
Solubility of metal–boron–hydrogen compounds†
Thi Phuong Thao
Nguyen
a,
Terry D.
Humphries
 a,
Hans
Hagemann
b,
Craig E.
Buckley
a,
Torben R.
Jensen
c and
Mark
Paskevicius
*a
a,
Hans
Hagemann
b,
Craig E.
Buckley
a,
Torben R.
Jensen
c and
Mark
Paskevicius
*a
aDepartment of Physics and Astronomy, Institute for Energy Transition, Curtin University, GPO Box U1987, Perth, WA 6845, Australia. E-mail: m.paskevicius@curtin.edu.au
bDépartement de Chimie Physique, Université de Genève, 30, quai E. Ansermet, CH-1211 Geneva 4, Switzerland
cDepartment of Chemistry, Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Langelandsgade 140, DK-8000 Aarhus, Denmark
First published on 25th November 2024
Abstract
Boron–hydrogen compounds are of increasing importance as electrolytes in solid state batteries, for hydrogen storage and possibly as high temperature super conductors. Solvent based methods are of increasing importance to obtain pure products, for purification of materials and also for the synthesis of novel compounds. In this context, the solubility information of several classes of metal–boron–hydrogen compounds such as borohydrides, closo-decahydridodecaborates, closo-dodecahydridododecaborates, arachno- and nido-hydridoborates in typical solvents is vital. This information is currently dispersed in the literature, hence the need to present a cohesive summary and comparison of these properties. This review collects, analyses and discusses the available data to provide inspiration for the future design of new synthesis routes and the discovery of novel materials.
Introduction
Metal borohydrides, based on the tetrahydridoborate complex, BH4−, and metal-polyhydridoborates, based on boron–hydrogen anions, BxHyn−, have found application in multiple fields as reducing agents, reagents in synthetic reactions, hydrogen-rich fuels, or novel optical, magnetic, medical, ion conductivity purposes, and as hydrogen storage materials.1–3 Metal–boron–hydrogen materials are a diverse class of compounds, but perhaps the archetypical example is metal borohydride (e.g. NaBH4). Aluminium borohydride, Al(BH4)3, was first discovered by Schlesinger et al. in 1939 in an attempt to prepare aluminium hydride (AlH3) using diborane and trimethyl aluminium.4 Today, the most well-known and large-scale industrially produced member of this class of compounds is sodium borohydride, NaBH4, which is typically used as a reducing agent in organic synthesis,5 and is now also considered as a hydrogen export vector.6 The current commercially practiced process to produce NaBH4 is the Brown–Schlesinger process, which is based on the reaction between sodium hydride (NaH) and trimethylborate (B(OMe)3),7 but there are also other alternatives such as metallothermic reduction, mechano-chemical synthesis, and electrochemical methods.8 The second most used metal borohydride is lithium borohydride (LiBH4). This compound was first synthesised by Schlesinger and Brown in 1940 using ethyl lithium and diborane9 however, because of difficulties and dangers in handling diborane gas as a synthetic agent, solvent-based and mechanochemical approaches have been developed.1Mechano-chemical synthesis is one of the most common synthetic routes for metal–boron–hydrogen compounds.10 This approach usually utilises solid–solid metathesis reactions, which produce the desired metal borohydride along with a relatively stable and inert metal halide by-product.11–13 Thus, normally a pure material cannot be obtained. Pure products can be obtained via mechano-chemical solid–gas reactions, for example where diborane is reacted with a metal hydride but it is challenging to scale-up owing to the use of toxic diborane gas.14 On the other hand, within the last decades, solvent-based synthesis has revealed numerous advantages in the production of selected materials with high purity, even in preferred structural polymorphs.15 This approach offers feasible isolation and purification of desired reaction products from by-products based on differences in solubility.16–22 A good choice of solvents can optimise synthetic reactions and produce pure metal borohydrides that are fundamental in their own right, or as building-blocks for derivatives. The success of solvent-based synthesis is closely related to the choice of solvent and the solubilities of reactants, products and unwanted by-products. This can lead to discovery of novel materials with unobserved properties, which may find technological applications in the future.
One aspect that inhibits large scale solvent-based synthesis and the application of metal–boron–hydrogen compounds, and many other materials, is the lack of knowledge about solubilities in solvents. A material's solubility relates closely to the dielectric constant (ε) of the solvent. Understanding of the correspondence between solubility and dielectric constant of the solvent may be useful in choosing reaction and extracting solvents to optimise the yield and purification of desired reaction products. Because metal borohydrides and metal–boron–hydrogen materials are known to be highly reactive, the chemical reactivity of the solvent is also of importance, where the most significant adverse reactivities are highlighted in the presented solubility figures. For example, metal borohydrides typically undergo hydrolysis in water, but water can still be used as a solvent, especially when a stabiliser, such as NaOH, is used to raise the pH.23 Therefore, in this study, we provide a coherent summary of available data on the solubility of these materials in various solvents charted as a function of dielectric constant of the solvent and cation charge density of the cations. We also highlight some reactions that have been reported in liquid media, including reactions with borane donor solvents (e.g. tetrahydrofuran borane, THF·BH3), functionalisation of closo-hydridoborates, solvothermal synthesis, mechano-chemistry using a solvent as grinding media, reactions in supercritical fluids, ionic liquids (IL) and low boiling point solvents, and solution-based electrochemistry. Solvent effects in each synthetic approach are analysed and discussed.
Solvent-based synthesis of metal borohydrides
Historically, solvent based metathesis reactions employed organometallic compounds, such as ethyl lithium, diethyl magnesium or trimethyl aluminium as precursors. However, a solvent based metathesis reaction can also be conducted using a metal halide MXn, X = Cl, Br or I, in conjunction with an industrially produced metal borohydride, such as NaBH4 or LiBH4, as a precursor.13,24 These reactions are then partially driven by the formation of a stable alkali metal halide and occur despite low solubility of the reactants. This is illustrated by the synthesis of manganese borohydride, Mn(BH4)2,25 although metathesis reactions have been used to form a variety of other borohydrides.13,26,27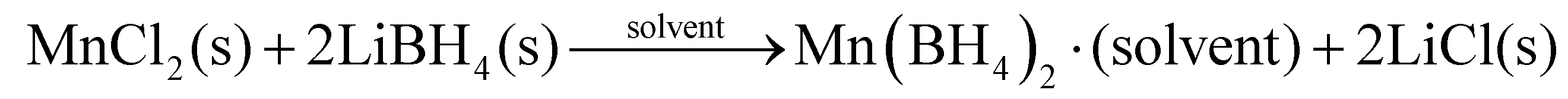 | (1) |
The obvious advantage of the above-mentioned strategy is that the product is dissolved in the selected solvent, which in the case of eqn (1) is a mixture of toluene and dimethylsulphide, (CH3)2S. The latter is denoted a co-solvent or extraction solvent, which coordinates to the metal and increases the solubility of the metal borohydride. This allows for the redundant metal halide by-product to be filtered away. In fact, the ionic metal halide is not necessarily an inert by-product. In some cases, secondary reactions can take place where the metal borohydride reacts with the metal halide and forms new (bi-)metal borohydride halide with ordered structures or as solid solutions.28–30
The removal of the solvent post-reaction often produces a solid solvated product, i.e. manganese borohydride hemidimethylsulfide, Mn(BH4)2·0.5((CH3)2S). The utilisation of relatively weakly coordinating co-solvents and/or extraction solvents allows for the removal of the solvent molecule at moderate conditions and production of a pure metal borohydride. Interestingly, optimisation of the exact physical conditions (time, temperature, pressure etc.) may allow formation of polymorphically pure crystalline products. This is illustrated by the treatment of Mn(BH4)2·0.5((CH3)2S) at room temperature (RT) in vacuum, which produces an open-structured polymorph, γ-Mn(BH4)2 (isostructural to γ-Mg(BH4)2), whereas solvent removal at elevated temperatures (RT < T < 100 °C) in vacuum provides a polymorph with a more dense crystal structure, α-Mn(BH4)2.
