
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicarbonate concentrations affect arsenic release from arsenopyrite and nanoscale iron(III) (hydr)oxide formation: importance of unconfined aquifer carbonate chemistry†
Ping-I Chou a,
Xuanhao Wuab,
Zhenwei Gaoa,
Yaguang Zhua and
Young-Shin Jun*a
a,
Xuanhao Wuab,
Zhenwei Gaoa,
Yaguang Zhua and
Young-Shin Jun*a
aDepartment of Energy, Environmental and Chemical Engineering, Washington University in St. Louis, One Brookings Drive, Campus Box 1180, St. Louis, Missouri 63130, USA. E-mail: ysjun@seas.wustl.edu; Web: https://encl.engineering.wustl.edu/ Fax: +(314) 696 1223; Tel: +(314) 935 4539
bCollege of Environment and Resource Sciences, Zhejiang University, Hangzhou, 310027, Zhejiang, China
First published on 18th March 2025
Abstract
Managed aquifer recharge (MAR) is an important engineering solution for achieving sustainable groundwater management. Unfortunately, if not operated properly, MAR can cause undesirable arsenic mobilization in groundwater. To avoid unexpected arsenic mobilization, we need a better understanding of the evolving water chemistry and nanoscale mineral–water interfaces in MAR systems. Bicarbonate is a ubiquitous groundwater component, but its effect on arsenic mobilization in MAR is not fully understood. Hence, we examined the effects of bicarbonate concentrations (0.01 mM, 0.1 mM, 1.0 mM, and 10 mM) on the dissolution of arsenopyrite and the nanoscale secondary mineral formation in both open systems (mimicking shallow unconfined aquifers) and closed systems (mimicking deep confined aquifers) over 7 days. In the open system, owing to pH evolution and the subsequent formation and growth of iron(III) (hydr)oxide nanoparticles, the arsenic mobilization decreased with increasing bicarbonate concentrations. However, the increase from 1.0 to 10 mM formed surface complexation and aqueous arseno–carbonate complexes and did not further reduce the arsenic mobilization. In the closed system, arsenic mobilization and iron(III) hydr(oxide) nanoparticle formation were similar for all conditions. This study highlights bicarbonate-controlled nanoparticle formation and arsenic mobilization in MAR systems, providing valuable insights for enabling safer and more sustainable MAR operations.
Environmental significanceManaged aquifer recharge (MAR) offers a promising approach to sustainable groundwater management by reusing water resources. However, one significant challenge associated with MAR is the unanticipated release of arsenic from arsenic-containing sulfide minerals, such as arsenopyrite. Our research delves into the mechanisms behind arsenic mobilization from arsenopyrite, as well as the roles of nanoscale secondary iron (hydr)oxides mineral formation. We examine the dissolution kinetics of arsenic and the formation of nanoscale secondary iron (hydr)oxides, including their morphology, oxidation state, and phase. These mechanistic findings of arsenic mobilization and nanoparticle formation is crucial for the control of groundwater quality. This study will be helpful for developing safer and more sustainable MAR practices and improving overall groundwater management strategies. |
1. Introduction
In recent years, rapid population growth and socioeconomic development have increased the demand for freshwater. The global groundwater withdrawal rate has increased by 1–3 percent annually.1 In particular, the total groundwater withdrawal rate has increased from 158 to 959 km3 per year from 1950 to 2017, and has been estimated to increase to 1100 km3 per year by 2050.2,3 Furthermore, global terrestrial water storage and drought severity have worsened recently.4 In October 2021, the California Department of Water Resources reported that California had experienced the driest year since 1924.5 The unfavorable combination of increased water demand and decreased water supply has resulted in groundwater over-extraction, which has caused significant land subsidence and irreversible seawater intrusion and consequent soil salinization.6,7 These problems urge us to manage groundwater storage and usage more sustainably. Managed aquifer recharge (MAR) is a viable engineering solution to achieve water storage, recycling, and reuse, and eventually to help in balancing groundwater extraction and supply.8,9 MAR operations recharge various types of source water (e.g. treated wastewater, run-off, or rainwater) into subsurface environments, such as shallow unconfined aquifers (dry wells or infiltration ponds) and deeper confined aquifers (aquifer storge recovery (ASR) or aquifer storage transfer and recovery (ASTR)).10,11While MAR can replenish groundwater in various subsurface strata, the injection can cause unfavorable water–mineral interactions, unexpectedly increasing arsenic concentrations in water recovered from MAR field sites.8 The recovered water arsenic concentrations can reach or even exceed the 10 μg L−1 maximum concentration level (MCL) for arsenic set by the Environmental Protection Agency.12 For example, the arsenic level in water from a MAR site in the South Central Florida groundwater basin, USA, was 10–130 μg L−1, while the injection water and native storage zone water contained less than 3 μg L−1.13 At another MAR site in Bolivar, Australia, the pre-injection water had 3 μg L−1 of arsenic, while the recovered water's arsenic level reached 22 μg L−1.14 The higher arsenic concentrations in the recovered water than in the injection water or the ambient groundwater before MAR brought significant concerns about arsenic mobilization during anthropogenic groundwater recharge,8,13–15 and highlight the importance of a better understanding of how water chemistry and nanoscale mineral–water interfaces evolve during MAR.
During MAR, arsenic is mobilized by the oxidative dissolution of arsenic-bearing pyritic minerals in the aquifer, such as arsenopyrite (FeAsS) and arsenian pyrite (0.5–10 wt% arsenic content).16,17 The water injected for MAR often contains many oxidants, such as dissolved oxygen and/or nitrate, triggering the oxidative dissolution of arsenic-bearing pyritic minerals and releasing arsenic into groundwater18,19 (as shown in eqn (1a) for arsenopyrite and eqn (1b) for arsenian pyrite):
| 4FeAsS(s) + 6H2O + 11O2(aq) → 4Fe2+(aq) + 4H3AsO3(aq) + 4SO42−(aq) | (1a) |
| 10Fe(As,S)2(s) + 22NO3−(aq) + 4H2O + 22H+(aq) → 10Fe2+(aq) + 10H3AsO3(aq) + 10SO42−(aq) + 11N2(g). | (1b) |
Common inorganic groundwater components, such as chloride, phosphate, silicate, and bicarbonate, can influence different magnitudes on arsenic mobilization.15,23,24 Previously we have observed that chloride ions can increase the mobilization of arsenic more significantly than nitrate by inhibiting the nucleation of iron(III) (hydr)oxide nanoparticles and promoting their phase transformation to less reactive iron(III) minerals (i.e., maghemite and hematite), and thus arsenic adsorption onto these secondary precipitates is decreased.15,23 Silicate increases arsenic mobilization by competitive adsorption between arsenic species and silicate, as well as by inhibiting iron(III) (hydr)oxide nanoparticle formation.24,25
Bicarbonate is a ubiquitous and environmentally important anion in many types of water. In the United States, the ambient bicarbonate concentration in typical groundwater is around 0.5 to 8 mM,26,27 and groundwater bicarbonate concentrations higher than 10 mM have been reported in India and China.28,29 Previous studies have shown that groundwater carbonate species such as carbonate or bicarbonate can affect the mobilization of arsenic in groundwater.24,27,30,31 Specifically, in an arsenic leaching experiment using core samples from the Marshall Sandstone aquifer in southeastern Michigan, the arsenic release rate from the core samples increased with increasing bicarbonate concentrations from 20 mM to 600 mM, and aqueous arseno-bicarbonate complexes have been proposed as the main cause of arsenic release from these aquifer rocks under an aerobic condition.30 Inhibition of arsenic mobilization by bicarbonate has also been found. Wu et al. (2020) focused on relatively low bicarbonate concentrations (i.e., 0.01 mM and 0.1 mM). Specifically, when the pH of in the 0.01 mM bicarbonate solution was reduced from 7.0 to 6.25, arsenic dissolved faster from arsenopyrite, whereas in the 0.1 mM bicarbonate solution the pH buffering effect maintained the reaction pH at 7.0, inducing more iron(III) (hydr)oxide precipitates once Fe had dissolved from arsenopyrite and slowing the mobilization of arsenic.24
Although the aforementioned studies have identified that bicarbonate's effects on arsenic mobilization are highly related to the bicarbonate concentration, both increasing and decreasing effects of bicarbonate on arsenic mobilization have been proposed in different ranges of bicarbonate concentrations. Additionally, MAR operations can alter groundwater bicarbonate concentrations. For instance, MAR injection water with a low bicarbonate concentration (<0.25 mM) could dilute the bicarbonate concentrations in groundwater,24,32 while the bicarbonate concentration can also be increased by dissolving carbonate minerals such as calcite14,33,34 or by injecting high alkalinity recharge water to prevent the dissolution of carbonate minerals.35 Moreover, previous studies have not examined bicarbonate concentrations relevant to groundwater, including MAR, in terms of arsenic mobilization from arsenic-bearing minerals.26,27,32,34 Deciphering complex nanoscale interfacial reactions caused by bicarbonate will advance our understanding of critical early stages of reactions under conditions relevant to MAR.21 Therefore, given a systematic study about the effects of bicarbonate concentrations on arsenic mobilization and secondary mineral formation in groundwater is still lacking, this work provided new comprehensive analyses for the effects of bicarbonate concentrations on arsenic mobilization from arsenic containing sulfide minerals in the settings relevant to groundwater.
Furthermore, MAR can create barriers to seawater intrusion in shallow unconfined aquifers or deep confined aquifers in coastal areas.36,37 Depending on the geological structures of aquifers, previous studies have emphasized the considerations of shallow unconfined aquifers (within tens of meters below land surface) and confined aquifers (deeper than tens or even hundreds of meters below land surface) in evaluating MAR implementation and groundwater quality.37,38 In shallow unconfined aquifers, the groundwater can be in equilibrium with air (i.e., open system with water pressure equals atmospheric pressure), and the concentrations of CO2 can change due to direct contact with atmosphere, whereas in deep confined aquifers, the concentrations of CO2 is relatively stable because these aquifers are not directly exposed to air.39–41 The carbonate equilibrium in groundwater can also alter other parameters of groundwater chemistry, such as pH.26,42 This change of pH may affect the arsenic mobilization. For example, Kim et al. (2000) found significant arsenic leaching from aquifer rock (i.e., Marshall Sandstone) in the extreme pH ranges of <1.9 and 8.0–10.4 in 40 mM NaHCO3.30 Hence, the current study aimed to study the effects of carbonate conditions in different aquifer settings (i.e., shallow unconfined aquifers and deep confined aquifers) on iron(III) (hydro) oxide formation and arsenic mobilization.
