
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-free Wacker-inspired oxidation: challenges and opportunities in catalysis
Inderpal
Yadav
 and
Rafael
Gramage-Doria
*
and
Rafael
Gramage-Doria
*
Univ Rennes, CNRS, ISCR–UMR6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
First published on 30th April 2025
Abstract
Palladium-catalysed Wacker-type oxidation of olefins to ketones or aldehydes is one of the most prominent homogeneous reactions globally, even performed at a multi-tonne scale. From a fundamental and an applied point of view, it is extremely appealing to replace palladium catalysts by other metal-based catalysts to increase the efficiency, selectivity and sustainability, particularly considering the reactivity of well-defined first-row transition metal complexes as catalysts. In this case, the ligand(s) coordinating to the metal(s) play a major role in controlling selectivity and activity, thanks to unique mechanistic considerations. This mechanistically-driven feature article emphasizes the advantages and disadvantages of currently existing approaches. Besides the main efforts devoted to homogeneous catalysis, heterogenous systems and biocatalysis have also been studied as they offer complementary strategies. Overall, this review presents an up-to-date analysis of key contributions while highlighting existing gaps for future developments in this important and exciting field.
 Inderpal Yadav | Inderpal Yadav was born in Khatod, Haryana, India. He received his master's degree in Chemistry from the University of Delhi (Hindu College), India in 2017. He did his PhD studies under the supervision of Prof. M. Sankar at the Indian Institute of Technology Roorkee, India. His doctorate research was focused on the synthesis of corroles for sensing, nonlinear optical, and catalytic applications. He is currently a postdoctoral researcher at the University of Rennes (France) in the group of Dr Rafael Gramage-Doria. His current research interests include porphyrinoid synthesis, organometallic, coordination and supramolecular chemistry, and homogeneous catalysis. |
 Rafael Gramage-Doria | Rafael Gramage-Doria has been a CNRS researcher at Institut des Sciences Chimiques de Rennes in the Université of Rennes (France) since 2015. He received his PhD from the University of Strasbourg, followed by postdoctoral appointments in the University of Amsterdam and Nagoya University. His research interests include transition metal catalysis for fine chemicals and green chemistry applications, C–H bond functionalization, supramolecular and coordination chemistry, and supramolecular and bio-inspired catalysis. He is a co-author of >60 research articles, 4 book chapters and 2 international patents. His contributions have been recognized by the French Chemical Society SCF (Jean-Pierre Sauvage DCO award, Young Investigator Catalysis DivCat award and SCF junior distinguished fellow) and by the Spanish Chemical Society RSEQ (Young Investigator GEQO award). |
Introduction
Palladium complexes are privileged catalysts, especially when considering the oxidation of olefins to ketones or aldehydes since the groundbreaking discoveries in the Wacker-Chemie company in the 1950s.1 In this case, the industrially-relevant ethylene starting material is oxidized to acetaldehyde in this now-famous process, which is catalysed by palladium with copper as a redox co-catalyst in an acidic environment with high oxygen pressure.2 This approach has been industrially refined over the years to achieve multi-tonne production of acetaldehyde as well as related carbonyl-containing fine-chemicals that are important building blocks and daily-life commodities.3 Importantly, these palladium-catalysed processes are highly selective since they avoid the formal cleavage of the carbon–carbon double bond olefin observed when using other metal catalysts under oxidizing conditions. As such, in the prototypical case of starting from terminal olefins, the reaction can produce either the ketone product via a Markovnikov-type intermediate or the terminal aldehyde via an anti-Markovnikov-type intermediate (Scheme 1).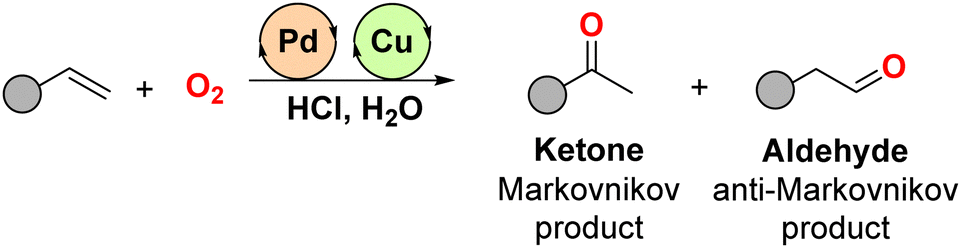 | ||
| Scheme 1 Well-established Pd-catalysed Wacker-type oxidation of olefins to ketones and aldehydes with product selectivity being controlled by the nature of the ligand attached to palladium and/or by the reaction conditions. | ||
The Wacker-type oxidation of olefins presents product selectivity challenges, which are largely governed by the electronic and steric bias at the olefin starting material and also by the nature of the palladium-coordinated ligand. In addition, product selectivity can eventually be tuned through careful control of reaction parameters such as oxygen pressure, temperature, solvent, reaction time, and additives.4 Generally, while there is a strong preference for Markovnikov selectivity when using standard palladium complexes as catalysts, a reversal of selectivity towards anti-Markovnikov products is feasible in the presence of nitrite salts.4 However, achieving full regioselective control in the oxidation of internal olefins is notably more difficult. This is a consequence of the fact that the bond dissociation energies of the relevant chemical bonds fall within a narrow range, making it almost impossible for the palladium catalyst to discriminate between both olefin carbon atoms. Additionally, Pd-catalysed oxidation of olefins to carbonyl derivatives may face competition from other transformations, particularly when sensitive functional groups are present in the starting olefin.
To overcome these limitations in Pd-catalysed olefin oxidation, alternative metal catalysts have been extensively explored over the past decades.5 Beyond addressing selectivity concerns, an important area of research focuses on conducting these transformations under environmentally benign conditions by either replacing or avoiding the use of palladium as well as simplifying the reaction setup to ensure compatibility with a broad range of functional groups. This feature article emphasizes the main contributions to homogeneous Wacker-type oxidation reactions using noble as well as 3d transition metal homogeneous catalysts beyond palladium. In particular, mechanistic studies are discussed herein as they are relevant for controlling and/or altering the fundamental steps of catalytic cycles, which may disclose more powerful catalysts for this unique transformation of strong interest in industry and academia. We also pay significant attention to catalytic approaches involving bio-, electro- and photo-catalysis as well as heterogeneous catalysis.