Diethylether (Et2O) has also been used as a solvent for the synthesis of α-Mn(BH4)2via a metathesis reaction between MnCl2 and LiBH4.31 After the reaction, diethylether was removed and replaced by an extraction solvent, (CH3)2S, but a phase-pure product was not obtained. In all cases the synthetic product was α-Mn(BH4)2 with varying amounts of Li2Mn(BH4)4. This was assumed to be due to a minor solubility of remaining LiBH4 in (CH3)2S or via formation of the coordination framework compound [(Li(Et2O)2)(Mn2(BH4)5)].32 This example demonstrates some of the complexity in solvent-choice and problems that can be associated with solubility. The use of a more strongly coordinating co-solvent such as tetrahydrofuran (THF) does not allow for the synthesis of pure Mn(BH4)2. The product from the synthesis is the solvate, Mn(BH4)2·3THF, and the solvent molecule (THF) is only released at T > 140 °C. Unfortunately, this temperature is above the thermal stability range and leads to instant decomposition of Mn(BH4)2 leaving an amorphous product.33
The examples provided above for Mn(BH4)2 are also largely true for many other borohydride systems. The selection of solvents is important for the synthesis of many metal borohydride materials due to the formation of strongly bonded complexes and the difficulty to break these bonds without decomposing the borohydride materials. Mg(BH4)2 has been shown to be soluble in a range of solvents such as diethyl ether, THF, dimethylsulphide and a range of amines, but some solvents such as tetramethylethylenediamine (TMEDA) coordinate too strongly to be removed.26,27,34,35 This illustrates the importance of careful evaluation of solubilities of by-products and the co-solvents ability to coordinate the metal and dissolve the wanted product. The solubilities of metal borohydrides are illustrated in Fig. 1.
 | ||
| Fig. 1 Solubility of borohydrides in solvents (listed by dielectric constant in brackets). Borohydride species are listed by cationic charge density (low to high).7,10,15,24,36–49 | ||
A fruitful approach is to utilise solid–liquid synthesis using metal hydrides and borane donating solvents as reactants. The metal hydride often needs to be mechanochemically activated to reduce the particle size and increase the surface area. This procedure significantly increases the kinetics of the reactions as compared to using hand-ground material. Then the metal hydride is suspended in an anhydrous non-coordinating solvent, e.g. toluene, or a coordinating solvent such as THF or diethyl ether. A borane donating complex such as dimethylsulfide borane complex (DMSB, (CH3)2SBH3) or triethylamine borane complex, (CH3CH2)3NBH3, typically with an excess of 50%, a concentration of 1–5 M and reaction time of 24–48 h under stirring. Thus, the reaction takes place between the borane donor complex in solution and a solid metal hydride surface. The reaction mechanism can be described as a nucleophilic addition mechanism where a hydride ion, H−, is transferred forming the borohydride complex, BH4−, and a cation, which can be solvated and removed from the surface and allows the reaction to proceed. Thus, the reaction involves the solvent mediated dissolution of M(BH4)x that is transported away from the reactive hydride interface.10 The selected solvent or combination of solvents will determine whether the metal borohydride formed stays in solution or precipitates in its pure form or as a solvate. A solution of dissolved metal borohydride will allow further purification by filtration to remove possible impurities of metal, metal hydrides or oxides. In a similar manner, borohydride synthesis can also be undertaken by solvent aided mechano-chemical methods, e.g. Na3(NH2)2BH4. This was achieved by ball milling NaNH2 and NaBH4 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, doped with a Co–Ni–B catalyst in cyclohexane.50 The solvent aids to absorb heat generated during the reaction and prevent oxidation.
:1 ratio, doped with a Co–Ni–B catalyst in cyclohexane.50 The solvent aids to absorb heat generated during the reaction and prevent oxidation.
As mentioned, the reaction mechanism for metal borohydride synthesis from a metal hydride and a borane complex is a nucleophilic addition mechanism, which is only possible for ionic or polar covalent M–H bonds, such as those formed by alkali- and earth alkali metals and rare earth metals. This is illustrated by the rare earth metal neodymium, which forms a metallic NdH2 hydride upon hydrogenation at moderate conditions and an ionic hydride at more harsh conditions, NdH3. By using the above mentioned synthetic strategy it was shown that the metallic NdH2 hydride cannot react with DMSB complex, but the ionic NdH3 readily formed the solvate, Nd(BH4)3·(CH3)2S.10 In this case, desolvation provides a trivalent neodymium(III) borohydride, α-Nd(BH4)3. In some cases, solvate formation stabilises trivalent metal borohydrides where a reductive desolvation leads to the formation of divalent M(BH4)2 and release of diborane gas. This is observed for the ytterbium(III) borohydride solvent complex Yb(BH4)3·x(CH3)2S, where desolvation leads to the formation of γ-Yb(BH4)2. However, reacting samarium(III) hydride in a solution of weak/non-coordination solvents, e.g. DMSB in toluene, leads directly to Sm(BH4)2, whereas with stronger coordination solvents, e.g. THF in toluene, forms Sm(BH4)3·1.5THF. A similar ‘solvent metal protection’ strategy was developed using ammonia in dimethylsulfide to produce [Fe(NH3)6](BH4)2 and [Co(NH3)6](BH4)2, where Fe2+ and Co2+ otherwise would be reduced by BH4−.51
Transition metal borohydrides, which are stable at room temperature can only be prepared with metals in a low transition state and with the d electron configurations, d0, d5 or d10.52 The transition metal borohydrides, M(BH4)2, M = Cr2+ (d4), Fe2+ (d6) and Co2+ (d7) are stable in solution only at T < −30 °C but not yet in the solid state at RT.51 The molecular titanium borohydride, Ti(BH4)3 with the d1 electron configuration has an in-between stability and decompose at T < 0 °C.
Solvent-based synthesis of polyhydridoborates
High temperature solvothermal synthesis of metal hydridoborates were typically performed in high boiling point solvents (e.g., glymes, dimethoxyethane) to synthesise so-called “polyhydridoborates” such as arachno-, nido- and closo-hydridoborates.2,53 The choice of solvent and reaction temperature may lead to different final products. For example, when triethylamine is used as the solvent for the reaction between B2H6 and NaBH4, Na2B12H12 is formed at 100–180 °C.54 When dimethoxyethane is used, NaB3H8 is formed at a reaction temperature of 25 °C and further heating the reaction to above 50 °C produces Na2B12H12 in a mixture with NaB11H14. However, one drawback of this approach is that the high boiling point solvents are experimentally difficult to remove from the final product. For this reason, solvothermal synthesis routes have also been explored and successfully applied to prepare compounds of B3H8−, B10H102− and B12H122−.43,55 Performing the reaction in a closed vessel, i.e. an autoclave, allows for the temperature to be raised above the solvent boiling point at autogenous pressures created by the vapour pressure of the solvent(s). The solubility of the reactants is often higher at elevated temperatures above the usual boiling point, which contributes to a higher yield of product. The solvothermal reactions typically involve one or more intermediates, e.g. B3H8, and then proceeds to form the thermodynamically more stable closo-hydridoborates, B10H102− and B12H122−. Here, a tetraalkylammonium borohydride (either tetrabutylammonium (TBA) or tetraethylammonium (TEA)) borohydride were used. These starting materials can be obtained commercially, but also by a metathesis reaction of NaBH4 with the either TBABr or TEACl by ball milling. Alternatively, the exchange reaction can also be performed using different solutions (e.g. acetonitrile (CH3CN) for TEACl and isopropylamine for NaBH4), which are then mixed to precipitate NaCl, while TEABH4 remains in the solution. The first reaction step is, for example:is performed at 80 °C for 1 h with TBA. It is interesting to note that in this reaction, the solvent dichloromethane is reduced to chloroform, i.e. it actively takes part in the reaction. The tetraalkyl salt can then either be used as obtained, purified or transformed into NaB3H8 using NaB(Ph)4 (sodium tetraphenylborate) to precipitate TEAB(Ph)4 or TBAB(Ph)4.

In the second step, TEAB3H8 was suspended in toluene and reacted at 185 °C in a closed container to produce an approximately equimolar mixture of (TEA)2B10H10 and (TEA)2B12H12. Converting this mixture into the corresponding sodium salts using NaB(Ph)4 in hot water produces, upon solvent evaporation, a mixture of Na2B10H10 and Na2B12H12. The recrystallization of this mixture in isopropanol yields the solid ionic conductor Na4(B10H10)(B12H12).43 The mixture of (TEA)2B10H10 and (TEA)2B12H12 can also be separated by fractional crystallisation in methanol, as (TEA)2B10H10 is more soluble in methanol than (TEA)2B12H12.
One further example is the thermolysis of KB3H8, which typically yields a mixture of products containing BH4−, B10H102−, B12H122− and B11H14. From this mixture, B10H102− is isolated and purified by first precipitating TEAB12H12 in MeOH, then TEAB11H14 is precipitated in water before recovering the water soluble TEAB10H10.56 Sequential extraction based on difference in solubility towards different extracting solvents can allow isolation of each boron–hydrogen product with high purity. The solubility of a wide range of metal closo-hydridoborates based on the complex anions, BnHn2−, provided in Fig. 2 can help designing effective purification sequences.
 | ||
| Fig. 2 Solubility of closo-hydridoborates and derivatives in solvents (listed by dielectric constant in brackets). The compounds are listed by cationic charge density (low to high).43,47,59,77–104 Abbreviation: EC-DMC: ethylene carbonate:dimethyl carbonate (1:1 vol%), BMIM-TFSI: 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide. | ||
Reactions of metal borohydrides with Lewis base borane-solvent adducts has been long used to synthesis polyhedral hydridoborate compounds. A good example is the synthesis of NaB12H12 and KB12H12 from reactions of NaBH4 or KBH4 with borane-trimethylamine and -triethylamine at 200–250 °C in high-boiling point alkane solvents.57,58 The formation mechanism of polyhydridoborate compounds is an aggregation or polycondensation of small borane complexes based on the nucleophilicity of B–H bonds.
Recently, a high-yield selective synthesis based on this approach has been developed and gained success in synthesising high purity alkali salts of some polyhydridoborate anions such as B12H122−, B3H8− and B11H14−. Solvothermal reactions of NaBH4 and KBH4 with DMSB in diglyme form Na2B12H12 and K2B12H12 at a yield greater than 80%.59 K2B12H12 can be directly isolated from the reaction solution due to its poor solubility in diglyme while solvent-free Na2B12H12 is obtained after removal of the solvent by heating under vacuum.
MB3H8 with M = Na, K, Rb, Cs can be synthesised by reactions of their respective metal borohydrides with DMSB in 1,4-dioxane at 90 °C for 48 h.60 The final product is a precipitate mixture of the metal octahydrotriborate and unreacted metal borohydride. Unsolvated MB3H8 salts of K, Rb and Cs can be purified by dissolution in THF followed by recrystallisation using dichloromethane (DCM) but NaB3H8 is obtained in a solvated form due to strong coordination of the solvent with Na+. Unsolvated NaB3H8 can be obtained by reaction with THF·BH3 in THF under reflux conditions.61 This method can also be used to synthesise LiB3H8 but the final product is solvated LiB3H8·1.5THF as a clear oil. LiB3H8 can be formed in the reaction of LiBH4 with DMSB in 1,4-dioxane but the formed LiB3H8 reacts with LiBH4, which is slightly soluble in 1,4-dioxane resulting in formation of B9H14− and B11H14−.60
Adjustment of parameters such as temperature, molar ratio of the reactants and reaction time of above reactions leads to the synthesis of nido-hydridoborates. Reaction of KBH4 and DMSB at a molar ratio of 1:10 in 1,4-dioxane at 90 °C for 5 d produces KB11H14 as the major product, which can be purified at 86% yield.16 Replacing KBH4 with LiBH4 or NaBH4 results in 1,4-dioxane solvated LiB11H14 and solvent-free NaB11H14, respectively. The alkali nido-hydridoborates can also be formed when using 1,2-dimethoxyethane as the solvent, but solvent-free products cannot be obtained because of strong coordination of the cations with 1,2-dimethoxyethane. Increasing reaction temperature to break cation-solvent bonds causes formation of more unwanted-products.
Solvent-based synthesis of closo-hydridoborate derivatives
Solvent based methods have successfully been developed and used during decades for the synthesis of novel metal–boron–hydrogen compounds and their derivatives.21,62 These methods have been previously reviewed.1,2,15,17,18,20,21,63 The functionalisation of closo-hydridoborates is an important field of boron chemistry to develop new chemicals and to modify the properties of this class of materials. In 1962 Knoth synthesised the first organic closo-hydridoborate derivative followed by a raft of closo-hydridoborate derivatives based on decahydrido-closo-decaborate(2-) (B10H102−) and dodecahydrido-closo-dodecaborate(2-) (B12H122−) anions, including the substitution of H groups for acyl, hydroxy, alkoxy, alkyl, mercapto, and alkylthio groups.64,65 The synthesis of these functionalised closo-hydridoborates was typically undertaken with solvent-based chemical approaches in a range of solvents and solvent mixtures. It is clear that the solubility and reactivity of the closo-hydridoborates and their products is integral to achieving high yield and purity.2The functionalisation of closo-hydridoborates can be achieved to varying degrees, whereby one, multiple, or the complete exchange of all terminal hydrogen atoms can occur for particular functional groups, e.g. formation of B12H11(OH)2− or B12(OH)122−.21,65,66 These functional groups can also be further modified to expand the class of closo-hydridoborates to an indefinite degree akin to organic chemistry, for instance, the conversion of B12(OH)122− to B12(OR)122−.66 Thus, a rich chemistry is possible based on the foundation of closo-hydridoborates, allowing fine tuning of chemical and physical properties. These compounds also form fascinating solvated complexes with a range of solvents.19
It is also possible to substitute a B–H group in the boron cages for another group, such as C–H, i.e. in the production of carbaborates such as CB11H12−.67,68 This class of derivatives has received enormous attention in recent years as promising solid-state electrolyte materials for battery applications. Solid solutions of closo-carbaborates, e.g. Na3(CB9H10)(B12H12), Na3(CB11H12)(B12H12), Na2(CB9H10)(CB11H12) and Na2−y(CB11H12)y(B12H12)1−y, have been shown to deliver high ionic conductivity at moderate temperatures and some have been utilised for fabrication of all-solid-state batteries.69–73 Typically, the synthesis of closo-carbaborates is undertaken from nido- or arachno-boranes or hydridoborates, which are not yet closo-hydridoborates and have accessible sites for deprotonation and subsequent insertion into the cage. They can be conveniently synthesised from decaborane (B10H14) reacted with formaldehyde (HCHO) in basic (KOH) solution, which initially form arachno-6-CH2B9H12−.74,75 Thermolysis at temperatures in the range 160 to 220 °C (in vacuum) provide mixed anion compounds NH(CH3)3(CB8H9)x(CB9H10)y(CB11H12)z, where the sample heated at 200 °C has the composition NH(CH3)3(CB8H9)0.26(CB9H10)0.66(CB11H12)0.08.76 Initially closo-2-CB9H10− is formed, which converts to the thermodynamically more stable configuration closo-1-CB9H10− after heating to 150 °C. At lower thermolysis temperatures (160 to 180 °C) the reaction resulted in minor amounts of unreacted NH(CH3)3(arachno-6-CH2B9H12), whereas higher temperatures facilitated the formation of closo-CB11H12− rather than closo-CB8H9−.76
A new and low-cost solvent-based synthesis of CB11H12− using common laboratory reagents68 has been recently proposed. This method avoids the use of the toxic, flammable and expensive decaborane, B10H14, precursor. (CH3)3NH[B11H14], which can be feasibly synthesised by thermal treatment of NaBH4 with bromopentane in diglyme, is used as the starting material. The nido-hydridoborate precursor is converted to Na2B11H13 in excess aqueous NaOH at 80 °C and then, to CB11H12− by the insertion of CCl2 from chloroform in dimethoxyethane with the presence of basic NaOH and K2CO3. Dimethoxyethane is chosen as the reaction solvent instead of THF because it is more polar, which improves the solubility of NaOH. Conducting the reaction in diethyl ether and dioxane produces no CB12H11− since Na2B11H13 does not dissolve in these solvents (Fig. 3).
 | ||
| Fig. 3 Solubility of arachno- and nido-hydridoborates in solvents (listed by dielectric constant in brackets). The compounds are listed by cationic charge density (low to high).53,56,60,68,78,92,103,118–132 | ||
Again, solvent-based synthesis is key to achieving high yields and purities of the closo-carbaborate products. With the rich chemistry of B–H bond, it is also possible to insert other groups or atoms into the boron-cage, such as the case of heavy metals like Pb or Sn, which cause modifications to the thermal, structural, and ionic conductivity properties of the solid-state compounds, e.g. B11H11Pb2− and B11H11Sn2−.105,106 In this case, the use of aqueous or non-aqueous synthesis is key to controlling the reaction pathways and formed products from a B11H14− precursor.
Ionic liquids (IL) have been demonstrated to provide new synthetic pathways for functionalised decaborane (B10H14) and ortho-carbaborane clusters.107 For instance, B10H14 reacts with an excess of RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 in a biphasic mixture of 1-butyl-3-methyl-imidiazolium (Bmim) or 1-butyl-4-methyl-pyridium (Bmpy)BF4 salts forming 6-R-B10H13.107 Hydrocarbons are insoluble in these IL's and so the decaborane is added to the IL and stirred vigorously with the olefin and toluene to form an emulsion. Separation is then relatively easy with high yields. Similar reactions can take place with a range of olefins. Decaborane reactions with alkynes in ILs do not yield hydroboration products, but instead result in alkyne insertion, thereby providing an important new route to ortho-carbaboranes.107 For example B10H14 reacts with 2 equivalents of a ligand (L) to form B10H12L2, where L is SEt2 or CH3CN. Alkyne insertion then can then be undertaken where B10H12L2 reacts with RC
CH2 in a biphasic mixture of 1-butyl-3-methyl-imidiazolium (Bmim) or 1-butyl-4-methyl-pyridium (Bmpy)BF4 salts forming 6-R-B10H13.107 Hydrocarbons are insoluble in these IL's and so the decaborane is added to the IL and stirred vigorously with the olefin and toluene to form an emulsion. Separation is then relatively easy with high yields. Similar reactions can take place with a range of olefins. Decaborane reactions with alkynes in ILs do not yield hydroboration products, but instead result in alkyne insertion, thereby providing an important new route to ortho-carbaboranes.107 For example B10H14 reacts with 2 equivalents of a ligand (L) to form B10H12L2, where L is SEt2 or CH3CN. Alkyne insertion then can then be undertaken where B10H12L2 reacts with RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH to form 1,2-RC2B10H11 + 2L. This is undertaken in a biphasic toluene/1-butyl-3-methylimidiazolium chloride (Bmim)Cl system. These reactions have been demonstrated for a wide range of functional alkynes.
CH to form 1,2-RC2B10H11 + 2L. This is undertaken in a biphasic toluene/1-butyl-3-methylimidiazolium chloride (Bmim)Cl system. These reactions have been demonstrated for a wide range of functional alkynes.
Dehydrocondensation reactions are key steps in many polyborane transformations leading to the formation of higher-order boron–hydrogen systems. For example, the thermally induced dissociation of DMS from 4-Me2S–B9H13 forms a Lewis acidic B9H13 fragment in xylene or in vacuo, which can then undergo fusion, accompanied by dehydrogenation, with another such fragment to produce n-B18H22.107 When carried out in an IL (in 1-butyl-3-methylimidazolium chloride (BmimCl) under biphasic conditions), only n-B18H22 is formed with a B10H14 side product.
Borohydride based ionic liquids have also been developed including 1-butyl-2,3-dimethylimidazolium (BMMIM), 1-ethyl-3-methylimidazolium (EMMIM), 1-propyl-1-methylpiperidinium (PropMPip), and 1-butyl-1-methylpyrrolidinium (BMP) borohydride. AmimBH4 and BmimBH4 were prepared and have a melting point below −60 °C.108 These have been successfully used for the selective reduction of carbon–carbon double bonds in conjugated alkenes as well as the α,β-carbon–carbon double bonds in highly activated α,β,γ,δ-unsaturated alkenes. In addition, ionic liquids of B3H8− have been developed including [EMMIM][B3H8], [PropMPip][B3H8], [N(Bu)4][B3H8] and [BMMIM][B3H8].109
A raft of organic cation salts of B12Cl122− show low melting points and interesting physicochemical properties and potential as ionic liquids for electrochemical applications. These salts also show favourable solubilities at room temperature with miscibility in MeCN, DMSO and Me2CO, whilst being non-miscible in H2O, EtOH and Et2O.110 Interestingly, the class of B12X122− (X = F, Cl, Br, I) also show quasi-reversible electrochemical oxidation to B12X12− and further non-reversible oxidation to B12X12 in liquid SO2, MeCN, or organic carbonates.111–113 Halogenated derivatives of sodium-dodecahydrido-closo-dodecaborate, Na2B12Cl12, Na2B12Br12, and Na2B12I12, have high thermal stability and high sodium ion conductivity at elevated temperatures.114,115 As such, the electrochemical properties of a wide array of boron compounds have been published and reviewed.116
Results and discussion
The solubility of inorganic salts in non-aqueous and organic solvents is of great interest across the fields of synthetic chemistry to industrial production. Of particular importance is the dielectric constant of these solvents, which dramatically range from very low (e.g. pentane at 1.8) to remarkably high (e.g. n-methylacetamide at 165) with water at 78.5.117 Despite solubility trends being shown across dielectric constant, the resulting solubility is not solely based on this one parameter and there is much complexity surrounding the prediction of the solubility in solvent classes that are often divided into protic, aprotic, and inert groups. One further consideration may be the solvent liquidus temperature range, its viscosity, coordinating nature, purity, and/or other physical and chemical (Lewis acidity) properties.Herein, we have provided graphical representations of the solubilities of well-known metal–boron–hydrogen compounds (and derivatives) in typical solvent systems, sourced from the available literature (Fig. 1–4). The data is classified and graphically presented in separate charts for borohydrides, closo-hydridoborates, arachno- and nido-hydridoborates and some of their derivatives, and neutral boranes. Although incomplete, these solubility charts do show certain trends and indications to predict unknown solubilities. To aid in discovering trends the solvents are listed by dielectric constant, the boron compounds are sorted by cationic charge density where appropriate. The solubility of each compound in a particular solvent is categorised qualitatively as insoluble, slightly soluble (<1 wt%), soluble (1–10 wt%), very soluble (>10 wt%), and reactive (denoting chemical incompatibility). A table of solubility information is also available in the ESI† that provides a mixture of qualitative and quantitative solubility information where available (including more detailed references).
