
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated polyanion-layered oxide cathodes enabling 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) 000 cycle life for sodium-ion batteries†
000 cycle life for sodium-ion batteries†
Zhiyu
Zou‡
ab,
Yongbiao
Mu‡
ab,
Meisheng
Han
*ab,
Youqi
Chu
ab,
Jie
Liu
 ab,
Kunxiong
Zheng
ab,
Qing
Zhang
ab,
Manrong
Song
ab,
Qinping
Jian
c,
Yilin
Wang
ab,
Hengyuan
Hu
ab,
Fenghua
Yu
ab,
Wenjia
Li
ab,
Lei
Wei
ab,
Lin
Zeng
*ab and
Tianshou
Zhao
*ab
ab,
Kunxiong
Zheng
ab,
Qing
Zhang
ab,
Manrong
Song
ab,
Qinping
Jian
c,
Yilin
Wang
ab,
Hengyuan
Hu
ab,
Fenghua
Yu
ab,
Wenjia
Li
ab,
Lei
Wei
ab,
Lin
Zeng
*ab and
Tianshou
Zhao
*ab
aShenzhen Key Laboratory of Advanced Energy Storage, Department of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: hanms@sustech.edu.cn; zengl3@sustech.edu.cn; zhaots@sustech.edu.cn
bSUSTech Energy Institute for Carbon Neutrality, Southern University of Science and Technology, Shenzhen 518055, China
cDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China
First published on 23rd January 2025
Abstract
The practical application of Na3V2(PO4)3, a polyanionic cathode for sodium-ion batteries, is constrained by its poor electronic conductivity, limited specific capacity, and slow kinetics. In this study, an integrated polyanion-layered oxide cathode embedded within a porous carbon framework is designed. This cathode features an intergrown biphasic heterostructure, consisting of a Na-rich polyanionic compound, Na3.5V1.5Fe0.5(PO4)3 (NVFP), and a layered oxide, V2O3 (NVFP–VO), which is optimized to enhance Na-ion storage performance. Fe doping reduces the bandgap of Na3V2(PO4)3 and activates its V4+/V5+ redox couple, enhancing both electronic conductivity and specific capacity. The porous carbon framework further improves the electronic conductivity of the integrated cathode and accommodates volume fluctuations during cycling. The heterostructure lowers ion transport barriers and accelerates reaction kinetics. Additionally, the low-strain V2O3 phase functions as a stabilizer, effectively buffering volume fluctuations and stress gradients in NVFP. The spontaneous activation of V2O3 further increases the capacity of the integrated cathode. Consequently, the cathode achieves a high reversible capacity of over 130 mA h g−1 at 0.1C and exhibits unprecedented cyclability, maintaining over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with 72.6% capacity retention at 100C in half-cells. This represents the longest cycle life reported among polyanion-based cathodes. In addition, our prepared Ah-level pouch cells exhibit a high energy density of 153.4 W h kg−1 and a long cycle life exceeding 500 cycles. This study demonstrates that synergistic effects in multiphase integrated cathodes promote the development of advanced cathode materials for high-energy-density, fast-charging, and long-life sodium-ion batteries.
000 cycles with 72.6% capacity retention at 100C in half-cells. This represents the longest cycle life reported among polyanion-based cathodes. In addition, our prepared Ah-level pouch cells exhibit a high energy density of 153.4 W h kg−1 and a long cycle life exceeding 500 cycles. This study demonstrates that synergistic effects in multiphase integrated cathodes promote the development of advanced cathode materials for high-energy-density, fast-charging, and long-life sodium-ion batteries.
Broader contextThe Earth's lithium reserves are both limited and unevenly distributed, posing significant challenges in meeting the growing demand driven by global electrification efforts. In light of the limitations of lithium-ion batteries (LIBs), exploring alternative battery technologies has become essential. Sodium-ion batteries (SIBs) represent a promising alternative, garnering attention for applications in energy storage systems and low-speed electric vehicles due to the abundance of sodium resources and their low cost. Sodium-containing transition metal layered oxides, Prussian blue analogues, and polyanionic compounds are the main categories of cathode materials for SIBs. Among them, polyanion-type cathodes with robust and stable P–O covalent bonds are of particular interest because of their inherent safety, high redox potential, and chemical and thermal stability. However, the heavy 3D framework and insulated characteristics of [PO4] result in limited capacity delivery (<110 mA h g−1), low electronic conductivity, and slow reaction kinetics, which inevitably result in poor electrochemical performances. As a result, the development of advanced cathode materials with high capacity, long cycling life, and fast reaction kinetics is of great significance, but it remains a huge challenge. Here, an integrated polyanion-layered oxide cathode embedded into a porous carbon framework is designed and optimized to enhance Na-ion storage performance, which shows capacities far exceeding the theoretical capacity of Na3V2(PO4)3 and outstanding fast-charging capability along with a long cycling life in half and Ah-level pouch full cells. Besides, we reveal the spontaneous activation and transport mechanism of this integrated cathode by combining advanced characterization techniques and theory calculations, such as in situ X-ray diffraction, spherical aberration-corrected transmission electron microscope techniques, X-ray absorption near-edge structure, density functional theory calculations, and COMSOL multiphysics simulation. This work demonstrates that synergistic effects in integrated cathodes can drive the development of advanced cathode materials for high-energy-density, fast-charging, and long-life sodium-ion batteries. |
1. Introduction
The Earth's lithium reserves are both limited and unevenly distributed, posing significant challenges in meeting the growing demand driven by global electrification efforts.1 In light of the limitations of lithium-ion batteries (LIBs), exploring alternative battery technologies has become essential. Sodium-ion batteries (SIBs) represent a promising alternative, garnering attention for applications in energy storage systems and mini electric vehicles due to the abundance of sodium resources and their low cost.2–4 Among the various cathode materials of SIBs, polyanion-type cathodes with robust and stable P–O covalent bonds are of particular interest because of three major advantages: (i) inherent safety, (ii) high redox potential, and (iii) chemical and thermal stability.5 However, the heavy 3D framework and insulated characteristics of [PO4] significantly limit capacity delivery (<117 mA h g−1), restrict electronic conductivity, and slow reaction kinetics, which inevitably result in poor electrochemical performances.6 To address these challenges, tremendous efforts have been focused on strategies, such as carbon coating,7,8 structure engineering,9 and cation substitution.10–14Although these strategies can partially improve electrochemical performance, the primary challenge for many researchers remains achieving both high energy density and long cycle life simultaneously. To ensure long cycling capability, cathode materials must accommodate significant volume changes during sodium-ion de/intercalation without inducing permanent crystal structure alterations. This is because large lattice variations during Na+ intercalation and deintercalation can degrade the structure, leading to irreversible phase transitions that ultimately shorten the cycle life. Conversely, achieving high energy density necessitates the activation of additional redox couples or the incorporation of new phases into the reaction. To the best of our knowledge, the strategy of multiphase construction that simultaneously improves both capacity and cycling stability in polyanion phosphate cathodes has not been reported, as a trade-off commonly exists between cycling stability and energy density in cathode materials.
Recently, phase regulation strategies involving multiphase structures have emerged, offering new avenues for designing advanced battery cathode materials.15,16 When materials with different band gaps interact, spontaneous charge redistribution occurs at the heterointerface until the Fermi levels equalize.17 This interaction results in charge accumulation at the interface, forming a built-in electric field that accelerates charge transfer. The potential created by accumulated electrons and ions near the heterointerface can therefore activate a specific phase within multiphase materials. For example, Xu et al. reported a new iron-based phosphate cathode with an intergrown heterostructure of NaFePO4 and Na4Fe3(PO4)2(P2O7) phases. With such biphasic structure design, the cathode provided an enhanced capacity over 130 mA h g−1 and maintained a discharge capacity of 93 mA h g−1 at 20C.18 Besides, the capacity of as-prepared cathode composites was beyond that of the single-phase Na4Fe3(PO4)2(P2O7) cathode, which demonstrated the promise of multiphase structure design. Similarly, Zhang et al. also showed that incorporating the Na6Fe(SO4)4 phase inside the particle bulk Na2.26Fe1.87(SO4)3 can improve Na+ migration and lower the energy barriers leading to excellent cycling performance, which delivered a high initial discharge capacity (101.3 mA h g−1 at 60 mA g−1), good rate performance (73.5 mA h g−1 at 1200 mA g−1) and long cycling-stability (80.69% capacity retention after 1300 cycles). These results on the iron-based phosphate and sulfate-based cathode underscore the potential of synergistic effects in integrated cathodes to drive the development of advanced cathode materials. Recently, V2O3 (VO) as a cathode material has been broadly utilized in zinc-ion batteries due to its excellent rate performance and long cycling life,19,20 which arise from its multiple oxidation states and large open crystal structures that facilitate the insertion and extraction of metal ions, with their abundant vanadium valence, easily deformable V–O polyhedrons, tunable chemical compositions, and stable crystal structure. Due to these advantages, VO chosen as another important part of intergrown biphasic structures is capable of improving the performance of the cathode in SIBs. However, the fabrication of intergrown biphasic structures between the vanadium-based phosphate cathode and VO is still a challenge and has not been previously reported.