Early studies with platinum-group metals (PGM)
In the 1970s and 1980s, independent contributions by the groups of Read,6 Mimoun,7 Drago,8 Bressan,9 Khan,10 and others11 focused on utilizing metal complexes isoelectronic to palladium(II) as prospective catalysts for the oxidation of olefins to ketones or aldehydes. Aside from one example with platinum,12 interesting results were observed when employing rhodium and ruthenium complexes (Fig. 1).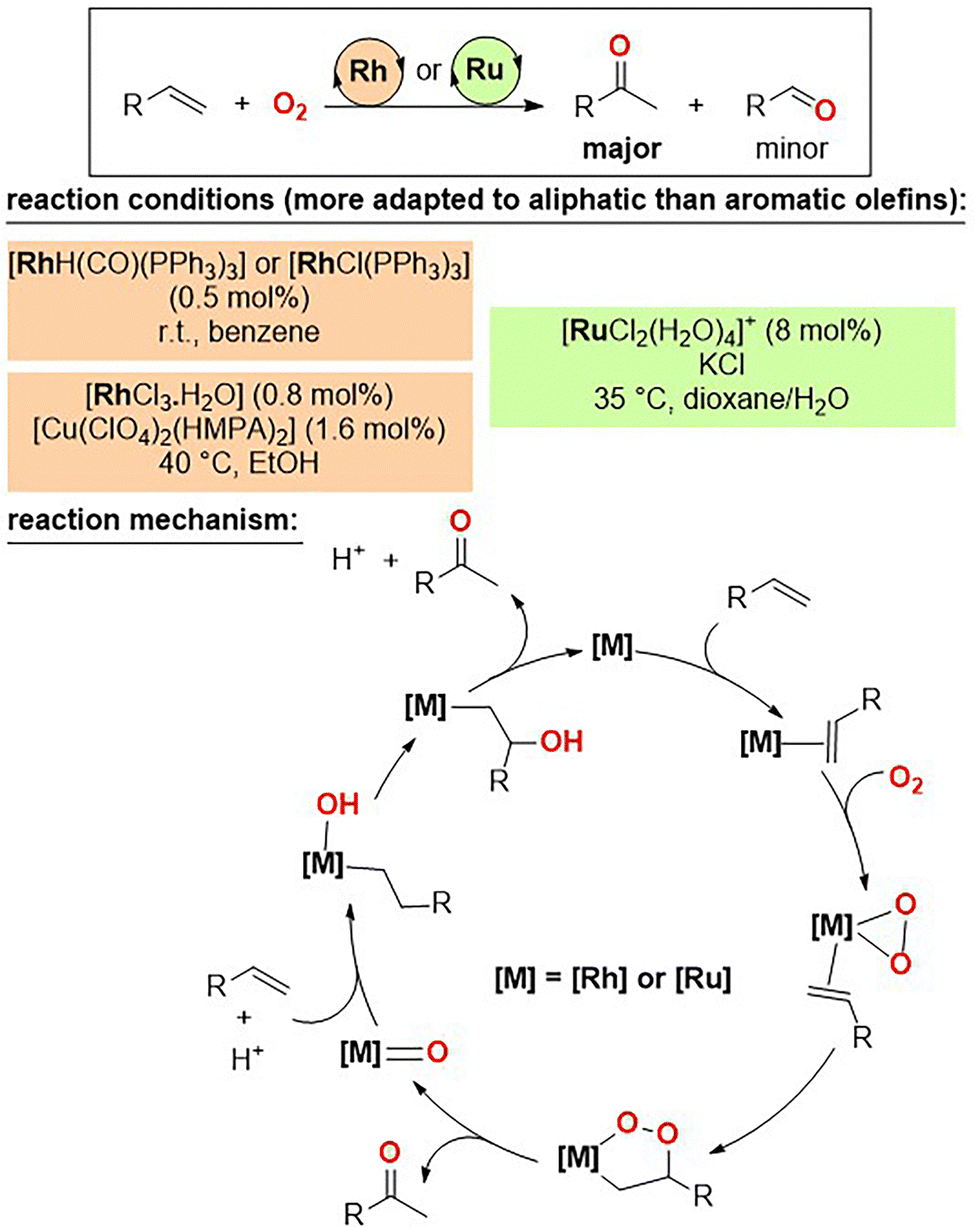 | ||
| Fig. 1 Selected examples of rhodium and ruthenium complexes as catalysts for the Wacker-type oxidation of olefins to ketones (top) and the postulated reaction mechanism (bottom). HMPA = hexamethylphosphoramide, R = alkyl or aryl. | ||
For instance, rhodium(I) complexes such as [RhH(CO)(PPh3)3] and [RhCl(PPh3)3] turned out to be catalytically active in the formation of methyl ketones from aliphatic olefins in the presence of a pure O2 atmosphere at room temperature, although minor formation of aldehydes resulting from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond scission was observed.6a,b The presence of catalytic amounts of copper salts such as [Cu(ClO4)2(HMPA)2] when using [RhCl3·H2O] as the pre-catalyst suppressed the formation of this side-product.7c Similarly, [RuCl2(H2O)4]+ enabled the oxidation of aliphatic olefins to methyl ketone derivatives reaching a turnover frequency (TOF) value of up to 62 h−1 for the formation of 2-hexanone from 1-hexene under mild conditions.10 In these examples, which appear more adapted to aliphatic substrates rather than aromatic ones, catalyst decomposition (i.e. phosphine oxidation) was a major issue. A number of selected mechanistic studies including kinetic analysis suggested the transient formation of metal-peroxo and metal-dioxetan species in the catalytic cycle (Fig. 1, bottom). Although further research should be carried out to identify the oxidation state and precise chemical structure of the reaction intermediates and transition states, the reaction mechanism here strongly differs from that accepted with palladium and one may wonder whether immobilization of such catalysts in heterogeneous systems could improve the current state-of-the-art.
C bond scission was observed.6a,b The presence of catalytic amounts of copper salts such as [Cu(ClO4)2(HMPA)2] when using [RhCl3·H2O] as the pre-catalyst suppressed the formation of this side-product.7c Similarly, [RuCl2(H2O)4]+ enabled the oxidation of aliphatic olefins to methyl ketone derivatives reaching a turnover frequency (TOF) value of up to 62 h−1 for the formation of 2-hexanone from 1-hexene under mild conditions.10 In these examples, which appear more adapted to aliphatic substrates rather than aromatic ones, catalyst decomposition (i.e. phosphine oxidation) was a major issue. A number of selected mechanistic studies including kinetic analysis suggested the transient formation of metal-peroxo and metal-dioxetan species in the catalytic cycle (Fig. 1, bottom). Although further research should be carried out to identify the oxidation state and precise chemical structure of the reaction intermediates and transition states, the reaction mechanism here strongly differs from that accepted with palladium and one may wonder whether immobilization of such catalysts in heterogeneous systems could improve the current state-of-the-art.
The epoxidation–isomerization (EI) pathway leads to anti-Markovnikov selectivity
A major breakthrough for accomplishing the selective anti-Markovnikov Wacker-type oxidation of olefins to aldehydes came from Che's laboratory in 2004.13 The strategy relies on a key oxygen atom transfer event from the metal catalyst to the olefin via formal epoxidation followed by isomerization (Fig. 2, bottom). As such, the use of metal complexes amenable to the formation of metal-oxo species was envisioned. For that, the combination of ruthenium porphyrin derivatives with over-stoichiometric amounts of oxygen atom sources (i.e. oxidants) was considered as a suitable catalytic system. Although a first generation of ruthenium porphyrin catalysts comprising an octa-chlorinated pattern (Ru-1, Fig. 2) operated with N-oxides as reagents (i.e. Cl2pyNO), improved ruthenium porphyrin catalysts (Ru-2 and Ru-3, Fig. 2) allowed the use of O2 from the air atmosphere even at room temperature. In these cases, the ruthenium active site is well buried and sterically protected in the porphyrin core due to the large and bulky mesityl groups placed in the meso position or the methyl and terphenyl patterns replacing the pyrrolic protons (Fig. 2). The reactions were highly selective and efficient for aldehyde formation from styrene derivatives as exemplified by the fact of reaching turnover number (TON) values of up to 1144 at 60 °C under a pure O2 atmosphere when employing 4-methoxystyrene as the substrate. The reactions were also compatible with conjugated dienes containing aromatic handles such as 1-phenyl-1,3-butadiene and related ones, thus significantly reducing the reactivity at the benzylic position, which is attainable with other strategies (vide infra). However, almost negligible reactivity was observed for the more demanding aliphatic olefins.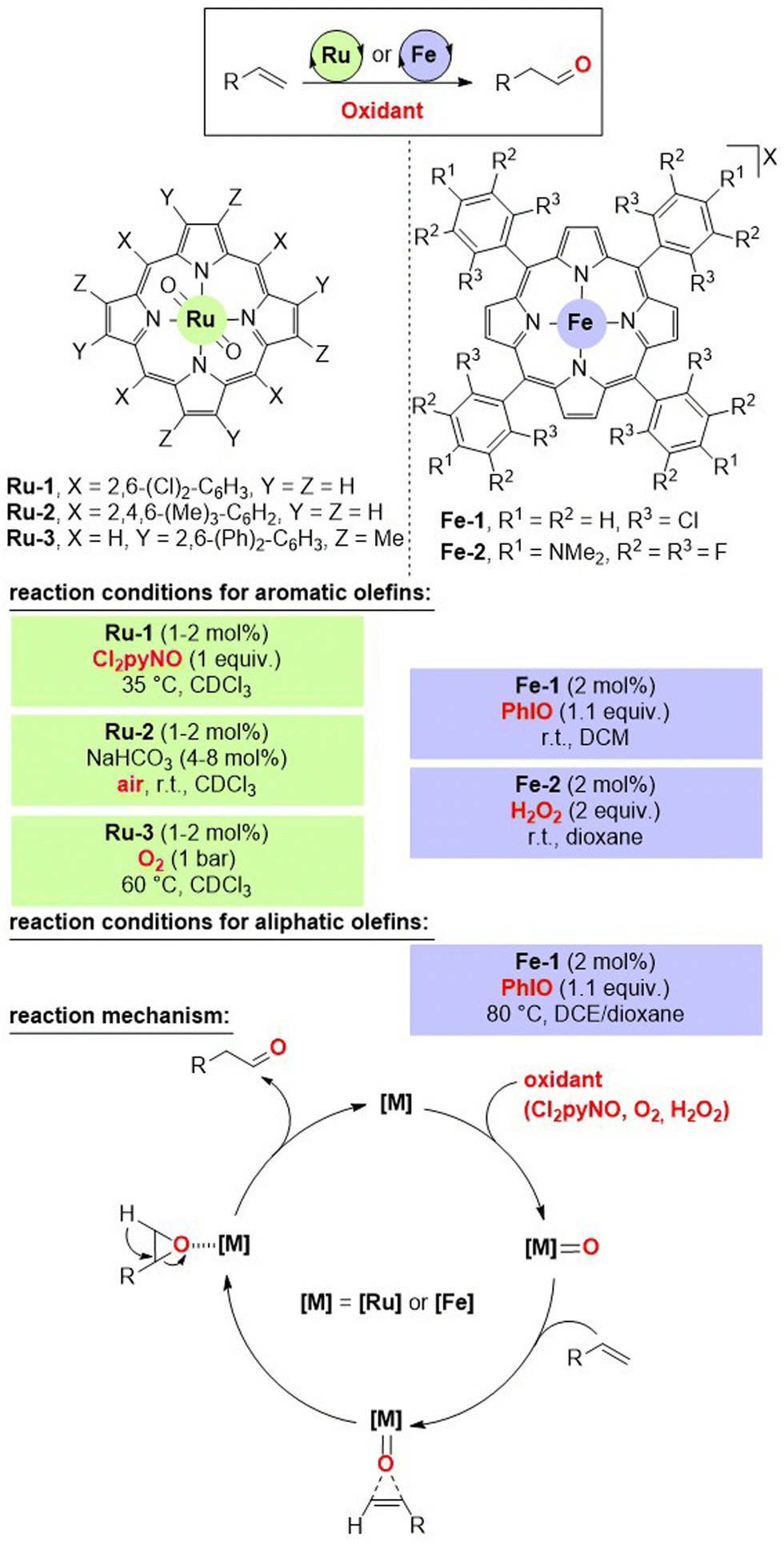 | ||
| Fig. 2 Wacker-type anti-Markovnikov oxidation of olefins to aldehydes using ruthenium- or iron-based porphyrin catalysts via the epoxidation/isomerization (EI) pathway. Cl2pyNO = 2,6-dichloropyridine N-oxide, R = aryl, alkyl or alkenyl, X = BArF, OTf, BF4, PF6, or SbF6. | ||
The same group realized in 2011 a significant advancement by replacing the catalytically active ruthenium(IV) centre by iron(III) while keeping a similar porphyrinoid environment as the ligand (Fig. 2).14 The fine-tuning of the structure of the porphyrin backbone has a strong impact on the reactivity of the iron centre. For instance, the octa-chlorinated iron catalyst Fe-1 was active for the selective oxidation of terminal olefins including aliphatic ones to aldehydes upon the use of PhIO, which is a relatively stronger oxidant than the ones used for ruthenium catalysis (Fig. 2). Interestingly, it was possible to use the highly benign H2O2 as an oxidant when using an iron porphyrin catalyst containing tetrafluoro-para-dimethylamino groups at all four meso positions (Fe-2, Fig. 2). In the case of the iron catalysis, it was necessary to use catalytic amounts of halide scavengers such as NaBArF [BArF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate] or AgX (X = OTf, BF4, PF6, SbF6) to ensure the presence of a cationic iron(III) species before entering the catalytic cycle. In fact, when the iron-porphyrin catalysts were not cationic, substantial amounts of epoxides were formed as side-products, thereby indirectly demonstrating the dual role of iron in both epoxide formation and isomerization. The iron-based catalytic systems proved so powerful that they enabled the so far elusive, at that time, oxidation of disubstituted terminal olefins to aldehydes at room temperature using catalyst loadings below 1 mol%.