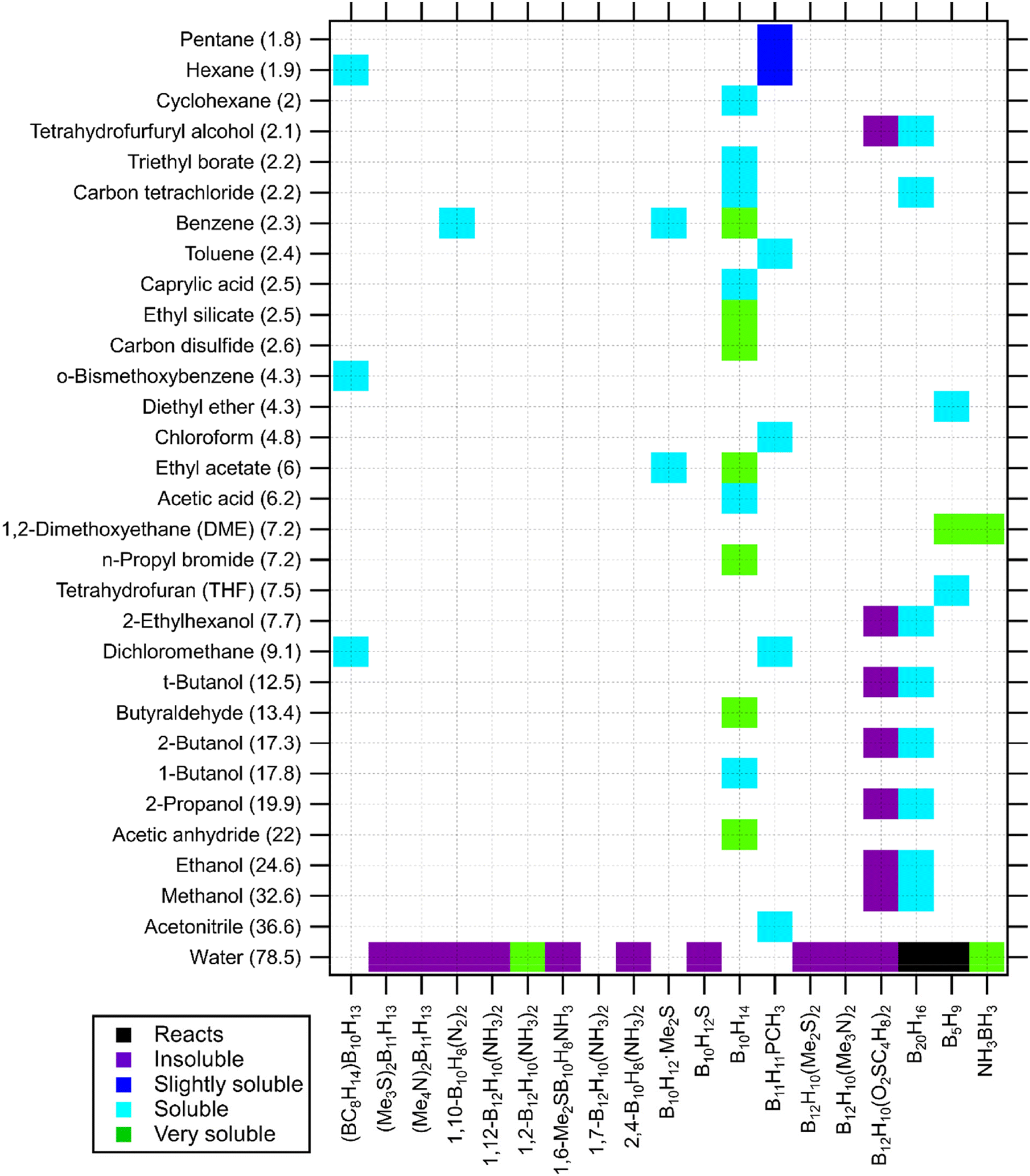 | ||
| Fig. 4 Solubility of neutral boranes in solvents (listed by dielectric constant in brackets). Borane species are listed alphabetically.39,78,90,119,121,123,127–129,138–143 | ||
Fig. 1 shows the solubility of borohydrides in water and a variety of non-aqueous organic solvents. In certain publications, metal borohydrides are stated to be soluble in water. This is true, but BH4− experiences hydrolysis when water is used as a solvent, forming B(OH)4− and releasing H2.49 This characteristic can be used to destroy borohydride impurities into forms that are removable from a product mixture.61 The hydrolysis of BH4− anion can be dramatically slowed down in alkaline solutions, e.g. NaOH. This is addressed as notation in the ESI.† NaBH4 has extensive solubility data in the literature as the most well-known metal borohydride. It is soluble in various organic solvents but insoluble in diethyl ether and 1,4-dioxane. This compound is reported to be only slightly soluble in alcohol solvents such as tert-butanol, 2-propanol and reacting with methanol and ethanol.
Fig. 2 provides solubility of closo-hydridoborates and their derivatives. Most of the data is reported for solubility in water. Alkaline and earth alkaline metal decahydrido-closo-decaborates and dodecahydrido-closo-dodecaborates and their halogenated relatives are soluble in water, except for Cs. Meanwhile, compounds of heavy metals such as Ag, Hg and Tl are not soluble in this solvent. closo-Hydridoborates of organic cations are also insoluble in water, except for a very few cases such as (Me4N)2B12H12 and (nBu4N)2B12H12. This may be related to the hydrophobic property of these cations.
Fig. 3 and 4 show solubility of archano- and nido-hydridoborates and some neutral boron–hydrogen compounds, respectively. Their solubility data is mostly hidden in synthesis procedures reported in the literature. Such little available data is not enough to establish specific trends for these branches of boron–hydrogen compounds but still, significant information can be drawn out from the charts. For instance, solubility data of alkaline octahydridotriborates (KB3H8 and NaB3H8 in Fig. 3) can aid in designing synthesis routes for borates with higher boron numbers, namely closo- and nido-hydridoborates.
Conclusions and outlook
The chemistry of boron hydrogen compounds is very rich as outlined by the examples cited above, while the necessity for knowledge about solubilities and solvent metal borohydride interactions (such as ability to coordinate the metals) is undeniable. Solvent-based purification of specific salts such as closo- and nido-hydridoborates formed from the thermolysis of BH4− or B3H8− salts may be the cheapest approach for the synthesis on a large scale due to the use of the least expensive starting materials. The present information of solubilities can also lead to other new synthesis strategies to prepare pure compounds in high yields. These compounds can be utilised directly or used as precursors for synthesis of derivatives. Currently there is much focus on development of novel types of batteries based on multivalent metals, such as magnesium and calcium. They can form the basis for more sustainable, safe and cheap solid-state batteries with higher energy contents to replace the well-known lithium ion battery. Most of the recently developed liquid state electrolytes for magnesium batteries are based on boron hydrides or fluorides.133 Recently, significant progress within solid state magnesium ionic conductors have been made by the discovery of derivatives of magnesium borohydride, Mg(BH4)2·xNH3 and Mg(BH4)2·xNH2CH3.134–136 They have very high Mg2+ conductivity at room temperature and show stability towards a metallic Mg anode. This has been demonstrated by development of a solid-state inorganic magnesium battery, which was charged and discharged several times.137 This illustrates that synthesis of boron-based compounds may have significant future societal impact, e.g. within efficient storage of renewable energy. Here we provide new inspiration for development of novel synthesis strategies, which may lead to novel compounds with unseen properties. Further synthetic innovations may be feasible by the exploration of sequential addition of solvents rather than utilising single or mixed solvent systems. This may improve the solubility of this class of compounds and further the range of reactions that may be possible.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Australian Research Council (ARC) for Linkage project LP190100297, Discovery project DP230100429, and Industrial Transformation Training Centre “GlobH2E” IC200100023 along with the Australian Renewable Energy Agency (ARENA) for a Transformative Research Accelerating Commercialisation (TRAC) Program grant (CB033). This work is supported in part by the Swiss National Science Foundation grant project 200020_182494. This work was supported by a research grant (40717) from VILLUM FONDEN and from the Independent research fund Denmark for technology and production (DFF - 0217-00327B).References
- M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Černý, D. B. Ravnsbæk, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565–1634 RSC.
- B. R. S. Hansen, M. Paskevicius, H.-W. Li, E. Akiba and T. R. Jensen, Coord. Chem. Rev., 2016, 323, 60–70 CrossRef CAS.
- E. M. Dematteis, M. B. Amdisen, T. Autrey, J. Barale, M. E. Bowden, C. E. Buckley, Y. W. Cho, S. Deledda, M. Dornheim and P. De Jongh, Prog. Energy, 2022, 4, 032009–032049 CrossRef CAS.
- H. Schlesinger, R. T. Sanderson and A. Burg, J. Am. Chem. Soc., 1939, 61, 536 CrossRef CAS.
- P. Rittmeyer and U. Wietelmann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- A. Ibrahim, M. Paskevicius and C. E. Buckley, Sustainable Energy Fuels, 2023, 7, 1196–1203 RSC.
- H. Schlesinger, H. C. Brown and A. Finholt, J. Am. Chem. Soc., 1953, 75, 205–209 CrossRef CAS.
- Y. Wu, M. T. Kelly and J. V. Ortega, Review of chemical processes for the synthesis of sodium borohydride, Millennium Cell Inc, 2004 Search PubMed.
- H. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1940, 62, 3429–3435 CrossRef CAS.
- B. Richter, J. B. Grinderslev, K. T. Møller, M. Paskevicius and T. R. Jensen, Inorg. Chem., 2018, 57, 10768–10780 CrossRef CAS.
- J. Huot, F. Cuevas, S. Deledda, K. Edalati, Y. Filinchuk, T. Grosdidier, B. C. Hauback, M. Heere, T. R. Jensen, M. Latroche and S. Sartori, Materials, 2019, 12, 2778–2821 CrossRef CAS PubMed.
- J. Huot, D. B. Ravnsbæk, J. Zhang, F. Cuevas, M. Latroche and T. R. Jensen, Prog. Mater. Sci., 2013, 58, 30–75 CrossRef CAS.
- C. Frommen, M. Sørby, M. Heere, T. Humphries, J. Olsen and B. Hauback, Energies, 2017, 10, 2115–2138 CrossRef.
- O. Friedrichs, A. Remhof, A. Borgschulte, F. Buchter, S. I. Orimo and A. Züttel, Phys. Chem. Chem. Phys., 2010, 12, 10919–10922 RSC.
- H. Hagemann and R. Černy, Dalton Trans., 2010, 39, 6006–6012 RSC.
- Y. Jing, X. Wang, H. Han, X.-R. Liu, X.-C. Yu, X.-M. Chen, D. Wei, L.-S. Wang and X. Chen, Sci. China: Chem., 2024, 67, 876–881 CrossRef CAS.
- S. Körbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef PubMed.
- X. Zhao, Z. Yang, H. Chen, Z. Wang, X. Zhou and H. Zhang, Coord. Chem. Rev., 2021, 444, 214042–214968 CrossRef CAS.
- V. V. Avdeeva, S. E. Korolenko, E. A. Malinina and N. T. Kuznetsov, Russ. J. Gen. Chem., 2022, 92, 393–417 CrossRef CAS.
- V. V. Avdeeva, E. A. Malinina and N. T. Kuznetsov, Coord. Chem. Rev., 2022, 469, 214636–214734 CrossRef CAS.
- P. Kaszynski, Collect. Czech. Chem. Commun., 1999, 64, 895–926 CrossRef CAS.
- V. I. Stanko, A. C. Yu, V. A. Brattsev and I. Z. Leonid, Russ. Chem. Rev., 1965, 34, 424 CrossRef.
- U. B. Demirci, O. Akdim, J. Andrieux, J. Hannauer, R. Chamoun and P. Miele, Fuel Cells, 2010, 10, 335–350 CrossRef CAS.
- T. D. Humphries, M. B. Ley, C. Frommen, K. T. Munroe, T. R. Jensen and B. C. Hauback, J. Mater. Chem. A, 2015, 3, 691–698 RSC.
- B. Richter, D. B. Ravnsbæk, N. Tumanov, Y. Filinchuk and T. R. Jensen, Dalton Trans., 2015, 44, 3988–3996 RSC.
- G. L. Soloveichik, M. Andrus, Y. Gao, J. C. Zhao and S. Kniajanski, Int. J. Hydrogen Energy, 2009, 34, 2144–2152 CrossRef CAS.
- K. Chłopek, C. Frommen, A. Léon, O. Zabara and M. Fichtner, J. Mater. Chem., 2007, 17, 3496–3503 RSC.
- M. B. Ley, S. Boulineau, R. Janot, Y. Filinchuk and T. R. Jensen, J. Phys. Chem. C, 2012, 116, 21267–21276 CrossRef CAS.
- R. Černý, N. Penin, V. D'Anna, H. Hagemann, E. Durand and J. Růžička, Acta Mater., 2011, 59, 5171–5180 CrossRef.
- C. Frommen, M. H. Sørby, P. Ravindran, P. Vajeeston, H. Fjellvåg and B. C. Hauback, J. Phys. Chem. C, 2011, 115, 23591–23602 CrossRef CAS.
- L. H. Jepsen, M. B. Ley, Y. Filinchuk, F. Besenbacher and T. R. Jensen, ChemSusChem, 2015, 8, 1452–1463 CrossRef CAS.
- N. A. Tumanov, D. A. Safin, B. Richter, Z. Łodziana, T. R. Jensen, Y. Garcia and Y. Filinchuk, Dalton Trans., 2015, 44, 6571–6580 RSC.
- V. Makhaev, A. Borisov, T. Gnilomedova, É. Lobkovskii and A. Chekhlov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 1582–1586 CrossRef.
- M. Bremer, H. Nöth and M. Warchhold, Eur. J. Inorg. Chem., 2003, 2003, 111–119 CrossRef.
- P. Zanella, L. Crociani, N. Masciocchi and G. Giunchi, Inorg. Chem., 2007, 46, 9039–9041 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, 88th edn, 2007 Search PubMed.
- C. R. Cloutier, Electrochemical recycling of sodium borohydride for hydrogen storage : physicochemical properties of sodium metaborate solutions, University of British Columbia, 2006 Search PubMed.
- H. Nöth and B. Wrackmeyer, Nuclear magnetic resonance spectroscopy of boron compounds, Springer Science & Business Media, 1978 Search PubMed.
- R. M. Adams, Boron, metallo-boron compounds and boranes, Interscience Publishers, New York, 1964 Search PubMed.
- Rohm and Haas, Sodium borohydride digest, Rohm and Haas Company, 2003 Search PubMed.
- M. D. Banus, R. W. Bragdon and T. R. Gibb Jr, J. Am. Chem. Soc., 1952, 74, 2346–2348 CrossRef CAS.
- D. J. Raber, W. C. Guida and D. C. Shoenberger, Tetrahedron Lett., 1981, 22, 5107–5110 CrossRef CAS.
- A. Gigante, L. Duchêne, R. Moury, M. Pupier, A. Remhof and H. Hagemann, ChemSusChem, 2019, 12, 4832–4837 CrossRef CAS PubMed.
- H. C. Brown, Y. M. Choi and S. Narasimhan, Inorg. Chem., 1982, 21, 3657–3661 CrossRef CAS.
- J. A. Heppert and D. F. Gaines, Inorg. Chem., 1983, 22, 3155–3161 CrossRef CAS.
- R. Černý, F. Murgia and M. Brighi, J. Alloys Compd., 2022, 895, 162659–162667 CrossRef.
- L. Titov and L. Gavrilova, Russ. J. Inorg. Chem., 1970, 15, 1509 Search PubMed.
- R. R. Rietz, A. Zalkin, D. H. Templeton, N. M. Edelstein and L. K. Templeton, Inorg. Chem., 1978, 17, 653–658 CrossRef CAS.
- E. Petit, F. Salles, D. Alligier and U. B. Demirci, Molecules, 2022, 27, 1975 CrossRef CAS.
- Z.-W. Pei, C. Wu, Y. Bai, X. Liu and F. Wu, Int. J. Hydrogen Energy, 2017, 42, 14725–14733 CrossRef CAS.
- E. Roedern and T. R. Jensen, Inorg. Chem., 2015, 54, 10477–10482 CrossRef CAS.
- L. H. Rude, T. K. Nielsen, D. B. Ravnsbæk, U. Bösenberg, M. B. Ley, B. Richter, L. M. Arnbjerg, M. Dornheim, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Phys. Status Solidi A, 2011, 208, 1754–1773 CrossRef CAS.
- D. H. Souza, K. T. Møller, S. A. Moggach, T. D. Humphries, A. M. D'Angelo, C. E. Buckley and M. Paskevicius, J. Mater. Chem. A, 2021, 9, 15027–15037 RSC.
- H. Miller, N. Miller and E. Muetterties, J. Am. Chem. Soc., 1963, 85, 3885–3886 CrossRef CAS.
- R. Moury, A. Gigante and H. Hagemann, Int. J. Hydrogen Energy, 2017, 42, 22417–22421 CrossRef CAS.
- J. M. H. Makhlouf, W. V. Hough and G. T. Hefferan, Inorg. Chem., 1967, 6, 1196–1198 CrossRef CAS.
- V. V. Volkov and I. S. Posnaya, Zh. Neorg. Khim., 1979, 24, 2824–2826 CAS.
- V. V. Volkov and I. S. Posnaya, Izv. Sib. Otd. Akad. Nauk SSSR, Ser. Khim. Nauk, 1979, 4, 88–92 Search PubMed.
- J. Wang, T. Steenhaut, H.-W. Li and Y. Filinchuk, Inorg. Chem., 2023, 62, 2153–2160 CrossRef CAS.
- X. Chen, X.-R. Liu, X. Wang, X.-M. Chen, Y. Jing and D. Wei, Dalton Trans., 2021, 50, 13676–13679 RSC.
- X.-M. Chen, N. Ma, Q.-F. Zhang, J. Wang, X. Feng, C. Wei, L.-S. Wang, J. Zhang and X. Chen, J. Am. Chem. Soc., 2018, 140, 6718–6726 CrossRef CAS.
- L. Wang, Y. Jiang, S. Duttwyler, F. Lin and Y. Zhang, Coord. Chem. Rev., 2024, 516, 215974–216007 CrossRef CAS.
- A. Grahame and K.-F. Aguey-Zinsou, Inorganics, 2018, 6, 106–143 CrossRef CAS.
- W. H. Knoth, H. C. Miller, D. C. England, G. W. Parshall and E. L. Muetterties, J. Am. Chem. Soc., 1962, 84, 1056–1057 CrossRef CAS.
- W. H. Knoth, J. C. Sauer, D. C. England, W. R. Hertler and E. L. Muetterties, J. Am. Chem. Soc., 1964, 86, 3973–3983 CrossRef CAS.
- A. I. Wixtrom, Y. Shao, D. Jung, C. W. Machan, S. N. Kevork, E. A. Qian, J. C. Axtell, S. I. Khan, C. P. Kubiak and A. M. Spokoyny, Inorg. Chem. Front., 2016, 3, 711–717 RSC.
- R. N. Grimes, Carboranes, Academic Press, 2016 Search PubMed.
- A. Berger, C. E. Buckley and M. Paskevicius, Inorg. Chem., 2021, 60, 14744–14751 CrossRef CAS.
- M. Brighi, F. Murgia and R. Černý, Cell Rep. Phys. Sci., 2020, 1, 100217–100233 CrossRef CAS.
- M. Brighi, F. Murgia and R. Černý, Adv. Mater. Interfaces, 2022, 9, 2101254–2101260 CrossRef CAS.
- F. Murgia, M. Brighi and R. Černý, Electrochem. Commun., 2019, 106, 106534–106539 CrossRef CAS.
- M. Brighi, F. Murgia, Z. Łodziana, P. Schouwink, A. Wołczyk and R. Cerny, J. Power Sources, 2018, 404, 7–12 CrossRef CAS.
- R. Asakura, D. Reber, L. Duchêne, S. Payandeh, A. Remhof, H. Hagemann and C. Battaglia, Energy Environ. Sci., 2020, 13, 5048–5058 RSC.
- B. Brellochs, J. Bačkovský, B. Štíbr, T. Jelínek, J. Holub, M. Bakardjiev, D. Hnyk, M. Hofmann, I. Císarová and B. Wrackmeyer, Eur. J. Inorg. Chem., 2004, 2004, 3605–3611 CrossRef.
- S. Li, Y. Zhang, Y. Ma, P. Qiu and X. Chen, Organometallics, 2021, 40, 3480–3485 CrossRef CAS.
- J. B. Grinderslev, L. N. Skov, D. R. Sørensen, I. Kantor, M. R. V. Jørgensen and T. R. Jensen, Dalton Trans., 2022, 51, 15806–15815 RSC.
- A. Blake, MS, Pittsburg State University, 2015.
- H. Miller, N. Miller and E. Muetterties, Inorg. Chem., 1964, 3, 1456–1463 CrossRef CAS.
- M. M. Green, MS, California State University, Northridge, 2023.
- D. V. Peryshkov, A. A. Popov and S. H. Strauss, J. Am. Chem. Soc., 2009, 131, 18393–18403 CrossRef CAS PubMed.
- W. H. Knoth, H. Miller, J. C. Sauer, J. Balthis, Y. Chia and E. Muetterties, Inorg. Chem., 1964, 3, 159–167 CrossRef CAS.
- E. Muetterties, J. Balthis, Y. Chia, W. Knoth and H. Miller, Inorg. Chem., 1964, 3, 444–451 CrossRef CAS.
- T. Peymann, C. B. Knobler, S. I. Khan and M. F. Hawthorne, J. Am. Chem. Soc., 2001, 123, 2182–2185 CrossRef CAS.
- A. Garcia, G. Müller, R. Černý, D. Rentsch, R. Asakura, C. Battaglia and A. Remhof, J. Mater. Chem. A, 2023, 11, 18996–19003 RSC.
- V. V. Avdeeva, E. A. Malinina, A. V. Churakov, I. N. Polyakova and N. T. Kuznetsov, Polyhedron, 2019, 169, 144–150 CrossRef CAS.
- V. Avdeeva, I. Polyakova, L. Goeva, G. Buzanov, E. Malinina and N. Kuznetsov, Inorg. Chim. Acta, 2016, 451, 129–134 CrossRef CAS.
- L. Zakharkin, V. Guseva and P. Petrovskii, Russ. J. Gen. Chem., 2001, 71, 1017–1018 CrossRef CAS.
- M.-C. Bay, R. Grissa, K. V. Egorov, R. Asakura and C. Battaglia, Mater. Futures, 2022, 1, 031001–031008 CrossRef CAS.
- O. Volkov, W. Dirk, U. Englert and P. Paetzold, Z. Anorg. Allg. Chem., 1999, 625, 1193–1200 CrossRef CAS.
- A. N. Bridges, J. Liu, R. G. Kultyshev, D. F. Gaines and S. G. Shore, Inorg. Chem., 1998, 37, 3276–3283 CrossRef CAS.
- J. W. Johnson and J. F. Brody, J. Electrochem. Soc., 1982, 129, 2213–2219 CrossRef CAS.
- X. Zheng, Y. Yang, F. Zhao, F. Fang and Y. Guo, Chem. Commun., 2017, 53, 11083–11086 RSC.
- A. Y. Bykov, A. Zhdanov, K. Y. Zhizhin and N. Kuznetsov, Russ. J. Inorg. Chem., 2016, 61, 1629–1648 CrossRef CAS.
- J. Forstner, T. Haas and E. Muetterties, Inorg. Chem., 1964, 3, 155–159 CrossRef CAS.
- N. Kuznetsov, L. Kulikova and S. Zhukov, Zh. Neorg. Khim. (USSR), 1976, 21, 96–99 CAS.
- W. Preetz and G. Peters, Eur. J. Inorg. Chem., 1999, 1999, 1831–1846 CrossRef.
- F. Klanberg, D. R. Eaton, L. J. Guggenberger and E. L. Muetterties, Inorg. Chem., 1967, 6, 1271–1281 CrossRef CAS.
- E. Muetterties, R. Wiersema and M. Hawthorne, J. Am. Chem. Soc., 1973, 95, 7520–7522 CrossRef CAS.
- F. Klanberg and E. Muetterties, Inorg. Chem., 1966, 5, 1955–1960 CrossRef CAS.
