
Catalytic evaluation of microwave-assisted copper cobaltite oxide (CuCo2O4) for propane oxidation†
Nidia Guadalupe
García-Peña
 ab,
Rocío
Redón
c,
Juan Ivan
Gomez-Peralta
d,
David
Díaz
*a,
Xim
Bohkimi
e and
Lucy-Caterine
Daza-Gómez
*fg
ab,
Rocío
Redón
c,
Juan Ivan
Gomez-Peralta
d,
David
Díaz
*a,
Xim
Bohkimi
e and
Lucy-Caterine
Daza-Gómez
*fg
aFacultad de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Coyoacán, Ciudad de México 04510, Mexico
bDepartmento de Física Aplicada, CINVESTAV-IPN, Antigua Carretera a Progreso km 6, A.P. 37, Mérida, Yucatán 97310, Mexico
cInstituto de Ciencias Aplicadas y Tecnología, Universidad Nacional Autónoma de México, Circuito Exterior S/N, Ciudad Universitaria, Coyoacán, 04510, Ciudad de México, Mexico
dLaboratorio Nacional de Nano y Biomateriales (LANNBIO), CINVESTAV-IPN, Antigua Carretera a Progreso km 6, A.P. 37, Mérida, Yucatán 97310, Mexico
eInstituto de Física, Universidad Nacional Autónoma de México, A.P. 20-364, Circuito de la Investigación Científica, Coyoacán, Ciudad de México 04510, Mexico
fDivisión de Ingeniería Mecánica e Industrial, Facultad de Ingeniería, Universidad Nacional Autónoma de México, Ciudad Universitaria, Coyoacán, Ciudad de México 04510, Mexico. E-mail: cgomez.nanoscience@gmail.com
gTecnologico de Monterrey, School of Engineering and Science, Ciudad de Mexico, Mexico
First published on 11th December 2024
Abstract
Attainment of a pristine copper cobaltite (CuCo2O4) phase by a fast and easy microwave (MW) assisted method is presented in this paper. The successful synthesis was supported by a series of characterization techniques, which confirmed the presence of a single phase and a possible inverted spinel crystal structure. Furthermore, catalytic performance evaluation of CuCo2O4 in propane oxidation demonstrated its selectivity towards carbon dioxide (CO2) formation, with negligible propanol production. Heating rate modification has a minimal impact on propane conversion, while catalyst stability tests indicate acceptable performance over multiple reaction cycles. X-ray diffraction analysis of the recycled catalyst suggests the formation of CuO, which seems to affect catalytic activity. Although the catalytic trials at lower temperatures resulted in lower efficiency, they effectively suppressed catalyst decomposition and allowed multiple recycling of the catalyst without any loss of catalytic activity.
1. Introduction
Emissions of volatile organic compounds (VOCs) prompt a series of environmental and human health impacts. Most of them contribute to the formation of atmospheric pollutants such as photochemical smog, tropospheric ozone and secondary aerosols.1 In the former case, photochemical smog forms when VOCs, combined with nitrogen oxides and sunlight, generate a harmful mix of gases and particles in the atmosphere, which can cause respiratory problems, eye irritation, and exacerbate pre-existing medical conditions in humans.2Propane is a compound included among the VOCs, that although it is a flammable gas that is not considered a greenhouse gas, its combustion generates negative impacts on the environment. Furthermore, propane is heavier than air and tends to accumulate in low areas,3 which may increase the risk of explosions or fires in confined spaces or poorly ventilated areas. Therefore, it may represent a hazard in some closed industrial zones.
It is in this scenario that high-efficiency thermal catalytic oxidation has emerged as a highly effective technique to remove a wide range of combustion-generated contaminants, including volatile organic compounds (VOCs), among them propane.4,5 This technique involves the use of catalysts that accelerate the oxidation reaction of contaminants at relatively low temperatures, compared to the reaction without a catalyst, which allows a complete and rapid removal of contaminants.6 This disposal method is considered relatively clean and energy efficient since it can be carried out at moderate temperatures, without the occurrence of a violent chemical reaction, and generally does not require the employment of additional chemicals. Therefore, this method results in an attractive option to mitigate contaminants such as propane.7
The most commonly used catalysts for high-efficiency thermal catalytic oxidation are those based on noble metals.8–11 Materials like Pt, Pd, and Rh have been notable for their exceptional catalytic activity, not only in the conversion of propane but also in a range of industrial applications.12–16 However, they present important drawbacks and restrictions in their application due to their high cost, shortage and limited stability.17 Therefore, exploration of more robust and cost-competitive non-noble metal catalysts is necessary. Among them, Co3O4 has attracted great interest as an alternative catalyst for non-precious metals due to its special characteristics such as its variability in valence states, rich in reactive oxygen species, different redox nature, abundant reserves and low cost in comparison with noble metals.18–21
Cobalt(II,III) oxide (Co3O4) is a material that belongs to a family of oxides with the AM2O4 spinel type structure, where A is a divalent cation (A2+) and M has a +3 oxidation state (M3+). Due to the presence of Co in two oxidation states, Co2+ and Co3+, this spinel material has gained a lot of interest in catalysis due to their redox properties.22–26 However, it remains of great interest to replace both cobalt ions with other transition cations to modulate its properties and, eventually, achieve a superior catalytic performance. In the present article, we focused on the substitution of Co2+ with other divalent transition metal cations (A2+). The choice to replace Co2+ rather than Co3+ is since Co3+ ions are critical for maintaining the redox activity and catalytic structure of the material, while Co2+ can be more easily replaced without compromising the structural integrity of the spinel-type oxide. By modifying the position of Co2+ with other divalent cations such as copper, the critical catalytic function of Co3+ is preserved. Among other oxides that may be used we found CuCo2O4, which has shown high activity in the oxidation of organic compounds, due to the abundant presence of Co3+ and more surface oxygen species.27 This spinel-type double oxide has not been widely reported in the oxidation of propane, so this work may serve to demonstrate its activity, and the degradation routes that this catalyst can follow. Moreover, copper cobaltite has shown excellent properties in different fields,28,29 consequently novel synthetic methods, where pure CuCo2O4 is obtained are highly desirable. However, in contrast to other MCo2O4 spinel-type cobaltites,18,30–32 this material has proven difficult to achieve in a pure phase. By-products such as copper oxides can be obtained if the preparation method is not accurately selected.33–37 Therefore, in the present article, a series of synthesis procedures were explored to obtain pristine oxide. Consequently, we developed a fast and simple microwave-assisted method, which has been scarcely used in the literature for CuCo2O4. Once obtained, this material was further analyzed to assure that we have obtained the desired single phase by techniques such as powder X-ray diffraction (XRD) Rietveld refinement, X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), electron dispersive spectroscopy (EDS), electron adsorption spectroscopy in the UV-vis region, and energy dispersive Raman spectroscopy. Finally, we examined the catalytic activity of copper cobaltite in the selective oxidation of propane to CO2. The catalytic performance evaluation of CuCo2O4 in propane oxidation demonstrated its selectivity towards carbon dioxide (CO2) formation, with negligible propanol production. Modification of the heating rate had minimal impact on propane conversion, while catalyst stability tests indicated acceptable performance over multiple reaction cycles.
2. Materials and methods
Co(NO3)2·6H2O (reagent grade, 98%), Cu(NO3)2·3H2O (purum p.a., 98–103%), urea (BioReagent, ≥98%), NaOH (ACS reagent, ≥97.0%, pellets), and H2O2 (30 wt%) were acquired from Sigma-Adrich; whereas ethanol (RA) and acetone (RA) were obtained from CTR scientific. Finally, propane (minimum purity 80%–99.0%) was purchased from Linde Brand. Deionized water was purified from a PURELAB™ Chorus 1 purification system from ELGA-VEOLIA. All reagents were used as received without further purification.2.1. Synthesis of CuCo2O4 (labeled as CoP)
Pure CuCo2O4 oxide was prepared by a dual-step synthesis method. Briefly, 17.46 g (60 mmol) of Co(NO3)2·6H2O and 7.25 g (30 mmol) of Cu(NO3)2·6H2O were dissolved in 100 mL of deionized (DI) water. Under agitation, 18.02 g (300 mmol) of urea, previously dissolved in 50 mL of DI water. The resulting mixture was transferred into two Teflon-lined microwave vessels, assembled, and introduced into a microwave digestion system with the following program: the sample was heated to 180 °C in 15 minutes, then it was maintained at that temperature during one hour with a power output of 600 W; finally, the mixture was allowed to cool down for 45 minutes. Once at room temperature, the resulting product was centrifuged for 10 minutes at 4500 rpm. The dark gray precipitate was washed with 3 × 50 mL of DI water, 3 × 50 mL of ethanol and 50 mL of acetone, to dry under vacuum for two hours at 60 °C. The final black product was obtained after an annealing step for two hours at 400 °C.Another reaction, with employment of half the amount of urea (9.01 g, 150 mmol), was performed to achieve the product Co5.
Synthesis techniques commonly used in literature that were employed to obtain the CuCo2O4 oxide:
Table 1 summarizes the products obtained in this work through different chemical routes. Among these chemical routes, the microwave assisted synthesis with urea excess was the unique that guaranteed the obtention of the CuCo2O4 phase as single product, which is named CoP in the current manuscript. This contrasts to other previously informed synthesis33–37 where secondary phases of copper oxides are present. Due to its single-phase composition, the product CoP was utilized for conducting the catalytic tests afterward.
| Product | Chemical route summary |
|---|---|
| CoP* | Microwave assisted reaction with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 urea content :5 urea content |
| Co1 | Coprecipitation followed by annealing |
| Co2 | Hydrothermal synthesis followed by annealing |
| Co3 | Hydrothermal synthesis with 1:2.5 urea concentration followed by annealing |
| Co4 | Similar to Co4 with 1:5 concentration of urea |
| Co5 | Microwave assisted with 1:2.5 urea content |
2.2. Catalytic activity of CuCo2O4 catalyst
The catalytic performance of propane oxidation was investigated in a fixed bed microreactor. A small amount of CuCo2O4 catalyst was introduced into a glass U-tube with an inner diameter of 10 mm. An oxidizing atmosphere, containing 0.5 vol% propane, 5 vol% O2, and 94.5% N2, was continuously supplied to the reactor at a constant rate of 100 mL min−1. The temperature was then gradually raised from 25 to 500 °C with a heating rate of 2 °C min−1 using a regulated electrical furnace. Effluent gas was analyzed for product concentrations (CO2, CO, and others) using a gas chromatograph (GC), particularly the Agilent Technologies 6890 network GC system equipped with a FID detector and a HP Plot Q column. The monoliths were reused for up to 5 consecutive reaction cycles. Thus, it was necessary to purge the reaction tubes with N2 and subject them to the same conditions as detailed above.2.3. Characterization techniques
The products were characterized with X-ray diffraction technique using a Brüker D-8 Advance diffractometer applying Cu Kα radiation (40 kV, 30 mA, l = 1.5418 Å), step size 0.02°, and time size 0.5 s. To perform the Rietveld refinement, additional diffractograms were recorded using a step-size of 0.010254° and a step-time of 134 ms. The Rietveld refinement was performed using the TOPAS software package. UV-visible electronic absorption spectroscopy studies were measured using a fiber optics arrangement with a deuterium-halogen light source (AvaLight DH-S-BAL), an integrating sphere (Labsphere USRS-99-010) interconnected with a spectrometer AvaSpec-2048 from Avantes Co.; an Ocean Optics WS-1-SL standard was used as a reference. Vibrational Raman spectroscopy was carried out using a Witec spectrometer, Alpha 300RA model using a 488 nm blue laser and a 100× objective lens; each spectrum was obtained as the average of 32 acquired spectra every 0.5 s. X-ray photoelectron spectroscopy analyses were measured in a Thermo Scientific equipment Fischer K-Alpha model equipped with a monochromatic Al Kα (1486.7 eV) X-ray source, and a spot diameter of 400 mm; to acquire high-resolution spectra, the pass energy employed was 10 eV; the base pressure of the analysis chamber was 10−9 Torr for data acquisition. The XPS core-level peaks were deconvoluted into their various components by using an interactive least-squares computer program AAanalyzer™, 2.04 version.38 Thermogravimetric analyses were carried out at a TA Instrument apparatus Discovery Series model; the TGA curves were acquired under a 25 mL min−1 air flow, from room temperature to 800 °C. Scanning electron microscopy (SEM) studies were performed at a JEOL microscope, JSM-7601F model operated at 15 kV and 10 kV.3. Results and discussion
3.1. Comparison among different synthesis methods
Achievement of a pure phase of CuCo2O4 has proven challenging due to multiple variables influencing the outcome. Issues such as the choice and the amount of the precipitant agent, as well as the heating method and reaction method, have been identified as significant factors. In literature it is possible to find examples where either cuprous oxide33 or cupric oxide34–37,39 are obtained as by-products. Their presences are typically detected in X-ray diffractograms by their distinct peaks at specific angles. Cupric oxide is easily distinguished, with its main diffraction peaks appearing between the main CuCo2O4 signal at 37.01°. However, in the former case, the presence of Cu2O is more difficult to detect, due to the proximity of the main signal of both species that overlap and hinder their identification by this characterization technique. In most products, the presence of cuprous oxide might be inferred by a slight shoulder on the left side of the main diffraction peak of the spinel, at 36.58°.To attain CuCo2O4 in a pure phase, we explored two commonly utilized methods along with a hardly explored synthesis approach. In the present section we will outline the methodologies implemented in this study to synthesize the pristine copper cobaltite and present their respective outcomes.
Conventional use of a co-precipitation technique, employing a strong base like sodium or potassium hydroxide, led to the formation of copper(II) oxide, as depicted in Fig. 1. Fig. 1 presents the theoretical patterns for the products CuCo2O4 (PDF # 073-2751, shown in black), Cu2O (PDF # 005-0667, portrayed in red), and CuO (PDF # 048-1548, depicted in green) at the bottom, in comparison it is presented the experimental pattern of product Co1, in blue. In this experimental pattern, besides the spinel product, other peaks at 32.53°, 35.56°, 38.75°, and 48.75° were visible. These signals correspond to (110), (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), (111), and (20
), (111), and (20![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) diffraction family planes, respectively, consistent with CuO in a monoclinic crystal structure, space group name C2/m, and space group number 15.
) diffraction family planes, respectively, consistent with CuO in a monoclinic crystal structure, space group name C2/m, and space group number 15.
 | ||
| Fig. 1 X-ray diffraction patterns of product Co1 (labeled in blue) synthesized by a coprecipitation technique with sodium hydroxide; Co2 (labeled in orange) solid obtained via hydrothermal synthesis in presence of sodium hydroxide; Co3 (labeled in violet) powder obtained with a hydrothermal method in presence of urea; and Co4 (labeled in cyan) product achieved via MW assisted heating in proportion 2:5 between Co(II) to urea. In the first (Co1) and second (Co2) products, the presence of CuO (PDF # 048-1548, which is depicted in the first theoretical pattern in green) is clearly visible, whereas in third (Co3) and fourth (Co4) products Cu2O (PDF # 005-0667, portrayed in red) was detected. | ||
As the screening of synthesis methods progressed, it became evident that even if the typical hydrothermal method was applied, the use of hydroxides generated CuO as by-product. This is evident in Fig. 1, where the diffractogram of Co2, is depicted, in orange. This product was obtained through a standard hydrothermal synthesis with the employment of sodium hydroxide. Once again, the presence of copper(II) oxide is discernible from observation of the aforementioned diffraction planes. Moreover, the addition of smaller amounts of NaOH had no impact on the formation of cupric oxide; it suppressed the formation of an initial blue precipitate, but the final mixture of products was the same. Similarly, variations in reaction time did not affect the presence of the products but influenced the size of the crystals obtained. Consequently, it was determined that an alternative precipitation agent must be used.
In literature, urea is frequently employed as an induced precipitation agent to facilitate the formation of various binary and ternary oxides.34,40–42 Among other benefits, urea is non-toxic, easy to manipulate, and tends to produce non-toxic by-products; hence it was decided to use it in the present research. With this reactant, a typical hydrothermal method was utilized to attain the desired oxide. The X-ray diffraction pattern of the resulting Co3 product, synthesized with urea in a 1:2.5 ratio, is displayed in Fig. 1, in purple. Upon comparing the diffraction pattern with the theoretical patterns of CuCo2O4 and Cu2O at the bottom, in black and red, respectively, it became apparent that cuprous oxide was present, as indicated by the peaks at 36.58°, 42.49°, and 61.65° 2theta degrees. These signals correspond to (1 1 1), (2 0 0), and (2 2 0) diffraction family planes from Cu2O in a cubic crystal structure (space group name Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, space group number 224, PDF # 00-005-0667). As previously discussed, the primary plane of Cu2O overlaps with the principal peak of copper cobaltite in a cubic spinel structure, which can sometimes hinder the identification of the former product in the mixture. However, in the present material, the presence of cuprous oxide is markedly discernible from the rest of the family planes. Moreover, comparison of Co3 diffractogram with theoretical CuO pattern, in red, seems to indicate the presence of a small amount of this oxide, as a small peak at 35.5° was observed.
m, space group number 224, PDF # 00-005-0667). As previously discussed, the primary plane of Cu2O overlaps with the principal peak of copper cobaltite in a cubic spinel structure, which can sometimes hinder the identification of the former product in the mixture. However, in the present material, the presence of cuprous oxide is markedly discernible from the rest of the family planes. Moreover, comparison of Co3 diffractogram with theoretical CuO pattern, in red, seems to indicate the presence of a small amount of this oxide, as a small peak at 35.5° was observed.