Similar reactivity was later found using a combination of the iron salt [Fe(BF4)2] and the pyridine-2,6-dicarboxylic acid ligand as the catalytically active system.15 Alternatively, it is worth noting that to overcome the poor reactivity and selectivity for internal olefins, Coates and coworkers performed a two-step reaction sequence involving epoxidation followed by isomerization using a hybrid aluminium/cobalt catalyst; however, this is beyond the scope of this feature article because it does not involve a formal oxidation reaction.16 In the same vein, processes enabling the selective ring-opening of terminal epoxide towards aldehydes (anti-Markovnikov products) or methyl ketones (Markovnikov products) by means of metal catalysts have been reported and reviewed elsewhere.17
Biocatalysis forms aldehydes via a carbocation intermediate
It is clear that the formation of metal-oxo species, in particular heme-like iron-oxo species, is decisive for the formation of aldehydes via the epoxidation/isomerization pathway. In this respect, and based on the promiscuous activity encountered in some cytochromes from the P450 family and related systems,18 the Arnold group pioneered the directed evolution of such biocatalysts in order to reach the anti-Markovnikov oxidation of olefins to aldehydes in the presence of air as the oxygen atom source and NADPH as the source of hydrogen atoms (Fig. 3).19 This biocatalyst approach, and further optimizations by the Hammer group in combination with computational calculations by Garcia-Borras, established a unique action mode, in which the carbonyl-leading pathway operates via a radical–carbocation transient formation and intramolecular electron transfer step followed by a final 1,2-hydride migration (Fig. 3, bottom). The former is controlled by enzymatic conformation and takes place in a stereoselective manner, thereby accessing enantiomerically-enriched aldehydes from prochiral olefins. Importantly, the electrostatic environment in the enzyme active site stabilizes this so far elusive type of carbocation intermediate. This biocatalytic approach is compatible with cascade and tandem transformations as exemplified in the sequential use of the alcohol dehydrogenase (ADH) enzyme en route to chiral alcohols. The system is highly efficient with world-record breaking TON values for styrene derivatives including 1,2-disubstituted ones and is significantly less productive for aliphatic olefins. | ||
| Fig. 3 Biocatalytic anti-Markovnikov oxidation of olefins to aldehydes under ambient conditions. NAD(P)H = reduced form of nicotinamide adenine dinucleotide (phosphate), ADH = alcohol dehydrogenase enzyme, R = aryl or alkyl, Z = H or alkyl. | ||
Merging hydrogen-atom transfer with metal-alkylperoxo catalysis for Markovnikov selectivity
Much inspired by the work of Baran and others disclosing the possibility to form iron-hydride species relevant for the formation of carbon radical intermediates upon reaction with olefins,20 Han and co-workers developed in 2017 the very first example of iron-catalysed oxidation of olefins to ketones (Fig. 4).21 The reactions required the use of, at least, stoichiometric amounts of a hydrosilane derivative as the hydrogen atom source and O2 from air. Han's initial contribution used FeCl2 as the catalyst in relatively high loadings (10–20 mol%) and polymethylhydrosiloxane (PMHS), which is a very affordable hydrosilane originating as a chemical waste from the silicon-based industry under air at 80 °C. In this way, the reaction was compatible with both aliphatic and aromatic olefins. Terminal olefins delivered mainly ketone products with trace amounts of aldehydes being observed in some cases. Later, the Knölker group reported a number of well-defined iron complexes that allowed similar reactivity leading to ketone products from olefins.22 The ligands play a substantial role in stabilizing the catalytically active iron species, making it possible to reduce the catalyst loading below 5 mol% while using different types of hydrosilanes (i.e. PhSiH3, Et3SiH) under ambient pressure and temperature conditions. Of particular relevance was the identification of phthalocyanine- (Fe-3), porphyrin- (Fe-4), phenanthroline- (Fe-5) and acac-type (Fe-6) ligands as suitable ones for these oxidation reactions (Fig. 4). Importantly, the reaction conditions developed by the groups of Han and Knölker were successfully applied to the synthesis of compounds of biological relevance.21,22 In parallel, our research group developed a tetracarboxylic acid iron-porphyrin (Fe-7) that enabled to perform such reactions at ppm levels of catalyst loading reaching high turnover numbers of ca. 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and making the catalysis compatible with the oxidation of unactivated, aliphatic olefins to ketones under ambient pressure and temperature conditions (Fig. 4).23 Mechanistic investigations allowed the identification of initial iron-hydride species via the reaction of the iron complex with the hydrosilane reagent whereas kinetic studies revealed a partial first order in catalyst concentration. As such, the μ-oxo bridged di-iron species are likely inactive for the catalysis. In the iron catalysts developed by Knölker,22 ourselves23 and others24 using well-defined iron complexes as catalysts, traces of alcohol side-products were observed in few selected cases, but mostly for aliphatic olefins. Unfortunately, these reactions are so far unselective for (unbiased) asymmetric, internal olefins because two different ketone products can readily form.
