
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The matere bond
Rosa M.
Gomila
 and
Antonio
Frontera
*
and
Antonio
Frontera
*
Department of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 6th January 2025
Abstract
This perpective delves into the emerging field of matere bonds, a novel type of noncovalent interaction involving group 7 elements such as manganese, technetium, and rhenium. Matere bonds, a new member of the σ-hole family where metal atoms act as electron acceptors, have been shown experimentally and theoretically to play significant roles in the self-assembly and stabilization of supramolecular structures both in solid-state and solution-phase environments. This perspective article explores the physical nature of these interactions, emphasizing their directionality and structural influence in various supramolecular architectures. Recent studies have expanded the understanding of matere bonds beyond classical metal–ligand coordination, highlighting their potential in crystal engineering and catalysis. This perspective article also examines the occurrence of matere bonds in biological systems, particularly within manganese-containing proteins, where they contribute to the structural integrity and catalytic activity. Theoretical and computational analyses, including molecular electrostatic potential surfaces and density functional theory, further elucidate the properties and applications of matere bonds, offering new insights for the design of advanced materials and biomimetic systems. This comprehensive overview underscores the versatility of matere bonds, paving the way for future innovations in supramolecular chemistry involving metals.
Introduction
Supramolecular chemistry,1 which extends beyond the traditional confines of covalent bond formation, is focused on the study of noncovalent interactions2 and the self-assembly3 processes that emerge from them. At the core of this discipline is crystal engineering,4 which leverages these interactions to design and synthesize new crystalline materials with tailored properties. Noncovalent forces, including hydrogen bonding,5 van der Waals interactions,6 π–π stacking,7 and electrostatic interactions,8 play crucial roles in the organization of molecular components within a crystal lattice. These interactions not only determine the structural integrity of supramolecular assemblies but also enable the creation of complex architectures that can be precisely tuned for various applications in materials science,9 catalysis,10 and molecular recognition.11 By harnessing these weak yet directional forces, researchers in supramolecular chemistry and crystal engineering continue to innovate in fields ranging from drug design to the development of novel functional materials.9–11The σ-hole concept has become a crucial framework for understanding and exploiting noncovalent interactions involving elements from groups 14 to 18.12 A σ-hole is an electron-deficient region that occurs along the extension of a covalent bond, leading to an anisotropic distribution of electron density and, in most cases, creating a region of positive electrostatic potential. This phenomenon is particularly prominent in heavier p-block elements, such as germanium and tin in group 14 (tetrel bonds),13 arsenic and antimony in group 15 (pnictogen bonds),14 selenium and tellurium in group 16 (chalcogen bonds),15 and bromine and iodine in group 17 (halogen bonds),16 all of which act as σ-hole donors. σ-Hole interactions, including halogen, chalcogen, pnictogen and tetrel bonding, are pivotal in supramolecular chemistry and crystal engineering, providing directionality and specificity in molecular assembly processes.17 These interactions are integral to the formation of highly ordered supramolecular structures and are increasingly being used in the design of functional materials, molecular recognition systems, and supramolecular catalysts. In catalysis, σ-hole interactions contribute to the stabilization of transition states and intermediates, enhancing both catalytic efficiency and selectivity.18 As our understanding of σ-hole interactions deepens, their applications continue to expand, offering new avenues for innovation in the design and synthesis of advanced materials and catalysts.19
Furthermore, σ- and π-hole interactions involving post-transition elements of groups 11 and 12, known as regium and spodium bonds,20,21 have gained significant attention, particularly from a computational perspective and for their unique role in crystal engineering.22 Regium bonds involve elements such as copper, silver, and gold (group 11),23 while spodium bonds are formed by zinc, cadmium, and mercury (group 12).24
These interactions arise from electropositive regions on the surface of these metal atoms, often located opposite a metal–ligand bond or perpendicular to a molecular framework (e.g., square planar Au(III) or trigonal Hg(II) complexes), leading to directional noncovalent interactions with electron-rich species such as lone pairs, anions, or π-electron systems. Regium and spodium bonds contribute to the stabilization and structural integrity of supramolecular assemblies, offering novel pathways for designing complex architectures with enhanced functionality.20–24 These interactions are particularly important in the development of materials with unique electronic, optical,25 and catalytic properties.26 As research in this area progresses, the ability to use these interactions opens new possibilities in the design of innovative supramolecular systems with applications across various fields, from materials science to molecular electronics.
Noncovalent interactions involving transition metals are a pivotal aspect of supramolecular chemistry, significantly contributing to the diversity and functionality of supramolecular assemblies.27 Transition metals, with their unique electronic configurations and variable oxidation states, participate in a wide range of noncovalent interactions, such as metal–π interactions28 and metallophilic interactions.29 In crystal engineering, these metal-mediated interactions allow precise control over molecular geometry and packing, facilitating the development of materials with tailored properties.30 In supramolecular catalysis, transition metal interactions are employed to activate substrates, mediate electron transfer, and stabilize reaction intermediates, thereby enhancing the efficiency and selectivity of catalytic processes.31 As we gain a deeper understanding of these interactions, their potential applications broaden, unlocking fresh avenues for innovation in materials science and molecular design.
Sigma-hole interactions involving metals are another critical component of supramolecular chemistry, especially in the context of specialized noncovalent bonds such as osme,32 matere,33 wolfium,34 and erythronium bonds.35 These interactions are characterized by electron-deficient regions (sigma-holes) on the metal atoms, enabling directional and specific noncovalent interactions with electron-rich species. For instance, osme bonds, primarily involving osmium, significantly influence the geometry and stability of cocrystals, particularly in complexes where osmium tetroxide interacts with nitrogen or oxygen nucleophiles, leading to short and linear Os⋯N/O contacts.32 Matere bonds, observed in compounds containing manganese, technetium, and rhenium, contribute to the stabilization of supramolecular structures and play a crucial role in fine-tuning catalytic properties.35b The study of these metal-centered σ-hole interactions continues to expand the possibilities for designing new materials with tailored properties.
It is worth noting that the diverse range of noncovalent interactions involving heavier main group elements and transition metals has been referred to as “secondary bonding” in the literature for many years. The term “secondary-bonding interactions” was first introduced by Alcock in 1972.36 to describe interactions that are longer than typical single bonds but shorter than the sum of van der Waals radii. These interactions have also been described using various terms, including soft–soft, closed-shell, nonbonding, semi-bonding, non-covalent, weakly bonding, and σ-hole interactions.
This perspective article provides a comprehensive overview of recent advances in the study of matere bonds, focusing on their physical nature and the progress made in understanding their role in both solid-state and solution-phase systems. We begin by exploring the fundamental principles governing matere bond formation, including the electronic and structural factors that contribute to their strength and directionality. This foundational understanding sets the stage for a detailed discussion of how matere bonds have been harnessed in various supramolecular architectures, with a particular emphasis on their applications in solid-state chemistry. The perspective article then shifts focus to the stability of matere bonds in solution, as proposed by DFT calculations. By examining these recent developments, this perspective article aims to shed light on the versatility and potential of matere bonds in advancing the field of supramolecular chemistry, offering insights that could pave the way for future innovations in material design, catalysis, and molecular recognition.
Discussion
Physical nature
Scheme 1a illustrates a comparison between σ-holes in typical p-block elements and those in the MaO4− molecule (Ma = matere atom). In elements of groups 17 to 15, the anisotropy of electron density arises inherently from the element itself, due to the coexistence of lone pairs and σ-holes within the same atom. The number of σ-holes increases from 1 to 3, while the number of lone pairs decreases from 3 to 1 as one moves from group 17 (halogens) to group 15 (pnictogens). In contrast, the anisotropy in the MaO4− anion results from the proximity of the oxygen atoms and their tetrahedral arrangement around the metal center, rather than being an intrinsic property of the matere atom. This distinction underscores a key difference between halogen, chalcogen, and pnictogen bonding compared to matere bonds.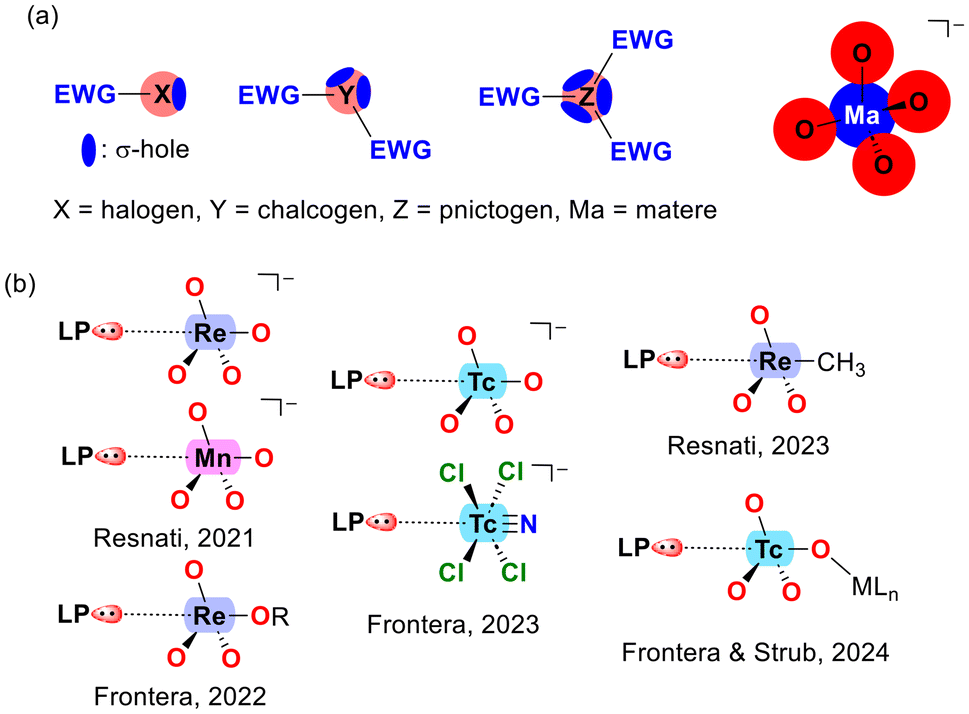 | ||
| Scheme 1 (a) Comparison of typical σ-hole distribution in elements from groups 17 to 15 with the anisotropy of electron density in MaO4− anions. (b) Previously reported noncovalent donor–acceptor interactions in matere derivatives. | ||
As mentioned earlier, matere bonds were initially described in the context of permanganate, pertechnetate, and perrhenate anions.33 However, subsequent research has extended the concept to include other oxidation states and geometries, as illustrated by the [TcNCl4]− compound shown in Scheme 1b.37
To understand the geometric requirements of matere bonds, visualizing molecular electrostatic potential (MEP) surfaces is an invaluable computational tool. Fig. 1 presents the MEP surfaces of several model systems. For the naked MaO4− anions (Ma = Mn, Tc, Re), the electron density distribution is anisotropic, revealing four regions (σ-holes) where the MEP is at a maximum. These negative σ-holes help explain the geometry adopted by the anions as they approach each other, working to minimize electrostatic repulsion. However, when the anion is coordinated to a metal center, such as Mg2+, the MEP at the σ-hole becomes positive, as demonstrated by the MgCl(ReO4) model system shown in Fig. 1b.
