
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the versatility of novel furan-based α,ω-diene carbonate monomers: synthesis, (co-)polymerization, and comparative study†
Beatriz
Chícharo
 ab,
Sami
Fadlallah
*a,
Giacomo
Trapasso
b,
Thomas
Gherardi
b,
Florent
Allais
*a and
Fabio
Aricò
*b
ab,
Sami
Fadlallah
*a,
Giacomo
Trapasso
b,
Thomas
Gherardi
b,
Florent
Allais
*a and
Fabio
Aricò
*b
aURD Agro-Biotechnologies Industrielles (ABI), CEBB, AgroParisTech, 3 Rue des Rouges-Terres, 51110 Pomacle, France. E-mail: florent.allais@agroparistech.fr; sami.fadlallah@agroparistech.fr
bDepartment of Environmental Sciences, Informatics and Statistics, Ca’ Foscari University of Venice, Via Torino 155, 30172 Venezia Mestre, Italy. E-mail: fabio.arico@unive.it
First published on 20th December 2024
Abstract
In this work, a novel family of α,ω-diene carbonate monomers was synthesized via the alkoxy carbonylation reaction of bis(hydroxymethyl)furan (BHMF) with dialkyl carbonates (DACs) of varying chain lengths, containing terminal olefins, in the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). These monomers were then subjected to acyclic diene metathesis (ADMET) polymerization with seven different ruthenium catalysts. The second-generation Hoveyda–Grubbs catalyst proved to be the most effective, yielding furan-based polycarbonates with molecular weights (Mn) up to 19 kDa. The resulting bio-based polymers exhibited thermal degradation temperatures (Td5%) ranging from 156 °C to 244 °C and glass transition temperatures (Tg) from −8 °C to −36 °C. NMR studies confirmed their polymeric structures and provided insights into the polymers organization, which influenced their properties. These novel polycarbonates were then compared to previously reported polyesters and polyethers derived from similar furan-based α,ω-diene monomers. Additionally, for the first time, co-polymerization studies were conducted on three families of furan-based α,ω-diene monomers—ester, ether, and carbonate—revealing the effect of incorporating different functional groups on the properties of the resulting materials. This unprecedented comparison and co-polymerization reactions highlight the versatility of furan-based monomers, but also underscores the possibility to expand their application in creating tailored bio-based materials for diverse applications.
Green foundation1. In this work, starting from a bio-based platform chemical, bis(hydroxymethyl)furan (BHMF), we report on the green synthesis of α,ω-diene carbonate monomers and their subsequent acyclic diene metathesis (ADMET) polymerization. This work highlights the versatility of furan-based monomers in ADMET polymerization, highlighting the possibility to expand their application in creating tailored bio-based materials for diverse applications.2. The synthesis of α,ω-diene carbonate monomers was developed following green chemistry principles. A catalytic amount of catalyst was used in the synthesis, and excess reagents were recovered for reuse whenever possible, minimizing waste. This approach led to highly selective reactions with excellent yields, requiring minimal purification. Additionally, the Ru-catalyzed ADMET polymerization was conducted under mild conditions, reducing energy consumption compared to other polymerization methods. Moreover, this type of polymerization demonstrates high atom economy. 3. A potential future development could involve the large-scale production of this type of monomer using continuous-flow apparatus. This would enable the preparation of larger quantities of the related bio-based polycarbonates, whose properties and potential applications could then be further explored. |
Introduction
Polycarbonates (PCs) are durable, lightweight polymers known for their optical clarity, weather resistance, and high thermal stability.1 Their unique properties, a consequence of the carbonate moieties, make them suitable for diverse applications, including packaging, optics, construction, automotive, electronics and in medical fields.2–4 The synthesis of polycarbonates can be accomplished via several approaches, such as step-growth polycondensation using organic carbonates and hydroxyl containing molecules, oxidative carbonylation, and ring-opening polymerization (ROP).2–4 Although ROP is known for its mild conditions,5 other methods often require harsher environmentally hazardous procedures, and/or offer limited control over the polymer structures, with side reactions frequently occurring.6 Moreover, while ROP is generally more desirable, it has limitations in terms of monomer structure and availability, and the existing monomers are not always tunable to allow precise control over polymer properties.In this context, there is significant interest in developing procedures that enable polycarbonate preparation under mild conditions, with stable catalysts that offer control over both monomer and polymer structures.
Acyclic diene metathesis (ADMET) polymerization represents a promising approach for achieving precise polymer control under mild, solvent-free conditions.7–9 In fact, this method was demonstrated to be suitable for large-scale industrial applications (i.e., synthesis of concrete superplasticizers) at Chryso Saint-Gobain facilities.10 Furthermore, ADMET polymerization and metathesis catalysts are highly tolerant to various functional groups, and they do not require strict anhydrous conditions, which facilitates their transfer to industrial processes.7