000 and making the catalysis compatible with the oxidation of unactivated, aliphatic olefins to ketones under ambient pressure and temperature conditions (Fig. 4).23 Mechanistic investigations allowed the identification of initial iron-hydride species via the reaction of the iron complex with the hydrosilane reagent whereas kinetic studies revealed a partial first order in catalyst concentration. As such, the μ-oxo bridged di-iron species are likely inactive for the catalysis. In the iron catalysts developed by Knölker,22 ourselves23 and others24 using well-defined iron complexes as catalysts, traces of alcohol side-products were observed in few selected cases, but mostly for aliphatic olefins. Unfortunately, these reactions are so far unselective for (unbiased) asymmetric, internal olefins because two different ketone products can readily form.
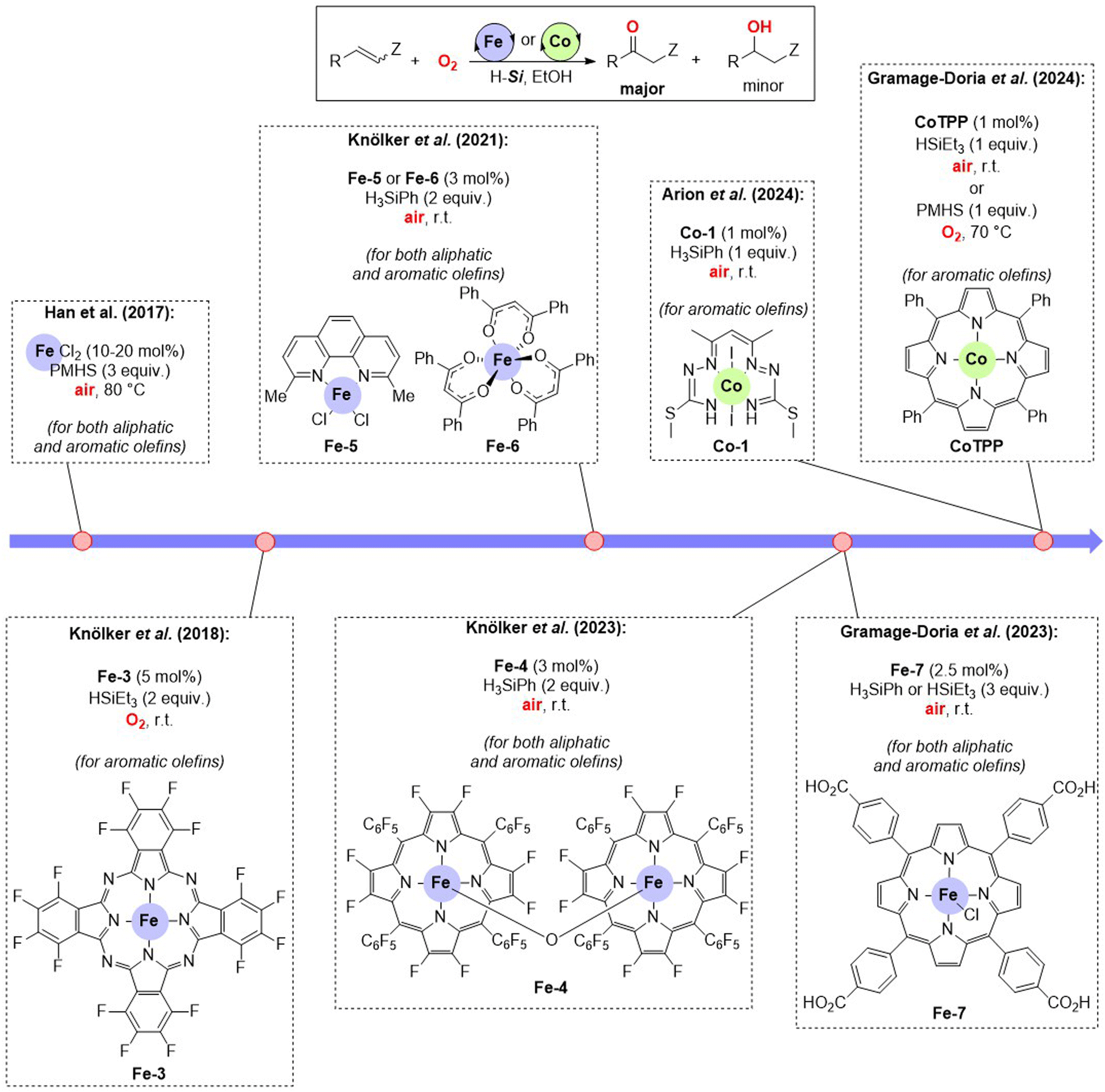 | ||
| Fig. 4 Markovnikov-selective oxidation of olefins to ketones under iron and cobalt catalysis, respectively. R = aryl or alkyl, Z = H or alkyl. | ||
The contributions by Knölker's group using Mössbauer spectroscopy including inverse magnetic susceptibility, radical trapping experimentation, and isotopically-labelled 18O-dioxygen as well as employing deuterated hydrosilanes represent the cornerstone regarding the fundamental understanding of the Markovnikov-selective iron-catalysed oxidation of olefins to ketones,25 whose simplified version is depicted in Scheme 2. It begins with iron(III) hydride species (the hydride originating from the hydrosilane reagent) followed by olefin addition towards the formation of the thermodynamically more stable internal iron(III)-alkyl species, which further undergoes radical-mediated dioxygen activation (with O2 from air) and insertion leading to alkyl-peroxo species that finally rearranges affording the ketone product and the iron(hydroxo) species. The alcohol side-products are believed to form via radical recombination in the final step, OH transfer, Russel fragmentation and/or hydride-mediated reduction of the ketone product. Our research group also demonstrated that the formation of other side-products originating from olefin reduction or olefin homo-coupling could be avoided by altering the order of addition of reagents into the reaction mixture.23
 | ||
| Scheme 2 Simplified reaction mechanism for the iron-, cobalt- or nickel-catalysed Markovnikov-selective oxidation of olefins to ketones following the HAT pathway. R = alkyl or aryl. | ||
Alternatively, pioneering contributions in the 1980s and 1990s by the research groups of Drago and Matsushita, independently, dealing with the use of cobalt catalysis for the oxidation of olefins demonstrated the feasibility of Markovnikov-selective ketone formation yet substantial amounts of hydroperoxide and alcohol side-products were present.26 This was somehow circumvented by applying two-step procedures,27 but obviously there was room for improvement. In 2024, our research group showed that cobalt(II)-tetraphenylporphyrin (CoTPP) is an excellent catalyst for the aerobic oxidation of styrenes to acetophenone derivatives using different hydrosilanes as hydrogen sources (Fig. 4).28 At room temperature and under an air atmosphere, the reactions were exceedingly fast occurring within 10 minutes with a very low catalyst loading (1 mol%) using Et3SiH as the hydrogen source while keeping excellent chemoselectivity as well as Markovnikov selectivity (up to 99% of ketone). High TOF values (up to 864 h−1) were achieved for the oxidation of aromatic olefins under these benign conditions. The same catalytic system also operates under stoichiometric amounts of PMHS as a hydrogen source in an oxygen atmosphere at 70 °C in less than 1 hour with TOF values of up to 530 h−1. Very similar observations were made by Arion and co-workers using a well-defined cobalt complex, Co-1, that features a pentane-2,4-dione bis(S-methylisothiosemicarbazone) ligand (Fig. 4).29 The chemical and redox non-innocence nature of the ligand was strongly supported by a combination of advanced spectroscopic techniques and computational calculations that indicated O2 activation by both the cobalt and the ligand, which is relevant for the catalysis to occur. Moreover, the reactions were highly chemoselective leaving unreacted allylic and alkyne functional groups as well as highly compatible with heteroaromatics and biologically-relevant motifs. However, achieving the reactions of aliphatic olefins under Markovnikov-selective cobalt catalysis remains elusive so far even with the recent development of salen-based cobalt catalysts.30 The reaction mechanism for the cobalt-catalysed Markovnikov-selective oxidation of olefins to ketones is most likely similar to that reported with iron catalysts based on a number of spectroscopic, kinetic and DFT calculations (Scheme 2). It is worth noting that this mechanism is reminiscent of the Mukaiyama-type hydration of olefins to yield alcohols,31 albeit the chemoselectivity is shifted towards ketone formation due to a different rate-determining step, thanks to the different nature of the ligand bound to cobalt. Interestingly, these methodologies share the fact that the catalysis is more efficient using ethanol as the solvent than others. The role of ethanol solvent in both cobalt and iron catalysis is very determinant because it appears to be involved in the stabilization of key intermediates via hydrogen bonding as shown by us recently.28 It is worth noting that a similar approach has been disclosed under nickel catalysis by Han and co-workers in 2019.32 Interestingly, the in situ formed phenanthroline-chelated nickel catalyst allows chain walking of the remote olefin double bond to reach the benzylic position in the substrate followed by a Markovnikov-selective oxidation leading to the corresponding acetophenone derivatives (Scheme 3). A catalytic system based on cobalt or nickel applicable to purely aliphatic olefins remains elusive so far.
