DOI:
10.1039/D4DT02857K
(Paper)
Dalton Trans., 2025,
54, 2069-2077
Structural and optical studies of fluoride ion binding using N-heteroaromatic ligands†
Received
12th October 2024
, Accepted 3rd December 2024
First published on 3rd December 2024
Abstract
8-Hydroxyquinoline and imidazole, two important N-heteroaromatic systems, have a strong affinity towards various anions via their acidic OH or NH protons. Three receptor ligands, 5-(1H-benzo[d]imidazol-2-yl)quinolin-8-ol (1), 5-(benzo[d]thiazol-2-yl)quinolin-8-ol (2), and 4-(1H-benzo[d]imidazol-2-yl)benzene-1,3-diol (3), were synthesized, and their fluoride (F−) ion binding properties were investigated. These ligands could selectively bind F− ions, and their respective F− complexes, namely, 1-TBAF, 2-TBAF, and 3-TBAF (TBAF = tetrabutylammonium fluoride), were characterized using single crystal X-ray analysis, NMR, UV-vis, Hirshfeld surface (HS) analysis and computational studies. Their solid-state structural analysis revealed that in each case, the F− ion is strongly bound within the receptor molecule scaffold through NH, OH, and aryl-CH hydrogen bonding interactions. In the case of 1-TBAF and 3-TBAF, F− binding is further strengthened by the inclusion of water molecules, forming fluoride-water cluster complexes. 1H NMR study exhibited the disappearance of NH and OH proton signals upon the addition of TBAF, indicating that the NH and OH protons are the primary sites for F− ion coordination. UV -vis absorbance spectra was used to study their lowest sensing limits towards F− ions. Ligands 1 and 2 showed good responsiveness towards F− ions at concentrations ≥0.5 μM, with obvious changes occurring at F− concentrations of 180 and 12 μM, respectively. HS analysis quantified non-covalent interatomic contacts between the atoms and fluoride ions, revealing that 1-TBAF contributed a maximum of 19.2% of all atoms⋯F contacts to the HS. Density functional theory studies were carried out on the 1-TBAF, 2-TBAF, and 3-TBAF crystal systems, and the band structure and total density of states of the systems were evaluated. The optical response of 2-TBAF was also investigated.
Introduction
Fluoride (F−) ions play a significant role in various fields of chemistry, biology, and health sciences.1–3 It is one of the essential trace elements whose deficiency and excess in the human body can cause serious health issues. The WHO has issued guidelines recommending lower fluoride intake levels of less than 1.5 ppm.4 Concentrations exceeding this value pose a higher risk of dental fluorosis, while concentrations much higher than this can lead to skeletal fluorosis. Therefore, there has been growing interest among researchers in monitoring and measuring the concentration of F− ions to protect human health and the environment owing to their widespread use in many chemical and pharmaceutical products5,6 as well as in uranium refineries.1 However, exclusive detection of F− ions at low levels has always been a difficult task considering their small ionic size, high electronegativity, and high Lewis basicity. Additionally, sensing F− ions in aqueous solutions is particularly challenging because of their strong affinity towards water molecules (hydration enthalpy of F− ions in water = −121 kcal mol−1).7 Recently, there has been significant interest in F− ion-sensing fluorescent and colorimetric probes owing to their selectivity, affordability, and, most importantly, high sensitivity at very low detection levels.1 These molecular probes can be small organic molecules8–13 or transition metal complexes.14–22 Their fluoride sensing mechanisms are based on several different interactions such as hydrogen(H)-bonding,13 Lewis acid–base,23 and anion–π interactions24,25 as well as, in some cases, desilylation of Si–C and Si–O bonds.2,26–28 Among the developed strategies, there is a significant interest in receptors that can attract F− ions through H-bonding interactions, such as N–H⋯F or O–H⋯F, and yield an optical response, and a colorimetric response in some cases.29,30
8-Hydroxyquinoline (8-HQ) and imidazole are two important heteroaromatic systems with a wide range of applications in biological, chemical, and materials sciences. Both molecules are amphoteric, meaning they can accept electrons by providing their acidic O–H or N–H protons to interact with anionic species.3,29,31 Additionally, they can donate a lone pair of electrons from one of the N atoms to coordinate with various metal ions.32,33 Furthermore, the strength of their acidity or basicity can be tuned by different substitutions. Notably, 8-HQ exhibits zwitterionic properties.34 However, the various aspects of the chemistry of these N-heteroaromatic molecules in selectively binding F− ions through N–H⋯F or O–H⋯F H-bonding interaction have not been extensively studied, despite the significant potential of this approach for understanding how F− ion sensing works in a complex environment. Some imidazole-based optical sensors have rapidly advanced in detecting F− ions over the past few decades, mostly relying on H-bonding or other possible non-covalent interactions with F− ions.17,18,35–42 We continued our research on the fundamental structural aspects of H-bonding interactions in the heteroaromatic systems mentioned above with different anionic species to collect new information on their F− ion sensing and/or coordination abilities. In this work, we analysed three different molecules (Fig. 1) that have been found to crystallize as strongly H-bonded networks with F− anions: 5-(1H-benzo[d]imidazol-2-yl)quinolin-8-ol (1), 5-(benzo[d]thiazol-2-yl)quinolin-8-ol (2), and 4-(1H-benzo[d]imidazol-2-yl)benzene-1,3-diol (3). We studied their crystallographic and electronic structures as well as their optical properties using both experiments and simulations to expand their potential use in F− ion detection for health and environmental protection.
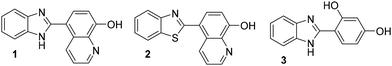 |
| | Fig. 1 Structures of N-heteroaromatic F− ion receptors studied in this work. | |
Results and discussion
Compounds 1–3 were synthesized according to a modified literature procedure;43,44 details about their synthesis and X-ray structural descriptions are given in ESI.†
Crystal structure of fluoride complexes
The F− ion binding behaviour of receptor molecules 1–3 in the solid state was investigated using SCXRD analysis. Well-formed single crystals were obtained by slowly evaporating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture solution of the receptor molecule and tetrabutylammonium fluoride (TBAF) in a minimal amount of methanol and dichloromethane (1:1). The resulting molecular structures of the F− complexes 1-TBAF–3-TBAF are shown in Fig. 2–4, and important H-bonding geometrical parameters are summarized in Table 1.
:1 mixture solution of the receptor molecule and tetrabutylammonium fluoride (TBAF) in a minimal amount of methanol and dichloromethane (1:1). The resulting molecular structures of the F− complexes 1-TBAF–3-TBAF are shown in Fig. 2–4, and important H-bonding geometrical parameters are summarized in Table 1.
 |
| | Fig. 2 (a) Molecular structures from the single crystal structure of 1-TBAF·2H2O (water molecules are removed for clarity). (b) A partial view of the crystal packing diagram, with dotted lines representing the H-bonding interactions (light green for –strong interactions, pink for –weak interactions). TBA+ ion is removed for clarity. C = grey, O = red, F = green, N = blue, and H = white. | |
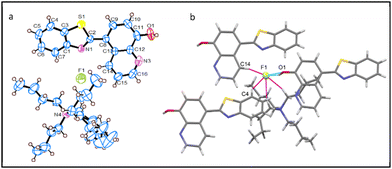 |
| | Fig. 3 (a) Molecular structures from the single crystal structures of 2-TBAF. (b) A partial view of the crystal packing diagram, and the dotted lines represent H-bonding interactions (light green = –strong; pink = –weak interaction). C = grey, O = red, F = green, N = blue, S = yellow, and H = white. | |
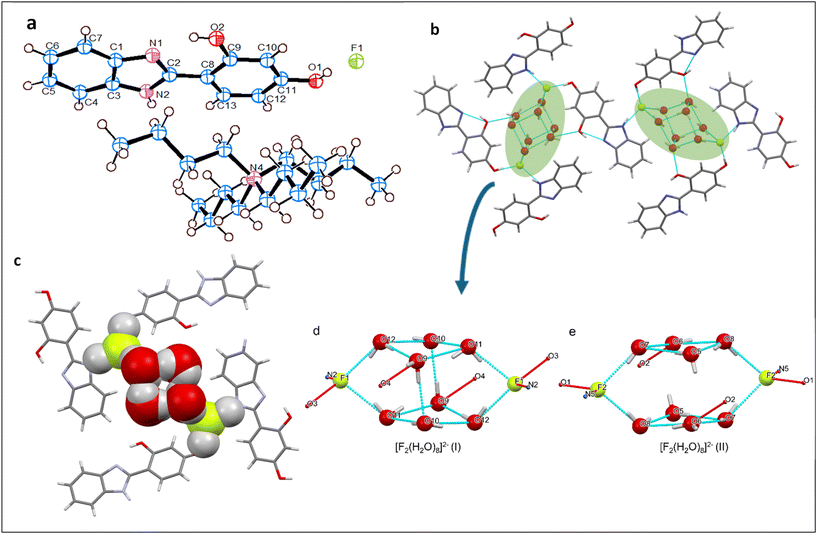 |
| | Fig. 4 (a) Molecular structures from the single crystal structures of 3-TBAF·4H2O, showing only one crystallographically inequivalent molecule within the asymmetric unit (water molecules are removed for clarity); (b) a partial view of the crystal packing diagram, with the blue dotted lines representing the O–H⋯F and N–H⋯F H-bonding interactions; (c) a partial packing diagram showing the encapsulation of a fluoride-water cluster within the molecular structure; (d) [F2(H2O)8]2−(I), and (e) [F2(H2O)8]2−(II) represent the two conformers of the fluoride-water cluster, present in the asymmetric unit. The atoms F2, N5, O3 and O4 are from the 2nd molecule in the asymmetric unit. C – grey, O – red, F – green, N – blue, H – white. | |
Table 1 Hydrogen bond D–H⋯F distances (Å) and angles (°) around the F− ion in 1-TBAF, 2-TBAF, and 3-TBAF
| |
D–H⋯F |
D–H |
H⋯F |
D⋯F |
∠D–H⋯F |
|
Two crystallographically independent molecules are present in the asymmetric unit. The atoms F2, N5, and O3 are from the 2nd molecule in the asymmetric unit, which are not shown here.
|
|
1-TBAF |
N2–H⋯F1 |
0.860 |
1.783 |
2.628(2) |
166.9 |
| O1–H⋯F1 |
0.820 |
1.610 |
2.411(2) |
164.5 |
| C9–H⋯F1 |
0.930 |
2.525 |
3.303(2) |
141.3 |
|
2-TBAF |
O1–H⋯F1 |
0.865 |
1.848 |
2.669(4) |
157.8 |
| C4–H⋯F1 |
0.930 |
2.628 |
3.438(5) |
145.9 |
| C14–H⋯F1 |
0.930 |
2.586 |
3.277(4) |
131.5 |
|
3-TBAFa |
N2–H⋯F1 |
0.860 |
1.769 |
2.593(4) |
159.9 |
| O1–H⋯F2 |
0.820 |
1.702 |
2.513(3) |
169.5 |
| N5–H⋯F2 |
0.859 |
1.879 |
2.631(4) |
145.2 |
| O3–H⋯F1 |
0.820 |
1.741 |
2.539(4) |
169.6 |
The crystal structures of 1-TBAF and 3-TBAF show the inclusion of water molecules in the complexes. This may be attributed to either hydration from TBAF·3H2O or to moisture in the air. These findings relating to the F− ion binding mode and the bond lengths of N–H⋯F and O–H⋯F are consistent with already reported literature data.29,45–50
Structural description of 1-TBAF
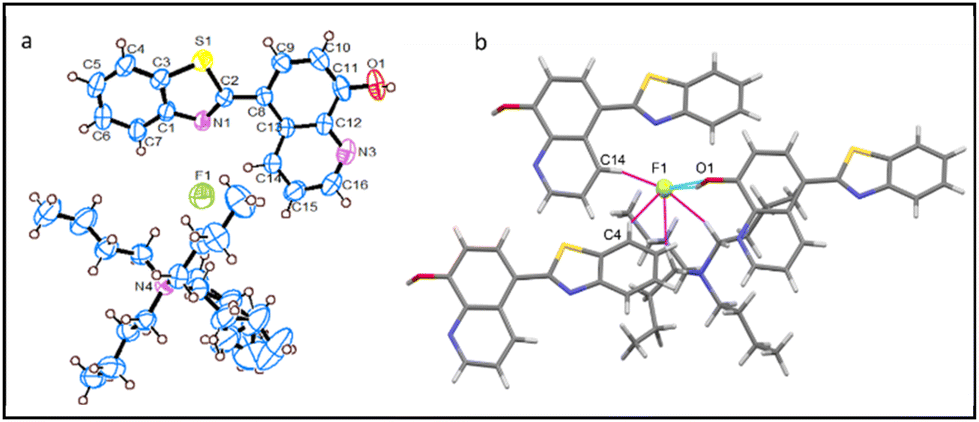
The asymmetric unit of the crystal structure of 1-TBAF possesses a single F− ion paired with a tetrabutylammonium ion (TBA+) as the counterion, accompanied with two water molecules and one receptor molecule (Fig. 2a). Within this structure, each receptor molecule encapsulates two F− ions. The interaction of the first F− ion involves 8-HQ's O1–H⋯F1 H-bond, while the second is engaged in H-bonding with imidazolyl, N2–H⋯F1, and benefits from 8-HQ's C9–H⋯F1 interactions. Moreover, each F− ion forms H-bonds with two adjacent receptor molecules, resulting in a total of three H-bonded contacts and one water molecule (O3), leading to four H-bonding interactions: three of these are strong interactions (N2–H⋯F1, O1–H⋯F1, and O3–H⋯F1), whereas one is a weaker (aryl C9–H⋯F1) interaction, as illustrated in Fig. 2b. The F− ion possesses a distorted square planar geometry, with an average bond angle of 89.5° around the central F− atom.
Structural description of 2-TBAF
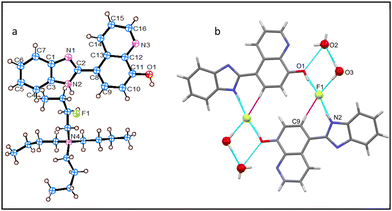
The crystallographic analysis of 2-TBAF reveals the presence of one F− ion paired with TBA+ as the counter ion, against with each receptor molecule 2 in the asymmetric unit (as depicted in Fig. 3a). Fig. 3b shows a partial view of the crystal packing diagram, emphasizing the encapsulation of the F− ion by three individual receptor molecules and a single TBA+ ion. Notably, the F− ion is penta-coordinated, comprising of four weak C–H⋯F interactions (pink dotted lines), with an average C⋯F bond length of 3.373 Å, and a C–H⋯F bond angle of 143.5°, as well as a strong O1–H⋯F (light blue dotted lines) H-bonding interaction. Further, each receptor molecule coordinates with only one F− ion in three different H-bonding interactions: a strong 8-HQ hydroxy group O1–H⋯F or two weak aryl-CH interactions, one from the imidazolyl (C4–H⋯F) and the other from the 8-HQ ring (C14–H⋯F). As shown in Table 1, although the C–H⋯F bond lengths are similar, the complex 1-TBAF holds a shorter O1–H⋯F bond length than 2-TBAF. This suggests that the former may be a stronger F− ion binding agent, which is further clarified in the subsequent sections.
Structural description of 3-TBAF
The crystal structure of 3-TBAF reveals the presence of two crystallographically inequivalent molecules in the asymmetric unit (Fig. 4). Each molecule comprises a single F− ion, accompanied with a TBA+ ion as the counterion, the receptor molecule 3, and four water molecules (Fig. 4a). Analysis of the crystal packing diagram (Fig. 4b) indicates that each receptor molecule forms hydrogen bonds with two individual F− ion via N–H⋯F and O–H⋯F bonds. The average N–H⋯F bond length (2.613 Å) is slightly shorter than that observed in 1-TBAF (Table 1). Alternatively, the average O–H⋯F bond length (2.526 Å) is longer than that in 1-TBAF but shorter than in 2-TBAF. Fig. 4b shows that each F− is H-bonded to two individual receptor molecules via imidazolyl NH and aryl OH contacts as well as two water molecules. The average O⋯F bond length is 2.742 Å, consistent with what has been reported in previous literatures.49,50 Thus, each F− ion possesses a distorted tetrahedral geometry, with an average bond angle of 107.1° around the central F-atom (Table S2†). However, despite these findings, 3-TBAF has not proven to be an effective F− ion binder, as discussed in the following section. Structural analysis further reveals that two water molecules that are H-bonded to the F− ion are part of a fluoride-water cluster [F2(H2O)8]2− (Fig. 4d and e), and their details are given in ESI.†
Solution state F− ion interaction studies using 1H NMR
We conducted 1H NMR experiments in DMSO-d6 to further validate the H-bonding interactions in solution between the NH/OH groups of the receptor molecules 1–3 and the F− ion. All three receptor molecules show a strong affinity towards the F− ion, with the most likely coordinating models depicted in Fig. 5a and b (1 and 2) and S2 (3). 1H NMR spectra were recorded for 1–3 after adding TBAF at various concentrations, and their partial views are shown in Fig. 5c and d (1 and 2) and S3 (3). The 1H NMR spectra clearly exhibit the signals for NH and OH protons. These proton signals rapidly disappeared after adding 0.1 equivalent of TBAF, indicating that NH and OH protons are the primary sites for F− ion binding in all the receptor molecules. By gradually increasing the concentration of TBAF, up to 3 equivalents, a new broad peak appeared at δ 15.90 (Fig. S4,†1) and δ 15.75 ppm (Fig. S5,†2), which is indicative of the formation of the deprotonated product HF2−.39,51 This suggests a more profound participation in F− recognition and a proton exchange process Dδ−⋯Hδ+⋯Fδ− (D = N/O). Notably, the signals for aryl-CH protons displayed almost no changes in the 1H NMR spectra of 1 and 3, indicating no interaction with F− ions. In contrast, 2 showed more significant changes in the aryl-CH peaks, with some being shielded and deshielded, as well as alterations in their shapes. This suggests a strong coordination of F− ions in solution via C–H⋯F interactions, like other studied molecules.41 Additionally, a new peak at δ 8.36 ppm (assigned to O⋯H–TBA+ H-bonding) appeared, which is more prominent in 2, leading to a significant change in fluorescence intensity and colour.
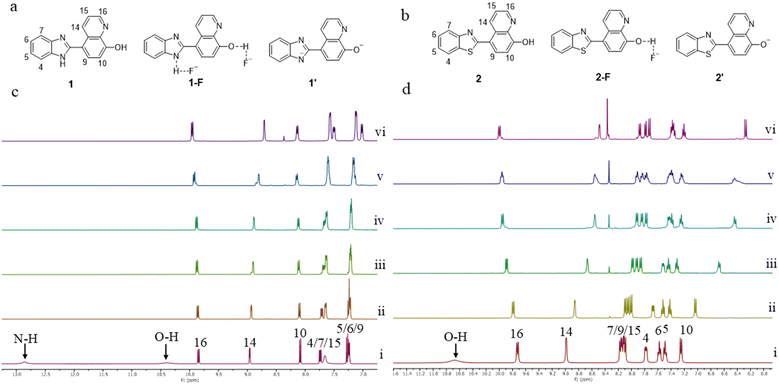 |
| | Fig. 5 (a and b) Binding modes of receptors 1 and 2, with the F− ion; (c and d) partial views of the 1H NMR of 1 and 2, with increasing concentration of the F− ion in DMSO-d6: (i) 0 eq. F−, (ii) 0.1 eq. F−, (iii) 0.2 eq. F−, (iv) 0.4 eq. F−, (v) 0.6 eq. F−, and (vi) 1.2 eq. F−. | |
UV absorbance studies
F− ion sensing ability and detection limits were investigated in DMSO solution using UV-vis spectroscopy, as shown in Fig. 6–8 and S6 (ESI)† for molecules 1–3. When TBA+ salts such as Cl−, Br−, I−, SCN−, HCO3−, and H2PO4− were added to DMSO solution containing 1–3, no noticeable changes were observed either under visual inspection or in the absorption spectra of the solutions (Fig. 6a, c (1), Fig. 6b, d (2), and S6† (3)). However, when TBAF was added to DMSO solutions containing 1 and 2, pronounced changes were observed both visually and in the absorption spectra. Meanwhile, no noticeable visual changes were observed for 3 upon addition of TBAF (Fig. S6b and S6c†); however, a slight change was observed in the absorption spectra at a high concentration of TBAF (1:100 equivalent) in DMSO (Fig. S6a†). F− ion sensing ability and the detection limits were investigated in DMSO solution using UV-vis spectroscopy, as shown in Fig. 6–8 and S6 (ESI)† for molecules 1–3. When TBA+ salts of Cl−, Br−, I−, SCN−, HCO3−, and H2PO4− were added to the DMSO solution containing compounds 1–3, no significant changes were noted in terms of visual appearances or absorption spectroscopy (Fig. 6a, c (1), Fig. 6b, d (2), and S6† (3)). In contrast, the addition of TBAF to the DMSO solutions with compounds 1 and 2, resulted in noticebale visual and spectroscopic changes. For compound 3, although there were no apparent changes after adding TBAF (Fig. S6b and c†), a minor alternation was detected in the absorption spectra at a higher concentration of TBAF (1:100 equivalent) in DMSO (Fig. S6a†).
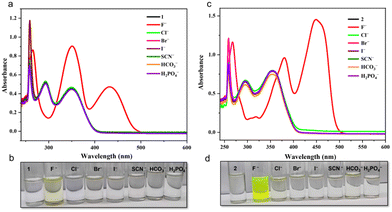 |
| | Fig. 6 (a and c) Changes in the UV-vis spectral features of 1 and 2 in DMSO upon the addition of various TBA+ salts at a ratio of 1:100. (b and d) Observation of color changes in visible light that occurred in 1 and 2 in DMSO after adding various TBA+ salts. [1] = 12 μM; [2] = 15 μM. | |
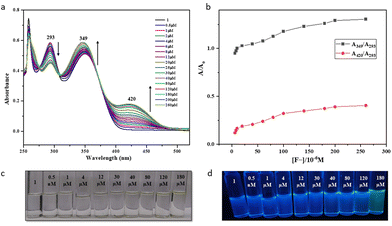 |
| | Fig. 7 (a) Changes the UV-vis spectral features of 1 in DMSO, with increasing concentrations of F− ions. (b) Plots of the ratiometric curves A349/A293 and A420/A293 as a function of F− ion concentration. (c and d) Observation of color changes that occurred in 1 in DMSO with the increasing concentration of F− ions shown in visible and UV light. [1] = 16 μM. | |
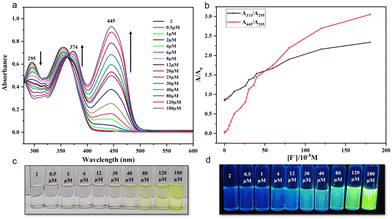 |
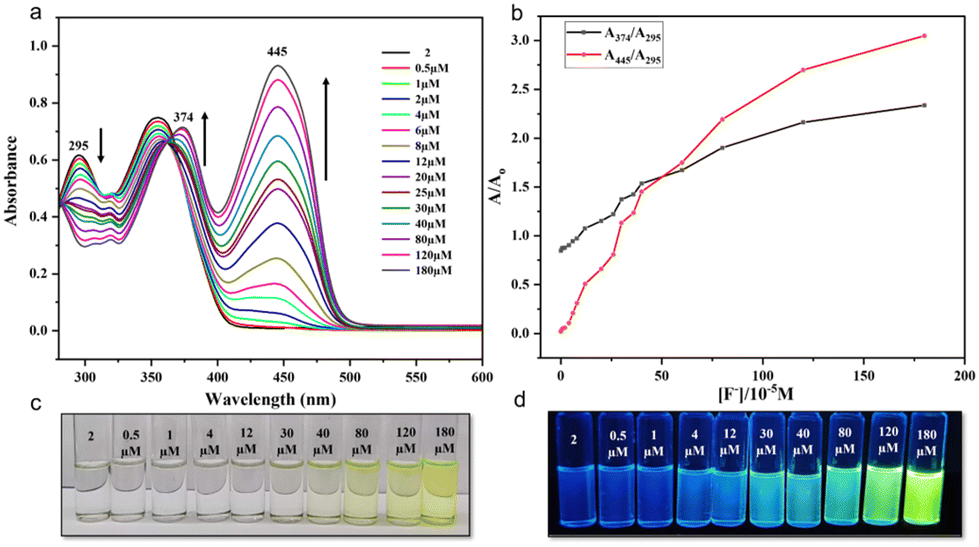
| | Fig. 8 (a) Changes in the UV-vis spectral features of 2 in DMSO, with increasing concentrations of F− ions. (b) Plots of the ratiometric curves A374/A295 and A445/A295 as a function of F− ion concentration. (c and d) Observation of color changes that occurred in 2 in DMSO with the increasing concentrations of F− ions shown in visible and UV light. [2] = 16 μM. | |
Having observed this high selectivity, we further investigated the sensitivity of 1 and 2 towards F− ions and established their low-level detection limits using absorption spectroscopy. The absorption spectra of 1 in DMSO showed characteristic absorption bands with λmax at 293 and 349 nm (Fig. 7), whereas 2 showed similar peaks with λmax at 295 and 354 nm (Fig. 8). Upon gradual addition of TBAF to the solutions of 1 and 2 in DMSO, we observed a significant bathochromic shift in the UV region for both molecules. This effect can be attributed to the formation of strong D–H⋯F (D = N/O) H-bonding. As a result, the intensity of absorption peaks at a λmax of 293 nm gradually decreased, whereas that of 349 nm gradually increased, and a new peak appeared at λmax of 420 nm for 1. In the case of 2, however, the intensity of peaks at λmax 295 and 354 nm gradually decreased, and new peaks appeared at 374 and 445 nm. These changes are further accompanied with the appearance of isosbestic points at 308 (for 1) and 365 nm (for 2). Both receptor molecules exhibit a shift of 75 nm upon the addition of TBAF, making it possible for to detect with the naked eye. Further, the UV-vis absorption spectra and respective ratiometric graphs for 1 and 2 show observable changes after adding 0.5 μM of TBAF. However, only in the case of 2, the changes are prominent both in visible and UV light, which may be attributed to the presence of the sulfur atom.41
Hirshfeld surface (HS) analysis
HS analysis is a useful tool for visualizing interatomic contacts and is often used for quantitative assessment of the nature and types of non-covalent interactions between atoms using different colors. Fig. 9a–c show the HS plots mapped with dnorm of the complexes 1-TBAF–3-TBAF and were generated using Crystal Explorer version 21.5.52dnorm is the normalized sum of di and de, where di and de are the distances from the HS to the nearest atom inside and outside the surface, respectively. It highlights some bright red regions with strong N–H⋯F and O–H⋯F interactions, and some white regions with weak C–H⋯F intermolecular interactions. In addition, 2D fingerprint plots were extracted from the respective complexes’ 3D HS plots to assess the nature and type of interatomic interactions experienced by individual molecules in the crystal packing. Fig. 9d–f present the corresponding 2D fingerprint plots, where distinct sharp spikes indicate the percentage contribution of contacts of all atoms with F− ions. Table 2 summarizes the contact contribution (%) of all atoms, as well as the contribution of each atom to the interactions with F− ions. The complex 1-TBAF shows the highest contribution towards coordinating an F− ion.
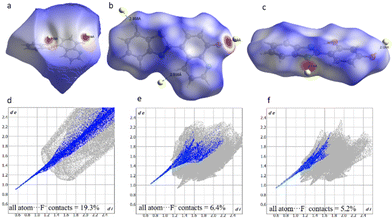 |
| | Fig. 9 (a), (b), and (c) show HS plots mapped with dnorm and (d), (e), and (f) show the 2D fingerprint plots of the percentage contribution of all atoms⋯F contacts to the HS of 1-TBAF–3-TBAF, respectively. Yellow dots = F. | |
Table 2 Percentage contributions of F− ion contacts with different atoms determined from the 2D fingerprint plots using HS analysis of complexes 1-TBAF–3-TBAF
| |
Contribution (%) |
| F− ion contacts |
| All atom⋯F |
H⋯F |
N⋯F |
O⋯F |
|
1-TBAF |
19.3 |
17.7 |
0 |
0.2 |
|
2-TBAF |
6.4 |
5.9 |
0 |
0 |
|
3-TBAF |
5.2 |
5.2 |
0 |
0 |
Computational details
Density functional theory (DFT) analysis for electronic and optical properties.
The three crystal systems were analyzed using the DFT approach, with all calculations performed using VASP. Electronic band structures (Fig. S12–S14†) show relatively discrete bands for all three crystals, with no dispersion, which is similar to bulk crystalline flat bands reported by Neves et al.53 A closer structural analysis is necessary to check the origin of such flat bands in our crystal systems. To eliminate the specific density functional with GGA-PBE functional, we performed the electronic structure studies of 2-TBAF with other functionals and approximations such as GGA + BLYP with Grimme's D2 correction54 to include dispersion, van der Waals density functional with rVV10L,55 van der Waals density functional with revPBE-vdW functional,56 the B3LYP hybrid functional with Grimme's D2 correction, and the hybrid GGA-PBE + Hartree–Fock functional. The band structure from all these various functionals show a flat band structure with large band gaps (Fig. S15–S17†). We have included the results from the GGA-PBE approximation for the rest of the computational results. Fig. 10 shows the total density of states (TDOS) estimated for the three crystal structures. All three crystals show large band gaps, which are reported in Table S7† as 2.52 eV for 1-TBAF, 1.71 eV for 2-TBAF, and 2.49 eV for 3-TBAF. A closer look at the partial density of states (PDOS) reveals that the primary contribution to the conduction band is generally located on F− ion receptor molecules involving p-orbitals. Conduction band contributions are mainly from the N, and C-atoms for both 1-TBAF (C11, C14, and N3) and 2-TBAF (C9, C11, C14, and N3), while for 3-TBAF, it is mostly from C-atoms (C2, C4, and C7), marked by arrows as shown in Fig. S18.† While valence band contributions are observed from the fluoride ion receptor molecules for 3-TBAF, F-atoms contribute solely to the valence band for 1-TBAF and 2-TBAF.
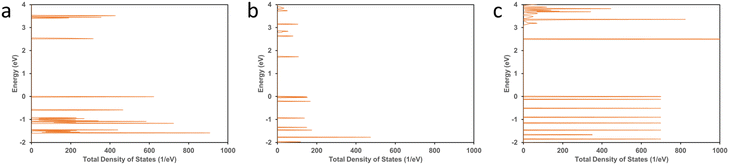 |
| | Fig. 10 Total density of states (TDOS) calculated for (a) 1-TBAF, (b) 2-TBAF, and (c) 3-TBAF crystals. | |
Since the 2-TBAF system showed the highest optical sensitivity towards F− ions, we performed a computational investigation on the optical response evaluation for 2-TBAF using DFT. To estimate the optical response, a complete optimization (atomic positions, supercell shape, and size) of 2-TBAF was performed until the Hellmann–Feynman force on each atom was less than 0.02 eV Å−1. The cell size changed and the cell volume expanded (∼7.25%) after optimization (Table S5†). The predicted absorption spectra for the 2-TBAF crystal are shown in Fig. 11 and S19†. The absorption response along the x-direction is observed to be the strongest, which is the receptor molecule stacking direction in the crystal (Fig. S20†). In addition, we computed the total optical response of the crystal (absorbance, transmittance, and reflectivity) and their directional dependence, which are presented in Fig. S19.† The computed absorption spectrum for 2 shows four bands below 600 nm: one relatively weak absorption band close to 280 nm, and the other three around 320, 370, and 540 nm. Interestingly, the experimentally observed UV-vis absorption spectrum shows three bands: a narrow band around 270 nm and two well-pronounced bands centred at 374 and 445 nm. The 320 and 540 nm bands from the computationally predicted absorption spectrum do not match the experimental data, where a third band at 445 nm in the UV-vis absorption spectrum is observed. However, two features in the computationally predicted absorption spectrum (∼280 and 370 nm) match fairly well with the experimentally observed spectrum (∼270 and 374 nm), indicating a partial match between the experimental and computational optical absorption results.
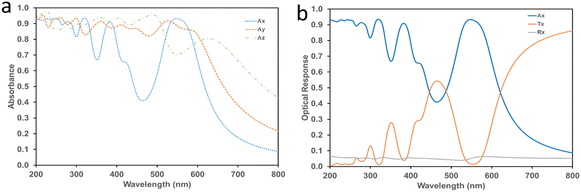 |
| | Fig. 11 (a) Absorption spectra of 2-TBAF calculated using DFT. (b) Total optical response of the optimized 2-TBAF crystal structure along the x-direction within the visible spectrum. | |
Conclusion
In conclusion, we report three receptor–ligand systems and studied their behaviour toward F− ion binding. SCXRD was used to characterize the ligands and their respective fluoride complexes 1-TBAF, 2-TBAF, and 3-TBAF. The solid-state structures of 1-TBAF–3-TBAF show similar features to other related fluoride complexes. Notably, in each case, the F− ion is H-bonded within the receptor molecule scaffold via NH, OH, and CH protons. Interestingly, in the molecular structures of 1-TBAF and 3-TBAF, the F− ion also coordinates with water molecules, resulting in fluoride-water complexation. 1H NMR in DMSO-d6 showed a rapid disappearance of NH and OH proton signals upon adding TBAF to the receptor molecule, thus suggesting the potential use of these ligand systems in F− ion recognition. However, UV-vis absorption spectra show that 2 is more responsive under UV and visible light at an F− ion concentration of about 12 μM, whereas 1 exhibits an obvious change only above 180 μM, and 3 does not show any sensing behavior, even at a concentration of 3 mM, beside a slight bathochromic shift in its UV-vis absorbance spectra. From HS analysis, all the F− complexes exhibit strong H-bonding interactions with the F− ion; however, 1-TBAF shows a maximum 19.2% contribution of contacts between all atoms and F− ions to the HS. Computational studies further support the observed optical absorption, although there is a partial matching between the observed and predicted spectra, leaving scope for further analysis of the absorption and luminescence aspects.
Author contributions
The manuscript was written with contributions from all authors. All authors have read and agreed to the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the free ligand compounds 1, 2 and 3 and their fluoride complexes 1-TBAF–3-TBAF have been deposited at the CCDC 2387451, 2387452, 2397411, and 2387453–2387455, respectively (Table S6†).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by the ICT-IOC start-up grant. The computational work was supported in part by the NYU IT high performance computing resources, services, and staff expertise. S. K. M. thanks the UGC for its support through the faculty recharge program (UGC-FRP) D.O No. F.4-5(155-FRP)/2014(BSR).
References
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- T. H. Kim and T. M. Swager, Angew. Chem., Int. Ed., 2003, 42, 4803–4806 CrossRef CAS.
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC.
-
J. K. Fawell, E. Ohanian, M. Giddings, P. Toft, Y. Magara and P. Jackson, Fluoride in Drinking-water Background document for development of WHO Guidelines for Drinking-water Quality, 2017, pp. 70–873 Search PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS.
- S. Saha, Acc. Chem. Res., 2018, 51, 2225–2236 CrossRef CAS PubMed.
- X. Zhou, R. Lai, H. Li and C. I. Stains, Anal. Chem., 2015, 87, 4081–4086 CrossRef CAS.
- S. Kumar, V. Luxami and A. Kumar, Org. Lett., 2008, 10, 5549–5552 CrossRef CAS PubMed.
- Q. S. Lu, L. Dong, J. Zhang, J. Li, L. Jiang, Y. Huang, S. Qin, C. W. Hu and X. Q. Yu, Org. Lett., 2009, 11, 669–672 CrossRef CAS PubMed.
- M. Bineci, M. Bağlan and S. Atilgan, Sens. Actuators, B, 2016, 222, 315–319 CrossRef CAS.
- Q. Lin, Q. P. Yang, B. Sun, J. C. Lou, T. B. Wei and Y. M. Zhang, RSC Adv., 2015, 5, 11786–11790 RSC.
- M. Delecluse, C. Colomban, B. Chatelet, S. Chevallier-Michaud, D. Moraleda, J. P. Dutasta and A. Martinez, J. Org. Chem., 2020, 85, 4706–4711 CrossRef CAS.
- M. Mu, X. Ke, W. Cheng, J. Li, C. Ji and M. Yin, Anal. Chem., 2022, 94, 11470–11475 CrossRef CAS.
- J. M. Stauber, G. E. Alliger, D. G. Nocera and C. C. Cummins, Inorg. Chem., 2017, 56, 7615–7619 CrossRef CAS PubMed.
- M. Hirai, M. Myahkostupov, F. N. Castellano and F. P. Gabbaï, Organometallics, 2016, 35, 1854–1860 CrossRef CAS.
- Y. Kang, J. W. Kampf and M. E. Meyerhoff, Anal. Chim. Acta, 2007, 598, 295–303 CrossRef CAS.
- Y. Cui, H. J. Mo, J. C. Chen, Y. L. Niu, Y. R. Zhong, K. C. Zheng and B. H. Ye, Inorg. Chem., 2007, 46, 6427–6436 CrossRef CAS PubMed.
- A. Sarkar, S. Bhattacharyya and A. Mukherjee, Dalton Trans., 2016, 45, 1166–1175 RSC.
- K. Yu, Q. Wang, W. Xiang, Z. Li, Y. He and D. Zhao, Inorg. Chem., 2022, 61, 13627–13636 CrossRef CAS PubMed.
- M. Toyama, T. Hasegawa and N. Nagao, RSC Adv., 2022, 12, 25227–25239 RSC.
- S. Naithani, T. Goswami, F. Thetiot and S. Kumar, Coord. Chem. Rev., 2023, 475, 214894 CrossRef CAS.