A wide range of polyfunctional hydroxyl compounds have been reported in the literature as suitable monomers for the synthesis of polycarbonates, however, the only commercially significant PC in the market as of now is derived from bisphenol A (BPA) or 2,2-bis-(4-hydroxyphenyl)propane.11
In a typical polymerization, BPA is reacted with phosgene via polycondensation leading to the desired PC.2 However, BPA is a non-renewable compound with harmful effects on human health, acting as an endocrine disruptor and a nephrotoxic agent. Moreover, phosgene toxicity is extensively documented in the literature.12 For this reason, the scientific community is actively seeking alternative feedstocks from renewable sources. Nevertheless, challenges remain, particularly concerning the sourcing of bio-based monomers, as well as the greenness of the synthetic procedures used to modify platform molecules. In fact, using renewable carbon is not enough and an assessment of synthetic procedures based on green chemistry principles and eventually on green metrics is necessary to ensure genuine sustainability.13
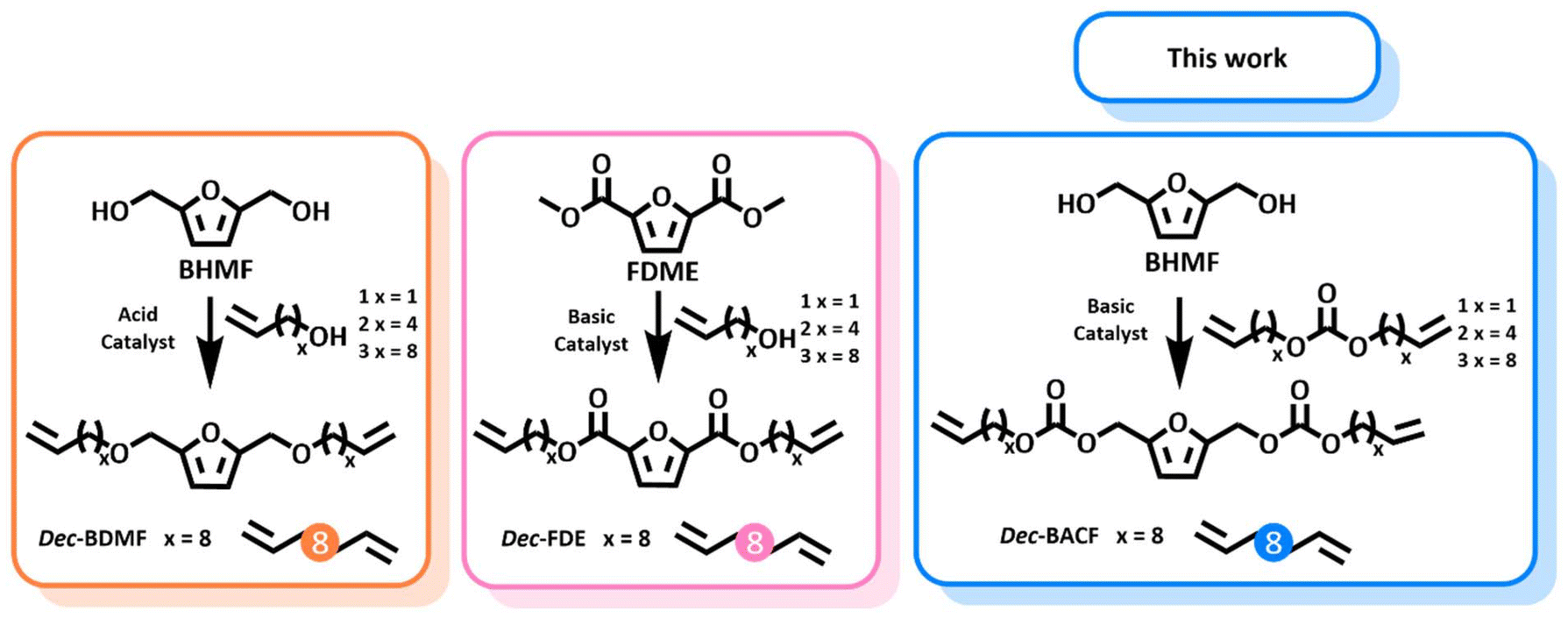
Recently, the use of terpenoids, such as limonene oxide, isosorbide carbonate, and vegetable oils, was explored for the synthesis of polycarbonates.14–17 On the other hand, furanic monomers – derived from hemicellulose and cellulose as second-generation biomass – have been scarcely studied for PCs production, with, to the best of our knowledge, only one report available in the literature to this date.18 Still, in this procedure the furan-based PC was achieved by step-growth polymerization of 2,5-bis(hydroxymethyl)furan (BHMF) with carbonyldiimidazole (CDI), which is a phosgene-derived chemical.18
Furan-based compounds have gained attention in recent years as monomers for the synthesis of high-performance renewable polymers19 such as polyethers and polyesters, that can potentially replace their petroleum-based counterparts, showing also improved mechanical and thermal properties.20–27
Developing polycarbonates from furan-based monomer is of particular interest as the presence of a rigid furan moiety could increase the thermal stability, rigidity, and crystallinity, which would broaden the applications for these bio-based materials.28 The ability to tailor carbonate-based monomers for ADMET polymerization would also lead to a new class of bio-based materials with precise control over polymer structure and properties. Additionally, while many studies have focused on synthesizing furan-based polymers, there is a gap in understanding how the incorporation of different functional groups (e.g., carbonate, ether, ester) affects polymer properties.
The herein proposed study aims at addressing all these challenges by reporting a novel class of furan-based α,ω-diene carbonate monomers and exploring their polymerization via ADMET using various ruthenium catalysts. This work also reports on the first detailed comparison of polycarbonates, polyethers,20 and polyesters21 derived from similar furan-based α,ω-diene monomers, demonstrating how the incorporation of different functional groups impacts polymer properties such as thermal stability, molecular weight, and crystallinity. Additionally, co-polymerization experiments revealed how blending different monomers can further expand the range of the polymer properties. The results collected highlight the versatility of these furan-based monomers, showing their potential as materials with tunable properties, while offering significant insights for the design of bio-based polymers.
Results and discussion
Furan-based α,ω-diene carbonate monomers
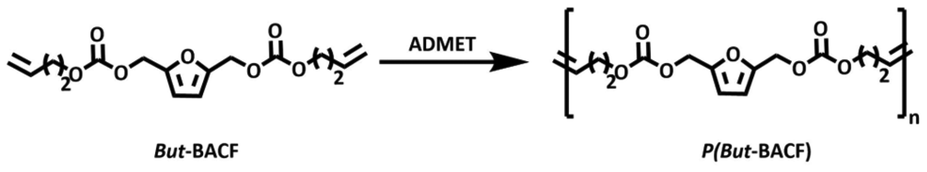
Furan-based α,ω-diene carbonates are within the family of bis(alkenylcarbonate)furans (BACFs) and for simplicity they will be indicated as x-BACFs where x indicates the length of the alkenyl chain (All = Allyl, But = Butenyl, Hex = Hexenyl, Dec = Decenyl).Two synthetic routes were explored for the preparation of these monomers:
i. The trans-carbonylation of dimethyl furan-2,5-di(methylene)bis(methylene)bicarbonate (DCMF) with terminal olefin-containing alcohols of different chain lengths in the presence of a base (Scheme 1, route i);
 | ||
| Scheme 1 Overview of the two routes investigated for synthesizing x-BACFs. | ||
ii. The alkoxy carbonylation reaction of BHMF with bis-alkyl carbonates containing terminal olefins catalyzed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (Scheme 1, route ii).
In the first approach (Scheme 1; route i), DCMF was prepared in almost quantitative yield adapting a previously reported procedure for the synthesis of dialkyl carbonates (DACs).29,30 DCMF (1.00 eq. mol) was then reacted with an excess of allyl alcohol (10.00 eq. mol) at 90 °C in the presence of a catalytic amount of different nitrogen-based catalyst such as, TBD, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or N-methylpyrrolidine (NMPy). Unfortunately, none of these reactions led to the formation of the desired product; the only compounds identified were BHMF and allyl methylcarbonate, as confirmed by 1H NMR spectra analyses. This unexpected result is mostly likely due to the thermodynamic nature of the reaction involving numerous concomitant equilibrium reactions.
BHMF alkoxy carbonylation reaction with DACs incorporating terminal olefin was next investigated (Scheme 1, route ii). For this procedure, BHMF – available in large amounts in our laboratory31 – and α,ω-diene DACs were used as starting material.
The latter compounds were prepared by adapting the aforementioned published approach to DACs.29 Briefly, DMC was reacted with an excess of commercially available alcohols containing a terminal olefin – allyl alcohol, but-3-en-1-ol, hex-5-en-1-ol, and dec-9-en-1-ol – in the presence of TBD (Table 1). The resulting α,ω-diene DACs were isolated as pure in good yield by fractional distillation under vacuum. For the synthesis of the x-BACFs, BHMF was reacted with the selected α,ω-diene DAC in the presence of TBD in a two-necked round bottom flask equipped with a Dean–Stark trap, a magnetic stirrer, and an inert gas flow to drive the reaction equilibrium forward. The progress of the reactions was monitored by TLC to ensure complete conversion.
|
|
|||||
|---|---|---|---|---|---|
| # | α,ω-Diene dialkyl carbonate | Cat. (% mol) | T (°C) | Conv.a (%) | Yieldb (%) |
| Reaction conditions: DMC was reacted with 10.00 eq. mol of different alcohols in the presence of 1.0% mol of TBD as a catalyst at DMC reflux temperature.a Conversion was calculated using 1H NMR spectroscopy.b Isolated yield. | |||||
| 1 | All-carbonate | 1.00 | 95 | 100 | 59 |
| 2 | But-carbonate | 1.00 | 95 | 100 | 74 |
| 3 | Hex-carbonate | 1.00 | 95 | 100 | 68 |
| 4 | Dec-carbonate | 1.00 | 95 | 100 | 70 |

Longer chains furanic monomers required either an excess of α,ω-diene DAC or greater amount of TBD (#2–4; Table 2). The purification of the shorter monomers, All-BACF and But-BACF involved filtration through acidic silica to remove the catalyst, followed by distillation of the excess of DACs. Purification via distillation was not feasible for the longer-chain carbonate monomers Hex-BACF and Dec-BACF that required column chromatography. However, it is worth noting that, for all the experiments, the excess DAC used was recovered and reused.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| # | BHMF (g) | DAC | DAC (eq. mol) | Cat. (% mol) | T (°C) | t (h) | Yielda (%) |
| Reaction conditions: BHMF and an excess of α,ω-diene dialkyl carbonate were reacted in the presence of TBD at a certain temperature. All the reported reactions achieved 100% selectivity towards the symmetrical products (x-BACF).a Isolated yield.b Product isolated via column chromatography. | |||||||
| 1 | 1.00 | All-Carbonate | 10.00 | 1.00 | 95 | 2 | 83 |
| 2 | 0.50 | But-Carbonate | 5.00 | 5.00 | 110 | 2 | 79 |
| 3 | 0.20 | Hex-Carbonate | 10.00 | 20.00 | 140 | 3 | 69b |
| 4 | 0.20 | Dec-Carbonate | 5.00 | 5.00 | 140 | 3 | 44b |

Some gram scale reactions were tested for But-BACF and Hex-BACF (Table S2, ESI†), leading to comparable results to the ones reported in Table 2.
The synthesis of the four furan-based α,ω-diene carbonate monomers required a two-step procedure – DACs synthesis followed by BHMF alkoxy carbonylation – both promoted by TBD. Thus, a one-pot reaction was also attempted for All-BACF and But-BACF (Scheme 2). In this synthetic approach, DMC was reacted with an excess of the starting alcohol in the presence of TBD for 8 hours; the excess of alcohol used was removed by distillation. BHMF was then added in an amount calculated based on the equivalent of DAC formed. The reaction was allowed to proceed until BHMF full conversion was achieved. It is important to note that the amount of TBD used was adjusted for each reaction based on the equivalent of DAC formed during the first step.
 | ||
| Scheme 2 One-pot synthesis of All-BACF and But-BACF. | ||
This one-pot reaction led to the desired product with excellent yields (Table 3). However, a slight decrease in yield was observed when attempting larger scale reaction (#2; Table 3).
| # | BHMF (g) | Alcohol | TBD (% mol) | T (°C) | Yielda (%) |
|---|---|---|---|---|---|
Reaction conditions: DMC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alcohol (1.0:10.0 mol eq.), 8 hours at reflux temperature. The calculated amount of BHMF and TBD was then added at a temperature between 95–110 °C for 2 hours. All the reported reactions achieved 100% selectivity towards the symmetrical products (x-BACF).a The reported isolated yields were calculated based on the amount of DMC used. :alcohol (1.0:10.0 mol eq.), 8 hours at reflux temperature. The calculated amount of BHMF and TBD was then added at a temperature between 95–110 °C for 2 hours. All the reported reactions achieved 100% selectivity towards the symmetrical products (x-BACF).a The reported isolated yields were calculated based on the amount of DMC used. |
|||||
| 1 | 0.3 | Allyl alcohol | 20 | 95 | 90 |
| 2 | 1.3 | Allyl alcohol | 16 | 95 | 69 |
| 3 | 0.2 | 3-Buten-1-ol | 13 | 110 | 81 |
ADMET polymerization
 | ||
| Fig. 1 Structures of the seven tested ruthenium-based catalysts. | ||
The polymerization was performed under solvent free conditions at 80 °C in the presence of 1.0% mol of Ru catalyst and 1,4-benzoquinone (3.0 eq. mol relative to the catalyst) to suppress double bond isomerization.32 A Schlenk line was used to maintain the reaction under constant vacuum (25 mbar) and inert atmosphere. It has been reported in the literature that, in ADMET polymerization, shorter α,ω-diene monomers tend to require longer reaction times for full conversion;20,21,33 thus the reaction time was set at 24 hours.
The success of the polymerizations was confirmed by 1H NMR, as indicated by the disappearance of the terminal olefin protons signals at 5.88–5.70 ppm and 5.18 ppm, along with the appearance of a new signal at 5.50 ppm (Fig. 2). Based on NMR analysis, no side products from monomer isomerization or ring-closing metathesis (RCM) were observed. All ADMET experiments were quenched with ethyl vinyl ether to halt the reaction, followed by precipitation in cold methanol to purify the formed polymer. Notably, purification of the P(But-BACF) synthesized was not feasible as the oligomers formed were dissolved in the solvent (e.g., methanol) used for the precipitation.
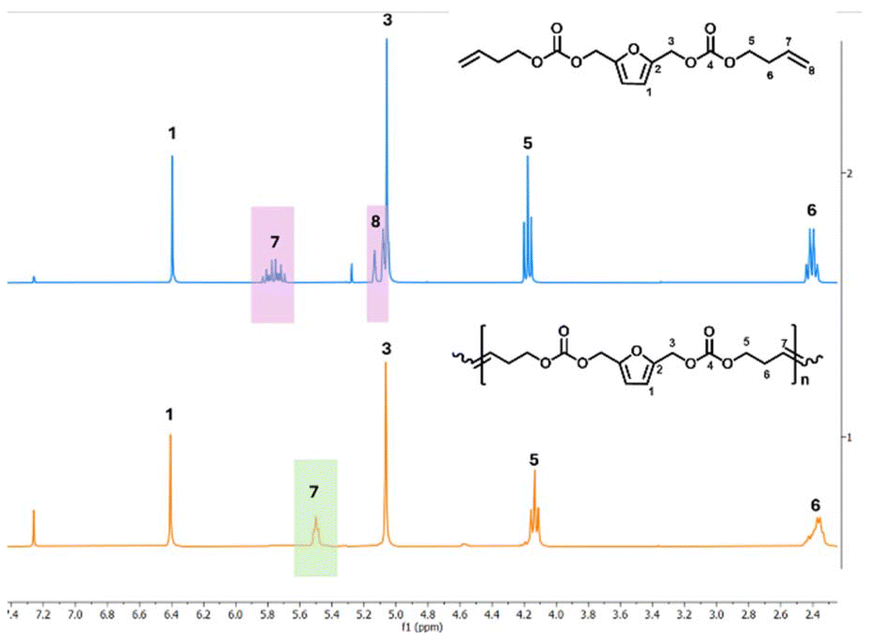 | ||
| Fig. 2 1H NMR (CDCl3) spectra of the polymerization of But-BACF to P(But-BACF) with assigned protons. | ||
In the case of But-BACF, 1H NMR analysis showed a conversion ranging from 21 to 100% depending on the Ru-catalyst used to initiate the metathesis. In particular, C2, C3, and C7 exhibited low conversion rates (#2, 3 and 7; Table 4), leading to the synthesis of oligomers with molecular weight (Mn) ranging from 0.8 to 2.4 kDa. On the other hand, four out of the seven tested catalyst – C1, C4, C5 and C6 – led to full conversion after 24 hours (#1, 4–6; Table 4).
|
|
||||||
|---|---|---|---|---|---|---|
| # | Catalyst | Conv.a (%) |
M
nb (kDa) |
Đ |
T
d5%d (°C) |
T
ge (°C) |
| Reaction conditions: 100 mg of But-BACF was added to a Schlenk flask in the presence of 1.0% mol of C1–C7 and 1,4-benzoquinone (3.0 eq. mol compared to catalyst) at 80 °C under 25 mbar of vacuum for 24 hours.a Conversion was calculated trough 1H NMR.b Determined by SEC in THF (10 mM LiBr) at 50 °C.c Đ = dispersity.d TGA degradation temperatures at which 5% mass loss (Td5%) was observed under nitrogen.e Glass transition temperature (Tg) determined by DSC, temperature ramp 10 °C min−1. n.a. = value not available. | ||||||
| 1 | C1 | 100 | 4.4 | 1.69 | 100 | −23 |
| 2 | C2 | 35 | 0.9 | 1.16 | 135 | −16 |
| 3 | C3 | 21 | 0.8 | 1.11 | 126 | n.a. |
| 4 | C4 | 100 | 13.0 | 1.36 | 159 | −8 |
| 5 | C5 | 100 | 8.5 | 1.77 | 169 | −10 |
| 6 | C6 | 100 | 0.8 | 1.00 | 133 | −12 |
| 7 | C7 | 38 | 2.4 | 1.09 | 171 | −13 |
Size-exclusion chromatography (SEC) was used to determine the molecular weight and homogeneity of the resulting polymers. The data showed that among the best performing catalysts, C1, C4 and C5 formed higher molecular weight products. When C4 was used the resulting polymer had a molecular weight of 13.0 kDa and Đ of 1.36, indicating a uniform composition (#4; Table 4).
The thermal properties of the bio-based polymers were analyzed using Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC). The results revealed that the materials were amorphous, with longer polymers being slightly more thermally stable than shorter ones (e.g., Td5% of 160 °C vs. 130 °C). All polymers displayed similar Tg around −10 °C except for the polymer obtained using C1 (#1; Table 4), that reached −23 °C. Based on the data collected, it was concluded that the optimal catalyst for the polymerization of furan-based α,ω-diene carbonate monomers was the second-generation Hoveyda–Grubbs catalyst (C4), which produced a homogeneous, high molecular weight polymer.
Different catalyst loading was evaluated for C4 to determine the effect on the polymerization. Reducing the catalyst amount from 1.0% mol to 0.5% mol resulted in a significant decrease of molecular weight from 13.0 kDa to 4.0 kDa (Fig. 3). Most probably a smaller amount of catalyst reduces the rate of polymerization, leading to less efficient polymer chain growth. Similarly, increasing the catalyst amount to 2.0% mol and 10.0% mol, resulted in lower molecular weight due to the higher active sites available for polymer propagation. This data confirms the importance of finding the optimal balance of catalyst loading. Additionally thermal analysis showed an increase of Td5% and a decrease Tg when decreasing the amount of catalyst, while when loading more catalyst (up to 10.0% mol) a decrease of both Td5% and Tg was observed (Table S3, ESI†). It can then be concluded that 1.0% mol of catalyst is necessary to achieve optimal polymerization condition, as illustrated in Fig. 3.
 | ||
| Fig. 3 Variation in P(But-BACF) molecular weight (Mn) and dispersity (Đ), with increasing of catalyst loading (Table S3, ESI†). | ||
 | ||
| Fig. 4 Furan-based α,ω-Diene carbonate monomers. | ||
As expected, the smallest monomer (All-BACF) showed no conversion even after 24 hours. This result is consistent with literature reports; longer monomers tend to be more reactive,20,21,33 as evident from the significantly faster polymerization rates observed for P(Hex-BACF) (2 hours) (#3; Table 5), and P(Dec-BACF) (30 minutes) (#4; Table 5). Additionally, a considerable increase in molecular weight was observed, from 13 kDa for P(But-BACF) (Đ = 1.4) to 19 kDa for P(Dec-BACF) (Đ = 2.28) (#2–#4; Table 5).
| # | Monomer | t (h) | Conv.a (%) | Yieldb (%) |
M
nc (kDa) |
Đ |
T
d5%e (°C) |
T
gf (°C) |
|---|---|---|---|---|---|---|---|---|
| Reaction conditions: 100 mg of α,ω-diene carbonate monomer was added to a Schlenk flask in the presence of 1.0% mol of second-generation Hoveyda–Grubbs catalyst (C4) and 1,4-benzoquinone (3.0 eq. mol compared to catalyst) at 80 °C under 25 mbar of vacuum for 24 hours.a Conversion was calculated trough 1H NMR.b Isolated yield after purification and drying, yield = (isolated mass/theoretical mass) × 100.c Determined by SEC in THF (10 mM LiBr) at 50 °C.d Đ = dispersity.e TGA degradation temperatures at which 5% mass loss (Td5%) was observed under nitrogen.f Glass transition temperature (Tg) determined by DSC, temperature ramp 10 °C min−1; n.a. = value not available. | ||||||||
| 1 | All-BACF | 24 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| 2 | But-BACF | 24 | 100 | n.a. | 13 | 1.4 | 159 | −8 |
| 3 | Hex-BACF | 2 | 100 | n.a. | 5 | 1.86 | 156 | n.a. |
| 4 | Dec-BACF | 0.5 | 100 | 85 | 19 | 2.28 | 244 | −36 |
The increase in polymer dispersity from more reactive monomers is most likely due to the less controlled nature of polymerization, which leads to a broader distribution of chain length. It is noteworthy that only P(Dec-BACF) could be successfully purified through cold methanol precipitation, resulting in a rubbery grey polymer recovered in 85% yield.
The structures of P(Hex-BACF) and P(Dec-BACF) were confirmed by NMR spectroscopy, with no evidence of RCM as for P(But-BACF) (Fig. 5).
 | ||
| Fig. 5 1H NMR (CDCl3) spectra of the polymerization of Hex-BACF to P(Hex-BACF) and Dec-BACF to P(Dec-BACF), with a molecular representation of the possible interactions occurring in P(Dec-BACF). | ||
Moreover, when analyzing the thermal properties of the polymers using TGA and DSC, a trend was observed. The thermal stability of the polymers, indicated by Td5%, increased from 159 °C to 244 °C as the monomer alkyl length increased. Additionally, the elasticity of the polymer chains, measured by Tg, also increased with longer alkyl chains, with Tg ranging from −8 °C to −36 °C.
Interestingly, DSC analysis demonstrated that not all polycarbonates were amorphous. P(Dec-BACF) exhibited a semi-crystalline structure, with a crystalline region having a melting point of −1 °C (see DSC thermogram of P(Dec-BACF), Fig. S34, ESI†). Crystallinity occurs when linear polymer chains are structurally oriented in a uniform, three-dimensional matrix. The higher degree of flexibility of P(Dec-BACF) in combination with larger presence of double bond cis-configuration most likely leads to a more crystalline structure, in comparison to the shorter monomers due to either a more folded configuration or the packing between two neighbor chains. The shorter alkyl chains might facilitate the formation of stronger intermolecular dipole–dipole interactions between carbonate–aromatic ring (CO–π) (see Fig. 5). This observation is also consistent with findings by Zhao et al., where solid NMR analysis demonstrated that the amount of cis configuration in a linear carbonate polymer influences its crystallinity by affecting chain–chain interactions.34 Additionally, it was previously reported evidence of a potential correlation between molecular weight and polymer crystallinity,35 which may also explain the behavior observed in P(Dec-BACF).
This hypothesis is corroborated by a substantial downfield shift (de-shielding) corresponding to the protons of the core of Dec-BACF monomer (Fig. 5). These shifts were not observed for other homopolymers obtained. The negative charge from the furan ring might be exerting an electronic effect on the protons in the core molecule of neighboring chains, de-shielding them, therefore shifting their signals to a higher frequency. Furthermore, the signals of the alkyl chain protons located between 1.88 to 1.23 ppm became broader after polymerization which might further support the aforementioned hypothesis.
It is interesting to note that previously reported long chain polyether20 and polyester21 achieved via ADMET from similar furanic monomers also presented a certain degree of crystallinity, observed by DSC. However, there was no shifting observed in the signals corresponding to the core molecule on the NMR analysis for the related polymers. This might be due to the higher charge of the carbonate group that allows this interaction to occur, while with the other polymers π–π stacking might be occurring.
Similarly to prior studies on polyester synthesis via ADMET polymerization,21 NMR deconvolution techniques36 revealed the presence of both cis and trans configuration in the linear polymeric chain of the longer monomers, while for P(But-BACF) the trans configuration is predominant, as it represents the lower-energy state. In P(Hex-BACF), approximately 13% of the polymer adopts the cis configuration, while in P(Dec-BACF), the cis content increases to 23%.
 | ||
| Fig. 6 Different Furan-Based α,ω-Diene monomers from different families. | ||
All these polymers were obtained via ADMET polymerization using 1.0% mol of C4 with 1,4-benzoquinone (3.0 eq. mol relative to the catalyst) at 80 °C under 25 mbar vacuum, for the time necessary to reach 100% conversion.
In terms of reactivity, the ester and the carbonate were slightly more reactive, requiring 30 minutes to form their respective homopolymers, while 2 hours were necessary for the ether-derivative. SEC revealed that the polyether had a lower molecular weight (9.5 kDa) when compared to the polycarbonate (19.0 kDa) and the polyester (26.4 kDa), further highlighting differences in reactivity (Fig. 7). The lower molecular weight of the polyether also affected its purification, as no precipitation occurred when cold methanol was used. On the other hand, both the polyester and the polycarbonate had yields were close to 90%.
 | ||
| Fig. 7 Results comparison of the homo-polymerization of different furan-based α,ω-diene monomers. | ||
Regarding thermal stability, polymers deriving from Dec-FDE and Dec-BDMF exhibited a Td5% value of 257 °C, while Dec-BACF led to a slightly lower value of 244 °C (Fig. 7). Additionally, DSC demonstrated that the polyether was more flexible, with a Tg of −53 °C, while the polycarbonate and the polyether had – as expected – a more rigid polymer structure, with similar Tg of −36 °C and −31 °C, respectively (Fig. 7).
:1.0, as previous studies have examined the effects of varying monomer ratios and lengths on thermal properties.21 As expected from the results obtained in the homo-polymerization trials, no conversion was observed after 24 hours for the co-polymerization of the two small monomers, All-BACF and diallyl furan-2,5-dicarboxylate (All-FDE). To address the challenges of incorporating a small monomer into a polymer chain, two further experiments were conducted using a longer, more reactive monomer, Dec-FDE. In the first experiment, Dec-FDE was co-polymerized with All-BACF, while in the second, Dec-FDE was co-polymerized with both All-FDE and All-BACF. 1H NMR analysis revealed that after 24 hours, no conversion was observed in both experiments. This result was unexpected, as we anticipated at least the formation of a homopolymer from the more reactive monomer Dec-FDE. Several factors might justify the lack of reactivity observed. One possibility is that the small monomers might have acted as solvents, diluting the reaction mixture and reducing overall reactivity. Another explanation, described by Hoveyda and co-workers,37 could be that the Ru-catalysts are prone to forming methylidene intermediates when reacting with terminal alkenes in small monomers. These intermediates are highly susceptible to rapid decomposition, leading to catalyst deactivation.38
Additional experiments were conducted by reacting longer monomer But-BACF with Dec-FDE. The reaction reached full conversion within 1 hour, yielding a copolymer with molecular weight 9.5 kDa (#1; Table 6). The thermal properties of the resulting polymer showed a slight decrease in comparison to P(Dec-FDE) (#7; Table 6) and an increase relative to P(But-BACF) (#5; Table 6), with values of 255 °C and −29 °C for both Td5% and Tg, respectively. DSC analysis demonstrated that the obtained polymer had an amorphous structure. NMR analysis allowed to confirm the structure of the synthesized co-polymer (Fig. 8) where the formation of a new double bond connecting both monomers is clearly visible.
 | ||
| Fig. 8 1H and 13C NMR analysis (CDCl3) of P(Dec-FDE : But-BACF). | ||
| # | Monomer (ratio) | t (h) | Conv.a (%) | Yieldb (%) |
M
nc (kDa) |
Đ |
T
d5%e(°C) |
T
gf (°C) |
T
cg (°C) |
T
mh (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions: Monomer was added to a Schlenk flask in the presence of 1.0% mol of second-generation Hoveyda−Grubbs catalyst (C4) and 1,4-benzoquinone (3.0 eq. mol compared to catalyst) at 80 °C under 25 mbar of vacuum util it reached full conversion.a Conversion was calculated trough 1H NMR.b Isolated yield after purification and drying, yield = (isolated mass/theoretical mass) × 100.c Determined by SEC in THF (10 mM LiBr) at 50 °C.d Đ = dispersity.e TGA degradation temperatures at which 5% mass loss (Td5%) was observed under nitrogen.f Glass transition temperature (Tg) determined by DSC, temperature ramp 10 °C min−1.g TC was determined by DSC and it stands for crystallization temperature.h Tm was determined by DSC and it stands for melting temperature.i The resulting polymer was so sticky that it was not possible to quantify the yield; n.a. = value not available. | ||||||||||
| 1 |
Dec-FDE:But-BACF (1:1) |
1 | 100 | n.a. | 9.5 | 1.61 | 225 | −29 | n.a. | n.a. |
| 2 |
Dec-BDMF:Dec-BACF (1:1) |
1 | 100 | n.a.i | 15.8 | 1.84 | 237 | −31 | −5 | 8 |
| 3 |
Dec-BACF:Dec-FDE (1:1) |
1 | 100 | 67 | 14.1 | 1.8 | 241 | −37 | −36 | −8 |
| 4 |
Dec-BDMF:Dec-FDE (1:1) |
1 | 100 | 62 | 14.3 | 1.9 | 275 | −45 | −13 | −1 |
| 5 | But-BACF | 24 | 100 | n.a. | 13.0 | 1.4 | 159 | −17 | n.a. | n.a. |
| 6 | Dec-BDMF20 | 2 | 100 | n.a. | 9.5 | 1.8 | 257 | −53 | 4.2 | 21 |
| 7 | Dec-FDE21 | 0.5 | 100 | 86 | 26.4 | 2.48 | 257 | −31 | 29 | 70 |
| 8 | Dec-BACF | 0.5 | 100 | 85 | 19.0 | 2.3 | 244 | −36 | −29 | −1 |
Furthermore, it was observed that the random formation of three different polymer dyads, AB, AA and BB (Fig. 8). The variation in the carbon signals corresponding to the double bond formed during polymerization can be attributed to the different lengths of the alkyl chains, which cause slight shifts in the vinylic hydrogen signals for each dyad combination (Fig. 8). To finish the co-polymerization experiments, three different long-chain monomers (Fig. 6) from different families were co-polymerized in a 1.0:1.0 ratio. All reactions reached 100% conversion after a 1 h reaction, resulting in molecular weights ranging from 14.1 k Da to 15.8 kDa (#2–4; Table 6).
Notably, the incorporation of Dec-BACF and Dec-FDE on Dec-BDMF led to a significant increase in molecular weight when compared with the homopolymer P(Dec-BDMF) (#5; Table 6), from 9.5 kDa to 15.8 kDa and 14.3 kDa, respectively (#2, 4; Table 6). This allowed the polymer to be purified through cold methanol precipitation.
P(Dec-BDMF:Dec-BACF) was a very sticky and a hard-to-handle polymer, while P(Dec-BDMF:Dec-FDE) was a more flexible and easier-to-handle material. Meanwhile, P(Dec-BACF:Dec-FDE) demonstrated a decrease in molecular weight when compared with its individual homopolymers. In terms of thermal properties, the co-polymerization of the polyether with the polycarbonate did not show significant changes on the resulting polymer thermal stability or elasticity. However, the incorporation of the polyether with the polyester (#4; Table 5) demonstrated an increase of polymer flexibility from −31 °C to −45 °C when compared to the polyester alone (#7; Table 5). Additionally, this co-polymerization (#4; Table 5) demonstrated an increase in thermal stability when compared to both polyether (#6; Table 5) and polyester (#7; Table 5) increasing from a Td5% of 257 °C to 275 °C. Copolymer structures were confirmed through NMR analyses (Fig. 9). From the signal intensity, it is evident that both monomers were equally incorporated into the copolymer chain. Additionally, the same shifts in the proton signal from the core of Dec-BACF monomer were observed on its co-polymers. Given that these polymers also exhibited crystalline structures, it is reasonable to suggest, as mentioned earlier, that these two observations are related. The success of these preliminary studies on co-polymerization of furan-based α,ω-diene monomers incorporating different moieties highlight their versatile nature in achieving bio-based materials with modulable physical–chemical properties.
