
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrosynthesis of nitriles from primary alcohols and ammonia on Ni catalysts†
Yiying
Xiao
 ab,
Chia Wei
Lim
b,
Linfeng
Gao
b and
Ning
Yan
*ab
ab,
Chia Wei
Lim
b,
Linfeng
Gao
b and
Ning
Yan
*ab
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China. E-mail: ning.yan@nus.edu.sg
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore
First published on 21st February 2025
Abstract
Despite increasing interest in electrocatalytic refineries for producing value-added chemicals, heterogeneous nitrile electrosynthesis from alcohols is still in the initial stages of investigation. Herein, we report the direct electrosynthesis of nitriles from primary alcohols and ammonia, with a simple nickel catalyst under benign conditions in aqueous electrolytes. The highest benzonitrile faradaic efficiency of 62.9% and a formation rate of 93.2 mmol m−2cat h−1 were achieved at room temperature. The reaction proceeds via a dehydrogenation–imination–dehydrogenation sequence, with the rate-determining step likely involving the cleavage of the α-carbon C–H bond of the alcohol. Based on electrochemical and in situ Raman analyses, we propose that the in situ formed Ni2+/Ni3+ redox species serves as the active site for converting alcohol to nitrile, while Ni2+ also exhibits capability for the oxidation of imine. Various aromatic, aliphatic and heterocyclic primary alcohols were transformed into the corresponding nitriles, exhibiting the broad feasibility of our strategy. This study offers a cost-effective catalyst-based electrocatalytic system for the synthesis of high-value nitriles under mild conditions.
Green foundation1. This work presents a benign electrocatalytic strategy for nitrile synthesis from primary alcohols and ammonia. Compared to conventional chemical routes that require toxic reagents and harsh conditions, and generate significant waste, our method eliminates the need for external oxidants and operates under mild conditions.2. Rather than relying on complex bimetallic catalysts or noble metals like Pd and Pt, a simple Ni catalyst is developed. The process enables catalyst reusability without significant loss of performance over multiple cycles. 3. Future research will focus on expanding this method to a broader range of alcohol feedstocks, including biomass-derived alcohols and ammonia from waste streams. |
Introduction
Due to the increased production of green electricity from renewable energy sources, there has been growing interest devoted to electrocatalytic refineries for the synthesis of value-added chemicals.1,2 The benefits afforded by electrocatalytic processes are multi-fold: apart from the fact that electrical energy inputs are readily available from renewable sources, water can be used as the proton source, and operation can be conducted under benign conditions at or near ambient temperature.3,4 In particular, C–N coupling reactions are an important class of reactions for manufacturing various bulk and fine chemicals, such as fertilizers, synthetic fibres, pigments and pharmaceuticals.5,6 These reactions involve the formation of carbon–nitrogen bonds between a carbon-based compound and a nitrogen source, giving rise to organonitrogen products. Notable works in electrocatalytic C–N coupling include the electrosynthesis of amines and amides from CO2 or CO.7–9 Interestingly, urea has been successfully synthesised from CO2 and various nitrogenous species.10,11 Recently, the valorisation of CO2-derived formic acid and methanol to formamide has also been demonstrated.12,13 Biomass-derived carbonyl compounds are also used for the electrocatalytic reductive amination (ERA) of the C–N coupling reaction.14,15 A special case of ERA, utilising various metal and carbon-based cathodes, uses α-keto acids as substrates to produce amino acids, which are of immense biological significance.16,17Despite extensive efforts made in developing the wider topic of electrocatalytic C–N coupling, the direct heterogeneous electrosynthesis of nitriles from primary alcohols is not common. Nitriles are versatile intermediates for producing higher value chemicals, including biological materials, pharmaceuticals and polymers.18,19 The conventional chemical methods for nitrile synthesis, such as the Sandmeyer20 and the Rosenmund–von Braun21 reactions, are not benign as they utilise toxic starting materials, require harsh reaction conditions and generate large amounts of chemical waste (Fig. 1a). Improved chemical routes, using alcohols and ammonia as substrates, via ammoxidation22–28 or oxidant-free dehydrogenation coupled with imination,29–31 have been recently reported (ESI Table S1†). However, they face certain issues, including the need for oxidants or high reaction temperatures. On the electrocatalysis front, the synthesis of hydrogen cyanide, an analogue of nitrile, has been demonstrated using methane and ammonia as the substrates,32 albeit at elevated temperatures of 800–1000 °C with solid electrolytes. The required temperature for hydrogen cyanide synthesis in solid electrolytes was decreased to 500–650 °C by replacing methane with methanol.33,34 In the past decade, nitriles have been synthesised from alcohols via homogeneous electrocatalysis using 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) or TEMPO-derived catalysts as mediators, but these methods necessitate additional separation steps for catalyst reuse.35,36 Thus far, only limited work has been conducted on nitrile electrosynthesis from primary alcohols using heterogeneous catalysts. Zhang and co-workers have developed an electro-oxidative coupling strategy for the synthesis of various nitriles with moderate to high selectivity from the corresponding alcohols and aqueous ammonia.37 Their bimetallic electrocatalyst consists of Cu and the noble metal Pd, which acted as the sites for the oxidation and coupling reactions, respectively. More recently, Ma et al. designed a NiCo nitride and oxide tandem catalyst for the electrosynthesis of benzonitrile from benzyl alcohol and ammonia.38 The oxide phase was said to enable the oxidation of alcohol to aldehyde, while the nitride phase allowed for the oxidation of the imine intermediate to the nitrile product. The catalysts in both of these works possess complex structures. Therefore, opportunities exist for developing an electrocatalytic nitrile synthesis from alcohols using cheaper and easily scalable catalysts.
 | ||
| Fig. 1 Schematic comparison of the nitrile syntheses. (a) Conventional chemical route. (b) Proposed electrochemical route. | ||
Here, we report a facile one-pot synthesis of various nitriles from primary alcohols and ammonia, in the presence of a simple Ni catalyst under ambient temperature in an aqueous electrolyte without the need for oxidants (Fig. 1b). Ten materials were first screened, and Ni was determined as being the optimal catalyst. Several control experiments and kinetic studies were performed to deduce the reaction pathway and rate-limiting step. To understand the metal sites contributing to the catalytic activity, we conducted electrochemical analyses and in situ experiments. The influence of different reaction parameters was also studied.
Experimental
Chemicals
Sodium perchlorate (NaClO4, ≥98.0%), sodium hydroxide (NaOH, ≥98.5%), potassium hydroxide (KOH, 90%), ammonia solution (∼25 wt% NH3 in water), benzyl alcohol (≥99%), benzyl-α,α-d2 alcohol (99%, D, 98%), benzaldehyde (≥99%), benzoic acid (≥99.5%), benzonitrile (≥99%), benzamide (99%), 4-methylbenzyl alcohol (98%), 4-methylbenzonitrile (98%), 4-methoxybenzyl alcohol (98%), 4-methoxybenzonitrile (99%), 2-hydroxybenzyl alcohol (99%), 2-hydroxybenzonitrile (99%), 4-hydroxybenzyl alcohol (99%), 4-cyanophenol (95%), 4-hydroxy-3-methoxybenzyl alcohol (98%), 4-hydroxy-3-methoxybenzonitrile (98%), 4-chlorobenzyl alcohol (99%), 4-chlorobenzonitrile (99%), 4-nitrobenzyl alcohol (99%), 4-nitrobenzonitrile (97%), 4-nitrobenzamide (98%), furfuryl alcohol (98%), 2-furonitrile (99%), acetamide (∼99%), 1-butanol (99.8%), butyronitrile (≥99%), 1-hexanol (≥99%), hexanenitrile (98%), 1,6-hexanediol (99%), adiponitrile (99%), 1-pentanol (≥99%), 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (97%), hydrochloric acid (HCl, 37%) and sulfuric acid (H2SO4, 95.0–98.0%) were purchased from Sigma-Aldrich. Nitric acid (HNO3, 65%), absolute ethanol (>99.7%) and ethyl acetate (≥99.8%) were provided by VWR Chemicals. Acetone (HPLC grade) and acetonitrile (HPLC grade) were purchased from Fisher Chemical. Deuterium oxide (D2O, D, 99.9%) and dimethyl sulfoxide-d6 (DMSO-d6, D, 99.9%) were purchased from Cambridge Isotope Laboratories. Nitrogen gas (N2, 99.9995%) was supplied by Air Liquide. Commercially available reagents were used as received without further purification. All aqueous solutions were prepared using ultra-pure water (Milli-Q®, resistivity of 18.0 MΩ cm).Preparation of electrodes
Manganese plate (Mn plate, ∼1 mm thickness, 99.9%) was purchased from Xingtai Xinnai Metal Materials Co., Ltd. Iron foam (Fe foam, 1.0 mm thickness, 99.9%), cobalt foam (Co foam, 1.6 mm thickness, 99.9%), nickel foam (Ni foam, 1.0 mm thickness, ≥99.9%) and copper foam (Cu foam, 1.0 mm thickness, ≥99.7%) were purchased from Kunshan Guangjiayuan New Materials Co., Ltd. Zinc foam (Zn foam, 1.0 mm thickness, 99.9%) was purchased from Kunshan Lvchuang Electronic Tech Co., Ltd. Ruthenium plate (Ru plate, 1 mm thickness, 99.95%) was purchased from Quanzhou Qijin New Material Tech Co., Ltd. Palladium plate (Pd plate, 0.1 mm thickness, ≥99.98%) was purchased from Wuxi Mini Chemistry Art Museum Co., Ltd. Platinum plate (Pt plate, 0.1 mm thickness, ≥99.99%) was purchased from Shanghai Chengxin Scientific Instrument Co., Ltd. Carbon paper (CP, TGP-H-060, 0.19 mm thickness) was purchased from Suzhou Sinero Tech Co., Ltd.Fe, Co, Ni, Cu, Zn and carbon paper electrodes used in this work were cut into 1.5 × 3 cm, while the dimensions of the used-as-received Mn, Ru, Pd and Pt plates were 1 × 1 cm. The cleaning and preparation of metal electrodes were carried out immediately prior to use in every experiment. Fe, Zn and Mn electrodes were mechanically polished using sandpaper, and then washed with ultra-pure water. Co, Ni and Cu electrodes were first sonicated with acetone for 30 min before washing with ultra-pure water, then pretreated by immersing in 2 M HCl for 30 min. Ru, Pd and Pt electrodes were stored in 33% HNO3 until use. The pretreatment of carbon paper was performed through sonicating it in 1 M HNO3 for 30 min. After that, the carbon paper was washed thoroughly using ultra-pure water and absolute ethanol, followed by drying overnight at 80 °C.
Material characterisation
Raman spectra were recorded using a Raman microscope (XploRA™ Plus, HORIBA Scientific) with a 638 nm excitation laser and a 100× objective lens. The chemical environments of the Ni foam were identified by X-ray photoelectron spectroscopy (XPS, Kratos AXIS UltraDLD, Kratos Analytical Ltd) with a mono-Al Kα X-ray source. All XPS data were calibrated to C 1s (C–C bond) at 284.50 eV and analysed using XPSPEAK Version 4.1 software. X-ray diffraction (XRD) patterns were obtained using a diffractometer (Bruker D8 Advance), with 2θ ranging from 10° to 80° at a scan rate of 5° min−1 and a step size of 0.02°. Scanning electron microscopy (SEM) images were taken with a field emission scanning electron microscope (JEOL JSM-7610F, 5 kV). Inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis was performed with a Thermo Scientific iCAP 6000 spectrometer.Electrochemical measurements
All the electrochemical measurements were carried out using a Gamry Interface 1010E potentiostat (Gamry Instruments Inc., USA). Similar to our previous study,16 a glass two-chamber (H-type) three-electrode- configured electrochemical cell, which was separated by a piece of Nafion 117 membrane (N117, DuPont, Xianfeng Instrument Tech Co., Ltd), was used for all experiments. The reference and counter electrodes were a Hg/HgO electrode (1 M KOH) with a double-salt bridge (Shanghai Yueci Electronic Tech Co., Ltd) and a platinum mesh (Pt mesh, 10 × 10 mm, ≥99.99%, Shanghai Chengxin Scientific Instrument Co., Ltd), respectively. For the H-cell with Fe, Co, Ni, Cu, Zn and carbon paper (4.5 cm2 area) as working electrodes, the electrolyte volume was 40 mL (50 mL in total volume) for both the anodic and cathodic chambers, while for Mn, Ru, Pd and Pt plates (1 cm2 area) as working electrodes, the electrolyte volume in each chamber was 9 mL (15 mL in total volume). This maintained the same ratio of electrode area to electrolyte volume, ensuring that the conditions were as close as possible for a fair comparison of electrocatalytic performance. In all cases, 15 mL min−1 purified nitrogen gas was purged through the anodic electrolyte for 30 min at the start of each experiment to exclude air. During the measurements, the electrolyte solution was stirred and bubbled with nitrogen gas continuously. The potentials applied against the Hg/HgO reference electrode were calibrated to the reversible hydrogen electrode (RHE) scale without iR compensation using the following equation:| ERHE (V) = EHg/HgO (V) + 0.098 V + 0.059 V × pH | (1) |
All current densities were calculated on the basis of the measured currents and the geometric areas of the working electrodes (4.5 cm2 for Fe, Co, Ni, Cu, Zn foams and carbon paper, 1 cm2 for Mn, Ru, Pd and Pt plates).
The electrosynthesis of nitriles was performed by chronoamperometry and investigated systematically under different working electrodes, applied potentials, pH levels, substrate concentrations, ammonia concentrations and substrate types. In a typical electrolysis procedure, certain concentrations of NH3, NaClO4 and KOH aqueous solutions with and without organic substances were used as the anolyte and catholyte, respectively. Specifically, the influence of pH on catalytic performance was carried out by varying the concentrations of KOH and/or NaClO4 to reach the desired pH values. For example, the electrolyte solution with pH 13 contained 0.1 M NaClO4 and around 0.1 M KOH, prepared by adding the required amount of KOH pellets while monitoring the solution pH with a pH meter.
The linear sweep voltammetry (LSV) measurements were performed at a scan rate of 5 mV s−1 or 10 mV s−1 under various conditions.
The cyclic voltammetry (CV) measurements were conducted with scan rates ranging from 5 to 200 mV s−1 under various conditions.
Product identification and quantification
During and after the chronoamperometry test, the electrolyte solution collected from the anodic chamber was immediately analysed by either high-performance liquid chromatography (HPLC), gas chromatography (GC) or 1H nuclear magnetic resonance (NMR) spectroscopy depending on the substrate type. The cathodic electrolyte was also analysed to make sure the membrane was in good condition. Before HPLC and GC analyses, the electrolyte solution was acidified to pH 5–6 by 2 M HCl and filtered through a polyethersulfone (PES) syringe filter (0.22 μm, Microlab Scientific).The aromatic and heterocyclic compounds were analysed by HPLC (Agilent, 1260 Infinity II), equipped with an InfinityLab Poroshell 120 EC-C18 column (3.0 × 150 mm, 2.7 micron, 1000 bar) and an ultraviolet-visible (UV-vis) detector (1260 Infinity II Refractive Index detector). 5 mM H2SO4 aqueous solution and acetonitrile with 0.2 mL min−1 flow rate (isocratic elution) were used as the A and B mobile phases, respectively. The column temperature was 30 °C and the injection volume was 3 μL.
The aliphatic substrates (except for ethanol and 1,6-hexanediol) and their derivatives were determined by GC (Agilent, 7890A), equipped with an HP-5 column (30 m × 0.320 mm, 0.25 micron) and a flame ionization detection (FID) detector. Nitrogen was used as the carrier gas. 1-Pentanol was added to the previously prepared 2 M HCl (used for acidifying the samples) as the internal standard to quantify the substrates, intermediates and products.
The qualitative and quantitative analyses of ethanol, 1,6-hexanediol and their derivatives were carried out by NMR spectroscopy (Bruker Ascend™ 400, 400 MHz) at room temperature with water suppression. In brief, 250 μL of the sampled electrolyte solution was mixed with 250 μL of an internal standard solution consisting of 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt in D2O.
The faradaic efficiency (FE) for target products, side products and intermediates was calculated as follows:
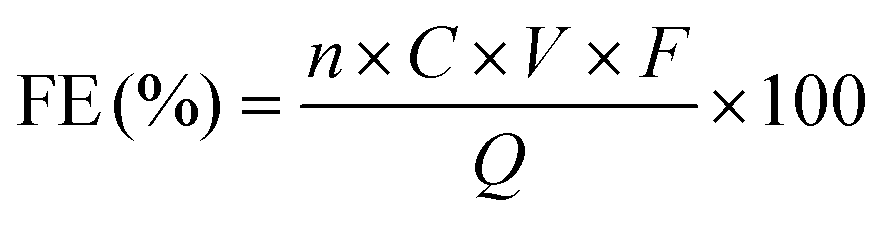 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Q represents the total charge passed during the electrolysis.
485 C mol−1), and Q represents the total charge passed during the electrolysis.
For the sample preparation for gas chromatography-mass spectrometry (GC-MS), and 1H NMR and 13C NMR analyses, the electrolyte solution (40 mL) from the chronoamperometry test was subjected to extraction with ethyl acetate (4 × 40 mL) to remove the benzoic acid side product. The organic phase was collected and most of the solvent was evaporated subsequently. GC-MS analysis was carried out on a GC system (Agilent, 7890B) fitted with an HP-5MS column (30 m × 0.250 mm, 0.25 micron) and a mass detector (Agilent, 5977B MSD analyser) using helium as the carrier gas. NMR spectroscopy measurements were conducted using DMSO-d6 as the deuterium reagent.
Kinetic modelling of the reaction
A set of time-dependent concentrations of benzyl alcohol, benzaldehyde, benzonitrile and benzoic acid were used to simulate the reaction rate constants. Reaction kinetic parameters were fitted according to the least squares fitting algorithm of MATLAB lsqcurvefit.39,40 Several constraints were used to confirm that the fitting curve was reasonable. The kinetic equations and fitting curves are shown in ESI Fig. S25.†In situ Raman spectroscopy measurements
A home-made electrochemical cell as shown in ESI Fig. S12† was used for in situ Raman spectroscopy experiments. In situ Raman spectra were recorded using the aforementioned Raman microscope (XploRA™ Plus, HORIBA Scientific), with a 638 nm excitation laser and a 10× objective lens, under controlled potentials by the electrochemical workstation (Gamry Interface 1010E potentiostat, Gamry Instruments Inc., USA). Further details of the setup and measurements are provided in ESI Fig. S12.†Results and discussion
Ni is the best catalyst among the screened materials
In the initial screening, nine monometallic catalysts, which were reported to be active in the thermocatalytic nitrile production from alcohols, including Zn, Mn, Fe, Co, Ni, Cu, Ru, Pd and Pt,41 as well as carbon paper, were studied using benzyl alcohol (BnOH) as a model compound (Fig. 2a). The experiment using Zn was not successful as Zn actively reacted with alkali to release hydrogen. For the other nine materials, Ni delivers benzonitrile (PhCN) as the main product with the highest faradaic efficiency (FE) of 49.4% and formation rate of 93.2 mmol m−2cat h−1, with the co-generation of benzaldehyde (PhCHO), benzoic acid (PhCOOH) and benzamide (PhCONH2). After extraction with ethyl acetate, the organic phase was analysed using GC-MS, 1H NMR and 13C NMR (Fig. 2b, c and ESI Fig. S1†), confirming the production of PhCN. Ru also has the capacity for PhCN production, but exhibits a significantly lower nitrile FE of 11.2%, and an unexpectedly low total FE for the organic products (25.6%) was observed. These results are possibly due to the competing ammonia oxidation reaction42 and the dissolution of Ru under oxidative potentials (as indicated by the dark green-coloured electrolyte). In sharp contrast, Mn, Fe, Co, Cu, Pd, Pt, and C show no activity for PhCN production. Among them, Mn, Co and Cu faced severe issues of metal oxidation and leaching under the reaction conditions. We further investigated the electrocatalytic properties of the catalysts using linear sweep voltammetry (LSV) and conducted the electro-oxidation of BnOH without the addition of ammonia. In addition to Ni, Ru, Co and Cu also exhibit activity in converting BnOH to the corresponding oxidative products, with a consumption rate of up to 946.7 mmol m−2cat h−1 on Ru (ESI Fig. S2†). However, the total FEs on Co, Cu and Ru are unsatisfactory, with only 50.3% for Ru and less than 15% for Co and Cu. In the absence of BnOH, Mn, Co, Ni, Cu and Ru exhibit anodic currents before the operating potentials (1.425 V or −0.265 V vs. RHE), likely due to metal oxidation (ESI Fig. S3a–e†). Contrary to Ni and Ru, which exhibit LSV current enhancements in the presence of BnOH, Mn, Co and Cu show decreased peak currents, suggesting that the BnOH oxidation rates are very slow on these metals and the metal oxidation is comparatively fast. For Fe, Pd, Pt and C, their LSV curves showed negligible current at 1.425 V vs. RHE (ESI Fig. S3f–i†), implying that these metals are not active towards any reactions, including BnOH oxidation, at this potential. We also conducted electrolysis using these four materials at higher oxidative potentials, but there was still negligible PhCN product formation (ESI Table S2†). For Mn, Co, and Cu, it may be beneficial to dope them with another element to stabilise the higher oxidation state metal species, thus reducing the metal dissolution rates.37,43,44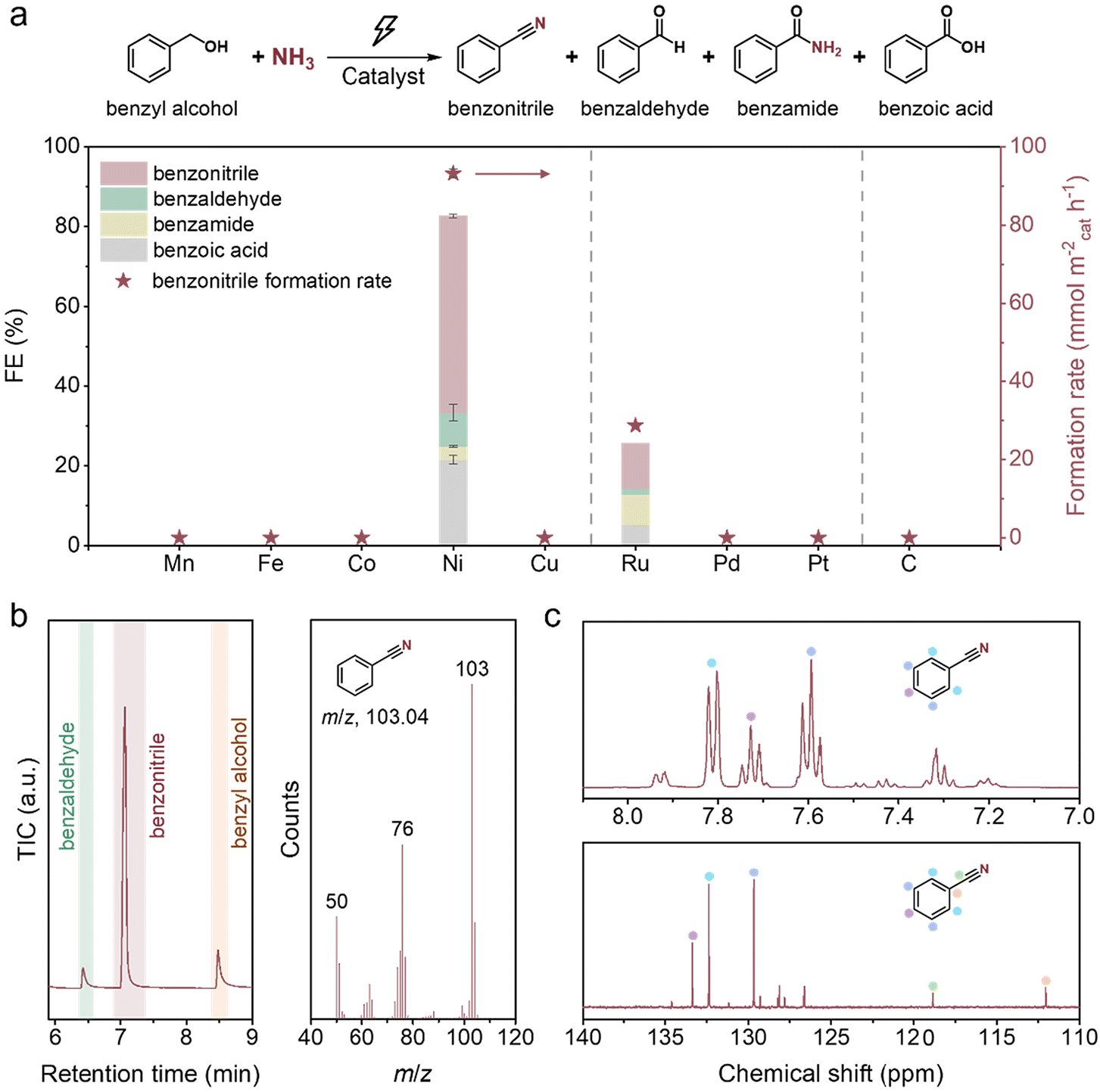 | ||
| Fig. 2 Catalyst screening and performance analysis. (a) FE values and PhCN formation rates of electro-oxidative coupling of BnOH and NH3 on various catalysts. The error bars denote the standard deviation of data from three independent experiments. Reaction conditions: 20 mM BnOH, 1 M NH3, pH 13, 1.425 V vs. RHE (0.675 V vs. RHE for Mn), 8 h reaction time (0.5 h for Co and Cu due to significant metal dissolution). Analysis of the extracted liquid carbonaceous compounds obtained from the reaction by (b) GC-MS (left: GC chromatogram, right: corresponding MS spectrum of PhCN) and (c) 1H NMR (top) and 13C NMR (bottom). Reaction conditions: Ni foam, 20 mM BnOH, 1 M NH3, pH 13, 1.425 V vs. RHE, 12 h reaction time. | ||
Nitrile synthesis follows a dehydrogenation–imination–dehydrogenation sequence
In thermocatalysis, there are two possible reaction pathways for nitrile synthesis from primary alcohols and ammonia (Scheme S1†).30 Pathway I starts with the dehydrogenation of the alcohol to an aldehyde, which then condenses with ammonia to form an imine intermediate. The imine is subsequently dehydrogenated to afford the nitrile product. In pathway II, a direct nucleophilic attack by an ammonia molecule on the α-carbon of the alcohol occurs. This results in an SN2 substitution of the –OH group with the –NH2 group to produce an amine intermediate, which then undergoes sequential dehydrogenations to generate the nitrile product.A series of control experiments were carried out to determine the main pathway (Table 1). No PhCN was detected in the absence of applied potential, BnOH or ammonia (entries 1–3), and only the oxidative products of BnOH were generated when ammonia was absent, confirming that PhCN originates from the electro-oxidative coupling of BnOH and ammonia. Notably, when BnOH was replaced by PhCHO, PhCN could be obtained with a similar FE of ∼50% (entry 5), demonstrating that PhCHO serves as the key intermediate for nitrile production. We also employed benzylamine (BnNH2) as the carbon source, considering that nitrile could be synthesised from the electrochemical dehydrogenation of amine on Ni-based catalysts.45–50 Indeed, the electro-oxidation of BnNH2 resulted in PhCN formation, although the FE (74.5%) was rather different from that when using BnOH as the carbon source (entry 6). Moreover, we were not able to detect the BnNH2 intermediate throughout the whole process of BnOH electrolysis (ESI Fig. S4 and S5a†). These results suggest that pathway II via the direct amination of BnOH to yield BnNH2 is highly unlikely.
| Entry | C source | N source | E/V vs. RHE | Main organic product (FE/%) |
|---|---|---|---|---|
| Reaction conditions: Ni foam, 20 mM C source (if present), 1 M N source (if present), pH 13, 1.425 V vs. RHE (if applicable), 8 h reaction time. N.D. = not detected. | ||||
| 1 | BnOH | NH3 | × | N.D. |
| 2 | × | NH3 | 1.425 | N.D. |
| 3 | BnOH | × | 1.425 | PhCOOH (94.0) |
| 4 | BnOH | NH3 | 1.425 | PhCN (49.4) |
| 5 | PhCHO | NH3 | 1.425 | PhCN (46.2) |
| 6 | BnNH2 | NH3 | 1.425 | PhCN (74.5) |
| 7 | PhCN | NH3 | 1.425 | PhCONH2 |
| 8 | PhCOOH | NH3 | 1.425 | N.D. |
| 9 | PhCONH2 | NH3 | 1.425 | N.D. |
To probe the possibility of the various side reactions, PhCN, PhCOOH and PhCONH2 were used to conduct the electrolysis. When PhCN was used as the carbon source, only a trace amount of PhCONH2 was detected, with a PhCONH2 to PhCN ratio of less than 4.5% (entry 7, ESI Fig. S5b and c†), which is smaller than the corresponding ratio during BnOH electrolysis (7–8%). This implies that PhCONH2 is probably produced from PhCN hydrolysis as well as hemiaminal (PhC(OH)NH2) dehydrogenation. Electrolysis using PhCOOH and PhCONH2 failed to generate any organic products (entries 8 and 9). Taken together, the electrocatalytic synthesis of nitrile using primary alcohol and ammonia follows a dehydrogenation–imination–dehydrogenation pathway (Scheme 1). The direct oxidation of aldehyde to form acid serves as the main competing reaction, and the dehydrogenation of the hemiaminal intermediate and hydrolysis of nitrile lead to the amide side product.
 | ||
| Scheme 1 Plausible reaction pathway in the Ni-foam-catalysed electro-oxidative coupling of primary alcohols and ammonia to nitriles. | ||
Ni2+/Ni3+ redox cycle and Ni2+ are active sites for nitrile synthesis
We initially used LSV to examine the electrochemical properties of Ni foam. In the absence of the organic compounds or ammonia, there is an oxidation peak with an onset at ∼1.35 V vs. RHE, which is ascribed to the transformation of Ni2+ to Ni3+ (Fig. 3a and b, eqn (3)):| Ni(OH)2 + OH− ⇌ NiOOH + H2O + e− | (3) |
 | ||
| Fig. 3 Investigation of the catalytic active sites. (a) LSV curves of Ni foam without (grey line) and with (green line) BnOH and NH3 at a scan rate of 10 mV s−1 without stirring (pH 13). (b) LSV curves of Ni foam without (grey line) and with (green line) PhCHO and NH3 at a scan rate of 10 mV s−1 without stirring (pH 13). (c) 2D spectra for the potential-dependent in situ Raman studies of Ni foam: (i) without BnOH, PhCHO or NH3; (ii) with BnOH and NH3; and (iii) with PhCHO and NH3. Further details for Raman spectra are given in ESI Fig. S14.† | ||
When ammonia is present, the current starts to climb in the same potential region (ESI Fig. S6†), attributable to the Ni3+-catalysed direct oxidation of ammonia.51 Upon the addition of BnOH, Ni foam exhibits an oxidative wave with enhanced current density (Fig. 3a), which is also observable in the cyclic voltammetry (CV) curves (ESI Fig. S7a†). Furthermore, the reduction peak corresponding to the conversion of Ni3+ to Ni2+ is weakened after introducing BnOH (ESI Fig. S7a†). A similar phenomenon was observed from the multi-potential chronoamperometry tests, showing that the reduction current of Ni3+ to Ni2+ disappeared when BnOH was injected during the open circuit state (ESI Fig. S8†). We used ex situ X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy to understand this process. As illustrated in the XPS spectra, the surface of the acid-treated Ni foam was mainly composed of metallic Ni and Ni(OH)2 (ESI Fig. S9b†). When the organic compounds and ammonia were absent, the characteristic peaks associated with Ni3+ were detected in both XPS and Raman spectra after applying a potential of 1.45 V vs. RHE (ESI Fig. S9c and S10†). Upon stirring in an electrolyte solution containing BnOH and ammonia, these Ni3+ peaks disappeared, while the Ni(OH)2 peak remained and a new peak attributed to NiO appeared (ESI Fig. S9d and S10†), indicating that Ni(OH)2 and NiO are participating species in the reaction. According to the above results, we deduce that the electrochemically generated Ni3+ triggers the C–N coupling of BnOH and ammonia to form PhCN, accompanied by the simultaneous reduction of Ni3+ to Ni2+ (ESI Fig. S11,†eqn (4a) and (4b)), which is likely the widely accepted Ni2+/Ni3+-mediated indirect oxidation of organic compounds:52
| 4NiOOH + BnOH + NH3 → 4Ni(OH)2 + PhCN + H2O | (4a) |
| 4NiOOH + BnOH + NH3 → 4NiO + PhCN + 5H2O | (4b) |
In situ Raman analyses (ESI Fig. S12†) were further carried out to verify the assumption through probing the changes in Ni foam during potential alterations. As shown in Fig. 3c(i), two peaks located at 473 and 553 cm−1 are observed around and above 1.40 V vs. RHE, which correspond to the Ni3+–O bending and stretching vibrations of NiOOH, respectively. The intensities of the NiOOH peaks increase progressively as the potential becomes more positive. The presence of ammonia has insignificant effects on the formation of NiOOH: it only results in slightly decreased peak intensities (ESI Fig. S13 and S14†), possibly due to the partial passivation of the electrode surface by ammonia. When BnOH was added, the NiOOH peaks only accumulate at potentials higher than 1.55 V vs. RHE, with distinctly decreased intensities (Fig. 3c(ii)). These results confirm that the in situ-formed Ni2+/Ni3+ redox species serves as the active site for PhCN production from BnOH and ammonia, which is consistent with reported computational results showing that the oxidation of alcohol is thermodynamically favourable on NiOOH sites.53
Interestingly, compared to that situation with BnOH and ammonia, the NiOOH peaks start to appear at a negatively shifted potential (∼1.45 V vs. RHE) with higher intensities when the PhCHO intermediate and ammonia are present (Fig. 3(iii)). This occurs despite a higher current recorded during the in situ Raman measurements (ESI Fig. S15†), which may imply that the reaction between PhCHO and NiOOH is faster (compared to that between BnOH and NiOOH) and the peaks may be expected to emerge at a more positive potential. Additionally, there is indeed an obvious increase in the Ni2+/Ni3+ oxidative current (Fig. 3b and ESI Fig. S7b†), as well as a decrease in the Ni3+/Ni2+ reductive current (ESI Fig. S7b†) after adding PhCHO. The characteristic peaks attributed to Ni3+ were also not discernible after mixing the oxidative-potential-treated Ni foam with PhCHO and ammonia (ESI Fig. S9e and S10†). Thus, these observations give us hints that the Ni2+/Ni3+ species may be just one of the several possible reactive sites for the C–N coupling of PhCHO and ammonia. Remarkably, given that the onset potential shifts largely in the negative direction to around 1.23 V vs. RHE when PhCHO is present (Fig. 3b), it is reasonable to speculate that Ni2+ also plays a key role in the oxidative coupling reaction (eqn (5)):
 | (5) |
To confirm this, we performed electrolysis in the presence of PhCHO and ammonia at 1.27 V vs. RHE (ESI Fig. S16†), where Ni3+ does not form (ESI Fig. S17 and S18†). Although at a relatively lower formation rate, PhCN is the only detectable product with a high FE of 74.8%, demonstrating the promising potential of the Ni2+ site in the production of nitriles from aldehydes and ammonia. We note that Ni2+ has only been hinted, based on LSV studies, as the active site for limited cases of electrochemical oxidative reactions, including cysteine (CySH) dimerisation54,55 and N-acetylglucosamine (NAG) oxidation56 reactions.
Nitrile synthesis depends on potential, pH and reactant concentrations
The effects of various applied potentials, pH values and ammonia/BnOH concentrations on the FEs and PhCN formation rate of the Ni foam-catalysed electrosynthesis of PhCN were systematically investigated. As the potential becomes more positive than ∼1.42 V vs. RHE, the PhCN formation rate increases sharply from around 40 to 90 mmol m−2cat h−1 and levels off (Fig. 4a), which could be rationalised by the earlier LSV results (Fig. 3a). As long as the NiOOH active phase could be rapidly regenerated under these sufficiently positive potentials, the rate of the Ni2+/Ni3+-mediated indirect oxidation of BnOH and ammonia is independent of the applied potentials, which also suggests that the rate-determining step (RDS) directly involves the PhCN formation as opposed to the generation of NiOOH. In the range of 1.35 to 1.50 V vs. RHE, the FE towards PhCN first increases to its highest value of 62.9% and shows a downward trend at potentials higher than 1.375 V vs. RHE. | ||
| Fig. 4 Influence of reaction conditions on PhCN electrosynthesis. FE values and PhCN formation rates of the electro-oxidative coupling of BnOH and NH3 on Ni foam for 8 h under different (a) applied potentials (20 mM BnOH, 1 M NH3, pH 13) and (b) pH values (20 mM BnOH, 1 M NH3, 1.425 V vs. RHE. c) FE of PhCN in the Ni-foam-catalysed electro-oxidative coupling of BnOH and NH3 for 8 h under different ammonia concentrations (20 mM BnOH, 1.425 V vs. RHE). (d) FE values and PhCN formation rates of the electro-oxidative coupling of BnOH and NH3 on Ni foam for 8 h under different BnOH concentrations (1 M NH3, pH 13, 1.425 V vs. RHE). The error bars denote the standard deviation of data from repeated experiments. | ||
Notably, the PhCN formation rate exhibits a strong pH dependence, increasing substantially from 0.66 mmol m−2cat h−1 (pH 12) to 93.2 mmol m−2cat h−1 (pH 13) and decreasing greatly thereafter (Fig. 4b). The very low PhCN formation rate at pH 12 is likely due to the lack of formation and/or regeneration of NiOOH caused by insufficient OH− as indicated in eqn (3). Possible reasons for the drops in PhCN formation rate and FE at pH 14 include the following: (1) imine formation being suppressed while the geminal diol formation being promoted on account of more prevalent nucleophilic attack by OH− on imine and aldehyde (Scheme S2†);57 and (2) the strongly alkaline electrolyte being beneficial for the Cannizzaro reaction and the hydrolysis of nitrile (Scheme S3†), leading to the enhancement of acid formation. The lower FE for PhCN at pH 14, in contrast to that at pH 13, is also observed regardless of the ammonia concentrations tested (Fig. 4c). As the ammonia concentration increases, the PhCN FE displays a significant increase, possibly owing to the shifted aldehyde–imine equilibrium favouring the imine side. There is a decline in FE for PhCN at high ammonia concentrations, probably because the ammonia oxidation reaction becomes more favourable. When the BnOH concentration increases, the formation rate and FE of PhCN show remarkable upward trends before fluctuating (Fig. 4d). The plateau in the PhCN formation rate at BnOH concentrations above 20 mM may be attributed to the saturation of the Ni3+ sites, whose rate of transformation from Ni2+ now limits the overall rate of PhCN formation.
Overall, we achieved a high PhCN FE (62.9%) and selectivity (59.8%) at a relatively low NH3 concentration (1 M) and overpotential (1.375 V vs. RHE) using a simple monometallic electrocatalyst, unlike existing reports that describe requirements of at least two distinct phases in their catalysts to achieve comparable selectivity (ESI Fig. S19†). We further performed a recycling test on the Ni electrode, which shows that the FE and yield of PhCN were stable over six reaction cycles (ESI Fig. S20†), demonstrating the good durability of our Ni foam catalyst. A very small amount of Ni dissolution occurred (less than 1‰ of the Ni foam, by mass) during the electrolysis, as quantified by inductively coupled plasma-optical emission spectroscopy (ICP-OES) (ESI Table S3†). The scanning electron microscopy (SEM) images and X-ray diffraction (XRD) patterns indicate that there are no obvious changes in the morphology and crystal phase of the spent Ni catalyst compared with its original state (ESI Fig. S21 and S22†).
The rate-determining step involves the alcohol α-carbon C–H bond cleavage
Kinetic analyses were carried out to further understand the RDS of the reaction at pH 13 and 1.425 V vs. RHE. The formation of PhCN was first order with respect to BnOH at low concentrations, attaining approximately zeroth-order dependence at BnOH concentrations beyond 20 mM (Fig. 5a). Similarly, a roughly first-order dependence on ammonia concentration was determined at lower concentrations, whereas a negative order was obtained above 1 M NH3 (Fig. 5b). The negative order could be rationalised by the ammonia poisoning effect, which is also revealed in the correlation between BnOH consumption rate and ammonia concentration (ESI Fig. S23†). | ||
| Fig. 5 Kinetic measurements of PhCN electrosynthesis. The dependences of PhCN formation rate on the concentration of (a) BnOH (1 M NH3) and (b) NH3 (20 mM BnOH) at pH 13, 1.425 V vs. RHE and conversion around 20%. | ||
On the basis of the proposed reaction pathway (Scheme 1) and observed reaction orders, we conducted kinetic modelling of the reaction (ESI Fig. S25†) to fit the experimental results as depicted in ESI Fig. S5a.† Due to the limited concentration of the PhCONH2 side product throughout the period of reaction (<3% yield), a simplified reaction scheme (Scheme 2) was used. We also performed an experiment to verify that the equilibrium between PhCHO and NH3 is established instantaneously (ESI Fig. S24†). The kinetic model predictions agree reasonably well with the experimental results and the optimised parameters are displayed in Table 2, where k1, k2 and k3 refer to the rate constants for the production of PhCHO, PhCN and PhCOOH, respectively, while K is the equilibrium constant for the reversible reaction between PhCHO and imine. For the synthesis of PhCN, the fact that k1 has the lowest value implies that the RDS may involve the dehydrogenation of BnOH to form PhCHO. We note that our deduction is different from that of a previous work demonstrating that the RDS is the coupling of surface-adsorbed PhCHO* and NH2* as the C–N coupling was said to occur at the Pd/CuO heterojunction in the system described.37 In contrast, our catalyst is monometallic, and a slow C–N coupling step occurring on our catalyst is very unlikely. Furthermore, the direct nucleophilic attack by free NH3 on PhCHO (adsorbed or free) is likely to be much faster than a surface C–N coupling.13 Thus, it is highly improbable that the RDS involves C–N coupling in our reaction.
 | ||
| Scheme 2 Simplified reaction pathway used for the modelling of the Ni-foam-catalysed benzonitrile synthesis from benzyl alcohol and ammonia. | ||
| k 1/h−1 | k 2/h−1 | k 3/h−1 | K/mM−1 |
|---|---|---|---|
| Reaction conditions: Ni foam, 20 mM BnOH, 1 M NH3, pH 13, 1.425 V vs. RHE. | |||
| 0.211 | 2.77 | 0.13 | 1.02 × 10−4 |
A kinetic isotope effect (KIE) study was performed to determine whether the RDS is the C–H bond cleavage at the α-carbon or the O–H bond breakage in the hydroxyl group of BnOH. The undeuterated substrate (PhCH2OH) and PhCD2OH (deuteration of both α-hydrogen atoms of benzyl alcohol) were transformed into PhCN under the same reaction conditions. As displayed in Table 3, the formation rate of PhCD2OH is smaller than that of PhCH2OH, yielding a KIE value of 1.73. The observed normal KIE value suggests that the α-hydrogen abstraction through a hydrogen atom transfer mechanism may indeed be the RDS for the overall reaction (eqn (6)):
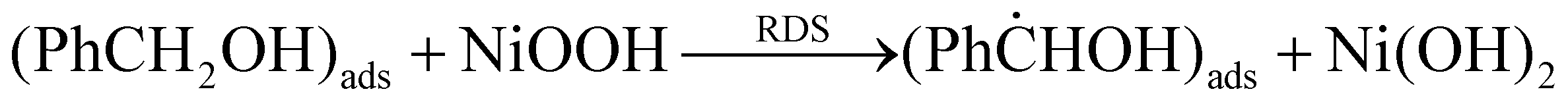 | (6) |
| Entry | Alcohol | PhCN formation rate/mmol m−2cat h−1 | KIE |
|---|---|---|---|
| Reaction conditions: Ni foam, 20 mM PhCH2OH or PhCD2OH, 1 M NH3, pH 13, 1.425 V vs. RHE, around 20% conversion. | |||
| 1 | PhCH2OH | 57.3 | |
| 2 | PhCD2OH | 33.2 | |
| 3 | k PhCH2OH/kPhCD2OH | 1.73 | |
It is worth mentioning that Choi et al. have demonstrated another novel mechanism involving hydride transfer from the α-hydrogen in alcohols to a Ni4+ site in NiOOH.58,59 Considering that this mechanism happens at more positive potentials (>1.5 V vs. RHE) and is potential-dependent with respect to the regeneration of the catalytically active species as the RDS, we rule out the possibility of this pathway in our case.
Electrosynthesis on Ni extends to other nitriles
A series of aromatic, aliphatic and heterocyclic primary alcohols were used as substrates to study the performance of our electrocatalytic system at synthesising the corresponding nitriles. Initially, we used aromatic substrates with electron-donating and withdrawing groups at the para-position or the ortho-position, which showed moderate to high performance (Fig. 6). A few substrates with electron-donating groups delivered very low to negligible conversions (ESI Table S4†). Based on the literature60,61 and our LSV analysis (ESI Fig. S26†), a probable reason for the inactivity is the passivation of the Ni foam brought about by a radical polymerisation process, as the substituents may be converted to negatively charged phenoxide ions under alkaline reaction conditions. Among the aromatic substrates para-substituted with electron-withdrawing groups (–Cl and –NO2), the conversions are relatively high (above 87%), although the nitrile selectivities are lower than those obtained using BnOH as the substrate (Fig. 6). This is mainly due to much higher amide selectivities (compared to those using BnOH as the substrate) for these substituted substrates (ESI Table S5†). Similar phenomena of depressed nitrile selectivity and high amide selectivity were seen when using furfuryl alcohol as the substrate. Interestingly, the 2-furonitrile (2i) was observed to convert rapidly and completely to the amide upon standing, within a relatively short period even without stirring. Remarkably, the Hammett plot (ESI Fig. S27†) exhibits a good linear correlation with a positive slope,62 implying that the rate-determining alcohol dehydrogenation step involves more proton-transfer than electron-transfer character.63,64 Meanwhile, the aliphatic alcohols could also be transformed into the corresponding nitriles, albeit with lower selectivities. As a majority of these substrates have been reported to be derivable from lignocellulose and/or CO2,65–69 our electrosynthetic strategy shows promising feasibility for valorising renewable feedstocks and/or waste materials to make organonitrogen products. | ||
| Fig. 6 Substrate scope of the electrosynthesis of nitriles from primary alcohols and ammonia. Reaction conditions: Ni foam, 20 mM primary alcohol (5 mM for 1b, 1c and 1d), 1 M NH3, pH 13, 1.425 V vs. RHE, 8 h reaction time. Conversions and selectivities are indicated below each compound, with the latter in parentheses. | ||
Electrosynthesis of nitriles is enhanced at higher temperatures
In principle, the reaction pathways leading to different products have different activation energies; thus varying the reaction temperature may alter the product distribution. We conducted the PhCN electrosynthesis from BnOH and NH3 on a Ni catalyst at temperatures between 0 and 60 °C and regulated the applied potential at 50 °C (ESI Fig. S28†). The LSV curves of the electrocatalytic system were obtained at a range of temperatures (ESI Fig. S29†), and the potential that gave the same current density as 1.45 V vs. RHE at 30 °C was identified for use at each non-room temperature point. Notably, both the yield and FE of PhCN increased sharply as the temperature increased from 0 to 50 °C, indicating that PhCN synthesis is favoured at higher temperatures (Fig. 7a). In contrast, as the temperature increased to 60 °C, the PhCN yield dipped slightly while the total FE values of the organic products decreased significantly, implying that a sizeable portion of the current is attributable to nickel oxidation. As 50 °C is the optimal temperature for PhCN production, we conducted further optimisation studies at this temperature, where several potentials were found to give PhCN yields exceeding 50% after 3 h (Fig. 7b). The highest PhCN yield of 61.1% was achieved using 20 mM BnOH and 1 M NH3 under an applied potential of 1.392 V vs. RHE at 50 °C after 5 h. The studied conditions at 50 °C that resulted in PhCN yields larger than 50% are summarised in Table 4.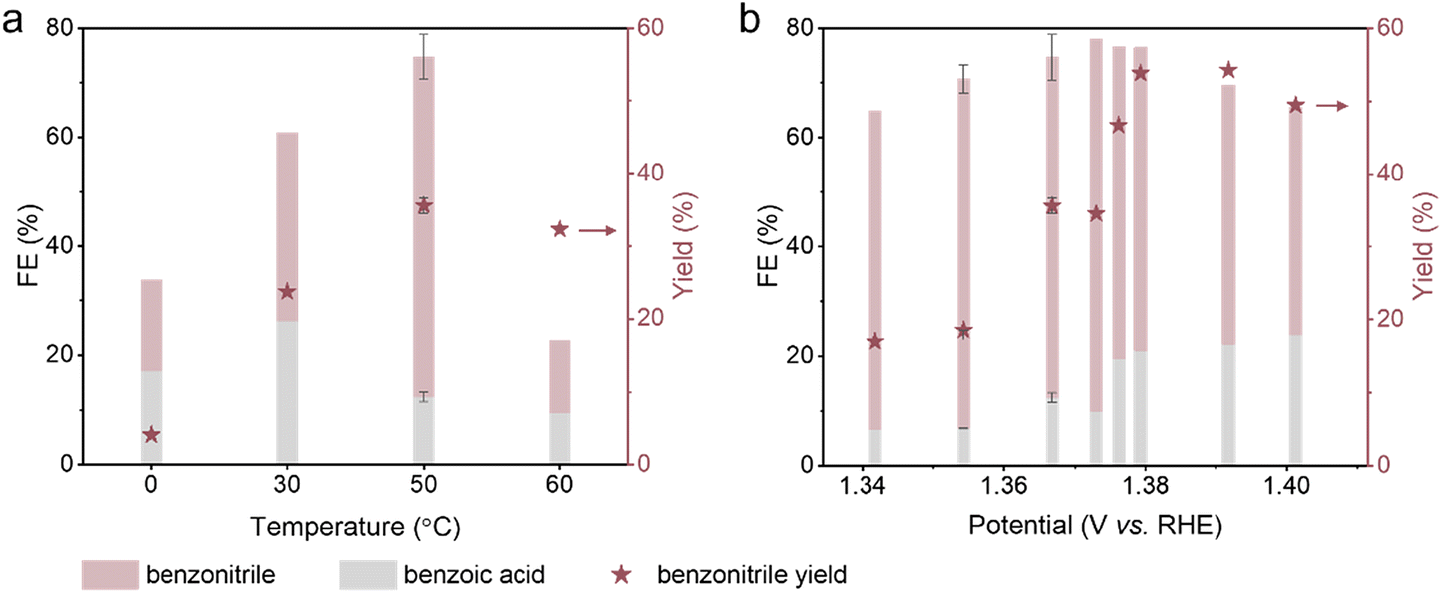 | ||
| Fig. 7 PhCN electrosynthesis over a range of temperatures. FE values and PhCN yields of the electro-oxidative coupling of BnOH and NH3 on Ni foam for 3 h under different (a) temperatures (20 mM BnOH, 1 M NH3, pH 13, the applied potentials were chosen based on LSV curves in ESI Fig. S29†) and (b) potentials (20 mM BnOH, 1 M NH3, pH 13, 50 °C). The error bars denote the standard deviation of data from two independent experiments. | ||
| Entry | Potential/V vs. RHE | Duration/h | PhCN yield/% | PhCN selectivity/% |
|---|---|---|---|---|
| Reaction conditions: Ni foam, 20 mM BnOH, 1 M NH3, pH 13, 50 °C. | ||||
| 1 | 1.367 | 8 | 54.1 | 65.8 |
| 2 | 1.376 | 6 | 55.8 | 65.1 |
| 3 | 1.379 | 6 | 59.9 | 67.9 |
| 4 | 1.392 | 5 | 61.1 | 62.6 |
Compared to existing thermal catalytic pathways, our system operates at more benign conditions, and the catalysts are based on an inexpensive metal with simple structure (a comparison is provided in ESI Table S1†). Relative to the demonstrated electrocatalytic approaches, our system provides comparable or better performance, in terms of nitrile yield and FE, while requiring a lower NH3 concentration, oxidative potential, similar or shorter reaction durations and a simpler monometallic non-noble metal catalyst (ESI Table S6†).
Conclusions
We have utilised Ni foam for the electrocatalytic synthesis of benzonitrile from benzyl alcohol, with the highest formation rate of 93.2 mmol m−2cat h−1 and FE of 62.9%. The reaction likely follows a dehydrogenation–imination–dehydrogenation pathway, with the oxidation of the aldehyde intermediate to carboxylic acid being the main competing reaction and amide as another side product. The Ni2+/Ni3+ redox species acts as a key site for the C–N coupling between benzyl alcohol and ammonia to produce benzonitrile. The kinetic studies revealed that the extraction of the α-hydrogen from the primary alcohol via a hydrogen atom transfer mechanism is likely the overall rate-limiting step. Notably, we showed that Ni2+ is a plausible site for oxidative coupling, specifically for the oxidation of imine to nitrile, which has not been reported so far. In all, we have demonstrated a noble-metal-free monometallic catalyst for the electrocatalytic nitrile synthesis from primary alcohols. Given the established electrocatalytic pathways for nitrogen-containing ions present in wastewater (NO3−, NO2−) to NH3, our work has the potential to enable the environmentally sustainable electrosynthesis of valuable nitrile compounds purely using waste materials (CO2, waste biomass, wastewater) as feedstocks.Author contributions
N. Y.: conceptualization, methodology, formal analysis, resources, writing – review & editing, supervision, funding acquisition. Y. X.: conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review & editing, visualization. C. W. L.: formal analysis, investigation, writing – original draft, writing – review & editing. L. G.: methodology, formal analysis, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Original data are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Research Foundation Singapore NRF Investigatorship (Award No. NRF-NRFI07–2021–0006) for supporting this work. We appreciate the help of Dr Zeliang Yuan for performing XPS measurements. We express our gratitude to Professor Kang Zhou for helping with the fabrication of the in situ Raman electrochemical cell. We thank Dr Hua An for assisting with the temperature-controlled experiments.References
- J. G. J. Olivier, J. A. H. W. Peters and G. Janssens-Maenhout, Trends in global CO2emissions. 2012 Report, Netherlands, 2012.
- C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- D. Bogdanov, M. Ram, A. Aghahosseini, A. Gulagi, A. S. Oyewo, M. Child, U. Caldera, K. Sadovskaia, J. Farfan, L. De Souza Noel Simas Barbosa, M. Fasihi, S. Khalili, T. Traber and C. Breyer, Energy, 2021, 227, 120467 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed.
- N. Kurig and R. Palkovits, Green Chem., 2023, 25, 7508–7517 Search PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- M. Jouny, J.-J. Lv, T. Cheng, B. H. Ko, J.-J. Zhu, W. A. Goddard and F. Jiao, Nat. Chem., 2019, 11, 846–851 CrossRef CAS PubMed.
- J. Li and N. Kornienko, Chem. Sci., 2022, 13, 3957–3964 RSC.
- Y. Wang, D. Chen, C. Chen and S. Wang, Acc. Chem. Res., 2024, 57, 247–256 CrossRef CAS PubMed.
- C. Yang, Z. Li, J. Xu, Y. Jiang and W. Zhu, Green Chem., 2024, 26, 4908–4933 RSC.
- C. Guo, W. Zhou, X. Lan, Y. Wang, T. Li, S. Han, Y. Yu and B. Zhang, J. Am. Chem. Soc., 2022, 144, 16006–16011 CrossRef CAS.
- N. Meng, J. Shao, H. Li, Y. Wang, X. Fu, C. Liu, Y. Yu and B. Zhang, Nat. Commun., 2022, 13, 5452 CrossRef.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC.
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC.
- Y. Xiao, C. W. Lim, J. Chang, Q. Yuan, L. Wang and N. Yan, Green Chem., 2023, 25, 3117–3126 RSC.
- M. Li, Y. Wu, B.-H. Zhao, C. Cheng, J. Zhao, C. Liu and B. Zhang, Nat. Catal., 2023, 6, 906–915 CrossRef.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- X. Chen, S. Song, H. Li, G. Gözaydın and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef.
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef.
- J. Lindley, Tetrahedron, 1984, 40, 1433–1456 CrossRef.
- T. Senthamarai, V. G. Chandrashekhar, N. Rockstroh, J. Rabeah, S. Bartling, R. V. Jagadeesh and M. Beller, Chem, 2022, 8, 508–531 Search PubMed.
- H. Wang, D. Xu, E. Guan, L. Wang, J. Zhang, C. Wang, S. Wang, H. Xu, X. Meng, B. Yang, B. C. Gates and F.-S. Xiao, ACS Catal., 2020, 10, 6299–6308 CrossRef.
- T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS PubMed.
- W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS.
- S. Shang, L. Wang, W. Dai, B. Chen, Y. Lv and S. Gao, Catal. Sci. Technol., 2016, 6, 5746–5753 RSC.
- J. He, P. Zhou, S. Zhang, J. C.-H. Lam, Y. Liao and Z. Zhang, Green Chem., 2024, 26, 1831–1845 RSC.
- Y. Hu, S. Jin, Z. Zhang, L. Zhang, J. Deng and H. Zhang, Catal. Commun., 2014, 54, 45–49 CrossRef.
- Y. Wang, S. Furukawa, Z. Zhang, L. Torrente-Murciano, S. A. Khan and N. Yan, Catal. Sci. Technol., 2019, 9, 86–96 RSC.
- Y. Wang, S. Furukawa and N. Yan, ACS Catal., 2019, 9, 6681–6691 Search PubMed.
- N. Kiratzis and M. Stoukides, J. Electrochem. Soc., 1987, 134, 1925–1929 CrossRef.
- N. Kiratzis and M. Stoukides, J. Catal., 1991, 132, 257–262 CrossRef.
- A. Raj, R. A. Rudkin and A. Atkinson, J. Electrochem. Soc., 2010, 157, B719 CrossRef.
- Z. Fan, X. Yang, C. Chen, Z. Shen and M. Li, J. Electrochem. Soc., 2017, 164, G54 CrossRef.
- R. M. Rodrigues, D. A. Thadathil, K. Ponmudi, A. George and A. Varghese, ChemistrySelect, 2022, 7, e202200081 CrossRef.
- Z. Fang, Y. Ding, M. Wang, L. Wang, F. Li, K. Fan, X. Wu, L. Sun and P. Zhang, Appl. Catal., B, 2023, 337, 122999 CrossRef.
- T. Mushiana, Z. Shao, M. Khan, N. Zhang and M. Ma, Chem. Eng. J., 2023, 477, 146742 CrossRef.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef.
- S. Song, Y. Wang and N. Yan, Mol. Catal., 2018, 454, 87–93 CrossRef.
- Y. Wang, S. Furukawa, X. Fu and N. Yan, ACS Catal., 2020, 10, 311–335 CrossRef.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130 CrossRef.
- J. Zhu, J. Shao, B.-A. Shen, J. Chen, Y. Yu, S. Song, X.-B. Zhang, B. Zhang and B.-H. Zhao, JACS Au, 2023, 3, 2987–2992 CrossRef PubMed.
- Y. Tang, X. Li, H. Lv, D. Xie, W. Wang, C. Zhi and H. Li, Adv. Energy Mater., 2020, 10, 2000892 CrossRef.
- P. M. Robertson, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 97–104 CrossRef.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef.
- W. Wang, Y. Wang, R. Yang, Q. Wen, Y. Liu, Z. Jiang, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2020, 59, 16974–16981 CrossRef PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 Search PubMed.
- M. T. Bender and K.-S. Choi, JACS Au, 2022, 2, 1169–1180 CrossRef.
- Y. Sun, H. Shin, F. Wang, B. Tian, C.-W. Chiang, S. Liu, X. Li, Y. Wang, L. Tang, W. A. Goddard III and M. Ding, J. Am. Chem. Soc., 2022, 144, 15185–15192 CrossRef.
- A. Kapałka, A. Cally, S. Neodo, C. Comninellis, M. Wächter and K. M. Udert, Electrochem. Commun., 2010, 12, 18–21 CrossRef.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef.
- L. Wei, M. D. Hossain, M. J. Boyd, J. Aviles-Acosta, M. E. Kreider, A. C. Nielander, M. B. Stevens, T. F. Jaramillo, M. Bajdich and C. Hahn, ACS Catal., 2023, 13, 4272–4282 CrossRef.
- D. Giovanelli, N. S. Lawrence, L. Jiang, T. G. J. Jones and R. G. Compton, Sens. Actuators, B, 2003, 88, 320–328 CrossRef.
- D. Jia, F. Li, L. Sheng, Q. Ren, S. Dong, S. Xu, Y. Mu and Y. Miao, Electrochem. Commun., 2011, 13, 1119–1122 CrossRef.
- H. Zhao, D. Lu, J. Wang, W. Tu, D. Wu, S. W. Koh, P. Gao, Z. J. Xu, S. Deng, Y. Zhou, B. You and H. Li, Nat. Commun., 2021, 12, 2008 CrossRef PubMed.
- H. J. Schäfer, Electrochemistry I, 1987 Search PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef PubMed.
- M. T. Bender, R. E. Warburton, S. Hammes-Schiffer and K.-S. Choi, ACS Catal., 2021, 11, 15110–15124 CrossRef.
- P. Parpot, A. P. Bettencourt, A. M. Carvalho and E. M. Belgsir, J. Appl. Electrochem., 2000, 30, 727–731 CrossRef.
- C. Z. Smith, J. H. P. Utley and J. K. Hammond, J. Appl. Electrochem., 2011, 41, 363–375 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef PubMed.
- M. J. Chalkley, P. Garrido-Barros and J. C. Peters, Science, 2020, 369, 850–854 CrossRef PubMed.
- J. Xu, J. Meng, Y. Hu, Y. Liu, Y. Lou, W. Bai, S. Dou, H. Yu and S. Wang, Research, 2023, 6, 0288 CrossRef.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef.
- I. Lauer, G. Philipps and S. Jennewein, Microb. Cell Fact., 2022, 21, 85 CrossRef PubMed.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DOCX file containing MS spectra, LSV curves, CV curves, HPLC chromatograms, XPS spectra, Raman spectra, in situ Raman setup, NMR spectra, electrochemical measurements, concentration profiles, kinetic modelling, Hammett plot, schemes, and tables. See DOI: https://doi.org/10.1039/d5gc00572h |
| This journal is © The Royal Society of Chemistry 2025 |