In this study, an in situ synthesis strategy was employed to achieve an intergrown heterostructure, which results in the electrochemical activation of V2O3, eliminating the need for extreme processing conditions (e.g., high-voltage constant charging, discussed later).17 With the nano-crystallized V2O3 embedded within the NVFP phase, the intergrown two-phase interfaces can optimize the heterostructure's band structure through a percolation process at the heterointerface. The optimized band structure facilitates Na+ transport through the V2O3 phase, enabling its electrochemical activation. Furthermore, the interfacial coupling effect promotes charge redistribution, accelerating ion and electron transport and increasing active sites, thereby enhancing both reversible capacity and rate capability. Additionally, further findings indicate that intercalation pseudocapacitance plays a crucial role in this integrated cathode.21 Encouragingly, this multiphase structure reduces the Na+ transport barrier, enhances reaction kinetics, and offers exceptional structural stability. Ultimately, the Na-rich integrated polyanion-layered oxide (NVFP–VO) cathodes deliver a high discharge capacity of over 130 mA h g−1 and long-term cyclability exceeding 100000 cycles at 100C which, to the best of our knowledge, represents the longest cyclability reported for polyanion phosphate cathodes. More importantly, the promising application of NVFP–VO cathodes is demonstrated by their scalable synthesis, and the pouch full cells deliver superior cycling performance over 500 cycles at 1C. This study may pave the way for the future development of advanced multiphase integrated cathodes for fast-charging and long-life SIBs.
2. Results and discussion
2.1 Material design and characterizations
A sol–gel-assisted solid-state method followed by high-temperature calcination in an argon atmosphere was employed to synthesize the NVFP and NVFP–VO compounds. Detailed synthesis procedures for both samples are provided in the Experimental section (ESI†). Fig. 1a provides the X-ray diffraction (XRD) Rietveld refinement result of NVFP–VO, showing a favorable weighted profile Rwp factor of 4.075%, validating the reliability of the structural analysis. Unlike the undoped NVP phase and single-phase NVFP structure (Fig. S1, ESI†), the NVFP–VO sample crystallized in a heterogeneous structure, consisting of the rhombohedral NVFP phase with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group (Fig. 1b) and a secondary phase of V2O3 with the same rhombohedral crystal system and Rc space group (Fig. 1c). Rietveld refinement reveals a phase ratio of approximately 90.7:9.3 by weight for NVFP and V2O3 structures, with detailed refinement factors and structural data on atomic coordinates, occupancy values, and cell parameters provided in Table S1 (ESI†).
c space group (Fig. 1b) and a secondary phase of V2O3 with the same rhombohedral crystal system and Rc space group (Fig. 1c). Rietveld refinement reveals a phase ratio of approximately 90.7:9.3 by weight for NVFP and V2O3 structures, with detailed refinement factors and structural data on atomic coordinates, occupancy values, and cell parameters provided in Table S1 (ESI†).
 | ||
| Fig. 1 Structural characterizations of NVFP–VO. (a) XRD Rietveld refinement of NVFP–VO. (b) and (c) Crystal structure illustration of NVFP, V2O3, and NaV2O3. (d) V 2p XPS and (e) Fe 2p XPS. (f)–(g) EELS of the V L-edge and Fe L-edge along different distances of the scanning direction. (h) HAADF-STEM image in the EELS scanning direction. (i) and (j) Representative SEM. (k) and (m) TEM images of the carbon layer and intergrown interfaces. (l) HAADF-STEM image of the heterostructure. (n) and (o) Atomic-resolution HADDF-STEM image enlargement of phase 1 and phase 2 viewed along the zone axis [−441]. | ||
To investigate further structural information of the as-prepared NVFP–VO, Raman and Fourier transform infrared (FTIR) analyses were conducted. The Raman spectra of all samples (Fig. S2, ESI†) clearly display two characteristic carbon peaks, corresponding to the D-band (1350 cm−1) and G-band (1590 cm−1). The peaks located at around 431 and 1000 cm−1 can be attributed to stretching vibrations of PO4 groups.11 Furthermore, the ID/IG value of NVFP–VO is 1.09, suggesting partial graphitization of the carbon skeleton formed from citric acid pyrolysis during calcination, which can efficiently improve the electronic conductivity of the composite cathode.22 Further chemical composition and specific bond information were analysed using FTIR as shown in Fig. S3 (ESI†). Two peaks at 579 and 1181 cm−1 are attributed to the PO4 tetrahedron (orange dashed line), while the broad band around 1012 cm−1 is also ascribed to PO4 units (orange area). Two additional signals near 538 and 630 cm−1 (blue dash line), are associated with the elastic vibrations M–O (M = Fe, V) bonds. Although similar signal peaks appear in the Raman and FTIR spectra for all samples, NVFP–VO shows lower signal intensity compared to NVP and NVFP. This reduction in intensity may be attributed to the high symmetric crystal structure of V2O3, which limits certain vibrational modes from being Raman- or IR-active due to symmetry constraints and the lack of a significant dipole moment.23
After removing the effect of adsorptive water and the oxidation of low-valence metal ions (Fe2+ and V3+), thermogravimetric (TG) analysis (Fig. S4, ESI†) shows that the carbon content of NVFP–VO is about 4.87 wt%, slightly lower than the traditional carbon content (>10 wt%) in NASICON-type cathode materials.24 A higher carbon content may reduce electrode compaction density and hinder Na+ ion migration across the electrode–electrolyte interface.7 Furthermore, specific surface area and pore size were measured using the nitrogen isothermal adsorption–desorption method (Fig. S5, ESI†). The presence of a hysteresis loop in NVFP–VO indicates a complex pore structure, containing both mesopores and macropores. According to the Brunauer–Emmett–Teller (BET) method, NVFP–VO displays a specific surface area of 22.452 m2 g−1 and a local pore size of about 8.423 nm, which facilitates electrolyte penetration and full capacity utilization in electrodes.25
Subsequently, X-ray photoelectron spectroscopy (XPS) was conducted to preliminarily identify the surface chemical/oxidation states of samples, as shown in Fig. 1d and e. The peaks at 516.1 eV (V 2p1/2) and 523.7 eV (V 2p3/2) in the V 2p spectrum align closely with the theoretical values for V3+, while the binding energies of Fe2+ and Fe3+ appearing simultaneously, indicate slight Fe oxidation and mixed valence state coexistence in NVFP–VO, similar to other reported Fe-based phosphate cathodes.26,27 Additionally, the full XPS survey with all identified elements and deconvoluted C 1s spectra are shown in Fig. S6 (ESI†). The element concentrations of all samples were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Table S2, ESI†), and the results show that the chemical composition of integrated NVFP–VO is acceptable, consistent with the designed chemical composition, with a higher vanadium content than single phase NVFP, as expected. Additionally, electron energy loss spectroscopy (EELS) was employed to examine the Fe and V atomic concentration distribution within a targeted area of NVFP–VO. The Fe content nearly disappears while V becomes concentrated along the scanning direction, indicating a heterogeneous elemental distribution transitioning from the NVFP phase to the V2O3 phase (Fig. 1f–g), with the scanning direction shown in Fig. 1h. Scanning electron microscopy (SEM) reveals that the morphological characteristics of NVFP–VO particles (Fig. 1i and j) differ slightly from those of NVP and NVFP samples (Fig. S7 and S8, ESI†). NVFP–VO particles exhibit larger particle sizes and a denser structure, which contribute to a higher compaction density and an optimal specific surface area. These features are likely to enhance the material's performance by improving ion transport pathways and facilitating better electrochemical activity. Energy-dispersive spectroscopy (EDS) mapping demonstrates even elemental distribution in most samples, with the exception of NVFP–VO. This variation is likely attributed to the heterogeneous phase composition, which may result in slight differences in Na distribution on the surface of NVFP–VO (Fig. S9, ESI†).