 | ||
| Scheme 3 Merging chain-walking processes with Wacker-type oxidation under nickel catalysis towards acetophenone derivatives. Ar = aryl. | ||
Dual catalysis: complexity at the service of sustainability
The possibility of altering the reactivity of well-defined transition metal catalysts by in situ combination with additional catalytic events including electro- or photocatalytic approaches represents a unique entry for achieving highly selective transformations.33 In this context, Lei and co-workers developed in 2016 a photocatalytic approach for the anti-Markovnikov oxidation of olefins to ketones, including tri-substituted olefins as well (Fig. 5).34 A cobalt complex behaved as a proton-reducing catalyst, whereas the acridinium photosensitizer participates in the blue LED-mediated generation of a radical cation in the olefin that further undergoes a reaction with water, which is the reagent sourcing the oxygen atom in the final ketone product. In this way, dihydrogen gas is produced as a side-product in this transformation that is conducted in acetonitrile solvent at room temperature. This major breakthrough represents the archetypical example of a reaction with 100% atom-economy. However, this methodology was not compatible with aliphatic olefins.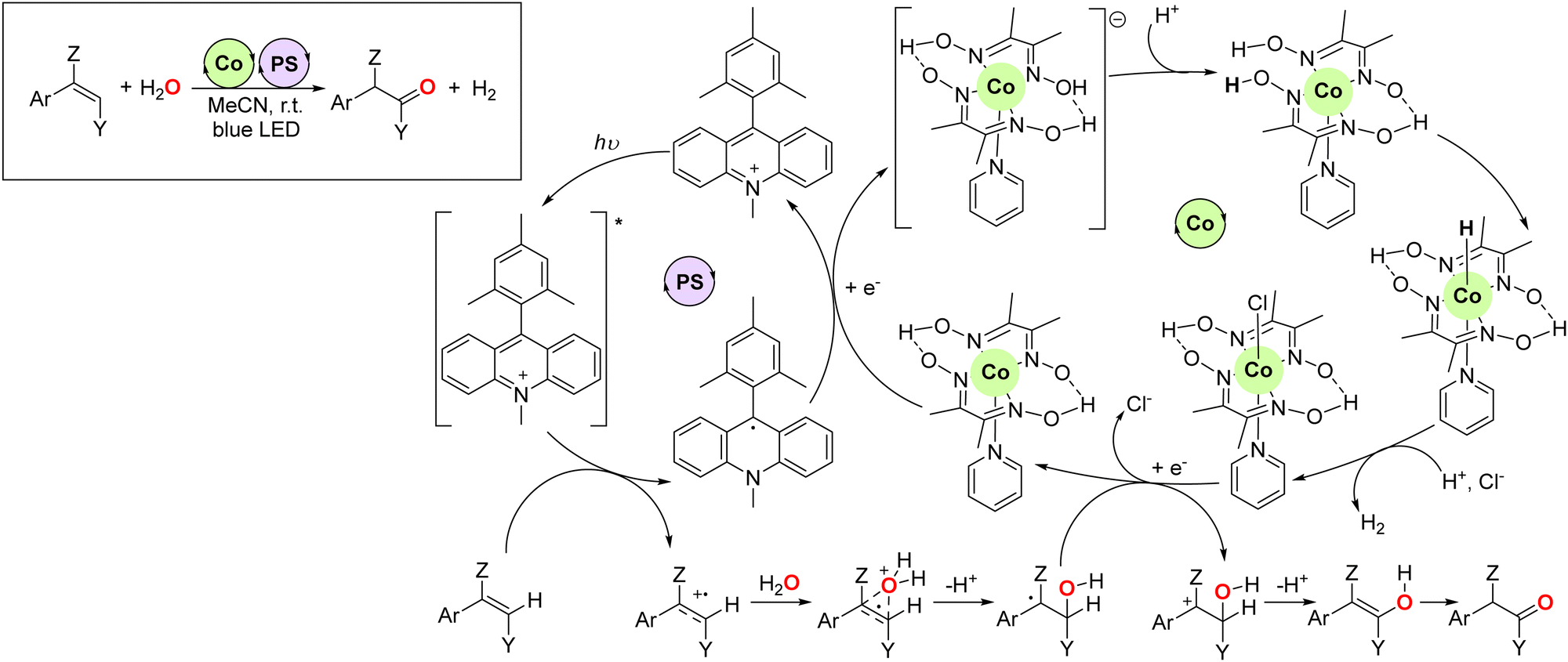 | ||
| Fig. 5 Anti-Markovnikov oxidation of olefins by means of photocatalysis and cobalt-catalysed proton reduction. PS = photosensitizer, Ar = aryl, Z = H or alkyl, Y = alkyl or H. | ||
In 2020, Pericas and co-workers developed an innovative electrochemical approach towards the sustainable oxidation of olefins to ketones by combining catalytic amounts of MnCl2 and CuCl2 in a water/acetonitrile mixture.35 Upon the application of a potential of 2.8 volts, mono- and 1,2-disubstituted aryl olefins were efficiently converted into the corresponding acetophenones with exclusive Markovnikov selectivity (Fig. 6). The catalysis is highly sensitive to steric effects as ortho-substituted olefins afforded lower yields compared to the para regioisomers and no reactivity was reported for purely aliphatic olefin substrates. Importantly, the reaction showed outstanding atom economy because the water from the solvent was formally acting as the source of both oxygen and hydrogen atoms in the final ketone product. The proposed mechanism showed that three parallel oxidative events were simultaneously taking place at the anode: the oxidation of MnII to MnIII, CuII to CuIII, and the acetonitrile molecule to its radical cation.
 | ||
| Fig. 6 Electrochemical Mn/Cu-catalysed Wacker-type oxidation of olefins with Markovnikov selectivity and its reaction mechanism. Ar = aryl, Z = H or alkyl. | ||
In the same year 2020, Milstein and co-workers integrated dual indium and ruthenium homogeneous catalysts for the environmentally friendly oxidation of olefins with excellent Markovnikov selectivity.36 The reaction design relied on the synergistic cooperation of an acid-catalysed hydration using In(OTf)3 as a catalyst and a pincer-Ru-catalysed acceptorless dehydrogenation catalyst (Fig. 7). The catalysis took place in a solvent mixture of water (acting as an oxidant) and dioxane with benign H2 being the sole side-product, thereby resulting in an atom-economical process. The reaction exhibited good compatibility with styrene derivatives containing multiple functional groups, albeit aliphatic olefins led to a mixture of ketones due to double-bond isomerization. On the other hand, strained cyclic olefins (i.e. norbornene, cycloheptene, cyclooctene) were well tolerated, affording the corresponding ketones. Mechanistic investigations pointed out that water serves as the oxygen source in the indium-based catalytic cycle, and dihydrogen is released during the catalytic cycle involving the ruthenium complex.
 | ||
| Fig. 7 Dual ruthenium- and indium-catalysed Wacker-type Markovnikov oxidation of olefins to ketones and mechanistic considerations. Z = H (for R = aryl) or Z = alkyl (for R = alkyl). | ||
Although iridium complexes were evaluated for the oxidation of terminal olefins to ketones and aldehydes, the reactivity was very low and due to the lack of mechanistic insights further research was abandoned until 2021.37 That year, Hong and co-workers envisioned boosting the reactivity of iridium complexes in the presence of additional copper ions. They reported an original bimetallic Ir–Cu catalytic system for the selective oxidation of water-soluble styrenes to the corresponding acetophenones in an aqueous solution (Fig. 8, framed).38 The catalysis was conducted in a formate buffer, leading to both high TON and high Markovnikov selectivity. The Ir–Cu catalyst comprised two metal-binding sites: one N,N-chelating arm was coordinated to [IrCp*Cl] (Cp* = pentamethylcyclopentadienyl), while the other site was coordinated to [Cu(dipic)] (dipic = pyridine-2,6-dicarboxylato). The initial oxidation state of iridium was +3 and that of copper was +2. The synergistic nature of the bimetallic system was demonstrated by the absence of the activity that was found in the individual mononuclear systems. Mechanistic investigations were carried out to detect the reaction intermediates by kinetic and spectroscopic studies along with DFT calculations. The proposed mechanism involved the formation of a substrate-to-catalyst complex via hydrophobic π–π interactions between the aromatic π-cloud of the Ir-coordinated pyridine moiety within the ligand and the aromatic moiety of the styrene reagents (Fig. 8, bottom), which was further supported indirectly by the absence of reactivity of purely aliphatic olefin substrates. The olefin insertion to generate the IrIII-alkyl species in the catalytic cycle is the rate-determining step. Additionally, an IrIII-peroxo species produced by oxygen-mediated insertion regenerated the active IrIII species and reacted with formic acid to create the ketone product through the release of H2O and CO2. The CuII center is thought to increase the reactivity of the IrIII-alkyl intermediate towards O2 in this acidic medium, demonstrating the cooperative action mode of this catalytic system, even though all bond-breaking and bond-forming events take place at the iridium site.