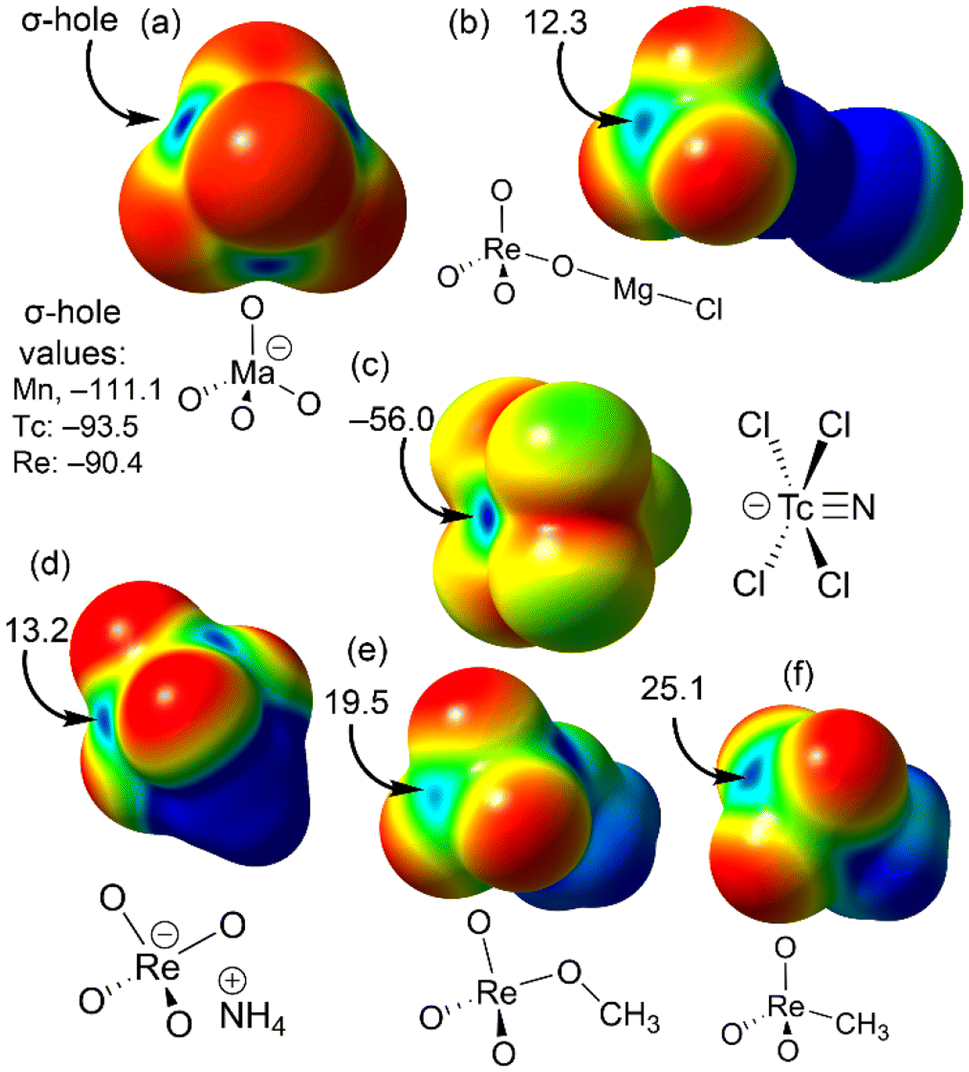 | ||
| Fig. 1 MEP surfaces of permanganate, pertechnetate and perrhenate (a), perrhenate coordinated to Mg2+ (MgCl(ReO4)) (b), [TcNCl4]− (c), perrhenate ammonium salt (d), perrhenate methyl ester (e) and methyltrioxorhenium(VII) (f). Energies in kcal mol−1. Red is used to represent nucleophilic regions and blue for electrophilic ones. | ||
Other rhenium(VII) compounds with positive σ-holes are illustrated in Fig. 1d–f. For instance, in the perrhenate ammonium salt, electron transfer from the anion to the cation dramatically impacts the σ-hole value, shifting from −90.4 kcal mol−1 in the naked anion to +13.2 kcal mol−1 in the salt. The perrhenate methyl ester exhibits positive σ-holes at the metal center, with a value of +19.5 kcal mol−1 opposite the Re–OCH3 bond. Similarly, methyltrioxorhenium(VII), shown in Fig. 1f, has a σ-hole value of +25.1 kcal mol−1 opposite the Re–CH3 bond. Fig. 1c depicts the MEP surface of the [TcNCl4]− anion, where negative values dominate, as expected. However, a σ-hole opposite the Tc![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, corresponding to the MEP maximum (−56 kcal mol−1), is clearly visible. This suggests that an electron-rich atom would likely approach opposite the TcN bond. Overall, the MEP surfaces in Fig. 1 indicate that matere bonds should exhibit strong directionality, as the σ-holes are small and flanked by electronegative atoms, necessitating precise alignment of the nucleophile along the σ-hole direction.
N bond, corresponding to the MEP maximum (−56 kcal mol−1), is clearly visible. This suggests that an electron-rich atom would likely approach opposite the TcN bond. Overall, the MEP surfaces in Fig. 1 indicate that matere bonds should exhibit strong directionality, as the σ-holes are small and flanked by electronegative atoms, necessitating precise alignment of the nucleophile along the σ-hole direction.
In addition to MEP surfaces, matere bonds are often characterized using Quantum Theory of Atoms in Molecules (QTAIM)38 and Noncovalent Interaction (NCI) plots.39 The presence of a bond critical point (BCP) and bond path connecting the nucleophilic atom with the group 7 element is a clear indication of interaction, whether attractive or repulsive. QTAIM analysis is typically paired with NCIplot analysis, which visualizes noncovalent contacts by representing regions of reduced density gradients (RDG). The RDG isosurfaces are colored according to the sign of the second Hessian eigenvalue (λ2), with negative values indicating attractive interactions and positive values indicating repulsive interactions. The color scale used commonly ranges from blue (strongly attractive) to green (weakly attractive), and from red (strongly repulsive) to yellow (weakly repulsive). Fig. 2 presents the QTAIM/NCIplot analysis of two matere anion⋯anion dimers, representing examples from the literature33,37 on Re and Tc. In both cases, a BCP (depicted as a small pink sphere) and a bond path (shown as dashed lines) connect the rhenium or technetium with the lone pair donor atom, confirming the existence of matere bonds. Additionally, green RDG isosurfaces, coinciding with the BCPs, further characterize the matere bonds. Notably, in the case of the tetrahedral ReO4− anion, part of the RDG isosurface appears yellow, indicating O⋯O repulsion, which correlates with the reduced accessibility of the σ-hole.
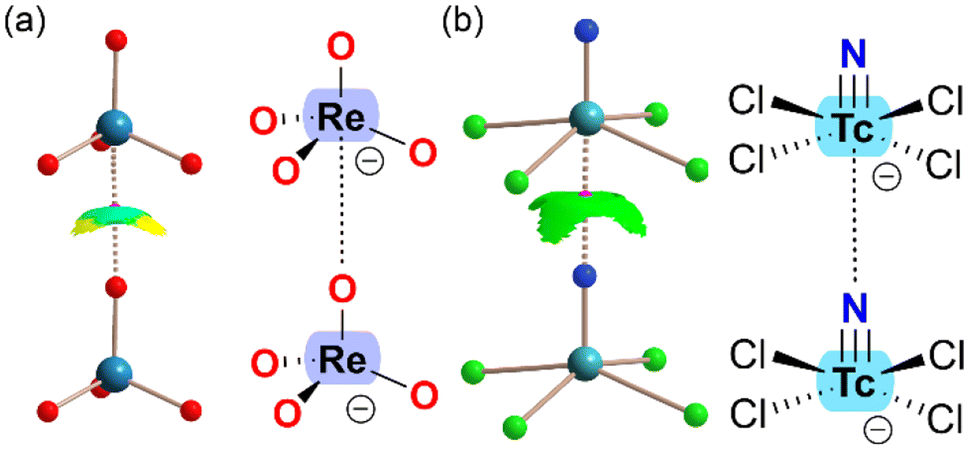 | ||
| Fig. 2 Combined QTAIM (BCPs in pink, bond paths as dashed bonds) and NCIplot analysis of the anion⋯anion dimers of ReO4− (a) and [TcNCl4]− (b). | ||
It is noteworthy that the dimerization energies are generally repulsive due to the strong coulombic repulsion between the anions. However, the matere bond itself is attractive, as evidenced by the negative sign of λ2 in both dimers.
