
Chromium-catalyzed sustainable C–C and C–N bond formation: C-alkylation and Friedländer quinoline synthesis using alcohols†
Vaishnavi
Atreya
,
Sachin
Jalwal
and
Subrata
Chakraborty
 *
*
Department of Chemistry, Indian Institute of Technology Jodhpur, Karwar, Jodhpur, 342037, Rajasthan, India. E-mail: subrata@iitj.ac.in
First published on 18th November 2024
Abstract
The synthesis of a novel phosphine-based pincer chromium(II) complex CrCl2(PONNH) (Cr-1) is reported in this study. The complex exhibited promising catalytic performance in C–C and C–N bond formation using the borrowing hydrogen methodology. Cr-1 catalyzed the α-alkylation of ketones using primary alcohols as alkyl surrogates in the presence of catalytic amount of a base. Cr-1 was also found to catalyze the β-alkylation of secondary alcohols using primary alcohols. In addition, the dehydrogenative annulation of 2-aminobenzyl alcohols with ketones to form quinolines was achieved using Cr-1 as the catalyst. Based on the mechanistic investigation, a plausible mechanism based on metal–ligand cooperation is proposed. The reactions are redox-neutral, atom-efficient, and produce water as the only by-product, thus contributing to green chemistry.
Introduction
The formation of carbon–carbon and carbon–nitrogen bonds is vital in organic synthesis as it lays the foundation for the synthesis of a large variety of pharmaceuticals, agrochemicals and fine and bulk industrial chemicals.1 Recently, the redox-neutral ‘borrowing hydrogen’ or the so-called ‘hydrogen auto-transfer’ strategy (BH/HA) has emerged as an atom-efficient and green protocol for the formation of C–C and C–N bonds by coupling non-activated alcohols with ketones and secondary alcohols with amines, respectively.2 This methodology has also been utilized for producing value-added heterocycles by multicomponent domino processes.3 Traditional C–C and C–N bond formation processes require expensive and/or hazardous starting materials, reagents, and additives, resulting in the generation of copious amounts of toxic wastes, thus limiting their sustainable application.4 The typical borrowing hydrogen route for C–C/C–N bond formation enables the dehydrogenative coupling of alcohols with ketones/amines, followed by reduction of the unsaturated intermediate (enone/imine) by the hydrogen produced from dehydrogenation of alcohol.5 The availability of starting materials from renewable resources, operational simplicity, and the generation of H2O as the only by-product render the borrowing hydrogen process sustainable, atom-economical, and environmentally benign.2,6 Nonetheless, precious late-transition metals Pt, Pd, Rh and Ru have been explored extensively for such types of reactions.7 Lately, 3d metal complexes have drawn considerable attention for the aforementioned tasks owing to their earth-abundance, cost-effectiveness and low toxicity.8 Numerous reports on homogeneous (de)hydrogenation reactions, including C- and N-alkylation reactions, by employing iron-,9 cobalt-,10 nickel-11 and manganese-based complexes have been reported.12 In this regard, chromium is also an attractive replacement to noble metal complexes in the realm of homogeneous (de)hydrogenation due to its earth-abundance and bio-compatibility.13 In fact chromium-catalyzed hydrogenation of ketones, aldehydes, and polycyclic aromatic hydrocarbons as well as the semi-hydrogenation of alkynes has been demonstrated by several groups.14 In recent years, well-defined Cr complexes have also been applied in dehydrogenative coupling reactions such as N-alkylation of amines via the borrowing hydrogen strategy and the dehydrogenative coupling of alcohols and amines as reported by Kempe and others.15 The growing interest towards the applications of chromium catalysis in (de)hydrogenative transformations has been reviewed by us recently.16In 2022, Kumar and coworkers17 reported the β-alkylation of secondary alcohols with primary alcohols using CrCl3 and the corresponding NNN pincer complex under conventional and microwave heating conditions. Very recently, Yinwu Li and Zhuofeng Ke18 have shown that an NHC ligand-based Cr(0) catalyst is very efficient in overcoming the alkoxide trap towards achieving the C-alkylation of secondary alcohols with primary alcohols in the presence of NaOH. To the best of our knowledge, there has been no report on the α-alkylation of ketones and Friedlander synthesis using well-defined chromium complexes. While this study was in progress, a parallel report on the α-alkylation of ketones using primary alcohols as the alkyl surrogates and Friedlander synthesis was published by Zhihua Peng et al., using the CrCl2/PPh3 combination and an over-stoichiometric amount of base with respect to the alcohol.19 Hence, there is ample scope for the development of an efficient catalytic system for achieving C–C and C–N bond formation via a green pathway.
Results and discussion
To achieve a sustainable, atom-economical and versatile catalytic system that can promote the formation of C–C and C–N bonds, we developed a pincer phosphine-based chromium(II) pre-catalyst Cr-1 (Table 1). The complex turned out to be effective in catalyzing the α-alkylation of a wide variety of ketones, including cyclic ketones and heteroaromatic ketones, and one-pot dehydrogenative annulation of 2-aminobenzyl alcohols with ketones to form a diverse range of substituted quinolines. Interestingly, the system also exhibited catalytic activity towards the β-alkylation of secondary alcohols.
