
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen evolution reaction mechanisms in thiosemicarbazone metal complexes: a combined theoretical and experimental investigation on the impact of proton source†
Jana Mehrez,
Alexandre Barrozo ,
Léa Delmotte,
Renaud Hardré,
Michael Papadakis* and
Maylis Orio*
,
Léa Delmotte,
Renaud Hardré,
Michael Papadakis* and
Maylis Orio*
Aix-Marseille Univ, CNRS, Centrale Med, iSm2, Marseille, France. E-mail: michail.papadakis@univ-amu.fr; maylis.orio@univ-amu.fr
First published on 5th February 2025
Abstract
In the pursuit of developping efficient and green methods for hydrogen production, a key focus is the development of the most energy-efficient catalysts. The analysis of various proton sources primarily aims at eliminating rate-limiting steps associated with protonation events and ensuring the stability of the catalyst. In this work, we report how two distinct proton sources can cause a mechanistic shift in the hydrogen evolution reaction. Specifically, we explore this reactivity change in the presence of triethylammonium and trifluoroacetic acid with two thiosemicarbazone-based complexes, using cobalt and nickel metal centers. Our combined experimental and theoretical results reveal that the complete sequence of steps leading to hydrogen release strongly depends on the proton source. This demonstrates the importance of thoroughly investigating the interactions between a catalyst and a proton source to optimize hydrogen evolution systems.
Introduction
With the increasing prominence of hydrogen as a key fuel source, the development of new catalysts able to perform the hydrogen evolution reaction (HER) continue to advance. Most of the focus has been put on molecules that either mimic the structure or the function of hydrogenases.1–13 However, the choice of proton sources is a rather overlooked area of study. Discussions often focus on finding acids that are strong enough to ensure that proton transfer does not become the rate-limiting step, while still maintaining the stability of the catalyst.14 Different proton sources, such as triethylammonium (Et3NH+) and trifluoroacetic acid (TFA) will interact with catalysts in different ways, leading to multiple hydrogen-bond interactions that can result in proton transfers to distinct protonation sites. Furthermore, it is worth noting that, as the redox state of the catalyst changes during the HER process, the proton affinities of each potential protonation site also evolve. Considering HER involves more than a single electrochemical event, the pKa of the proton source and the catalyst's nature can influence the sequence and location of proton transfer steps. This versatility can lead to different catalytic cycles, depending on the specific conditions at play.We have recently reported comprehensive experimental and theoretical studies investigating the mechanisms of homogeneous HER catalysts based on non-innocent thiosemicarbazone (TSC) ligands which can actively participate in the catalytic process by storing both electrons and protons.15–22 These studies focused on a Ni-based TSC complex (NiTSC-OCH3) in the presence of trifluoroacetic acid (TFA),19,21,22 and an analogous Co-based complex (CoTSC-OCH3) using triethylammonium (Et3NH+).20,23 In these studies, the nature of both the metal centre and the proton source differs, resulting in distinct reaction pathways. Interestingly, our complementary study on a series Ni-based complexes explored how chemical modifications to the non-innocent TSC ligand might influence the reaction pathway, considering three substituents of different electron-donating abilities placed at the para position on the phenyl rings.21 Our findings indicated that these changes had no significant impact on the catalytic cycle and only affected the HER catalysis parameters.24 However, the precise factors responsible for the variations in catalytic performance remain unclear. Consequently, further exploration was needed to unravel how parameters such as electronic properties of the metal, proton transfer dynamics, and ligand interactions contribute to the observed mechanistic discrepancies and influence the overall efficiency of the catalysts in the HER process. In this work, we aimed to conduct a more in-depth comparison between two TSC metal-based catalysts, namely NiTSC-OCH3 and CoTSC-OCH3 (Scheme 1) by examining their behavior using TFA and Et3NH+ as proton sources.
 | ||
| Scheme 1 Structures of nickel and cobalt thiosemicarbazone complexes considered in this work. | ||
Our study reveals that the catalytic mechanism can shift depending on the choice of acid. While it is usually assumed that a stronger proton source will enhance HER catalytic performance by facilitating proton transfer, our results indicate that the interaction between the acid and the catalyst is more nuanced, with different acid-catalyst pairs leading to distinct catalytic pathways. This is an important finding, as any attempt to optimize a given catalyst needs now to account for its exact interaction with the acid and distinct rate-limiting steps.
Results and discussion
Synthesis and characterization
Syntheses of complexes NiTSC-OCH3, CoTSC-OCH3 and Et3NHBF4 salt were performed following protocols previously reported in literature.22,25,26Electrocatalytic studies and benchmarking of performances
Cyclic voltammograms (CV) in catalytic conditions were recorded using experimental conditions similar to those employed in our previously reported studies for electrocatalytic proton reduction combining NiTSC-OCH3 with trifluoroacetic acid (TFA)19 and CoTSC-OCH3 with triethylammonium tetrafluoroborate (Et3NHBF4).20 In this work, we considered switching proton sources for each complex to investigate the reactivity of NiTSC-OCH3 with Et3NHBF4 and that of CoTSC-OCH3 with TFA. The redox behaviour of the complexes was first investigated in the potential range between 0 and −2.6 V vs. Fc+/0. The experiments were performed at room temperature, with a glassy carbon electrode in a nitrogen-purged anhydrous DMF solution, containing tetrabutylammonium hexafluorophosphate (NBu4PF6) as a supporting electrolyte. In the absence of acid, two quasi-reversible processes are detected in the cathodic region for both complexes (Table 1).| Complex | E° 1st reduction | E° 2nd reduction | E° 3rd reduction |
|---|---|---|---|
| CoTSC-OCH3 | −0.60 | −1.48 | −2.48 |
| NiTSC-OCH3 | −1.57 | −2.20 | — |
To study the capability of the two complexes to mediate proton reduction catalysis, we recorded their cyclic voltammograms under nitrogen in the presence of a proton source using TFA (pKa = 6.0 in DMF)27 with CoTSC-OCH3 and Et3NHBF4 (pKa = 9.2 in DMF)14 with NiTSC-OCH3 (Fig. 1 and S1†). Focusing on the cobalt complex with TFA, the addition of acid triggers the appearance of three irreversible waves indicative of potential-dependent mechanisms and consistent with previous observations for NiTSC-OCH3 with TFA. The first wave displays a peak potential at −1.42 V vs. Fc+/0 while the peak potential of the next two waves are shifted to more negative values upon successive additions of TFA (from −1.79 to −1.89 V, and from −2.09 to −2.37 V vs. Fc+/0, with 10 and 60 mM of acid, respectively). The mid-wave potential of the process occurring at more positive potentials is of −1.28 V vs. Fc+/0 and its intensity is correlated with acid concentration in the solution (Fig. S2†). A different response in current is obtained for NiTSC-OCH3 in the presence of Et3NH+ with the appearance of two irreversible waves upon acid addition (Fig. S2†). The first wave with a peak potential at −1.65 V vs. Fc+/0 is mostly unaffected when raising acid concentration, while the second one slightly shifts with increasing amounts of Et3NH+ (from −2.28 to −2.42 V vs. Fc+/0 with 20 and 100 mM of acid, respectively). This catalytic wave displays a mid-wave potential of −2.08 V vs. Fc+/0 with a current response proportional to acid concentration.
 | ||
| Fig. 1 Successive cyclic voltammograms of a DMF solution (0.1 M NBu4PF6) with 1 mM of (top) CoTSC-OCH3 in the presence of increasing amounts of TFA and (bottom) NiTSC-OCH3 in the presence of increasing amounts of Et3NHBF4. Scan rate 500 mV s−1 using a glassy carbon working electrode. | ||
Dip and rinse tests were performed to confirm that the current response observed upon adding a proton source was not due to the absorption of any species on the electrode surface.
After each measurement, the working electrode was transferred to a fresh DMF solution (0.1 M NBu4PF6). Following the addition of either TFA or Et3NH+, no catalytic wave was recorded, confirming that these systems are homogeneous in nature (Fig. S3†). To confirm that the observed catalytic waves resulted from the electrocatalytic reduction of protons into hydrogen, bulk electrolysis experiments were conducted alongside gas analysis.
Solutions were prepared in 8 mL of DMF (0.1 M NBu4PF6) containing 1 mM of each complex. For NiTSC-OCH3, 100 mM of Et3NH+ was added, while for CoTSC-OCH3, 60 mM of TFA was used to match the conditions of our previous reported works. Continuous in-line GC analysis was used to monitor the gas products from each reaction. A mercury pool working electrode was employed to ensure that any nanoparticle formed during the catalysis would be absorbed into the mercury, thereby maintaining the systems’ homogeneity.
For the cobalt catalyst with TFA, bulk electrolysis experiments were performed by applying a controlled potential of −1.6 V vs. Fc+/0 for 16 hours and the complex demonstrated catalytic activity for proton reduction (Fig. 2 and S4†).
 | ||
| Fig. 2 Coulometry of CoTSC-OCH3 with 60 mM TFA during bulk electrolysis experiment at −1.6 V vs. Fc+/0 (top blue line) and blank experiment of 60 mM TFA in the absence of CoTSC-OCH3 (top grey line) and NiTSC-OCH3 with 100 mM Et3NHBF4 during bulk electrolysis experiment at −2.1 v vs. Fc+/0 (bottom red line) and blank experiment of 100 mM Et3NH+ in the absence of NiTSC-OCH3 (bottom grey line). The electrolytic solutions contain 0.1 M NBu4PF6 in DMF and 1 mM of each complex. Bulk electrolysis experiments were performed using a mercury pool working electrode. | ||
Both catalysts produce hydrogen with TFA using similar applied potential values (−1.6 and −1.7 V vs. Fc+/0 for CoTSC-OCH3 and NiTSC-OCH3 respectively). While the turnover frequency (TOF) of CoTSC-OCH3 is higher at early-stage catalysis (96 s−1 vs. 11 s−1 for NiTSC-OCH3), they eventually yield fairly similar TOF values after 16 h of catalysis (117 and 90 s−1, respectively). However, the cobalt complex provides enhanced H2 production compared to the nickel analogue with higher turnover number (TON) and faradaic yield (FY, 35 and 88% vs. 21 and 80% for NiTSC-OCH3, Tables S1 and S2†) and displays a decreased overpotential value, η (0.294 V vs. 0.344 V for NiTSC-OCH3, Table S3†).19
For the nickel catalyst with Et3NH+, measurements were performed at a fixed potential of −2.1 V vs. Fc+/0 over 4 hours (Fig. 2 and S5†). Comparing the catalytic performances of the two complexes with Et3NH+, the Co complex significantly outperforms the Ni analogue in every aspect. Indeed, CoTSC-OCH3 mediates proton reduction into hydrogen at a lower applied potential (−1.6 V vs. −2.1 V vs. Fc+/0 for NiTSC-OCH3) to provide increased FY (65 vs. 58% for NiTSC-OCH3) and TON (9 vs. 2 for NiTSC-OCH3) leading to a higher efficiency with an enhanced TOF value (130 s−1 vs. 24 s−1 for NiTSC-OCH3) and a lower overpotential requirement (0.311 V vs. 0.908 V for NiTSC-OCH3, Tables S1–S3†).20 For both proton source, the cobalt-based TSC complex seems to be a better catalyst for hydrogen evolution that its nickel counterpart. However, the mechanistic implications of the proton source strength cannot be firmly established based only on the current set of experimental data. The main insight we can get originates from the UV-vis data of the complexes recorded both in the presence and absence of proton sources. Considering no change is observed in the spectral signatures with and without acid (Fig. S6†), these data indeed support our previous analysis suggesting that the reaction mechanisms involving our nickel and cobalt complexes do not involve an initial protonation step.22–24
To better understand how acid strength influences the HER pathway, we have conducted an extensive theoretical investigation of the possible catalytic cycles involving CoTSC-OCH3 and NiTSC-OCH3 using density functional theory (DFT) calculations. Our computational studies will allow us to probe the reaction energetics of both thiosemicarbazone metal-based complexes in the presence of each proton source, providing valuable insights into the underlying mechanistic differences. Through theoretical modelling of the reaction pathways, we aim at identifying key steps in the catalytic cycle where the proton source could have significant effect.
DFT Calculations: pKa values of protonation sites
Scheme 1 shows the structure of the two complexes analyzed in this work. In our study of CoTSC-OCH3 with Et3NH+,23 calculations showed that HER proceeds via a metal-centered pathway, with the ligand playing mainly the role of electron relay (Fig. S7†). The reaction under investigation usually proceeding via the transfer of two electrons (electrochemical step, E) and two protons (chemical step, C) to form H2, the catalytic mechanism involving CoTSC-OCH3 with Et3NH+ was found to be an (E) ECEC-type cycle with the first step (E) being an activation step associated with the release of an axial NCS ligand (Scheme 2).23 In contrast, HER for NiTSC-OCH3 with TFA has a ligand-centered reactivity, with protons and electrons being stored in the ligand (Fig. S8†). In this case, we showed that the catalytic cycle proceeds via two pathways, both involving two subsequent proton-coupled electron transfer reactions, [EC][EC]. The difference between these pathways lies with the difference in protonation states, with one of the mechanisms requiring the presence of a third proton, this latter step one being rate-limiting.24 | ||
| Scheme 2 Structure of the two complexes studied in this work. M represents either Ni or Co, NM is the N atom coordinated to the metal center, Nd, the distal N atom and NCS the axial ligand in CoTSC-OCH3 dissociating after the first reduction. | ||
Our main question is whether these differences in mechanism are metal-dependent, acid-dependent, or an interplay between both. We first need to review the relative pKas for protonating the possible sites in both complexes following the methodology used in our previous studies (Tables 2 and 3).23,24 Four possible sites were considered: M (Ni or Co), S, NM and Nd (Scheme 2). Other sites were excluded from this analysis based on previous studies.19 We then performed free energy calculations for the protonated complexes and compared the free energy differences of the distinct protonation states. For the two complexes, with both acids, we are assuming that the complexes have already been reduced once. For NiTSC-OCH3, this is in line with cyclic voltammograms and UV-vis spectra in the presence of TFA,21 and since TFA is a stronger acid than Et3NH+, we would expect the same behavior with the latter. In the case of CoTSC-OCH3, a first reduction step is necessary to activate the complex for the beginning of the catalytic cycle, regardless of the proton source.20 Thus, for investigating the first protonation event, we are only looking at one-electron reduced species. For NiTSC-OCH3, we are not considering two subsequent reduction events, as those would require larger potentials to be applied (−2.29 vs. Fc+/0)24 and the same is true for three subsequent reduction reactions for CoTSC-OCH3 (−2.36 V vs. Fc+/0).23 Taking this into account, we see that Nd is the most favourable site for protonation for both complexes after a single reduction. Table 3 suggests that the preferred protonation site can become the metal center for CoTSC-OCH3, depending on the oxidation state of the complex. On the second protonation, it is important to consider the presence of TFA, as a possible double H-bond interaction between this proton source and the complex can take place.
 |
 |
This interaction involves the protonated Nd, the conjugated base TFA− and either the adjacent protonated NM atom for NiTSC-OCH3,24 or the metal centre for CoTSC-OCH3. Such interaction can affect the pKas of the adjacent NM or M atoms. Since it is not the case with Et3NH+, free energy and relative pKa calculations in the presence of the conjugated base TFA− were performed only for the stronger proton source. Our results suggest that, for NiTSC-OCH3 with TFA, protonation could occur either on the second Nd atom or on the NM atom adjacent to the previously protonated Nd atom. This leads to two possible catalytic pathways, involving two and three protons.23
In the case of Et3NH+, Table 2 suggests that we would most likely go through the two-proton mechanism for NiTSC-OCH3, with first and second protonation events occurring at both Nd atoms. Thus, the main change in the catalytic mechanism for NiTSC-OCH3 when considering TFA and Et3NH+ as proton sources lies in the fact that TFA enables a second pathway.
While the complexity in the catalytic mechanism of NiTSC-OCH3 lies with the possibility of three protons, for CoTSC-OCH3 we have to consider the transfer of three electrons.20,23 Previous studies on the HER for CoTSC-OCH3 with Et3NH+ show that, after an initial reduction step, the catalytic cycle follows an ECEC sequence, with the metal center being the preferred protonation site (Table 3). However, this is the only mechanism available in the presence of Et3NH+. With a stronger acid such as TFA, protonation could occur right after the activation step, and if that is the case, Co is no longer the preferred protonation site, but rather Nd. In the next section, we will show how this order of steps can be changed, leading to distinct pathways being accessed.
DFT calculations: the catalytic cycles
From the data presented in Tables 2 and 3, we can focus the study of the catalytic cycle on the most relevant protonation sites according to the oxidation state of the complexes. Fig. 3 displays the pathways that can be taken for NiTSC-OCH3 and CoTSC-OCH3 as function of the proton source strength. We herein report relevant redox potentials, proton transfer free energy differences, as well pKa differences between the complex and the proton source.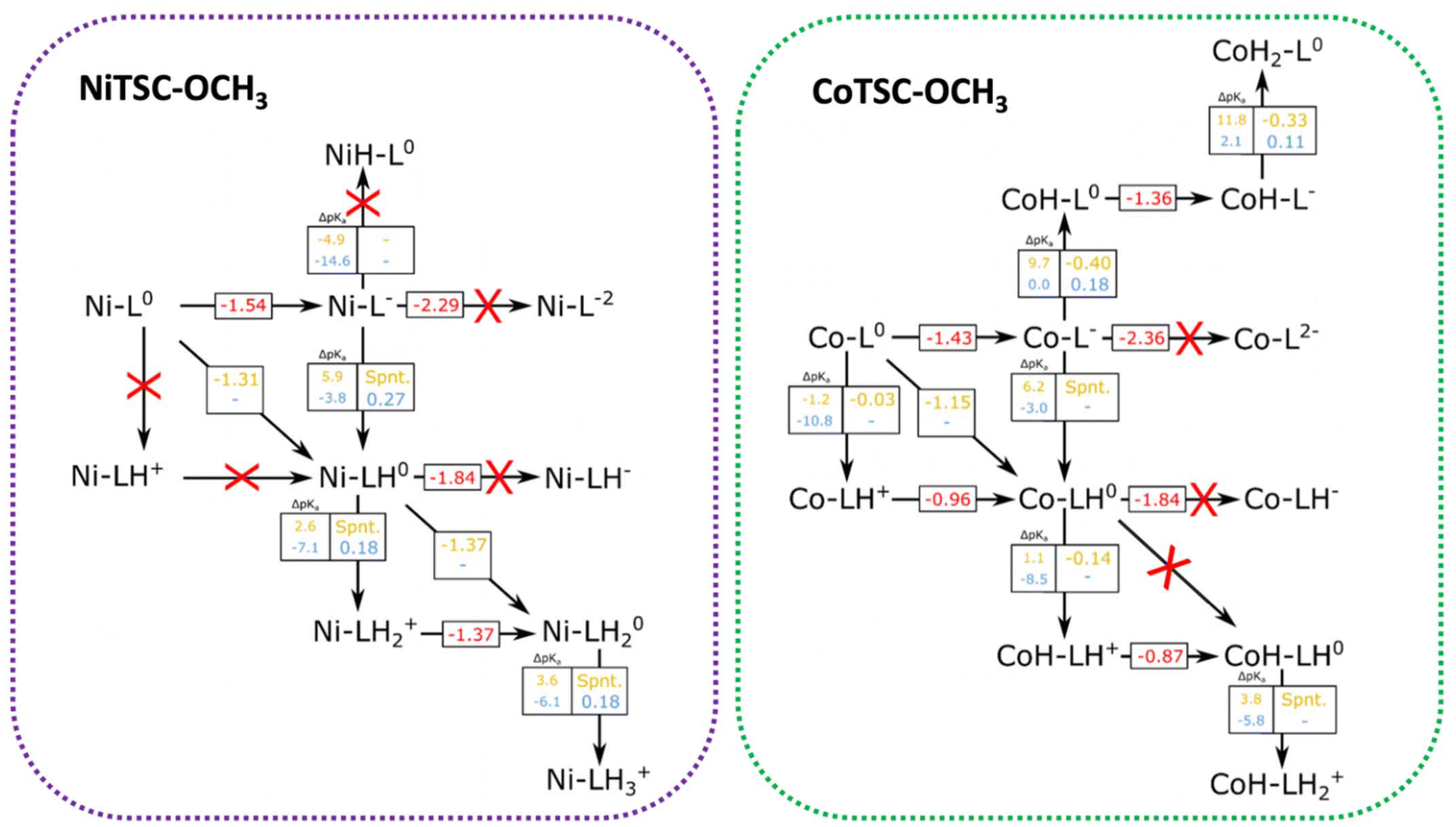 | ||
| Fig. 3 Possible pathways of HER for NiTSC-OCH3 and CoTSC-OCH3. Protonation is marked as being either on the metal or the ligand, with the specific protonation sites in the ligand determined by Tables 2 and 3. Numbers in red (V vs. Fc+/0) correspond to reduction potentials. Numbers in yellow (TFA) and blue (Et3NH+) correspond to pKa differences between the complexes and the proton source, as well as reaction-free energy differences, all in eV. Red crosses correspond to processes unlikely to occur due to either too negative potential requirements, or large unfavourable pKa differences (negative numbers are unfavourable). | ||
Starting with NiTSC-OCH3, since the metal center is unlike to be a proton relay, all the process focuses on the ligand, with the first two protonation events taking place at the Nd atoms, and the third one to the NM site. The main striking difference between the catalytic cycles for the two proton sources is that TFA can induce proton-coupled electron transfer reactions, whereas for Et3NH+, the cycle undergoes stepwise electron and proton transfers. For TFA, we previously reported that two possible mechanisms were at stake: one involving two protons and two electrons, and another with three protons and two electrons. Both catalytic pathways consisted in two successive PCET providing Ni-LH0 and NiLH20 and following an [EC][EC] sequence for the first mechanism while the third protonation event involved in the second mechanism provided NiLH3+ following an [EC][EC]C sequence. Particularly for Et3NH+, UV-Vis experiments show that the absence and presence of the proton source does not affect the spectrum (Fig. S6†). This finding suggest that we can discard a mechanism starting with a chemical step (CE and CC) and consider the first part of the cycle being either a stepwise EC process, or a single and concerted [EC] step. As opposed to TFA, our calculations do not predict that a proton-coupled electron transfer is feasible, leaving only the possibility of a reduction as first step (−1.54 V vs. Fc+/0) providing NiL−. From there, the arrival of a second electron is unlikely (−2.29 V) while a proton transfer would be able to occur since the ΔpKa is rather small (−3.8). In this case, such protonation in the presence of Et3NH+ would occur at Nd (Table 2), leading to Ni-LH0 through a stepwise electron–proton transfer, EC. The second part of the cycle with Et3NH+ is predicted to be a stepwise proton–electron, CE, considering the unfavourable energy requirement for a reduction step following the EC sequence (−1.84 V). In this case, the second protonation would occur at Nd leading the formation of NiLH2+ before a second electron is transferred (−1.37 V) to form the catalytic active species NiLH20. It is worth noting that pKa differences between the catalyst and the acid are negative, and reaction free energy differences are substantially positive for all proton transfer events, meaning that the proton transfer reactions are energetically unfavourable, and thus are likely to become rate-limiting steps in the HER process, in contrast with TFA, where the final H2 bond formation is the rate-limiting step. These results thus indicate that the reaction mechanism for NiTSC-OCH3 with Et3NH+ proceeds through an ECCE sequence.
In the case of CoTSC-OCH3, the situation is more complex, especially since the metal center is now playing an active role in the catalytic cycle, holding both protons and electrons. Our previous theoretical study focused on the reaction mechanism of CoTSC-OCH3 with Et3NH+ showed the catalytic cycle can be best described as a (E) ECEC sequence with a first reduction as the activation step providing CoL0 and two successive stepwise electron–proton transfers. The first EC steps provided CoL− and CoH-L0, while the subsequent EC steps lead to formation of CoH-L− and CoH2-L0, respectively. Considering the catalytic cycle of CoTSC-OCH3 with TFA and CoL0 as the starting species, either a reduction or a protonation event can occur at this stage. From Fig. 3, we can see that pKa differences for an early proton transfer are negative regardless of the acid (ΔpKa = −1.2 and −10.8), meaning that the process is energetically unfavourable. We thus consider that a mechanism starting with a chemical step (CE and CC) is unlikely to occur which points at the first two steps being either an EC or an [EC] process. From CoL0, a subsequent reduction step predicted at −1.43 V vs. Fc+/0 is possible and from there, only a proton transfer would be able to occur. In such case, proton transfer seems to be feasible with TFA, since the ΔpKa values are both favorable (9.7 and 6.2, respectively). Such protonation in the presence of TFA could occur either at Co or at Nd (Table 3), providing thus CoH-L0 and Co-LH0. It is worth noting that a proton-coupled electron transfer, occurring at −1.15 V and providing Co-LH0, is also predicted according to our calculations. This reduction process being more than 0.25 eV less negative than in the absence of TFA (−1.43 V), such stabilization effect strongly suggests a PCET phenomenon, just like for NiTSC-OCH3 with TFA also featuring [EC] as the first steps of the catalytic cycle. A change of similar magnitude in reduction potentials is also observed in the cyclic voltammograms of CoTSC-OCH3 (Fig. 1) featuring a mid-wave catalytic potential of −1.28 V vs. Fc+/0 compared to the 2nd reduction potential of −1.48 V vs. Fc+/0 (Table 1). From there, the arrival of a third electron would be unlikely to occur (−1.84 V), meaning the second protonation event would take place at the metal center, in the presence of TFA, providing CoH-LH+ (Table 3). This second proton transfer is energetically favourable and will be followed by a third electron transfer (−0.87 V) providing CoH-LH0.
The last step would involve a third proton transfer, which, according to Table 3, would leave both Nd and Co protonated providing CoH-LH2+. The three protons are not in optimal distance for H2 bond formation, requiring an intramolecular proton transfer to take place. Table 3 shows that the second-best candidate for the three proton-mechanism would be to have both Nd and one NM atom protonated, the same protonation pattern seen for NiTSC-OCH3, which allows for proton tunneling for H2 formation.24 The pKa difference between these two protonation states in CoTSC-OCH3 is of 1.3, a rather small gap that would not prevent such rearrangement from happening. Our results show that two catalytic pathways can occur for CoTSC-OCH3 with TFA, involving the transfer of either two or three protons, and proceeding through either [EC]CE or [EC]CEC sequences, respectively.
To summarize, the first steps of the catalytic cycles of both catalysts with Et3NH+ are stepwise electron and proton transfers, EC, but involve different protonation sites e.g. the metal for CoTSC-OCH323 and the ligand for NiTSC-OCH3.24 Our calculations indicate that the steps following the first EC sequence are different with protonation at the ligand being favored over the reduction for NiTSC-OCH3, leading to sequential CE steps, while reduction remains favored over protonation for the cobalt, providing stepwise EC steps. This mechanistic shift is consistent with the previously reported metal-assisted ligand-based reactivity for nickel-based TSC catalysts.19,24 When comparing the behavior of the two complexes with TFA, a proton-coupled electron transfer, [EC], is the first step of both catalytic cycles. However, the subsequent stepwise proton and electron transfer, CE, occurring for CoTSC-OCH3 differs from NiTSC-OCH3 undergoing a second proton-coupled electron transfer, [EC]. Such a change in the reaction pathway is attributed to the metal nature, based on Table 3 which indicates that the cobalt center is a facile protonation site for any investigated redox state. This is also in line with the observed ligand-assisted metal centered-reactivity of the cobalt-based TSC catalyst.20,23
The previous section showed that two possible reaction mechanisms can occur concomitantly when using TFA as proton source. One involves two protons and two electrons, while another one requires three protons and two electrons with the transfer of the third proton being the probable rate-limiting steps for both catalysts.24 For the three proton-mechanism, the relevant nickel and cobalt species display two adjacent N atoms protonated (Nd and NM), so that two protons are in close proximity for H2 evolution. In addition, TFA− is bound by two H-bonds and the second Nd atom is also protonated (Scheme 3). We thus performed a potential energy surface (PES) scan by probing H–H distances in the [2.35–0.75 Å] range and corresponding to values larger than that for complex equilibrium and to H2 bond length, respectively (Table S4†). The scan was performed by re-optimizing the structure at each point, allowing the remaining atoms to relax while keeping the H⋯H bond distance constrained.
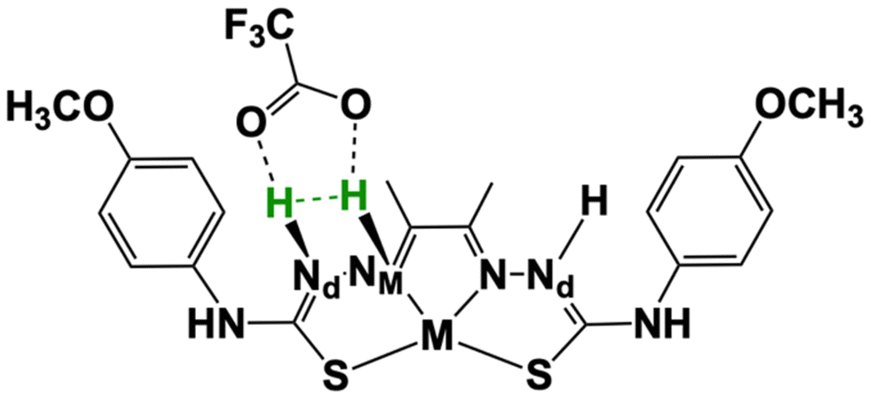 | ||
| Scheme 3 Schematic representation of the intramolecular proton tunnelling coordinate (green labels) used for potential energy surface scan. | ||
The calculated PES is presented in Fig. 4. In the [2.35–1.05 Å] range, no noticeable change is observed from the interaction between the two protons, the complexes and the conjugated base TFA−. The distance between the complex and TFA− starts increasing when the H–H distance gets shorter leading to the binding of the Nd proton to the adjacent NM proton. There is an drastic energy drop after the H–H distance reaches 1.10 Å, as H2 is close to form, and thus dissociates from the complex, together with TFA−. As previously observed with our series of nickel thiosemicarbazone complexes and other related intramolecular proton transfer processes,24,36–38 our results provide high activation barriers which would lead to slow reaction rates. In the above cases as well in ours, the energy profiles are rather thin, making tunnelling the main mechanism. We then calculated the rate constant for proton tunneling for the three-proton configuration occurring with TFA, using the PES together with WKB approximation (see details in Experimental section).23,36–38 The differences in zero-point vibrational energy (ZPVE) between the ground state and the highest energy states were determined through harmonic analysis, which provides the energy at which protons are likely to tunnel through the barrier.
 | ||
| Fig. 4 Potential energy surface scan for the intramolecular proton tunnelling, leading to H2 formation for the three-proton, two electron mechanisms. | ||
We computed the vibrational frequencies for the two protons (Scheme 3 and Table S4†), specifically for the mode that would bring them together by fitting a parabola to the PES near the equilibrium H–H distance (1.9–2.35 Å).24 These frequencies will indicate the rate at which tunnelling attempts will occur. The tunnelling probability is finally calculated using the WKB approximation,36–38 and the kinetic rates are derived by multiplying this probability with the calculated frequency of vibration between the two protons. Table 4 compare the kinetics and energetics of the tunneling process for CoTSC-OCH3 and NiTSC-OCH3, showing that CoTSC-OCH3 would be a better catalyst for this peculiar step. The barrier from the potential energy surface of CoTSC-OCH3 is slightly thinner and together with a slightly larger zero-point vibrational energy, this leads to a gain of about one order of magnitude in the tunneling rate. These data indicate that there is no apparent correlation between the activation barriers and calculated rates. This aligns with our previous study, where we suggested that the calculated rates resulted from an interplay of all relevant parameters and the contour of the energy barrier.24 However, we observe a qualitative agreement between the calculated rates and the kinetic experimental data, with the difference in magnitude between the nickel and cobalt catalysts being well reproduced by our calculations. This is consistent with our findings on the series of nickel thiosemicarbazone catalysts for which an agreement was also found from comparing the computed H2 turnover rates for the three-proton, two-electron mechanism with the experimental TOF values.24
Conclusions
This study provides an in-depth investigation into the HER mechanisms of two metal-based thiosemicarbazone complexes, integrating experimental work with theoretical studies to provide a comprehensive understanding of their catalytic behaviour. Our results confirm that the strength of the proton source significantly influences the reaction pathways, with the stronger proton source, TFA, favouring a proton-coupled electron transfer (PCET) as the dominant initial step in the catalytic process for both catalysts. This finding is in sharp contrast with the step-by-step reduction and protonation events that predominate when using a weaker proton source, such as Et3NH+. We also showed the existence of two competing mechanisms with TFA, involving two electrons and either two or three protons while a single reaction pathway was identified with Et3NH+. Irrespective of the proton source, our calculations identified mechanistic shifts between the catalysts following the first electron–proton transfers that were attributed to the metal-centered reactivity of CoTSC-OCH3 as opposed to the ligand-centered one for NiTSC-OCH3. Part of these mechanistic distinctions align with previous findings reported by the group of Artero who highlighted the gap in understanding how acid strength impacted the mechanism of cobalt-based complexes for HER.14 Their work identified distinct catalytic pathways, with either stepwise or concerted events, featuring different hydride intermediates. Isolating the latter can be challenging, which complicates further characterization. As a result, the complete sequence of steps leading to hydrogen release remains elusive when relying solely on experimental approaches. In this context, theoretical studies can provide a detailed framework for analyzing the pivotal role of proton source strength in shaping the reaction mechanism. In this study, DFT calculations allowed to propose plausible reaction pathways, pinpoint key intermediates, and identify potential rate-limiting steps within the catalytic cycle. The computed values derived from these calculations could be further compared with experimental data, providing valuable insights into the HER mechanism. For instance, our calculations indicated that the PCET process is energetically less demanding compared to the stepwise process which nicely correlates with the lower overpotential requirements of both complexes when using the stronger proton source. Our results also revealed the presence of two catalytic pathways with the stronger proton source which can be correlated to prolonged HER activity of the catalysts with TFA when compared to that with Et3NH+. Our computations also indicated the favorable formation of reactive hydride species with the cobalt catalyst which can be ascribed with the enhanced catalytic efficiency of CoTSC-OCH3 compared to NiTSC-OCH3 with both proton sources at early-stage catalysis. From the above correlations, our study lays the groundwork for future research targeting the design of next-generation metal-based HER catalysts with redox-active ligands. Our findings suggest the preferential use of cobalt systems with softer proton sources to enhance catalyst activity and efficiency. By improving our mechanistic understanding of thiosemicarbazone metal-based complexes for HER, this study highlights the potential of a multidisciplinary approach to accelerate the optimization of catalytic systems.Experimental section
All chemical compounds were purchased from Sigma Aldrich and used without further purification. UV-vis spectra were recorded on a Varian Cary 60 spectrophotometer.Cyclic voltammetry experiments were performed using an Origaflex OGF01A potentiostat and a three-electrode set-up consisting of a glassy carbon working electrode, a platinum wire counter electrode and a Leakless (Ag/AgCl) reference electrode. Ferrocene was used as an internal standard with E° (Fc+/0) = 0.5 V versus Ag/AgCl for each measurement. All studies were performed in deoxygenated DMF containing NBu4PF6 (0.1 M) as supporting electrolyte. Controlled potential electrolysis experiments were carried out in a two-compartment cell. The volume of solution (DMF, 0.1 M NBu4PF6) used in the working compartment of the cell was 8 mL. The working electrode used was a pool of mercury, separated from the coiled platinum wire counter electrode by a porous frit. Bulk electrolysis solutions were purged with N2 gas for at least 1 hour prior to electrolysis and stirred throughout bulk electrolysis experiment. During the experiment, the cell was continuously purged with nitrogen (5 mL min−1) and the output gas was analysed at ten-minute intervals in a Shimadzu GC-2010 pro gas chromatographer.
DFT calculations
We used the same computational protocol from out previous studies.23,24 Calculations were performed with the ORCA program package,28,29 with BP86/def2-TZVP(-f) as our level of theory.30–33 Following ORCA's convention, we used ‘Grid4’ and ‘TightSCF’ as our integration grid and convergence criteria. Solvent effects from DMF were included via CPCM.34 Free energy differences were obtained from numerical frequencies calculations (NumFreq in ORCA). We obtained the Gibbs free energy difference for the ferrocene reduction in DMF, to allow for direct comparison of reduction potentials reported experimentally against such reference, with ΔG(Fc+/0) = 4.87 eV. We used Chemcraft to create images for the 3D chemical structures and orbitals.35Tunneling calculations
We used the WKB approximation to obtain approximate reaction rates for H2 formation.36–38 The probability of tunnelling is given by:where a and b are the initial and final H–H distances for the protons to tunnel from one well to another of the PES. This interval is determined based on zero-point vibration energy differences between the ground and transition states of the proton tunnelling PES, which give an offset to the ground state, reflecting the vibration of the protons following the transfer through the PES. We calculated the difference between the ground and highest-energy states. As previously reported for NiTSC-OCH3,24 we took the highest-energy state as an approximation to the transition state. Although it possessed two imaginary frequencies, both are associated with the proton vibrations, as well as with the movement of TFA−. In the equation, V is the potential energy, E is the ground state energy, which includes ZPVE, and mp is the mass of the proton. We are considering twice the mass since both protons are attempting tunnelling through the barrier at once. To perform the PES, we took 33 points for distinct H–H distances, from 2.35 Å to 0.75 Å with intervals of 0.05 Å in between. Finally, the PES curve is obtained using a cubic spline, and the integral is solved numerically using the Fortran library QUADPACK, as implemented in the SciPy library.
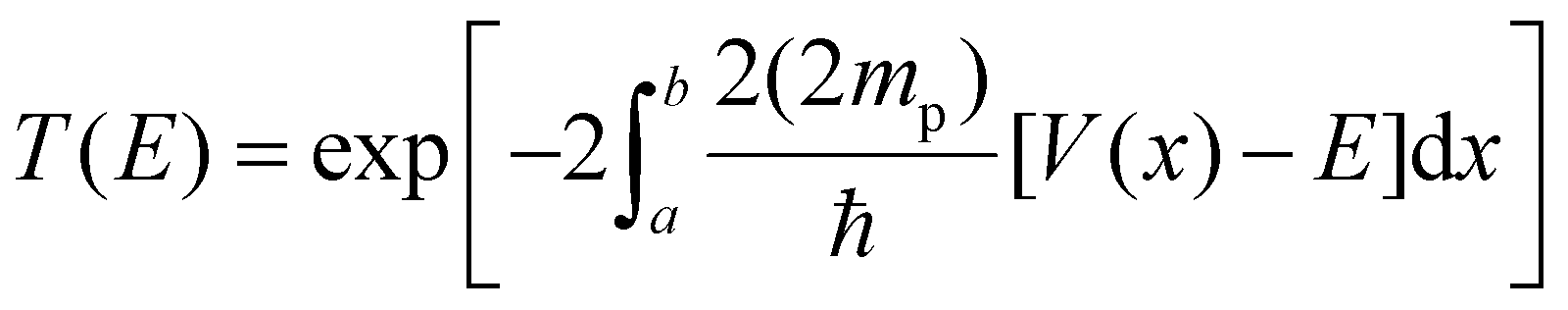
Author contributions
J. M., L. D. and M. P. carried out the experimental work and A. B. carried out the theoretical calculations. R. H., M. P. and M. O. analysed and interpreted the experimental and theoretical data. M. O. conceived and designed the project. M. P. and M. O. prepared the manuscript. All authors reviewed and contributed to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for NiTSC-OCH3 and CoTSC-OCH3 are deposited at the CCDC 1497214 and 1576527, respectively and can be obtained from https://www.ccdc.cam.ac.uk/.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge research support of this work by the French National Research Agency (ANR-19-CE05_0030_01).References
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, Coord. Chem. Rev., 2000, 206–207, 533–561 CrossRef CAS.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107–2116 CrossRef CAS.
- Z. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950–8951 CrossRef CAS PubMed.
- K. Weber, T. Krämer, H. S. Shafaat, T. Weyhermüller, E. Bill, M. Van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745–20755 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pécaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS.
- M. Razavet, V. Artero and M. Fontecave.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- E. J. Thompson and L. A. Berben, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS PubMed.
- N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274–282 CrossRef CAS.
- X. Jing, P. Wu, X. Liu, L. Yang, C. He and C. Duan, New J. Chem., 2015, 39, 1051–1059 RSC.
- C. F. Wise, D. Liu, K. J. Mayer, P. M. Crossland, C. L. Hartley and W. R. McNamara, Dalton Trans., 2015, 44, 14265–14271 RSC.
- A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, J. Am. Chem. Soc., 2016, 138, 7844–7847 CrossRef CAS.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS PubMed.
- T. Straistari, J. Fize, S. Shova, M. Réglier, V. Artero and M. Orio, ChemCatChem, 2017, 9, 2262–2268 CrossRef CAS.
- T. Straistari, R. Hardré, J. Fize, S. Shova, M. Giorgi, M. Réglier, V. Artero and M. Orio, Chem. – Eur. J., 2018, 24, 8779–8786 CrossRef CAS PubMed.
- M. Papadakis, A. Barrozo, T. Straistari, N. Queyriaux, A. Putri, J. Fize, M. Giorgi, M. Réglier, J. Massin, R. Hardré and M. Orio, Dalton Trans., 2020, 49, 5064–5073 RSC.
- M. Papadakis, J. Mehrez, I. Wehrung, L. Delmotte, M. Giorgi, R. Hardré and M. Orio, ChemCatChem, 2024, e202400426 CrossRef CAS.
- A. Barrozo and M. Orio, ChemPhysChem, 2022, 23, e202200056 CrossRef CAS.
- A. Barrozo and M. Orio, RSC Adv., 2021, 11, 5232–5238 RSC.
- M. Papadakis, G. Landrou, M. Poisson, L. Delmotte, K. Achileos, S. Bertaina, R. Hardré, K. Ladomenou, A. G. Coutsolelos and M. Orio, Eur. J. Inorg. Chem., 2023, 26, e202300352 CrossRef CAS.
- B. D. McCarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Inorg. Chem., 2014, 53, 8350–8361 CrossRef CAS.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B:Condens. Matter Mater. Phys., 1986, 34, 7406–7406 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- Chemcraft – Graphical software for visualization of quantum chemistry computations, https://www.chemcraftprog.com.
- P. R. Schreiner, H. P. Reisenauer, D. Ley, D. Gerbig, C.-H. Wu and W. D. Allen, Science, 2011, 332, 1300–1303 CrossRef CAS.
- M. Schäfer, K. Peckelsen, M. Paul, J. Martens, J. Oomens, G. Berden, A. Berkessel and A. J. H. M. Meijer, J. Am. Chem. Soc., 2017, 139, 5779–5786 CrossRef PubMed.
- H. Quanz and P. R. Schreiner, J. Comput. Chem., 2019, 40, 543–547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03507k |
| This journal is © The Royal Society of Chemistry 2025 |