Another valuable computational tool for analyzing matere bonds is natural bond orbital (NBO) analysis,40 which characterizes the interaction from an orbital perspective and provides insights into charge-transfer effects. NBO analysis also helps confirm the σ-hole nature of the interaction. Unlike typical coordination bonds, where the d-orbitals of the metal center participate in metal–ligand bonding, noncovalent interactions involve the antibonding σ-orbital as the acceptor and the lone pair (LP) of the nucleophile as the donor orbital. Fig. 3 illustrates two examples evidencing the LP → σ* nature of the matere bond.
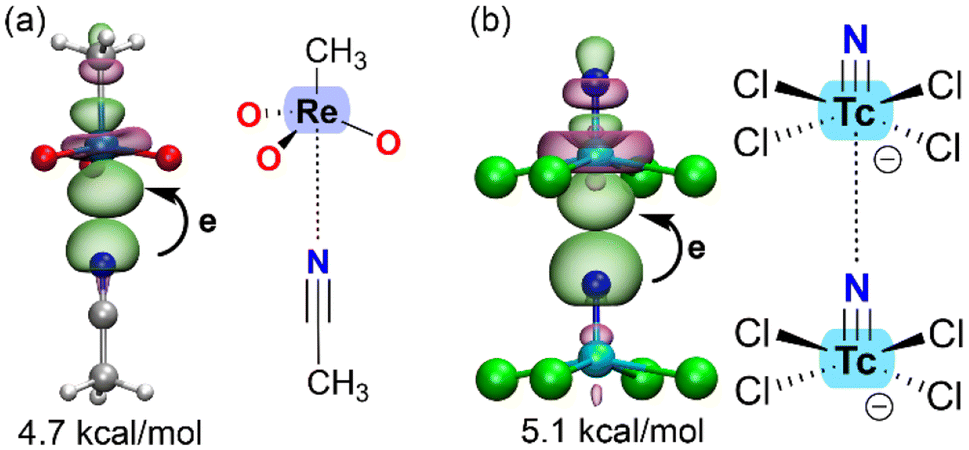 | ||
| Fig. 3 Plot of the NBOs involved in the donor–acceptor interaction in ReO3CH3⋯acetonitrile (a) adduct and [TcNCl4]− dimer (b) with indication of the E(2) energies. | ||
In addition to NBO analysis, another effective methodology for identifying the electron donor and acceptor atoms in any pairwise interaction involves examining the electron density (ED) and electrostatic potential (ESP) functions along the bond path connecting the two atoms.41 It has been demonstrated that the minimum of the electron density function is displaced toward the electron acceptor atom, while the minimum of the electrostatic potential function is displaced toward the electron donor atom. Fig. 4 presents two examples corresponding to the same complexes shown in Fig. 3, allowing for direct comparison. In both cases, the ReO3CH3⋯ACN adduct and the [TcNCl4]− dimer, the minimum of the ESP is displaced toward the nitrogen atom, and the ED minimum (position of the bond critical point, BCP) is displaced toward the transition metal. This confirms the electron donor role of the nitrogen atom and the electron acceptor role of the transition metal. It is worth noting that this computational tool is particularly useful in confirming the roles of interacting atoms in anion⋯anion complexes, where the role of the transition metal as an electron acceptor might seem counterintuitive.
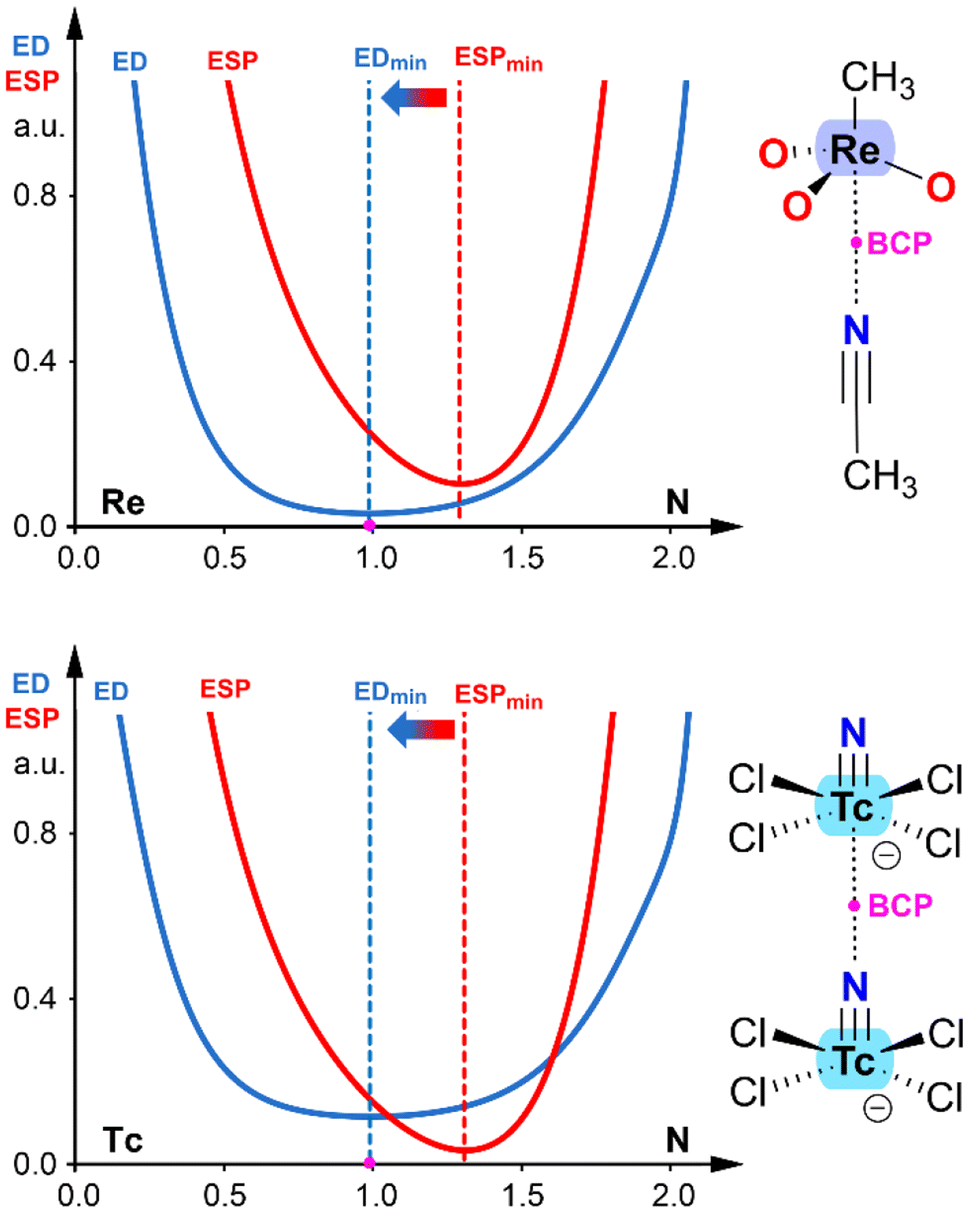 | ||
| Fig. 4 ED vs. ESD plots in ReO3CH3⋯ACN adduct (top) and [TcNCl4]− dimer (bottom). The plots are computed along the bond path connecting the N-atom to the metal. | ||
Matere bond in crystal engineering
In a 2021 study by Daolio et al.,33 the concept of matere bonds was introduced as a novel type of noncovalent interaction involving group 7 tetroxide anions, such as perrhenate, pertechnetate, and permanganate. The research provided both experimental and theoretical evidence supporting the existence of these σ-hole interactions, where the metal atom in the Mn/ReO4− anion acts as an electron acceptor. The study utilized single-crystal X-ray analyses, MEP surfaces, QTAIM, and NCI plots to demonstrate that oxygen atoms from adjacent anions can act as electron donors, forming supramolecular anionic dimers or polymers. This discovery expanded the understanding of σ-hole interactions and introduced the term “matere bond” to categorize these interactions, distinguishing them from classical metal–ligand coordination bonds.Fig. 5 illustrates the X-ray structures reported by Daolio et al.,33 highlighting the geometric characteristics of matere bonds. In the MnO4− salt (NAPWAG), self-assembled anion dimers are formed, with two Mn⋯O matere bonds observed, measuring 3.447 Å, significantly longer than typical coordination bonds. In the NAPWEK structure, the matere bond is notably directional, with an angle of 177.1°, formed between the rhenium (Re) atom of the ReO4− anion and the oxygen atom of the carboxylic group in the betaine counterion. In perrhenate melaminium salt (NAPVUZ), the nucleophile is the oxygen atom of another anion and the Re⋯O matere bond forms quite linear polyanionic infinite chains (the O–Re⋯O angle is 177.7°).
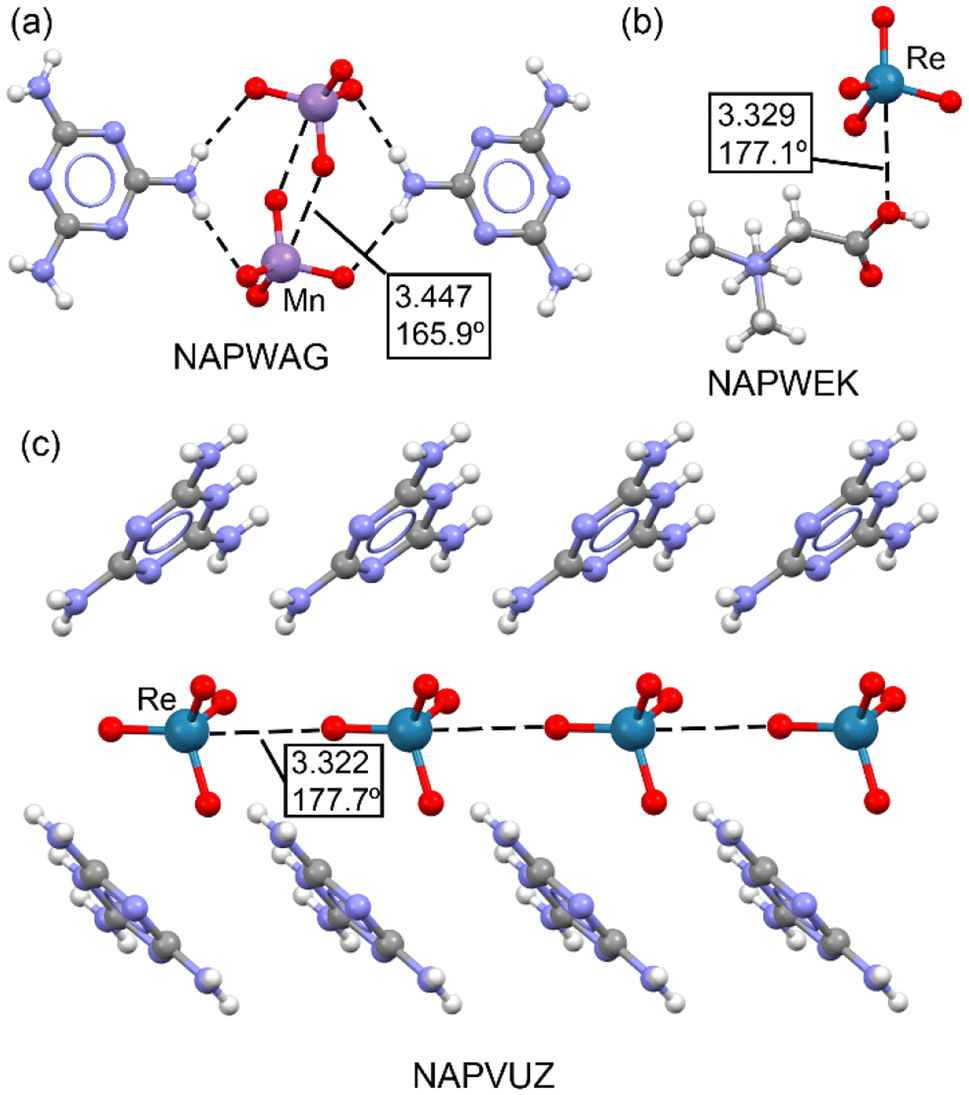 | ||
| Fig. 5 Partial view of the X-ray structures of NAPWAG (a), NAPWEK (b) and NAPVUZ (c). Distances in Å. | ||
This study is particularly relevant as it underscores the significance of matere bonds in forming anion–anion interactions strong enough to drive the self-assembly of supramolecular structures, even in the presence of electrostatic repulsion. The researchers observed that heavier elements, such as rhenium, form shorter and more directional matere bonds compared to lighter elements like manganese, consistent with general trends in σ-hole interactions. Additionally, theoretical calculations suggested that matere bonds might exist in solution, particularly at high concentrations, despite the repulsive forces between like-charged anions.
In a 2022 study, Gomila and Frontera42 explored the concept of matere bonds in comparison with multivalent halogen and chalcogen bonds through three case studies (see Scheme 2). The study employed X-ray crystallography and DFT calculations to analyze matere bonds, using a neutral Re(VII) system as the matere bond donor, rather than the perrhenate anion used in previous work. This research compared matere bonds with well-established σ-hole interactions, such as halogen and chalcogen bonds, highlighting both the structural similarities and the unique electronic properties of the σ-holes involved. The σ-hole nature of the matere bonds in the three examples from Scheme 2 was confirmed using various computational tools, including MEP surfaces, NCI plots, and NBO analysis, underscoring their significance in the solid state.
 | ||
| Scheme 2 Self-assembled dimers dictated by matere (HETRUT),43 chalcogen (WABPAR)44 and halogen (KUCRAD)45 bonds. The dimerization energies are indicated. | ||
This study further demonstrates that matere bonds in neutral Re(VII) compounds are structure-directing and have a strength comparable to multivalent and chalcogen bonds, and stronger than halogen bonds (see Scheme 2). The study also demonstrated that the σ-holes present opposite to the Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Re–O bonds significantly contribute to the directionality of the interaction, dictating the formation of two-dimensional assemblies. This research further shows that these interactions are not merely a consequence of crystal packing but have an inherent structural influence, as evidenced by the consistency between the experimental and theoretical geometries.
O and Re–O bonds significantly contribute to the directionality of the interaction, dictating the formation of two-dimensional assemblies. This research further shows that these interactions are not merely a consequence of crystal packing but have an inherent structural influence, as evidenced by the consistency between the experimental and theoretical geometries.
In a 2023 study by Burguera et al.,37 the significance of matere bonds in technetium compounds across various oxidation states (+7, +6, +5, and +3) was thoroughly explored. The study extends the concept of matere bonds, initially associated with rhenium and manganese in the +7 oxidation state, to technetium derivatives. Using a combination of the Cambridge Structural Database (CSD) and density functional theory (DFT) calculations, the researchers demonstrated the structure-directing role of matere bonds in X-ray structures of technetium, particularly in cases involving anion–anion interactions. Some of these X-ray structures are illustrated in Fig. 6. For instance, the solid-state structure of AZEMUO46 (see Fig. 6a) revealed several N–H⋯O hydrogen bonds (HBs) connecting the pertechnetate to pyridinium cations, similar to the NAPWAG perrhenate structure in Fig. 5a. In addition to these charge-assisted HBs, linear O–Tc⋯O contacts were established. Specifically, two symmetrically equivalent O–Tc⋯O matere bonds form a parallelepiped-shaped dimer with an O–Tc⋯O angle of 168.9° and a distance almost identical to the sum of the van der Waals radii [∑RvdW(Tc + O) = 3.60 Å].
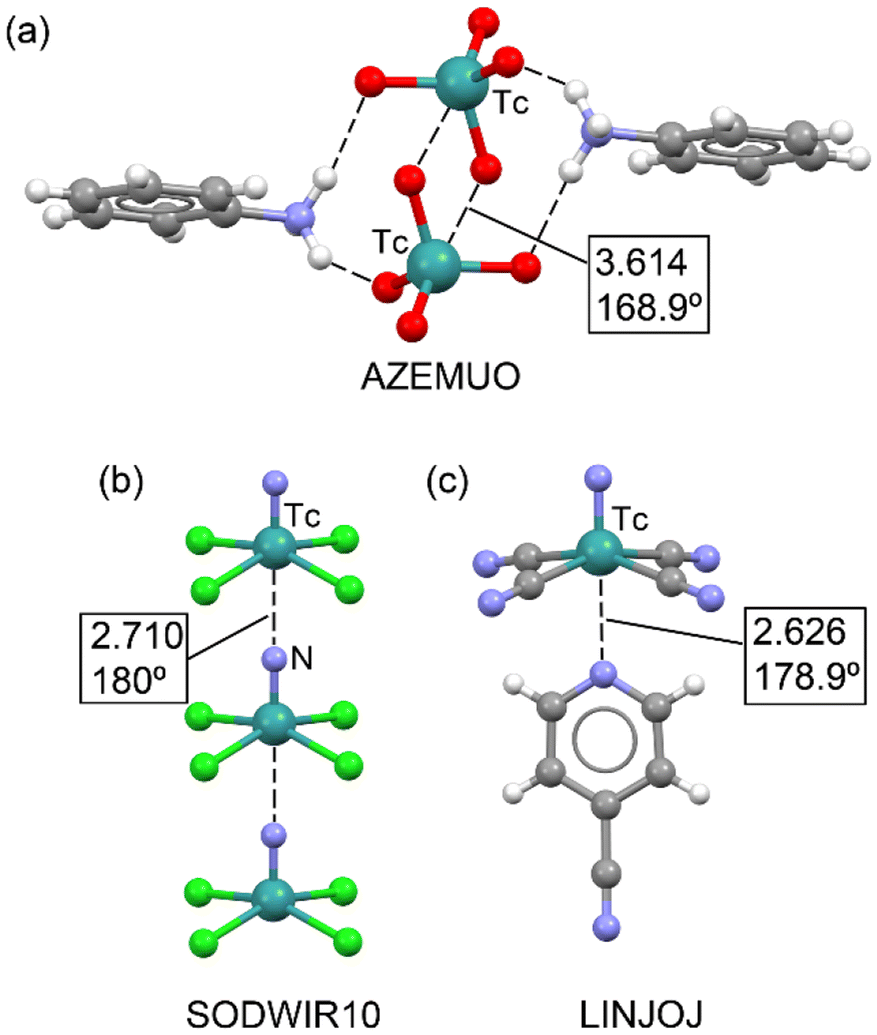 | ||
| Fig. 6 Partial view of the X-ray structures of AZEMUO (a), SODWIR (b) and LINJOJ (c). Distances in Å. | ||
An interesting structure is represented in Fig. 6b, where anion⋯anion matere bonds propagate the [TcNCl4]− anion into an infinite 1D assembly.47 In this example, the lone pair on the N-atom of one tetrachloro-nitrido-technetate anion points to the Tc-atom of the next anion, opposite the TcN bond. This 1D assembly is flanked by catena-((μ2-18-crown-6)-cesium) counterions, which are not shown for clarity. Similar 1D anion⋯anion assemblies have recently been observed in nitrido-osmium(VI) complexes,32b which present similar square-pyramidal geometries. Another noteworthy X-ray structure is shown in Fig. 6c, where tetracyanidonitridotechnetium(V) interacts with 4-cyanopyridine (refcode LINJOJ),48 establishing a directional Tc⋯N matere bond. In this case, the dianion [TcN(CN)4]2− acts as an electron acceptor. In the original work, the authors considered the Tc atom to be six-coordinated, with the 4-cyanopyridine as an axial ligand. However, the Tc⋯N separation is longer than the sum of the covalent radii of technetium and nitrogen atoms (∑Rcov = 2.18 Å), and the N(nitride)–Tc–C angles are approximately 100°, supporting the predominantly noncovalent nature of the Tc⋯N(py) interaction. This assumption was further confirmed theoretically using QTAIM and NCIplot analyses.
The study concludes that matere bonds play a crucial role in directing the structure of technetium compounds, even across different oxidation states. The research provides compelling evidence that these noncovalent interactions significantly influence the assembly of supramolecular structures, particularly through anion–anion interactions. This work not only broadened the scope of matere bonds beyond rhenium but also opened new avenues for their application in materials science and coordination chemistry.
In a 2023 study by Calabrese et al.,35 the role of σ-hole interactions in the catalytic behavior of methyltrioxorhenium(VII) (MTO), a widely used organometallic catalyst, was investigated. The research highlights how Re⋯N/O matere bonds contribute to the catalytic efficiency of MTO in various reactions, including olefin epoxidation. The study combines experimental data from X-ray crystallography with theoretical analyses to demonstrate the presence and significance of these interactions. Four crystalline adducts of MTO with nitrogen and oxygen Lewis bases were synthesized and characterized, revealing short Re⋯N/O contacts consistent with strong σ-hole interactions. These findings suggest that the catalytic performance of MTO is not solely dependent on its traditional coordination chemistry but is also significantly influenced by noncovalent interactions, which enhance the stability and reactivity of the catalyst under various conditions.
Fig. 7 presents the X-ray structures of three representative co-crystals, corresponding to trimeric adducts assembled via short Re⋯N/O matere bonds. In co-crystals XEXWIK and XEXXEH, these adducts are assembled through Re⋯N matere bonds formed opposite to the H3C–Re bond (Fig. 7a and c). These interactions are much shorter than the ∑RvdW of the involved atoms (∼2.45 Å). The C–Re⋯N angles are almost linear, as is typical in σ-hole interactions. Co-crystal XEXWOQ displays the matere bond at the extension of an O–Re bond, with a much shorter distance (2.286 Å), consistent with the higher electronegativity of the O-atom compared to the methyl group. In XEXWOQ, the O–Re⋯O angle (169.2°) is less linear than in the other two cases. Importantly, the formation of these adducts does not result in a pronounced distortion of the O–Re–O angles, so the tetrahedral geometry is substantially maintained.
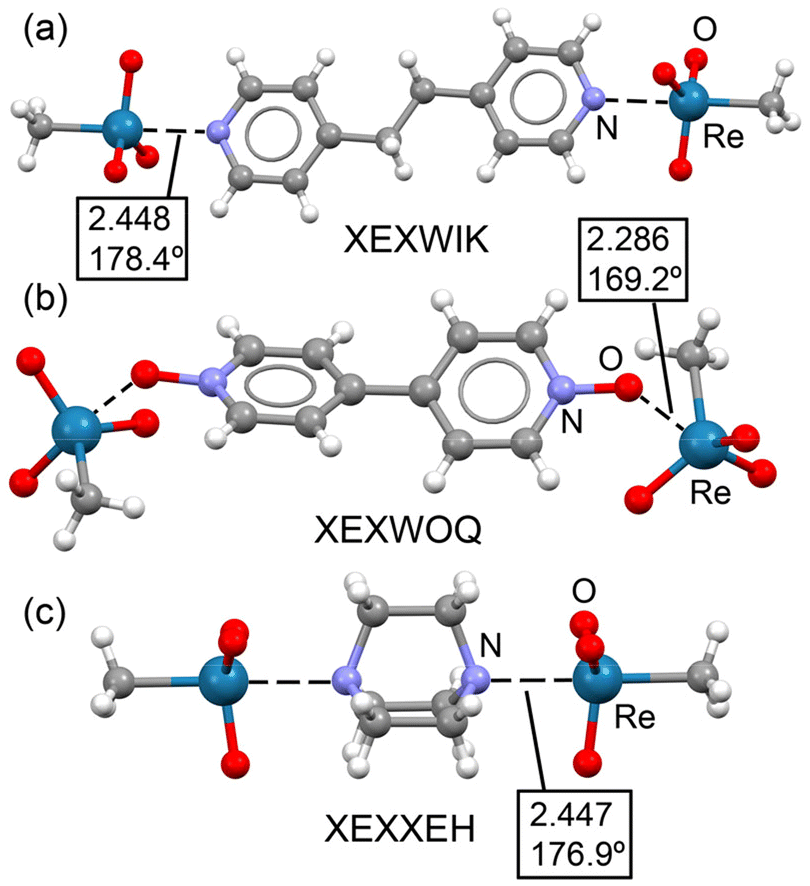 | ||
| Fig. 7 Partial view of the X-ray structures of XEXWIK (a), XEXWOQ (b) and XEXXEH (c). Distances in Å. | ||
The conclusions drawn from this study underscore the crucial role of σ-hole interactions in the catalytic mechanisms of methyltrioxorhenium(VII) (MTO). The Re⋯N/O matere bonds observed in the crystalline adducts play a key role in stabilizing the catalyst and promoting selective self-assembly processes, even in competitive environments where other noncovalent interactions are present. DFT analysis supported the σ-hole nature of these matere bonds, revealing significant orbital contributions that suggest partial covalent character, consistent with the short matere bond distances. This research indicates that these interactions are not merely structural artifacts but are fundamental to the catalytic activity of MTO, particularly in processes such as olefin epoxidation, where ligand-accelerated catalysis is critical.
In 2023, Xu et al.49 conducted an in-depth investigation of non-covalent matere bonds in a series of organic perrhenates, utilizing advanced spectroscopic techniques such as ultrahigh-field rhenium-185/187 solid-state NMR (SSNMR) and zero-field nuclear quadrupole resonance (NQR). The study aimed to directly probe and characterize matere bonds in perrhenates, where the rhenium atom acts as an electron acceptor in a σ-hole interaction. The researchers focused on three perrhenate salts, i.e. melaminium perrhenate, betaine perrhenate, and methylimidazolium perrhenate, known to exhibit matere bonds (see some of them in Fig. 5), and compared them with two control samples, pyridinium perrhenate and acetylcholine perrhenate, which do not exhibit these interactions.
The findings revealed that the quadrupolar coupling constants of rhenium are lower in the matere-bonded samples compared to the control samples, suggesting a significant influence of matere bonds on the electronic environment of the rhenium centers. Additionally, the study shows that matere bonds contribute to structural distortions in the ReO4− tetrahedra, which in turn affect the observed NMR parameters. Despite the complexity of the spectra due to large quadrupolar interactions, the researchers successfully demonstrated the potential of NMR and NQR spectroscopy as powerful tools for studying non-covalent interactions, even in systems with strong quadrupolar coupling. The study provided new insights into the role of matere bonds in determining the structural and electronic properties of perrhenates, paving the way for further exploration of these interactions in other systems using NMR and NQR instead of more conventional X-ray analysis.
In 2024, Grödler et al.50 investigated the structural and electronic properties of matere bonds in a series of newly synthesized pertechnetate complexes with the general formula [M(H2O)4(TcO4)2], where M = Mg, Co, Ni, Cu, or Zn. The study also included a perrhenate compound, [Zn(H2O)4(ReO4)2], for comparative purposes. These complexes feature octahedral coordination geometries, with pertechnetate or perrhenate ions serving as axial ligands. The key finding is that these complexes exhibit strong and directional Tc⋯O matere bonds, which play a crucial role in forming one-dimensional (1D) supramolecular polymers in the solid state (see Scheme 3, green-colored parallelepipeds). These 1D polymers are further linked into two-dimensional (2D) layers through additional matere bonds (see Scheme 3, pink parallelepipeds), highlighting the structure-directing influence of matere bonds in these systems.
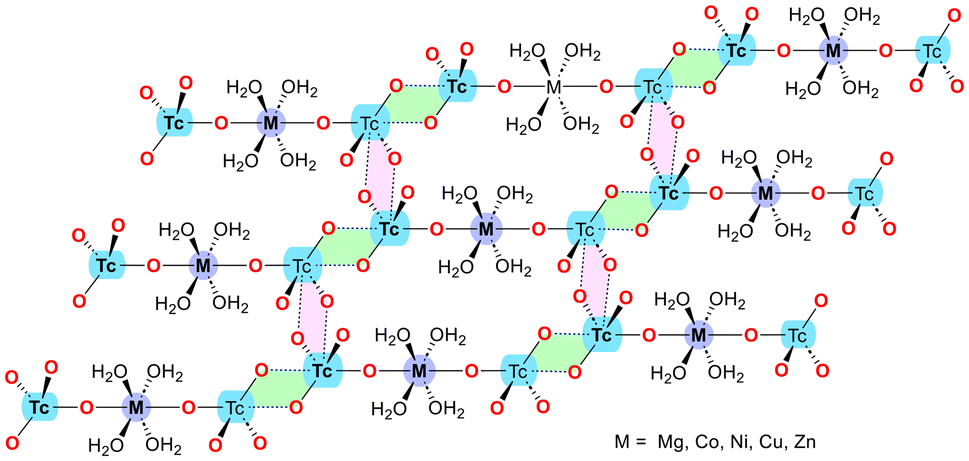 | ||
| Scheme 3 Schematic representation of the 2D layer observed in the solid state of [M(H2O)4(TcO4)2] compounds, where M = Mg, Co, Ni, Cu, Zn. | ||
The study utilized various theoretical and computational approaches to characterize the nature of these interactions. The results revealed that matere bonds are best described as σ-hole interactions, with their strength varying based on the metal center, which influences the σ-hole intensity at the Tc or Re atoms.