To assess the effect of urea addition on the hydrothermal synthesis of CuCo2O4 spinel, an additional sample (Co4) was synthesized by adding urea in a 1:5 ratio. The corresponding diffractogram, displayed in Fig. 1 in magenta, indicates that this powder exhibits better crystalline compared to the previously obtained sample, Co3. This is evident from the decreased noise and more defined crystal planes in the diffractogram, despite both products being synthesized under the same conditions. However, the presence of CuO is more pronounced, with a small shoulder indicative of Cu2O remains detectable. Therefore, we may conclude that increasing the urea addition resulted in improved crystallinity and possibly a higher proportion of copper(II) oxide in relation to copper(I) oxide. Nonetheless, this strategy did not yield pure CuCo2O4.
Thus far, conventional synthesis methods have generated CuO and Cu2O by-products, casting doubt on the successful synthesis of CuCo2O4 instead of Co3O4. Consequently, an alternative heating method was deemed necessary to ensure the successful synthesis of CuCo2O4. Among multiple choices, microwave-assisted (MW) heating emerged as a promising option. This technique has proven beneficial across various domains such as organic and inorganic chemistry, material synthesis, or catalysis.43–46 In the MW method, the heating of reaction mixtures arises from the interaction of dipole moments attempting to align with the oscillating electric field of the microwave radiation.47 As a result, increased friction and collisions occur, leading to accelerated reactions compared to conventional heating methods. Consequently, reaction times were significantly reduced from hours to minutes, enabling the attainment of our desired product in just two hours instead of twelve. Moreover, the localized heating generated by MW radiation, creating hot spots, facilitates the formation of products that are challenging to achieve using conventional methods.43
The first trial product achieved using the MW-assisted method, labeled Co5, is depicted in Fig. 1d, which was obtained with a 1:2.5 ratio of Co(II) to urea. This pattern closely resembles the theoretical pattern of CuCo2O4, with only a minor peak at 42.49° 2theta degrees being distinctly visible. However, as previously mentioned a small shoulder was detected to the left of the primary spinel diffraction peak. These two peaks led us to suspect the presence of Cu2O. The result resembles the product obtained by hydrothermal method with the same Co(II):urea ratio (1:2.5), where cuprous oxide was obtained as the main by-product. However, the amount of Cu2O seems to be present in less concentration, since the product presented a smaller intensity in its main diffraction peak. Moreover, the synthetic method positively suppressed the formation of CuO.
Nevertheless, the formation of cuprous oxide was effectively suppressed when the amount of urea was doubled to a 1:5 ratio of Co(II) to urea. The diffractogram obtained from the product under this revised condition, labeled as CoP, is illustrated in Fig. 2a. Analysis of the pattern confirms the successful attainment of the pure CuCo2O4 phase with the increased urea concentration. The pattern exhibits all the diffraction peaks corresponding to a spinel structure with a cubic crystal lattice (space group name Fdm, a = 8.0869 Å), as compared with the theoretical diffractogram (black lines). Fig. 2 depicts the typical cubic spinel crystal cell, highlighting the distribution of Co(III) ions, shown deep blue, occupying the octahedral sites (Wyckoff sites 16c). Whereas copper(II) ions, displayed in cyan, are located in the tetrahedral sites (Wyckoff sites 8b). This result contrasts with the outcomes obtained when hydrothermal method was used, where the increment in the amount of urea used rendered into an increment in the formation of CuO. This difference must arise from the synthesis method, since microwave assisted methods tend to achieve products that are not thermodynamically stable. However, to claim this effect, further analysis must be carried out that is outside the scope of the present research.
 | ||
| Fig. 2 (a) X-ray diffraction pattern of CoP product, obtained from a MW assisted heating method with a proportion 1:5 of Co(II) to urea. The achievement of a pure phase, corresponding to a typical spinel with a cubic crystal lattice (space group name Fdm, space group number 227, a = 8.0869 Å), is presented. The scheme of the crystal unit cell is depicted as an inset in the figure. (b) X-ray diffraction pattern of CuCo2O4 after Rietveld refinement. The recorded (in gray) and calculated (black line) diffraction pattern are shown in the graph at the top. The difference between the observed and calculated diffraction pattern is shown at the bottom (Rwp = 2.36%). | ||
3.2. Characterization of CuCo2O4 spinel unique-phase sample (CoP)
So far, we have been able to obtain a pure phase that may allow us to study its behavior in propane oxidation. However, this product was further analyzed before being used in catalysis.X-ray photoelectron spectroscopy (XPS) analyses on the CoP product were carried out. The results obtained are presented in Fig. 3. From the initial analysis of the survey spectrum in the whole spectral window (Fig. 3a), it can be concluded beyond any reasonable doubt that copper was present in the product, since all the spectral lines 3s, 3p, and 2p, along some Auger lines, informed for copper element could be detected.48 Beside these copper signals, peaks from Co, C and O were present. Moreover, the determination of elemental concentrations revealed values of 21.88, 39.03, and 26.06 % for Cu, Co, and O, respectively; with carbon (C) accounting for the remaining 13.02%. These Cu and Co percentages correspond to a Cu:Co ratio of approximately 1:2, consistent with the desired CuCo2O4 spinel composition, thereby confirming the purity of our ternary oxide.
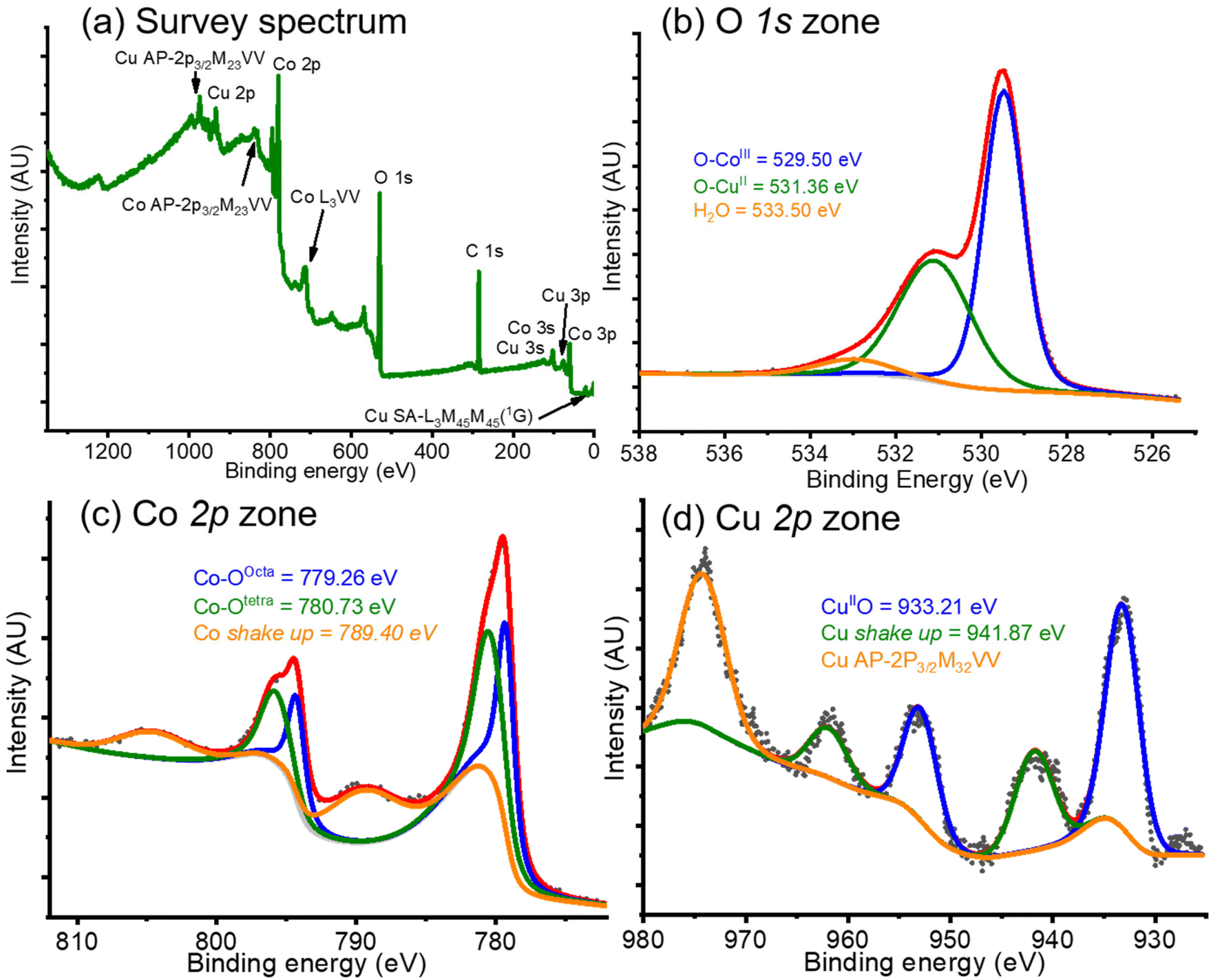 | ||
| Fig. 3 X-ray photoelectron spectroscopy studies on CuCo2O4 (CoP). Survey spectrum (a) shows the informed spectral lines for Cu, Co, C and O elements, thus confirming the attainment of the desired product. High-resolution spectra on the O 1s (b), Co 1s (c), and Cu 1s (d) zones are presented, all of them supporting the achievement of CuCo2O4. Noteworthy, Co 2p spectrum (c) seems to indicate the presence of an inversion in the octahedral and tetrahedral sites. | ||
Additionally, high resolution XPS studies were conducted. In the O 1s zone, the experimental spectrum (black dotted line, in Fig. 3b), was deconvoluted into three signals at 529.49 eV, 531.36 eV, and 533.50 eV. The first signal (blue line, in Fig. 3b) was assigned to Co–O bond coming from a spinel crystal structure;49 whereas the second peak (green line, in Fig. 3b) may come from a CuII–O bond in a spinel structure;50 finally, the peak at higher binding energy (orange line, in Fig. 3b) comes from adsorbed water51 or adsorbed ethanol.52 It is known that the areas under the curve are not directly related to concentrations since factors such as density of the materials and intensity of the signals, among other factors, must be accounted for. However, as an empirical approximation, Co–O area was approximately twice the area of CuII–O. This corresponds to the Co:Cu mol ratio present in the CuCo2O4 oxide (2:1) and seems to indicate that the desired product was obtained.
In the Co 2p zone, the experimental spectrum (black dotted line, in Fig. 3c) presented two doublets coming from the p3/2 and p1/2 lines, which were deconvoluted into three double signals at 779.26 eV and 794.29 eV, 780.73 eV and 796.20 eV, and 789.40 eV and 804.28 eV. The first doublet (blue line in Fig. 3c) was assigned to Co–O in the octahedral sites in the Co3O4 spinel crystallite;49,53–55 while the second peak (green line in Fig. 3c), was attributed to Co–O in tetrahedral sites in the Co3O4 spinel architecture;49,53–55 finally, the last signal (orange line in Fig. 3c) was designated as shake-up line due to the paramagnetic nature of Co.49 The areas under the curve for the first and second deconvoluted signals were calculated and established as an approximate 1:1 ratio. Since these two doublets seem to indicate a difference in the cobalt geometry, we may use them to estimate an inversion in the spinel geometry. Hence, these values seem to indicate that just half cobalt centers are located at the octahedral sites, whereas the other half Co are placed at the tetrahedral sites. Therefore, in contrast to the crystal structure depicted in Fig. 2a, Cu(II) must be located in half octahedral sites (depicted in dark blue), whereas cobalt ions must be placed at the tetrahedral sites (labeled in light blue).
Likewise, in the Cu 2p zone (shown in Fig. 3d), a series of signals can be observed in the experimental spectrum (black dotted line), which were deconvoluted into two doublets at 933.21 and 952.98 eV, and 941.87 and 961.96 eV, and a singlet centered at 974 eV. The first doublet (blue line in Fig. 3d) comes from a CuII–O bond,56,57 perhaps in octahedral sites;58 whereas the second signal (green line in Fig. 3d) was attributed to a shake-up line coming from the paramagnetic nature of Cu(II). Finally, the singlet (orange line in Fig. 3d) should arise from an Auger line from copper.48 From these results, we may conclude that CuCo2O4 was obtained as desired with a pure phase, although inversion in the cobalt-octahedral and copper-tetrahedral sites must exist. This behavior was previously observed in literature for a copper cobaltite sample synthesized by Farag and colleagues.59 In their investigation, the authors inform that in the CuCo2O4 structure, almost all the Cu(II) is located at the octahedral sites, whereas some Co(II) is positioned at the tetrahedral sites. This result is not uncommon, since a series of inverted spinel structures were previously studied.60–62 Nonetheless, it should be interpreted with caution. Spinel compounds exhibit behavior like that of a solid solution, where A2+ and M3+ cations in the AM2O4 spinel structure may interchange between tetrahedral and octahedral sites. This interchange depends on several factors, including temperature, the size and nature of the ions, or their covalency.63 Additionally, since Cu2+ and Co3+ are similar in nature, accurately determining their specific positions is challenging.
A Rietveld refinement of the CoP sample was performed using the crystallographic information reported for CuCo2O4 (CIF number 5910155 from the Crystallography Open Database). The refined diffraction pattern exhibited good agreement with the experimental data (Rwp = 2.23%). It is important to mention that the CIF used for the refinement considers that Cu and Co atoms occupy the tetrahedral and octahedral sites, respectively, which corresponds to the Wyckoff sites 8b and 16c.
When the refinement is done considering the inversion suggested by XPS analysis, there was not a change in the residual of the refinement. This outcome aligns with expectations, as the atomic numbers of the involved elements are similar, resulting in comparable contributions to the diffraction pattern. Consequently, the degree of inversion could not be quantified through Rietveld refinement. However, the results strongly support the presence of a single spinel phase incorporating Co atoms into its structure, and which is obtained with our novel chemical route based on microwave assistance.
In order to establish the obtained morphology in the powder, scanning electron microscopy (SEM) analyses were carried out on CoP. The micrographs obtained are shown in Fig. 4. Noteworthy, the synthesized material exhibited two very different morphologies: bundles, some of them resembling truncated cuboctahedrons (as can be seen in Fig. 4b), and rectangular sheets were observed, as depicted in Fig. 4c. The first shape presented a mean size of 198.2 nm, determined on 100 particles, which were agglomerated. The size distribution histogram is presented in Fig. 4d. Whereas the sheets presented a mean diameter of 5.2 mm, calculated over 100 measured layers; some of them appear to be squared, while others had rectangular shapes. Both structures seem to be intercalated. For the layers, it was not possible to measure a mean thickness since the value was less than 20 nm, below the resolution of the available SEM equipment. The size distribution histogram of these structures is shown in Fig. 4e. The attained structures may correspond to the synthesis method employed, which allowed the formation of two very distinct morphologies with a very different anisotropic behavior, as shown previously by Yang and collaborators.64
 | ||
| Fig. 4 Scanning electron micrographs of pure copper cobaltite (CoP). The material presented two very distinct morphologies: (a) some bundles with a mean size of 198.2 nm; and (b–c) sheets with a mean size of 5.2 μm. (d) and (e) Present the size distribution histograms of sphere and sheet particles, respectively. | ||
To assure that both morphologies have the same chemical identity (although so far, all characterization techniques have supported the presence of a single product), additional electron dispersive spectroscopy (EDS) studies were carried out, and the resulting analyses are depicted in Fig. S1.† From the micrograph observed in Fig. S1a,† EDS was performed on two very distinct zones: the first one was located on agglomerated bundles, while the second was located on a group of layers. An EDS spectrum typical of both regions is depicted in Fig. S1b,† where signals corresponding to copper, cobalt and oxygen were detected. Finally, semiquantitative concentrations for both regions were calculated (c and d), the two of them showing concentrations near the theoretical concentrations for Co and Cu in CuCo2O4 compound (25.9% and 48.3%, respectively).
Additionally, electronic adsorption UV-vis spectroscopy measurements were performed on CoP. The resulting spectrum can be observed in Fig. S2.† From this spectrum, the Kubelka–Munk graphic was calculated for direct band gaps. The resulting graphic is shown in the inset in Fig. S2.† From this graph it was possible to calculate two direct band gaps at 1.90 eV and 2.59 eV. These values are in full concordance with the two band gaps informed for this material, where two similar energies are reported.59,65,66
Thermogravimetric analysis was carried out on the already formed CuCo2O4 powder (CoP). The result can be seen in Fig. S3.† From this thermogram, we can determine that the product is thermally stable up to 800 °C. The only change can be seen in the 200–334 °C range, where a slight loss of 5.03% was observed, perhaps due to some dehydration of surface hydroxyl groups formed during storage. Nonetheless, this hydrated material existed barely in a 5% composition.
The CoP product was also analyzed by Raman scattering spectroscopy and the obtained spectrum is shown in Fig. S4.† In the analysis, the material presented five signals at 197, 481, 522, 618, and 687 cm−1. According to literature, these bands may be assigned to F2g, Eg, F2g, F2g, and A1g Raman-active modes from MCo2O4 spinel structures.67,68 For a laser power below 10 mW, no other signals were detected. Otherwise, when higher power incidence was used, it revealed signals beyond 1000 cm−1, which might come from decomposition of the ternary oxide.
Finally, gas adsorption–desorption analyses were conducted to determine the specific surface area of the catalyst. To obtain accurate results, the sample was heated to 260 °C for five hours to ensure complete dehydration. Following this, measurements were taken at 77 °C. The BET (Brunauer–Emmett–Teller) model was used to calculate the specific surface area, which was determined as 44.59 m2 g−1.
3.3. Evaluation of the catalytic performance
To evaluate the catalytic activity of CuCo2O4 in propane oxidation, a dynamic study was performed over a temperature range of 25 to 500 °C, with a heating rate of 2 °C min−1, under an oxygen atmosphere (Fig. 5(a and b)). Chromatographic monitoring during this experiment indicated that carbon dioxide formation began at around 87 °C (Fig. 6). This observation aligned with the detection of propanol (0.02%), suggesting its role as an intermediate in the complete oxidation of propane. The concentration of propanol remained low, but gradually increased, peaking at 1.0 % at 255 °C, and then decreased, disappearing entirely at 399 °C. Although the propanol percentage was consistently much lower than that of CO2, the latter increased steadily from 0.13% to 94.45% over the same temperature range. These results demonstrate a strong selectivity toward CO2 formation, indicating a clear preference for the complete oxidation of propane. This trend is illustrated in Fig. 5a, where the propane composition decreases proportionally as the CO2 composition increases, maintaining a close stoichiometric relationship. Between 400 and 500 °C, only CO2 formation is detected, reaching 98.8%. Based on chromatographic monitoring, it can be assumed that CuCo2O4 could follow a Langmuir–Hinshelwood kinetic model (Fig. 5(c)). Since the formation of propanol was detected in the range between 110 and 390 °C, in a small amount, the main product was carbon dioxide. Propanol is a proposed intermediate in this model, when other spinel-type oxides are used.69 The first steps proposed in this model involve the adsorption of O2 and C3H8, and their subsequent dissociation to generate species such as C3H8*, O*, OH* and C3H7O*. This last intermediate can lead to the formation of propanol that can be further oxidized to CO2. Other possible intermediates in this model could be formaldehyde (CH2O) or acetic acid (CH3COOH); however, they were not detected in the oxidation dynamics. After 390 °C, propanol is not detected.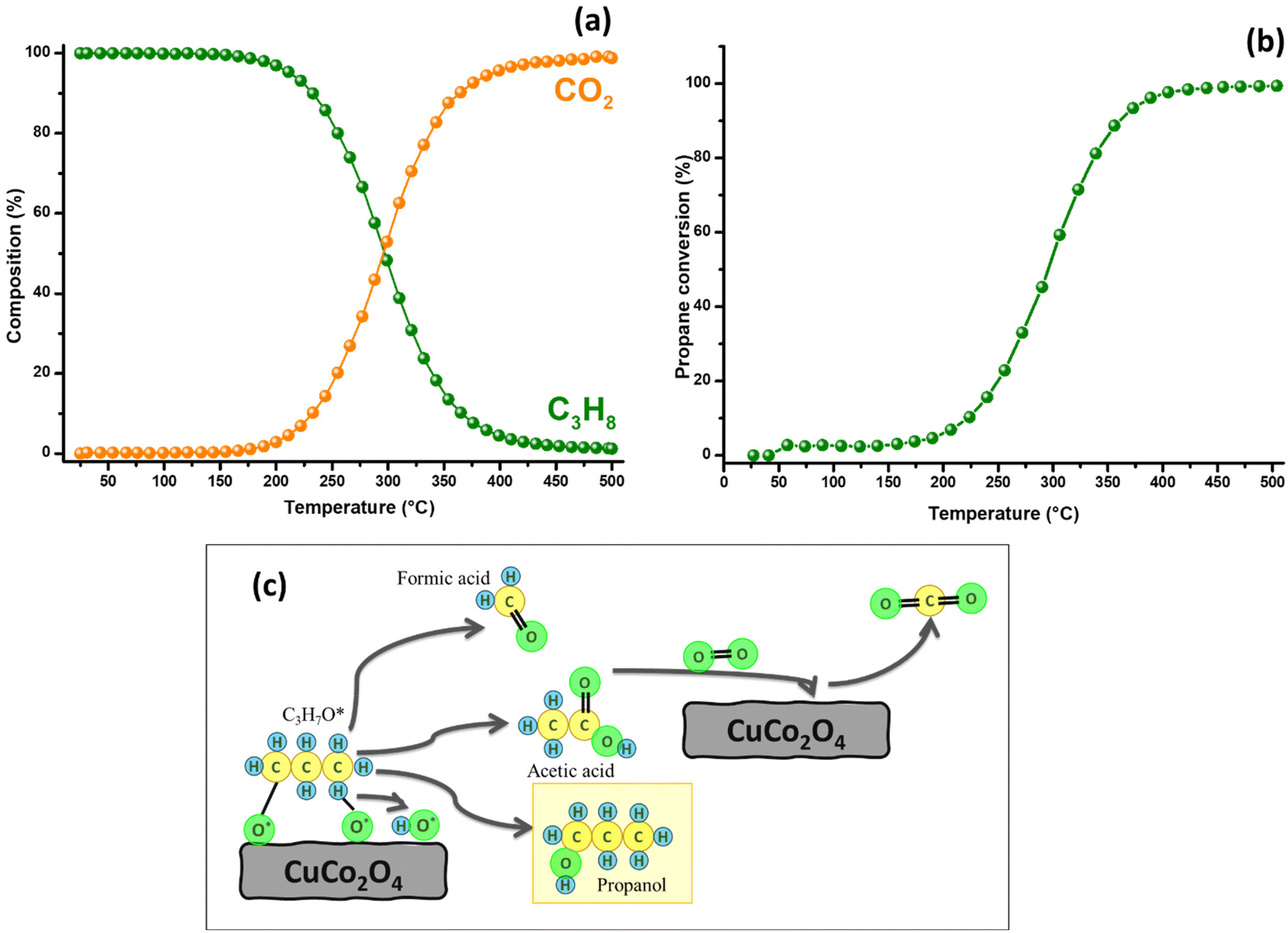 | ||
| Fig. 5 Catalytic performance in the oxidation of propane was evaluated over a temperature range between 25 and 500 °C, employing 10 mg of copper cobaltite with a heating rate of 2 °C min−1 (a and b) and proposed diagram of the reaction mechanism (c). | ||
 | ||
| Fig. 6 Catalytic performance in the oxidation of propane was evaluated over a temperature range between 25 and 180 °C, employing 10 mg of CoP sample with a heating rate of 2 °C min−1. | ||
The heating rate generally influences the conversion of reactants in catalytic reactions, leading to adjustments in the heating rate to evaluate its effect on propane conversion. The objective was to ascertain if this adjustment enhanced the catalyst's selectivity for complete oxidation, particularly the formation of CO2, while avoiding the production of propanol. As depicted in Fig. S4,† increasing the heating rate does not significantly impact propane conversion. At lower temperatures, propane conversion is slightly higher with a 3 °C min−1 ramp compared to a 2 °C min−1 ramp. However, from 250 °C onwards, both ramps show similar conversion rates, indicating that the catalyst's performance is not notably affected by the heating rate. In terms of propanol formation, the 3 °C min−1 ramp results in its presence at low concentrations, but this intermediate disappears at 400 °C. This suggests that altering the heating rate does not significantly change the catalyst's behavior in this aspect. Comparative tests were also performed with CuO and Co3O4 obtained by the microwave-assisted synthesis method (Fig. S5†). CuCo2O4 shows a higher activity than CuO and comparable to Co3O4, reaching complete propane conversion at a lower temperature than CuO. This highlights that the spinel structure of CuCo2O4, with a synergy between the redox sites of Cu and Co, provides a higher density of active sites compared to monophase catalysts such as CuO.
Furthermore, the presence of CuO in CuCo2O4, when not properly controlled during synthesis, can be detrimental to catalytic performance, as CuO exhibits lower activity and can block the active sites of the spinel phase. However, in our synthesis method, we managed to minimize this secondary phase, ensuring higher purity of CuCo2O4 and maximizing its catalytic efficiency. Therefore, the microwave-assisted method not only allows for faster and more efficient synthesis, but also superior control over catalyst composition and structural properties, resulting in improved performance in propane oxidation.
The stability of the catalytic performance of the copper cobaltite catalyst was further assessed in relation to temperature and time. Fig. 7 illustrates propane conversion across a temperature range of 25 to 500 °C, over five reaction cycles. The propane conversion exhibits abrupt fluctuations between cycles without a consistent trend during catalyst recycling. Notably, from the first to the second cycle, conversion drops sharply from a peak of 99.4% to 71.6%. Although CO2 production begins at 90 °C in both cycles, it increases much more slowly in the second cycle with rising temperature. This decrement in catalytic activity was expected, as the unsupported catalyst leads to nanoparticle agglomeration, reducing catalytic efficiency. This behavior was expected in our catalyst due to the presence of the previously shown morphologies of nanometric layers (Fig. 4) that are more prone to collapsing. Interestingly, in the third and fourth cycles, conversion recovers somewhat, though it does not reach the initial level observed in the first cycle. This recovery might be due to the second cycle acting as a pretreatment for the subsequent cycles. In the final cycle, conversion decreases again, though propane conversion remains at an acceptable level. Throughout all cycles, propanol forms consistently in small amounts, disappearing at 400 °C.
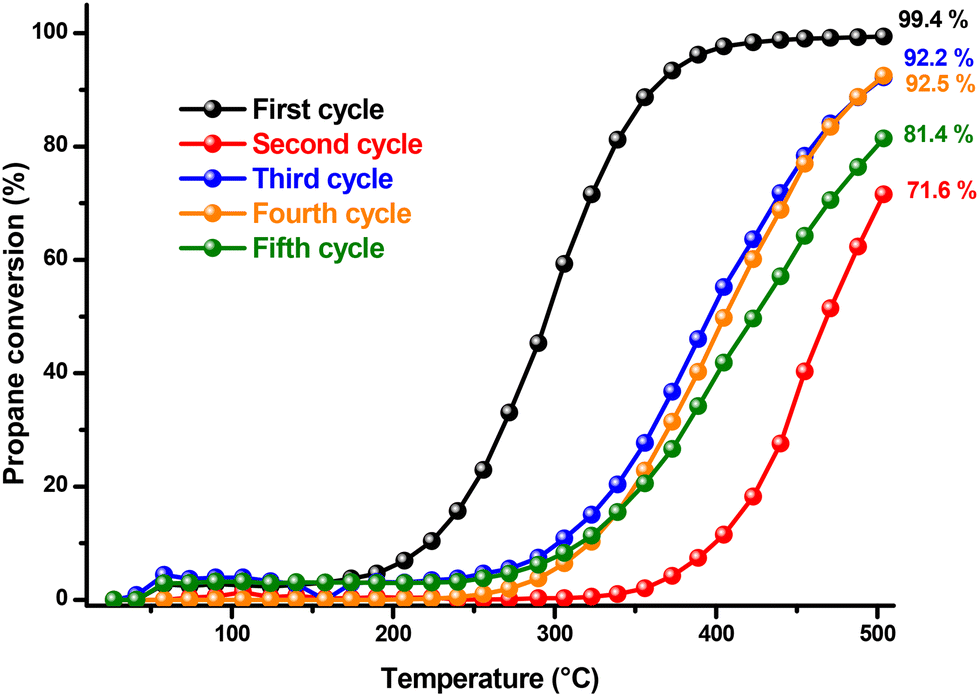 | ||
| Fig. 7 Catalytic performance in the oxidation of propane was evaluated over a temperature range between 25 and 500 °C, employing 10 mg of CuCo2O4 with heating rate of 3 °C min−1 in five reaction cycles. | ||
The stability of the catalyst overtime demonstrated improved performance, as shown in Fig. 8. Propane conversion remained consistent throughout the 180 minute reaction period, with no significant decline observed over time in any of the reaction cycles. Unlike the recycling experiments depicted in Fig. 5, the decrease in conversion from one cycle to the next was less pronounced. From the first to the fifth cycle, propane conversion decreased from 98.5 % to 87.5 %. At this temperature, the reduction in catalytic activity is likely due to a decrease in the catalyst's surface area caused by nanoparticle agglomeration or due to the catalyst irreversible conversion into other products. Intermediate products such as carbon monoxide and propanol can easily oxidize or detach from the catalyst surface.
 | ||
| Fig. 8 Recycling of the catalyst for propane oxidation at 500 °C was conducted using 10 mg of CuCo2O4. | ||
To suppress the decomposition of the catalyst, catalytic cycles were carried out at a lower temperature (Fig. 9). At 180 °C, propane conversion is low; however, this temperature decrement effectively allows determining the catalyst's capability to be reused in several reaction cycles. Over five reaction cycles, the copper cobaltite catalyst maintained its catalytic activity between 3.0 and 6.0 % propane conversion, indicating that this catalyst can be reused in multiple reaction cycles.
 | ||
| Fig. 9 Recycling of the catalyst for propane oxidation at 180 °C was conducted using 10 mg of copper cobaltite. | ||
The use of CuCo2O4 as a catalyst in propane oxidation has been scarcely explored in the literature. Some studies report the synthesis of CuCo2O4 by the combustion method, but as a supported composite material (Pd–CuCo2O4/Al2O3),70 evaluating its catalytic activity in the oxidation of propane, CH4 and NOx. In that work, the temperatures required to reach 100% conversion of CH4 and propane were 415 °C and 323 °C, respectively. However, most reports related to spinel-type oxides focus on Co3O4 and NiCo2O4.71–73 Applications of CuCo2O4 in oxidation reactions have been mostly focused on the oxidation of compounds such as toluene, methane and carbon monoxide.74 These antecedents reinforce the need to explore and understand the behavior of pure CuCo2O4 in the oxidation of propane, since it has not been directly studied so far. Furthermore, our microwave-assisted approach aims not only to address this gap in the literature, but also to propose a simple, rapid and efficient synthesis method to obtain CuCo2O4 in pure phase, which has proven to be a significant challenge compared to other MCo2O4 spinels. This approach offers a promising starting point for future comparisons with catalysts obtained by other methods.
3.4. Characterization of used CoP catalyst
Since the CoP catalyst has lost nearly 15% of its activity after five cycles, it is essential to investigate the cause of this activity loss. To this end, a previously used sample of the product was analyzed using powder X-ray diffraction. The Rietveld refinement of the obtained diffraction pattern is displayed in Fig. 10. This analysis revealed the presence of two phases: CuO and Co3O4, depicted in blue and green lines, respectively. The detection of both compounds in the diffraction pattern is significant and indicates the decomposition of the original CuCo2O4 spinel phase under the propane oxidation conditions.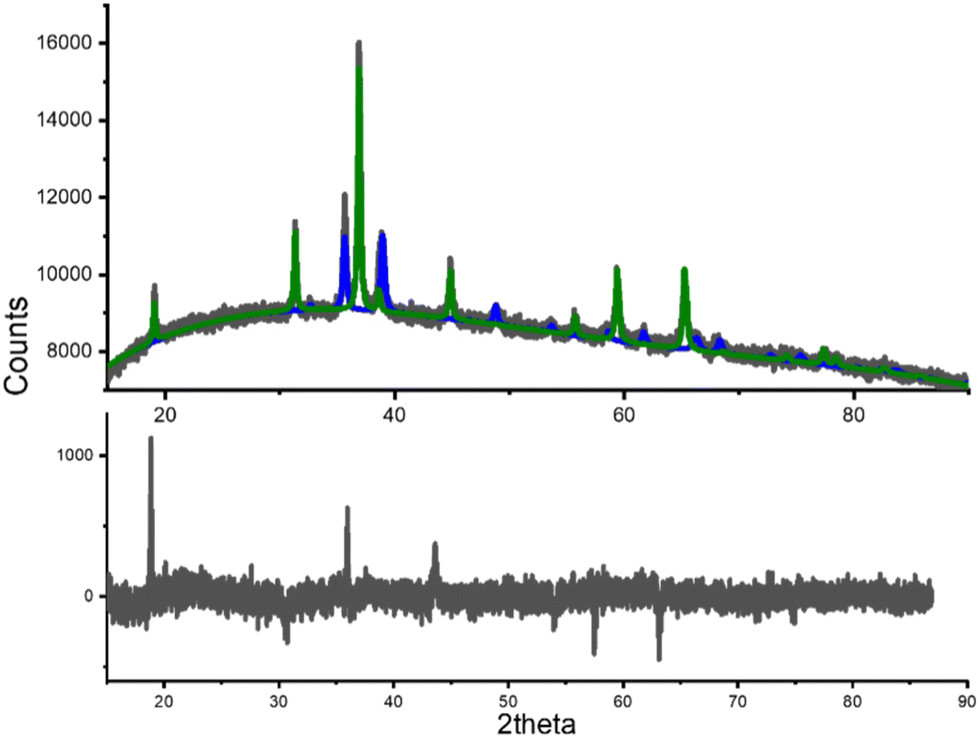 | ||
| Fig. 10 X-ray diffraction pattern of the degraded catalyst after Rietveld refinement. The graphic at the top shows the calculated profiles for CuO (blue line) and Co3O4 (green line). The recorded diffraction pattern is shown in gray dots. The graph at the bottom shows the difference between the observed and the calculated diffraction patterns (Rwp = 1.41%). | ||
Calculated CuO phase was compared with the CIF 1528838 (space group C2/m), while calculated Co3O4 was compared with the CIF 1538531 (space group Fdm, a = 8.0881 Å), both obtained from of the Crystallography Open Database. The estimated weight concentration of CuO after Rietveld refinement was 30.16 %; whereas the remaining 69.84 % of the concentration in weight corresponded to Co3O4. The weighted-pattern residual (Rwp) for the refined diffraction pattern was 1.41 %. Therefore, we may suggest that the decrement in the catalytic activity is due to the decomposition of CuCo2O4 spinel. The activity is not completely lost, since Cobalt(II,III) oxide is also useful in propane oxidation; however, copper(II) oxide formation seems to be detrimental on propane oxidation.
A sample of the used catalyst was sent for SEM analysis. The resulting micrographs are presented in Fig. 11. In all obtained images, in contrast to the as-synthesized product, very few sheet structures were detected. Conversely, a series of new tile-like assemblies were observed, as observed on the left side of Fig. 11a. These new structures may have resulted from the collapse of the original sheets. Additionally, the bundle-like structures visible in the pristine CoP have transformed into intertwined cubes, which can be seen at the bottom right of Fig. 11a and on the right side of Fig. 11b. These changes reflect the decomposition the catalyst underwent under the catalytic conditions applied at 500 °C.
 | ||
| Fig. 11 Scanning electron micrographs of already used CoP catalyst. The sample presents two types of morphologies: intertwined cubes, in bottom right in (a) micrograph and right in (b) micrograph; and intertwined tile-like structures at the center of (a) and at the left in (b). | ||
Moreover, the specific surface area of the used catalyst was measured under the same conditions as the one for the as-synthesized CoP catalyst. The resulting value was 33.77 m2 g−1, which is lower than the initial amount of 44.59 m2 g−1. This decrease appears to be due to the collapse of the sheets present in the CoP oxide as well as the appearance of two phases, as was previously observed in SEM and XRD analyses.
Nonetheless, the reduction in the reaction temperature suppresses the loss in catalytic activity, making it possible to recycle the catalyst without loss in efficiency.
4. Conclusions
In summary, we presented a novel synthetic route based on microwave-assisted heating to obtain CuCo2O4 as unique phase. Other synthesis methods based on co-precipitation and hydrothermal synthesis failed to produce the pristine copper cobaltite spinel, since copper oxide phases were obtained as by-products. Characterization studies include encompassing X-ray diffraction, scanning electron microscopy, electronic adsorption spectroscopy, Raman scattering spectroscopy, and X-ray photoelectron spectroscopy (XPS) provided comprehensive insights into the purity, crystal structure, morphology, and thermal stability of the synthesized CuCo2O4. Furthermore, evaluation of the catalytic performance of as-synthesized copper cobaltite in propane oxidation demonstrated its remarkable selectivity towards CO2 formation, indicating its potential application in catalytic processes. The CuCo2O4 catalyst demonstrated stability under different conditions modifications of the heating rate and to reaction cycles. Challenges such as catalyst degradation, indicated by the formation of CuO after extended reaction cycles, underscore the need for further research to elucidate the mechanism of catalytic activity and address stability issues. Nonetheless, the decrement of temperature in the catalytic cycles allowed the recycling of the catalyst in five sequences without loss of catalytic activity.Author contributions
Nidia Guadalupe García-Peña and Lucy-Caterine Daza-Gómez contributed to the conception, experimental design, carrying out measurements, as well as manuscript composition. Rocío Redón, Juan Ivan Gomez-Peralta, David Díaz and Xim Bohkimi contributed to experimental design, analysis tools, performed the analysis, and manuscript composition.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. All relevant datasets generated and/or analyzed during this study, including the characterization data for CuCo2O4 (X-ray diffraction, scanning electron microscopy, catalytic performance tests, and stability assessments), are stored in institutional repositories and can be provided for further validation or replication.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by Universidad Nacional Autónoma de México (UNAM) Postdoctoral Program (POSDOC), PAPIIT-UNAM IG100220 and CONAHCyT (Consejo Nacional de Humanidades, Ciencias y Tecnologías) 140617. J. I. G.-P. acknowledges CONAHCyT for the postdoctoral stipend (CVU: 620161). The authors are grateful for the use of the facilities at the National Laboratory for Nano and Biomaterials (LANNBIO) of CINVESTAV-Mérida (Centro de Investigación y de Estudios Avanzados). Technical assistance of Daniel Aguilar, Wilian Cauich, and Adriana Franco is especially appreciated. M. en C. Viridiana Maturano Rojas (Instituto de Ciencias Aplicadas y Tecnología, ICAT) is acknowledged for its participation in propane oxidation reactions. Antonio Morales Espino (Instituto de Física, UNAM), for recording the diffraction patterns of the catalyst at the end of the life cycle. David Díaz wants to express his gratefulness to Facultad de Química-UNAM, DGAPA-UNAM (Dirección General de Asuntos del Personal Académico) and CONAHCyT, for the financial support (PAIP 5000-9039), (PAPIIT IN 101009) and (SEP-CB-132094) projects.References
-
E. Sher, Handbook of Air Pollution From Internal Combustion Engines, 1998, pp. 27–41 Search PubMed
.
- X. Zhou, X. Zhou, C. Wang and H. Zhou, Chemosphere, 2023, 313, 137489 CrossRef CAS PubMed
- J. A. Vílchez, D. Villafañe and J. Casal, J. Loss Prev. Process Ind., 2014, 29, 1–7 CrossRef
- L. Nie, S. Li, S. Chai, N. Han and Y. Chen, Appl. Surf. Sci., 2022, 605, 154567 CrossRef CAS
- H. Zhang, C. Li, Q. Lu, M. J. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2021, 143, 3967–3974 CrossRef CAS PubMed
- W. Tang, J. Weng, X. Lu, L. Wen, A. Suburamanian, C. Y. Nam and P. X. Gao, Appl. Catal., B, 2019, 256, 117859 CrossRef CAS
- S. Wu, D. Ruan, Z. Huang, H. Xu and W. Shen, Inorg. Chem., 2024, 63, 10264–10277 CrossRef CAS PubMed
- C. Feng, X. Liu, T. Zhu and M. Tian, Environ. Sci. Pollut. Res., 2021, 28, 24847–24871 CrossRef CAS PubMed
- H.-S. Kim, H.-J. Kim, J.-H. Kim, S.-H. Kang, J.-H. Ryu, N.-K. Park, D.-S. Yun, J. Bae, H.-S. Kim, H.-J. Kim, J.-H. Kim, J.-H. Kim, S.-H. Kang, J.-H. Ryu, N.-K. Park, D.-S. Yun and J.-W. Bae, Catalysts, 2022, 12, 63 CrossRef CAS
- J. Guo, C. Lin, C. Jiang and P. Zhang, Appl. Surf. Sci., 2019, 475, 237–255 CrossRef CAS
- S. Chu, E. Wang, F. Feng, C. Zhang, J. Jiang, Q. Zhang, F. Wang, L. Bing, G. Wang, D. A. Han, S. Chu, E. Wang, F. Feng, C. Zhang, J. Jiang, Q. Zhang, F. Wang, B. Liancheng, G. Wang and D. Han, Catalysts, 2022, 12, 1543 CrossRef CAS
- L. H. Vieira, L. F. Rasteiro, C. S. Santana, G. L. Catuzo, A. H. M. da Silva, J. M. Assaf and E. M. Assaf, ChemCatChem, 2023, 15, e202300493 CrossRef CAS
- C. Li and J. B. Baek, ACS Omega, 2020, 5, 31–40 CrossRef CAS PubMed
- S. Yae, Y. Morii, N. Fukumuro and H. Matsuda, Nanoscale Res. Lett., 2012, 7, 1–5 CrossRef PubMed
- A. A. Komarova and D. S. Perekalin, Organometallics, 2023, 42, 1433–1438 CrossRef CAS
- S. Wahid and B. J. Tatarchuk, Ind. Eng. Chem. Res., 2013, 52, 15494–15503 CrossRef CAS
- W. E. Armstrong, T. J. Jennings and H. H. Voge, J. Catal., 1972, 24, 502–509 CrossRef CAS
- M. Zhang, X. Sui, X. Zhang, M. Niu, C. Li, H. Wan, Z. A. Qiao, H. Xie and X. Li, Appl. Surf. Sci., 2022, 600, 154040 CrossRef CAS
- Z. Hu, L. Hao, F. Quan and R. Guo, Catal. Sci. Technol., 2022, 12, 436–461 RSC
- C. Kong, F. Liu, H. Sun, Z. Zhang, B. Zhu and W. Li, Water Cycle, 2021, 2, 15–22 CrossRef
- R. Sanchis, A. García, F. Ivars-Barceló, S. H. Taylor, T. García, A. Dejoz, M. I. Vázquez and B. Solsona, Materials, 2021, 14, 7120 CrossRef CAS PubMed
- J. Zhu and Q. Gao, Microporous Mesoporous Mater., 2009, 124, 144–152 CrossRef CAS
- T. H. Lim, S. Bin Park, J. M. Kim and D. H. Kim, J. Mol. Catal. A: Chem., 2017, 426, 68–74 CrossRef CAS
- C. Zhang, J. Wang, S. Yang, H. Liang and Y. Men, J. Colloid Interface Sci., 2019, 539, 65–75 CrossRef CAS PubMed
- J. Liu, X. Ji, C. Wang, L. Wang and P. Jian, Inorg. Chem., 2023, 62, 18750–18757 CrossRef CAS PubMed
- L. Yang, C. Zhang, J. Xiao, P. Tu, Y. Wang, Y. Wang, S. Tang and W. Tang, Inorg. Chem., 2024, 63, 6854–6870 CrossRef CAS PubMed
- T. H. Lim, S. Bin Park, J. M. Kim and D. H. Kim, J. Mol. Catal. A: Chem., 2017, 426, 68–74 CrossRef CAS
- A. K. Das, N. H. Kim, S. H. Lee, Y. Sohn and J. H. Lee, Composites, Part B, 2018, 150, 269–276 CrossRef CAS
- X. Xiao, Z. Zhang, L. Cai, Y. Li, Z. Yan and Y. Wang, J. Alloys Compd., 2019, 797, 548–557 CrossRef CAS
- A. Manalu, K. Tarigan, S. Humaidi, M. Ginting, K. Sebayang, M. Rianna, M. Hamid, A. Subhan, P. Sebayang and I. P. Manalu, Int. J. Electrochem. Sci., 2022, 17, 22036 CrossRef CAS
- H. Liu and J. Wang, Electrochim. Acta, 2013, 92, 371–375 CrossRef CAS
- A. Arumugam, S. Perumal, G. Muthusamy, S. Murugesan and K. J. Ganesan, , Tetrabutylammonium Perchlorate electrolyte on electrochemical properties of spinel MgCo2O4 nanoparticles, Nano Dimens., 2020, 11(1), 26–31 Search PubMed
- S. M. N. Jeghan and M. Kang, Mater. Res. Bull., 2017, 91, 108–113 CrossRef CAS
- D. Zhu, X. Sun, J. Yu, Q. Liu, J. Liu, R. Chen, H. Zhang, R. Li, J. Yu and J. Wang, J. Colloid Interface Sci., 2019, 557, 76–83 CrossRef CAS PubMed
- C. Zhang, J. Wang, S. Yang, H. Liang and Y. Men, J. Colloid Interface Sci., 2019, 539, 65–75 CrossRef CAS PubMed
- S. Samanta and R. Srivastava, J. Colloid Interface Sci., 2016, 475, 126–135 CrossRef CAS PubMed
- D. Lan, M. Qin, R. Yang, S. Chen, H. Wu, Y. Fan, Q. Fu and F. Zhang, J. Colloid Interface Sci., 2019, 533, 481–491 CrossRef CAS PubMed
- A. Herrera-Gomez, J. Vac. Sci. Technol., A, 2020, 38, 033211 CrossRef CAS
- A. A. Yadav, Y. M. Hunge, S. B. Kulkarni, C. Terashima and S. W. Kang, J. Colloid Interface Sci., 2020, 576, 476–485 CrossRef CAS PubMed
- M. Lan, B. Liu, R. Zhao, M. Dong, X. Wang, L. Fang and L. Wang, J. Colloid Interface Sci., 2020, 566, 79–89 CrossRef CAS PubMed
-
V. Štengl, J. Šubrt, P. Bezdička, M. Maříková and S. Bakardijeva, in Solid State Phenomena, Trans Tech Publications Ltd, 2003, vol. 90–91, pp. 121–126 Search PubMed
- H. S. Jadhav, A. Roy, D. C. M. Tiongco, H. Kim and J. G. Seo, Ceram. Int., 2021, 47, 3322–3328 CrossRef CAS
- S. Dąbrowska, T. Chudoba, J. Wojnarowicz and W. Łojkowski, Crystals, 2018, 8, 379 CrossRef
- L. Kustov and K. Vikanova, Metals, 2023, 13, 1714 CrossRef CAS
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef CAS PubMed
-
A. de la Hoz, A. Díaz-Ortiz and P. Prieto, in Alternative Energy Sources for Green Chemistry, ed. G. Stefanidis and A. Stankiewicz, The Royal Society of Chemistry, 2016, pp. 1–33 Search PubMed
- P. Priecel and J. A. Lopez-Sanchez, ACS Sustainable Chem. Eng., 2019, 7, 3–21 CrossRef CAS
- NIST X-ray Photoelectron Spectroscopy Database, https://srdata.nist.gov/xps/SpectralByElm/Cu, (accessed 5 April 2024).
-
T. J. Chuang, C. R. Bridle and D. W. Rice, Interpretation of the X-Ray Photoemission Spectra of Cobalt Oxides and Cobalt Oxide Surfaces, 1976, vol. 59 Search PubMed
- G. Ertl, R. Hierl, H. Knozinger, N. Thiele and H. P. Urbach, XPS Study of Copper Aluminate Catalysts, Appl. Surf. Sci., 1980, 5(1), 49–64 CrossRef CAS
- C. D. Wagner and D. A. Zatko, Use of the oxygen KLL Auger lines in identification of surface chemical states by electron spectroscopy for chemical analysis, Anal. Chem., 1980, 52(9), 1445–1451 CrossRef CAS
- H. G. Jenniskens, P. W. F. Dorlandt, M. F. Kadodwala and A. W. Kleyn, Surf. Sci., 1996, 357–358, 624–628 CrossRef CAS
- W. Meng, X. Song, L. Bao, B. Chen, Z. Ma, J. Zhou, Q. Jiang, F. Wang, X. Liu, C. Shi, X. Li and H. Zhang, Chem. Eng. J., 2024, 494, 153028 CrossRef CAS
- H. Wu, X. Sui, Y. Lei, L. Liu, W. Xu, G. Liang, C. Li and X. Li, Fuel, 2024, 369, 131786 CrossRef CAS
- W. Meng, S. Sun, D. Xie, S. Dai, W. Shao, Q. Zhang, C. Qin, G. Liang and X. Li, Mol. Catal., 2024, 553, 113768 CrossRef CAS
- A. Gauzzi, H. J. Mathieu, J. H. Jamest and B. Kellettt, Vacuum, 1990, 41(4–6), 870–874 CrossRef CAS
- S. W. Gaarenstroom and N. Winograd, J. Chem. Phys., 1977, 67, 3500–3506 CrossRef CAS
- R. Nakhowong and R. Chueachot, J. Alloys Compd., 2017, 715, 390–396 CrossRef CAS
- N. M. Farag, M. A. Deyab, A. M. El-Naggar, A. M. Aldhafiri, M. B. Mohamed and Z. K. Heiba, J. Mater. Res. Technol., 2021, 10, 1415–1426 CrossRef CAS
- G. Concas, G. Spano, C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, J. Magn. Magn. Mater., 2009, 321, 1893–1897 CrossRef CAS
- S. Roy and S. Roy, Sustainable Energy Fuels, 2024, 8, 3854–3864 RSC
- M. Gaudon, L. Robertson, E. Lataste, M. Duttine, M. Ménétrier and A. Demourgues, Ceram. Int., 2014, 40(4), 5201–5207 CrossRef CAS
- V. Jagadeesha Angadi, A. T. Kozakov, A. V. Nicolski, Y. V. Rusalev, I. A. Ahmed and S. P. Kubrin, Inorg. Chem. Commun., 2023, 158, 111583 CrossRef CAS
- Z. Yang, G. Wang, Y. Guo, F. Kang, Y. Huang and D. Bo, Mater. Res. Bull., 2012, 47, 3965–3970 CrossRef CAS
- R. Rahmatolahzadeh, M. Mousavi-Kamazani and S. A. Shobeiri, J. Inorg. Organomet. Polym. Mater., 2017, 27, 313–322 CrossRef CAS
- Z. K. Heiba, M. B. Mohamed, N. M. Farag and A. Badawi, Z. Naturforsch., A:Phys. Sci., 2022, 77, 291–304 CrossRef CAS
- V. Venkatachalam, A. Alsalme, A. Alghamdi and R. Jayavel, J. Electroanal. Chem., 2015, 756, 94–100 CrossRef CAS
- V. Venkatachalam, A. Alsalme, A. Alghamdi and R. Jayavel, Ionics, 2017, 23, 977–984 CrossRef CAS
- L. Daza-Gómez, K. Y. Pérez Salas, L. Ruiz-Huerta, N. G. García Peña, V. Maturano Rojas and R. Redón, ChemistrySelect, 2024, 9, 1–10 CrossRef
- U. F. Zavjalova, V. F. Tretjakov, T. N. Burdeynaya and P. G. Tsytulnikov, Chem. Sustainable Dev., 2005, 13, 751–754 Search PubMed
- X. He, F. Dong, W. Han, Z. Tang and Y. Ding, J. Mater. Chem. A, 2024, 12, 7470–7507 RSC
- K. Chen, W. Li, Z. Zhou, Q. Huang, Y. Liu and Q. Duan, Catal. Sci. Technol., 2020, 10, 2573–2582 RSC
- M. Zhang, X. Sui, X. Zhang, M. Niu, C. Li, H. Wan, Z.-A. Qiao, H. Xie and X. Li, Appl. Surf. Sci., 2022, 600, 154040 CrossRef CAS
- X. Zhuge, J. Zhou, Z. Chen, S. Liu and K. Du, Colloids Surf., A, 2023, 677, 132340 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02828g |
| This journal is © The Royal Society of Chemistry 2025 |