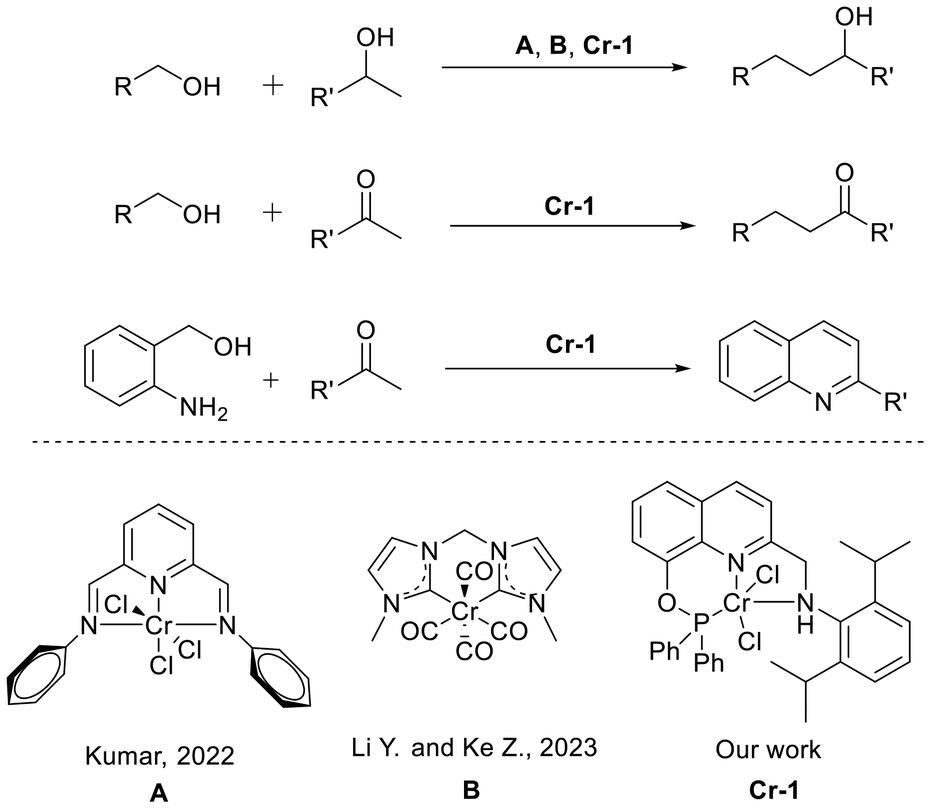
|
Initially, we designed an 8-hydroxy-quinoline-based novel pincer phosphinite ligand (N-((8-((diphenylphosphaneyl)oxy)naphthalen-2-yl)methyl)-2,6-diisopropylaniline) (PONNH). Phosphinite ligands have been proven to display unique reactivities in homogeneous catalysis.20a–d However, insights on 8-hydroxyquinoline-derived phosphinite complexes in homogeneous (de)hydrogenation reactions are elusive despite their usage in the hydroalkoxylation of terminal alkynes catalyzed by Rh complexes and other related reactions.20e–k
Hence, in this work, we have probed quinoline-phosphinite ligand-based 3d-metal complexes in homogeneous dehydrogenation reactions. Thus, the pincer PONNH ligand was prepared by treating chlorodiphenylphosphine with an amine precursor in the presence of triethylamine (yield = 58%, Scheme 1). The 31P NMR of the synthesized ligand displayed a sharp signal at 117.3 ppm, indicating the presence of one phosphorus ligand attached to oxygen. The N–H moiety and the CH2 arm of the synthesized PONNH ligand are expected to display metal–ligand cooperation (MLC).21
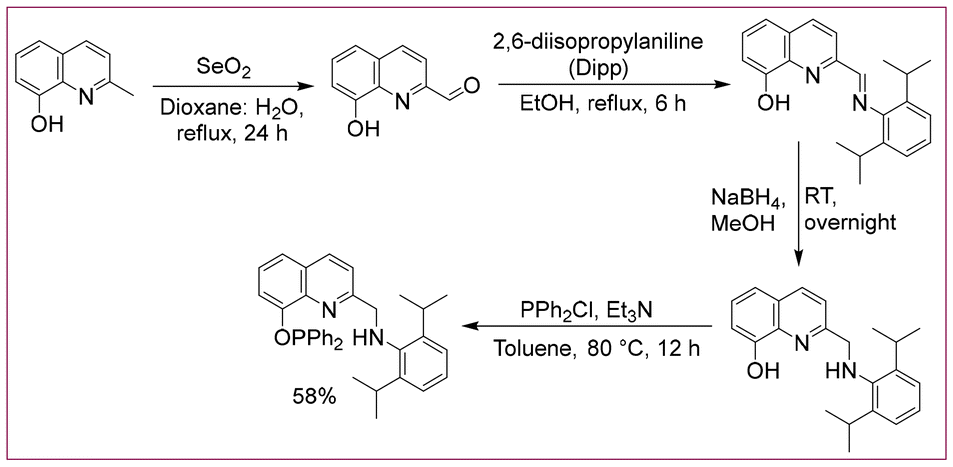 | ||
| Scheme 1 Synthetic process of the PONNH ligand. | ||
The reaction of CrCl2 with the PONNH ligand at room temperature led to the formation of a dark green complex CrCl2(PONNH) (Cr-1) with a 68% yield (Scheme 2). The chromium complex Cr-1 was characterized using IR and HRMS. The attempt to obtain single crystals using various solvent combinations failed. As the compound was paramagnetic in nature, magnetic susceptibility was measured using the Evans method and, its EPR spectrum was also recorded (Fig. S3 and S7; see ESI†).22
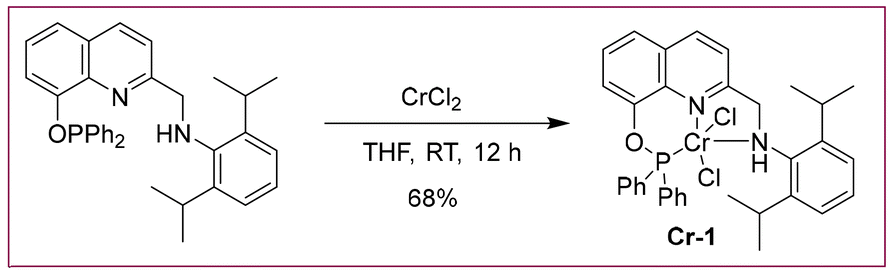 | ||
| Scheme 2 Preparation of the Cr-1 complex. | ||
To evaluate the redox properties of the complex, a cyclic voltammetry (CV) experiment was conducted, which showed two oxidation potential peaks at −0.615 V and +1.143 V, respectively, corresponding to a reversible Cr(II)/Cr(III) and an irreversible Cr(III)/Cr(IV) redox couples (Fig. 1).23,24
 | ||
| Fig. 1 Cyclic Voltammetry study of Cr-1. Conditions: 1.0 mM analyte in 0.1 M TBAPF6/MeCN, under a dry argon atmosphere; glassy carbon working electrode, platinum wire counter electrode, and Ag/AgCl reference electrode; scan rate = 0.02 V s−1. | ||
Next, the newly synthesized well-defined complex Cr-1 was tested in the α-alkylation of ketones using primary alcohols. We reacted acetophenone (0.5 mmol) with benzyl alcohol (0.5 mmol) using KOtBu (20%) and 3 mol% Cr-1 in a closed system using toluene as the solvent under a nitrogen atmosphere at 135 °C for 24 h. The alkylated product 1,3-diphenylpropan-1-one was formed with a 76% yield (Table S1, entry 2†). Interestingly, p-methylbenzyl alcohol furnished a better yield under similar conditions (88%, Table S1, entry 3†). Hence, for further optimization, p-methylbenzyl alcohol was used as the benchmark alkylating partner with acetophenone. Various bases were employed to explore the effect of cation in this reaction. The results are summarized in Table S1† (entries 5–9, 20; NaOH, 84%, KOH, 80%, NaOtBu 78%, LiOtBu 84% Cs2CO3, 86%, and LiOH, 87%). Changing the base had minimal effect on the alkylation reaction, and hence KOtBu was chosen for further studies. However, in the absence of a base, a trace amount of product formation was observed (Table S1, entry 10†). When the reaction was carried out at a lower temperature (120 °C), a decrease in conversion (62%) of alcohol was noticed (Table S1, entry 1†). Further, the evaluation of different solvents was performed. Tertiary amyl alcohol resulted in the lowering of conversion efficiency (50%, Table S1, entry 11†). Surprisingly, in the presence of dioxane and THF, only trace amounts of products were formed (Table S1, entries 12–14†). 45% of the desired product was obtained in the presence of chlorobenzene solvent. When mesitylene was used as the solvent in place of toluene, a 71% yield was observed (Table S1, entries 16 and 17†). To our delight, when the optimized reaction was carried out for 36 h, a yield of up to 98% was achieved (Table S1, entry 15†). When CrCl2 was used as the catalyst, only 20% of the corresponding product was formed, whereas, with 20% KOtBu as the base and in the absence of catalyst Cr-1, only a trace amount of product was observed (Table S1, entries 18 and 19†). To compare the catalytic applicability of our Cr(II) complex Cr-1 with the Cr(III) complex, we treated the PONNH ligand with anhydrous CrCl3. This resulted in the formation of a golden-brown powder with a 62% yield, which was confirmed by HRMS as the CrCl3(PONNH) complex Cr-2 (see ESI, Scheme S3†). The α-alkylation of acetophenone with benzyl alcohol was carried out using the freshly prepared Cr-2 complex, which yielded 59% of the desired product after heating for 24 h (Table S1, entry 21†), indicating its inferior catalytic efficiency in comparison with Cr-1 (76%).
Thus, under the optimized conditions (135 °C, 3 mol% Cr-1, and 20 mol% KOtBu), α-alkylation reactions of a wide array of primary alcohols and arylketones were explored. The reaction was compatible with benzyl, heteroaromatic and even allyl alcohols. An isolated yield of 74% 1,3-diphenylpropan-1-one was obtained after the careful separation of the product from the reactants through column chromatography (Scheme 3; 3d).
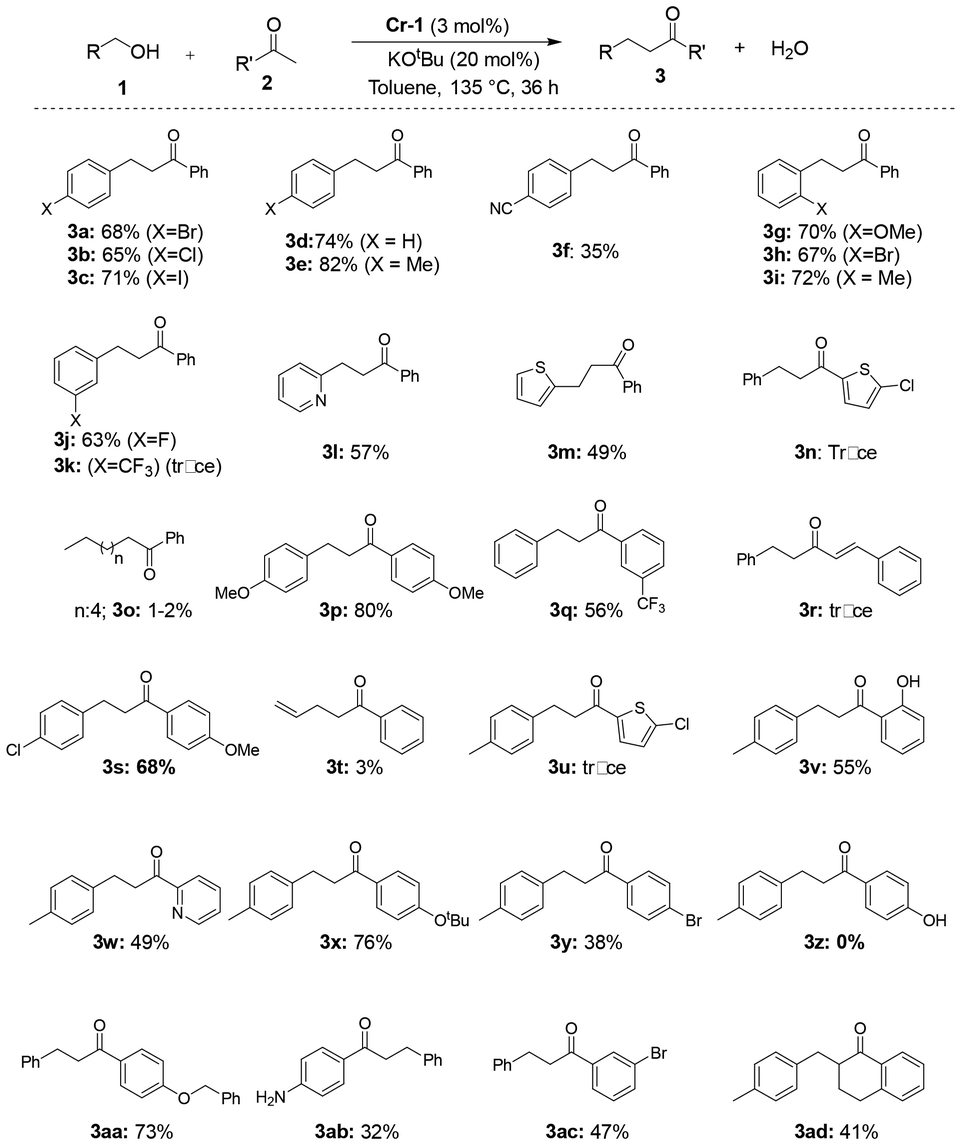 | ||
| Scheme 3 Cr-1-catalyzed α-alkylation reactions of ketones with a variety of primary alcohols. Reaction conditions unless otherwise specified: 0.5 mmol alcohol and 0.5 mmol ketone, 20 mol% KOtBu, 3 mol% cat. Cr-1 and toluene (1.5 mL) were heated at 135 °C for 36 h. The mol% values are with respect to the total alcohol content. Yields are isolated (purified by silica gel column chromatography) using 0–5% ethyl acetate in n-hexane as the eluent. | ||
The reaction was found to tolerate electron-withdrawing halides (4-Cl, Br, I) at the para position of benzyl alcohol, producing decent yields of the corresponding coupled products (Scheme 3; 3a–3c). Electron-donating p-methylbenzyl alcohol furnished an 82% yield of the corresponding coupled product with acetophenone (Scheme 3; 3e). When the reaction was conducted using the strong electron-withdrawing cyano group at the para position of the alcohol under optimized conditions, a decrease in yield to 35% was observed (Scheme 3; 3f). ortho-Substituted benzyl alcohols bearing electron-donating and -withdrawing groups (2-OMe, 2-Br, 2-Me) afforded good yields of the C–C coupled products with acetophenone under standard conditions (Scheme 3; 3g–3i). While meta-fluorobenzyl alcohol yielded 63% of the product, meta-trifluoromethylbenzyl alcohol produced only a trace amount of the desired alkylated product (Scheme 3; 3j and 3k). The catalytic performance of Cr-1 in the dehydrogenative reaction of heteroaromatic alcohols like 2-picolylamine and 2-thiopheneethanol was also decent, furnishing moderate yields (57% and 49%; Scheme 3; 3l and 3m) of the desired products. Nevertheless, in the cases of allylic alcohol and straight-chain alcohols, the yields of the alkylated products were rather unsatisfactory (3–5%, Scheme 3; 3o and 3t). Several substituted arylketones bearing various functional groups were also employed in the coupling reactions. In the case of para-substituted methylketones and primary alcohols, moderate to good yields of the corresponding alkylated products were obtained (Scheme 3; 3p and3s). Additionally, meta-CF3-substituted acetophenone gave a 56% yield when coupled with p-methylbenzyl alcohol (Scheme 3; 3q). Arylmethylketones bearing 4-OtBu and 4-OBn groups at the para position were also tested for C–C bond formation with alcohols, which generated products with decent yields of 76% and 73%, respectively (Scheme 3; 3x and 3aa). Nonetheless, in the case of 4-aminoacetophenone, only 32% product yield was isolated, whereas, with p-OH acetophenone, no product formation was observed, as revealed by 1H NMR (Scheme 3; 3ab, 3z). Hetero-aromatic 2-acetyl-5-chlorothiophene produced only trace amounts of the desired products (3n and 3u), but 3-acetylpyridine gave a moderate yield of about 49% for the desired alkylated ketone (Scheme 3; 3w). The scope of the alkylation reaction was also expanded to a cyclic ketone, which furnished a satisfactory yield of 41% (3ad, Scheme 3), whereas, an α,β unsaturated ketone yielded no desired product when coupled with benzyl alcohol (Scheme 3; 3r ).
Next, we applied the catalyst in the synthesis of N-heterocycles, particularly substituted quinolines, which are essential components in natural products and biologically active molecules that are widely used in pharmaceuticals, medicinal chemistry, agrochemicals, and functional materials.25
Traditional methods of quinoline synthesis usually suffer from multi-step processes, harsh reaction conditions and low yields. The Friedländer reaction, though versatile, too faces limitations like instability of starting materials (2-aminobenzaldehyde derivatives). A modified Friedländer method of quinoline synthesis utilizing amino alcohols as aldehyde surrogates has been explored using 3d metals;26 however, only one study has reported the use of chromium metal for such a transformation and was published during the preparation of this manuscript.19 Pleasingly, Cr-1 was found to display quite efficient activity in the formation of quinoline derivatives via Friedländer annulation using 2-aminobenzyl alcohol and various aryl ketones. On reacting acetophenone (0.5 mmol) with 1 equiv. 2-aminobenzyl alcohol in the presence of 3 mol% of Cr-1 and 20 mol% of KOtBu in toluene, 2-phenylquinoline with an 84% isolated yield was obtained after 36 h at 135 °C. Several other quinoline derivatives were synthesized using substituted acetophenones. Electron-withdrawing groups at the para or meta position (4-Br, 4-CF3, 3-Br, and 3-CF3) produced substituted quinolines with slightly lower yields (43–64%) than electron-donating groups, such as 4-OCH2Ph (81%) and 4-OtBu (70%) (Scheme 4; 6b–6g). While 4-hydroxy acetophenone did not lead to product formation, a lower yield (27%) of the corresponding quinoline derivative was obtained when p-aminoacetophenone was used as the coupling partner (Scheme 4; 6h and 6i). With heteroaromatic ketones, rather poor yields were observed (18%, 6k, Scheme 4). Interestingly, we were able to obtain polycyclic heteroaromatic compounds with moderate yields using cyclic ketones (6l and 6m, Scheme 4).
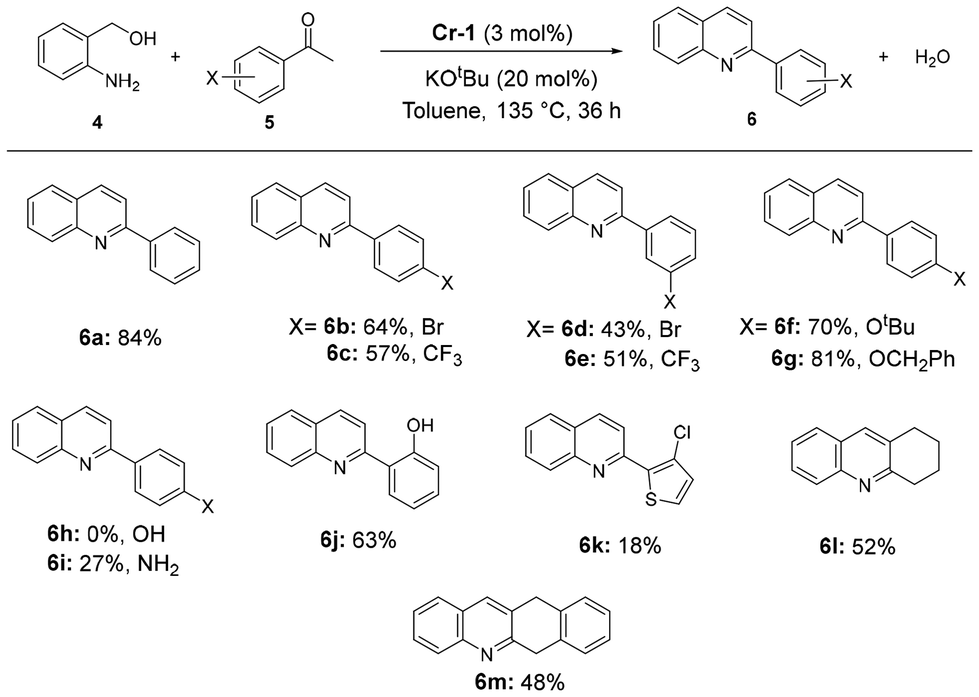 | ||
| Scheme 4 Cr-1-catalyzed Friedländer quinoline synthesis using 2-aminobenzyl alcohol and substituted methylketones. Reaction conditions unless otherwise specified: 0.5 mmol of 2-aminobenzyl alcohol and 0.5 mmol ketones, 20 mol% of KOtBu, 3 mol% of Cr-1 and toluene (1.5 mL) were heated at 135 °C for 36 h. The mol% values are with respect to total alcohol content. Product yields are isolated (purified by silica gel column chromatography using 5–10% ethyl acetate in n-hexane as the eluent). | ||
To further establish the significance of this Cr(II) pre-catalyst, we explored the scope of β-alkylation of secondary alcohols using benzyl alcohol derivatives. Pleasingly, Cr-1 presented a convincing catalytic performance in this direction as well. However, using the standard α-alkylation reaction and catalytic amount of a base, a low conversion rate (20%) was achieved with the benchmark substrates benzyl alcohol and 1-phenylethanol. By increasing the amount of base to 1 equiv. relative to the alcohols, a 74% yield of desired 1,3-diphenylpropan-1-ol was observed, as determined by NMR spectroscopy using 1,1,2,2-tetrachloroethane as the internal standard (isolated yield = 70%).
We also examined the potential of the newly synthesized Cr(III) complex Cr-2 in the β-alkylation reaction. When the reaction was performed under analogous conditions to those reported by Kumar et al., 0.005 mol% Cr-2 and 2 mmol each of benzyl alcohol and 1-phenylethanol along with 5 mol% of NaOtBu base yielded only 6% of the desired β-alkylated secondary alcohol (Table S1, entry 22†), indicating inferior performance to the system developed by Kumar.17 However, a higher loading of complex Cr-2 (3 mol%) and 20 mol% NaOtBu at 135 °C furnished a yield of 54% in 24 h (entry 23, Table S1†).
Further experiments to evaluate the scope of β-alkylation of 1-phenylethanol using various benzyl alcohol substrates revealed that electron-withdrawing groups afforded much higher rates of conversion to desired products compared with electron-donating groups, such as methyl and methoxy functional groups (Scheme 5; 8b–8d). The position of the functional groups also apparently affected product formation; functional groups at the para position resulted in higher product yields than that those at the ortho and meta positions (Scheme 5; 8a–8h). Heteroaromatic primary alcohols did not exhibit satisfactory conversion to alkylated products (Scheme 5; 8i and 8j). The scope of the reaction was further expanded to a variety of secondary alcohols bearing electron-withdrawing and -donating groups, which furnished the desired β-alkylated secondary alcohols at good to excellent yields (64–82%; Scheme 5; 8k–8n).
 | ||
Scheme 5
Cr-1-catalyzed β-alkylation of substituted 1-phenylethanol using alcohols. Reaction conditions unless otherwise specified: 0.5 mmol of primary alcohol and 0.5 mmol 1-phenylethanol derivatives, 1 equiv. KOtBu and 3 mol% of catalyst Cr-1 and toluene (1.5 mL) were heated at 135 °C for 40 h. The mol% values are with respect to the total primary alcohol content. Yields are isolated (purified by silica gel column chromatography) using 0–5% ethyl acetate in n-hexane as the eluent. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NMR yield with respect to 1,1,2,2 tetrachloroethane as the internal standard. NMR yield with respect to 1,1,2,2 tetrachloroethane as the internal standard. | ||
To investigate the mechanism of the C-alkylation reactions catalyzed by Cr-1, we performed stoichiometric experiments and monitored the changes by HRMS, 1H NMR and IR spectroscopy. The addition of 3 equiv. KOtBu to a dark-green toluene solution of complex Cr-1 changed the colour of the reaction mixture to greenish brown. This could be due to the formation of an unstable dearomatized (Cr-1a) or amido complex (Cr-1a′) formed via the deprotonation of the methylene arm proton or N–H moiety (Scheme 7). The air sensitivity and high reactivity of the formed complex precluded us from characterizing the reaction mixture spectroscopically. In a similar context, the Milstein group developed a PNNH pyridyl-based Ru system having the potential for dual-mode MLC via aromatization–dearomatization or the amine–amido route.28 In fact, they could crystallographically characterize the monoanionic de-aromatized enamido Ru(II) species by deprotonating both the N–H and methylene protons of the N-arm of the PNNH pincer ligand.28 Their VT NMR studies after the addition of base at low temperatures suggested that the deprotonation of the methylene arm proton takes place initially, and they finally obtained the double-deprotonated monoanionic Ru(II) complex at room temperature. However, in our case, the double dehydrohalogenation of Cr-1via deprotonation of the methylene arm and the N–H moiety might lead to a very unstable low-coordinated Cr(II) species, which is also less likely to be formed. Thus, the formation of a double-deprotonated dearomatized enamido complex could not be confirmed at this stage. Nevertheless, the colour of the reaction mixture further turned brown with the addition of 3 equiv. benzyl alcohol at room temperature, affording the alkoxy complex Cr-1b (Scheme 6a). The formation of Cr-1b was confirmed by HRMS and IR spectroscopy analyses of the brown reaction mixture (Fig. S14 and 17, see ESI†). However, in the 1H NMR spectra, we did not observe any peak for the alkoxy species because of the paramagnetic nature of the complex. Alternatively, complex Cr-1b could also be prepared by reacting benzyl alcohol with tBuOK, which generated a benzyloxy species after the addition of Cr-1 (Fig. S14, 17 and 18; see ESI†). We also conducted an experiment with benzyl alcohol and acetophenone under the optimized reaction conditions for 3 h, and the formation of various intermediates was analysed by 1H NMR and HRMS (Scheme 6b). As expected, a peak at δ = 9.96 was detected for benzaldehyde due to the dehydrogenation of benzyl alcohol along with the formation of the desired product, 1,3-diphenylpropan-1-one. Interestingly, the formation of a small amount of 1,3-diphenylpropan-1-ol was also observed in the 1H NMR spectra (Fig. S8, see ESI†).