 | ||
| Fig. 8 Bimetallic Ir–Cu-catalysed Wacker-type Markovnikov oxidation of water-soluble aromatic olefins to acetophenones and the postulated reaction mechanism. | ||
Very recently, Reiser and co-workers developed an intriguing concept enabling the selective oxidation of terminal olefins to methyl ketone derivatives by exploiting a copper catalyst that behaves as both a photooxidant and a photoreductant (Fig. 9).39 The reaction proceeded under light irradiation (365 nm) with catalytic amounts of CuCl2 in the presence of KBr, water, and trichloroisocyanuric acid (TCCA) in acetone solvent at mild temperature (50 °C). The reaction mechanism, strongly supported by a large number of control experiments and isotope labelling, involves a TCCA-mediated copper-catalysed oxidation of the olefin starting material to a chlorohydrin intermediate, followed by a reduction to the ketone product merging key hydrogen atom transfer (HAT) and spin-center shift (SCS) steps prior to protodemetallation. While the reductive cycle can occur in the dark, the oxidative one is light-dependent. In this reaction design, water (and not O2 from air) is the source of the oxygen atom in the final product. However, the reaction was only tolerant to aromatic olefins with no examples reported for aliphatic olefin substrates. This contribution brings some light to complement previous studies dealing with copper-catalysed Wacker-type oxidation of very specific olefinic substrates under acidic conditions that were reported as mere curiosities in the past with radical-based intermediates being claimed to form at some stage in the catalytic cycle.40
 | ||
| Fig. 9 Copper-catalysed oxidation of styrene derivatives to acetophenones using a dual photooxidation/photoreduction process with a copper catalyst. Ar = aryl. | ||
Heterogeneous catalysis: a single-atom-based approach delivering Markovnikov selectivity
Heterogeneous catalysts are typically regarded as the ultimate goal for large-scale production of chemical commodities from raw materials. This has also been investigated for the Wacker-type oxidation of olefins using palladium-containing heterogeneous catalysts.41 However, only a couple of examples have demonstrated the possibility of Wacker-type oxidation reaction of olefins under a pure heterogenous regime in the absence of palladium. For instance, the groups of Gao and An presented the very first example of a heterogeneous cobalt-based catalytic system for Wacker-type oxidation (Fig. 10, framed).42 Utilizing a unique type of single-atom dispersed Co–N/C catalyst with isopropanol as both the hydrogen donor and the solvent under an oxygen atmosphere, styrene derivatives were transformed into acetophenone products mainly with variable formation of secondary alcohols as side-products. This Markovnikov-selectivity was explained from a mechanistic point of view with bond-breaking and bond-forming processes taking place in the single-atom cobalt ion (Fig. 10) based on an extensive number of control experiments as well as in situ and operando studies (electron microscopy, XANES and FT spectroscopy, etc.). First, Co(III)-superoxide species are generated via electron transfer from the Co-3d orbitals to the oxygen, where oxygen is chemically adsorbed onto the single cobalt centres, while iPrOH is deprotonated with the assistance of K2CO3, thus forming KHCO3. Co(III)-hydroperoxide species is then produced from Co(III)-superoxide species through proton abstraction by KHCO3, facilitated by intermediate Co(III)-bicarbonate superoxide species. Next, the synergistic addition of styrene to Co(III)-hydroperoxide species leads to the formation of Co(III)-alkylperoxy species, which subsequently rearranges into the ketone (and alcohol) products. However, it cannot be ruled out that acetophenones were formed through the oxidation of the alcohol side-products. Finally, the catalytic cycle is completed through the continuous oxidation of the solvent, resulting in the formal removal of one water molecule. Although no recyclability studies were reported, the reaction conditions reported (150 °C and 40 bar O2) might be compatible with an eventual industrial implementation. | ||
| Fig. 10 Wacker-type oxidation of olefins with Markovnikov selectivity with a heterogenous cobalt-based catalyst and the proposed mechanistic scenario. Ar = aryl. | ||
The second and last example of heterogeneous palladium-free Wacker-type oxidation of olefins corresponds to the contribution of Chen and co-workers, who reported a copper-based single-atom catalyst (Scheme 4).43 The catalyst reactivity was only evaluated for the conversion of 4-vinylanisole into the corresponding ketone product as a result of the Markovnikov selectivity observed. The reaction required the presence of over-stoichiometric amounts of CH3NHOH·HCl as the oxygen donor at 90 °C under aerobic conditions in dioxane and water. Mechanistic investigations by isotope labelling revealed an intriguing hydrogen shift key step in a reaction mechanism that remains rather speculative at this stage. Nevertheless, this research direction offers a potential pathway to substitute noble metal catalysts in conventional homogeneous reactions with SACs. In both cobalt- and copper-based heterogenous catalysis highlighted here, the use of less reactive aliphatic olefin substrates has not been discussed so far.44
 | ||
| Scheme 4 Wacker-type oxidation of olefins with Markovnikov selectivity (only one example) with a heterogenous copper-based catalyst. | ||
Conclusion and outlook
In view of the above discussions, it seems coherent to anticipate a new avenue for the timely development of Wacker-type catalysed processes avoiding the use of palladium. While PGM catalysts were abandoned relatively soon, one can speculate that revisiting such pioneering contributions while considering new catalyst action modes should provide access to interesting discoveries. On the other hand, the use of carefully engineered molecular catalysts has found tremendous attention for achieving full selectivity control in Markovnikov versus anti-Markovnikov oxidation reactions. For instance, the epoxidation–isomerization pathway disclosed with ruthenium- and iron-based porphyrins represents a standard entry towards aldehyde formation from terminal olefins. On the other hand, iron-, cobalt- and nickel-based complexes appear to be excellent catalysts for the formation of methyl ketones via the HAT pathway under an oxygen atmosphere. In this regard, high TON and TOF values have been obtained through the fundamental understanding of the underlying reaction mechanisms in each case. That being said, controlling the selectivity for internal olefins remains elusive at this stage. For that, the development of new biocatalysts or novel concepts exploiting electro- or photo-catalysis could be envisaged in the future considering the promising results reported in the last ten years using these approaches in which novel fundamental steps are nowadays accessible. A common observation from all the different approaches is the relatively poor reactivity of aliphatic olefins when compared to the aromatic ones with the exception of selected examples with ruthenium and iron catalysts, respectively. The real applicability of the palladium-free Wacker-type reactions towards industrial processes still remains to be investigated. In this case, heterogeneous catalysts or other types of reactor engineering (i.e. flow chemistry, mechanochemistry) could represent an important advancement to completely avoid the use of palladium as a catalyst in this industrially, important reaction.Author contributions
IY and RGD: writing, review & editing.Data availability
No primary research results, software, or code was included and no new data were generated for this feature article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Rennes and CNRS are acknowledged for financial support.References
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger and H. Kojer, Angew. Chem., 1959, 71, 176–182 CrossRef CAS.
- (a) R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034–9037 CrossRef CAS; (b) W. Hafner, R. Jira, J. Sedlmeier and J. Smidt, Chem. Ber., 1962, 95, 1575–1581 CrossRef CAS; (c) J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier and A. Sabel, Angew. Chem., Int. Ed. Engl., 1962, 1, 80–88 CrossRef.