Furthermore, the morphology and heterostructure of NVFP–VO were examined using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and high-resolution transmission electron microscopy (HR-TEM). Fig. 1k shows a uniform carbon layer coating on the particle surface, with a thickness of 5–8 nm. Spherical aberration-corrected TEM (STEM) and HR-TEM images clearly reveal the presence of a porous matrix structure, along with biphasic intergrown interfaces and heterostructures between NVFP and V2O3 (Fig. 1l and m). A similar porous matrix structure is also observed in another particle (Fig. S10, ESI†). This porous matrix structure may be attributed to the thermal decomposition of citric acid during calcination, which generates significant gas, acting as a pore-forming agent. In locally magnified STEM images, two distinct parallel lattice fringes with d-spacings of 4.42 Å and 4.36 Å are observed, corresponding to the (104) and (110) planes of rhombohedral NVFP (Fig. 1n). Additional lattice fringes with d-spacings of 2.71 Å and 2.47 Å also correspond to the (104) and (110) planes of the V2O3 phase (Fig. 1o).
The above results indicate the successfully synthesis of an integrated Na-rich polyanion Na3.5V1.5Fe0.5(PO4)3, low-strain layered-oxide V2O3, and a porous carbon framework as a heterogeneous structural cathode material. Such biphasic intergrown interfaces facilitate Na+ transportation and storage on the surface or near-surface region of the V2O3 phase, reducing polarization and optimizing electrode material utilization during rapid charge/discharge processes. This is essential for the pseudo-capacitance process and the further activation of the entire V2O3 particle in the composite cathode.18,28
2.2 Sodium storage performances
Initially, the electrochemical stability of the electrolyte was evaluated using linear sweep voltammetry (LSV, Fig. S11, ESI†). The results indicate that the electrolyte begins to decompose at approximately 3.8 V, with the decomposition rate accelerating after 4.8 V. Consequently, the electrochemical performance of NVFP–VO was measured and compared with that of NVP across various voltage windows (2–3.8 V, 2–4.1 V, and 2–4.3 V), as shown in Fig. S12 (ESI†). As the charging cut-off voltage increases, NVFP–VO electrodes achieve higher specific capacities, likely due to the activation of V2O3 and the V4+/V5+ redox couple, while no similar trend is observed for NVP. On the one hand, side reactions and electrolyte decomposition intensify as the operating voltage increases. On the other hand, to remain within the power converter's operating range, the minimum battery voltage should not fall below 50–55% of its maximum voltage, according to the U.S. Advanced Battery Consortium.29 To balance these factors, most electrochemical performance tests were conducted within the 2–4.1 V range. As shown in the charge–discharge curves of NVFP–VO (Fig. 2a), which deliver initial discharge capacities of 128.3 mA h g−1 at 0.2C (20 mA g−1) within the 2.0–4.1 V range. | ||
| Fig. 2 Electrochemical properties of NVFP–VO at the voltage window of 2–4.1 V. (a) Charge–discharge profiles and (b) CV curves at various scan rates from 0.1 to 2.0 mV s−1. (c) log(v) vs. log(i) plots for the different peaks in the CV curves. (d) Cycling stability at 0.2C and (e) rate capability from 0.1 to 100C. (f) Ragone plots and (g) long cycling stability at 100C. (h) 3D plots for comparison between NVFP–VO and other reported polyanionic cathodes. (i) Cycling performance of the NVFP/HC pouch cell at 1C in the range of 1.7–4.1 V at 25 °C. Note that reducing the low cut-off voltage from 2 V to 1.7 V is mainly attributed to the higher cut-off voltage of the HC anode. Inset: Photo of the as-synthesized lab-scale NVFP–VO products and an assembled pouch cell. | ||
To investigate the capacity differences among NVP, NVFP, and NVFP–VO cathodes, we analyzed the charge–discharge profiles and dQm/dV curves after ten cycles (Fig. S13, ESI†). The charge–discharge curves of both NVFP and NVP cathode materials exhibit three similar voltage plateaus around 2.5, 3.4, and 4.1 V, while NVFP–VO curves lack a prominent voltage plateau in the high-voltage region compared to NVFP. This suggests that the intergrown heterostructure effectively suppresses deleterious phase transitions and accompanying lattice mismatches in the high-voltage region, as confirmed by in situ XRD analysis discussed later. The results indicate that the in situ activation of V2O3 nanoparticles in NVFP–VO composites contributes to the incremental capacity. The broader peaks in the dQm/dV curves of NVFP–VO suggest a wider distribution of reaction potentials, potentially reflecting the pseudocapacitive behavior in V2O3.20 Additionally, the multiphase integrated strategy also influences the doping of Fe, as confirmed by ICP-OES (Table S2, ESI†), which enhances the capacity and reversibility of Fe2+/3+ reactions.
To further investigate the electrochemical behavior of V2O3, we conducted standalone electrochemical tests on V2O3 (Fig. S14, ESI†). Before spontaneous activation, V2O3 delivers a reversible capacity of ∼4.5 mA h g−1. After spontaneous activation, its capacity increases to ∼154.3 mA h g−1 but stabilizes at ∼14.8 mA h g−1 due to rapid decay. The dQm/dV curves confirm that incremental capacity is associated with the activated V2O3 phase. Furthermore, both physically mixed NVP-V2O3 and NVFP-V2O3 cathodes (9:1 by weight) were subjected to electrochemical testing (Fig. S15, ESI†). Similarly, the simply mixed NVP-V2O3 cathode undergoes spontaneous activation, showing a discharge capacity of ∼110 mA h g−1 after activation (137.5% higher than the pristine material) with no significant decay. Similar trends were observed for physically mixed NVFP-V2O3 cathodes, with incremental capacity contributed to the activation of the V2O3 phase. However, the discharge capacity of both physically mixed cathodes were significantly lower than that of the in situ synthesized NVFP–VO with the intergrown NVFP and V2O3 heterostructure. Notably, during spontaneous activation, additional Na-ion insertion is required for the phase transition from V2O3 to NaV2O3, resulting in lower Coulombic efficiency during the activation process. These results highlight the distinct mechanisms of the in situ activated V2O3 phase in NVFP–VO composites compared to the ex situ activated V2O3 in mixed cathodes. The superior performance of NVFP–VO is attributed to the stable intergrown heterostructure and uniform carbon encapsulation achieved during in situ synthesis.
To investigate the pseudocapacitive characteristics of NVFP–VO, cyclic voltammetry (CV) measurements were conducted at various scan rates within a voltage window of 2.0–4.1 V vs. Na+/Na. At a scan rate of 0.1 mV s−1, three pairs of broad redox peaks are observed at 2.59/2.23 V, 3.56/3.29 V, and 4.06/3.96 V, corresponding to the Fe2+/Fe3+, V3+/V4+, and V4+/V5+ redox couples, respectively, indicating a three-step Na+ (de)intercalation process (Fig. 2b). Additionally, kinetic information was obtained through CV sweep voltammetry. The calculated b-values for NVFP–VO electrodes (Fig. 2c) are significantly higher than those of NVP (Fig. S16, ESI†), indicating that capacitive processes dominate the charge–discharge behavior of NVFP–VO electrodes, driven by the pseudocapacitive characteristics of V2O3, as demonstrated in previous studies.21,30,31Fig. 2d shows the cycling capabilities of NVFP–VO electrodes at 0.2C. Using 1 M NaClO4 in PC and 5 vol% FEC electrolytes in half-cells, the initial discharge capacity of the NVFP–VO cathode is 128.3 mA h g−1 with an initial Coulombic efficiency exceeding 100% due to the Na-rich design strategy. However, a relatively low Coulombic efficiency is observed during the first 40 cycles, attributed to the activation of V2O3, as discussed in Fig. S15 (ESI†). The rate performance comparison highlights the superior electrochemical performance of NVFP–VO (Fig. 2e and Fig. S17, and S18, ESI†). Even at an ultra-high current density of 100C, the NVFP–VO electrode maintains excellent rate performance, with a specific capacity of ∼81.7 mA h g−1 (∼60% retention compared to at 0.1C) and recovers to 128.6 mA h g−1 (∼97% retention of the pristine value) when the rate is reduced back to 0.1C. The Ragone plot in Fig. 2f highlights the outstanding energy and power densities of NVFP–VO. The calculated energy density, based on the average voltage for standard comparisons, is approximately 403.2 W h kg−1 over a voltage range of 2.0–4.1 V at 0.1C, exceeding that of most V-based, Fe-based, and other reported polyanionic cathodes within a similar voltage window (Table S3, ESI†). Following activation at 1C for the first ten cycles, NVFP–VO demonstrates excellent long-term cycling performance, with 99.3% capacity retention after 10000 cycles at 50C (Fig. S19, ESI†). Even more impressively, NVFP–VO exhibits a gradually increasing capacity at a current density of 100C, rising from 60.4 mA h g−1 to 83.2 mA h g−1 with 92.2% retention after 60000 cycles, outperforming NVP (80.9% retention after 10000 cycles), NVFP (28.8% retention after 60000 cycles), and most advanced polyanionic cathodes (Fig. 2g). Notably, 60000 cycles is the maximum range of our test instrument, thus we restarted the test until the capacity retention dropped below 80%, eventually reaching 100000 cycles. To the best of our knowledge, NVFP–VO exhibits the longest cycling performance among various advanced cathodes (Fig. 2h), highlighting its potential for fast-charging and long-life SIBs. Furthermore, achieving higher loading densities is crucial for practical applications. Remarkably, even at a high rate of 20C, the NVFP–VO composite cathode demonstrates a considerable capacity of approximately 70 mA h g−1, coupled with outstanding capacity retention of 95.36% after 7000 cycles at a high loading mass of ∼5.0 mg cm−2 (Fig. S20, ESI†). This result highlights the exceptional cycling stability of NVFP–VO, even in thick electrode configurations.