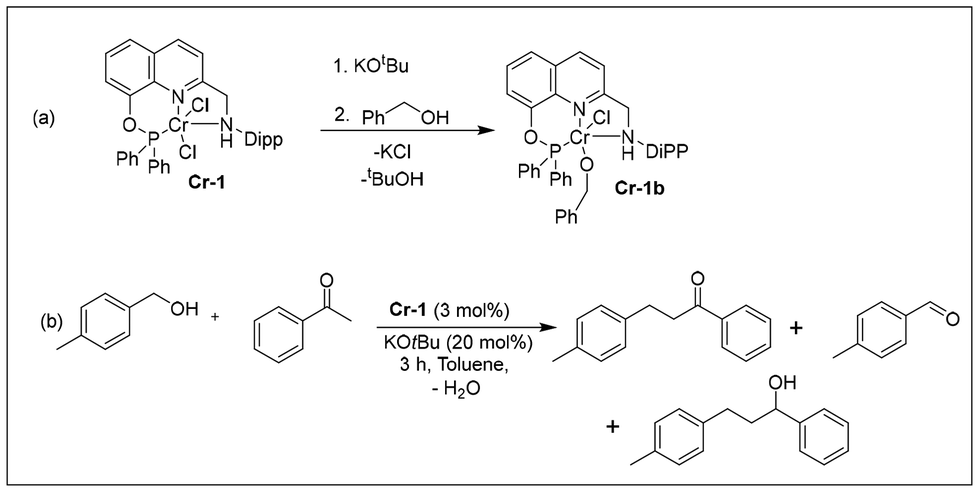 | ||
| Scheme 6 Stoichiometric reactions and intermediate detection study. | ||
 | ||
| Scheme 7 Proposed mechanism of the Cr-catalyzed alkylation reactions. | ||
Based on these observations and the literature,28 a mechanism for the alkylation reaction involving metal–ligand cooperation is proposed.21,27 The pre-catalyst Cr-1 is activated by a base, forming the initial dearomatized kinetic product Cr-1avia dehydrohalogenation of Cr-1. Cr-1a is expected to be in equilibrium with the amido complex Cr-1a′. This unsaturated species reacts with alcohol to form a stable alkoxy-metal complex Cr-1b. Subsequently, β-hydrogen elimination from the alkoxy species results in the formation of aldehyde along with a hydride complex C-1c. The aldehyde undergoes an aldol condensation step in the presence of a base, producing α–β unsaturated ketone, which is concomitantly hydrogenated by the hydrogen released from complex C-1c, thus closing the catalytic cycle.
Experimental section
General information
All experiments were performed under an inert nitrogen atmosphere using standard Schlenk techniques and inside a glove box unless stated otherwise. Tetrahydrofuran (THF), toluene, chloroform (CHCl3), diethyl ether (Et2O), hexane and ethyl acetate were heated under reflux over a suitable drying agent (sodium/benzophenone for THF, Et2O, hexane; calcium hydride for CHCl3 and kept under freshly dried molecular sieves), distilled, and stored under a nitrogen atmosphere. All solvents were degassed and kept in the glovebox over activated 4 Å molecular sieves. Deuterated solvents were purged with nitrogen and kept in a glovebox. Unless otherwise stated, commercial reagents were used without purification; only benzyl alcohol and acetophenone were purified via vacuum distillation. Chromium chloride (CrCl2) was purchased from Sigma-Aldrich; calcium hydride (CaH2) and all other materials were purchased from Avra, BLDpharma, TCI and Sigma-Aldrich, stored under nitrogen and used as received. The reaction temperatures reported indicate the temperature of the oil bath. All the reactions carried out for ligand synthesis and substrate scope analysis were monitored by TLC, and the products were isolated by column chromatography or flash chromatography (silica gel 60–120). Then, each product was condensed with the help of a rotary evaporator and characterized by 1H NMR and 13C NMR performed on a Bruker 500 MHz spectrometer using CDCl3 or C6D6. All spectra were recorded at room temperature, unless otherwise noted. ATR-IR spectra were recorded on a PerkinElmer FT-IR spectrometer. The mass spectra were recorded on an Agilent spectrometer. The EPR analysis was performed on a JES-FA200 ESR Spectrometer in the X-band region (8.75–9.65 GHz). The elemental analysis was carried out on a Flash smart V CHNS/O. Solution-state UV–visible absorption spectra were recorded on a Cary 4000 UV-vis spectrophotometer using dichloromethane (DCM) as the solvent.Synthesis of the PONNH ligand
(2-(((2,6-Diisopropylphenyl)amino)methyl)quinolin-8-ol)29 (1004 mg, 3 mmol) was dissolved in 20 mL toluene in a 100 mL Schlenk tube under a nitrogen atmosphere, and an equimolar amount of triethyl amine (418 μL, 3 mmol) was added to it dropwise with vigorous stirring. After 5 minutes, 3 mmol (540 μL) chlorodiphenylphosphine was also added dropwise, while stirring the solution vigorously at room temperature. Then, the Schlenk tube was closed tightly and heated at 80 °C overnight. The reaction mixture was cooled to room temperature, and the solvent was evaporated under a vacuum. The residue was extracted with 20 mL of toluene and filtered through Celite. Again, the solvent was evaporated under a vacuum. This product was again washed with dried hexane, and finally, a colourless oily liquid with a 58% yield was obtained after drying in a vacuum. The purity of the ligand was checked by 31P NMR, and the sample was used without further purification. 31P NMR (203 MHz, Chloroform-d) δ 117.3. 1H NMR (500 MHz, Chloroform-d) δ 7.93 (d, J = 8.4 Hz, 1H), 7.70 (dd, J = 11.5, 7.6 Hz, 1H), 7.63 (dd, J = 13.7, 7.5 Hz, 1H), 7.51 (t, J = 7.9 Hz, 6H), 7.29 (dt, J = 8.3, 4.1 Hz, 1H), 7.22 (d, J = 7.3 Hz, 1H), 7.16 (t, J = 7.3 Hz, 6H), 7.10 (d, J = 7.7 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.00 (d, J = 7.7 Hz, 1H), 6.84 (s, 1H), 4.78 (s, 1H), 3.60 (p, J = 6.6 Hz, 2H), 1.06 (d, J = 6.6 Hz, 12H). 13C NMR (126 MHz, Chloroform-d) δ 152.78, 143.44, 135.40 (d, J = 7.1 Hz), 132.59 (d, J = 2.8 Hz), 131.85, 131.66, 131.51, 131.42–131.17 (m), 130.79, 130.70, 130.38, 129.61, 128.97, 128.87, 128.64 (d, J = 7.1 Hz), 128.45 (dd, J = 14.3, 9.7 Hz), 119.95, 117.53, 113.03, 45.82, 28.34, 24.49. IR (ATR, cm−1): 3380 (N–H), 2962 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 2861 (C–C–H), 1589 (aryl C–H).
C–H), 2861 (C–C–H), 1589 (aryl C–H).
Synthesis of CrCl2(PONNH) (Cr-1)
To a suspension of 5 mmol CrCl2 (615 mg) in THF, the PONNH ligand (5 mmol, 2.6 g) dissolved in THF was added dropwise and stirred at room temperature overnight. The dark green suspension formed was washed twice with hexane and then with ether and dried in vacuo to get a green crystalline powder. 1H NMR (500 MHz, benzene-d6) δ 7.32 (s, 2H), 7.08–7.02 (m, 44H), 7.00 (s, 3H), 4.79 (s, 0H), 4.27 (s, 1H), 2.25 (s, 1H), 2.11 (s, 1H), 1.55 (s, 2H). Selected IR (ATR, cm−1): 3054 (N–H), 2959 (C–H). UV–vis (DCM) λ/nm: 374 (broad). HRMS calcd for C34H35Cl2CrN2OP + Na+: 663.1161, observed: 663.4545. Room temperature X-band EPR, g value: 2.002. Chemical formula: C34H35Cl2CrN2OP. Elemental analysis (calculated): C, 63.66; H, 5.50; Cl, 11.05; Cr, 8.10; N, 4.37; O, 2.49; P, 4.83. Observed: C, 59.205; H, 5.888; N, 3.736. μeff = 2.86 (Evans, CDCl3).Synthesis of CrCl3(PONNH) (Cr-2)
To a suspension of 80 mg (0.5 mmol) of CrCl3 in THF, 1 equiv. PONNH ligand (260 mg) in THF was added dropwise and stirred at room temperature overnight. The brown suspension formed was dried in vacuo and washed twice with hexane and ether to get a golden-brown powder with a 62% yield. HRMS calcd, for C34H35Cl3CrN2OP + 2Na+: 721.0958; observed: 721.0955.General procedure for the catalytic reactions (Table S1†)
Typically, 0.5 mmol of p-methylbenzyl alcohol and 0.5 mmol of acetophenone were added to 0.015 mmol of the pre-catalyst Cr-1 and 20 mol% of tBuOK (or other bases, as mentioned in Table S1†), and the mixture was dissolved in 1.5 mL toluene (or t-amyl alcohol, 1,4-dioxane or THF; Table S1, entries 11, 12, 13, and 14,† respectively) and placed in a 25 mL Schlenk tube under N2. The tube was heated at 120–135 °C with stirring for the mentioned duration. The reaction mixture was then cooled down, and the products formed were monitored using TLC. The products were analyzed by NMR spectroscopy either based on substrate consumption or using 1,1,2,2-tetrachloroethane as the internal standard.For Scheme 3
A 25 mL flame-dried Schlenk tube was charged with Cr-1 (9.6 mg, 0.015 mmol, 3 mol%), and KOtBu (11.2 mg, 0.10 mmol, 20 mol%) dissolved in 1.5 mL toluene under a nitrogen atmosphere. Subsequently, 0.5 mmol of alcohol and 0.5 mmol of ketone were added and stirred for 5 minutes. Then, the Schlenk flask was closed tightly and kept at 135 °C for 36 h, followed by cooling to room temperature. Then, an aliquot was taken for the determination of conversion by NMR analysis. Purification was accomplished by column chromatography, and NMR was employed for the characterization of the isolated compound.For Scheme 4
A flame-dried Schlenk tube (25 mL) under a nitrogen atmosphere was charged with Cr-1 (9.6 mg, 0.015 mmol, 3 mol%) and potassium tert-butoxide (11.2 mg, 0.10 mmol, 20 mol%) dissolved in 1.5 mL toluene. Then, 0.5 mmol of 2-aminobenzyl alcohol and 0.5 mmol (1 equiv.) of various acetophenone derivatives were added to the reaction mixture and stirred for 5 minutes at room temperature. After the reaction mixture became homogeneous, the reaction vessel was heated at 135 °C for 36 h. After that, the reaction mixture was cooled to room temperature, and the products were analyzed by NMR after detection by TLC and separation by column chromatography.For Scheme 5
A flame-dried Schlenk tube (25 mL) under a nitrogen atmosphere was charged with Cr-1 (9.6 mg, 0.015 mmol, 3 mol%) and potassium tert-butoxide (56 mg, 0.5 mmol, 100 mol%) dissolved in 1.5 mL toluene. Then, 0.5 mmol of 1-phenylethanol and 0.5 mmol (1 equiv.) of corresponding benzyl alcohol derivatives were added to the reaction mixture and stirred for 5 minutes at room temperature. After the reaction mixture became homogeneous, the reaction vessel was heated at 135 °C for 40 h. After that, the reaction mixture was cooled to room temperature, and product formation was monitored by TLC and analyzed by NMR spectroscopy either based on substrate consumption or using 1,1,2,2-tetrachloroethane as the internal standard.General procedure for the stoichiometric reactions
The chromium complex Cr-1 (25 mg, 0.039 mmol,) was dissolved in toluene, forming a dark green solution. To this reaction mixture, 3 equiv. KOtBu (13 mg, 0.1171 mmol) was added at room temperature and stirred for 5 min. The colour of the reaction mixture gradually changed to greenish brown. After 5 min, 3 equiv. benzyl alcohol (12 µL) was added to this mixture and stirred at room temperature. It was observed that the colour of the mixture changed to brown. This mixture was examined by HRMS, which confirmed the formation of alkoxy species Cr-1b (exact mass = 712.2077 u, observed mass = 712.2745 u). However, in the 1H NMR spectra, we did not observe any distinct peak for the alkoxy complex even after heating the reaction mixture at 135 °C for 6 h because of the paramagnetic nature of the complex. Cr-1b could also be obtained by the reaction of Cr-1 with potassium benzyloxy species obtained from benzyl alcohol and tBuOK under heating conditions, as confirmed by the IR spectrum: IR (ATR, cm−1) = 3109 (N–H) (Fig. S14, 17, and 18; see ESI†).Conclusions
We have demonstrated the fabrication of a novel pincer PONNH ligand-based multi-tasking chromium complex Cr-1, which was effectively employed in the α-alkylation of ketones, as well as β-alkylation of secondary alcohols using primary alcohols as alkyl surrogates. The catalyst could also accomplish Friedländer quinoline synthesis by the dehydrogenative annulation of alcohols and 2-aminobenzylalcohol. A plausible mechanism based on metal–ligand cooperation is proposed based on the borrowing hydrogen methodology. The one-pot redox-neutral reaction is found to be atom-efficient, releasing water as the only byproduct. This work focuses on developing a new catalytic system using the earth-abundant and inexpensive chromium metal. The usage of a low amount of pre-catalyst (3 mol% Cr-1) and catalytic amount of tBuOK base (20 mol%) for α-alkylation and Friedländer quinoline synthesis in this study is advantageous in contrast to the contemporary protocol developed by Peng's group19 using an over stoichiometric amount of base and higher amounts of Cr(II) and ligands (5 mol% CrCl2, 10 mol% PPh3, and 1.2 equiv. LiOH). Thus, this method demonstrates an upper hand in terms of sustainability and atom economy, obviating copious waste generation. We believe this work will open up new avenues for the sustainable catalysis of relatively less-explored non-noble middle-transition (group six) metals.Data availability
Data supporting the manuscript are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is financially supported by SERB (Project No. SRG/2020/001047) and IIT Jodhpur (seed grant). Vaishnavi is thankful to IIT Jodhpur for research fellowship. Sachin Jalwal would like to thank CSIR, New Delhi for research fellowship. We are grateful to the CRDSI, IIT Jodhpur, for NMR facility. SC acknowledges DST-FIST [SR/FST/CS-II/2019/119(C)] for the HRMS facility at IIT Jodhpur.References
- (a) F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef CAS; (b) D. A. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS; (c) M.-J. Zhang, X.-L. Ge, D. J. Young and H.-Xi Li, Tetrahedron, 2021, 93, 132309 CrossRef CAS.