- (a) J. Tsuji, Synthesis, 1984, 369–384 CrossRef CAS; (b) W. H. Clement and C. M. Selwitz, J. Org. Chem., 1964, 29, 241–243 CrossRef CAS; (c) J. M. Takacs and X.-T. Jiang, Curr. Org. Chem., 2003, 7, 369–396 CrossRef CAS; (d) C. N. Cornell and M. S. Sigman, Inorg. Chem., 2007, 46, 1903–1909 CrossRef CAS; (e) Y. Ura, Chem. Rec., 2021, 21, 3458–3469 CrossRef CAS PubMed; (f) J. Muzart, Tetrahedron, 2007, 63, 7505–7521 CrossRef CAS; (g) J. Muzart, Tetrahedron, 2021, 87, 132024 CrossRef CAS; (h) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- (a) J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef CAS; (b) B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 2944–2948 CrossRef CAS PubMed; (c) B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9751–9754 CrossRef CAS PubMed; (d) Z. K. Wickens, B. Morandi and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 11257–11260 CrossRef CAS; (e) P. Kočovský and J.-E. Bäckvall, Chem. – Eur. J., 2015, 21, 36–56 CrossRef PubMed; (f) X. Qi, D. G. Kohler, K. L. Hull and P. Liu, J. Am. Chem. Soc., 2019, 141, 11892–11904 CrossRef CAS PubMed; (g) Y. Ura, Synthesis, 2020, 848–860 Search PubMed; (h) M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874–884 CrossRef CAS PubMed; (i) J. J. Dong, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 734–744 CrossRef CAS; (j) T. V. Baiju, E. Gravel, E. Doris and I. N. N. Namboothiri, Tetrahedron Lett., 2016, 57, 3993–4000 CrossRef CAS; (k) P. Teo, Z. K. Wickens, G. Dong and R. H. Grubbs, Org. Lett., 2012, 14, 3237–3239 CrossRef CAS PubMed; (l) Z. K. Wickens, K. Skakuj, B. Morandi and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 890–893 CrossRef CAS PubMed; (m) C. K. Chu, D. T. Ziegler, B. Carr, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2016, 55, 8435–8439 CrossRef CAS PubMed; (n) K. E. Kim, J. Li, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 13179–13182 CrossRef CAS.
- (a) R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448–7470 RSC; (b) P. Rajeshwaran, J. Trouvé, K. Youssef and R. Gramage-Doria, Angew. Chem., Int. Ed., 2022, 61, e202211016 CrossRef CAS PubMed.
- (a) C. W. Dudley, G. Read and P. J. C. Walker, J. Chem. Soc., Dalton Trans., 1974, 1926–1931 RSC; (b) G. Read and P. J. C. Walker, J. Chem. Soc., Dalton Trans., 1977, 883–888 RSC; (c) G. Read, J. Mol. Catal., 1988, 44, 15–33 CrossRef CAS; (d) L. Carlton, G. Read and M. Urgelles, J. Chem. Soc., Chem. Commun., 1983, 586–588 RSC; (e) M. T. Atlay, L. Carlton and G. Read, J. Mol. Catal, 1983, 19, 57–68 CrossRef CAS.
- (a) F. Igersheim and H. Mimoun, J. Chem. Soc., Chem. Commun., 1978, 559–560 RSC; (b) H. Mimoun, Pure Appl. Chem., 1981, 53, 2389–2399 CrossRef CAS; (c) H. Mimoun, M. M. Perez Machirant and I. Seree de Roch, J. Am. Chem. Soc., 1978, 100, 5437–5444 CrossRef.
- (a) R. S. Drago, A. Zuzich and E. D. Nyberg, J. Am. Chem. Soc., 1985, 107, 2898–2903 CrossRef CAS; (b) E. D. Nyberg, D. C. Pribich and R. S. Drago, J. Am. Chem. Soc., 1983, 105, 3538–3544 CrossRef CAS.
- (a) A. Morvillo and M. Bressan, J. Mol. Catal., 1986, 37, 63–74 CrossRef CAS; (b) A. Morvillo and M. Bressan, Inorg. Chim. Acta, 1986, 121, 219–222 CrossRef CAS; (c) M. Bressan, F. Morandini and P. Rigo, J. Organomet. Chem., 1983, 247, c8–c10 CrossRef CAS; (d) M. Bressan and A. Morvillo, Inorg. Chim. Acta, 1989, 166, 177–179 CrossRef CAS; (e) M. Bressan, F. Morandini, A. Morvillo and P. Rigo, J. Organomet. Chem., 1985, 280, 139–146 CrossRef CAS.
- M. M. T. Khan and A. P. Rao, J. Mol. Catal., 1988, 44, 95–105 CrossRef CAS.
- (a) A. Fusi, R. Ugo, F. Fox, A. Pasini and S. Cenini, J. Organomet. Chem., 1971, 26, 417–430 CrossRef CAS; (b) O. Bortolini, F. Di Furia, G. Modena and R. Seraglia, J. Mol. Catal., 1984, 22, 313–317 CrossRef CAS; (c) J. E. Lyons and J. O. Turner, Tetrahedron Lett., 1972, 13, 2903–2906 CrossRef; (d) C. Martin, M. Faraj, J. Martin, J.-M. Brégeault, J. Mercier, J. Fillaux and P. Dizabo, J. Mol. Catal., 1986, 37, 201–212 CrossRef CAS; (e) B. El Ali, J.-M. Brégeault and J. Martin, J. Organomet. Chem., 1987, 327, C9–C14 CrossRef CAS; (f) M. Faraj, J. Martin, C. Martin, J.-M. Bregeault and J. Mercier, J. Mol. Catal., 1985, 31, 57–71 CrossRef CAS; (g) P. Müller and H. Idmoumaz, J. Organomet. Chem., 1988, 345, 187–199 CrossRef; (h) M. Faraj, J.-M. Brégeault, J. Martin and C. Martin, J. Organomet. Chem., 1984, 276, c23–c26 CrossRef CAS; (i) K. Januszkiewicz and H. Alper, Tetrahedron Lett., 1983, 24, 5163–5164 CrossRef CAS.
- J. M. Brown, R. A. John and A. R. Lucy, J. Organomet. Chem., 1985, 279, 245–257 CrossRef CAS.
- (a) J. Chen and C.-M. Che, Angew. Chem., Int. Ed., 2004, 43, 4950–4954 CrossRef CAS PubMed; (b) Z.-M. Wang, X.-L. Sang, C.-M. Che and J. Chen, Tetrahedron Lett., 2014, 55, 1736–1739 CrossRef CAS; (c) G. Jiang, J. Chen, H.-Y. Thu, J.-S. Huang, N. Zhu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 6638–6642 CrossRef CAS; (d) L.-L. Zhang, X.-Y. Wang, K.-Y. Jiang, B.-Y. Zhao, H.-M. Yan, X.-Y. Zhang, Z.-X. Zhang, Z. Guo and C.-M. Che, Dalton Trans., 2018, 47, 5286–5297 RSC.
- (a) G.-Q. Chen, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2011, 47, 10963–10965 RSC; (b) Y.-D. Du, C.-W. Tse, Z.-J. Xu, Y. Liu and C.-M. Che, Chem. Commun., 2014, 50, 12669–12672 RSC.
- A. D. Chowdhury, R. Ray and G. K. Lahiri, Chem. Commun., 2012, 48, 5497–5499 RSC.
- J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 15049–15054 CrossRef CAS PubMed.
- (a) W. W. L. See and Z. Li, Chem. – Eur. J., 2023, 29, e202300102 CrossRef CAS; (b) J. L. Jat and G. Kumar, Adv. Synth. Catal., 2019, 361, 4426–4441 CrossRef CAS; (c) Y. Tian, E. Jürgens, K. Mill, R. Jordan, T. Maulbetsch and D. Kunz, ChemCatChem, 2019, 11, 4028–4035 CrossRef CAS; (d) A. Cabré, J. Cabezas-Giménez, G. Sciortino, G. Ujaque, X. Verdaguer, A. Lledós and A. Riera, Adv. Synth. Catal., 2019, 361, 3624–3631 CrossRef; (e) T. Caneva, L. Sperni, G. Strukul and A. Scarso, RSC Adv., 2016, 6, 83505–83509 RSC; (f) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1994, 59, 7195–7196 CrossRef CAS.