The exceptional electrochemical performance of NVFP–VO in half-cells motivates further exploration of its practical applications. Before fabricating pouch cells, comprehensive characterization of commercial hard carbon anodes (Kuraray Co. type 2) was carried out (Fig. S21, ESI†). The Na storage performance of the hard carbon anode was evaluated using the same electrolyte as in the half-cells. Fig. S22 (ESI†) illustrates the charge/discharge curves of hard carbon, which exhibit an initial Coulombic efficiency of 67.5%, along with its rate performance. At high current densities exceeding 10C (1C = 300 mA h g−1), the capacity of hard carbon significantly decreases. When tested at 1C, only a reversible capacity of approximately 70 mA h g−1 is retained. Notably, the electrochemical performance of hard carbon is highly dependent on the electrolyte used. When the same commercial hard carbon anodes were tested with different ester and ether electrolytes, without any additives, markedly different performance was observed (Fig. S23, ESI†). However, to maintain consistency with previous studies and the half-cell tests, the electrolyte was not altered.14,18,32
As shown in Fig. 2i, Ah-level pouch cells measuring 130 × 50 mm were assembled to examine cycle stability (Table S4, ESI†). After industrial pretreatment of the electrodes (battery formation and aging), the pouch cell demonstrated a reversible capacity of approximately 0.74 Ah at 1C between 1.7 and 4.1 V. Moreover, it maintained an impressive retention rate of 77.38% after 500 cycles, highlighting the significant application potential of the NVFP–VO cathode. Additionally, the charge–discharge profile of the NVFP–VO pouch cell at 0.2C (Fig. S24, ESI†) shows a reversible capacity of ∼1.2 Ah, achieving an energy density of 153.4 W h kg−1 based on the active materials of both the cathode and anode. The Na-rich cathode design leads to a slight capacity increase during the initial cycling process, compensating for the Na+ consumed during SEI layer formation on the hard carbon anode, improving the initial Coulombic efficiency. It should be noted that pouch cell degradation over 500 cycles is not attributed to structural failure of the NVFP–VO cathode, but rather to the limitations of the hard carbon anode and electrolyte system. Further optimization of the hard carbon and electrolyte is expected to significantly improve rate capability and cycling performance, which will be explored in our future work.
Based on these electrochemical results, the integrated polyanion-layered oxide cathode embedded in a porous carbon framework effectively enhances structural stability and kinetic performance. To further optimize the cathode materials, the integrated cathodes were synthesized at different calcination temperatures and with varying initial vanadium content, as shown in Fig. S25 and S26 (ESI†). Through this optimization process, we found that thermodynamically stable biphasic compounds were only produced at calcination temperatures of 700 °C. At lower sintering temperatures, the crystallinity was insufficient, and fewer intergrown biphasic heterostructures were formed, as shown in Fig. S25 (ESI†). On the other hand, increasing the sintering temperature beyond 700 °C led to the formation of impurity phases (highlighted with blue markers). We also varied the initial vanadium content in the precursors. However, we observed that further increasing the vanadium content did not significantly increase the V2O3 content in the biphasic compounds. In contrast, this change negatively affected the doping of iron. This suggests that the system reached a saturation point in terms of V2O3 incorporation, as confirmed by ICP-OES (Table S2, ESI†). Besides, excess V2O3, beyond the optimal content, may adversely impact the overall electrochemical performance by introducing inactive phases.
Generally, electrostatic repulsion between Na+ ions and the transition metal (TM) configuration significantly influences the cathode's electrochemical behavior. The partial substitution of low-cost iron at vanadium sites not only reduces material costs but also decreases local electrostatic repulsion when iron atoms occupy octahedral sites.11,14 Additionally, the Kelvin probe force microscope (KPFM) analysis reveals distinct differences between NVFP and NVFP–VO samples. As shown in Fig. S27 (ESI†), NVFP–VO exhibits a heterogeneous surface potential distribution, with significant variations compared to the uniform potential in NVFP. The local surface potential profile of NVFP–VO shows a sharp decline, indicating that the heterostructure modification significantly alters the electronic structure and enhances surface charge distribution. This finding confirms the presence of a built-in electric field, which may improve electrochemical performance by reducing electrostatic interactions between the host material and ions, accelerating reaction kinetics and facilitating the reversible storage of sodium ions.17 This integrated design endows the NVFP–VO cathode with excellent fast-charging capability and cycling stability. Given the limited reports on integrated cathodes with heterostructure applications in SIBs, the storage mechanism of this novel cathode will be the focus of the following discussion.
2.3 Sodium storage mechanism
To further elucidate the sodium-ion storage mechanisms, in situ XRD patterns of NVFP–VO during the initial two charge/discharge cycles within a voltage range of 2.0–4.1 V were recorded to investigate the structural evolution during de/sodiation. Fig. 3a shows that all diffraction peaks can be attributed to the heterogeneous structure of rhombohedral NVFP (phase 1) [e.g., (012), (104), (113), (024), (211), (116), and (300) crystal faces] and rhombohedral V2O3 (phase 2) [e.g., (012), (104), and (110) faces], which undergo a series of reversible changes during de/sodiation, including peak shifts, splitting, and merging. Upon charging to 3.4 V, the peaks assigned to the NVFP phase shift to higher degrees as Na+ is extracted and the Fe2+ is oxidized to Fe3+. Within this voltage range, all diffraction peaks reversibly return to their initial positions during subsequent discharge, demonstrating a quasi-solid solution electrochemical reaction.11 As charging progresses around the voltage plateau at 3.4 V, the splitting of the (104) peak into two new peaks and the disappearance of (024), (211), and (300) peaks indicate a typical biphasic reaction mechanism associated with the oxidation process of V3+/4+, which usually involves additional kinetic barriers for phase transition initiation and boundary propagation.14 With further charging, the V4+/V5+ redox process occurs, and most diffraction peaks exhibit continuous and symmetric shifts during both charging and discharging. This behavior is particularly evident in the intensity and position of the (113) diffraction peak, which changes abruptly within the narrow voltage range of 3.6–4.1 V (sites 1 and 2). However, as shown in the contour maps, the characteristic shoulder peaks of the (012), (104), and (110) crystal planes of V2O3 remain nearly unchanged in both sodiated and desodiated states, indicating typical pseudocapacitive responses, where sodium ions intercalate into V2O3 tunnels through faradaic charge transfer and without crystallographic phase changes.21,33 Specifically, during V2O3 pseudocapacitive responses in the redox reactions, V2O3 serves as a stabilizer, effectively buffering volume changes and stress gradients caused by NVFP's phase transformations, thereby contributing to the cathode's strong cycling stability.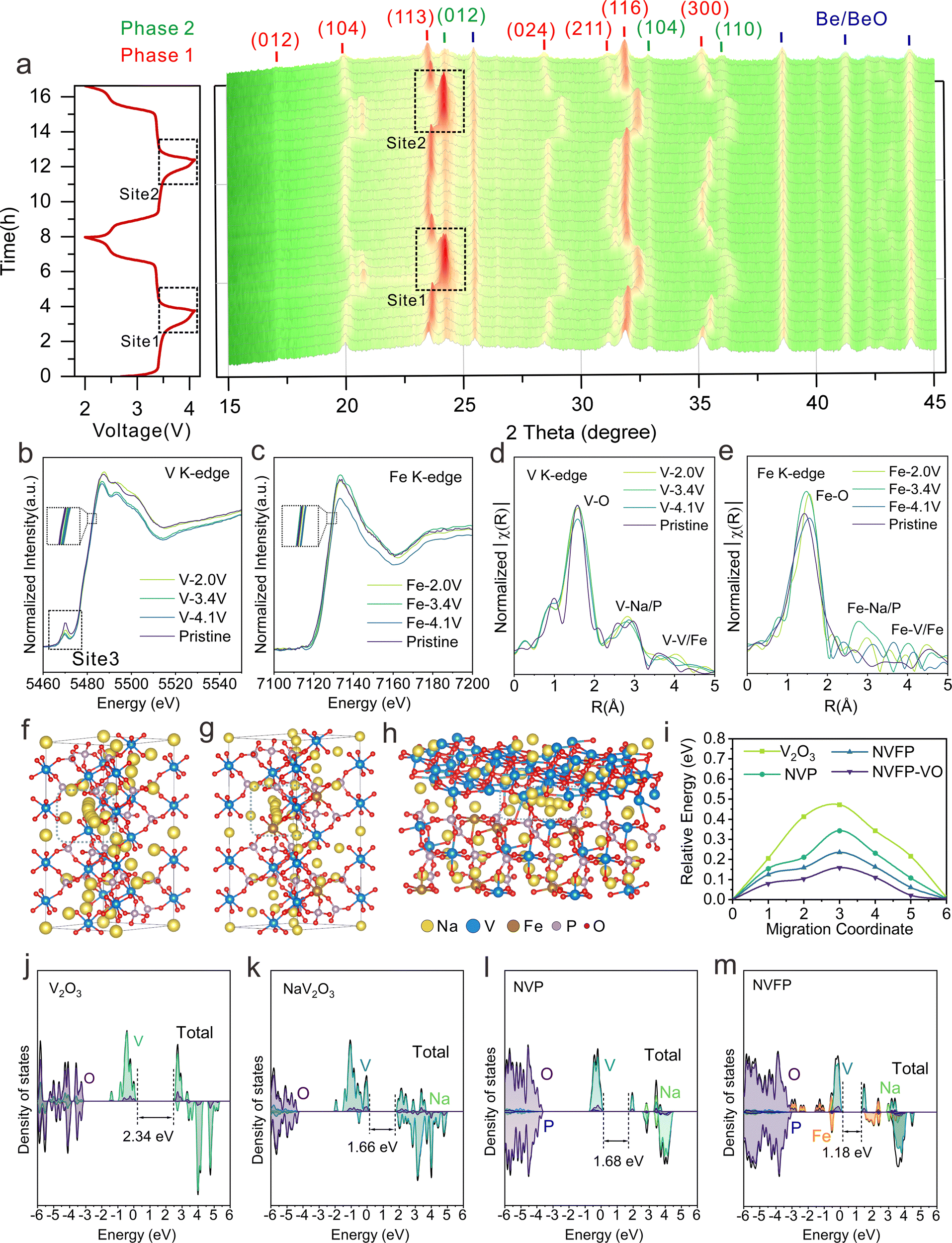 | ||
| Fig. 3 Sodium storage mechanism of NVFP–VO. (a) in situ XRD contour map and corresponding charge/discharge curves of the NVFP–VO electrode collected during the first two cycles at 0.1C. Ex situ XANES spectra at the (b) V K-edge and (c) Fe K-edge. The corresponding normalized ex situ EXAFS spectra at the (d) V K-edge and (e) Fe K-edge. Schematic diagram of the Na+ migration trajectory in (f) NVP, (g) NVFP, and (h) NVFP–VO heterostructures and (i) the corresponding migration energy barriers. Total and partial DOS of (j) V2O3, (k) NaV2O3, (l) NVP and (m) NVFP. | ||
Additionally, the detailed structural evolution and stepwise redox reactions of the NVFP–VO cathode were systematically investigated using ex situ X-ray absorption near-edge structure (XANES) spectra and ex situ XPS. Compared with NVP and NVFP, the XANES spectrum of the V K-edge of the pristine NVFP–VO electrode (Fig. 3b, site 3, and Fig. S28, ESI†) shows a highly distorted environment with the highest pre-edge intensity, which may be attributed to varying crystal configurations of the biphasic design in the NVFP–VO electrode, with structural distortions around V atoms resulting from the heterostructure.34 Besides, compared with its pristine state, the absorption spectrum of the V K-edge for NVFP–VO shifts to a higher energy during charging to 4.1 V, indicating a progressive oxidation from V3+ to V4+ and even partial V5+. After discharging to 2 V, the spectrum is back to the initial energy, proving high reversibility of the evolution of the V oxidation state. The spectra of Fe K-edge also show similar evolution processes. As shown in Fig. 3c, the XANES spectra of Fe K-edge first shift to a higher energy position of Fe3+ upon charging to 4.1 V, then return to the initial energy position upon subsequent discharging to 2.0 V, indicating a reversible Fe2+/Fe3+ redox process during the de/sodiation of the NVFP–VO electrode. Additionally, extended X-ray absorption fine structure (EXAFS) spectroscopy indicates that the EXAFS signal intensity in NVFP–VO electrodes decreases upon charging to 4.1 V, as shown in Fig. 3d and e, with reductions in the interatomic distances of V–O and Fe–O bonds attributed to Na+ extraction from the crystal lattice during oxidation. Other prominent peaks between 2–5 Å correspond to TM-Na/P and TM–TM (TM = V or Fe) shells.14,34 Notably, the EXAFS data (Fig. 3e) show irregularities in Fe–metal shells compared to the Fe–O shell, which may be ascribed to the following points. First, the local structural amorphization (confirmed below) around Fe during cycling may result in irregular scattering paths and broader distributions of Fe–Fe bond lengths, thus causing the irregularities in Fe–metal shells. Second, the presence of multiple coordination environments involving metals (V and Na) or nonmetals (P and O) around Fe may introduce diverse scattering pathways and increase irregularities. Third, the electrochemical cycling-induced stress due to repeated (de)sodiation may result in the change of Fe coordination environments such as bond lengths and coordination numbers. Such stress-induced heterogeneity might lead to the irregularities. Last, the relative regularity of Fe–O bonds compared to the disorder in Fe–metal shells may indicate the selective breaking of metallic bonds in localized regions. This could occur due to structural rearrangements that preserve oxygen coordination around Fe while disrupting metallic interactions, thus causing irregularities in Fe–metal shells. For cathode materials, the dissolution of transition metals during cycling is common, often leading to a decrease in cycling stability. To further demonstrate the advantages of the constructed heterostructure, the V and Fe dissolution amounts of NVFP and NVFP–VO electrodes after 10000 cycles at 100C were determined through ICP-OES. Specifically, the disassembled electrodes were immersed in propylene carbonate solvent for 24 hours and subsequently diluted to ensure accurate concentration measurements. As shown in Table S5 (ESI†), the dissolution amounts of V and Fe of the NVFP–VO electrode are lower than those of NVFP, indicating that the biphasic heterostructure can effectively suppress V and Fe dissolution, thus stabilizing the structure and enhancing cycling stability. The stepwise Fe2+/Fe3+ redox process was further characterized through ex situ XPS analysis (Fig. S29, ESI†). By integrating XPS and XANES findings, we offer a comprehensive view of the oxidation states in both surface and bulk regions. Based on these studies, it can be concluded that the NVFP–VO electrode undergoes highly reversible V3+/V4+/V5+ and Fe2+/Fe3+ redox processes during electrochemical reactions, ensuring structural stability, which favors the enhancement of cycling stability.
To further explore the origins of NVFP–VO's exceptional performance, galvanostatic intermittent titration technique (GITT) measurements were conducted to study the Na+ ion diffusion coefficients (DNa+) of the electrodes after initial activation cycles. As shown in Fig. S30 (ESI†), the DNa+ of NVFP–VO demonstrates favorable Na storage kinetics, particularly in the low-voltage region, where the overpotential significantly decreases. The DNa+ of NVFP–VO is approximately 10−8 cm2 s−1, significantly higher than that of NVP, which is around 10−12 cm2 s−1, at the V3+/V4+ plateau, with calculation details provided in Fig. S31 (ESI†). Additionally, electrochemical impedance spectroscopy (EIS) was conducted to assess the electronic and ionic conductivities of the electrodes. As shown in Fig. S32 and Table S6 (ESI†), the fitting results reveal a lower charge transfer resistance for the NVFP–VO cathode, confirming that the integrated cathodes’ design strategy accelerates both ion and electron transport.