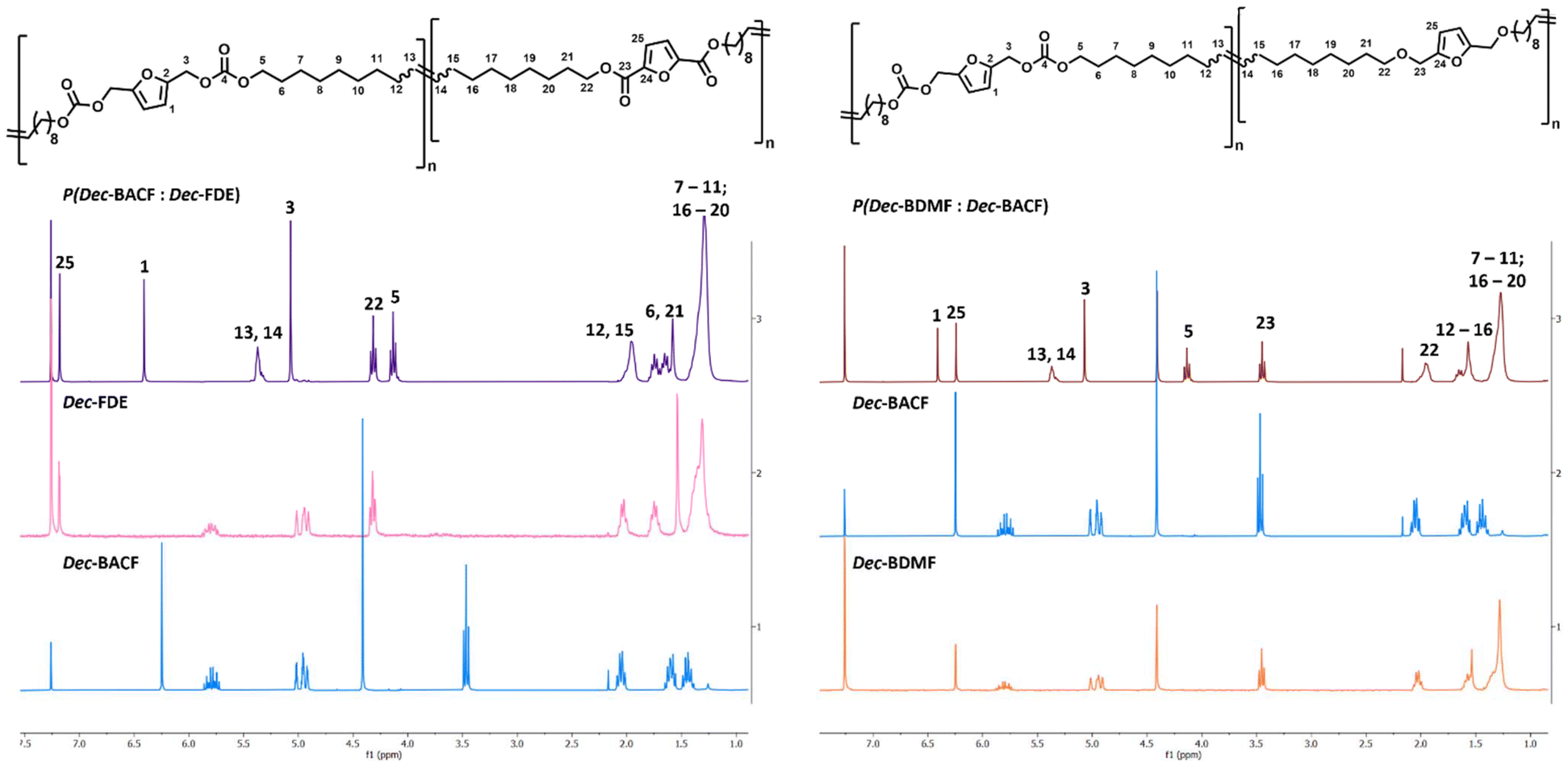 | ||
| Fig. 9 Co-polymer NMR analysis (CDCl3) for P(Dec-BACF : Dec-FDE) and P(Dec-BDMF : Dec-BACF). | ||
Conclusions
A novel class of furan-based α,ω-diene carbonate monomers of varying chain lengths was successfully synthesized via an alkoxy carbonylation reaction, reacting BHMF with bis-carbonates containing terminal olefins. The reaction conditions were optimized, scaled up, and a one-pot reaction was also achieved with high yield.These novel furan-based α,ω-diene carbonate monomers underwent ADMET polymerization using seven different ruthenium catalysts, with the second-generation Hoveyda–Grubbs catalyst (C4) emerging as the most efficient. This catalyst produced polymers with molecular weights up to 19.0 kDa, exhibiting Td5% ranging from 156 °C to 244 °C and Tg from −8 °C to −36 °C, depending on the monomer length. Notably, only Dec-BACF showed a crystalline structure, while But-BACF and Hex-BACF were amorphous.
NMR analysis revealed a shift in the proton signals associated with the core structure of Dec-BACF compared to its homopolymer, P(Dec-BACF), suggesting either folding or stacking between polymeric chains, potentially enhancing the structural organization and crystallinity of the polymer. Additionally, NMR deconvolution techniques detected an increase in cis-configuration, from 0% for But-BACF to 23% for Dec-BACF, indicating a more folded polymer chain configuration with longer monomers.
When comparing homopolymers derived from different furan-based α,ω-diene monomers (carbonate, ester, and ether), the polyether exhibited lower reactivity than the polyester and polycarbonate, while enhancing polymer flexibility without compromising thermal stability.
Co-polymerization experiments between Dec-BDMF, Dec-BACF, Dec-FDE, and But-BACF were also performed. The incorporation of a small allyl monomer with a long monomer such as Dec-FDE was unsuccessful due to catalyst deactivation. However, co-polymerization with But-BACF reached full conversion in 1 hour. The thermal properties of the resulting polymer showed a slight decrease compared to P(Dec-FDE), and an improvement compared to P(But-BACF), with Td5% and Tg values of 255 °C and −29 °C, respectively. 13C NMR analysis revealed three different configurations of the monomer in the polymer chain, suggesting a random organization. The co-polymerization between long monomers Dec-BDMF, Dec-BACF, and Dec-FDE was successful, resulting in molecular weights up to 15.9 kDa. It was noted that incorporating monomers with different functional groups can alter the properties of the resulting polymer, thus expanding the potential applications of these novel polymeric materials.
Author contributions
B. Chícharo: investigation, data curation, writing – original draft; S. Fadlallah: conceptualization, validation, supervision, writing – original draft, writing – review & editing; G. Trapasso: methodology, writing – review & editing; T. Gherardi: methodology; F. Allais and F. Aricò: conceptualization, supervision, funding acquisition, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DoE 2023–2027 (MUR, AIS.DIP.ECCELLENZA2023_27.FF project). We are grateful to Arnaud Haudrechy for the support in the designing of molecular interactions of Fig. 5.References
- D. G. Legrand and J. T. Bendler, Handbook of Polycarbonate Science and Technology, CRC Press, Boca Raton, FL, 1st edn, 1999 Search PubMed.
- D. Kyriacos, in Brydson's Plastics Materials, Elsevier, 2017, pp. 457–485 Search PubMed.
- H. Wang, F. Xu, Z. Zhang, M. Feng, M. Jiang and S. Zhang, RSC Sustain., 2023, 1, 2162–2179 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- Z. Abdel Baki, H. Dib and T. Sahin, Polymers, 2022, 14, 2031 CrossRef CAS.
- L. Caire Da Silva, G. Rojas, M. D. Schulz and K. B. Wagener, Prog. Polym. Sci., 2017, 69, 79–107 CrossRef CAS.
- H. Li, L. Caire Da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- M. D. Schulz and K. B. Wagener, Macro Chemistry & Physics, 2014, 215, 1936–1945 Search PubMed.
- D. Breilly, S. Fadlallah, A. Flourat, P. Boustingorry, V. Froidevaux and F. Allais, ACS Sustainable Chem. Eng., 2024, 12, 10701–10712 CrossRef CAS.
- D. J. Brunelle, in Encyclopedia of Polymer Science and Technology, 2006 Search PubMed.
- J. Dai, Y. Peng, N. Teng, Y. Liu, C. Liu, X. Shen, S. Mahmud, J. Zhu and X. Liu, ACS Sustainable Chem. Eng., 2018, 6, 7589–7599 CrossRef CAS.
- S. Fadlallah, P. Sinha Roy, G. Garnier, K. Saito and F. Allais, Green Chem., 2021, 23, 1495–1535 RSC.
- M. Brodin, M. Vallejos, M. T. Opedal, M. C. Area and G. Chinga-Carrasco, J. Clean. Prod., 2017, 162, 646–664 CrossRef CAS.
- C. Zhang, T. F. Garrison, S. A. Madbouly and M. R. Kessler, Prog. Polym. Sci., 2017, 71, 91–143 CrossRef CAS.
- F. Pei, L. Liu, H. Zhu and H. Guo, Polymers, 2023, 15, 829 CrossRef CAS.
- W. Qian, X. Ma, L. Liu, L. Deng, Q. Su, R. Bai, Z. Zhang, H. Gou, L. Dong, W. Cheng and F. Xu, Green Chem., 2020, 22, 5357–5368 RSC.
- E. H. Choi, J. Lee, S. U. Son and C. Song, J. Polym. Sci. Part A: Polym. Chem., 2019, 57, 1796–1800 CrossRef CAS.
- A. S. Amarasekara, in Renewable Polymers, ed. V. Mittal, Wiley, 1st edn, 2011, pp. 381–428 Search PubMed.
- A. L. Flourat, M. Annatelli, S. Fadlallah, F. Aricò and F. Allais, Macromolecules, 2023, 56, 8845–8855 CrossRef CAS.
- B. Chícharo, S. Fadlallah, F. Allais and F. Aricò, ChemSusChem, 2024, 17, e202301311 CrossRef.
- M. Vannini, P. Marchese, A. Celli and C. Lorenzetti, Green Chem., 2015, 17, 4162–4166 RSC.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci. A Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- A. Kayishaer, M. Annatelli, C. M. Hansom, L. M. M. Mouterde, A. A. M. Peru, F. Aricò, F. Allais and S. Fadlallah, Macromol. Rapid Commun., 2024, 45, 2300483 CrossRef CAS.
- M. Annatelli, J. E. Sánchez-Velandia, G. Mazzi, S. V. Pandeirada, D. Giannakoudakis, S. Rautiainen, A. Esposito, S. Thiyagarajan, A. Richel, K. S. Triantafyllidis, T. Robert, N. Guigo, A. F. Sousa, E. García-Verdugo and F. Aricò, Green Chem., 2024, 26, 8894–8941 RSC.
- J. Deng, X. Liu, C. Li, Y. Jiang and J. Zhu, RSC Adv., 2015, 5, 15930–15939 RSC.
- A. Gandini, in Biopolymers – New Materials for Sustainable Films and Coatings, ed. D. Plackett, Wiley, 1st edn, 2011, pp. 179–209 Search PubMed.
- G. Odian, Principles of polymerization, John Wiley and Sons, 4th edn, 2004, pp. 144–146 Search PubMed.
- G. Trapasso, M. Annatelli, D. Dalla Torre and F. Aricò, Green Chem., 2022, 24, 2766–2771 RSC.
- A. G. Sathicq, M. Annatelli, I. Abdullah, G. Romanelli and F. Aricò, Sustainable Chemistry and Pharmacy, 2021, 19, 100352 CrossRef CAS.
- G. Trapasso, G. Mazzi, B. Chícharo, M. Annatelli, D. Dalla Torre and F. Aricò, Org. Process Res. Dev., 2022, 26, 2830–2838 CrossRef CAS.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160–17161 CrossRef CAS.
- N. Zeaiter, S. Fadlallah, A. L. Flourat and F. Allais, ACS Sustainable Chem. Eng., 2022, 10, 17336–17345 CrossRef CAS.
- Z. Zhao, P. Ren, Y. Liu, K. Zhao, X.-B. Lu and W. Zhang, J. Energy Chem., 2018, 27, 361–366 CrossRef.
- A. Fernández-Tena, R. A. Pérez-Camargo, O. Coulembier, L. Sangroniz, N. Aranburu, G. Guerrica-Echevarria, G. Liu, D. Wang, D. Cavallo and A. J. Müller, Macromolecules, 2023, 56, 4602–4620 CrossRef.
- S. Fadlallah, J. Jothieswaran, F. Capet, F. Bonnet and M. Visseaux, Chem. – Eur. J., 2017, 23, 15644–15654 CrossRef CAS PubMed.
- M. J. Koh, R. K. M. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, Nature, 2015, 517, 181–186 CrossRef CAS PubMed.
- T.-W. Hsu, S. J. Kempel, A. P. F. Thayne and Q. Michaudel, Nat Chem., 2023, 15, 14–20 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05132g |
| This journal is © The Royal Society of Chemistry 2025 |