- (a) E. Sansiaume, R. Ricoux, D. Gori and J.-P. Mahy, Tetrahedron: Asymmetry, 2010, 21, 1593–1600 CrossRef CAS; (b) A. M. d’A. Rocha Gonsalves and A. C. Serra, J. Chem. Soc., Perkin Trans. 2, 2002, 715–719 RSC; (c) J. T. Groves and R. S. Myers, J. Am. Chem. Soc., 1983, 105, 5791–5796 CrossRef CAS; (d) J. P. Collman, T. Kodadek and J. I. Brauman, J. Am. Chem. Soc., 1986, 108, 2588–2594 CrossRef CAS; (e) D. Mansuy, J. Leclaire, M. Fontecave and M. Momenteau, Biochem. Biophys. Res. Commun., 1984, 119, 319–325 CrossRef CAS PubMed; (f) D. Mansuy, J. Leclaire, M. Fontecave and P. Dansette, Tetrahedron, 1984, 40, 2847–2857 CrossRef CAS; (g) D. C. Liebler and F. P. Guengerich, Biochemistry, 1983, 22, 5482–5489 CrossRef CAS PubMed; (h) R. E. Miller and F. P. Guengerich, Biochemistry, 1982, 21, 1090–1097 CrossRef CAS PubMed; (i) Y.-C. Yin, H.-L. Yu, Z.-J. Luan, R.-J. Li, P.-F. Ouyang, J. Liu and J.-H. Xu, ChemBioChem, 2014, 15, 2443–2449 CrossRef CAS; (j) S. P. de Visser, D. Kumar and S. Shaik, J. Inorg. Biochem., 2004, 98, 1183–1193 CrossRef CAS; (k) O. Vakuliuk, F. G. Mutti, M. Lara, D. T. Gryko and W. Kroutil, Tetrahedron Lett., 2011, 52, 3555–3557 CrossRef CAS.
- (a) S. Gergel, J. Soler, A. Klein, K. H. Schülke, B. Hauer, M. Garcia-Borràs and S. C. Hammer, Nat. Catal., 2023, 6, 606–617 CrossRef CAS; (b) J. Soler, S. Gergel, C. Klaus, S. C. Hammer and M. Garcia-Borràs, J. Am. Chem. Soc., 2022, 144, 15954–15968 CrossRef CAS PubMed; (c) S. C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges and F. H. Arnold, Science, 2017, 358, 215–218 CrossRef CAS PubMed.
- (a) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed; (b) J. C. Lo, D. Y. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. H. Gui, T. Qin, S. Gutierrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed; (c) S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS PubMed; (d) H. Jiang, W. Lai and H. Chen, ACS Catal., 2019, 9, 6080–6086 CrossRef CAS; (e) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 RSC; (f) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 RSC; (g) P. V. Kattamuri and J. G. West, J. Am. Chem. Soc., 2020, 142, 19316–19326 CrossRef CAS PubMed.
- B. Liu, F. Jin, T. Wang, X. Yuan and W. Han, Angew. Chem., Int. Ed., 2017, 56, 12712–12717 CrossRef CAS PubMed.
- (a) F. Puls and H.-J. Knölker, Angew. Chem., Int. Ed., 2018, 57, 1222–1226 CrossRef CAS; (b) F. Puls, P. Linke, O. Kataeva and H. J. Knölker, Angew. Chem., Int. Ed., 2021, 60, 14083–14090 CrossRef CAS; (c) T. Schuh, O. Kataeva and H.-J. Knölker, Chem. Sci., 2023, 14, 257–265 RSC.
- (a) J. Trouvé, K. Youssef, S. Kasemthaveechok and R. Gramage-Doria, ACS Catal., 2023, 13, 4421–4432 CrossRef; (b) J. Trouvé, K. Youssef, S. Kasemthaveechok and R. Gramage-Doria, FR Pat., EP4375266A1 and INT. Pat., WO2024110633A1, 2024 Search PubMed.
- (a) C. Jiang, Y. Wu, Y. Zhang, J. Zong, N. Wang, G. Liu, R. Liu and H. Yu, Angew. Chem., Int. Ed., 2025, 64, e202413901 CrossRef CAS; (b) T. Hashimoto, T. Maruyama, T. Ishimaru, M. Matsugaki, K. Shiota and Y. Yamaguchi, ChemistrySelect, 2021, 6, 5534–5537 CrossRef CAS; (c) Y. Liu, K. Qiao, J. Yang, W. Jiang, R. Zhang and L. Shi, Tetrahedron Lett., 2025, 158, 155492 CrossRef CAS.
- F. Puls, F. Seewald, V. Grinenko, H.-H. Klauß and H.-J. Knölker, Chem. – Eur. J., 2021, 27, 16776–16787 CrossRef CAS.
- (a) A. Zombeck, D. E. Hamilton and R. S. Drago, J. Am. Chem. Soc., 1982, 104, 6782–6784 CrossRef CAS; (b) Y. Matsushita, T. Matsui and K. Sugamoto, Chem. Lett., 1992, 1381–1384 CrossRef CAS.
- (a) Y.-I. Matsushita, K. Sugamoto and T. Matsui, Chem. Lett., 1992, 2165–2168 CrossRef CAS; (b) M. Nagatomo, K. Hagiwara, K. Masuda, M. Koshimizu, T. Kawamata, Y. Matsui, D. Urabe and M. Inoue, Chem. – Eur. J., 2016, 22, 222–229 CrossRef CAS PubMed; (c) M. Nagatomo, M. Koshimizu, K. Masuda, T. Tabuchi, D. Urabe and M. Inoue, J. Am. Chem. Soc., 2014, 136, 5916–5919 CrossRef CAS PubMed.
- (a) N. Abuhafez, A. W. Ehlers, B. de Bruin and R. Gramage-Doria, Angew. Chem., Int. Ed., 2024, 63, e202316825 CrossRef CAS; (b) N. Abuhafez and R. Gramage-Doria, ChemCatChem, 2024, 16, e202400333 CrossRef CAS.
- V. Porte, M. N. M. Milunovic, U. Knof, T. Leischner, T. Danzl, D. Kaiser, T. Gruene, M. Zalibera, I. Jelemenska, L. Bucinsky, S. A. V. Jannuzzi, S. DeBeer, G. Novitchi, N. Maulide and V. B. Arion, JACS Au, 2024, 4, 1166–1183 CrossRef CAS.
- S. N. Pandey, A. Sengupta, R. Bera, S. Ali and S. Yadav, Catal. Sci. Technol., 2024, 14, 4487–4495 RSC.
- (a) T. Mukaiyama, S. Isayama, S. Inoki, K. Kato, T. Yamada and T. Takai, Chem. Lett., 1989, 449–452 CrossRef CAS; (b) S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 569–572 CrossRef CAS; (c) S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 573–576 CrossRef CAS; (d) S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 1071–1074 CrossRef CAS.
- B. Liu, P. Hu, F. Xu, L. Cheng, M. Tan and W. Han, Commun. Chem., 2019, 2, 5 CrossRef.
- (a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS; (b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (c) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (d) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- J. Lai and M. A. Pericàs, Org. Lett., 2020, 22, 7338–7342 CrossRef CAS.
- S. Tang, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142, 5980–5984 CrossRef CAS.
- M. T. Atlay, M. Preece, G. Strukul and B. R. James, J. Chem. Soc., Chem. Commun., 1982, 406–407 RSC.
- Y. Shimoyama, Y. Kitagawa, Y. Ohgomori, Y. Kon and D. Hong, Chem. Sci., 2021, 12, 5796–5803 RSC.
- Y. Abderrazak, A. Chinchole and O. Reiser, Catal. Sci. Technol., 2024, 14, 4068–4074 RSC.
- (a) S.-L. Zhang, X.-J. Wang and Z.-L. Yu, Org. Lett., 2017, 19, 3139–3142 CrossRef CAS; (b) X.-H. Li, C. Mi, X.-H. Liao and X.-G. Meng, Catal. Lett., 2017, 147, 2508–2514 CrossRef CAS.
- (a) T. Kubota, F. Kumada, H. Tominaga and T. Kunugi, Int. Chem. Eng., 1973, 13, 539–545 Search PubMed; (b) P. H. Espeel, M. C. Tielen and P. A. Jacobs, J. Chem. Soc., Chem. Commun., 1991, 10, 669–671 RSC; (c) K. Choi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32, 180–181 CrossRef CAS; (d) S. Donck, E. Gravel, N. Shah, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2015, 7, 2318–2322 CrossRef CAS; (e) K. Nowińska, D. Dudko and R. Golon, Chem. Commun., 1996, 277–279 RSC.
- G. Huang, L. Wang, H. Luo, S. Shang, B. Chen, S. Gao and Y. An, Catal. Sci. Technol., 2020, 10, 2769–2773 RSC.
- J. Song, J. Liu, K. P. Loh and Z. Chen, Chem. Res. Chin. Univ., 2022, 38, 1239–1242 CrossRef CAS.
- C. Chang, Y. Hou and J. Li, Mol. Catal., 2025, 577, 114959 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |