DOI:
10.1039/D4DT03001J
(Paper)
Dalton Trans., 2025, Advance Article
Reactivity of copper(I) complexes supported by tripodal nitrogen-containing tetradentate ligands toward gaseous diatomic molecules, NO, CO and O2†
Received
28th October 2024
, Accepted 13th February 2025
First published on 13th February 2025
Abstract
Series of Cu(I) complexes supported by nitrogen-based tetradentate ligands were examined for their reactivity toward nitric oxide (NO). The copper complexes generated the corresponding Cu(II)–nitrite complexes in the presence of an excess molar amount of NO. A higher reactivity of the Cu(I) complexes toward NO was observed with a more negative Cu(I/II) redox potential, same as their reactivity toward O2 and CO, while [CuI(tepa)]+ with the most positive oxidation potential only reacted with NO among the diatomic gaseous molecules (NO, O2, and CO) examined in this study. DFT studies explained that the reactivity of the Cu–NO complex was the key to its selectivity rather than its coordination bond stability.
Introduction
Nitric oxide (NO) plays significant roles in many bio-physiological processes.1 Bacteria generate NO from nitrite ions (NO2−) and further reduce this to nitrous oxide (N2O) in the course of their anaerobic respiration, which is part of the geochemical nitrogen cycle.2 In mammals, NO is synthesized via the oxidation of L-arginine as a secondary messenger, neurotransmitter, or cytotoxic agent.3,4 In these NO-related processes, metalloenzymes containing iron, copper, or molybdenum play essential roles.5 Copper-containing nitrite reductase (CuNiR) plays a pivotal role in the conversion of NO2− to NO and the reverse reaction.6–10 Furthermore, CuNiR has been reported to catalyse NO reduction to N2O.11 Such divergent reactivity of the same enzyme is derived from the redox flexibility of NO, which can act as either an electron donor or electron acceptor in metal/NO interactions. Thus, it would be of great interest to clarify the mechanistic details of these processes occurring at the copper centre of the enzyme. On a related note, copper-ion-loaded zeolites have been reported to effectively decompose NOx, suggesting that the reaction involving N2O formation from NO likely shares a common mechanism.12
To understand details of the NO chemistry in enzymatic systems, several model studies of CuNiR have been conducted over the last few decades. For instance, Tolman and co-workers reported the reaction between NO and Cu(I) complexes supported by a Tp (Tp: hydrotris(pyrazolyl)borate) ligand to form N2O and NO2−, which is also seen in CuNiR.13 They also succeeded in isolating an intermediate species of the reaction, i.e., the Cu–NO adduct. Cu(I) complexes of tmpa (tris(pyridin-2-ylmethyl)amine) and tacn (1,4,7-triazacyclononane) were also found to show reactivity toward NO. However, there has been little exploration to date regarding the factors that control NO reactivity.14
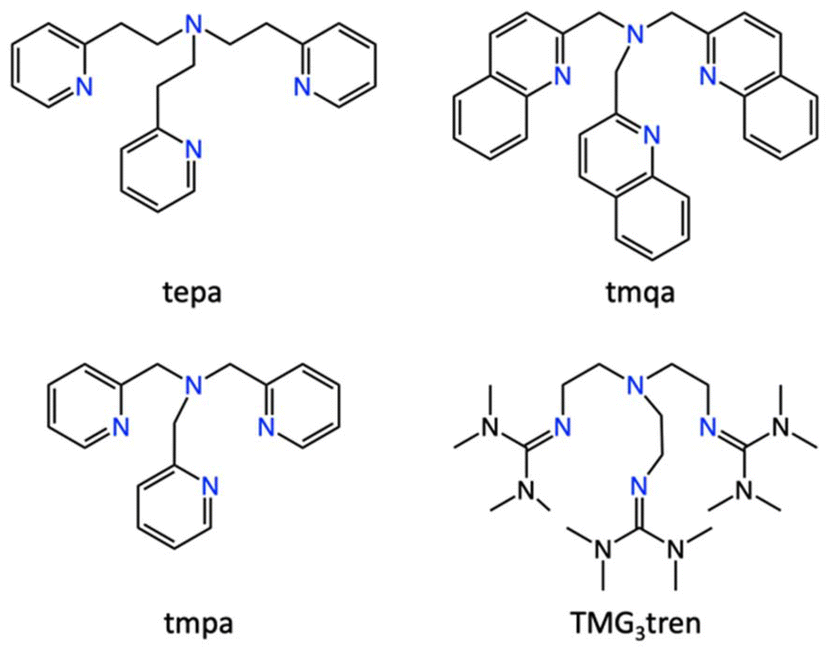
In this study, we systematically investigated the reactivity of Cu(I) complexes supported by a series of tripodal nitrogen-containing tetradentate ligands (Fig. 1) towards NO. All these carefully designed neutral tripodal ligands feature a central alkylamine nitrogen and three sp2 nitrogen donor groups, providing a well-defined platform to elucidate structure–reactivity relationships. To gain comprehensive insights into the NO chemistry of these copper complexes, we also examined their reactivity towards O2 and CO, allowing for a comparative analysis.
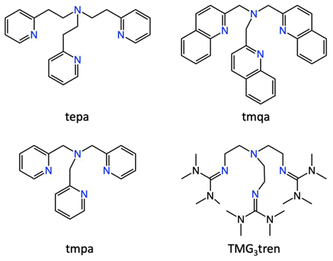 |
| | Fig. 1 Structures of the supporting ligands employed in this work. tepa: tris(2-(pyridin-2-yl)ethyl)amine, tmqa: tris(quinolin-2-ylmethyl)amine, tmpa: tris(pyridine-2-ylmethyl)amine, and TMG3tren: tris(2-(N-tetramethylguanidyl)ethyl)-amine. | |
Results and discussion
Reactivity of Cu(I) complexes toward NO
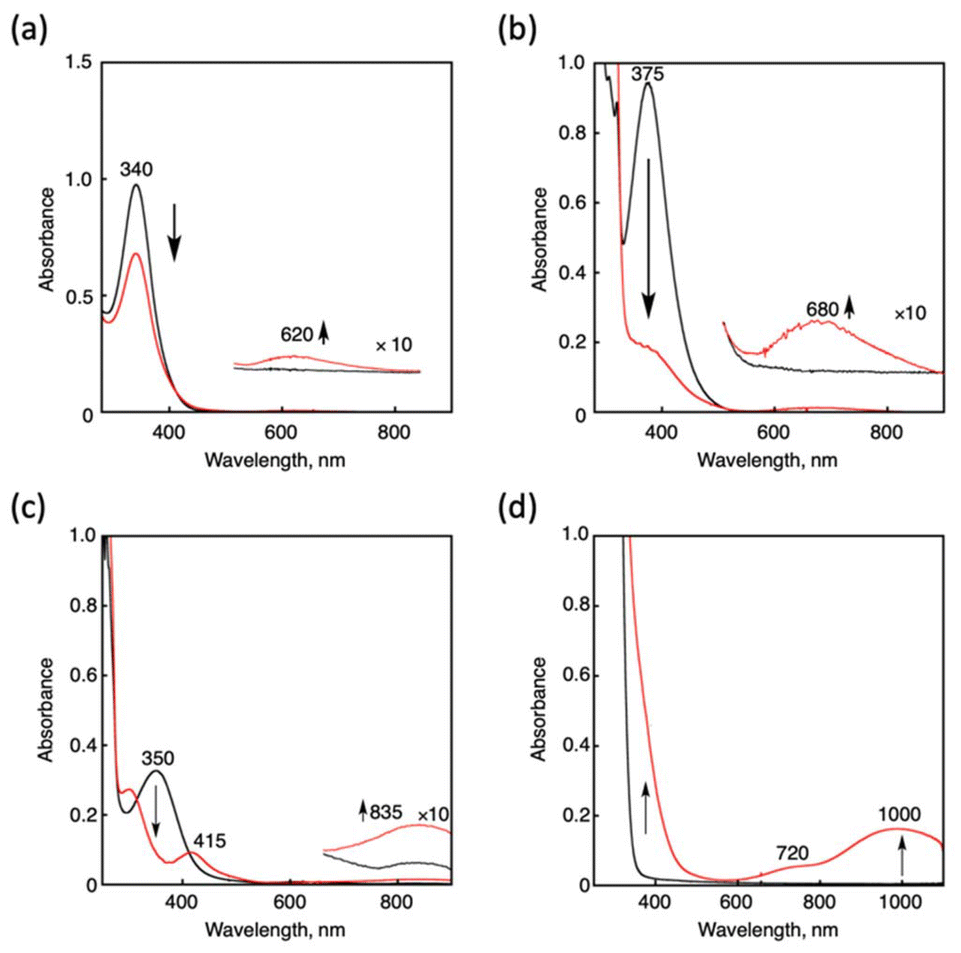
Cu(I) complexes supported by tripodal nitrogen-containing tetradentate ligands, namely, [CuI(tepa)](OTf),15 [CuI(tmqa)](OTf),16 [CuI(tmpa)(CH3CN)](OTf),17 and [CuI(TMG3tren)](OTf),18 were prepared according to the reported procedures. Reactions of the Cu(I) complexes and NO were followed by UV-vis spectroscopy (Fig. 2). Upon an addition of excess NO(g) into a THF solution of [CuI(tepa)]+, the MLCT band of the Cu(I) complex (340 nm, ε = 9800 M−1 cm−1) decreased, and the d-d band at 620 nm (ε = 170 M−1 cm−1) concomitantly increased, also showing a clean isosbestic point at 410 nm (Fig. 2a). However, decay of the Cu(I) absorption band remained incomplete due to the reactivity of the generated Cu(II) complex toward NO.§ Even in the presence of an excess molar amount of NO to Cu(I), the time course did not obey first-order kinetics. The generated absorption band at 620 nm was identical to that of [CuII(tepa)(ONO)]+, which was synthesized separately, and the yield was determined to be 40% based on a comparison of the absorption intensity. The formation of [CuII(tepa)(ONO)]+ was also confirmed by X-ray crystallography of a single crystal obtained from the resulting solution. Fig. 3a shows the crystal structure of [CuII(tepa)(ONO)]+, while its crystallographic data are summarized in Table S1 in the ESI.† It was found that the nitrite ion (NO2−) was coordinated to the copper centre with a κ2-O,O binding mode having weak interaction with the distal oxygen atoms (Cu1–O1, 2.027(2) Å; Cu1–O2, 2.644(2) Å). Thus, the copper centre exhibited a square pyramidal geometry (τ5 = 0.05), where the basal plane consisted of two pyridine nitrogen atoms (N2 and N4), the alkylamine nitrogen (N1) and the oxygen atom of NO2− (O1), with the axial position occupied by the remaining pyridine atom (N3).19
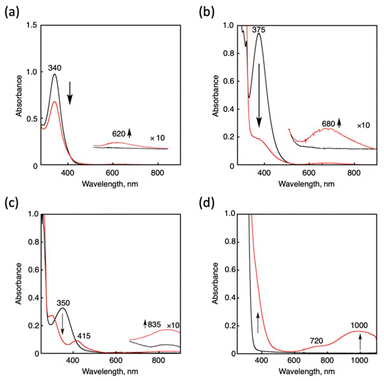 |
| | Fig. 2 UV-vis spectral changes observed after NO gas bubbling into deaerated THF solution containing the Cu(I) complexes ([CuI(tepa)](OTf) (0.10 mM) (a), [CuI(tmqa)](OTf) (0.10 mM) (b), [CuI(tmpa)(CH3CN)](OTf) (0.10 mM) (c), and [CuI(TMG3tren)](OTf) (0.50 mM) (d)) at 25 °C. Black and red lines show the initial and resulting spectra, respectively, measured after 5000 s (a), 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (b), 10 s (c), and 300 s (d). 000 s (b), 10 s (c), and 300 s (d). | |
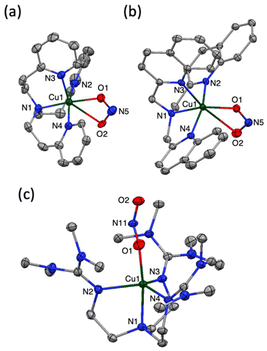 |
| | Fig. 3 ORTEP drawings of (a) [CuII(tepa)(ONO)](OTf), (b) [CuII(tmqa)(ONO)] (OTf), and (c) [CuII(TMG3tren)(ONO)](OTf) showing 50% probability thermal ellipsoids. The counter anion, solvent molecule, and hydrogen atoms are omitted for clarity. | |
Reactivities of [CuI(tmqa)]+ and [CuI(TMG3tren)]+ toward NO were also examined in THF at 25 °C. Upon the addition of NO gas into the solution of each Cu(I) complex, there was a decrease in the MLCT band (λmax = 375 nm, ε = 9400 M−1 cm−1 and around 370 nm) and an increase in the d–d band (λmax = 680 nm, ε = 120 M−1 cm−1 and λmax = 720 nm and 1000 nm, ε1000 = 320 M−1 cm−1), as shown in Fig. 2b and d.¶ The products were determined as Cu(II) nitrite complexes, namely, [CuII(tmqa)(ONO)]+ and [CuII(TMG3tren)(ONO)]+, respectively, by comparison of the solution spectra of the authentic samples and X-ray analysis of the crystals obtained from the resulting solution as per the tepa system described above (Fig. 3b and c).|| [CuII(tmqa)(ONO)]+ and [CuII(TMG3tren)(ONO)]+ also included a nitrite ion coordinated to the Cu centre in κ2-O,O and κ1-O binding modes, respectively. The distal oxygen atom of NO2−interacts with the Cu centre in [CuII(tmqa)(ONO)]+ as well as [CuII(tepa)(ONO)]+, whereas that of [CuII(TMG3tren)(ONO)]+ was located outside of the copper coordination sphere because of the bulkiness of the TMG group. It was found that [CuII(tmqa)(ONO)]+ has a square pyramidal structure (τ5 = 0.03), whereas [CuII(TMG3tren)(ONO)]+ has a slightly distorted trigonal bipyramidal structure with τ5 = 0.83, which is a typical geometry of tren ligands. Karlin and co-workers reported that the reaction of [CuI(tmpa)]+ and NO in THF gave [(tmpa)CuII-ONO]2+.20 We also confirmed the formation of [CuII(tmpa)(ONO)]+ in THF with absorption maxima at 300, 415 and 835 nm (Fig. 2c). The reaction of [CuI(tepa)]+ and [CuI(tmqa)]+ with NO took more than 10000 s, whereas [CuI(tmpa)(CH3CN)]+ and [CuI(TMG3tren)]+ were readily converted into the Cu(II) complexes (vide infra). GC-MS analysis of the generated gas product revealed the formation of N2O gas in the reaction of [CuI(TMG3tren)]+, whereas the [CuI(tepa)]+ and [CuI(tmqa)]+ system did not generate N2O, which was possibly due to the inefficiency of the reaction.
The reaction between Cu(I) complexes and NO has been reported to form N2O and NO2−, though the exact mechanism remains elusive.14a,21 Karlin's group identified the formation of [(tmpa)2CuII2(μ-N2O22−)]2+, supported by crystallographic evidence.
The formation of the CuII2(μ-N2O22−) complex is outlined as a stepwise process involving (i) Cu–NO bond formation, (ii) dinuclear complex formation and (iii) N–N bond formation. Several potential intermediates were incorporated in Scheme 1, including [LCu(N2O2)]+, [LCu(NO)2]+ and [LCu2(μ-NO)2]2+, proposed by Tolman's group.14a,21 Our experimental investigation of [CuI(TMG3tren)]+ suggests that pathways (b) and (c), involving the coordination of two NO molecules on a copper centre are unlikely to occur. This conclusion arises from the steric constraints imposed by the three tetramethylguanidino groups, which may hinder the simultaneous binding of two NO molecules within the confined coordination environment. In the later calculational study section, we discuss the formation process and conversion process of the CuII2(μ-N2O22−) complex to form CuII(ONO−) based on the chemical stability of each intermediate.
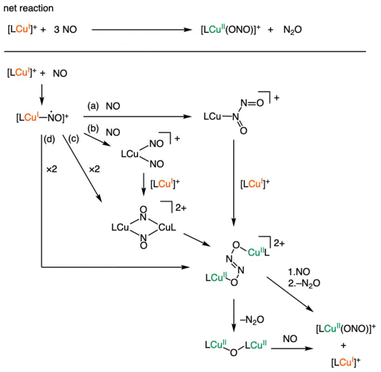 |
| | Scheme 1 Possible reaction pathways between the Cu(I) complexes and NO. | |
Reactivity of Cu(I) complexes toward O2 and CO
Cu(I) complexes exhibit distinct reactivity toward O2 and CO due to their different coordination modes. Typically, O2 binds to the copper center as an X-type ligand, whereas CO acts as an L-type ligand, characterized by strong π-back donation. We analyzed the reactivity of Cu(I) complexes toward O2 and CO from the perspective of the oxidation potential, incorporating previously reported findings as outlined below. As shown in Fig. 4, [CuI(tepa)]+ reacted with neither O2 nor CO, as also reported by Karlin and co-workers (Fig. 4a and b).15,22** Notably, [CuI(tepa)]+ only reacted with NO without showing any reactivity toward O2 and CO. Such a copper complex has never been reported so far. [CuI(tmqa)]+ showed sluggish reactivity toward O2 in THF at 25 °C and was oxidized to a Cu(II) complex slowly under an O2 atmosphere in two days (Fig. 4c).16 Conversely, [CuI(tmqa)]+ immediately reacted with CO to generate a Cu-CO adduct (Fig. 4d). In the case of tmpa and TMG3tren systems, the Cu(I) complexes showed high reactivity toward O2 and CO under identical experimental conditions to those used for the tepa and tmqa systems (Table 1). As reported, [CuI(tmpa)(CH3CN)]+ and [CuI(TMG3tren)]+ reacted with O2 even at a low temperature (−80 °C) to provide a dinuclear Cu(II) end-on peroxide complex17,23 and a mononuclear Cu(II) end-on superoxide complex,24 respectively. These complexes also showed high reactivities toward CO to provide Cu-CO adducts.17,25 From the results, it became clear that these Cu(I) complexes showed different reactivities toward gaseous molecules though they had similar tetradentate-tripod ligands. The difference in reactivity may have resulted from their difference in electronic structures (vide infra).
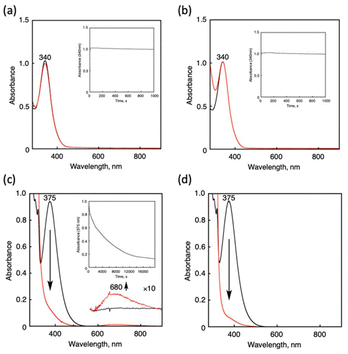 |
| | Fig. 4 UV-vis spectral changes of [CuI(tepa)]+ (0.1 mM) in the presence of excess (a) O2 and (b) CO gas in THF at 25 °C. Black line: initial spectra of the Cu(I) complexes; red line: spectra obtained after the addition of O2 (1000 s) or CO gas (1000 s). Inset: time course of the UV-vis spectral changes followed at 340 nm. UV-vis spectral changes of [CuI(tmqa)]+ (0.10 mM) in the presence of excess (c) O2 and (d) CO gas in THF at 25 °C. Black line: initial spectra of the Cu(I) complexes; red line: spectra obtained after the addition of O2 (200000 s) or CO gas (10 s). Inset: time course of the UV-vis spectral changes followed at 375 nm. | |
Table 1 Correlation between the reactivity and redox potentials of Cu(I) complexes
| Complex |
E1/2a |
Reactivityb |
| NO |
O2 |
CO |
| Versus SCE. Evaluated by the half-life time of the reaction. Half-life time is less than 50 s. Not reacted. |
| [CuI(TMG3tren)]+ |
–0.28 |
Fastc |
Fastc |
Fastc |
| [CuI(tmpa)(CH3CN)]+ |
–0.02 |
Fastc |
Fastc |
Fastc |
| [CuI(tmqa)]+ |
0.35 |
2 × 104 |
4 × 104 |
Fastc |
| [CuI(tepa)]+ |
0.44 |
3 × 103 |
N.R.d |
N.R.d |
Electrochemical property
Oxidation potentials of the Cu(I) complexes were determined by cyclic voltammetry (CV) to get insights into the differences in their reactivities (Fig. 5). The cyclic voltammograms of [CuI(TMG3tren)]+, [CuI(tmpa)(CH3CN)]+, [CuI(tmqa)]+, and [CuI(tepa)]+ exhibited reversible Cu(I)/Cu(II) redox couples at E1/2 = −0.28 V, −0.02 V, 0.35 V and 0.44 V vs. SCE in CH3CN, respectively. The oxidation potentials of [CuI(tmpa)(CH3CN)]+ and [CuI(tepa)]+ showed a significant gap of 0.46 V, regardless of their same ligand-donor sets. The more positive oxidation potential of [CuI(tepa)]+ was ascribed to its tetrahedral geometry (τ4 = 0.840), which could stabilize the Cu(I) state, also having an impact on the MLCT energy and its coefficient (Fig. 2a and c). As the redox potentials of the copper complexes became negative, the reactivities of the Cu(I) complexes toward the diatomic molecules became higher (Table 1). It is well known that the reactions between transition-metal complexes and O2 or CO are mainly governed by the electron-donation ability of the metal to O2 or CO.26 The similar tendency in reactivity toward NO suggests that the reactivity of the Cu(I) complexes with NO also depended on the electron-donation ability of the Cu(I) complexes when tripodal tetradentate ligands are employed. Judging from the very negative one-electron reduction potential of NO (−1.05 V vs. SCE),27,28 the reaction between the Cu(I) complex and NO could not be an outer sphere electron-transfer reaction but requires significant orbital overlaps.
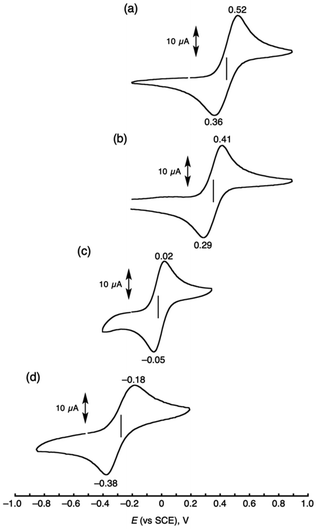 |
| | Fig. 5 Cyclic voltammograms of [CuI(tepa)]+ (a), [CuI(tmqa)]+ (b), [CuI(tmpa)(CH3CN)]+ (c), and [CuI(TMG3tren)]+ (d) (1.0 mM) in dry acetonitrile containing TBAPF6 (0.10 M) under a N2 atmosphere; working electrode: glassy carbon, counter electrode: Pt, and reference electrode: Ag/AgNO3 (10 mM). Scan rate was 100 mV s−1. | |
DFT calculations
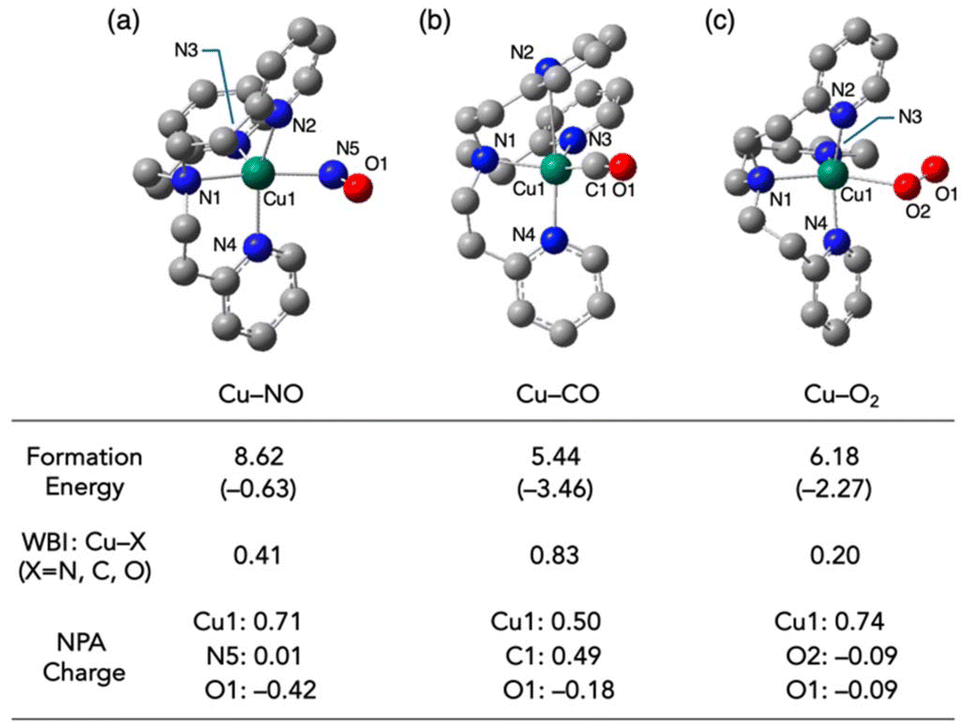
To elucidate the reaction selectivity of [CuI(tepa)]+ complex toward NO over CO and O2, the electronic structures of [Cu(tepa)(NO)]+, [Cu(tepa)(CO)]+ and [Cu(tepa)(O2)]+ were investigated using DFT calculations at the UB3LYP-D3BJ functional level with the def2-SVP basis set. Optimized structures of the NO and O2 adducts of the Cu(I)-tepa complex were obtained as penta-coordinate complexes with distorted square pyramidal geometries. The diatomic molecules coordinated to the transposition of the tert-alkyl amine nitrogen had the highest donor ability. Alternatively, the CO complex took a tetrahedral geometry, where one of the pyridine nitrogens was detached from the copper center (Cu–N = 3.04 Å) but still had an interaction via a π-coordination mode. The Gibbs free energy changes by the coordination of [CuI(tepa)]+ with NO, CO and O2 were evaluated to be endothermic, with values of 7.75, 4.86 and 4.84 kcal mol−1, respectively. In all the cases, the electronic energies were stabilized by the coordination of the gaseous molecules, while the entropy term made the coordination endothermic. In contrast to the experimental results, NO binding to [CuI(tepa)]+ was the most unfavourable process. Therefore, the reaction selectivity of [CuI(tepa)]+ toward NO can be explained by the reaction heat of the subsequent reaction of [CuI(tepa)(NO)]+ with another NO molecule, as shown in Scheme 1.
As mentioned above, CO and O2 interact with Cu(I) complexes in different ways, although both ligands prefer more electron-rich Cu(I) centres. The Cu(I)–CO complex is primarily stabilized through π-back donation. By contrast, in the end-on copper–O2 complex, the main interaction involved σ-donation from the superoxide (O2˙−) to the Cu(II) centre produced by electron transfer from Cu(I) to O2. The NO ligand, which has an intermediate number of electrons and orbital energy, has the potential to interact with Cu(I) complex via both mechanisms. Electrons in the π* orbitals of NO and O2 occupy anti-bonding character orbitals between the copper centre and the ligand, weakening the coordination bond (Fig. S1a–c†). This weakening is reflected in the Wiberg bond index (WBI);29 whereby the Cu–CO, Cu–NO, and Cu–O2 complexes had bond orders of 0.83, 0.41 and 0.20, respectively. Despite the decrease in WBI for the Cu–CO, Cu–NO and Cu–O2 complexes, the lowest unoccupied Kohn–Sham orbital energy levels of the diatomic molecules (−0.81, −2.84 and −3.02 eV, respectively) approached the HOMO energy level of [CuI(tepa)]+ (−7.99 eV), facilitating the formation of more stable coordination bonds. A comparison of the NPA (natural population analysis) charges suggests that electrostatic interaction between the copper center and the ligand are also critical in determining the stability of the complexes. Among the systems, the Cu–O2 complex had the most positively charged copper center (NPA charge: 0.74) and the most negatively charged coordinating atom (NPA charge on O2: −0.09). The valence of two factors, namely, π-back donation and σ-donation, resulted in the Cu–NO complex being the most unstable among the three complexes. In the Cu–NO complex, the NO ligand moiety accepted the largest amount of electron density among the three systems (NPA charge on NO: −0.41). Structural parameters (∠Cu–N–O = 118.5°, dN–O = 1.17 Å, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) N–O = 1814 cm−1) were assignable to the NO in a reduced state. The electron transfer from the Cu(I) centre to the NO ligand may control the reaction process, as suggested by the clear correlation observed between the oxidation potential of the Cu(I) complexes and their reactivities toward NO (Fig. 6).
N–O = 1814 cm−1) were assignable to the NO in a reduced state. The electron transfer from the Cu(I) centre to the NO ligand may control the reaction process, as suggested by the clear correlation observed between the oxidation potential of the Cu(I) complexes and their reactivities toward NO (Fig. 6).
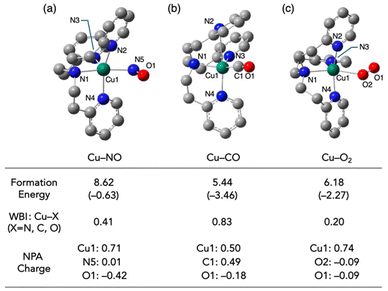 |
| | Fig. 6 Optimized structures of (a) [Cu(tepa)(NO)]+, (b) [Cu(tepa)(CO)]+ and (c) [Cu(tepa)(O2)]+ calculated by DFT at the B3LYP-D3BJ/def2-SVP level. Formation energies [electronic energy (shown in parenthesis) with the thermal energy correction] from their precursors, namely, [Cu(tepa)]+ and diatomic molecule, Wiberg bond index values between the copper center and the coordinating atom, and NPA charges of the selected atoms are shown together. | |
We also performed computational evaluations on the plausible reaction pathway shown in Scheme 1. Herein, the dinitrogen dioxide complex [LCu–N(O)–NO], formed by the second addition of NO to the Cu–NO complex, was calculated to be more unstable than the Cu–NO complex (8.00 kcal mol−1, Fig. 7). The Cu–N and N–N bond lengths were found to be 2.86 and 1.93 Å, respectively, indicating the absence of strong interactions between the copper centre and the nitrogen atoms. The distal nitrogen atom was positioned at a distance of 3.01 Å from the copper centre, indicating that the optimized structure was indistinguishable from a Cu-dinitrosyl complex [LCu–(NO)2]. The second addition of nitrosyl ligand was thus thermodynamically unfavourable. The dimerization of the [LCu–(NO)] complex to form di(μ-nitroso-k1N)dicopper [LCu(NO)2CuL] was also estimated to be an energetically unfavourable process with 24.93 kcal mol−1. By contrast, μ-hyponitritodicopper [LCu(ONNO)CuL] was estimated to be a thermodynamically favourable intermediate with a stabilization energy of −10.30 kcal mol−1 relative to LCu–NO. This intermediate was −2.55 kcal mol−1 lower in energy and thus more stable than the starting materials. The dissociation of nitrous oxide from [LCu(ONNO)CuL] to yield the μ-oxidodicopper complex (LCu–O–CuL) was calculated to be an exothermic process (−10.55 kcal mol−1). Furthermore, the final nitritocopper(II) complex was more stable than LCu–O–CuL by −30.55 kcal mol−1. Our computational results suggest that Path d in Scheme 1 is the most reasonable. Given the sufficient reaction exothermicity to release N2O, the stepwise mechanism via the μ-oxidodicopper complex appears to be the most plausible pathway.
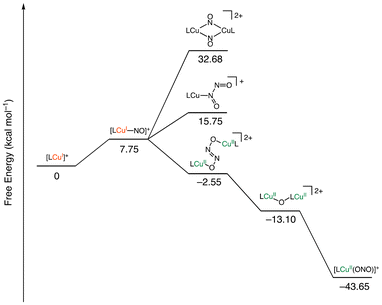 |
| | Fig. 7 Calculated free energies for the plausible intermediates in the reaction of [LCuI]+ (L = tepa) with NO to form [LCuII(NO2)]+ and N2O. | |
Conclusion
A series of Cu(I) complexes supported by tripodal tetradentate ligands was employed to investigate the controlling factors in the reaction between the Cu(I) complexes and NO. The reactivity of the complexes were also examined toward CO and O2 to understand the characteristics of NO. All the Cu(I) complexes reacted with NO, and the formation of Cu(II)–nitrite complexes were confirmed in the tepa, tmqa and TMG3tren systems. The Cu(I) complexes also showed reactivity toward O2 and CO, except for [CuI(tepa)]+. Generally, Cu(I) complexes of TMG3tren and tmpa showed higher reactivity toward diatomic molecules than the Cu(I) complexes supported by tmqa and tepa ligands. Such a tendency was consistent with the order of the oxidation potentials of the Cu(I) complexes. Among the Cu(I) complexes examined in this study, [CuI(tepa)]+, having a very positive E1/2 value, was found to exhibit unique reaction selectivity toward NO over O2 and CO. Based on the DFT study, the NO molecule binding to the copper centre was estimated to be weaker than the binding of CO and O2; however, in the following reaction, to form thermodynamically favourable product, N2O and NO2− facilitated the conversion of Cu(I) complex to the corresponding Cu(II) complex. This explanation is consistent with the slower reaction of NO with [CuI(tmqa)]+ than that of CO and O2. This is the first report to demonstrate that the oxidation potential of Cu(I) complexes controls their reactivity toward NO when the copper complexes have a similar coordination geometry. This finding provides a valuable insight into understanding the role of copper-containing NO reductase and the NO conversion reaction involving Cu(I) ions existing in biological systems.
Author contributions
YM led the conceptualization, funding acquisition, quantum calculation and review & editing of the work, KI conducted data curation and wrote the original draft and SI supervised the work and finalized the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2391963, 2392088 and 2392146† contain crystallographic data for [CuII(tepa)(ONO)](OTf), [CuII(tmqa)(ONO)](OTf), and [CuII(TMG3tren)(ONO)](OTf), respectively.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (JP22H02095) and Grant-in-Aid for Young Scientists (16K17878) from JSPS, as well as ENEOS TonenGeneral Research/Development Encouragement & Scholarship Fundation (all for Y. M.). Additional support was provided by JST-CREST (JPMJCR16P1) and a Grant in Aid for Scientific Research (B) (JSPS 23K26669) for S. I.
References
-
(a) A. R. Butler and D. L. H. Williams, The physiological role of nitric oxide, Chem. Soc. Rev., 1993, 22(4), 233–241 RSC;
(b) L. J. Ignarro and B. Freeman, Nitric Oxide: Biology and Pathobiology, Elsevier Science, 2017 Search PubMed.
-
(a) L. I. Hochstein and G. A. Tomlinson, The Enzymes Associated with Denitrification, Annu. Rev. Microbiol., 1988, 42, 231–261 CrossRef CAS PubMed;
(b) B. A. Averill, Dissimilatory Nitrite and Nitric Oxide Reductases, Chem. Rev., 1996, 96(7), 2951–2964 CrossRef CAS PubMed;
(c) W. J. Payne, Denitrification, Wiley, 1981 Search PubMed.
- R. M. Palmer, D. D. Rees, D. S. Ashton and S. Moncada, L-arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation, Biochem. Biophys. Res. Commun., 1988, 153(3), 1251–1256 CrossRef CAS PubMed.
- M. W. Radomski, R. M. J. Palmer and S. Moncada, Characterization of the L-arginine: nitric oxide pathway in human platelets, Br. J. Pharmacol., 1990, 101(2), 325–328 CrossRef CAS PubMed.
- A. Sigel, H. Sigel and R. K. O. Sigel, Biogeochemical cycles of elements; Metal ions in biological systems, Taylor & Francis, 2005, vol. 43 Search PubMed.
- J. W. Godden, S. Turley, D. C. Teller, E. T. Adman, M. Y. Liu, W. J. Payne and J. LeGall, The 2.3 Angstrom X-Ray Structure of Nitrite Reductase from Achromobacter cycloclastes, Science, 1991, 253(5018), 438–442 CrossRef CAS PubMed.
- S. Suzuki, K. Kataoka and K. Yamaguchi, Metal Coordination and Mechanism of Multicopper Nitrite Reductase, Acc. Chem. Res., 2000, 33(10), 728–735 CrossRef CAS PubMed.
- M. E. P. Murphy, S. Turley and E. T. Adman, Structure of Nitrite Bound to Copper-containing Nitrite Reductase from Alcaligenes faecalis: Mechanistic Implications, J. Biol. Chem., 1997, 272(45), 28455–28460 CrossRef CAS PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Side-On Copper-Nitrosyl Coordination by Nitrite Reductase, Science, 2004, 304(5672), 867–870 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper Active Sites in Biology, Chem. Rev., 2014, 114(7), 3659–3853 CrossRef CAS PubMed.
-
(a) C. L. Hulse, J. M. Tiedje and B. A. Averill, Evidence for a copper-nitrosyl intermediate in denitrification by the copper-containing nitrite reductase of Achromobacter cycloclastes, J. Am. Chem. Soc., 1989, 111(6), 2322–2323 CrossRef CAS;
(b) S. Ghosh, A. Dey, O. M. Usov, Y. Sun, V. M. Grigoryants, C. P. Scholes and E. I. Solomon, Resolution of the Spectroscopy versus Crystallography Issue for NO Intermediates of Nitrite Reductase from Rhodobacter sphaeroides, J. Am. Chem. Soc., 2007, 129(34), 10310–10311 CrossRef CAS PubMed.
-
(a) F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139(13), 4935–4942 CrossRef CAS PubMed;
(b) P. Granger and V. I. Parvulescu, Catalytic NOx Abatement Systems for Mobile Sources: From Three-Way to Lean Burn after-Treatment Technologies, Chem. Rev., 2011, 111(5), 3155–3207 CrossRef CAS PubMed;
(c) J. H. Kwak, D. Tran, J. Szanyi, C. H. F. Peden and J. H. Lee, The Effect of Copper Loading on the Selective Catalytic Reduction of Nitric Oxide by Ammonia Over Cu-SSZ-13, Catal. Lett., 2012, 142(3), 295–301 CrossRef CAS.
- S. M. Carrier, C. E. Ruggiero, W. B. Tolman and G. B. Jameson, Synthesis and structural characterization of a mononuclear copper nitrosyl complex, J. Am. Chem. Soc., 1992, 114(11), 4407–4408 CrossRef CAS.
-
(a) P. P. Paul and K. D. Karlin, Functional modeling of copper nitrite reductases: reactions of NO2− or nitric oxide with copper(I) complexes, J. Am. Chem. Soc., 1991, 113(16), 6331–6332 CrossRef CAS;
(b) S. Mahapatra, J. A. Halfen and W. B. Tolman, Catalytic reduction of nitric oxide to nitrous oxide by alcohols mediated by copper(I) complexes, J. Chem. Soc., Chem. Commun., 1994,(14), 1625–1626 RSC.
- K. D. Karlin, J. C. Hayes, J. P. Hutchinson, J. R. Hyde and J. Zubieta, Synthesis and X-ray structural characterization of Cu(I) and Cu(II) derivatives of a new symmetric tripodal ligand N(CH2CH2-py)3, (py = 2-pyridyl), Inorg. Chim. Acta, 1982, 64, L219–L220 CrossRef CAS.
- N. Wei, N. N. Murthy, Q. Chen, J. Zubieta and K. D. Karlin, Copper(I)/Dioxygen Reactivity of Mononuclear Complexes with Pyridyl and Quinolyl Tripodal Tetradentate Ligands: Reversible Formation of Cu:O2 = 1:1 and 2:1 Adducts, Inorg. Chem., 1994, 33(9), 1953–1965 CrossRef CAS.
- R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, A copper-oxygen (Cu2–O2) complex. Crystal structure and characterization of a reversible dioxygen binding system, J. Am. Chem. Soc., 1988, 110(11), 3690–3692 CrossRef CAS.
- V. Raab, J. Kipke, O. Burghaus and J. Sundermeyer, Copper Complexes of Novel Superbasic Peralkylguanidine Derivatives of Tris(2-aminoethyl)amine as Constraint Geometry Ligands, Inorg. Chem., 2001, 40(27), 6964–6971 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Structural Variation in Copper(I) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-coordinate Geometry Index, τ4, Dalton Trans., 2007,(9), 955–964 RSC.
- G. B. Wijeratne, S. Hematian, M. A. Siegler and K. D. Karlin, Copper(I)/NO(g) Reductive Coupling Producing a trans-Hyponitrite Bridged Dicopper(II) Complex: Redox Reversal Giving Copper(I)/NO(g) Disproportionation, J. Am. Chem. Soc., 2017, 139(38), 13276–13279 CrossRef CAS PubMed.
- C. E. Ruggiero, S. M. Carrier and W. B. Tolman, Reductive Disproportionation of NO Mediated by Copper Complexes: Modeling N2O Generation by Copper Proteins and Heterogeneous Catalysts, Angew. Chem., Int. Ed. Engl., 1994, 33(8), 895–897 CrossRef.
- S. Itoh and Y. Tachi, Structure and O2-reactivity of copper(I) complexes supported by pyridylalkylamine ligands, Dalton Trans., 2006,(38), 4531–4538 RSC.
- Z. Tyeklar, R. R. Jacobson, N. Wei, N. N. Murthy, J. Zubieta and K. D. Karlin, Reversible reaction of dioxygen (and carbon monoxide) with a copper(I) complex. X-ray structures of relevant mononuclear Cu(I) precursor adducts and the trans-(.mu.-1,2-peroxo)dicopper(II) product, J. Am. Chem. Soc., 1993, 115(7), 2677–2689 CrossRef CAS.
- C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler, Crystallographic Characterization of a Synthetic 1:1 End-on Copper Dioxygen Adduct Complex, Angew. Chem., Int. Ed., 2006, 45(23), 3867–3869 CrossRef PubMed.
- H. C. Fry, H. R. Lucas, A. A. Narducci Sarjeant, K. D. Karlin and G. J. Meyer, Carbon Monoxide Coordination and Reversible Photodissociation in Copper(I) Pyridylalkylamine Compounds, Inorg. Chem., 2008, 47(1), 241–256 CrossRef CAS PubMed.
-
(a) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Structure and Spectroscopy of Copper−Dioxygen Complexes, Chem. Rev., 2004, 104(2), 1013–1046 CrossRef CAS PubMed;
(b) E. A. Lewis and W. B. Tolman, Reactivity of Dioxygen−Copper Systems, Chem. Rev., 2004, 104(2), 1047–1076 CrossRef CAS PubMed.
- M. D. Bartberger, W. Liu, E. Ford, K. M. Miranda, C. Switzer, J. M. Fukuto, P. J. Farmer, D. A. Wink and K. N. Houk, The reduction potential of nitric oxide (NO) and its importance to NO biochemistry, Proc. Natl. Acad. Sci.
U. S. A., 2002, 99(17), 10958–10963 CrossRef CAS PubMed.
- A. S. Dutton, J. M. Fukuto and K. N. Houk, Theoretical Reduction Potentials for Nitrogen Oxides from CBS-QB3 Energetics and (C)PCM Solvation Calculations, Inorg. Chem., 2005, 44(11), 4024–4028 CrossRef CAS PubMed.
- I. Mayer, Bond order and valence indices: A personal account, J. Comput. Chem., 2007, 28(1), 204–221 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2392146, 2392088 and 2391963. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03001j |
| ‡ Current Address: Department of Chemistry, School of Science, Institute of Science Tokyo, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. |
| § [CuII(tepa)]2+ separately prepared was found to react with NO to form [CuI(tepa)]+. Details of the reaction will be described elsewhere. |
| ¶ The time courses of reactions did not follow the first-order kinetics, preventing quantitative determination of the reaction rate constants. |
| || ESI-MS analyses of the LCu(II)–nitrite complexes were hampered by the instability of the complex under mass spectrometry conditions, resulting in peaks corresponding only to LCu(II) complexes. |
| ** A small increase in absorption around 300 nm can be ascribed to the formation of the CO complex, although we cannot detect any other spectroscopic evidence using 1H NMR and IR measurement. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Keisuke Inoue and
Shinobu Itoh
*,
Keisuke Inoue and
Shinobu Itoh