2.4 Density functional theory calculations
Density functional theory (DFT) calculations offer a comprehensive understanding of the roles of Fe and V2O3 within NVFP–VO from an electronic structure perspective, with detailed calculations provided in the ESI.† Using the nudged elastic band (NEB) method, four possible Na+ migration pathways were constructed in NVP, NVFP, NVFP–VO and V2O3 (Fig. 3f–h and Fig. S33, ESI†), with calculated maximum energy barriers of 0.472, 0.342, 0.235, and 0.159 eV, respectively (Fig. 3i). This gradual decrease in energy barriers aligns with the electrochemical results, and two key factors contribute to this trend. First, the lower migration energy barriers of Na+ near Fe suggest that Fe ions influence adjacent sodium ion diffusion, thereby enhancing ionic conductivity. Second, the biphasic heterostructure interface integrates the mechanical forces generated by electrochemical reactions in each phase, intertwining their Na+ transport mechanisms and leading to enhanced Na+ transport properties. Therefore, the low diffusion energy barrier underpins the material's exceptional rate performance. Furthermore, the intrinsic electronic band structures of V2O3, NaV2O3, NVP and NVFP were analyzed using DFT, as shown in Fig. 3j–m. The total density of states (TDOS) and partial density of states (PDOS) indicate that the NaV2O3 phase has a smaller band gap than V2O3. This phenomenon likely arises from a Mott transition of the electronic structure, transitioning from a localized to a delocalized state, due to unique percolation at the heterogeneous interfaces. This reduces the band gap through the built-in electric field at these interfaces. Additionally, the calculated band gaps for NVFP demonstrate that Fe doping contributes to a reduced forbidden band gap. As shown in Fig. 3m, the unpaired 3d electrons of Fe contribute significantly near the Fermi energy level and shift the DOS of V toward the conduction band. This suggests that Fe doping introduces additional electronic states, which alters the electronic structure and enhances the electrical conductivity of the material. Consequently, the bandgap of NVFP is reduced to 1.18 eV, compared to the undoped NVP (1.68 eV). Moreover, discrepancies and asymmetry in the spin-up and spin-down spectra near the Fermi energy level indicate the presence of spin polarization and magnetism, likely due to local magnetic moments introduced by Fe doping. To further substantiate this conclusion, we also performed ultraviolet photoelectron spectroscopy (UPS) tests, and the results support the observed changes in the electronic structure. As shown in Fig. S34 (ESI†), the valence band maximum energy (EVBM) was determined by observing the initial rise of the UPS curve. Comparison of the UPS spectra reveals that the EVBM of NVFP and NVFP–VO are closer to the Fermi energy level, indicating easier electronic transitions and potentially better conductivity. The left-shifted peaks observed for NVFP and NVFP–VO suggest a higher and more concentrated valence band electron density. These results align with the DFT calculations, further confirming the beneficial impact of Fe doping on the electronic structure of NVP.112.5 COMSOL multiphysics simulation
To elucidate the mechanisms enhancing electrode kinetics, conductivity, and mechanical stability in the NVFP–VO cathode, multiphysics simulations were employed to analyze ion concentration distribution, sodiation-induced stress/strain, and electrode current density (with detailed computational models provided in the ESI†). Prior to simulation, the Young's modulus and electrical conductivity of all cathode materials were measured using the single particle crush test and powder conductivity test (PCT). To minimize complications arising from system-level interfaces between active materials, conductive agents, binders, and electrolytes during the single-particle crush test, appropriate simplifications were made, wherein only NVP, NVFP, and NVFP–VO particles were tested. By doing so, the results only reflect the intrinsic mechanical behavior and structural stability of the active materials alone.Fig. 4a–c and Fig. S35, and Video S1 (ESI†) demonstrate that NVFP–VO undergoes secondary particle fragmentation, indicating a complex internal biphasic structure.35 PCT results indicate that the conductivity of NVFP–VO increases more rapidly than that of other cathodes as compaction density increases, suggesting that the heterostructure effectively enhances electronic conductivity (Fig. S36, ESI†). In the multiphysics simulations, three idealized models were constructed: the NVP model, the NVFP model, and the NVFP–VO model. Notably, the NVFP–VO model exhibits a higher ion concentration distribution compared to the NVP and NVFP models, owing to its faster ion and electron transfer rates (Fig. 4d1–f1 and Fig. S37, ESI†). The stress/strain distribution is coupled with ion concentration distribution and is influenced by the electrode's electronic conductivity and ion transport characteristics.36 Following sodiation, the stress/strain distribution in the NVP and NVFP models becomes significantly uneven, with higher values at the center and lower values near the edges (Fig. 4d2–e2). In contrast, no stress escalation is observed in the NVFP–VO model (Fig. 4f2), indicating that V2O3 not only enhances ion transfer but also acts as a buffer, dispersing residual stress and alleviating volume expansion. Further analysis reveals that the NVP and NVFP models exhibit relatively low current densities (Fig. 4d3 and e3). In contrast, the NVFP–VO model exhibits enhanced current density with localized charge accumulation, particularly at the heterostructure interfaces (Fig. 4f3), which improves the electrode's electrical conductivity and mitigates interface-related issues.
 | ||
| Fig. 4 Single particle crush test at different times. (a) NVP, (b) NVFP, and (c) NVFP–VO. The COMSOL multiphysics simulation results of the electrode. (1) Ion concentration distribution, (2) stress/strain distribution, and (3) current distribution of (d) NVP, (e) NVFP, and (f) NVFP–VO. | ||
Collectively, these results demonstrate that the superior capacity retention of NVFP–VO can primarily be ascribed to its enhanced mechanical stability, as confirmed by the single-particle crush test (Fig. 4a–c), and a more uniform stress/strain distribution, demonstrated by multiphysics simulations (Fig. 4d–f), and the electrochemical activation and amorphization of VO during cycling (Fig. S14, S38 and S39, ESI†), which can all improve structure stability, thus obtaining more excellent cycling stability.
2.6 Ex situ characterization
To perform a more in-depth analysis of the changes in cathode materials after cycling, we extracted the electrodes from the coin cells and conducted ex situ XRD, SEM, and TEM and XPS tests to gather more detailed information. From the XRD spectra (Fig. S38, ESI†), it can be observed that most crystalline facets of NVFP–VO still exist even after 100000 high-rate cycles, indicating that the NVFP–VO materials largely retain their crystallinity. However, the diffraction peaks of VO basically disappear. This finding is further corroborated by the HR-TEM images and SAED patterns (Fig. S39, ESI†), which only show clear lattice fringes and diffraction rings of NVFP. These observations suggest a gradual amorphization of VO, a phenomenon commonly observed in transition metal oxides and certain alloy materials. Amorphization typically results from the loss of long-range structural order, resulting in weakened or completely vanished XRD signals. The structure amorphization can yield beneficial effects, such as increased ionic conductivity and reduced volume expansion, which effectively explain the observed improvements in capacity and cycling stability in our tests.
Furthermore, we analyzed the morphology and volume changes of NVFP–VO electrodes. As shown in Fig. S40 (ESI†), the pristine uncycled electrodes are flat. After 10000 cycles at 100C, the electrode remains intact without obvious cracks. However, after 100000 cycles at 100C, some cracks are observed clearly. With the accumulation of stresses over such long-term cycling, the active materials may undergo crushing or rearrangement, leading to localized changes in density and the formation of cracks, which may also contribute to the capacity decay observed in electrochemical performance (Fig. 2g). Regarding the volume change of the electrode, we also conducted SEM cross-sectional analysis. Compared to the pristine electrode, the thickness of the cycled electrode increases by only 0.2% and 6.0% after 10000 and 100000 cycles, respectively. The small thickness change further confirms the high stability of the electrode structure, which is favorable for maintaining stability under high current densities.
The electrode/electrolyte interfaces, as essential part, considerably influence the cycle lifespan and rate capability of the active materials. Ionic-transfer kinetics are closely linked to the interphase structure and components within the bulk material. A stable cathode electrolyte interphase (CEI) can isolate direct contact between electrolytes and cathode materials, thereby inhibiting electrolyte oxidation and reduction, as shown in the LSV results (Fig. S11, ESI†). In contrast, an uneven CEI can lead to excessive electrolyte decomposition and poor ionic conductivity, hampering fast and reversible Na+ transfer across interfaces.37
To investigate the effect of electrode structure on optimized CEI formation, in-depth composition distribution analysis was conducted using high-resolution XPS on NVP and NVFP–VO cathodes by different etching depths. Ex situ in-depth XPS measurements of all electrodes (Fig. 5a–d) show that the peaks at 684.3 and 687.7 eV in the F 1s spectra correspond to Na–F and C–F bonds, while the peaks at 284.5, 286.4, and 289.5 eV in the C 1s spectra are attributed to C–C, C–O, and CO32− bonds, respectively. In-depth C 1s spectra of NVP and NVFP–VO electrodes reveal a similar trend. The CO32− content decreases with etching in both cathodes, indicating a reduction in the inorganic components (Na2CO3) in the residual CEI (Fig. 5b and d). Meanwhile, the relative content of CO32− in NVFP–VO is higher than that in NVP and is distributed throughout the whole depth during the etching process, indicating that the CEI of NVFP–VO is richer in inorganic components, namely sodium carbonate (Na2CO3). Besides, the inorganic Na–F is totally dominant along with the disappearance of an organic C–F component in the NVFP–VO electrode (Fig. 5c) throughout the whole depth etching process, while in the NVP electrode (Fig. 5a) there are still a few amounts of C–F representing organic components, further indicating the richer inorganic components (NaF) in the CEI of the NVFP–VO electrode than that of NVP. Similarly, in-depth O 1s spectra of NVP and NVFP–VO electrodes exhibit a similar trend with C 1s and F 1s. Clearly, the content of inorganic component (Na2O) in the CEI of NVFP–VO is higher than that of NVP at different etching depths (Fig. S41, ESI†). Additionally, in-depth F 1s spectra of NVFP–VO are significantly broader than those of NVP and are split into two peaks centered at 683.2 and 684.8 eV, indicating the formation of two kinds of Na–F bonds (Fig. 5c). Obviously, the peak located at 684.8 eV is similar to that of NVP. The appearance of the peak at a lower binding energy (683.2 eV) may be ascribed to the variation of the coordination environment of the Na–F bonds in NVFP–VO. A richer inorganic component and the existence of intergrown biphasic heterostructure may favor the formation of the peak at a lower binding energy. The porous carbon skeleton further promotes the adsorption of ClO4− anions and fluoroethylene carbonate (FEC) molecules from the electrolyte, resulting in the preferential oxidative decomposition into an inorganic-rich interphase at the cathode surface.37 Meanwhile, ex situ HAADF-STEM and the corresponding STEM-EDS images are used to observe the thickness and component uniformity of the CEI layer. Clearly, the NVP cathode (Fig. 5e) exhibits an uneven CEI with a thickness ranging from 5 to 15 nm. In contrast, the NVFP–VO cathode (Fig. 5f) is fully covered with a uniform CEI of approximately 5 nm thickness.
 | ||
| Fig. 5 Ex situ characterizations of NVP and NVFP–VO cathodes. In-depth element distributions and high-resolution XPS spectra of F 1s and C 1s. (a) and (b) NVP, and (c) and (d) NVFP–VO. HAADF-STEM and STEM-EDS mappings of (e) NVP and (f) NVFP–VO. | ||
Based on the XPS and STEM-EDS results, it can be concluded that the CEI of NVFP–VO has a richer inorganic component such as Na2CO3, NaF, and Na2O than that of NVP with different etching depths, a thinner thickness (∼5 nm), and a more homogeneous composition distribution. Compared with organic species, inorganic species possess a higher ionic conductivity and robustness, which can ensure rapid Na+ transfer and stabilize the interface structure, thus contributing to high Na+ diffusion coefficients38 and minimizing adverse interfacial side reactions, thus improving rate performance and cycling stability. Obviously, a thinner CEI thickness can shorten the diffusion distance of sodium-ion, thus enhancing rate performance. A more homogeneous CEI composition can prevent electrolyte decomposition at high voltages, maintain structural integrity during long-term cycling, and promote Na+ transfer across the interface, as demonstrated by EIS and GITT analysis (Fig. S30–S32, ESI†). Consequently, the NVFP–VO composite cathode exhibits excellent rate performance and cycling stability.
This work introduces significant innovations in sodium-ion battery cathode materials through the integration of an intergrown biphasic heterostructure of polyanion and layered oxide for the first time. This structure enables a novel activation mechanism for layered oxides, achieving the electrochemical activation without extreme processing conditions and subsequent amorphization of V2O3, thereby enhancing capacity and cycling stability. The main contributions of this work are threefold: first, the structural innovation of integrating intergrown biphasic heterostructures in SIBs; second, the mechanism innovation revealing a new activation pathway for layered oxides; and third, comprehensive characterizations—including in situ XRD, ex situ XANES spectra, ex situ TEM, ex situ XPS, DFT calculations, and COMSOL simulations—to elucidate the synergistic effects of the integrated NVFP–VO cathode. Notably, the exceptional cycling stability of over 100000 cycles at 100C marks an unprecedented milestone for cathode materials in sodium-ion batteries. These findings underscore the potential of synergistic effects in integrated cathodes to drive advancements in cathode materials and provide valuable insights to broaden the compositional space for future research on advanced SIB cathodes.
3. Conclusions
In summary, an integrated polyanion-layered oxide cathode embedded in a porous carbon framework was designed and optimized to enhance Na-ion storage performance. Fe doping reduces the bandgap of NVP and activates its V4+/V5+ redox couple. The porous carbon framework further enhances the electronic conductivity of the integrated cathode and accommodates volume changes during cycling. The heterostructure reduces ion transport barriers and enhances reaction kinetics. Additionally, the low-strain V2O3 phase acts as a stabilizer, effectively buffering volume changes and stress gradients in NVFP, while the spontaneous activation mechanism further enhances the capacity of the integrated cathode. Consequently, the integrated cathode demonstrates unprecedented cyclability, achieving over 100000 cycles with 72.6% capacity retention at 100C in half-cells. To the best of our knowledge, this cycling performance represents the longest cycle life reported among advanced cathodes. Additionally, it delivers a high reversible specific capacity of 130 mA h g−1 at 0.1C, corresponding to an energy density of approximately 405 W h kg−1, surpassing most polyanionic cathodes for sodium-ion batteries. Laboratory scale-up synthesis was conducted to validate the practical application of the integrated NVFP–VO cathode. Ah-level pouch full cells demonstrated a high energy density of 153.4 W h kg−1 with a long cycling life exceeding 500 cycles. This work bridges the two primary families of sodium-ion battery cathodes – polyanion and layered oxide cathode, which demonstrates that synergistic effects in integrated cathodes can drive the development of advanced cathode materials for high-energy-density, fast-charging, and long-life SIBs.
Author contributions
Zhiyu Zou, Yongbiao Mu, and Meisheng Han conceived the idea, designed the experiments, conducted the characterizations and co-wrote the paper. Youqi Chu, Jie Liu, and Kunxiong Zheng assisted in the material synthesis and conducted the characterizations. Qing Zhang performed the AC-TEM test. Manrong Song, Qinping Jian, and Yilin Wang provided important advice on experiments and the manuscript. Hengyuan Hu and Fenghua Yu contributed to part of the electrochemical measurements. Wenjia Li and Lei Wei revised the manuscript and triggered helpful discussion. Tianshou Zhao, Meisheng Han and Lin Zeng supervised the project. All the authors discussed the results, and provided comments and revisions to the manuscript.Data availability
The data that support the findings of this study have been included in this article and its ESI.† Source data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Shenzhen Science and Technology Plan Project (No. SGDX20230116091644003), Guangdong Major Project of Basic and Applied Basic Research (No. 2023B0303000002), the Shenzhen Key Laboratory of Advanced Energy Storage (No. ZDSYS20220401141000001) and High level of special funds (G03034K001). The TEM study utilized the resources of the Pico Center at SUSTech Core Research Facilities, which is supported by the Presidential Fund and the Development and Reform Commission of Shenzhen Municipality. The authors would also like to acknowledge technical support from the SUSTech Core Research Facilities and thank Xueyanhui (www.xueyanhui.com) for plotting the diagrams.References
- P. Greim, A. A. Solomon and C. Breyer, Nat. Commun., 2020, 11, 4570 CrossRef CAS PubMed.
- A. Rudola, R. Sayers, C. J. Wright and J. Barker, Nat. Energy, 2023, 8, 215–218 CrossRef.
- Y. Sun, J.-C. Li, H. Zhou and S. Guo, Energy Environ. Sci., 2023, 16, 4759–4811 RSC.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- P. Barpanda, L. Lander, S. Nishimura and A. Yamada, Adv. Energy Mater., 2018, 8, 1703055 CrossRef.
- R. Thirupathi, V. Kumari, S. Chakrabarty and S. Omar, Prog. Mater. Sci., 2023, 137, 101128 CrossRef CAS.
- R. Huang, D. Yan, Q. Zhang, G. Zhang, B. Chen, H. Y. Yang, C. Yu and Y. Bai, Adv. Energy Mater., 2024, 14, 2400595 CrossRef CAS.
- H. Xiong, G. Sun, Z. Liu, L. Zhang, L. Li, W. Zhang, F. Du and Z.-A. Qiao, Angew. Chem., Int. Ed., 2021, 60, 10334–10341 CrossRef CAS.
- R. Liu, S. Zheng, Y. Yuan, P. Yu, Z. Liang, W. Zhao, R. Shahbazian-Yassar, J. Ding, J. Lu and Y. Yang, Adv. Energy Mater., 2021, 11, 2003256 CrossRef CAS.
- Z. Cai, B. Ouyang, H.-M. Hau, T. Chen, R. Giovine, K. P. Koirala, L. Li, H. Ji, Y. Ha, Y. Sun, J. Huang, Y. Chen, V. Wu, W. Yang, C. Wang, R. J. Clément, Z. Lun and G. Ceder, Nat. Energy, 2024, 9, 27–36 CrossRef CAS.
- Y. Zhou, G. Xu, J. Lin, Y. Zhang, G. Fang, J. Zhou, X. Cao and S. Liang, Adv. Mater., 2023, 35, 2304428 CrossRef CAS.
- S. Park, J.-N. Chotard, D. Carlier, F. Fauth, A. Iadecola, C. Masquelier and L. Croguennec, Chem. Mater., 2023, 35, 3181–3195 CrossRef CAS.
- W. Zhang, Y. Wu, Z. Xu, H. Li, M. Xu, J. Li, Y. Dai, W. Zong, R. Chen, L. He, Z. Zhang, D. J. L. Brett, G. He, Y. Lai and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS.
- C. Xu, J. Zhao, Y.-A. Wang, W. Hua, Q. Fu, X. Liang, X. Rong, Q. Zhang, X. Guo, C. Yang, H. Liu, B. Zhong and Y.-S. Hu, Adv. Energy Mater., 2022, 12, 2200966 CrossRef CAS.
- Y. Huang, Y. Dong, Y. Yang, T. Liu, M. Yoon, S. Li, B. Wang, E. Y. Zheng, J. Lee, Y. Sun, Y. Han, J. Ciston, C. Ophus, C. Song, A. Penn, Y. Liao, H. Ji, T. Shi, M. Liao, Z. Cheng, J. Xiang, Y. Peng, L. Ma, X. Xiao, W. H. Kan, H. Chen, W. Yin, L. Guo, W.-R. Liu, R. Muruganantham, C.-C. Yang, Y. Zhu, Q. Li and J. Li, Nat. Energy, 2024, 9, 1497–1505 CrossRef CAS.
- R. Liu, W. Huang, J. Liu, Y. Li, J. Wang, Q. Liu, L. Ma, G. Kwon, S. N. Ehrlich, Y. Wu, T. Liu, K. Amine and H. Li, Adv. Mater., 2024, 36, 2401048 CrossRef CAS PubMed.
- E. Gabriel, C. Ma, K. Graff, A. Conrado, D. Hou and H. Xiong, eScience, 2023, 3, 100139 CrossRef.
- C. Xu, L. Zhou, T. Gao, Z. Chen, X. Hou, J. Zhang, Y. Bai, L. Yang, H. Liu, C. Yang, J. Zhao and Y.-S. Hu, J. Am. Chem. Soc., 2024, 146, 9819–9827 CrossRef CAS PubMed.
- K. Zhu, S. Wei, H. Shou, F. Shen, S. Chen, P. Zhang, C. Wang, Y. Cao, X. Guo, M. Luo, H. Zhang, B. Ye, X. Wu, L. He and L. Song, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- H. Luo, B. Wang, F. Wang, J. Yang, F. Wu, Y. Ning, Y. Zhou, D. Wang, H. Liu and S. Dou, ACS Nano, 2020, 14, 7328–7337 CrossRef CAS.
- T. Jin, H. Li, Y. Li, L. Jiao and J. Chen, Nano Energy, 2018, 50, 462–467 CrossRef CAS.
- J. Xu, J. Jiang, H. Tang, Z. Chen, J. Chen, Y. Zhang and C.-S. Lee, Adv. Power Mater., 2024, 3, 100169 Search PubMed.
- P. Hu, P. Hu, T. D. Vu, M. Li, S. Wang, Y. Ke, X. Zeng, L. Mai and Y. Long, Chem. Rev., 2023, 123, 4353–4415 CrossRef CAS PubMed.
- H. Li, Y. Wang, X. Zhao, J. Jin, Q. Shen, J. Li, Y. Liu, X. Qu, L. Jiao and Y. Liu, ACS Energy Lett., 2023, 3666–3675 CrossRef CAS.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O’Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M.-M. Titirici, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- S. Xiao, J. Jiang, Y. Zhu, J. Zhang, H. Li, R. Wu, X. Niu, J. Qin and J. S. Chen, Adv. Power Mater., 2023, 2, 100120 Search PubMed.
- M. Hadouchi, N. Yaqoob, P. Kaghazchi, M. Tang, J. Liu, P. Sang, Y. Fu, Y. Huang and J. Ma, Energy Storage Mater., 2021, 35, 192–202 CrossRef.
- H. Kwak, J.-S. Kim, D. Han, J. S. Kim, J. Park, G. Kwon, S.-M. Bak, U. Heo, C. Park, H.-W. Lee, K.-W. Nam, D.-H. Seo and Y. S. Jung, Nat. Commun., 2023, 14, 2459 CrossRef CAS PubMed.
- J. P. Christophersen, Battery Test Manual For Electric Vehicles, Revision 3, United States, 2015 Search PubMed.
- M. Han, Z. Zou, J. Liu, C. Deng, Y. Chu, Y. Mu, K. Zheng, F. Yu, L. Wei, L. Zeng and T. Zhao, Small, 2024, 20, 2312119 CrossRef CAS PubMed.
- Z. Li, M. Han, J. Wang, L. Zhang, P. Yu, Q. Li, X. Bai and J. Yu, Adv. Funct. Mater., 2024, 34, 2404263 CrossRef CAS.
- C. Xu, W. Hua, Q. Zhang, Y. Liu, R. Dang, R. Xiao, J. Wang, Z. Chen, F. Ding, X. Guo, C. Yang, L. Yang, J. Zhao and Y.-S. Hu, Adv. Funct. Mater., 2023, 33, 2302810 CrossRef CAS.
- Q. Wei, X. Chang, D. Butts, R. DeBlock, K. Lan, J. Li, D. Chao, D.-L. Peng and B. Dunn, Nat. Commun., 2023, 14, 7 CrossRef PubMed.
- D. Wang, X. Bie, Q. Fu, D. Dixon, N. Bramnik, Y.-S. Hu, F. Fauth, Y. Wei, H. Ehrenberg, G. Chen and F. Du, Nat. Commun., 2017, 8, 15888 CrossRef CAS PubMed.
- J. Moon, J. Y. Jung, T. D. Hoang, D. Y. Rhee, H. B. Lee, M.-S. Park and J.-S. Yu, J. Power Sources, 2021, 486, 229359 CrossRef CAS.
- M. Han, J. Liu, K. Zheng, C. Deng, Y. Mu, J. Guo, Y. Chu, Z. Zou, F. Yu, W. Li, L. Wei, L. Zeng and T. Zhao, Adv. Energy Mater., 2024, 14, 2401065 CrossRef CAS.
- J. Zhang, Y. Yan, X. Wang, Y. Cui, Z. Zhang, S. Wang, Z. Xie, P. Yan and W. Chen, Nat. Commun., 2023, 14, 3701 CrossRef CAS PubMed.
- Y. Liao, L. Yuan, Y. Han, C. Liang, Z. Li, Z. Li, W. Luo, D. Wang and Y. Huang, Adv. Mater., 2024, 36, 2312287 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05110f |
| ‡ These authors contributed equally: Zhiyu Zou and Yongbiao Mu. |
| This journal is © The Royal Society of Chemistry 2025 |