- (a) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed; (b) B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS; (c) A. J. Watson and J. M. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- (a) M. Peña-López, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 7826–7830 CrossRef; (b) N. Deibl, K. Ament and R. Kempe, J. Am. Chem. Soc., 2015, 137, 12804–12807 CrossRef CAS PubMed; (c) D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012–4015 CrossRef CAS PubMed; (d) S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326–6329 CrossRef CAS; (e) D. M. D'Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC; (f) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS; (g) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (h) R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS and references therein.
- (a) M. T. Reetz, Angew. Chem., Int. Ed. Engl., 1982, 21, 96–108 CrossRef; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- (a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS.
- (a) D. Yang, H. Wang and C. Chang, Adv. Synth. Catal., 2022, 364, 3100–3121 CrossRef CAS; (b) R. Sharma, A. Samanta, B. Sardar, M. Roy and D. Srimani, Org. Biomol. Chem., 2022, 20, 7998–8030 RSC.
- (a) S. Genç, B. Arslan, S. Gülcemal, S. Günnaz, B. Çetinkaya and D. Gülcemal, J. Org. Chem., 2019, 84, 6286–6297 CrossRef PubMed; (b) D. Wei, V. Dorcet, C. Darcel and J. Sortais, ChemSusChem, 2019, 12, 3078–3082 CrossRef CAS; (c) A. Maji, A. Singh, N. Singh and K. Ghosh, ChemCatChem, 2020, 12, 3108–3125 CrossRef CAS; (d) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (e) Y. Obora, ACS Catal., 2014, 4, 3972–3981 CrossRef CAS.
- (a) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed; (b) M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619–5621 CrossRef CAS; (c) S. Jalwal, V. Atreya, T. Singh and S. Chakraborty, Tetrahedron Lett., 2021, 82, 153362 CrossRef CAS.
- (a) G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (b) T. Irrgang and R. Kempe, Chem. Rev., 2018, 119, 2524–2549 CrossRef PubMed; (c) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2018, 119, 2681–2751 CrossRef PubMed; (d) S. Elangovan, J. B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 48, 14483–14486 CrossRef PubMed; (e) L. Bettoni, S. Gaillard and J. L. Renaud, Org. Lett., 2020, 22, 2064–2069 CrossRef CAS PubMed; (f) C. Seck, M. D. Mbaye, S. Coufourier, A. Lator, J. F. Lohier, A. Poater and J. L. Renaud, ChemCatChem, 2017, 23, 4410–4416 CrossRef; (g) M. S. Abdallah, N. Joly, S. Gaillard, A. Poater and J. L. Renaud, Org. Lett., 2022, 30, 5584–5589 CrossRef PubMed; (h) T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS.
- For cobalt catalyzed alkylation see: (a) G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 5, 1080–1083 CrossRef PubMed; (b) P. Chakraborty, M. K. Gangwar, B. Emayavaramban, E. Manoury, R. Poli and B. Sundararaju, ChemSusChem, 2019, 15, 3463–3467 CrossRef PubMed.
- For nickel catalyzed alkylation see: (a) S. P. Midya, J. Rana, J. Pitchaimani, A. Nandakumar, V. Madhu and E. Balaraman, ChemSusChem, 2018, 22, 3911–3916 CrossRef; (b) D. Wu, Y. Wang, M. Li, L. Shi, J. Liu and N. Liu, Appl. Organomet. Chem., 2022, 2, e6493 CrossRef; (c) S. Genç, B. Arslan, D. Gülcemal, S. Gülcemal and S. Günnaz, Org. Biomol. Chem., 2022, 48, 9753–9762 RSC.
- (a) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS; (b) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS; (c) T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS; (d) B. Maji and M. Barman, Synthesis, 2017, 3377–3393 CAS; (e) P. Chandra, T. Ghosh, N. Choudhary, A. Mohammad and S. M. Mobin, Coord. Chem. Rev., 2020, 411, 213241 CrossRef CAS; (f) M. K. Barman, A. Jana and B. Maji, Adv. Synth. Catal., 2018, 360, 3233–3238 CrossRef CAS; (g) S. Das, D. Maiti and S. De Sarkar, J. Org. Chem., 2018, 83, 2309–2316 CrossRef CAS; (h) S. Das, S. Sinha, D. Samanta, R. Mondal, G. Chakraborty, P. Brandao and N. D. Paul, J. Org. Chem., 2019, 84, 10160–10171 CrossRef CAS; (i) S. Donthireddy, V. K. Pandey and A. Rit, J. Org. Chem., 2021, 86, 6994–7001 CrossRef CAS; (j) S. Jalwal, A. Regina, V. Atreya, M. Paranjothy and S. Chakraborty, Dalton Trans., 2024, 53, 3236–3243 RSC.
- M. S. Holzwarth and B. Plietker, ChemCatChem, 2013, 5, 1650–1679 CrossRef CAS.
- (a) E. N. Frankel, E. Selke and C. A. Glass, J. Am. Chem. Soc., 1968, 90, 2446–2448 CrossRef CAS; (b) E. N. Frankel, J. Org. Chem., 1972, 37, 1549–1552 CrossRef CAS; (c) B. J. Gregori, M. Nowakowski, A. Schoch, S. Pöllath, J. Zweck, M. Bauer and A. Jacobi Von Wangelin, ChemCatChem, 2020, 12, 5359–5363 CrossRef CAS; (d) L. Ling, C. Hu, L. Long, X. Zhang, L. Zhao, L. L. Liu, H. Chen, M. Luo and X. Zeng, Nat. Commun., 2023, 14, 990 CrossRef CAS PubMed; (e) T. Vielhaber, K. Faust, T. Bögl, W. Schöfberger and C. Topf, J. Catal., 2022, 416, 352–363 CrossRef CAS; (f) B. Han, P. Ma, X. Cong, H. Chen and X. Zeng, J. Am. Chem. Soc., 2019, 141, 9018–9026 CrossRef CAS.
- (a) F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS; (b) Y. Miao, S. V. Samuelsen and R. Madsen, Organometallics, 2021, 40, 1328–1335 CrossRef CAS.
- (a) T. Singh, V. Atreya, S. Jalwal, A. Anand and S. Chakraborty, Chem. – Asian J., 2023, 18, e202300758 CrossRef CAS.
- H. Narjinari, N. Tanwar, L. Kathuria, R. V. Jasra and A. Kumar, Catal. Sci. Technol., 2022, 12, 4753–4762 RSC.