- M. J. Bayer and M. F. Hawthorne, Inorg. Chem., 2004, 43, 2018–2020 CrossRef CAS PubMed.
- W. H. Knoth, J. C. Sauer, J. H. Balthis, H. C. Miller and E. L. Muetterties, J. Am. Chem. Soc., 1967, 89, 4842–4850 CrossRef CAS.
- X. Mu, J. C. Axtell, N. A. Bernier, K. O. Kirlikovali, D. Jung, A. Umanzor, K. Qian, X. Chen, K. L. Bay and M. Kirollos, Chem, 2019, 5, 2461–2469 CAS.
- M. A. Nelson and G. Kodama, Inorg. Chem., 1981, 20, 3579–3580 CrossRef CAS.
- V. Mustyatsa, N. Votinova, I. Dudenkov, K. Solntsev and N. Kuznetsov, Russ. J. Coord. Chem., 1998, 24, 847–850 CAS.
- T. A. Hales, K. T. Møller, T. D. Humphries, A. M. D'Angelo, C. E. Buckley and M. Paskevicius, J. Phys. Chem. C, 2023, 127, 949–957 CrossRef CAS.
- T. A. Hales, K. T. Møller, T. D. Humphries, A. M. D'Angelo, C. E. Buckley and M. Paskevicius, Phys. Chem. Chem. Phys., 2023, 25, 31249–31256 RSC.
- Y. Li, U. Kusari, P. J. Carroll, M. G. Bradley and L. G. Sneddon, Pure Appl. Chem., 2006, 78, 1349–1355 CrossRef CAS.
- S. Li, H. Gao and J. N. M. Shreeve, Angew. Chem., 2014, 126, 3013–3016 CrossRef.
- M. Bürchner, A. M. T. Erle, H. Scherer and I. Krossing, Chem. – Eur. J., 2012, 18, 2254–2262 CrossRef.
- N. Zhou, G. Zhao, K. Dong, J. Sun and H. Shao, RSC Adv., 2012, 2, 9830–9838 RSC.
- R. T. Boeré, J. Derendorf, C. Jenne, S. Kacprzak, M. Keßler, R. Riebau, S. Riedel, T. L. Roemmele, M. Rühle, H. Scherer, T. Vent-Schmidt, J. Warneke and S. Weber, Chem. – Eur. J., 2014, 20, 4447–4459 CrossRef.
- R. T. Boeré, S. Kacprzak, M. Keßler, C. Knapp, R. Riebau, S. Riedel, T. L. Roemmele, M. Rühle, H. Scherer and S. Weber, Angew. Chem., Int. Ed., 2011, 50, 549–552 CrossRef.
- S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, J. Am. Chem. Soc., 2003, 125, 4694–4695 CrossRef CAS PubMed.
- B. R. S. Hansen, M. Paskevicius, M. Jørgensen and T. R. Jensen, Chem. Mater., 2017, 29, 3423–3430 CrossRef CAS.
- E. V. Bukovsky, D. V. Peryshkov, H. Wu, W. Zhou, W. S. Tang, W. M. Jones, V. Stavila, T. J. Udovic and S. H. Strauss, Inorg. Chem., 2017, 56, 4369–4379 CrossRef CAS PubMed.
- J. H. Morris, H. J. Gysling and D. Reed, Chem. Rev., 1985, 85, 51–76 CrossRef CAS.
- A. Covington, Physical chemistry of organic solvent systems, Springer Science & Business Media, 2012 Search PubMed.
- B. Chamberland and E. Muetterties, Inorg. Chem., 1964, 3, 1450–1456 CrossRef CAS.
- V. Aftandilian, H. Miller, G. Parshall and E. Muetterties, Inorg. Chem., 1962, 1, 734–737 CrossRef CAS.
- L. Titov and P. Petrovskii, Russ. J. Inorg. Chem., 2011, 56, 1032–1033 CrossRef CAS.
- S. G. Shore and O. S. U. R. F. COLUMBUS, Studies on Syntheses and Derivatives of Boron Hydride Systems, 1988 Search PubMed.
- L. Titov, L. Gavrilova and P. Petrovskii, Russ. J. Inorg. Chem., 2008, 53, 565–567 CrossRef.
- S. Pylypko, S. Ould-Amara, A. Zadick, E. Petit, M. Chatenet, M. Cretin and U. B. Demirci, Appl. Catal., B, 2018, 222, 1–8 CrossRef CAS.
- G. B. Dunks, K. Barker, E. Hedaya, C. Hefner and P. Remec, Inorg. Chem., 1981, 20, 1692–1697 CrossRef CAS.
- A. C. Dunbar, J. A. Macor and G. S. Girolami, Inorg. Chem., 2014, 53, 822–826 CrossRef CAS PubMed.
- H. Beall and D. F. Gaines, Inorg. Chim. Acta, 1999, 289, 1–10 CrossRef CAS.
- W. R. Hertler, F. Klanberg and E. L. Muetterties, Inorg. Chem., 1967, 6, 1696–1706 CrossRef CAS.
- W. Knoth and E. Muetterties, J. Inorg. Nucl. Chem., 1961, 20, 66–72 CrossRef CAS.
- D. F. Gaines and T. V. Iorns, J. Am. Chem. Soc., 1967, 89, 3375–3376 CrossRef CAS.
- S. Payandeh, D. Rentsch, Z. Łodziana, R. Asakura, L. Bigler, R. Černý, C. Battaglia and A. Remhof, Adv. Funct. Mater., 2021, 31, 2010046 CrossRef CAS.
- D. H. Souza, A. M. D'Angelo, T. D. Humphries, C. E. Buckley and M. Paskevicius, Dalton Trans., 2022, 51, 13848–13857 RSC.
- V. Brice, H. Johnson, D. Denton and S. G. Shore, Inorg. Chem., 1972, 11, 1135–1137 CrossRef CAS.
- M. R. Palacin, P. Johansson, R. Dominko, B. Dlugatch, D. Aurbach, Z. Li, M. Fichtner, O. Lužanin, J. Bitenc and Z. Wei, J. Phys.: Energy, 2024, 6, 031501 CAS.
- M. B. Amdisen, J. B. Grinderslev, L. N. Skov and T. R. Jensen, Chem. Mater., 2023, 35, 1440–1448 CrossRef CAS.
- Y. Yan, J. B. Grinderslev, M. Jørgensen, L. N. Skov, J. R. Skibsted and T. R. Jensen, ACS Appl. Energy Mater., 2020, 3, 9264–9270 CrossRef CAS.
- Y. Yan, W. Dononelli, M. Jørgensen, J. B. Grinderslev, Y.-S. Lee, Y. W. Cho, R. Černý, B. Hammer and T. R. Jensen, Phys. Chem. Chem. Phys., 2020, 22, 9204–9209 RSC.
- L. N. Skov, J. B. Grinderslev, A. Rosenkranz, Y.-S. Lee and T. R. Jensen, Batteries Supercaps, 2022, 5, e202200163 CrossRef CAS.
- W. Hertler and M. Raasch, J. Am. Chem. Soc., 1964, 86, 3661–3668 CrossRef CAS.
- E. V. Bukovsky, PhD, Colorado State University, 2015.
- N. Miller, J. Forstner and E. Muetterties, Inorg. Chem., 1964, 3, 1690–1694 CrossRef CAS.
- H. Miller, W. Hertler, E. Muetterties, W. Knoth and N. Miller, Inorg. Chem., 1965, 4, 1216–1221 CrossRef CAS.
- W. Knoth, J. Am. Chem. Soc., 1966, 88, 935–939 CrossRef CAS.
- S. H. Lawrence, J. R. Wermer, S. K. Boocock, M. A. Banks, P. C. Keller and S. G. Shore, Inorg. Chem., 1986, 25, 367–372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: List of borohydrides and their solubility. See DOI: https://doi.org/10.1039/d4dt02256d |
| This journal is © The Royal Society of Chemistry 2025 |