
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into copper coordination in O2 reduction by water-soluble cytochrome c oxidase models†
Mathilde
Berthe
a,
Corinne
Boudon
b,
Nolwenn
Le Breton
c,
Hiroaki
Kitagishi
 d,
Koji
Oohora
e,
Takashi
Hayashi
e,
Jennifer A.
Wytko
*a and
Jean
Weiss
*a
d,
Koji
Oohora
e,
Takashi
Hayashi
e,
Jennifer A.
Wytko
*a and
Jean
Weiss
*a
aCLIC, Institut de Chimie de Strasbourg, UMR 7177 CNRS-Unistra, 4 rue Blaise Pascal, 67000 Strasbourg, France. E-mail: jweiss@unistra.fr
bLECPCS, Institut de Chimie de Strasbourg, UMR 7177 CNRS-Unistra, 4 rue Blaise Pascal, 67000 Strasbourg, France
cPOMAM Teams, Institut de Chimie de Strasbourg, UMR 7177 CNRS-Unistra, 4 rue Blaise Pascal, 67000 Strasbourg, France
dDepartment of Molecular Chemistry and Biochemistry, Doshisha University, Kyotanabe City, Kyoto 610-0321, Japan
eDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan
First published on 16th January 2025
Abstract
Iron–copper complexes have been extensively studied in the search for efficient cytochrome c oxidase models. Whereas most dinuclear materials usually focus on fine-tuning the coordination of heme-Fe, this work shows that the coordination of copper in cytochrome c oxidase models should be carefully taken into consideration. A β-cyclodextrin dimer was built around a bipyridine linker and combined with Fe-tetraphenylsulfonatoporphyrinate (FeTPPS) to generate a self-assembled hydrosoluble cytochrome c oxidase model. Cyclic voltammetry and rotating ring disk electrode experiments showed that this model with a tetrahedral coordination of copper(I) is efficient for the reduction of molecular oxygen with an average of 3.6 electrons indicating a preference and efficiency for the four-electron reduction to water.
Introduction
The reduction of molecular dioxygen by cytochrome c oxidase (CcO) is the terminal step of the respiratory process. The active center responsible for the 4-electron reduction of oxygen comprises an iron (Fea3) and a copper (CuB) in various oxidation states.1 As shown by the original solid-state structure reported by Yoshikawa, the iron is bound to a heme topped by a binding site for copper that contains three histidines, one of which is coupled to a tyrosine.2–4 Although several models have successfully reproduced either the function or the structure of various states of the enzyme, non-systematic variation of the molecular architectures used to position a copper ion above a iron porphyrinate hampers the rationalization of the parameters governing the efficiency of the models.5–8 Our groups previously reported a porphyrin-cyclodextrin scaffold [FeIIITPPS/CuIITerpyCD2]− (Fig. 1) which, after reduction to its FeIICuI derivative, is able to reduce oxygen in aqueous medium with a 50% preference for the 4-electron pathway.9 At that time, a terpyridine was chosen as the ligand for copper at the distal site. This ligand was introduced as a linker between two permethylated β-cyclodextrins (CDs) that encapsulate an iron tetraphenylsulfonateporphyrin (FeTPPS). Although terpyridine forms stable complexes with CuII, its CuI complexes are less stable because the tridentate, planar terpyridine ligand does not allow CuI to adopt its preferred tetrahedral geometry, rendering it more susceptible to disproportionation10 or oxidation. We suspected that the poor stability of the CuI complex might influence O2 reduction. Hence, the terpyridine bridge between two CDs has now been replaced with a 2,2′-bipyridine, which is capable of forming stable complexes with both oxidation states of copper. Once combined with an FeTPPS core, the resulting supramolecular assembly [FeTPPS/BipyCD2]3− allows a direct comparison between bi- and tridentate ligands for copper in CcO models.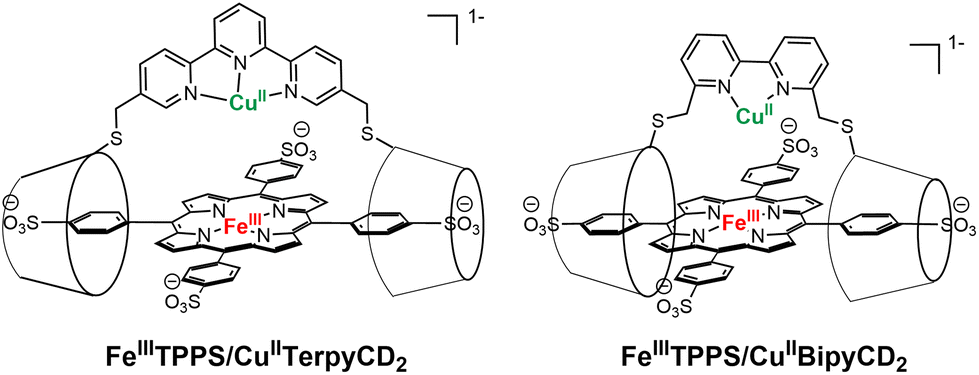 | ||
| Fig. 1 Porphyrin-β-CD2 assemblies previously reported (left) and studied (right). | ||
Results and discussion
The permethylated β-cyclodextrin dimer (BipyCD2) was prepared by reacting dithiol derivative11,121 with the epoxy derivative13,142 of the permethylated β-cyclodextrin to yield the cyclodextrin dimer BipyCD2 (Scheme 1). This dimer was purified by column chromatography and then by steric exclusion chromatography and isolated in 43% yield. | ||
| Scheme 1 Synthesis of the bipyridine-bridged CDs. | ||
The ability of the BipyCD2 to bind FeTPPS3−, as schematically illustrated in Fig. 1, was probed by titration in an aqueous phosphate buffer (pH = 7). As shown in the UV-visible spectra in Fig. 2, addition of the BipyCD2 dimer to FeTPPS3− induces a significant red shift of the Soret band of the porphyrin, from 406 to 419 nm. This change is concomitant with the appearance of an absorption at 296 nm, which is typical of the CD-dimer, and with a red shift of the Q bands from 566 to 574 nm and from 608 to 620 nm. A hyperchromic effect is also observed for the Q bands. These changes continue until the equivalence point is reached. Further addition of BipyCD2 resulted only in the growth of the absorption band of BipyCD2 at 296 nm.
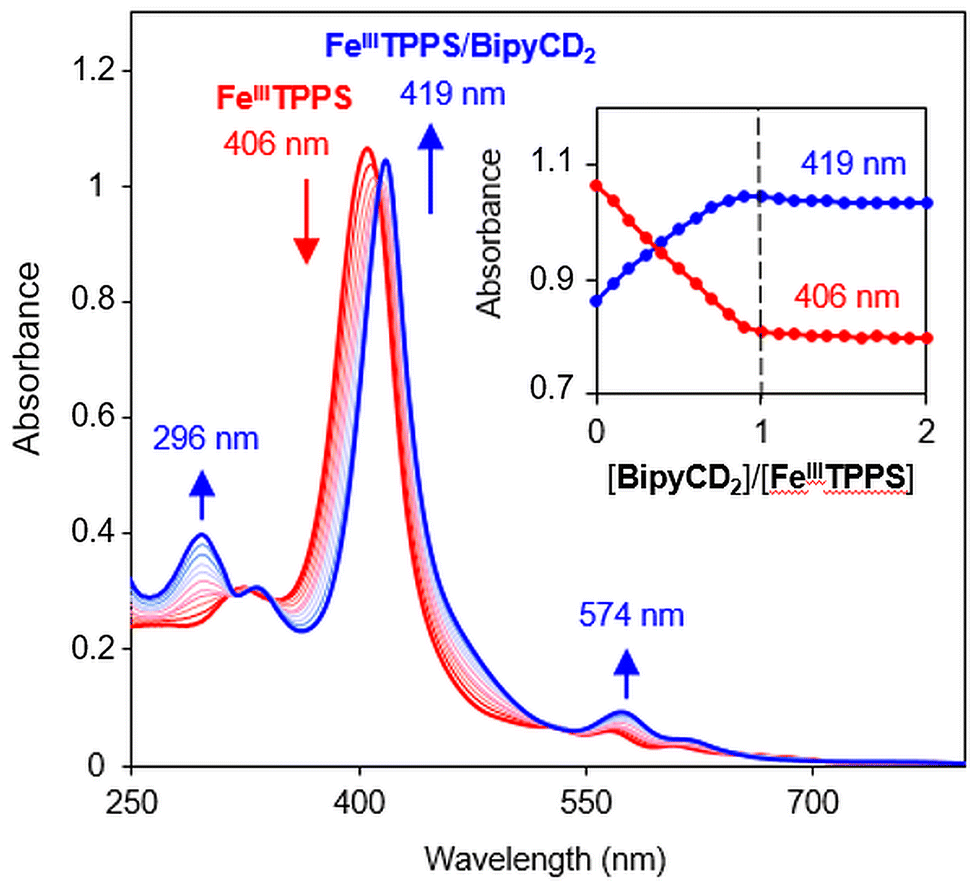 | ||
| Fig. 2 UV-visible titration of FeIIITPPS (10 μM) in 0.05 M phosphate buffer with the stepwise addition of BipyCD2 in phosphate buffer. The inset shows changes in absorbance as a function of [BipyCD2]/[FeIIITPPS] concentrations. | ||
All of the changes in the absorption spectrum of FeTPPS are consistent with the reported formation of an inclusion complex in CD-dimers.14 The association constant for the formation of the inclusion complex [FeTPPS/BipyCD2]3− was too high to be determined from the UV-vis titration. A rough estimation based on reported methods15 yields a value in the 107 M−1 range. This inclusion complex is extremely stable in aqueous solutions. In the negative electrospray ionization (ESI−) spectrum of the inclusion complex, the isotopic profile of the major peak at m/z = 1004.0844 is consistent with a species in which an axial hydroxyl ligand is bound to the iron [Fe(OH)TPPS/BipyCD2]4− (see ESI†). Additionally, a peak at m/z = 1333.1348 corresponding to the same compound without the axial ligand [FeTPPS/BipyCD2]3− was observed. These results suggest that, in aqueous medium, the iron core in [Fe(OH)TPPS/BipyCD2]4− is probably in a pentacoordinated high-spin state with an axial hydroxyl ligand. X-band electron paramagnetic resonance (EPR) confirmed this hypothesis, showing effective g values of g⊥ = 5.68 and g‖ = 1.99 at 25 K at pH = 7 in a phosphate buffer (vide infra). These results fully agree with literature reports.16–18 The ligands in the coordination sphere for the iron core of the porphyrin can be pH dependent. Thus, UV-visible monitored pH titration studies were performed on [FeTPPS/BipyCD2]3− in a sodium perchlorate buffer to explore a wider range of pH values (2 to 12) with non-coordinating counter ions (Fig. 3). Initially, the pH of the solution of [FeTPPS/BipyCD2]3− was adjusted to 2 by the addition of 0.1 M perchloric acid. Then, the pH was varied by stepwise addition of a NaOH solution. At pH higher than 5.2, a spectrum identical to that of [Fe(OH)TPPS/BipyCD2]4− in Fig. 2 (blue line) was observed and is consistent with literature data.18 According to the same reports, the species present in solution at pH < 5.2 display a Soret band at 400 nm that is characteristic of the low spin, hexacoordinated bis-aquo complex [Fe(OH2)2TPPS/BipyCD2]3−. In contrast to the spectrum of the complex in phosphate buffer (Fig. 2, blue line), at pH 2, only one absorption band at 328 nm is observed in the UV region. This band was attributed to a protonated bipyridine, for which a pKa of 2.7 was estimated. The reversible nature of this spectral change in the UV upon addition of base (comparison of the spectra in blue in Fig. 2 and 3) indicates that the thioether bipyridine-CD linkage remains intact at low pH.
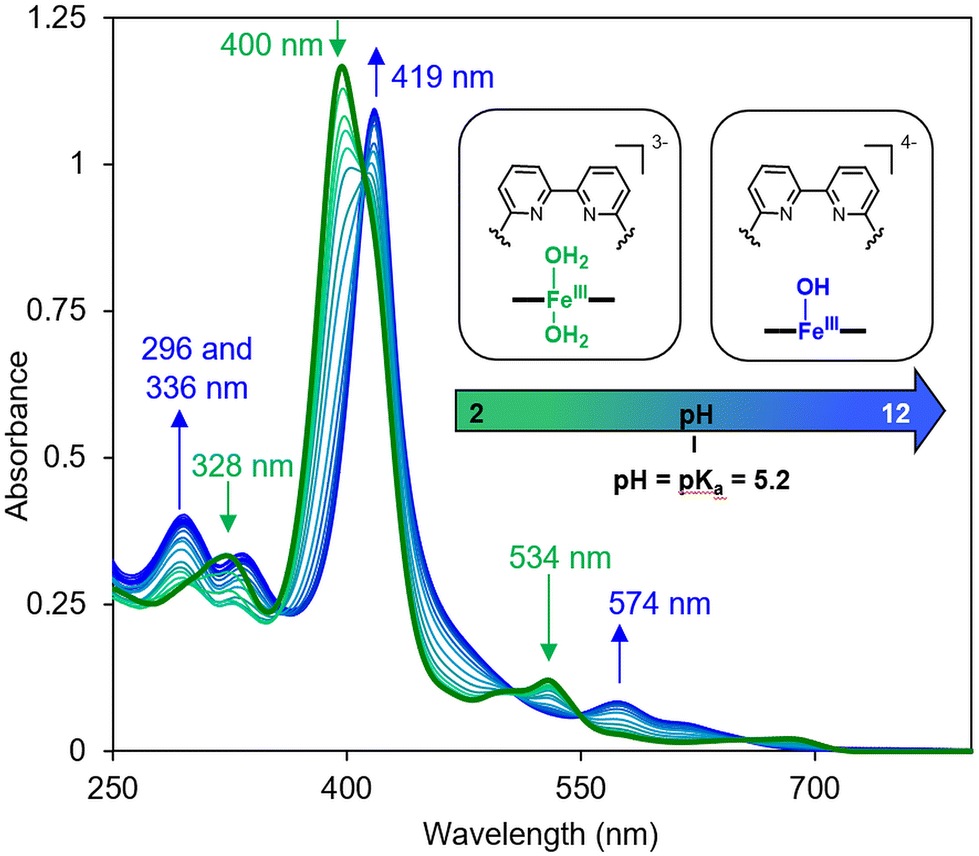 | ||
| Fig. 3 UV-visible spectral changes of FeIIITPPS/BipyCD2 (10 μM) in aqueous 0.1 M NaClO4 as a function of the pH of the solution from 2 to 12. The pH was adjusted by addition HClO4 and NaOH. | ||
Having confirmed the formation of the inclusion complex with the porphyrin heme model, the insertion of copper in the CcO model was then studied. Two approaches were possible to generate iron–copper complexes. Unfortunately, the first strategy consisting of insertion of copper(II or I) in the previously described inclusion complex [FeTPPS/BipyCD2]3− was unsuccessful. On one hand, the instability of CuI in aqueous media makes experiments with CuI difficult to reproduce. On the other hand, although the addition of CuII to the inclusion complex [FeTPPS/BipyCD2]3− seemed appropriate, the use of a phosphate buffer was precluded because interactions between phosphate anions and CuII gave a water-insoluble ion pair that precipitated. Although no precipitation occurs in the absence of phosphate buffer, the approach described in the next paragraph was preferred.
A second method was thus carried out in which FeTPPS in phosphate buffer was added in small aliquots to a solution of pre-formed [CuIIBipyCD2]2+ in pure water. First, the [CuIIBipyCD2]2+ complex was formed by adding one equivalent of a solution of CuSO4·5H2O in Milli-Q water to a solution of BipyCD2 in Milli-Q water. The formation of [CuIIBipyCD2]2+ was monitored by absorption spectroscopy in the UV region (see ESI†), where spectral changes were consistent with the stoichiometric binding of CuII to the bipyridyl bidentate with an approximate binding constant of 3 × 106 M−1 in water at pH 7. Mass spectrometry of a stoichiometric mixture of BipyCD2 and CuSO4·5H2O in Milli-Q water also confirmed the coordination of copper to the bipyridine (see ESI†).
With the copper bipyridine complex in hand, the [FeIIITPPS/CuIIBipyCD2]− inclusion complex could be formed in phosphate buffer, and no precipitation occurred. From the titration curves in Fig. 4, a binding constant greater than 107 M−1 was determined, thus confirming the stability of the supramolecular scaffold. In addition to confirming the formation of a heme-copper complex, mass spectrometry provided complementary information regarding the coordination spheres of both metals. ESI in negative mode showed two major peaks that correspond to the unbound porphyrin [FeIII-TPPS]3− and to the porphyrin bearing an axial hydroxyl ligand [FeIII(OH)-TPPS]4− (see ESI†). Several other peaks suggested the formation of a μ-oxo FeIII–CuII derivative. This type of bridged species has been observed in organic solvents by many authors in the past.19–22
 | ||
| Fig. 4 UV-visible titration of FeIIITPPS (10 μM) in 0.05 M phosphate buffer with the stepwise addition of CuIIBipyCD2 in water. The inset shows changes in absorbance as a function of ratio of the concentrations [CuIIBipyCD2]/[FeIIITPPS]. | ||
Because this work aimed at O2 reduction under physiological conditions, it was necessary to determine the predominant species at neutral pH. The existence of μ-oxo species is pH dependent; therefore, a pH titration of the FeIII–CuII species was performed. A solution of the [FeIIITPPS/CuIIBipyCD2]− complex in a 0.1 M perchlorate buffer was prepared and the pH was adjusted to 2 with a dilute perchloric acid solution. Then, the pH was varied by addition of an aqueous solution of NaOH. Four species were identified by comparison with literature data9,18,21–23 (Fig. 5).
 | ||
| Fig. 5 UV-visible spectral changes of [FeIIITPPS/CuIIBipyCD2] (14 μM) in aqueous 0.1 M NaClO4 as a function of the pH of the solution from 2 to 12. The pH was adjusted by addition HClO4 and NaOH. | ||
At acidic pH, a first species, with a Soret band at 398 nm, is assigned to the low spin inclusion complex [μ-(H2O)-FeIIITPPS/CuII(H2O)-BipyCD2]− bearing two molecules of water in axial positions. This assignment is in agreement with a recent report describing a μ-aquo FeIII–CuII species in organic solvent formed in the presence of [H(OEt2)2][B(C6F5)4 ].23 This species is dominant over the pH range of 2 to 5.3 but could not be characterized by paramagnetic NMR due to the complexity of the spectrum. Above pH 5.3, a new species appears (blue curves, Fig. 5) that is very similar to that of the [HO-FeIIITPPS/BipyCD2]4− and can be assigned to the formation of [μ-(HO)-FeIIITPPS/CuII(H2O)-BipyCD2]2− by the loss of one proton from the bridging water molecule. This species dominates from pH 5.3–7.6. Above pH 7.6, a new species appears (red curves, Fig. 5) and shows a Soret band at 445 nm, which, according to the work of Karlin and coworkers, is typical of μ-oxo species.21,22 This species is dominant in solution in the range from 7.6 < pH < 9. At pH higher than 9, a species is observed with a Soret band at 418 nm and two other absorption bands at 296 and 336 nm (purple curves, Fig. 5). The two bands in the UV region are typical of the bipyridine ligand without copper, confirming that at pH > 9 the bipyridine strap releases the copper ion. In conclusion, the dominant species at pH 7 is the [μ-(HO)-FeIIITPPS/CuII(H2O)-BipyCD2]2− and at lower pH the bridging hydroxyl may be protonated.
The paramagnetic nature of both the ferric and cupric ions prevented structural investigations by routine NMR, therefore EPR experiments were performed. To characterize the coordination sphere of CuII in our supramolecular assembly, two samples of [CuIIBipyCD2]2+, either in pure Milli-Q water or in buffer solution were studied by X-band EPR spectroscopy at 25 K (Fig. 6a). Each sample was prepared by mixing stoichiometric amounts of [FeIIITPPS/BipyCD2]3− and CuSO4·5H2O in water in the absence of phosphate buffer. In Milli-Q water, the pH was adjusted to 7 with dilute acidic and alkaline solutions, whereas for the buffered solution, the stoichiometric mixture was evaporated to dryness and redissolved in a 0.025 M phosphate buffer.
 | ||
| Fig. 6 EPR spectra at 25 K of (a) [CuIIBipyCD2]2+ (0.5 μM) in water (purple line) or in 0.025 μM phosphate buffer at pH 7 (green line) and (b) [FeIIITPPS/BipyCD2]3− (red line), [CuIIBipyCD2]2+ (green line) and [FeIIITPPS/CuIIBipyCD2]− (blue line) in 0.025 phosphate buffer at pH 7. | ||
In pure water, a CuII species with a rhombic symmetry is in very good agreement with a simulated spectrum for gx = 2.18, gy = 2.06 and gz = 2.03. The fact that gz > gx and gy indicates that the lone electron is located in the dx2−y2 orbital. According to the literature, these parameters are in agreement with elongated octahedral (Jahn–Teller distortion), square pyramidal or square planar geometries.24 Considering that the bipyridine is a bidentate ligand, we can deduce that two to four molecules of water surround the cupric ion in the complex. In buffered solution, the two species observed correspond to the previously observed species of rhombic symmetry and a second species of axial or rhombic symmetry. The latter species must correspond to a species with a phosphate coordinated to CuII. No hyperfine coupling constants were extracted from the data.
Similar experiments were then recorded for the FeIII–CuII species [μ-(HO)-FeIIITPPS/CuII(H2O)-BipyCD2]2− in phosphate buffer (Fig. 6b). The EPR spectrum differs strikingly from the EPR-silent behavior of cytochrome c oxidase in its oxidized state.25 Indeed, two signals corresponding to the ferric and the cupric ions are observed in the spectrum of [μ-(HO)-FeIIITPPS/CuII-(H2O)BipyCD2]2−. However, the superposition of the separate Cu and Fe complexes with that of the binuclear species at identical concentrations (Fig. 6b) clearly shows the weakening of both the CuII and the FeIII signals, suggesting that these two ions undergo an antiferromagnetic coupling through a μ-hydroxo ligand. Such a coupling was also observed for the terpyridine analogue.9 Thus, the formation of the supramolecular CcO model [FeIIITPPS/CuIIBipyCD2]− has been established and, at pH = 7, the dominant species is a μ-hydroxo bridged dinuclear species [μ-(HO)-FeIIITPPS/CuII(H2O)-BipyCD2]2−.
Once the nature of the resting state present at physiological pH was established, the reactivity of the reduced species was investigated. First, the copper-free model [FeIIITPPS/BipyCD2]3− was reduced with an excess of sodium dithionite in a degassed 0.05 M phosphate buffer under argon. The UV-visible spectrum of [FeIITPPS/BipyCD2]4− (black spectrum, Fig. 7) showed a hypsochromic shift of the Q band from 573 nm to 555 nm characteristic of the formation of a reduced, iron(II) complex.26 In addition, a strong hyperchromic effect and bathochromic shift of the Soret band, from 420 to 432 nm, are consistent with the formation of a pentacoordinated, high spin iron(II) complex, as seen in previous porphyrin/cyclodextrin dimer inclusion complexes.14 Overall, the UV-vis spectrum is very similar to that of deoxymyoglobin, which indicates that the FeII center in the model without copper is a pentacoordinated, high-spin complex, for which the fifth ligand is probably a water molecule.
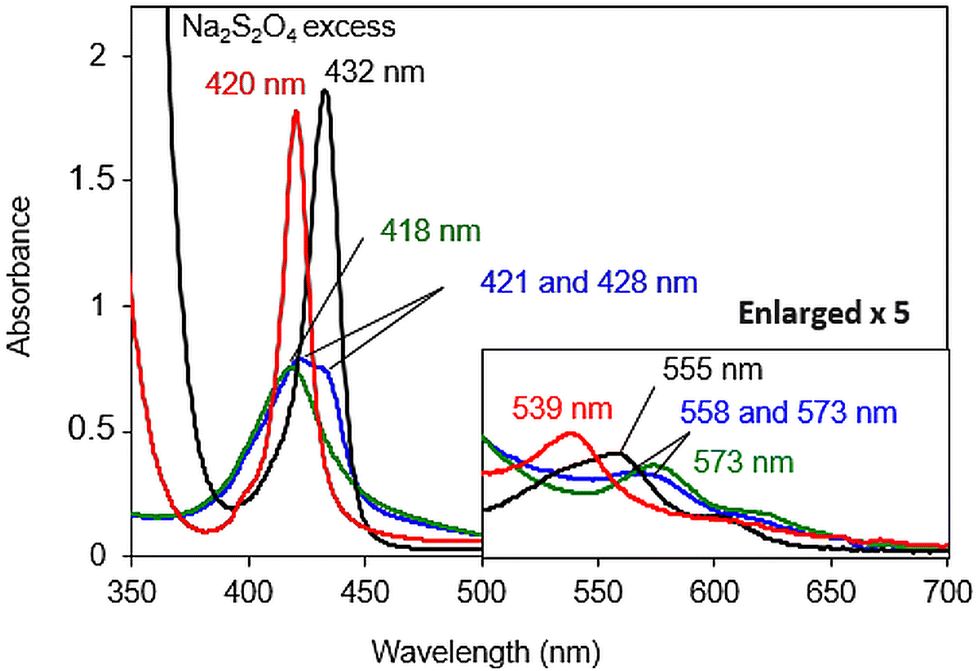 | ||
| Fig. 7 UV-visible spectra of 8 μM solutions of the [FeIIITPPS/BipyCD2]− complex (green line), its reduced FeII (black line), oxygenated FeIII–O2˙− (blue line) and carbonylated FeII–CO (red line) products in 0.05 M phosphate buffer at pH 7. The inset shows an enlargement of the Q-band region. | ||
The carbonmonoxy species was generated by bubbling CO into a solution of the iron(II) complex [FeIITPPS/BipyCD2]4−. A spectrum similar to that of carboxymyoglobin is observed, with a blue-shifted Soret band at 420 nm and a blue-shifted Q band at 539 nm (red spectrum, Fig. 7). These spectral changes correspond to a hexacoordinated low spin FeII in which a CO as well as a hydroxyl ligand are coordinated to the iron center.9,14
To form an oxygenated species, the excess sodium dithionite was first removed from a solution of [FeIITPPS/BipyCD2]4− by chromatography of the solution over Sephadex G-25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9,27 under argon. The reduced sample was then mixed in a UV-visible cell with an oxygen-saturated buffer at 10 °C. Under these conditions, the spectrum obtained shows the existence of two species with Soret bands at 421 (major) and 430 nm (minor), (blue spectrum, Fig. 7 top), suggesting that the reduced species (Soret at 430 nm) is only partially oxygenated. The presence of two Q bands at 558 and 573 nm provides further confirmation of a mixture of these two species.
9,27 under argon. The reduced sample was then mixed in a UV-visible cell with an oxygen-saturated buffer at 10 °C. Under these conditions, the spectrum obtained shows the existence of two species with Soret bands at 421 (major) and 430 nm (minor), (blue spectrum, Fig. 7 top), suggesting that the reduced species (Soret at 430 nm) is only partially oxygenated. The presence of two Q bands at 558 and 573 nm provides further confirmation of a mixture of these two species.
The influence of copper at the distal site of the heme was also studied by UV-visible spectroscopy. The spectra of reduced species [FeIITPPS/CuIBipyCD2]3− (Fig. 8 black line) and its CO adduct (Fig. 8 red line) are similar to those observed in the copper-free complex (Fig. 7) indicating the same coordination numbers and spins of the FeII centers in the absence or presence of copper. For the CO adduct [FeIITPPS-CO/CuIBipyCD2]3−, a 3 nm blue shift of its highest energy Q band (536 nm) compared to that of the copper-free complex (539 nm, shown in Fig. 7) suggests a distortion of the CO coordination to the iron, possibly due to a hindered distal site28 in the presence of copper. The oxygenated species, prepared as described above, displays a Soret band at 420 nm but no distinct Q bands. The absence of a distinct Q band was attributed to the presence of a mixture of several species including the reduced complex [FeIITPPS/CuIBipyCD2]3−, the superoxo species [FeIIITPPS-O2˙−/CuIBipyCD2]3− and/or the peroxo species [FeIIITPPS-O2/CuIBipyCD2]3−. Nevertheless, the Soret band at 420 nm appears at a wavelength similar to that of the superoxo terpyridine-bridged complex9[FeIIITPPS-O2˙−/CuITerpyCD2]3−.
 | ||
| Fig. 8 UV-visible spectra of 6 μM solutions of the [FeIIITPPS/CuIIBipyCD2]− complex (green line), its reduced FeII/CuI (black line), oxygenated FeIII–O2˙−/CuI (blue line) and carboxylated FeII–CO/CuI (red line) products in 0.05 M phosphate buffer at pH 7. The inset shows an enlargement of the Q-band region. | ||
The electrochemical properties of the complexes were investigated by cyclic voltammetry (CV) in homogeneous, neutral aqueous solution with a phosphate buffer at pH 7 with Na2SO4 as supporting electrolyte and deposited on a rotating ring disk electrode (RRDE) and rotating disk electrode (RDE). In solution, as previously observed with an inclusion complex of Mn(TPPS) bound within two permethylated-β-CDs,29 no signal was observed for the FeTPPS once inserted in the cyclodextrin dimer BipyCD2. With the iron/copper complex [FeIIITPPS/CuIIBipyCD2]−, cyclic voltammetry showed a weak reduction signal for the CuII/I couple and the FeIII/II couple. A broad reduction peak (Epa = −0.06 V vs. SCE) of the copper complex indicates an ill-defined coordination sphere around the CuII, as seen earlier with the coordination of multiple water molecules for example. A sharper signal for the re-oxidation of CuI to CuII suggests a better-defined coordination sphere for CuI in [FeIITPPS/CuIBipyCD2]3−. Nevertheless, the voltammograms were reproducible and showed no change after 10 cycles. This reproducibility confirms that the supramolecular model withstands the redox processes without decomposition or precipitation at the electrode.
In an O2-saturated electrolyte, the cyclic voltammetry shows a catalytic wave that is facilitated for [FeIITPPS/CuIBipyCD2]3− (Epc = −0.55 V vs. SCE) compared to [FeIITPPS/BipyCD2] (Epc = −0.75 V vs. SCE) (see ESI†). In the presence of copper, this gain of potential clearly demonstrates the contribution of copper in the reduction of O2. Compared to the previous [FeIITPPS/CuITerpyCD2]3− inclusion complex (see Table 1), the new iron/copper [FeIITPPS/CuIBipyCD2]3− model shows a lower onset potential (Eponset = −0.1 V vs. SCE), suggesting that O2 reduction is facilitated because of a better adapted coordination sphere for copper in the latter model.4 As this work aimed at studying the effects of the copper coordination sphere on the efficiency of the catalysis, RRDE experiments were performed to determine the number of electrons involved in the catalytic reduction of O2.
| Catalyst | E pc (V vs. SCE) | E ponset (V vs. SCE) | n RRDE | n KL | K app (M s)−1 |
|---|---|---|---|---|---|
| a Number of electrons exchange during the reduction of O2 by the catalyst at −1 V determined from eqn (1). b Number of electrons exchange during the reduction of O2 by the catalyst at −0.6 V determined from eqn (2). c Determined from eqn (3). d Taken from ref. 9; calculated at −1.2 V. | |||||
| [FeIIITPPS/BipyCD2] | −0.75 | −0.20 | 3.5 | 3.47 | 2.1 × 10−5 |
| [FeIIITPPS/CuIIBipyCD2] | −0.55 | −0.10 | 3.6 | 3.55 | 3.0 × 10−5 |
| [FeIIITPPS/TerpyCD2] | — | −0.24d | — | 1.63d | — |
| [FeIIITPPS/CuIITerpyCD2] | — | −0.20d | — | 3.03d | — |
In RRDE experiments, the 4-electron reduction of oxygen is performed by the catalyst deposited on the central, edge oriented pyrolytic graphite disk while the potential of the peripheral ring, towards which the reduction products diffuse due to the rotation of the electrode, is set at 0.8 V per SCE to re-oxidize H2O2 should it be produced by a competing 2-electron process. To prevent desorption and dissolution of the water-soluble catalysts, the catalysts were fixed on the pyrolytic graphite disk with Nafion® as in our previous work.9 Using the RRDE setup, the number of electrons exchanged can be calculated from eqn (1):30
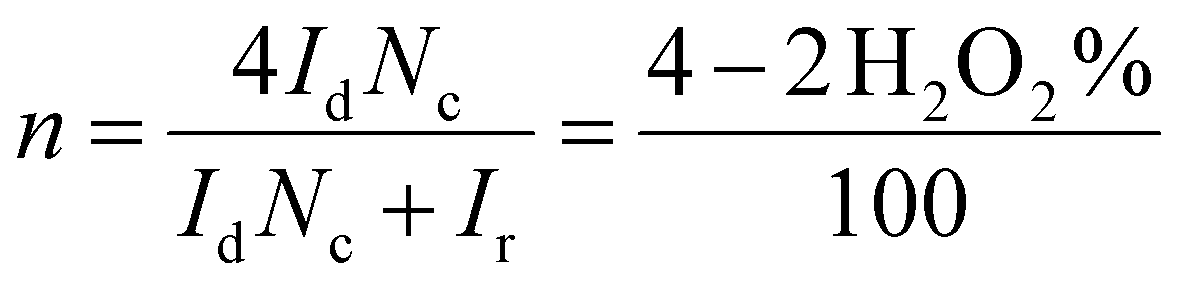 | (1) |
All RRDE experiments were performed by depositing 2.5 nmol of the supramolecular models [FeIIITPPS/BipyCD2]3− and [FeIIITPPS/CuIIBipyCD2]− as their sodium salts on the disk and covering them with a 10 μL Nafion membrane in an oxygen-saturated phosphate buffer containing 0.5 M Na2SO4 as supporting electrolyte. The currents recorded for O2 as a reference, [FeIIITPPS/BipyCD2]3− and [FeIIITPPS/CuIIBipyCD2]− are plotted in Fig. 9 for both the disk (left traces) and the ring (right traces) currents. The RRDE experiments clearly show that, despite the minor gain in terms of potential, the reduction of oxygen by the catalysts immobilized on the disk produces a minimum amount of hydrogen peroxide. From the ratio of ring and disk currents, the number of electrons (nRDDE in Table 1) involved in the reduction of oxygen is respectively 3.5 with the iron-only complex and 3.6 with the iron–copper complex. These numbers compare favorably with the 3.03 average number of electrons measured with the reported terpyridine analogue (Table 1).9 This first observation suggests that the reduction of oxygen occurs through a preferred 4-electron pathway but complementary RDE experiments were necessary to confirm the number of electrons exchanged at the electrode.
 | ||
| Fig. 9 Voltammograms measured by RRDE experiments of the various complexes (10 μL of a 2.5 mM solution) coated with Nafion (10 μL) on the disk electrode in O2-saturated solution, measured at pH 7 in 0.05 M phosphate buffer containing 0.5 M Na2SO4 at a scan rate of 0.02 V s−1 and 100 rpm. | ||
In these complementary experiments, for a given amount of catalyst deposited on the disk, the rotation speed of the electrode was systematically varied, and the reciprocal intensity of the corresponding current Im was plotted against a function of the angular rotation rate of the electrode: Im−1 = f(ω−1/2). The resulting electrochemical response and the associated Koutecký–Levich plots are represented in Fig. 10. For comparison, a 2-electron reference plotted from the experimental reduction of oxygen on the graphite electrode without catalyst, and a 4-electron reference plot simulated from the previous reference are also shown. For both catalysts, the average number of electrons (nKL) involved in the electrocatalytic process was determined from the slope of Koutecký–Levich plots at −0.6 V (see Table 1 and ESI†). These averages are consistent with the number of electrons (nRDDE) previously determined by RRDE experiments. In addition, Koutecký–Levich can provide information regarding the kinetics of the process at the electrode. In the Koutecký–Levich equation (eqn (2)):31
 | (2) |
| Ik = nFAΓcat[O2]kapp | (3) |
 | ||
| Fig. 10 (a–c) Stationary voltammograms at various rotation speeds on a RDE modified with (a) O2 reference with no catalyst, (b) [FeIIITPPS/BipyCD2]3− and coated with Nafion or (c) [FeIIITPPS/CuIIBipyCD2]− and coated with Nafion. Medium: phosphate buffer pH 7 + 0.5 M Na2SO4. (d and e) Koutecký–Levich plots for [FeIIITPPS/CuIIBipyCD2]−. Values observed at −0.6 V vs. SCE were used to determine the average number of electrons used for the reduction of O2. Values measured at −1 V vs. SCE were used to determine kinetic parameters. | ||
The difference in these values is significant whereas the number of electrons exchanged is almost identical, which suggests that the presence of copper in these models mostly influences the kinetics but does not play a significant role in the reduction mechanism. The number of electrons exchanged is significantly higher than with the terpyridine-based model, which also suggests that the efficiency of the electrocatalysts strongly depends on the stability and/or denticity of the ligand for the cuprous ion versus the cupric ion. Thus, the synergy between the FeII and CuI in models seems to reside in the orientation of the bound oxygen molecule and the affinity of oxygen for the distal site of the models, as suggested by the work of Karlin21,22,35,36 or Collman37,38 in which the copper-iron scaffold is organized around a μ-peroxide or superoxide, respectively. These results also suggest that to augment the preference for the 4-electron pathway, a tetrahedral cuprous ion is ideal.
Conclusions
A water-soluble supramolecular cytochrome c oxidase model consisting of a FeTPPS bound within a copper-bipyridine-bridged cyclodextrin dimer was prepared and characterized. The electrocatalytic properties of this supramolecular CcO model were investigated and compared with a terpyridine analogue. This work is a rare example of the direct comparison of iron–copper electrocatalysts built on a similar supramolecular inclusion complex in the 4-electron reduction of oxygen. Previous results from our group and others demonstrated that three N-ligands provide an ideal coordination sphere around CuI but, as demonstrated by the comparison of [FeIIITPPS/CuII-Terpy-CD2]− with [FeIIITPPS/CuIIBipyCD2]−, co-planarity of the three nitrogen atoms is prejudicial to the 4-electron efficiency of the resulting electrocatalysts. Future work will tackle the preparation of CD-dimers organized around TREN caps to provide a perfect fit in order to stabilize CuI intermediates.Author contributions
JW, JAW and HK designed and supervised the project. MB performed the synthesis, characterization and all titrations. CB and MB designed, performed and analyzed the electrochemical experiments. NLB performed and analyzed the EPR experiments. TH and KO designed and supervised the gas binding experiments performed by MB. MB, JW and JAW wrote the manuscript and all authors contributed to its preparation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
MB gratefully acknowledges the CNRS for a PhD fellowship. JW, JAW, CB and MB are indebted to the CNRS for supporting staff and student exchanges through the IRP “SUPRHEME” grant. Funding was also provided by grant JP18KK0156 (TH, and KO) and KAKENHI grant number 24K01640 (HK).References
-
R. Boulatov, in N4-Macrocyclic Metal Complexes, ed. J. H. Zagal, F. Bedioui and J. P. Dodelet, Springer, New York, NY, 2006, pp. 1–40 Search PubMed
.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1995, 269, 1069 CrossRef CAS
- S. Ferguson-Miller and G. T. Babcock, Chem. Rev., 1996, 96, 2889 CrossRef CAS
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936 CrossRef CAS
- E. Kim, E. E. Chufán, K. Kamaraj and K. D. Karlin, Chem. Rev., 2004, 104, 1077 CrossRef CAS
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 56 CrossRef PubMed
- T. Ogura, S. Hirota, D. A. Proshlyakov, K. Shinzawa-Itoh, S. Yoshikawa and T. Kitagawa, J. Am. Chem. Soc., 1996, 118, 5443 CrossRef CAS
- C. Kahlfuss, J. A. Wytko and J. Weiss, ChemPlusChem, 2017, 4, 584 CrossRef PubMed
- H. Kitagishi, D. Shimoji, T. Ohta, R. Kamiya, Y. Kudo, A. Onoda, T. Hayashi, J. Weiss, J. A. Wytko and K. Kano, Chem. Sci., 2018, 9, 1989 RSC
- C. S. Day and R. Martin, Chem. Soc. Rev., 2023, 52, 6601 RSC
- G. R. Newkome and D. K. Kohli, Heterocycles, 1981, 15, 739 CrossRef CAS
- A. P. Smith, J. J. S. Lamba and C. L. Fraser, Org. Synth., 2002, 78, 82 CrossRef CAS
- T. Jiang, D. K. Sukumaran, S.-D. Soni and D. S. Lawrence, J. Org. Chem., 1994, 59, 5149 CrossRef CAS
- K. Kano, H. Kitagishi, M. Kodera and S. Hirota, Angew. Chem., Int. Ed., 2005, 44, 435 CrossRef CAS PubMed
- J. R. Miller and G. D. Dorough, J. Am. Chem. Soc., 1952, 74, 3977 CrossRef CAS
- F. A. Walker, D. Reis and V. L. Balke, J. Am. Chem. Soc., 1984, 106, 6888 CrossRef CAS
- C. T. Watson, S. Cai, N. V. Shokhirev and F. A. Walker, Inorg. Chem., 2005, 44, 7468 CrossRef CAS PubMed
- K. Kano, H. Kitagishi, S. Tamura and A. Yamada, J. Am. Chem. Soc., 2004, 126, 15202 CrossRef CAS PubMed
- K. D. Karlin, A. Nanthakumar, S. Renard, N. N. Murthy, N. Ravi, B. Hanh Huynh, R. D. Orosz and E. P. Day, J. Am. Chem. Soc., 1994, 116, 4753 CrossRef CAS
- S. Renard, A. Nanthakumar, M. Wikström, K. D. Karlin and N. J. Blackburn, J. Am. Chem. Soc., 1996, 118, 24 CrossRef
- S. Hematian, I. Garcia-Bosch and K. D. Karlin, Acc. Chem. Res., 2015, 48, 2462 CrossRef CAS
- H. Kim, S. K. Sharma, A. W. Schaefer, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2019, 58, 15423 CrossRef CAS
- M. C. Carrasco, K. J. Dezarn, F. S. T. Khan and S. Hematian, J. Inorg. Biochem., 2021, 225, 111593 CrossRef CAS
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229 CrossRef CAS
- B. F. Van Gelder and H. Beinert, Biochim. Biophys. Acta, Bioenerg., 1969, 189, 1 CrossRef CAS PubMed
- W. J. Bowen, J. Biol. Chem., 1949, 179, 235 CrossRef CAS
- T. Hayashi, H. Dejima, T. Matsuo, H. Sato, D. Murata and Y. Hisaeda, J. Am. Chem. Soc., 2002, 124, 11226 CrossRef CAS
- M. Momenteau, B. Loock, C. Tetreau, D. Lavalette, A. Croisy, C. Schaeffer, C. Huel and J.-M. Lhoste, J. Chem. Soc., Perkin Trans. 2, 1987, 249 RSC
- K. Kano, H. Kitagishi, Y. Sone, N. Nakazawa and M. Kodera, Eur. J. Inorg. Chem., 2006, 4043 CrossRef CAS
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720 CrossRef CAS
-
W. Xing, G. Yin and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, Amsterdam, 2014, ch. 5, p. 171 Search PubMed
-
C. Dua, Y. Suna, T. Shena, G. Yina and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Amsterdam, Elsevier, 2014, ch. 7, p. 231 Search PubMed
-
W. Xing, M. Yin, Q. Lv, Y. Hu, C. Liu and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, Amsterdam, 2014, ch. 1, p. 1 Search PubMed
- R. Boulatov, J. P. Collman, I. M. Shiryaeva and C. J. Sunderland, J. Am. Chem. Soc., 2002, 124, 11923 CrossRef CAS
- A. W. Schaefer, M. A. Ehudin, D. A. Quist, J. A. Tang, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 4936 CrossRef CAS PubMed
- M. A. Ehudin, A. W. Schaefer, S. M. Adam, D. A. Quist, D. E. Diaz, J. A. Tang, E. I. Solomon and K. D. Karlin, Chem. Sci., 2019, 10, 2893 RSC
- J. P. Collman, C. J. Sunderland, K. E. Berg, M. A. Vance and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 6648 CrossRef CAS
- J. P. Collman, R. A. Decréau, Y. Yan, J. Yoon and E. I. Salomon, J. Am. Chem. Soc., 2007, 129, 5794 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, mass spectra, UV-visible titration with copper, cyclic voltammograms and parameters determined from Koutecky–Levich plots. See DOI: https://doi.org/10.1039/d4dt03188a |
| This journal is © The Royal Society of Chemistry 2025 |