- R. S. Sathish, U. Sujith, G. N. Rao and C. Janardhana, Spectrochim. Acta, Part A, 2006, 65, 565–570 CrossRef PubMed.
- Y. Liu, Y. Zhou, H. Li, J. Gao, M. Yang, Z. Yuan and X. Li, ACS Omega, 2022, 7, 34317–34325 CrossRef CAS.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS.
- T. Chen, L. Wang, S. Li, L. Dong and L. Tan, Org. Lett., 2023, 25, 5774–5778 CrossRef CAS PubMed.
- X. Wu, H. Wang, S. Yang, H. Tian, Y. Liu and B. Sun, ACS Omega, 2019, 4, 4918–4926 CrossRef CAS PubMed.
- X. Wu, Q. Niu and T. Li, Sens. Actuators, B, 2016, 222, 714–720 CrossRef CAS.
- Y. Bao, B. Liu, H. Wang, J. Tian and R. Bai, Chem. Commun., 2011, 47, 3957–3959 RSC.
- U. Manna, G. Das and M. A. Hossain, Coord. Chem. Rev., 2022, 455, 214357 CrossRef CAS.
- S. Kundu, T. K. Egboluche and M. A. Hossain, Acc. Chem. Res., 2023, 56, 1320–1329 CrossRef CAS.
- S. Kashyap, R. Singh and U. P. Singh, Coord. Chem. Rev., 2020, 417, 213369 CrossRef CAS.
- R. Kumar, A. Thakur, Sachin, D. Chandra, A. K. Dhiman, P. K. Verma and U. Sharma, Coord. Chem. Rev., 2024, 499, 215453 CrossRef CAS.
- T. Kumari, R. Meena, L. Giri, B. Sarma, P. R. Angarkhe, J. M. Jacob, J. Tripathy, J. Joshi, M. K. Ravva, R. K. Behera and S. K. Mohapatra, J. Mol. Struct., 2023, 1273, 134253–134263 CrossRef CAS.
- D. B. Naik, P. Dwibedy, G. R. Dey, K. Kishore and P. N. Moorthy, J. Phys. Chem. A, 1998, 102, 684–688 CrossRef CAS.
- Y. C. Wu, J. Y. You, K. Jiang, J. C. Xie, S. L. Li, D. Cao and Z. Y. Wang, Dyes Pigm., 2017, 140, 47–55 CrossRef CAS.
- B. Chetia and P. K. Iyer, Sens. Actuators, B, 2014, 201, 191–195 CrossRef CAS.
- Y. C. Wu, J. P. Huo, L. Cao, S. Ding, L. Y. Wang, D. Cao and Z. Y. Wang, Sens. Actuators, B, 2016, 237, 865–875 CrossRef CAS.
- A. L. Brazeau, K. Yuan, S. B. Ko, I. Wyman and S. Wang, ACS Omega, 2017, 2, 8625–8632 CrossRef CAS PubMed.
- Z. A. Tabasi, E. A. Younes, J. C. Walsh, D. W. Thompson, G. J. Bodwell and Y. Zhao, ACS Omega, 2018, 3, 16387–16397 CrossRef CAS.
- A. Kushwaha, S. K. Patil and D. Das, New J. Chem., 2018, 42, 9200–9208 RSC.
- A. Kumari, W. Dehaen, D. Chopra and S. Dey, New J. Chem., 2022, 46, 10628–10636 RSC.
- S. Gharami, K. Aich, P. Ghosh, L. Patra, N. Murmu and T. K. Mondal, Dalton Trans., 2019, 49, 187–195 RSC.
- A. Shaikh, S. Sahoo, S. R. Marder, S. Barlow and S. K. Mohapatra, Org. Biomol. Chem., 2024, 22, 2115–2123 RSC.
- S. K. Mohapatra, K. Al Kurdi, S. Jhulki, G. Bogdanov, J. Bacsa, M. Conte, T. V. Timofeeva, S. R. Marder and S. Barlow, Beilstein J. Org. Chem., 2023, 19, 1651–1663 CrossRef CAS PubMed.
- S. K. Dey, B. K. Datta and G. Das, CrystEngComm, 2012, 14, 5305–5314 RSC.
- S. Saha, B. Akhuli, I. Ravikumar, P. S. Lakshminarayanan and P. Ghosh, CrystEngComm, 2014, 16, 4796–4804 RSC.
- W. Liu, A. G. Oliver and B. D. Smith, J. Org. Chem., 2019, 84, 4050–4057 CrossRef CAS PubMed.
- U. Manna, R. Chutia and G. Das, Cryst. Growth Des., 2016, 16, 2893–2903 CrossRef CAS.
- Q. Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119–2123 CrossRef CAS PubMed.
- M. Arunachalam and P. Ghosh, Chem. Commun., 2011, 47, 6269–6271 RSC.
- E. Mulugeta, R. Dutta, Q. He, V. Lynch, J. Sessler and C. H. Lee, Eur. J. Org. Chem., 2017, 2017, 4891–4895 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- P. M. Neves, J. P. Wakefield, S. Fang, H. Nguyen, L. Ye and J. G. Checkelsky, npj Comput. Mater., 2024, 10, 39 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. Peng and J. P. Perdew, Phys. Rev. B, 2017, 95, 81105 CrossRef.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural description, experimental details, crystallographic data, NMR spectra, absorption spectra, HS 2D fingerprints plots and computational data. CCDC 2387451–2387455 and 2394711. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02857k |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Manav
Upadhyay
c,
Manav
Upadhyay