To further advance our understanding about the specific roles of bicarbonate in arsenic mobilization and secondary mineral formation in aquifer-relevant settings, the objective of this study is to examine molecular-scale arsenopyrite–water interactions. Specifically, we elucidated the effects of bicarbonate concentrations ranging from 0.01 mM to 10 mM on arsenic mobilization under conditions relevant to groundwater where MAR is operated. Arsenopyrite (FeAsS) was chosen as a model arsenic-bearing pyrite mineral because it is a common arsenic-bearing mineral in aquifers and has uniform chemical compositions, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio of iron, sulfur, and arsenic,18,43 allowing well-controlled experiments to carefully examine dissolution and nanoscale secondary mineral formation. To understand the effects of carbonate conditions on different aquifer settings, arsenopyrite dissolution experiments were conducted in both open and closed systems, mimicking conditions in MAR recharged in unconfined and confined aquifers, respectively. To further elucidate the roles of bicarbonate in the secondary mineral formation at nanoscale, the morphologies, phases, and extent (i.e., the amount of secondary mineral formed) of secondary mineral nanoparticles were examined. Both qualitative and quantitative molecular-scale analyses in this study provide useful information about the fate of arsenic mobilized from arsenopyrite and the nanoscale morphology and mineralogy of secondary iron(III) (hydr)oxide precipitation that can form during MAR. The results of this study offer valuable insights into managing the impacts of bicarbonate on arsenic mobilization and the correlated reaction pH and nanoscale secondary mineral formation in different MAR operations.
:1:1 molar ratio of iron, sulfur, and arsenic,18,43 allowing well-controlled experiments to carefully examine dissolution and nanoscale secondary mineral formation. To understand the effects of carbonate conditions on different aquifer settings, arsenopyrite dissolution experiments were conducted in both open and closed systems, mimicking conditions in MAR recharged in unconfined and confined aquifers, respectively. To further elucidate the roles of bicarbonate in the secondary mineral formation at nanoscale, the morphologies, phases, and extent (i.e., the amount of secondary mineral formed) of secondary mineral nanoparticles were examined. Both qualitative and quantitative molecular-scale analyses in this study provide useful information about the fate of arsenic mobilized from arsenopyrite and the nanoscale morphology and mineralogy of secondary iron(III) (hydr)oxide precipitation that can form during MAR. The results of this study offer valuable insights into managing the impacts of bicarbonate on arsenic mobilization and the correlated reaction pH and nanoscale secondary mineral formation in different MAR operations.
2. Material and methods
2.1 Arsenopyrite powder and coupon preparation
Arsenopyrite samples from Gold Hill, Tooele County, UT, were purchased from Mineralogical Research Company (San Jose, CA). The mineral phases of powdered arsenopyrite samples were characterized by an X-ray diffractometer (XRD, Bruker d8 Advance) (Fig. S1A†), and the results indicated mainly arsenopyrite and quartz phases, consistent with previous publications.15,23 To conduct batch dissolution experiments and facilitate the dissolution of arsenopyrite, powdered arsenopyrite samples with particle sizes ranging from 300 to 500 μm were used. The Brunauer–Emmett–Teller (BET) specific surface area of the arsenopyrite powder, prepared using the same sample preparation method, has been reported to be 0.116–0.555 m2 g−1,15 and the isoelectric point (pHiep) was around 3.6.24 To investigate the morphologies and phases of nanoscale secondary mineral precipitation on arsenopyrite mineral surfaces, we used 1 mm thick flat arsenopyrite coupons prepared by Burnham Petrographics, LLC (Rathdrum, Idaho, USA) from the same arsenopyrite. The surface morphology of an unreacted coupon was characterized by tapping mode atomic force microscopy (AFM, Veeco Inc.) and environmental scanning electron microscopy (ESEM, Thermo Scientific Quattro S) (Fig. S1B and C,† respectively). More detailed information related to the sample preparation, cleaning, and characterization of the arsenopyrite powdered samples and arsenopyrite coupons is in the ESI.†2.2 Water chemistries and batch experiments in open and closed systems
All chemicals used in this study were at least American Chemical Society grade, and all solutions were prepared using ultrapure deionized (DI) water (resistivity ≥18.2 MΩ cm, Barnstead Ultrapure Water System, MA). To determine the dissolution of arsenopyrite, we conducted a series of batch reactor experiments with different concentrations of bicarbonate. First, to mimic groundwater, the pH of the solutions was adjusted to 7.0 ± 0.2, using diluted hydrochloric acid and sodium hydroxide. Then 0.01 mM, 0.1 mM, 1.0 mM, and 10 mM bicarbonate concentrations were tested by adding sodium bicarbonate, which are relevant concentrations to common groundwater environments as well as to MAR operations.26,32,34 10 mM nitrate was added as a commonly found oxyanion and oxidant arsenopyrite dissolution by adding sodium nitrate.15,44 Because secondary iron(III) (hydr)oxide mineral precipitation could be affected by the salinity of water, the ionic strength (IS) was set to 100 mM by adding sodium chloride, which will allow us to test the bicarbonate concentration effects without changing the background IS. This ionic strength condition (equivalent to 6375 ± 150 mg L−1 of the total dissolved solids, TDS) can provide the salinity relevant to moderately saline groundwater (TDS concentrations between 3000 and 10000 mg L−1), based on the definition from The National Ground Water Association.45,46 The water chemistry equilibrium values were calculated using Visual MINTEQ thermodynamic modeling software.47 The initial target water chemistry and the calculated equilibrium water chemistry of the open and closed systems are summarized in Table S1.† The full calculated components concentrations at equilibrium of the open and closed systems are summarized in Table S2.†
In the open system, prior to any reaction, 250 mL aqueous solutions with the desired water chemistry were prepared in polypropylene (PP, VWR International, PA) batch reactors. To initiate the reaction, 0.050 ± 0.001 g of arsenopyrite powder was added to each solution, and the open-to-air reactors were stirred continuously for the desired reaction time. Immediately after the arsenopyrite powder addition, 2 mL aliquots of solution were taken from the reactors every hour for 6 hours and every day for 7 days (i.e., 168 hours). As in our previous studies,15,48 the short-term (first 6 hours) reaction time was chosen mainly to focus on dissolution of arsenopyrite, while the long-term (7 days) reaction time allowed us to examine the formation of nanoscale secondary mineral precipitates and their roles in arsenic mobilization. Three arsenic concentrations were quantified: Aqueous arsenic, adsorbed arsenic onto nanoscale secondary iron(III) (hydr)oxide mineral precipitates, and the total dissolved arsenic concentration (i.e., aqueous arsenic + adsorbed arsenic). At specific elapsed time, aqueous samples were taken, immediately filtered using a 0.2 μm polytetrafluoroethylene (PTFE) membrane syringe filter, and acidified to 2% w/w acid with nitric acid. Arsenic concentrations, defined as aqueous arsenic in this work, for these samples were measured using inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer NexION 2000). At each sampling time over the 7 day reaction period, pHs and oxidation–reduction potentials (ORP) for each system were recorded using a pH electrode (VWR 89231-604, with an Ag/AgCl internal reference) and an ORP electrode (VWR 89231-642, with an Ag/AgCl internal reference), respectively. Triplicate batch experiments were conducted for each condition.
In addition to aqueous arsenic concentrations, we evaluated the adsorbed arsenic concentration (i.e., arsenic adsorption onto nanoscale secondary iron(III) (hydr)oxide mineral precipitates) and the total dissolved arsenic concentration (i.e., aqueous arsenic + adsorbed arsenic). To quantify the total dissolved arsenic concentration, the same reaction conditions as in the batch dissolution experiments were used. Immediately after the dissolution reaction, the reacted solution was then added to a sodium hydroxide (0.5 M) solution for 1 hour to extract adsorbed arsenic. As reported in previous publications, this extraction process recovers >95% of the adsorbed arsenic.49,50 This solution was immediately filtered using a 0.2-μm PTFE membrane syringe filter, and acidified to 2% w/w acid with nitric acid. The total arsenic concentrations (aqueous + adsorbed) released from arsenopyrite were then determined by ICP-MS measurements. It should be noted that this extraction method is specific to the quantification of surface adsorbed arsenic rather than the incorporation of As into secondary iron(III) (hydr)oxides.48–50 Once the total dissolved arsenic concentrations were obtained from the extraction method, the concentrations of adsorbed arsenic can be calculated based on the difference between total arsenic and aqueous arsenic concentrations.
To investigate the role of bicarbonate in arsenic mobilization in the closed system, the same experimental procedures were conducted as in the open system, but 250 ml Boston bottles (VWR) with a septum cap were used as the reactors, enabling sample collection by syringes while minimizing the gas exchange during sampling. In the closed system experiments, the same initial water chemistry was used as in the open system, while the bicarbonate concentrations of 0.1 mM, 1.0 mM, and 10 mM provided well-controlled pH values, as shown in the control experiments (Fig. S2†). To understand the arsenic speciation (i.e., As(III) and As(V)), a column packed with anion-exchange resin in chloride form (Dowex, Sigma-Aldrich) was used and the detailed procedure is described in ESI† (Fig. S5). Triplicate batch experiments were conducted for this measurement.
To identify the chemical bonds on the arsenopyrite surface, we analyzed the powders before and after reaction, using attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy (Thermo Scientific, Nicolet iS10, equipped with a diamond crystal). For each FTIR measurement, the scanning range was from 600 cm−1 to 1650 cm−1 with an average of 400 scans with a resolution of 4 cm−1. At least duplicate measurements were conducted for each condition. To prepare the samples for FTIR measurements, the arsenopyrite powders were collected by filtration right after the 7 day reaction, rinsed with DI water, and then dried.
2.3 Characterization and quantification of secondary mineral nanoparticles
Arsenopyrite coupons were used to examine the extents, phases, and morphologies of nanoscale secondary mineral precipitates on arsenopyrite surfaces because the concentrations of newly formed nanoparticles in bulk solutions were too low to characterize their mineral phases and sizes in solution. Before a reaction, arsenopyrite coupons (5 mm × 5 mm × 1 mm, W × L × H) were cut using a dicing saw (DISCO Corporation, DAD323). To initiate the reaction, four coupons were horizontally placed in 250 mL of reaction solution along with 0.05 ± 0.001 g of arsenopyrite powders to maintain the same water chemistry and solid-to-water ratio as in the PP batch reactors. At 6 hours and again at 7 days, one coupon was removed, rinsed with deionized water, dried with high purity nitrogen gas, and stored in an anaerobic chamber (Coy Laboratory Products, Inc.) prior to characterizations.To examine the heterogenous secondary mineral formation, the morphologies and the heights of nanoscale secondary mineral precipitates on coupons surfaces were characterized with tapping mode atomic force microscopy (AFM, Veeco Inc.), with a probe (Bruker, model: RTESP, part: MPP-11100-10), and were analyzed using Nanoscope 7.20 software (Veeco). Each coupon was measured at five or more locations on the substrate surface. To identify the phases of the heterogeneous secondary mineral precipitates, we used an InVia Raman Microscope (Renishaw, UK), with a 514 nm laser (∼4 mW) and a grating of 1800 lines per mm. A 20× objective and a decreased power of 50% was used because this operation condition did not induce mineral phase transformation based on our previous work.15,24,48 Iron(III) (hydr)oxide standards and unreacted arsenopyrite coupons were also measured. To determine the oxidation states of iron (Fe 2p) on the arsenopyrite coupons, X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe II, Ulvac-PHI with monochromatic Al Kα radiation (1486.6 eV)) was utilized. For XPS data analysis, the binding energies were referenced to the C 1s line at 284.8 eV.24 The binding energy peaks of Fe(II) are at 710.43, 713.61, 723.45, and 728.54 eV,51,52 and the peaks for Fe(III) are at 711.70, 719.05, and 725.89 eV.51,52 Gaussian–Lorentzian curve fitting was utilized to derive the absolute peak areas of Fe 2p, which were used to obtain the percentages of Fe(II) and Fe(III) and the ratio of Fe(II)/Fe(III). Table S3† summarizes the Fe 2p reference binding energies, absolute values of the areas for each peak, and the calculated percentages for the oxidation states.
To quantify the extent of secondary iron(III) oxide precipitates formed on arsenopyrite, a citrate–bicarbonate–dithionite (CDB) extraction method was employed.23,53 This method can selectively dissolve iron(III) oxide from the samples and quantify the amount of iron(III) (hydr)oxide precipitated on the arsenopyrite surface during the reaction. This information provides a basis for comparing the extent of nanoscale iron(III) (hydr)oxide precipitation under different bicarbonate concentrations. Detailed experimental procedures are described in the ESI (S2).†
3. Results and discussion
3.1 Bicarbonate controls arsenic dissolution in the open system
Arsenic mobilization decreased with increasing bicarbonate concentrations that encompassed the range relevant to groundwater and MAR systems. The arsenic concentrations dissolved from the arsenopyrite mineral samples in the open system are shown in Fig. 1A (long term, 7 days) and Fig. S3A† (short term, 6 hours). Specifically, after 7 days, the 0.01 mM bicarbonate batches showed the highest dissolved arsenic concentration (497 μg L−1, or 6.6 μM), followed by the 0.1 mM bicarbonate batches. Both the 1 mM and 10 mM bicarbonate batches have the slowest arsenic dissolution rates. A t-test result showed a significant difference, with p < 0.05, among all the 7 day dissolved arsenic concentrations except for 1 mM and 10 mM bicarbonate. In the first 6 hours, however, the dissolved arsenic concentrations were not significantly different among the four different bicarbonate concentrations. This finding highlighted that the aqueous systems with low bicarbonate concentrations can cause more arsenic release from arsenopyrite in the open system, especially in a long period of time (i.e., more than 1 day).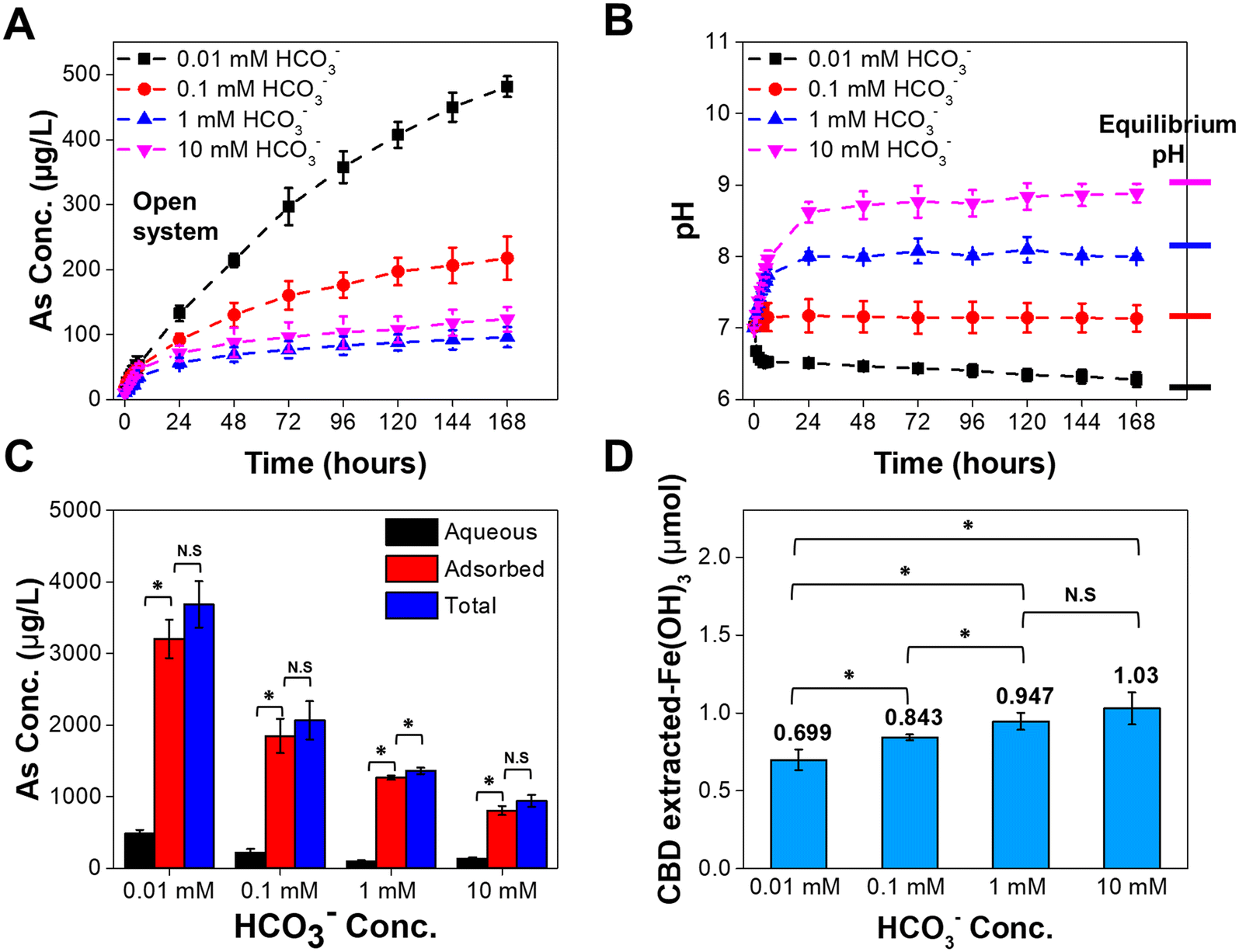 | ||
| Fig. 1 Dissolution of arsenopyrite in 0.01 mM, 0.1 mM, 1 mM, and 10 mM bicarbonate concentrations in the open system. (A) Dissolved arsenic concentrations at 7 days. (B) Trend of pH over 7 days; horizontal bars on the right axis represent the equilibrium pH values. (C) Total dissolved arsenic concentrations, including aqueous arsenic and adsorbed arsenic, at 7 days. (D) Quantification of secondary iron(III) (hydr)oxide mineral on arsenopyrite mineral powder at 7 days. Symbols: “*” denotes that a t-test found statistical significance with a p value <0.05; “N.S.” means no significant difference. Triplicate batch experiments were conducted for each condition. | ||
To further investigate the mechanisms that cause the inverse relation between bicarbonate concentration and dissolved arsenic (except for 10 mM), the pH of each system was recorded and are shown in Fig. 1B. Compared to the initial pH (7.0 ± 0.2), after the 7 day reactions, the reaction pH values of the 0.01 mM, 0.1 mM, 1 mM, and 10 mM bicarbonate concentrations were 6.3, 7.1, 8.0, and 8.9, respectively. When we compared the experimental pH values and the modeled equilibrium pH values (marked as horizontal bars on the right side of Fig. 1B and Table S1†), the reaction pH values of all bicarbonate concentrations approach the modeled equilibrium pH values. Because a similar trend in pH can be found in the control experiment (i.e., without arsenopyrite) (Fig. S3B†) of the open system, we can conclude that the reaction pH is driven by the carbonate equilibrium. In addition, the oxidative dissolution of arsenopyrite is pH sensitive. Previous studies have suggested that protons can increase arsenopyrite dissolution.8,44 The lower pH in the lower concentration bicarbonate systems can, thus, enhance the arsenopyrite dissolution in our systems. Although the pH has a nearly one-unit difference (8.0 and 8.9) in the 1 mM and 10 mM solutions, respectively, no significant difference in dissolved arsenic was found. Thus, we hypothesized that there could be other controlling factors, such as complexation, including surface complexation or aqueous complexation of arsenic and carbonate, which promote the dissolution of arsenopyrite, thereby offsetting the effect of pH.
To test this hypothesis, we conducted ATR-FTIR measurements to provide information about chemical bonds before and after the reaction (Fig. S4A and B†). The unreacted arsenopyrite has band positions at 695 and 777–796 cm−1, which we attribute to the sulfate adsorption and the S–O/S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations, respectively.54 These sulfur-oxygen bonds can form during the sample preparation process through oxidation with the atmospheric oxygen air. Comparing the unreacted arsenopyrite and the arsenopyrite reacted with different bicarbonate concentrations, new band formation can be observed. The bands at ∼1390 and ∼1510 cm−1 are attributed to CO-stretching in the monodentate carbonate,55,56 while the bands at ∼1340 and ∼1560 cm−1 are attributed to CO-stretching in the bidentate carbonate.57,58 The FTIR spectra in Fig. S4† confirm the occurrence of carbonate surface complexation on arsenopyrite, and these surface complexes can induce higher arsenic mobilization by competitive adsorption.31,59
O vibrations, respectively.54 These sulfur-oxygen bonds can form during the sample preparation process through oxidation with the atmospheric oxygen air. Comparing the unreacted arsenopyrite and the arsenopyrite reacted with different bicarbonate concentrations, new band formation can be observed. The bands at ∼1390 and ∼1510 cm−1 are attributed to CO-stretching in the monodentate carbonate,55,56 while the bands at ∼1340 and ∼1560 cm−1 are attributed to CO-stretching in the bidentate carbonate.57,58 The FTIR spectra in Fig. S4† confirm the occurrence of carbonate surface complexation on arsenopyrite, and these surface complexes can induce higher arsenic mobilization by competitive adsorption.31,59
Moreover, aqueous complexation of arsenic and carbonate can occur in the experimental systems. A Visual MINTEQ calculation (Table S4A†) was performed by inputting the stability constants of As(CO3)2− and AsCO3+ derived in a previous study.30 The calculation was based on the 7-day dissolved arsenic aqueous chemistries (Fig. 1), and the results in Table S4A† indicated the formation of arseno-carbonate aqueous complexes, i.e., As(CO3)2− and AsCO3+, with AsCO3+ being the dominant species in most conditions (except for the 10 mM bicarbonate concentration). Altogether, arseno-carbonate aqueous complexation was favored at all four different bicarbonate concentrations, and the total concentrations of the arseno-carbonate complex increased with increasing bicarbonate concentrations. The formation of these complexes can facilitate the oxidative dissolution of arsenopyrite by consuming the dissolved arsenic.30,60 The calculation of arseno–carbonate aqueous complexation suggests that it can promote arsenopyrite dissolution, especially at a high bicarbonate concentration (e.g., 10 mM in this study). However, comparing the dissolved arsenic concentrations and the aqueous complex concentrations, the number of aqueous complexes could be negligible (in a range of 10−17–10−25 M, Table S4†). Therefore, we concluded that surface complexation is the main mechanism driving arsenopyrite dissolution at a high bicarbonate concentration.
Furthermore, as shown in Fig. S3C,† the measured redox potentials, EH (mV), for all four bicarbonate concentrations are within 250–450 mV, indicating an oxidizing condition for all concentrations. Compared to the other three bicarbonate concentrations, 10 mM bicarbonate has a relatively low redox potential. This oxidizing condition can also be reflected in the arsenic speciation. Based on the anion exchange column tests, we found that, from day one to day seven, the As(V)/As(total) increased from 92% to 96% in 0.01 mM bicarbonate, and from 83% to 89% in 10 mM bicarbonate (Fig. S5†). The predominant arsenate and the increasing trend of As(V)/As(total) could be resulting from oxidizing condition as reflected in redox potentials.
Fig. 1C shows the fate of arsenic after arsenopyrite dissolution, which includes aqueous arsenic, adsorbed arsenic, and total dissolved arsenic (aqueous + adsorbed) under different bicarbonate concentrations at 7 days. Compared to aqueous arsenic, adsorbed arsenic comprises most of the total dissolved arsenic (with percentages from 86% to 93%). A similar trend of predominant adsorbed arsenic can also be found in 6 hours reaction (Fig. S6†), indicating that secondary nanoparticle formation can immobilize the majority of dissolved arsenic, both in the short term (6 hours) and over the long term (7 days). Higher concentrations of bicarbonate cause smaller amounts of adsorbed arsenic, owing to the smaller amount of the total dissolved arsenic released from the arsenopyrite mineral surface into the aqueous solution, and the less arsenic available for adsorption onto the nanoscale iron(III) (hydr)oxide mineral surface. This result suggests that bicarbonate concentration is important for controlling mobile arsenic in environmental systems. If we consider the mass balance of the arsenic released from arsenopyrite solids, the percentages of total dissolved arsenic from the solids ranged from 0.47% to 1.84%, and the percentages of aqueous arsenic from the solids ranged from 0.05% to 0.24%. These small percentages of dissolved arsenic can explain the dissolution has not reached the equilibrium based on eqn (1) and can also be observed in Fig. 1A.
3.2 Bicarbonate promotes nanoscale secondary mineral precipitation in the open system
Secondary mineral precipitation can critically affect arsenic mobilization by adsorbing dissolved arsenic onto newly formed reactive surfaces or by covering the pre-existing mineral surfaces, then changing the number of active mineral surface sites for dissolution.24 The differences in the secondary mineral precipitation in the four bicarbonate concentrations could offer useful insight into arsenic mobilization. Based on a citrate–bicarbonate–dithionite (CDB) extraction, we found that higher bicarbonate concentrations promote the formation and growth of iron(III) (hydr)oxide nanoparticles (Fig. 1D) as a result of the higher pH at higher bicarbonate concentrations. Higher pH can increase the saturation index (log(IAP/Ksp)—where IAP is the ion activity product, and Ksp is the solubility product of the iron(III) (hydr)oxide minerals—and therefore increase the nucleation rate of iron(III) (hydr)oxides on arsenopyrite surfaces. A balance between slower dissolution of arsenic from arsenopyrite and the increased iron(III) (hydr)oxide formation at higher bicarbonate concentrations can explain the non-stoichiometric dissolution of arsenopyrite reported in previous studies.17,19 Based on 7 day dissolved arsenic aqueous chemistries and thermodynamic calculations by using Visual MINTEQ (ver. 3.1), the experimental conditions in the open system are supersaturated with respect to several iron(III) (hydr)oxides, including ferrihydrite (Saturation Indices, SI = 2.62–5.16), lepidocrocite (SI = 4.46–6.99), goethite (SI = 5.34–7.87), maghemite (SI = 5.27–10.34), and hematite (SI = 13.07–18.14). Hence, in the following Section, we will discuss specific mineral phases formed in our experimental systems.To examine the morphologies, particle concentrations, and surface properties of secondary mineral formation on arsenopyrite surfaces, AFM was used to measure the samples after both 6 hours and 7 days under different bicarbonate concentrations. At the lowest bicarbonate concentration (0.01 mM bicarbonate), in the 6-hour image (Fig. 2A1), scattered secondary precipitated particles formed on the coupon surface, but most of the surface remains uncovered. However, in the 7 day image (Fig. 2A2), most of the coupon surface is covered by newly formed nanoparticles, some of which have even formed large aggregates. Over the bicarbonate concentration range from 0.01 mM to 10 mM (Fig. 2A1–D1 and Fig. 2A2–D2), clear increases in particle coverage and particle height are observed. Regarding the reaction time, for all bicarbonate concentrations, the coupon surfaces at 7 days show more particle coverage and a greater particle height than those at 6 hours. The increases in bicarbonate concentration and reaction time also increased the surface root-mean-square roughness (Rq) and average particle size (height of at least 50 particles in each 5 × 5 μm2 image), confirming the formation and aggregation of secondary mineral precipitates. In 6 hours, the tenfold stepwise increases in bicarbonate concentrations increased Rq by amounts ranging from 6.1 ± 1.0 nm to 29 ± 2.8 nm and the height of precipitates from 27 ± 6.3 nm to 195 ± 24 nm. In 7 days, the increases in bicarbonate concentrations increased Rq from 9.0 ± 1.8 nm to 59 ± 3.7 nm and the height of precipitates from 72 ± 9.9 nm to 321 ± 64 nm. The findings from 7 day AFM images are consistent with the findings from CBD extraction, suggesting that higher bicarbonate concentrations will induce more heterogeneously precipitated iron(III) (hydr)oxide nanoparticles. Then, we characterized the morphology and covering of the secondary mineral precipitates at a larger scale using ESEM (Fig. S7A–D†). The ESEM images show a trend of secondary precipitate formation similar to that in the AFM images, corroborating the observation that higher bicarbonate concentrations create more particle coverage on the arsenopyrite surface and that, among all the bicarbonate concentrations, the 10 mM concentration yields the largest particles and the most significant aggregation.
 | ||
| Fig. 2 Nanoscale secondary mineral phase formation. Representative AFM height images for arsenopyrite coupons after 6 hours (A1–D1) and 7 days (A2–D2) in the 0.01 mM, 0.1 mM, 1 mM, and 10 mM bicarbonate concentrations, respectively, at room temperature (22 °C) and in an open-to-air condition. At least three different spots were measured on each coupon. The scan size of these image was 5 μm, and Rq is the root-mean-square surface roughness of the 5 × 5 μm2 images on a 100 nm height scale. | ||
To examine the oxidation state of the iron phases of arsenopyrite coupons after 6 hour and 7 day experiments, XPS analyses were conducted (Fig. 3). The relative proportions of the Fe(II)/Fe(III) ratio provide semiquantitative information about the extent of iron(III) formation on coupon surfaces. Unreacted arsenopyrite coupons showed the least amount of iron(III) formation on coupon surfaces. The Fe(II)/Fe(III) ratios dropped between 6 hours and 7 days in all bicarbonate concentrations, suggesting that longer reaction times allow more iron(III) (hydr)oxide mineral formation. As the bicarbonate concentration was increased from 0.1 mM to 10 mM, the Fe(II)/Fe(III) ratio decreased from 2.01 to 1.34 in 6 hours and from 1.08 to 0.89 in 7 days. The higher bicarbonate concentrations cause more iron(III) (hydr)oxide nanoparticle formation, which is consistent with the findings of the CBD extraction.
 | ||
| Fig. 3 XPS spectra of Fe 2p obtained from unreacted arsenopyrite coupons (A) and the arsenopyrite coupons in 0.1 mM (B), 1 mM (C), and 10 mM (D) bicarbonate concentrations in an open-to-air condition. Dotted lines are the positions of two different Fe(III) 2p peaks: Fe(III) 2p3/2 and Fe(III) 2p1/2. Blue and green peaks represent Fe3+ and Fe2+, respectively. The red curves are the fitting results from Gaussian–Lorentzian curve-fitting. Triplicate samples were measured for calculating the Fe(II)/Fe(III) ratios of each system (error ≈ ±0.1). | ||
Summarizing the findings from CBD extraction, AFM images, and XPS measurements of the open system experiment, it can be concluded that more iron(III) secondary nanoparticles formed heterogeneously at higher bicarbonate concentrations and decreased the arsenic mobilization. Two mechanisms could be responsible: (1) the greater amount of iron(III) (hydr)oxide nanoparticles can create more reactive sites for arsenic adsorption.24 Thus, more arsenic can be removed by adsorption on or incorporation into nanoscale iron(III) (hydr)oxides. (2) The more extensive coating of newly formed nanoparticles on arsenopyrite surface slowed down arsenopyrite dissolution.27,61 These two mechanisms also support the findings in Fig. 1C. Although iron(III) (hydr)oxide nanoparticles can adsorb arsenic from the aqueous solution and affect the extent and fate of dissolved arsenic, interestingly, we found more nanoscale iron(III) (hydr)oxide precipitates but less adsorbed arsenic in high bicarbonate concentrations (i.e., higher pH values). This could result from the low available total dissolved arsenic concentrations at high bicarbonate concentrations, limiting the amount of arsenic available for adsorption. Based on the findings in Fig. 1–3, we have found that the reduced arsenic mobilization was correlated to the increased bicarbonate concentrations that elevated the reaction pH and promoted formation of secondary iron(III) (hydr)oxide nanoparticles.
3.3 Nanoscale secondary mineral phase identification in the open system
The phases of secondary iron(III) (hydr)oxide nanoparticles were identified by Raman microscopy. As shown in Fig. 4, for the unreacted arsenopyrite coupons, the peaks at 333, 827, and 1368 cm−1 were exhibited, which are attributed to vibrations of the As–S, As–O, and Fe–O bonds, respectively.62,63 At low bicarbonate concentrations (0.01 mM and 0.1 mM, Fig. 4A and B respectively), not many peaks appear in either the 6 hour or 7 day spectra, suggesting that either the mineral phase of the coupons is still mostly arsenopyrite or the amounts of secondary mineral precipitates are not sufficient to be detected. However, at both 1.0 mM and 10 mM bicarbonate concentrations (Fig. 4C and D), more peaks appear than at either of the lower concentrations or for the unreacted arsenopyrite coupons. For both 1 mM and 10 mM bicarbonate, significant peaks at 665, 1330 and 1600 cm−1, corresponding to maghemite,64 can be found in both the short term (6 hours) and long term spectra (7 days). Interestingly, peaks related to siderite formation (1085 cm−1, and 1729 cm−1),64 and a peak related to magnetite (670 cm−1)64 appear on the 6 hour spectra, but do not appear in the 7 day spectra. This result indicates the phase transformation of siderite and magnetite into a more thermodynamically stable iron(III) phase, maghemite. The more diverse mineral phases found at higher bicarbonate concentrations can result from the higher solution pH driven by carbonate equilibrium in the open system. In the optical microscope images to the left of each Raman spectrum in Fig. 4, more particles can be seen at longer reaction times and higher bicarbonate concentrations. Overall, the higher bicarbonate concentrations (1.0 mM and 10 mM) can develop more phases of secondary iron-containing minerals (siderite, magnetite, and maghemite) on arsenopyrite mineral surfaces than the two low bicarbonate concentrations. Phase transformation of iron(III) (hydr)oxide nanoparticles, from siderite and magnetite into maghemite, also occurred in the 1.0 mM and 10 mM bicarbonate concentrations.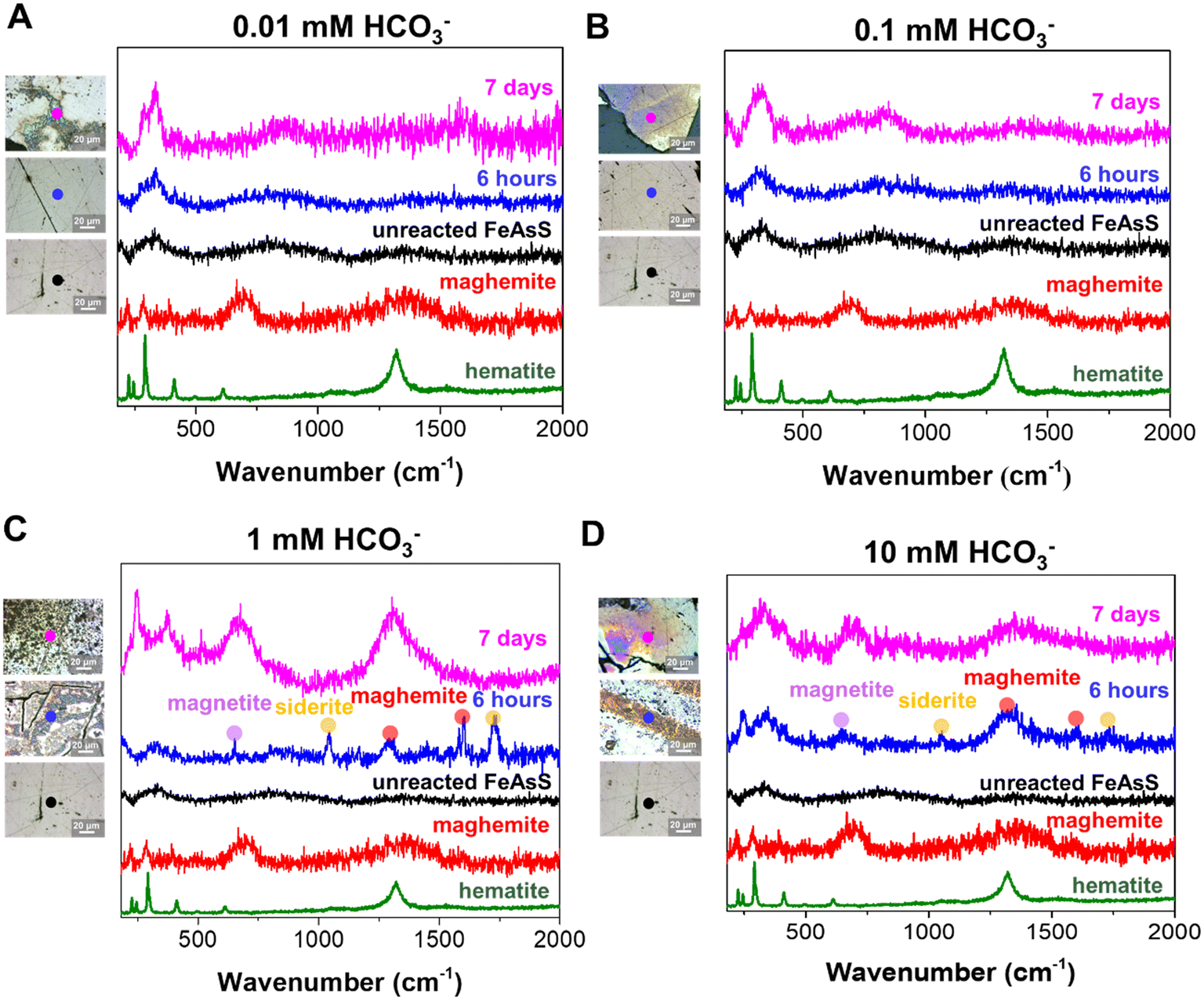 | ||
| Fig. 4 Mineral phase characterization of nanoscale secondary mineral particulates on arsenopyrite. Raman spectra and optical microscope images obtained from arsenopyrite coupons reacted for 6 hours and 7 days in 0.01 mM (A), 0.1 mM (B), 1 mM (C) and 10 mM (D) in an open-air condition. The colored dots indicate the mineral phases of the Raman spectra. At least triplicate samples were observed for each condition, and they are representative. | ||
The possible phase transformation is important in arsenic mobilization because it can influence the arsenic adsorption behaviors on iron (hydr)oxide minerals and their potential reverse mobilization.15,65–67 For example, iron oxides sub-micron sized particles phase transformation from magnetite to maghemite increased the adsorption performance of arsenic due to higher surface adsorption sites in maghemite.66 In addition, the phase transformation of siderite to goethite can enhance the arsenic adsorption by increasing the number of iron atoms coordinated with arsenic.65
3.4 Comparison of open and closed systems
In previous sections, we have primarily discussed the results in open systems where it shows distinct arsenopyrite dissolution behaviors. In this section, we presented the dissolution kinetics of arsenopyrite in a closed system as shown in Fig. 5. Over 7 days, the dissolved arsenic increases with time, but there is no significant difference among the three bicarbonate concentrations. In both the open and closed systems, the 0.1 mM bicarbonate solution contains similar dissolved arsenic amounts at 7 days (open, 218 ± 33 μg L−1; closed, 202 ± 32 μg L−1). However, the 1 mM bicarbonate solution contains different dissolved arsenic amounts at 7 days (open, 96 ± 16 μg L−1; closed, 181 ± 15 μg L−1), as does the 10 mM bicarbonate solution (open: 123 ± 19 μg L−1; closed: 168 ± 15 μg L−1). In the closed system, for all three bicarbonate concentrations, the dissolved arsenic concentrations are similar within the range of error because of their similar equilibrium pH values. From the calculated and experimental values of the pH, in the closed system, bicarbonate concentrations from 0.1 mM to 10 mM have equilibrium pH values around 7 (Fig. S8†), similar to the pH values for 0.1 mM bicarbonate in the open system. The disparities of dissolved arsenic concentration and pH in the open and closed systems were controlled by the concentrations of bicarbonate, as shown in the thermodynamically-calculated percentages of total dissolved carbonate in Table S1.† In the open system, high bicarbonate concentrations caused slow arsenic dissolution kinetics (Fig. 5B, yellow zone), while in the closed system the arsenic mobilization was not significantly affected by bicarbonate (Fig. 5B, gray zone). Although ATR-FTIR revealed surface complexation (Fig. S4B†), and the calculated results indicated the formation of aqueous complex (Table S4B†), the similar dissolution behaviors in the closed system not only indicate the primary driving force of unchanged reaction pH, but also rule out the influence of both aqueous and surface complexation. Notably, in a control experiment with different initial pH conditions using 0.1 mM bicarbonate in the closed system, inhibited dissolution kinetics was found from pH 7 to pH 8, while no significant difference was found from pH 8 to pH 8.9 (Fig. S9†). This trend corroborates our hypothesis in the open system that although pH can be the dominant driving force of arsenopyrite dissolution, if pH was changed from 8 to 8.9, aqueous and surface complexation could offset the effect of pH. Additionally, Fig. S9† shows that at 7 days in the closed system, 0.1 mM bicarbonate at pH 8 and pH 8.9 has arsenic concentrations of 76 ± 11 μg L−1 and 87 ± 20 μg L−1, respectively. In contrast, Fig. 1A shows that in the open system, arsenic concentrations at 7 days are 96 ± 16 μg L−1 for 1 mM bicarbonate (pH 8) and 123 ± 19 μg L−1 for 10 mM bicarbonate (pH 8.9). Although the differences in arsenic concentrations are not significantly large, the relatively higher arsenic concentrations observed in the open system could be attributed to the higher bicarbonate concentrations (1 mM and 10 mM) compared to the 0.1 mM bicarbonate in the closed system, supporting the complexation mechanism. Furthermore, the bicarbonate concentrations in the closed system have only a minor effect on secondary mineral formation. AFM images in Fig. S10A–C† show a root-mean-square surface roughness, Rq, comparable to that of the 0.1 mM bicarbonate concentration in the open system (Fig. 2B2). ESEM images (Fig. S7E–G†) of the closed system coupon surface are all similar, without significant aggregated mineral formation, and are similar to the 0.1 mM bicarbonate images in the open system (Fig. S7B†). The similarity of the secondary mineral coverages would provide similar reactive surface areas, which can explain the minor impact of bicarbonate concentrations on arsenic mobilization.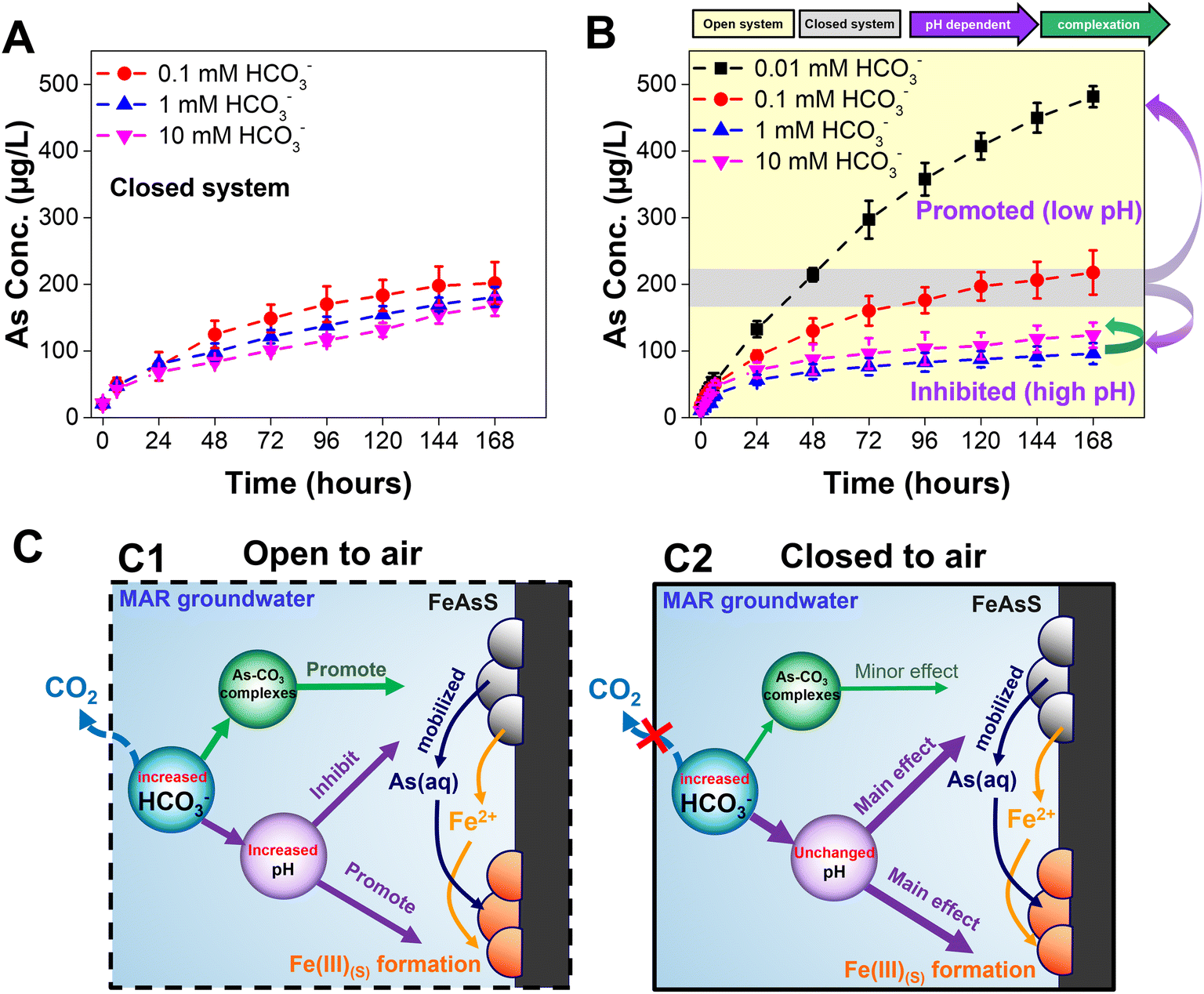 | ||
| Fig. 5 Dissolution of arsenopyrite in 0.1 mM, 1 mM, and 10 mM bicarbonate concentrations reacted for 7 days in a closed system. A well-controlled pH was not achieved by the 0.01 mM bicarbonate, so no data for this concentration is reported for the closed system. (A) Dissolved arsenic concentrations. (B) Comparison of arsenic mobilization in open and closed systems at 7 days. (C) Proposed mechanisms of arsenopyrite dissolution in open (C1) and closed systems (C2). | ||
4. Conclusions
Bicarbonate is a ubiquitous groundwater component that regulates the fate and transport of many earth elements. In MAR, injected water can alter the groundwater chemistry, triggering different extents of bicarbonate and mineral dissolution. Here, we found that high bicarbonate concentrations relevant to groundwater environments and MAR, resulted in lower arsenic dissolution and mobilization in comparison to lower bicarbonate concentrations in an open system. This reduced arsenic mobilization was attributed to the extent of CO2 (i.e., bicarbonate) that increased the reaction pH and promoted formation of secondary iron(III) (hydr)oxide nanoparticles. This finding suggests that, in an open system, increasing the bicarbonate concentrations or alkalinity of groundwater during MAR can attenuate arsenic mobilization. However, this attenuation can be specific to the range of bicarbonate concentrations between 0.01 mM to 1 mM. Higher bicarbonate concentrations (10 mM in this work) did not further decrease arsenic mobilization, and the arsenic concentrations at 10 mM bicarbonate were even less than those at 0.01 mM and 0.1 mM bicarbonate concentrations. This trend reflects the presence of aqueous and surface complexation that promotes the release of arsenic in open systems (Fig. 5B, green arrow). In an open system, quantitative and qualitative observations showed that 1.5 times more secondary mineral precipitates, based on the CBD extraction results in Fig. 1D, formed with higher bicarbonate concentrations, and greater coverage of the arsenopyrite surface by nanoscale secondary iron(III) (hydr)oxide minerals could reduce the number of reactive sites, based on the AFM images in Fig. 2. Therefore, newly formed iron(III) (hydr)oxide nanoparticles hindered the dissolution of arsenopyrite. The quantification of dissolved arsenic in this work also pointed out the importance of nanoscale secondary iron(III) (hydr)oxide mineral formation, because a large portion of the dissolved arsenic was immobilized by adsorption. On the other hand, in the closed system, arsenic mobilization and secondary mineral formation was not affected by different bicarbonate concentrations. These similar behaviors of arsenopyrite dissolution and secondary nanoparticle formation were controlled mainly by the unchanged reaction pH while aqueous and surface complexation had minor influence.In this study, building from the current understanding of the correlation between bicarbonate and arsenic mobilization,24,27,30,31 we have extended this knowledge and found that arsenic mobilization depended not only on the bicarbonate concentration but also on systems specific to aquifers open or closed to air (Fig. 5C). In the open system (Fig. 5C1), the increased bicarbonate concentrations can increase reaction pH and therefore inhibit arsenic mobilization and promote secondary iron (hydr)oxide mineral formation. At high bicarbonate concentration (i.e., 10 mM in this work), the aqueous and surface complexation were also found to attenuate the inhibited arsenic mobilization. In the closed system (Fig. 5C2), the arsenic mobilization and secondary minerals were not significantly affected by bicarbonate concentrations which could be mainly controlled by unchanged reaction pH. This finding has important implications for the engineered applications. MAR has been used to mitigate seawater intrusion, which can infiltrate both shallow unconfined aquifers and deep confined aquifers.37 When MAR is applied to shallow unconfined aquifers (e.g., a dry well, percolation tank, or infiltration basin),11,68 recharging with low bicarbonate water can decrease the bicarbonate concentration and increase the arsenic mobilization in the aquifer. Further, the simple infiltration of reclaimed water can decrease pCO2, unbalancing the bicarbonate concentration in groundwater.69 With regard to MAR, because arsenic mobilization can be higher in shallow unconfined aquifers with low bicarbonate concentrations, pretreatment to adjust the alkalinity of the recharge water can help reduce it. In the case of deep confined aquifers, where bicarbonate equilibrium is likely a minor factor in arsenic mobilization, the pH and the concentrations of other common groundwater oxyanions (e.g., phosphate, chloride, and sulfate) should receive more attention for accurate controlling arsenic mobilization.
Other than bicarbonate (an inorganic component) we studied in this work, microorganisms and organic molecules can also play important roles in the oxidative dissolution of arsenopyrite. For example, arsenite-oxidizing bacteria can directly transform and mobilize arsenic.70 Sulfur- and iron-oxidizing bacteria can also indirectly mobilize arsenic from sulfide minerals.71 As for organic molecules, dissolved organic matter (DOM) can decrease the particle sizes and growth rates of iron(III) (hydr)oxide nanoparticles and DOM with a high molecular weight can also increase arsenic mobilization.48 Thus far, the individual roles of inorganic ions, microorganisms, and organic compounds in arsenic mobilization have been identified by previous studies.48,70,71 Future studies focusing on MAR can more systematically examine arsenic mobilization involving the co-existence of commonly encountered inorganic and organic components.
Moreover, the dominance of adsorbed arsenic in the total dissolved arsenic content also suggests that the fate of arsenic, such as adsorption by nanoscale secondary iron(III) (hydr)oxide precipitates or other mineral surfaces, should be considered in evaluating the total dissolution of arsenic from arsenic-bearing sulfide minerals. Our findings here can help in developing more accurate and comprehensive reactive transport models to predict MAR's long-term groundwater quality impacts and eventually achieve safe and sustainable MAR designs to protect the environment.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful for the support received from the National Science Foundation (EAR-1424927). P.-I. Chou acknowledges a fellowship from the McDonnell International Scholars Academy. The authors would like to acknowledge the Nano Research Facility (NRF) of Washington University in St. Louis for the use of the ICP-MS, and ICP-OES, Institute of Materials Science & Engineering (IMSE) for the use of XPS and ESEM, and Professor Srikanth Singamaneni for the use of Raman spectroscopy. The authors thank Professor James C. Ballard for carefully reviewing the manuscript and also thank the Environmental NanoChemistry Group members for valuable discussions.References
- V. G. Gude, Desalination of deep groundwater aquifers for freshwater supplies – Challenges and strategies, Groundw. Sustain. Dev., 2018, 6, 87–92 CrossRef.
- A. Boretti and L. Rosa, Reassessing the projections of the World Water Development Report, npj Clean Water, 2019, 2, 15 CrossRef.
- UN, The United Nations World Water Development Report 2022: groundwater: making the invisible visible, UN, 2022 Search PubMed.
- Y. Pokhrel, F. Felfelani, Y. Satoh, J. Boulange, P. Burek, A. Gädeke, D. Gerten, S. N. Gosling, M. Grillakis, L. Gudmundsson, N. Hanasaki, H. Kim, A. Koutroulis, J. Liu, L. Papadimitriou, J. Schewe, H. Müller Schmied, T. Stacke, C.-E. Telteu, W. Thiery, T. Veldkamp, F. Zhao and Y. Wada, Global terrestrial water storage and drought severity under climate change, Nat. Clim. Change, 2021, 11, 226–233 CrossRef.
- California Department of Water Resources, Water Year 2021: An Extreme Year, California Department of Water Resources, 2021 Search PubMed.
- T. Hayashi, T. Tokunaga, M. Aichi, J. Shimada and M. Taniguchi, Effects of human activities and urbanization on groundwater environments: an example from the aquifer system of Tokyo and the surrounding area, Sci. Total Environ., 2009, 407, 3165–3172 CrossRef CAS.
- J. Hoffman, H. A. Zebker, D. L. Galloway and F. Amelung, Seasonal subsidence and rebound in Las Vegas valley, Nevada, observed by synthetic aperture radar interferometry, Water Resour. Res., 2001, 37, 1551–1566 CrossRef.
- C. W. Neil, Y. J. Yang and Y. S. Jun, Arsenic mobilization and attenuation by mineral-water interactions: implications for managed aquifer recharge, J. Environ. Monit., 2012, 14, 1772–1788 RSC.
- S. Alam, A. Borthakur, S. Ravi, M. Gebremichael and S. K. Mohanty, Managed aquifer recharge implementation criteria to achieve water sustainability, Sci. Total Environ., 2021, 768, 144992 CrossRef CAS PubMed.
- S. Fakhreddine, H. Prommer, B. R. Scanlon, S. C. Ying and J. P. Nicot, Mobilization of Arsenic and Other Naturally Occurring Contaminants during Managed Aquifer Recharge: A Critical Review, Environ. Sci. Technol., 2021, 55, 2208–2223 CrossRef CAS PubMed.
- S. Toze, E. Bekele, D. Page, J. Sidhu and M. Shackleton, Use of static Quantitative Microbial Risk Assessment to determine pathogen risks in an unconfined carbonate aquifer used for Managed Aquifer Recharge, Water Res., 2010, 44, 1038–1049 CrossRef CAS PubMed.
- EPA, Arsenic in drinking water rule economic analysis, EPA, 2000 Search PubMed.
- G. W. Jones and T. Pichler, Relationship between Pyrite Stability and Arsenic Mobility During Aquifer Storage and Recovery in Southwest Central Florida, Environ. Sci. Technol., 2007, 41, 723–730 CrossRef CAS PubMed.
- J. L. Vanderzalm, P. J. Dillon, K. E. Barry, K. Miotlinski, J. K. Kirby and C. Le Gal La Salle, Arsenic mobility and impact on recovered water quality during aquifer storage and recovery using reclaimed water in a carbonate aquifer, Appl. Geochem., 2011, 26, 1946–1955 CrossRef CAS.
- C. W. Neil, Y. J. Yang, D. Schupp and Y. S. Jun, Water chemistry impacts on arsenic mobilization from arsenopyrite dissolution and secondary mineral precipitation: implications for managed aquifer recharge, Environ. Sci. Technol., 2014, 48, 4395–4405 CrossRef CAS PubMed.
- G. Simon, H. Huang, J. E. Penner-Hahn, S. E. Kesler and L.-S. Kao, Oxidation state of gold and arsenic in gold-bearing arsenian pyrite, Am. Mineral., 1999, 84, 1071–1079 CrossRef CAS.
- M. A. McKibben, B. A. Tallant and J. K. del Angel, Kinetics of inorganic arsenopyrite oxidation in acidic aqueous solutions, Appl. Geochem., 2008, 23, 121–135 CrossRef CAS.
- D. K. Nordstrom, Worldwide Occurrences of Arsenic in Ground Water, Science, 2002, 296, 2143 CrossRef CAS PubMed.
- Y. Yu, Y. Zhu, Z. Gao, C. H. Gammons and D. Li, Rates of Arsenopyrite Oxidation by Oxygen and Fe(III) at pH 1.8−12.6 and 15−45 °C, Environ. Sci. Technol., 2007, 41, 6460–6464 CrossRef CAS PubMed.
- R. Gubler and L. K. ThomasArrigo, Ferrous iron enhances arsenic sorption and oxidation by non-stoichiometric magnetite and maghemite, J. Hazard. Mater., 2021, 402, 123425 CrossRef CAS PubMed.
- Y.-S. Jun, Catalyst: The roles of chemistry in clean water for all, Chem, 2023, 9, 1335–1339 Search PubMed.
- Y.-S. Jun, D. Kim and C. W. Neil, Heterogeneous Nucleation and Growth of Nanoparticles at Environmental Interfaces, Acc. Chem. Res., 2016, 49, 1681–1690 CrossRef CAS PubMed.
- C. W. Neil and Y.-S. Jun, Fe3+Addition Promotes Arsenopyrite Dissolution and Iron(III) (Hydr)oxide Formation and Phase Transformation, Environ. Sci. Technol. Lett., 2015, 3, 30–35 CrossRef.
- X. Wu, S. Burnell, C. W. Neil, D. Kim, L. Zhang, H. Jung and Y.-S. Jun, Effects of Phosphate, Silicate, and Bicarbonate on Arsenopyrite Dissolution and Secondary Mineral Precipitation, ACS Earth Space Chem., 2020, 4, 515–525 CrossRef CAS.
- R. Kaegi, A. Voegelin, D. Folini and S. J. Hug, Effect of phosphate, silicate, and Ca on the morphology, structure and elemental composition of Fe(III)-precipitates formed in aerated Fe(II) and As(III) containing water, Geochim. Cosmochim. Acta, 2010, 74, 5798–5816 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, Wiley, New York, 1996 Search PubMed.
- T. Radu, J. L. Subacz, J. M. Phillippi and M. O. Barnett, Effects of Dissolved Carbonate on Arsenic Adsorption and Mobility, Environ. Sci. Technol., 2005, 39, 7875–7882 CrossRef CAS PubMed.
- S. Krishna Kumar, S. Hari Babu, P. Eswar Rao, S. Selvakumar, C. Thivya, S. Muralidharan and G. Jeyabal, Evaluation of water quality and hydrogeochemistry of surface and groundwater, Tiruvallur District, Tamil Nadu, India, Appl. Water Sci., 2016, 7, 2533–2544 CrossRef.
- N. Zhan, Y. Huang, Z. Rao and X.-L. Zhao, Fast Detection of Carbonate and Bicarbonate in Groundwater and Lake Water by Coupled Ion Selective Electrode, Chin. J. Anal. Chem., 2016, 44, 355–360 CAS.
- M.-J. Kim, A. J. Nriagu and S. Haack, Carbonate Ions and Arsenic Dissolution by Groundwater, Environ. Sci. Technol., 2000, 34, 3094–3100 CrossRef CAS.
- C. A. J. Appelo, M. J. J. Van Der Weiden, C. Tournassat and L. Charlet, Surface Complexation of Ferrous Iron and Carbonate on Ferrihydrite and the Mobilization of Arsenic, Environ. Sci. Technol., 2002, 36, 3096–3103 CrossRef CAS PubMed.
- S. Fakhreddine, J. Dittmar, D. Phipps, J. Dadakis and S. Fendorf, Geochemical Triggers of Arsenic Mobilization during Managed Aquifer Recharge, Environ. Sci. Technol., 2015, 49, 7802–7809 CrossRef CAS.
- J. Greskowiak, H. Prommer, G. Massmann, C. D. Johnston, G. Nützmann and A. Pekdeger, The impact of variably saturated conditions on hydrogeochemical changes during artificial recharge of groundwater, Appl. Geochem., 2005, 20, 1409–1426 CrossRef CAS.
- S. Fakhreddine, H. Prommer, S. M. Gorelick, J. Dadakis and S. Fendorf, Controlling Arsenic Mobilization during Managed Aquifer Recharge: The Role of Sediment Heterogeneity, Environ. Sci. Technol., 2020, 54, 8728–8738 CrossRef CAS PubMed.
- A. Antoniou, F. Smits and P. Stuyfzand, Quality assessment of deep-well recharge applications in the Netherlands, Water Supply, 2017, 17, 1201–1211 CrossRef CAS.
- D. Perrone and M. Merri Rohde, Benefits and Economic Costs of Managed Aquifer Recharge in California, San Franc. Estuary Watershed Sci., 2016, 14(2) DOI:10.15447/sfews.2016v14iss2art4.
- S. Jasechko, D. Perrone, H. Seybold, Y. Fan and J. W. Kirchner, Groundwater level observations in 250,000 coastal US wells reveal scope of potential seawater intrusion, Nat. Commun., 2020, 11, 3229 CrossRef CAS PubMed.
- S. R. Maples, G. E. Fogg and R. M. Maxwell, Modeling managed aquifer recharge processes in a highly heterogeneous, semi-confined aquifer system, Hydrogeol. J., 2019, 27, 2869–2888 CrossRef CAS.
- C. M. Wicks and J. S. Herman, The effect of a confining unit on the geochemical evolution of ground water in the Upper Floridan aquifer system, J. Hydrol., 1994, 153, 139–155 CrossRef CAS.
- S. Gjengedal, L. A. Stenvik, P.-T. S. Storli, R. K. Ramstad, B. O. Hilmo and B. S. Frengstad, Design of Groundwater Heat Pump Systems. Principles, Tools, and Strategies for Controlling Gas and Precipitation Problems, Energies, 2019, 12, 3657 CrossRef CAS.
- M. C. Hidalgo and J. Cruz-Sanjulián, Groundwater composition, hydrochemical evolution and mass transfer in a regional detrital aquifer (Baza basin, southern Spain), Appl. Geochem., 2001, 16, 745–758 CrossRef CAS.
- J. Choi, S. M. Hulseapple, M. H. Conklin and J. W. Harvey, Modeling CO2 degassing and pH in a stream-aquifer system, J. Hydrol., 1998, 209, 297–310 CrossRef CAS.
- K. Pi, Y. Wang, X. Xie, T. Ma, Y. Liu, C. Su, Y. Zhu and Z. Wang, Remediation of arsenic-contaminated groundwater by in-situ stimulating biogenic precipitation of iron sulfides, Water Res., 2017, 109, 337–346 CrossRef CAS PubMed.
- I. Wallis, H. Prommer, C. T. Simmons, V. Post and P. J. Stuyfzand, Evaluation of Conceptual and Numerical Models for Arsenic Mobilization and Attenuation during Managed Aquifer Recharge, Environ. Sci. Technol., 2010, 44, 5035–5041 CrossRef CAS PubMed.
- A. National Ground Water, Brackish Groundwater, National Ground Water Association, Westerville, OH, Information Brief, 2010 Search PubMed.
- M. Kang and R. B. Jackson, Salinity of deep groundwater in California: Water quantity, quality, and protection, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7768–7773 CrossRef CAS.
- J. P. Gustafsson, Visual MINTEQ 3.1 user guide, 2014 Search PubMed.
- X. Wu, B. Bowers, D. Kim, B. Lee and Y. S. Jun, Dissolved Organic Matter Affects Arsenic Mobility and Iron(III) (hydr)oxide Formation: Implications for Managed Aquifer Recharge, Environ. Sci. Technol., 2019, 53, 14357–14367 CrossRef CAS PubMed.
- J.-H. Jang and B. A. Dempsey, Coadsorption of Arsenic(III) and Arsenic(V) onto Hydrous Ferric Oxide: Effects on Abiotic Oxidation of Arsenic(III), Extraction Efficiency, and Model Accuracy, Environ. Sci. Technol., 2008, 42, 2893–2898 CrossRef CAS PubMed.
- G. Qiu, T. Gao, J. Hong, W. Tan, F. Liu and L. Zheng, Mechanisms of arsenic-containing pyrite oxidation by aqueous arsenate under anoxic conditions, Geochim. Cosmochim. Acta, 2017, 217, 306–319 CrossRef CAS.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Use of XPS in the determination of chemical environment and oxidation state of iron and sulfur samples: constitution of a data basis in binding energies for Fe and S reference compounds and applications to the evidence of surface species of an oxidized pyrite in a carbonate medium, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- O. P. Mehra and M. L. Jackson, in Clays and Clay Minerals, ed. E. Ingerson, Pergamon, 2013, pp. 317–327, DOI:10.1016/B978-0-08-009235-5.50026–7.
- L. Zhang, Y. Zhu, X. Wu and Y.-S. Jun, Effects of sulfate on biotite interfacial reactions under high temperature and high CO2 pressure, Phys. Chem. Chem. Phys., 2019, 21, 6381–6390 RSC.
- H. Wijnja and C. P. Schulthess, ATR–FTIR and DRIFT spectroscopy of carbonate species at the aged γ-Al2O3/water interface, Spectrochim. Acta, Part A, 1999, 55, 861–872 CrossRef.
- P. Roonasi and A. Holmgren, An ATR-FTIR study of carbonate sorption onto magnetite, Surf. Interface Anal., 2010, 42, 1118–1121 CrossRef CAS.
- L. F. Liao, C. F. Lien, D. L. Shieh, M. T. Chen and J. L. Lin, FTIR Study of Adsorption and Photoassisted Oxygen Isotopic Exchange of Carbon Monoxide, Carbon Dioxide, Carbonate, and Formate on TiO2, J. Phys. Chem. B, 2002, 106, 11240–11245 CrossRef CAS.
- V. K. Díez, C. A. Ferretti, P. A. Torresi, C. R. Apesteguía and J. I. Di Cosimo, Effect of MgO activation conditions on its catalytic properties for base-catalyzed reactions, Catal. Today, 2011, 173, 21–27 CrossRef.
- T. Hiemstra, R. Rahnemaie and W. H. Van Riemsdijk, Surface complexation of carbonate on goethite: IR spectroscopy, structure and charge distribution, J. Colloid Interface Sci., 2004, 278, 282–290 CrossRef CAS PubMed.
- R. T. Nickson, J. M. McArthur, P. Ravenscroft, W. G. Burgess and K. M. Ahmed, Mechanism of arsenic release to groundwater, Bangladesh and West Bengal, Appl. Geochem., 2000, 15, 403–413 CrossRef CAS.
- Y. Arai, D. L. Sparks and J. A. Davis, Effects of Dissolved Carbonate on Arsenate Adsorption and Surface Speciation at the Hematite−Water Interface, Environ. Sci. Technol., 2004, 38, 817–824 CrossRef CAS PubMed.
- Y. Jia, L. Xu, Z. Fang and G. P. Demopoulos, Observation of Surface Precipitation of Arsenate on Ferrihydrite, Environ. Sci. Technol., 2006, 40, 3248–3253 CrossRef CAS PubMed.
- A. Courtin-Nomade, H. Bril, J. M. Beny, M. Kunz and N. Tamura, Sulfide oxidation observed using micro-Raman spectroscopy and micro-X-ray diffraction: The importance of water/rock ratios and pH conditions, Am. Mineral., 2010, 95, 582–591 CrossRef CAS.
- M. Hanesch, Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
- H. Guo, Y. Ren, Q. Liu, K. Zhao and Y. Li, Enhancement of arsenic adsorption during mineral transformation from siderite to goethite: mechanism and application, Environ. Sci. Technol., 2013, 47, 1009–1016 CrossRef CAS PubMed.
- M. E. Mejia-Santillan, N. Pariona, J. Bravo-C, M. Herrera-Trejo, F. Montejo-Alvaro, A. Zarate, D. L. Perry and A. I. Mtz-Enriquez, Physical and arsenic adsorption properties of maghemite and magnetite sub-microparticles, J. Magn. Magn. Mater., 2018, 451, 594–601 CrossRef CAS.
- S. Dixit and J. G. Hering, Comparison of arsenic (V) and arsenic (III) sorption onto iron oxide minerals: implications for arsenic mobility, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- R. J. Rossi, R. A. Tisherman, J. M. Jaeger, J. Domen, S. B. C. Shonkoff and D. C. Digiulio, Historic and Contemporary Surface Disposal of Produced Water Likely Inputs Arsenic and Selenium to Surficial Aquifers, Environ. Sci. Technol., 2023, 57, 7559–7567 CrossRef CAS PubMed.
- Q. Xia, J. He, B. Li, B. He, J. Huang, M. Guo and D. Luo, Hydrochemical evolution characteristics and genesis of groundwater under long-term infiltration (2007—2018) of reclaimed water in Chaobai River, Beijing, Water Res., 2022, 226, 119222 CrossRef CAS PubMed.
- E. D. Rhine, K. M. Onesios, M. E. Serfes, J. R. Reinfelder and L. Y. Young, Arsenic Transformation and Mobilization from Minerals by the Arsenite Oxidizing Strain WAO, Environ. Sci. Technol., 2008, 42, 1423–1429 Search PubMed.
- K. Tomczyk-Żak, S. Kaczanowski, Ł. Drewniak, Ł. Dmoch, A. Sklodowska and U. Zielenkiewicz, Bacteria diversity and arsenic mobilization in rock biofilm from an ancient gold and arsenic mine, Sci. Total Environ., 2013, 461–462, 330–340 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00805g |
| This journal is © The Royal Society of Chemistry 2025 |