A recent study investigated the formation and characteristics of matere bonds in four newly synthesized pentacoordinated manganese(III) complexes.51 These complexes, derived from Schiff base ligands, feature chloride, azide, or thiocyanate ligands in the apical position. The research combined single-crystal X-ray diffraction, Hirshfeld surface analysis, and density functional theory (DFT) calculations to explore the supramolecular assemblies in the solid state and characterize the Mn⋯O interactions. These interactions, identified as matere bonds, are noncovalent interactions driven by σ-hole phenomena, with the manganese atom acting as an electron acceptor. This type of interaction is unprecedented in square pyramidal manganese complexes and highlights the unique behavior of group 7 elements in forming directional and noncovalent bonds that influence the self-assembly of these complexes into supramolecular dimers.
The study also emphasized that matere bonds significantly contribute to the stabilization of supramolecular dimers, with interaction energies ranging from −8 to −11 kcal mol−1, depending on the nature of the axial ligand (see Fig. 8). While these bonds are weaker than coordination bonds, they play a crucial role in defining the structural arrangement of the complexes. These findings open up new avenues for the design of supramolecular materials using noncovalent interactions involving pentacoordinated manganese(III) complexes.
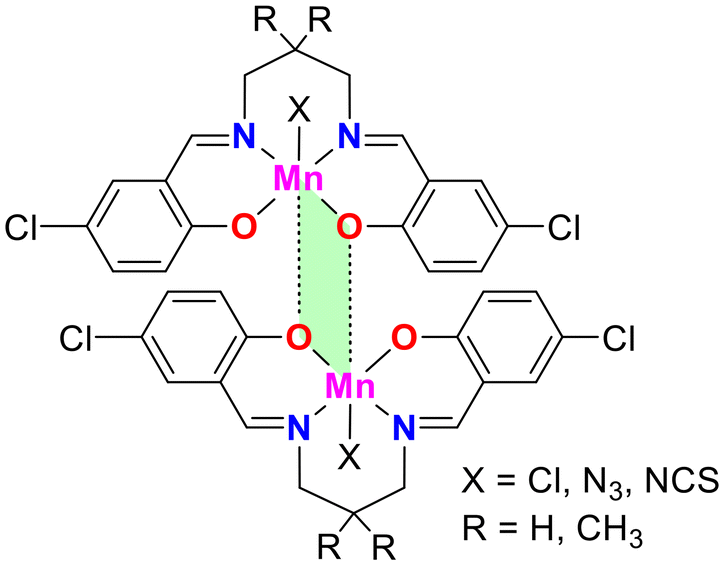 | ||
| Fig. 8 Representation of the self-assembled observed in the solid state of pentacoordinated Mn(III) compounds, where X = Cl, N3, NCS. | ||
In 2024, Burguera et al.52 investigated the occurrence and characteristics of matere bonds involving manganese centers within biological systems. The research was based on an extensive search of the Protein Data Bank (PDB) and complemented by DFT calculations. The study identified noncovalent interactions between Mn centers and protein residues, particularly focusing on interactions with tyrosine (TYR), serine (SER), and histidine (HIS) residues. These interactions are characterized as matere bonds, where the Mn atoms serve as σ-hole donors, interacting with electron-rich lone pairs on oxygen or nitrogen atoms from the amino acids.
One selected example, shown in Fig. 9, corresponds to the pyruvate carboxylase (PC) enzyme from Staphylococcus aureus, as studied by Xiang and collaborators.53 PC catalyzes the biotin-dependent production of oxaloacetate, playing a key role in gluconeogenesis and other cellular processes. It consists of biotin carboxylase (BC), carboxyltransferase (CT), and biotin-carboxyl carrier protein (BCCP) domains. The CT domain, which contains a bound Mn2+ ion, facilitates the transfer of an activated carboxyl group to pyruvate, forming oxaloacetate. This Mn2+ ion is coordinated to HIS771, HIS773, LYS741, and ASP572 residues in a seesaw configuration. A nearby pyruvate molecule interacts noncovalently with the Mn2+ ion, specifically through the oxygen of its carbonyl group at a distance of 3.371 Å, with an interaction angle of 161.1°.
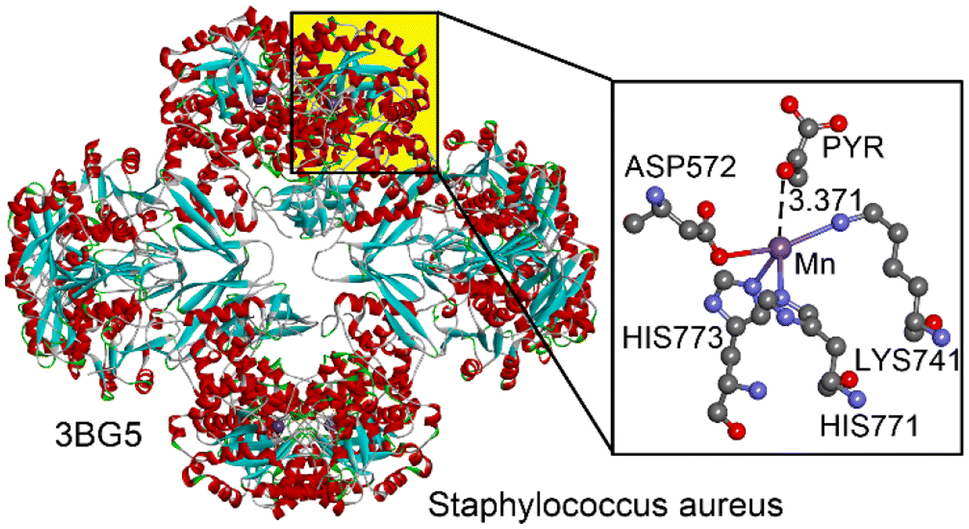 | ||
| Fig. 9 Left: Ribbon representation of the PDB structure 3BG5. Right: Amplification of the active site with indication of the matere bond interaction between a Mn(II) centre and a pyruvate (PYR) molecule. Distance in Å. | ||
The overall findings combining the PDB search and theoretical calculations revealed that matere bonds in proteins are geometrically distinct compared to those in the X-ray structures shown in Fig. 5. Regardless, the analysis of the entire PDB, focusing on preferred distances and angles, suggests that matere bonds play a significant role in stabilizing protein structures. The study particularly highlights the presence of matere bonds in various biologically relevant Mn-containing proteins, including manganese superoxide dismutase (MnSOD), where these bonds may influence the enzyme's catalytic activity and stability. Theoretical analyses further supported the noncovalent nature of these interactions, demonstrating that matere bonds contribute to the overall supramolecular architecture of proteins. This research provides valuable insights into the role of transition metal-based noncovalent interactions in bioinorganic chemistry, with implications for understanding protein–metal interactions and designing biomimetic materials.
Matere bond in solution
Several interactions enable the formation of anion⋯anion adducts by effectively balancing the coulombic repulsion between like charges. For instance, it has been shown that the attractive force localized in the hydrogen bond region between protic hydroxyanions (e.g., HCO3−, HSO4−, and H2PO4−) facilitates the formation of dimers that can be stable.54 Additionally, halogen bonding is responsible for the presence of infinite chains in crystalline solids formed through anion self-assembly.55 The stability of anion⋯anion adducts formed by polyatomic anions from groups 2, 11–13, 15, and even 18 has been explained by the anisotropic distribution of electron density within the anions.56For Group 7, two anion⋯anion dimers have been analyzed in the literature. One example is the ReO4− dimer,33 with the computed energy profile in water (interaction energy vs. distance) shown in Fig. 10 (top). For comparison, the energy profile in the gas phase is also included. In the absence of counterions, the dimer is not stable in the gas phase, and the monomers separate to infinity (inset graph in Fig. 10, top). However, in water, the ReO4−⋯ReO4− dimer is energetically favorable, as the two separate monomers are less stable than the dimer by 0.7 kcal mol−1. The minimum occurs at a distance of approximately 4 Å, with a small barrier of 1.4 kcal mol−1. These results suggest that the electrostatic repulsion in anion⋯anion interactions can be balanced by a suitable environment, allowing these interactions to exist in solution even without counterion participation, as demonstrated in saturated phosphate solutions.
