped through carbon cloths at a flow rate of 30 mL min−1. In galvanostatic tests, the cell was charged to 10 mA h cm−2 at 20 mA cm−2 and discharged at various current densities (Fig. S9, ESI†). The round-trip energy efficiency dropped from 85% to 70% as the discharging current density was increased from 20 to 60 mA cm−2. The high-frequency ASR of the battery was about 1.7 Ω cm2 while the polarization ASR was 6.8 Ω cm2 due to the slow kinetics of the redox couples. Linear sweep voltammetry tests of the charged cell demonstrated an OCV about 1.9 V and a peak power density of 280 mW cm−2 (Fig. 4b). Crossover of hydroxide was about 10 nmol s−1 cm−2, due to the utilization of high concentration of NaOH (>2 M) for supporting Na2Zn(OH)4.19 Such crossover rates can cause cycling instability in the long run by decreasing the pH difference between negolyte and posolyte. This can be mitigated by driving water splitting across the cell. As shown in Fig. 4c and d, by applying 3 V during charging for about 1 min while cutting the flow rate to 2 mL min−1, the pH difference between the posolyte and the negolyte increased. While water splitting serves as an ideal proton pump, forced OER and hydrogen evolution reaction (HER) can be problematic. Although they produce protons, unfavored side reactions like the oxidation of iodine and acetates could also happen. A large voltage applied on the cell also drove fast growth of Zn dendrites on the negolyte side, leading to a faster cell failure. Passive reactions like ORR and HOR might be safer and more controllable proton pumps in some cases. ex situ HOR can also serve as a proton source for posolytes when suitable. We provided an external source of hydrogen gas to react with posolyte, catalyzed by Pt, to generate protons, as shown in Fig. S10 (ESI†).
 | ||
| Fig. 4 pH recovery in a pH-decoupling electrodepositing battery. (a) Chemistry and properties of the cell. Reactions during charging are marked with red arrows, and reactions during discharging are marked with blue arrows. Iodide was added to accelerate and complete the reduction of MnO2. Mn(OAc)2, HOAC and other small ions exhibit unwanted crossover through the CEM. (b) Cell voltage and power density of the cell during discharge. (c) Reactions during over-charging driven water splitting. (d) pH of the cell during charging (green shade) and over-charging (red-shade). | ||
Runing proton-coupled electrochemical reactions as proton pumps to balance acid/base crossover can be an in situ, inexpensive and convenient method to maintain operational stability of pH-decoupling ARFBs. However, as demonstrated, the rebalancing reactions need to be carefully chosen to avoid unwanted side reactions. pH-decoupling cells demonstrated in this work all have unwanted side-reactions or operational challenges to some extent. There are ways to suppress side-reactions. For example, employing a sub-cell with catalysts for reactions that function as proton pumps, or directly integrating catalysts in the main cell, can possibly increase the selectivity of reactions (Fig. 5a).37 Yet the key is to find proton-coupled electrochemistry that matches the redox couples utilized in the cell. As long as the cell remains balanced, self-discharging reactions can be ideal for maintaining pH, involving only water chemistry (2e ORR, 4e ORR, OER, peroxide oxidation, HER, HOR), resulting in the proton pumping rate matching the acid–base crossover rate at steady-state (Table S2, ESI†).29,32,38
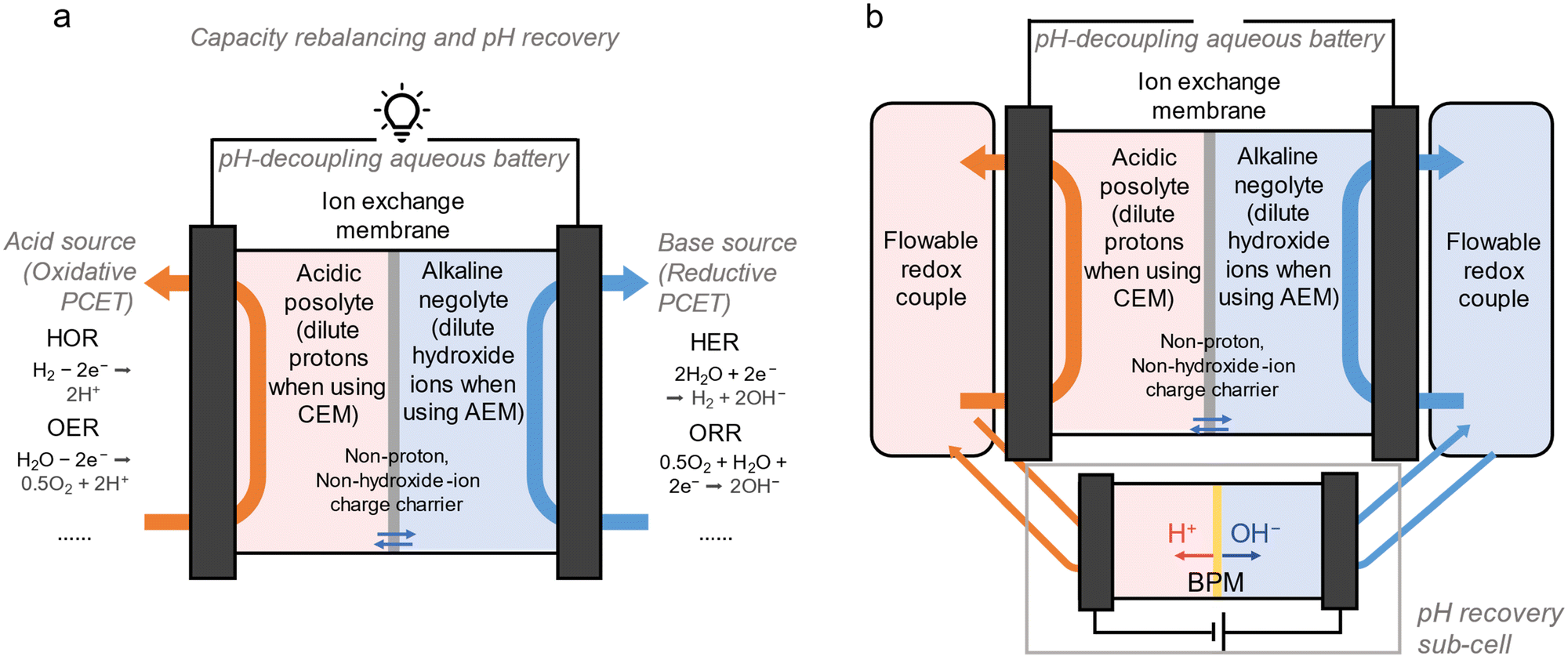 | ||
| Fig. 5 pH-decoupling ARFBs with different pH recovery strategies. (a) A pH-decoupling cell using proton-coupled electrochemistry for capacity rebalancing and pH recovery. The reactions functioning as proton pumps, identified as acid source and base source, may occur in the cell as in situ pH recovery or outside the cell as ex situ pH recovery. Catalysts for proton pumps can be introduced to lower the overpotential of these PCET reactions. (b) A pH-decoupling cell using a BPM sub-cell for pH recovery. The cell must contain flowable redox molecules to drive water dissociation in the BPM cell. | ||
Using water dissociation as a proton pump in BPM sub-cells is more compatible with less robust redox couples and is more controllable because it only requires normal discharging and generates acid and base symmetrically.19 But it can be difficult to use in redox couples involving electrodeposition. To utilize a BPM sub-cell for pH recovery in pH-decoupling electrodeposition batteries, redox molecules of which both redox states are soluble must be added (Fig. 5b). For example, in the case of MnO2/Mn(OAc)2 that uses iodide/iodine as a redox mediator, iodine can serve as a flowable redox couple to be discharged in the BPM cell. Generally, a small concentration of always-soluble redox mediators that have a lower redox potential than that of the posolyte species, but a higher potential than that of the negolyte species, can be deliberately added into electrolytes to realize BPM pH recovery (Note S3, ESI†).
Proton-coupled electrochemistry and water dissociation as proton pumps are interconnected strategies. In instances where the rate of acid–base generation through proton-coupled electrochemistry fails to match the acid–base crossover rate,3 the utilization of a BPM sub-cell can help stabilize the pH. In some special cases, BPM sub-cells can be used to maintain a uniform pH in ARFBs that have ideally zero acid–base crossover rate but have proton coupled self-discharging reactions. If the cell goes out of balance, however, due to asymmetric self-discharge, the BPM sub-cell cannot rebalance the cell. But if self-discharging reactions are proton-coupled water electrochemistry, e.g. the HER, then proton-coupled water electrochemistry can be applied to one side as a proton pump, balancing the cell capacity and acid-bass loss. For example, one can drive the OER when the cell is over-oxidized due to negolyte self-discharging, or one can drive the ORR when the cell is over-reduced due the posolyte self-discharging. Through coupled generation or neutralization of protons and hydroxide, maintaining the cell balance with proton-coupled electrochemistry helps adjust the pH of posolyte and negolyte symmetrically.
Conclusion
With suitable redox couples, pH-decoupling can be realized in a conventional single membrane cell architecture, exhibiting acceptably low acid–base crossover flux and lower ohmic resistance compared to multi-membrane cells. In CEM pH-decoupling ARFBs, it's possible to employ a high concentration of strong base in the negolyte against a posolyte with a dilute proton concentration. AEM type pH-decoupling ARFBs, if outfitted with a highly selective AEM, can use highly concentrated strong acid posolyte against a negolyte with a dilute hydroxide concentration. In order to avoid rapid pH drifting during cell operation, buffer salts are used on the mild pH side to increase the proton buffer capacity under working pH. With proton-coupled electrochemistry functioning as proton pumps, pH drifting can be recovered and the pH difference between the posolyte and the negolyte can be maintained. This strategy can also be applied to pH-decoupling electrodeposition batteries, due to their potential for in situ pH recovery. We found water-related proton-coupled chemistry, if not introducing unwanted side reactions, to be ideal as a proton pump. In comparing in situ and ex situ pH recovery strategies and considering the relationship between cell capacity balance and pH balance, we found that proton-coupled electrochemistry can work together with BPM based proton pumps to stabilize both the cell capacity balance and pH. Despite limitations associated with the stability, dendrite formation, metal ion crossover, buffer molecule interference, slow kinetics, or unwanted side reactions of most redox couples examined in this study, the concept of single membrane pH-decoupling with engineering of acid–base crossover and pH recovery can greatly broaden the scope for development of electrolyte chemistry and guide future works looking into pH-decoupling ARFBs in greater depth.Author contributions
D. X., Z. Y., and A. M. A. designed and conducted cell tests and electrochemical experiments. D. X. conceived the idea. M. J. A. supervised the project. All authors drafted and edited the manuscript.Data availability
The datasets analyzed and generated during the current study are included in the paper, its ESI† and uploaded to Figshare at https://doi.org/10.6084/m9.figshare.25962982.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by U. S. DOE award DE-AC05-76RL01830 through PNNL subcontract 654799 and by the Harvard Climate Change Solution Fund. A. M. A acknowledges the MSE department at KFUPM and the Ministry of Education of Saudi Arabia for doctoral scholarship. The authors thank James MacArthur from Harvard Electronic Instrument Design Lab for developing multi-channel pH sensors.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- Y. Liang and Y. Yao, Nat. Rev. Mater., 2022, 8, 109–122 CrossRef.
- Y. Yao, J. Lei, Y. Shi, F. Ai and Y.-C. Lu, Nat. Energy, 2021, 6, 582–588 Search PubMed.
- R. Dmello, J. D. Milshtein, F. R. Brushett and K. C. Smith, J. Power Sources, 2016, 330, 261–272 CAS.
- L. Zhang, R. Feng, W. Wang and G. Yu, Nat. Rev. Chem., 2022, 6, 524–543 CrossRef PubMed.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Haupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater, 2016, 2, 1–18 Search PubMed.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 RSC.
- M. L. Perry, K. E. Rodby and F. R. Brushett, ACS Energy Lett., 2022, 7, 659–667 CrossRef CAS.
- Y.-h Zhu, Y.-f Cui, Z.-l Xie, Z.-b Zhuang, G. Huang and X.-b Zhang, Nat. Rev. Chem., 2022, 6, 505–517 CrossRef CAS PubMed.
- C. Lin, S.-H. Kim, Q. Xu, D.-H. Kim, G. Ali, S. S. Shinde, S. Yang, Y. Yang, X. Li, Z. Jiang and J.-H. Lee, Matter, 2021, 4, 1287–1304 CrossRef CAS.
- Z. Yan, R. J. Wycisk, A. S. Metlay, L. Xiao, Y. Yoon, P. N. Pintauro and T. E. Mallouk, ACS Cent. Sci., 2021, 7, 1028–1035 CrossRef CAS PubMed.
- G.-M. Weng, C.-Y. V. Li and K.-Y. Chan, J. Electrochem. Soc., 2013, 160, A1384 CAS.
- S. Gu, K. Gong, E. Z. Yan and Y. Yan, Energy Environ. Sci., 2014, 7, 2986–2998 RSC.
- K. Gong, X. Ma, K. M. Conforti, K. J. Kuttler, J. B. Grunewald, K. L. Yeager, M. Z. Bazant, S. Gu and Y. Yan, Energy Environ. Sci., 2015, 8, 2941–2945 RSC.
- A. S. Metlay, B. Chyi, Y. Yoon, R. J. Wycisk, P. N. Pintauro and T. E. Mallouk, ACS Energy Lett., 2022, 7, 908–913 CrossRef CAS.
- M. Park, E. S. Beh, E. M. Fell, Y. Jing, E. F. Kerr, D. Porcellinis, M. A. Goulet, J. Ryu, A. A. Wong, R. G. Gordon, J. Cho and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900694 CrossRef.
- D. Xi, A. M. Alfaraidi, J. Gao, T. Cochard, L. C. I. Faria, Z. Yang, T. Y. George, T. Wang, R. G. Gordon, R. Y. Liu and M. J. Aziz, Nat. Energy, 2024, 9, 479–490 CrossRef CAS.
- F. Yu, L. Pang, X. Wang, E. R. Waclawik, F. Wang, K. K. Ostrikov and H. Wang, Energy Storage Mater., 2019, 19, 56–61 CrossRef.
- J. Lei, Y. Yao, Y. Huang and Y.-C. Lu, ACS Energy Lett., 2022, 8, 429–435 CrossRef.
- C. Li, W. Wu, P. Wang, W. Zhou, J. Wang, Y. Chen, L. Fu, Y. Zhu, Y. Wu and W. Huang, Adv. Sci., 2019, 6, 1801665 CrossRef.
- X. Yuan, X. Wu, X. X. Zeng, F. Wang, J. Wang, Y. Zhu, L. Fu, Y. Wu and X. Duan, Adv. Energy Mater., 2020, 10, 2001583 CrossRef CAS.
- M. Mousavi, H. Dou, H. Fathiannasab, C. J. Silva, A. Yu and Z. Chen, Chem. Eng. J., 2021, 412, 128499 CrossRef CAS.
- J. Zhang, G. Jiang, P. Xu, A. G. Kashkooli, M. Mousavi, A. Yu and Z. Chen, Energy Environ. Sci., 2018, 11, 2010–2015 RSC.
- J. Gao, K. Amini, T. Y. George, Y. Jing, T. Tsukamoto, D. Xi, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2022, 12, 2202444 CrossRef CAS.
- Y. Zhu, Y. Li, Y. Qian, L. Zhang, J. Ye, X. Zhang and Y. Zhao, J. Power Sources, 2021, 501, 229984 CrossRef CAS.
- Y. Jing, K. Amini, D. Xi, S. Jin, A. M. Alfaraidi, E. F. Kerr, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2023, 9, 3526–3535 CrossRef.
- T. Kong, J. Liu, X. Zhou, J. Xu, Y. Xie, J. Chen, X. Li and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214819 CAS.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Joule, 2019, 3, 2942–2954 CrossRef CAS.
- Y. Ji, M. A. Goulet, D. A. Pollack, D. G. Kwabi, S. Jin, D. Porcellinis, E. F. Kerr, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900039 CrossRef.
- B. H. Robb, J. M. Farrell and M. P. Marshak, Joule, 2019, 3, 2503–2512 CrossRef CAS.
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W.-H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 4412 CrossRef CAS.
- J. Lei, Y. Zhang, Y. Yao, Y. Shi, K. L. Leung, J. Fan and Y.-C. Lu, Nat. Energy, 2023, 1–10, DOI:10.1038/s41560-023-01370-0,.
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng, N. Zhao and W. Hu, Nat. Energy, 2020, 5, 440–449 CrossRef CAS.
- R. Thamizhselvan, R. Naresh, M. Ulaganathan, V. G. Pol and P. Ragupathy, Electrochim. Acta, 2023, 441, 141799 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- S. E. Waters, B. H. Robb, S. J. Scappaticci, J. D. Saraidaridis and M. P. Marshak, Inorg. Chem., 2022, 61, 8752–8759 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00279b |
| This journal is © The Royal Society of Chemistry 2024 |