- P. Su, Z. Chen, J. Ni, Z. Yang, Y. Li and Z. Ke, ACS Catal., 2023, 13, 12481–12493 CrossRef CAS.
- D. Wei, B. Feng, Q. Chen, W. Yue, Y. Wang and Z. Peng, Org. Chem. Front., 2024, 11, 1955–1962 RSC.
- (a) Rajanbabu and T. V. Babu, Phosphinite and Phosphonite Ligands, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, 2012, pp. 159–232 Search PubMed; (b) S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS; (c) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed; (d) A. Adhikary and H. Guan, ACS Catal., 2015, 5, 6858–6873 CrossRef CAS; (e) M. Kondo, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2011, 133, 32–34 CrossRef CAS; (f) F. Kakiuchi, S. Takano and T. Kochi, ACS Catal., 2018, 8, 6127–6137 CrossRef CAS; (g) Y. Morimoto, T. Kochi and F. Kakiuchi, J. Org. Chem., 2021, 86, 13143–13152 CrossRef CAS; (h) E. Cook, J. D. Masuda and A. Xia, Can. J. Chem., 2010, 88, 99–103 CrossRef CAS; (i) B. Crociani, S. Antonaroli, M. Burattini, F. Benetollo, A. Scrivanti and M. Bertoldini, J. Organomet. Chem., 2008, 693, 3932–3938 CrossRef CAS; (j) H. Firouzabadi, N. Iranpoor and M. Gholinejad, J. Organomet. Chem., 2010, 695, 2093–2097 CrossRef CAS; (k) R. Favela-Mendoza, E. Rufino-Felipe, H. Valdés, R. A. Toscano, S. Hernandez-Ortega and D. Morales-Morales, Inorg. Chim. Acta, 2020, 512, 119920 CrossRef CAS.
- (a) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (b) M. A. Stevens and A. L. Colebatch, Chem. Soc. Rev., 2022, 51, 1881–1898 RSC; (c) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (d) B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS.
- (a) A. Cornia, L. Rigamonti, S. Boccedi, R. Clérac, M. Rouzières and L. Sorace, Chem. Commun., 2014, 50, 15191–15194 RSC; (b) A. Cornia, A.-L. Barra, V. Bulicanu, R. Clérac, M. Cortijo, E. A. Hillard, R. Galavotti, A. Lunghi, A. Nicolini, M. Rouzières, L. Sorace and F. Totti, Inorg. Chem., 2020, 59, 1763–1777 CrossRef CAS; (c) J. C. Ott, D. Isak, J. J. Melder, H. Wadepohl and L. H. Gade, Inorg. Chem., 2020, 59, 14526–14535 CrossRef CAS.
- (a) S. Wyantuti, R. A. Hafidza, S. Ishmayana and Y. W. Hartati, J. Phys.: Conf. Ser., 2017, 812, 012006 CrossRef.
- (a) R. N. Bose, B. Fonkeng, G. Barr-David, R. P. Farrell, R. J. Judd, P. A. Lay and D. F. Sangster, J. Am. Chem. Soc., 1996, 118, 7139–7144 CrossRef CAS.
- (a) J. A. Joule, K. Mills and G. F. Smith, Heterocycl. Chem, CRC Press, 3rd edn, 2020 Search PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315–7324 CrossRef CAS.
- (a) A. Maji, S. Gupta, M. Maji and S. Kundu, J. Org. Chem., 2022, 87, 8351–8367 CrossRef CAS PubMed; (b) Z. Wang, Q. Lin, N. Ma, S. Liu, M. Han, X. Yan, Q. Liu, G. A. Solan and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 8026–8036 RSC; (c) M. Tuan Ha, N. Thi Nguyen, N. Huyen Tran, Q. Viet Ho, N. Thi Son, V. H. Nguyen, H. Nguyen, D. Van Do, T. Quang Hung, B. Khanh Mai and T. Thanh Dang, Chem. – Asian J., 2022, 17, e202200909 CrossRef CAS; (d) R. Sharma, A. Mondal, A. Samanta, N. Biswas, B. Das and D. Srimani, Adv. Synth. Catal., 2022, 364, 2429–2437 CrossRef CAS; (e) Z. Hao, X. Zhou, Z. Ma, C. Zhang, Z. Han, J. Lin and G.-L. Lu, J. Org. Chem., 2022, 87, 12596–12607 CrossRef CAS; (f) D. Pal, A. Mondal, R. Sarmah and D. Srimani, Org. Lett., 2024, 26, 514–518 CrossRef CAS; (g) G. Sivakumar, M. Subaramanian and E. Balaraman, ACS Sustainable Chem. Eng., 2022, 10, 7362–7373 CrossRef CAS; (h) M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2016, 138, 15543–15546 CrossRef CAS; (i) S. Elangovan, J. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed; (j) K. Patra, A. Bhattacherya, C. Li, J. K. Bera and H. S. Soo, ACS Catal., 2022, 12, 15168–15180 CrossRef CAS; (k) X.-B. Lan, Z. Ye, M. Huang, J. Liu, Y. Liu and Z. Ke, Org. Lett., 2019, 21, 8065–8070 CrossRef CAS; (l) S. Shee, K. Ganguli, K. Jana and S. Kundu, Chem. Commun., 2018, 54, 6883–6886 RSC; (m) G. Chakraborty, R. Sikari, S. Das, R. Mondal, S. Sinha, S. Banerjee and N. D. Paul, J. Org. Chem., 2019, 84, 2626–2641 CrossRef CAS PubMed; (n) J. Das, K. Singh, M. Vellakkaran and D. Banerjee, Org. Lett., 2018, 20, 5587–5591 CrossRef CAS PubMed; (o) A. K. Bains, V. Singh and D. Adhikari, J. Org. Chem., 2020, 85, 14971–14979 CrossRef CAS; (p) S. Das, D. Maiti and S. De Sarkar, J. Org. Chem., 2018, 83, 2309–2316 CrossRef CAS; (q) S. Donthireddy, V. K. Pandey and A. Rit, J. Org. Chem., 2021, 86, 6994–7001 CrossRef CAS; (r) R. Mondal, G. Chakraborty, A. K. Guin, S. Pal and N. D. Paul, Tetrahedron, 2021, 100, 132479 CrossRef CAS; (s) S.-Q. Zhang, B. Guo, Z. Xu, H.-X. Li, H.-Y. Li and J.-P. Lang, Tetrahedron, 2019, 75, 130640 CrossRef; (t) F. Doraghi, F. Gilaninezhad, S. Karimian, A. M. Mahdavian, B. Larijani and M. Mahdavi, Adv. Synth. Catal, 2024, 366, 2142–2164 CrossRef CAS; (u) A. Mondal, R. Sharma, D. Pal and D. Srimani, Eur. J. Org. Chem., 2021, 2021, 3690–3720 CrossRef CAS; (v) M. Maji, D. Panja, I. Borthakur and S. Kundu, Org. Chem. Front., 2021, 8, 2673–2709 RSCand references therein; (w) S. Parua, R. Sikari, S. Sinha, S. Das, G. Chakraborty and N. D. Paul, Org. Biomol. Chem., 2018, 16, 274–284 RSC.
- (a) H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed; (b) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- (a) E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem. – Eur. J., 2014, 20, 15727–15731 CrossRef CAS; (b) S. Kar, M. Rauch, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2020, 10, 5511–5515 CrossRef CAS; (c) S. Kar, Y. Xie, Q. Q. Zhou, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 7383–7393 CrossRef CAS PubMed.
- P. Hu, Y.-L. Qiao, J.-Q. Wang and G.-X. Jin, Organometallics, 2012, 31, 3241–3247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01481b |
| This journal is © The Royal Society of Chemistry 2025 |