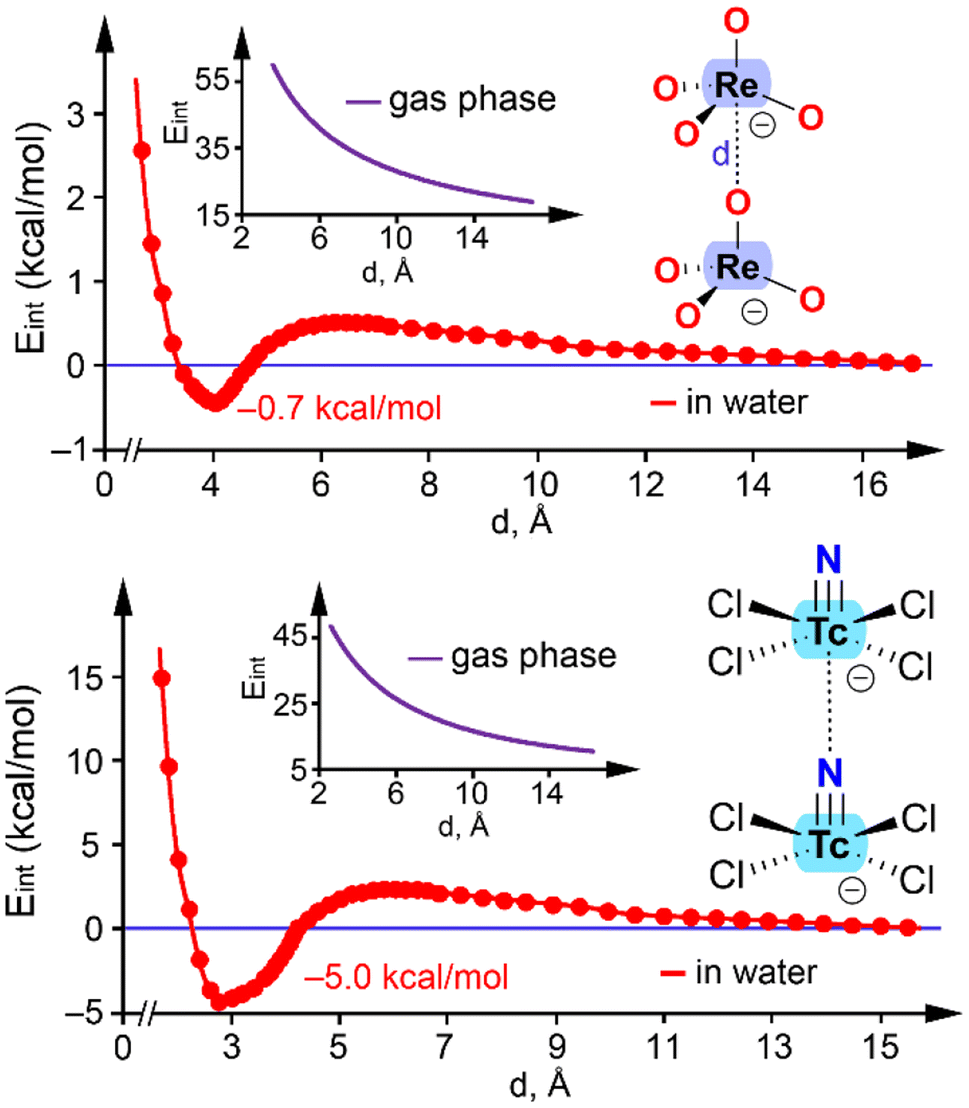 | ||
| Fig. 10 Energetic profiles obtained by varying the interatomic distance (d) in the ReO4−⋯ReO4− (top) and [TcNCl4]−⋯[TcNCl4]− (bottom) dimers at the PBE0-D3/def2-TZVP level of theory. | ||
The second example corresponds to the [TcNCl4]− dimer (Fig. 10, bottom).37 Similar to the ReO4− case, the dianionic [TcNCl4]−⋯[TcNCl4]− dimer is not stable in the gas phase, and the monomers separate to infinity. It is important to note that the [TcNCl4]−⋯[TcNCl4]− polymeric chain in the solid state is stabilized by surrounding cations, which influence the 1D assembly. This effect is mimicked by the continuum solvation model and the dielectric constant (ε) of water. As a result, the energy profile reveals the existence of a minimum with a Tc⋯N distance of 2.670 Å, close to the experimental distance of 2.710 Å (Fig. 6b). In this case, the dimer is more stable (−5.0 kcal mol−1) compared to the ReO4− dimer. This result confirms that the electrostatic repulsion in the [TcNCl4]−⋯[TcNCl4]− dimer can be counterbalanced by a suitable environment, indicating that this matere bond is also possible in solution. Similar results have been reported for the [AuI4]−⋯[AuI2]− dimer57 and the [I3]−⋯[I3]− dimer.58
Summary and outlook
This perspective explores the emerging field of matere bonds, a novel type of σ-hole interaction involving group 7 elements, such as manganese, technetium, and rhenium. These bonds, characterized by their directionality and strength, have been shown to play significant roles in stabilizing supramolecular architectures in both solid-state and solution-phase environments. Furthermore, their potential applications in catalysis and material design have been highlighted, demonstrating their versatility and relevance in supramolecular chemistry.Future research should address several key areas to further advance our understanding and utilization of matere bonds. First, experimental challenges in studying matere bonds in biological systems, particularly within manganese-containing enzymes and proteins, must be overcome. Second, the application of matere bonds in novel catalytic designs offers significant potential. Investigating their ability to stabilize transition states, mediate electron transfer, or enhance substrate binding could pave the way for innovative catalytic processes.
Additionally, expanding the scope of matere bonds to other group 7 derivatives with diverse oxidation states and geometries could uncover new interactions with unique properties. Further theoretical and computational studies are crucial to refine the understanding of their electronic and structural features, particularly in solution-phase systems where their behavior remains less explored. By addressing these challenges and opportunities, future work will undoubtedly unlock new possibilities for matere bonds in materials science, catalysis, and biological applications.
Author contributions
R. M. G. and A. F.: conceptualization, investigation, writing initial draft. A. F.: supervision, funding acquisition, project management.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank “Ministerio de Ciencia, Investigacion y Universidades/Agencia Estatal de Investigación” (MICIU/AEI/10.13039/501100011033) of Spain (projects PID2020-115637GB-I00 and PID2023-148453NB-I00, ERDF a way of making Europe).References
- (a) K. Mali, N. Pearce, S. De Feyter and N. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC; (b) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC; (c) G. Fukuhara, J. Photochem. Photobiol., C, 2020, 42, 100340 CrossRef CAS.
- (a) J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 17397–17478 CrossRef PubMed; (b) I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS; (c) K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef.
- (a) D. Pochan and O. Scherman, Chem. Rev., 2021, 121, 13699–13700 CrossRef CAS PubMed; (b) L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed.
- (a) A. Nangia and G. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4105 CrossRef CAS PubMed; (b) D. Braga, Chem. Commun., 2003, 2751–2754 RSC.
- S. J. D. Lugger, S. J. A. Houben, Y. Foelen, M. G. Debije, A. P. H. J. Schenning and D. J. Mulder, Chem. Rev., 2022, 122(5), 4946–4975 CrossRef CAS.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- (a) C. J. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (b) C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS PubMed.
- S. R. Gadre, C. H. Suresh and N. Mohan, Molecules, 2021, 26, 3289 CrossRef CAS.
- A. Haque, K. M. Alenezi, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2023, 52, 454–472 RSC.
- (a) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC; (b) A. Frontera and A. Bauza, Int. J. Mol. Sci., 2021, 22, 12550 CrossRef CAS PubMed.
- (a) L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS; (b) Y. C. Pan, J. H. Tian and D. S. Guo, Acc. Chem. Res., 2023, 56, 3626–3639 CrossRef CAS.
- (a) R. Siddiqui, J. Rani, H. M. Titi and R. Patra, Coord. Chem. Rev., 2024, 517, 215994 CrossRef CAS; (b) N. Tarannam, R. Shukla and S. Kozuch, Phys. Chem. Chem. Phys., 2021, 23, 19948–19963 RSC; (c) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed; (d) P. Politzer, J. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed; (b) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Rec., 2016, 16, 473–487 CrossRef PubMed.
- G. Resnati, D. L. Bryce, G. R. Desiraju, A. Frontera, I. Krossing, A. C. Legon, P. Metrangolo, F. Nicotra, K. Rissanen, S. Scheiner and G. Terraneo, Pure Appl. Chem., 2024, 96, 135–145 CrossRef CAS.
- (a) C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS; (b) I. Vargas-Baca, Chapter 1.11 - Computational modeling and characterization of secondary bonding in compounds of late p-block elements, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 3rd edn, 2023, pp. 572–585. ISBN 9780128231531 Search PubMed; (c) S. Kolb, G. A. Oliver and D. B. Werz, Chapter 1.13 - Chalcogen bonding in supramolecular structures, anion recognition, and catalysis, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 3rd edn, 2023, pp. 602–651. ISBN 9780128231531 Search PubMed.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (b) J. S. Ward, K.-N. Truong, M. Erdélyi and K. Rissanen, Chapter 1.12 - Halogen-bonded halogen(I) ion complexes, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 3rd edn, 2023, pp. 586–601. ISBN 9780128231531 Search PubMed.
- (a) R. Shukla and D. Chopra, Curr. Sci., 2021, 120, 12 CrossRef; (b) L. Brammer, A. Peuronen and T. M. Roseveare, Acta Crystallogr., Sect. C: Struct. Chem., 2023, 79, 377–388 Search PubMed; (c) N. Liu, Q. Li and S. A. McDowell, Front. Chem., 2020, 8, 608486 CrossRef CAS PubMed; (d) R. M. Gomila and A. Frontera, CrystEngComm, 2020, 22, 7162–7169 RSC.
- (a) Y. Li, L. Meng, C. Sun and Y. Zeng, J. Phys. Chem. A, 2020, 124, 3815–3824 CrossRef CAS PubMed; (b) D. Jovanovic, M. P. Mohanan and S. M. Huber, Angew. Chem., Int. Ed., 2024, 63, e202404823 CrossRef CAS PubMed; (c) L. Lu, Y. Lu, Z. Zhu and H. Liu, J. Mol. Model., 2020, 26, 1–12 CrossRef PubMed.
- (a) A. Gini, M. Paraja, B. Galmés, C. Besnard, A. I. Poblador-Bahamonde, N. Sakai and S. Matile, Chem. Sci., 2020, 11, 7086–7091 RSC; (b) H. Chen, A. Frontera, M. Ángeles Gutiérrez López, N. Sakai and S. Matile, Helv. Chim. Acta, 2022, 105, e202200119 CrossRef CAS.
- (a) M. N. Piña, A. Frontera and A. Bauzá, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef PubMed; (b) A. Terrón, J. Buils, T. J. Mooibroek, M. Barceló-Oliver, A. García-Raso, J. J. Fiol and A. Frontera, Chem. Commun., 2020, 56, 3524–3527 RSC; (c) A. Frontera and A. Bauzá, in Supramolecular Assemblies Based on Electrostatic Interactions, Springer International Publishing, Cham, 2022, pp. 243–265 Search PubMed; (d) S. Burguera, A. Frontera and A. Bauza, Inorg. Chem., 2023, 62, 6740–6750 CrossRef CAS PubMed.
- (a) A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef PubMed; (b) H. S. Biswal, A. Kumar Sahu, A. Frontera and A. Bauzá, J. Chem. Inf. Model., 2021, 61, 3945–3954 CrossRef CAS PubMed.
- (a) E. Priola, A. Giordana, P. P. Mazzeo, G. Mahmoudi, R. M. Gomila, F. I. Zubkov, K. M. Pokazeev, K. S. Valchuk, A. Bacchi, E. Zangrando and A. Frontera, Dalton Trans., 2021, 50, 16954–16960 RSC; (b) J. Li, Q. Feng, C. Wang and Y. Mo, Phys. Chem. Chem. Phys., 2023, 25, 15371–15381 RSC.
- (a) A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., 2021, 133, 14506–14510 CrossRef; (b) A. Pizzi, M. Calabrese, A. Daolio, M. Ursini, A. Frontera and G. Resnati, CrystEngComm, 2022, 24, 3846–3851 RSC.
- (a) S. Burguera, A. Bauzá and A. Frontera, ChemPhysChem, 2023, 24, e202300585 CrossRef CAS PubMed; (b) G. Mahmoudi, A. Masoudiasl, M. G. Babashkina, A. Frontera, T. Doert, J. M. White and D. A. Safin, Dalton Trans., 2020, 49, 17547–17551 RSC; (c) R. M. Gomila, E. R. Tiekink and A. Frontera, Inorganics, 2023, 11, 468 CrossRef CAS.
- S. Hazra, D. Majumdar, A. Frontera, S. Roy, B. Gassoumi, H. Ghalla and S. Dalai, Cryst. Growth Des., 2024, 24(17), 7246–7261 CrossRef CAS.
- (a) M. Karmakar, A. Frontera, S. Chattopadhyay, T. J. Mooibroek and A. Bauzá, Int. J. Mol. Sci., 2020, 21, 7091 CrossRef CAS PubMed; (b) T. Basak, R. M. Gomila, A. Frontera and S. Chattopadhyay, CrystEngComm, 2021, 23, 2703–2710 RSC; (c) G. Mahmoudi, S. E. Lawrence, J. Cisterna, A. Cárdenas, I. Brito, A. Frontera and D. A. Safin, New J. Chem., 2020, 44, 21100–21107 RSC; (d) S. Burguera, A. K. Sahu, A. Frontera, H. S. Biswal and A. Bauza, Inorg. Chem., 2023, 62, 18524–18532 CrossRef CAS PubMed; (e) J. Yan, Y. Zeng, L. Meng, X. Li and X. Zhang, Phys. Chem. Chem. Phys., 2023, 25, 29155–29164 RSC; (f) I. Alkorta, C. Trujillo, G. Sánchez-Sanz and J. Elguero, Crystals, 2020, 10, 137 CrossRef CAS.
- Y. Cornaton and J. P. Djukic, Acc. Chem. Res., 2021, 54, 3828–3840 CrossRef CAS PubMed.
- E. S. Shubina, N. V. Belkova and L. M. Epstein, J. Organomet. Chem., 1997, 536, 17–29 CrossRef.
- (a) Q. Zheng, S. Borsley, G. S. Nichol, F. Duarte and S. L. Cockroft, Angew. Chem., 2019, 131, 12747–12753 CrossRef; (b) L. H. Doerrer, C. Del Rosario and A. Fan, Chapter 1.15 - Metallophilic interactions, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 3rd edn, 2023, pp. 665–739. ISBN 9780128231531 Search PubMed.
- (a) M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC; (b) A. Vellé, L. Rodríguez-Santiago, M. Sodupe and P. J. Sanz Miguel, Chem. – Eur. J., 2020, 26, 997–1002 CrossRef PubMed.
- (a) I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS; (b) L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 230–240 CrossRef CAS PubMed.
- (a) M. Calabrese, A. Pizzi, A. Daolio, R. Beccaria, C. Lo Iacono, S. Scheiner and G. Resnati, Chem. – Eur. J., 2024, 30, e202304240 CrossRef CAS PubMed; (b) R. M. Gomila and A. Frontera, Inorganics, 2022, 10, 133 CrossRef CAS; (c) X. Wang, Q. Li and S. Scheiner, Molecules, 2023, 29, 79 CrossRef PubMed; (d) A. Pizzi, A. Daolio, M. Calabrese, G. Terraneo, A. Frontera and G. Resnati, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, C800–C800 Search PubMed; (e) A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., 2021, 133, 20891–20895 CrossRef.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS PubMed.
- (a) A. Bauzá and A. Frontera, Chem. – Eur. J., 2022, 28, e202201660 CrossRef PubMed; (b) M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2024, 26, 5836–5847 RSC.
- (a) M. Calabrese, R. M. Gomila, A. Pizzi, A. Frontera and G. Resnati, Chem. – Eur. J., 2023, 29, e202302176 CrossRef CAS PubMed; (b) M. Calabrese, A. Pizzi, A. Daolio, A. Frontera and G. Resnati, Dalton Trans., 2023, 52, 1030–1035 RSC.
- N. W. Alcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58 CrossRef CAS.
- S. Burguera, R. M. Gomila, A. Bauzá and A. Frontera, Crystals, 2023, 13, 187 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- E. R. Chakalov, E. Y. Tupikina, D. M. Ivanov, E. V. Bartashevich and P. M. Tolstoy, Molecules, 2022, 27, 4848 CrossRef CAS PubMed.
- R. M. Gomila and A. Frontera, Molecules, 2022, 27, 6597 CrossRef CAS PubMed.
- H.-J. Gosink, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, E. Irmer and R. Herbst-Irmer, Organometallics, 1994, 13, 3420–3426 CrossRef CAS.
- L. Richtera, J. Taraba and J. Touzin, Z. Anorg. Allg. Chem., 2003, 629, 716–721 CrossRef CAS.
- N. V. Yashin, E. B. Averina, Y. K. Grishin, V. B. Rybakov, T. S. Kuznetsova and N. S. Zefirov, Russ. Chem. Bull. Int. Ed., 2016, 65, 451–455 CrossRef CAS.
- A. Y. Maruk, M. S. Grigor'ev and K. E. German, Koord. Khim., 2010, 36, 381 CrossRef CAS.
- J. Baldas, J. F. Boas, S. F. Colmanet, A. D. Rae and G. A. Williams, Proc. R. Soc. London, Ser. A, 1993, 442, 437 CrossRef CAS.
- H. Ikeda, A. Ito, E. Sakuda, N. Kitamura, T. Takayama, T. Sekine, A. Shinohara and T. Yoshimura, Inorg. Chem., 2013, 52, 6319 CrossRef CAS PubMed.
- Y. Xu, M. Calabrese, N. Demitri, A. Pizzi, T. Nag, I. Hung, Z. Gan, G. Resnati and D. L. Bryce, Chem. Commun., 2023, 59, 12609–12612 RSC.
- D. Grödler, S. Burguera, A. Frontera and E. Strub, Chem. – Eur. J., 2024, 30, e202400100 CrossRef PubMed.
- M. Karmakar, R. M. Gomila, A. Frontera and S. Chattopadhyay, Cryst. Growth Des., 2024, 24, 5990–6000 CrossRef CAS.
- S. Burguera, A. K. Sahu, M. J. Chávez Romero, H. S. Biswal and A. Bauzá, Phys. Chem. Chem. Phys., 2024, 26, 18606–18613 RSC.
- S. Xiang and L. Tong, Nat. Struct. Mol. Biol., 2008, 15, 295 CrossRef CAS PubMed.
- (a) W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC; (b) I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2015, 119, 183–194 CrossRef CAS PubMed.
- (a) J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed; (b) Y. Li, L. Meng and Y. Zeng, ChemPlusChem, 2021, 86, 232–240 CrossRef CAS PubMed.
- (a) W. Zierkiewicz, R. Wysokiński, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed; (b) R. Wysokiński, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 4818–4828 RSC; (c) S. Scheiner, R. Wysokiński, M. Michalczyk and W. Zierkiewicz, J. Phys. Chem. A, 2020, 124, 4998–5006 CrossRef CAS PubMed; (d) A. Grabarz, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Molecules, 2021, 26, 2116 CrossRef CAS PubMed; (e) R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef PubMed; (f) R. Wysokiński, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2021, 22, 818–821 CrossRef PubMed; (g) A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef CAS PubMed.
- L. Andreo, R. M. Gomila, E. Priola, A. Giordana, S. Pantaleone, E. Diana, G. Mahmoudi and A. Frontera, Cryst. Growth Des., 2022, 22, 6539–6544 CrossRef CAS PubMed.
- F. Groenewald, C. Esterhuysen and J. Dillen, Comput. Theor. Chem., 2016, 1090, 225–233 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |