DOI:
10.1039/D4LF00395K
(Review Article)
RSC Appl. Interfaces, 2025, Advance Article
A review on research progress of double perovskite oxides for oxygen evolution reaction electrocatalysts and supercapacitors†
Received
25th November 2024
, Accepted 9th January 2025
First published on 10th January 2025
Abstract
In the past decade, the rapidly increasing global demand for energy and extensive concerns about the greenhouse effect and environmental problems from fossil fuels have stimulated intensive research interest in developing sustainable and clean energies and new electrochemical energy storage systems. Practical utilization of clean energies requires energy conversion involving different processes such as electricity-driven water splitting facilitating the storage of electrical energy in the form of hydrogen gas, and energy storage devices such as fuel cells and supercapacitors. A key issue to realize a high-efficiency conversion process is to find stable, low-cost and environment-friendly functional materials. Due to their extreme structural and compositional flexibilities, double perovskite (DP) oxides have gained much attention in the fields of electrocatalysis and supercapacitors. Recently, high-level theoretical studies have led to significant progress in the atomic-scale understanding of the catalytic mechanism of the DP oxide-driven oxygen evolution reaction (OER) and the electrochemical energy storage mechanism in DP oxide-based supercapacitors. In parallel, numerous experimental studies have been carried out to explore novel catalytic materials with advanced properties and kinetics, and more promising pseudocapacitive DP oxides have been developed. This review first introduces the structural and compositional flexibilities of DP perovskite oxides, and their prepared methods are described. Several strategies (e.g., nanostructure designs, elemental doping, tuning morphologies, crystallinity and surface defect engineering for improving oxygen vacancies) for modulating their electrochemical performance are also described. The recent progress of their applications in the electrochemical OER and supercapacitors is summarized. Finally, we conclude this review by giving some challenges and future perspectives of DP oxides in renewable energy conversion and energy storage devices.
 Liangdong Chen | Liangdong Chen is currently a master's student at the School of Physics, Nanjing University, majoring in condensed matter physics. In 2022, he completed his bachelor's degree at Dalian Maritime University, Dalian, China. His research activities are primarily focused on the synthesis and structural characterization of double perovskite oxide nanostructures, as well as their applications in spintronics, energy storage, and electrocatalysis. |
 Xinhua Zhu | Xinhua Zhu is currently a Full Professor at Nanjing University. He received his BS (1989), MS (1992), and PhD (1995) degrees in Electronic Materials Science from Xi'an Jiaotong University. He worked as Academic Consultant at KAUST (Kingdom of Saudi Arabia) in 2012 and 2013, Queen's University of Belfast (United Kingdom) from 2004 to 2006, Max-Planck-Inst Mikrostrukturphys (Halle, Germany) from 2003 to 2004 as an Alexander von Humboldt (AvH) Research Fellow, and The Hong Kong Polytechnic University from 2000 to 2001. His current research interests focus on the nanoscale fabrication of multiferroic (double) perovskite nanostructures and their structural characterization by (HR)TEM techniques. |
1. Introduction
With the development of science and technology, the increasing demands for energy supply and energy crisis have become a worldwide concern.1–3 The global energy crisis coupled with environmental issues has driven scientists to search for renewable energy resources to replace fossil fuels.4,5 Primary renewable energy sources (e.g., wind, solar power, and tidal energy) have the advantages of being sustainable and relatively benign in terms of impact on the environment and human health. However, they are intermittent on daily, seasonal and also regional scales with unpredictable supply in nature, restricting their widespread application in the global energy mix.6 In this framework, water electrolysis is one of the most efficient and reliable methods for producing hydrogen from renewable energy sources (e.g., solar, wind, and hydropower) because hydrogen can store greater energy per unit weight or volume due to its high energy density and large-scale uninterrupted energy storage. Therefore, water electrolysis plays a vital role in the development of a sustainable energy system.7 Generally, a water electrolysis cell (i.e., electrolyzer) is composed of three basic components: a hydrogen electrode (cathode) for the hydrogen evolution reaction (HER), an oxygen electrode (anode) for the oxygen evolution reaction (OER), and an electrolyte for ionic transport from one electrode to another, as schematically shown in Fig. 1.3 The HER and OER are two half-reactions of water electrolysis. At the cathode and anode, electrocatalysts are required for the HER and OER, respectively. When compared with the HER, the OER is more kinetically sluggish because the OER is a sequential multistep reaction and 4-e transfer reaction, while the HER needs only two electrons. Therefore, the OER is the crucial process that governs the overall efficiency of electrochemical water splitting. At the early stage of OER research, noble-metals (e.g., Pt, Pd, Ru, Ir and Rh) and their related oxides (e.g., IrO2 and RuO2) were considered as active OER electrocatalysts under acidic and alkaline conditions due to their efficient catalytic activity and stability.8 However, their low-reserves and high-cost limit their large-scale commercial utilization. Thus, it is very desirable to develop cheap and earth-abundant electrocatalysts with high activity for clean-energy technologies. During the search for their alternatives, low-cost transition metal oxides have gained much attention to be developed as effective electrocatalysts,9–12 but unfortunately, the low conductivity, surface passivation, and limited catalytic performance of transition metal oxides restrict their practical applications in clean energy conversion. Thus, to solve the above issues, much effort has been devoted to non-precious metal-based materials such as perovskite oxides with a chemical formula of ABO3 (where A is an alkaline- and/or rare-earth element and B is a transition-metal element), to explore new efficient non-precious metal-based electrocatalysts.13,14 Perovskite oxides have demonstrated remarkable catalytic OER activity due to their chemical and structural flexibilities.15,16 However, perovskite oxides still have a problem in generating high current density for a longer duration due to the issue of structural instability. Thus, the development of durable perovskite oxides is very much required in the pursuit of their future commercial-grade applications. Double perovskite (DP) oxides with a chemical formula of A2BB′O6 deliver significant advantages over standard single ABO3-perovskites, such as easier oxygen ion diffusion, faster exchange of surface oxygen, and higher electrical conductivity. They display intriguing electrochemical performance due to the enhanced coupling effects via intervening oxygen bridging every B′ and B atom pairs.17–19 Profitting from ordered B-site ion arrangement and physical correlation effects are prominent in DP oxides, which result in a novel modulation of their electronic structures and electric transport property. This paves a distinct avenue to modulate OER performance and to provide a platform for investigating the OER mechanism in DP oxide-based electrocatalysts.20,21 In recent years, DP oxides have been extensively investigated as promising electrode materials for the OER process at the anode due to their unique structural and chemical flexibilities and high structural stability. High-level theoretical tools and computational studies have led to significant progress in understanding of the OER and electrocatalyst behavior at the atomic-level. In parallel, numerous experimental studies have been performed to search for new catalytic materials with advanced properties and kinetics at a technically relevant level. This contribution summarizes the previous and the most recent theoretical predictions and experimental results in the field of DP oxide-based electrocatalysts for the OER, both operated in acidic and alkaline environments.
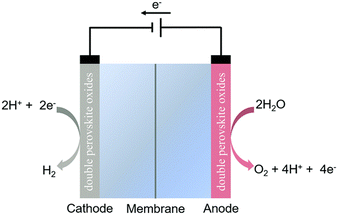 |
| | Fig. 1 Schematic illustration of a water electrolyzer.3 Copyright 2020, the Royal Society of Chemistry. | |
Supercapacitors, fuel cells, and batteries are the main energy conversion and energy storage devices. Among them, supercapacitors have the advantage of rapid charge and discharge kinetics as compared to batteries. Supercapacitors have the advantages of high-power density and long cycle life as compared to conventional dielectric capacitors.22 But the issue with supercapacitors is their energy density, which leads to a new research area for developing appropriate electrode materials and electrolytes for supercapacitor applications. An ideal supercapacitor electrode material should be highly electrically conductive and nanoporous for enhancing electron/ion transport. Perovskite oxides are recognized as promising materials for supercapacitor applications due to their high crystallinity, excellent ionic and electrical conductivity, high charge storage capacity, good electrochemical activity, ease of synthesis and cost effectiveness.23 DP oxides are considered as a subclass of perovskite materials with a different chemical formula of A2BB′O6, which are recognized as efficient pseudocapacitive materials due to their unique structural flexibility and compositional flexibility as well as unique coupling effects.24,25 The recent reviews on DP oxides used as OER electrocatalysts are available for readers.22,26–28 However, in spite of the high charge storage performance of perovskite oxides, leaching of cations during electrochemical measurement is a major pitfall. To circumvent this issue, it is highly desirable to develop a new paradigm for synthesis of highly stable perovskite oxides that can be employed for a longer time with stable crystal structure.29 In the cation-ordered DP structure, B sites are alternatively occupied by B- and B′-site cations, providing a short reaction path and paving a distinct avenue to tailor the OER performance. In the past decade, much effort has been made to synthesize DP oxides and to investigate their crystal structures, structural stability, electrochemical kinetics, and charge storage mechanisms, to improve the electrochemical performance of DP oxides. Currently, there are only two reviews available on the recent progress of DP oxides in supercapacitor application.25,30 Out of them only one review has a complete focus on DP oxides as supercapacitor electrodes.30 However, to date, no comprehensive reviews on DP oxides for energy conversion and storage applications are available in the literature.
This review focuses on DP oxides and their applications in the fields of electrocatalysts and supercapacitors. The structural and compositional flexibilities of DP perovskites, and the strategies for tuning their electrochemical performance are described. Recent progress on DP oxides as high-performance electrocatalysts and supercapacitors is summarized. The pros and cons of DP oxides when used as electrocatalytic and pseudocapacitive materials are discussed and the prospective avenues for future research in the fields of OER electrocatalysts and supercapacitors are presented.
2. Structural and compositional flexibilities of double perovskite (DP) oxides
2.1. Structural flexibility
DP oxides are described as the perovskites with the A or B site occupied by two different types of cations, forming the formula of AA′B2O6 (double A-site) or A2BB′O6 (double B-site). Since A-site cations usually act as electron donors to the [BO6] framework and the physical properties of ABO3 perovskites are highly dependent on B-site cations, DP oxides are usually indicated as double B-site perovskites. The crystal structure of A2BB′O6 DP oxide is determined by the arrangement of B and B′ cations in the B-site sublattice. Due to the charge (size) difference between B and B′ cations, the Madelung (strain) energy of A2BB′O6 DP oxide can be reduced by the particular ordering on the B-site. There are mainly three kinds of B-cation sublattices including random, rock salt and layered structures as illustrated in Fig. 2 (ref. 24) and Table 1.31 The arrangement types are generally determined by the charge difference (ΔQ) between B and B′. Based on the existing double perovskites, the random order dominates when ΔQ = 1, but the rock-salt order dominates when ΔQ > 2. This could be explained by the larger Madelung energy for the random order and a larger ΔQ. Since ΔQ ≥ 2 in most of the double perovskites, the rock-salt order is preferred in the majority of double perovskites. In the rock-salt order, the crystal symmetries of double perovskites are reduced compared to their corresponding single perovskites due to the difference in B ions. For example, the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry of single perovskite is reduced to Fmm symmetry in DP oxides, as shown in Table 2. A literature survey showed that the same four kinds of Glazer tilts seen in single perovskite also dominate in double perovskites.32 The corresponding crystal symmetries of dominating single and double perovskite phases are shown in Table 2 (ref. 33) with their crystal structures shown in Fig. 2. When the distortion is large enough, some B–O bonds and the three-dimensional (3D) [B–O] framework are broken, leading to one-dimensional (1D) and two-dimensional (2D) perovskite derivatives. These can be considered as low-dimensional perovskites as classified by the spatial arrangements of the octahedral units within their crystal structures, as depicted in Fig. 2. In 1D perovskites, the [BO6] octahedra can be corner-sharing, edge-sharing, or face-sharing to form a 1D nanowire with a linear or zigzag configuration,34 while 2D perovskites typically consist of stacked layers with mainly edge-sharing octahedra.35 These low dimensional perovskite derivatives exhibit much higher surface to bulk ratios than their 3D counterparts, thus, they have promising applications in catalysis.
m symmetry of single perovskite is reduced to Fmm symmetry in DP oxides, as shown in Table 2. A literature survey showed that the same four kinds of Glazer tilts seen in single perovskite also dominate in double perovskites.32 The corresponding crystal symmetries of dominating single and double perovskite phases are shown in Table 2 (ref. 33) with their crystal structures shown in Fig. 2. When the distortion is large enough, some B–O bonds and the three-dimensional (3D) [B–O] framework are broken, leading to one-dimensional (1D) and two-dimensional (2D) perovskite derivatives. These can be considered as low-dimensional perovskites as classified by the spatial arrangements of the octahedral units within their crystal structures, as depicted in Fig. 2. In 1D perovskites, the [BO6] octahedra can be corner-sharing, edge-sharing, or face-sharing to form a 1D nanowire with a linear or zigzag configuration,34 while 2D perovskites typically consist of stacked layers with mainly edge-sharing octahedra.35 These low dimensional perovskite derivatives exhibit much higher surface to bulk ratios than their 3D counterparts, thus, they have promising applications in catalysis.
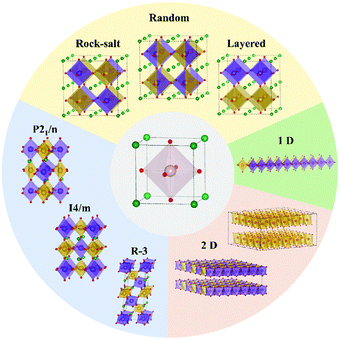 |
| | Fig. 2 The structural flexibility of perovskite oxides. From single cubic perovskite (center) to double cubic perovskite with B-site ordering of rock-salt, random and layered structures (see Table 1 for detailed information). Through [BO6] octahedral tilt, cubic phases can transform to different tilted phases with three dominating ones shown on the bottom left. Under large distortion of [BO6] octahedra, the three-dimensional (3D) connection of octahedra can be broken, leading to 2D and 1D perovskite derivatives.24 Copyright 2019, The Royal Society of Chemistry. | |
Table 1 Crystallographic information for common double perovskites31
| Sublattice type |
Cell size |
Crystal system |
Space group |
| Random |
1ap × 1ap × 1ap |
Cubic |
Pmm |
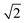 ap × ap × 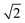 ap × ap × 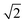 ap ap |
Orthorhombic |
Pbnm |
| Rock salt |
2ap × 2ap × 2ap |
Cubic |
Fmm |
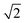 ap × ap × 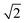 ap × ap × 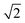 ap ap |
Monoclinic |
P21/n |
| Layered |
2ap × 2ap × 2ap |
Monoclinic |
P21/m |
Table 2 Four common phases for single and double perovskite oxides and the number of compounds reported for each phase.32,33 The data are from ref. 32 and 33
| Glazer notation |
Single perovskite |
Double perovskite |
| Space group |
Number of compounds |
Space group |
Number of compounds |
| a−a−b+ |
Pnma |
119 |
P21/n |
168 |
| a0a0a0 |
Pmm |
21 |
Fmm |
94 |
| a0a0c− |
I4/mcm |
9 |
I4/m |
27 |
| a−a−a− |
Rc |
24 |
R3 |
15 |
2.2. Compositional flexibility
A2BB′O6 DP oxides have an important feature in their crystal structure, which is the extremely large tolerance due to different elemental combinations of (B, B′). To maintain the charge balance, A2BB′O6 must satisfy eqn (1):where Qi (i = A, B, and B′) indicates the charges distributed at the A-, B- and B′-site, respectively. Based on Fig. 3 and Table 3,24 the number of DP candidates is about 105. However, only about one thousand DP oxides have been experimentally synthesized since the 1950s.17,36 As is evident, there are still many DP oxides that have not been extensively studied yet, as shown in Fig. 3, which provides several potential applications.
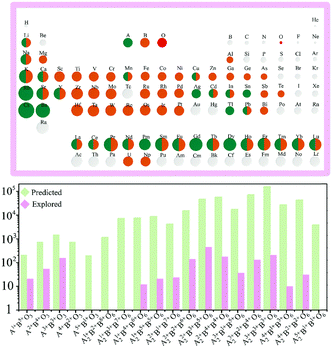 |
| | Fig. 3 Compositional flexibility of single and double perovskite oxides. A- and B-site cations are marked according to the existing single and double perovskite oxides. The bottom figure shows the ideal amount (green bar) and experimentally synthesized amount (pink bar) of perovskite oxides categorized by difference combinations of cation oxidation states. The considered oxidation states ranging from 1+ to 8+ are listed in Table 3.24 Copyright 2019, The Royal Society of Chemistry. | |
Table 3 Possible cations in the periodic table with valence states from 1+ to 8+ (ref. 24)
| Valence |
Elements |
| 1+ |
Li+ |
Na+ |
K+ |
Rb+ |
Cs+ |
Cu+ |
Ag+ |
Au+ |
In+ |
Tl+ |
Hg+ |
Pd+ |
|
|
|
|
|
| 2+ |
Ag2+ |
Am2+ |
Ba2+ |
Be2+ |
Ca2+ |
Cd2+ |
Co2+ |
Fe2+ |
Cr2+ |
Cu2+ |
Dy2+ |
Eu2+ |
Sr2+ |
Sn2+ |
Ti2+ |
Mg2+ |
Tm2+ |
| G2+ |
Hg2+ |
Mn2+ |
Nd2+ |
Ni2+ |
No2+ |
Np2+ |
Pb2+ |
Pd2+ |
Pt2+ |
Ra2+ |
Sm2+ |
V2+ |
Yb2+ |
Zn2+ |
|
|
| 3+ |
As3+ |
B3+ |
Br3+ |
Cf3+ |
Cm3+ |
N3+ |
Np3+ |
P3+ |
Pa3+ |
Pd3+ |
U3+ |
Sc3+ |
Mo3+ |
Mn3+ |
Fe3+ |
Co3+ |
Ni3+ |
| Y3+ |
La3+ |
Ti3+ |
V3+ |
Nb3+ |
Ta3+ |
Cr3+ |
Sb3+ |
Bi3+ |
Ru3+ |
Rh3+ |
Ir3+ |
Sm3+ |
Eu3+ |
Gd3+ |
Tb3+ |
Lu3+ |
| Cu3+ |
Ag3+ |
Au3+ |
Al3+ |
Ga3+ |
In3+ |
Tl3+ |
Ho3+ |
Er3+ |
Tm3+ |
Yb3+ |
Dy3+ |
Pr3+ |
Nd3+ |
Pm3+ |
Bk3+ |
Ce3+ |
| Ac3+ |
Pu3+ |
Am3+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 4+ |
Am4+ |
Bk4+ |
C4+ |
Ce4+ |
Cf4+ |
Cm4+ |
Co4+ |
Cr4+ |
Fe4+ |
Ge4+ |
Hf4+ |
Ir4+ |
Pb4+ |
Pd4+ |
Po4+ |
Pr4+ |
Pt4+ |
| Mn4+ |
Mo4+ |
Nb4+ |
Ni4+ |
Np4+ |
Os4+ |
Pa4+ |
Ti4+ |
U4+ |
V4+ |
W4+ |
Zr4+ |
Ta4+ |
Tb4+ |
Tc4+ |
Te4+ |
Th4+ |
| Pu4+ |
Re4+ |
Rh4+ |
Ru4+ |
S4+ |
Se4+ |
Si4+ |
Sn4+ |
|
|
|
|
|
|
|
|
|
| 5+ |
As5+ |
Au5+ |
Bi5+ |
Br5+ |
Cl5+ |
Cr5+ |
I5+ |
Ir5+ |
Mn5+ |
Mo5+ |
N5+ |
Nb5+ |
Rh5+ |
Ru5+ |
Sb5+ |
Ta5+ |
Tc5+ |
| Np5+ |
Os5+ |
P5+ |
Pa5+ |
Pt5+ |
Pu5+ |
Re5+ |
U5+ |
V5+ |
W5+ |
|
|
|
|
|
|
|
| 6+ |
Cr6+ |
Fe6+ |
Mn6+ |
Mo6+ |
Np6+ |
Os6+ |
Po6+ |
Pu6+ |
Re6+ |
S6+ |
Se6+ |
Te6+ |
U6+ |
W6+ |
Ir6+ |
|
|
| 7+ |
At7+ |
Br7+ |
Cl7+ |
F7+ |
I7+ |
Mn7+ |
Np7+ |
Os7+ |
Re7+ |
Ru7+ |
Tc7+ |
|
|
|
|
|
|
| 8+ |
Os8+ |
Ru8+ |
Xe8+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3. Synthesis methods of DP oxides used for electrocatalysts and supercapacitors
The development of highly active electrocatalysts to increase the activity of the OER and the development of nanostructured DP oxide electrodes for supercapacitors are the most promising topics in the past several years. In this section, we highlight some of the major synthesized methods reported in the literature for OER electrocatalysts and pseudocapacitive materials.
3.1. Solid-state reaction (SSR) method
The SSR method is widely used to synthesize DP oxides, which involves mixing and high-temperature calcination of oxides or carbonate precursors to form polycrystalline bulk samples. For example, a series of Pr1−xBa1+xCo2O6−δ (x = 0.05, 0.10, 0.15, and 0.20) DP oxides were synthesized by the SSR method,37 which exhibited catalytic activity for both the OER and ORR. Pr0.90Ba1.10Co2O6−δ displayed excellent OER activity among all the catalysts with a specific capacitance of 598.40 F g−1, while Pr0.95Ba1.10Co2 O6−δ exhibited potential behavior for the ORR. Sr2CoWO6 DP oxides (Co/W = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:2, and 1:4) were also synthesized by the SSR method.38 It is found that the Sr2CoWO6 (Co/W = 1:1, 3:2) samples could only drive water oxidation under visible light irradiation, whereas the W-rich Sr2CoWO6 (Co/W = 1:4) samples exhibited both photocatalytic OER and HER activities with the presence of sacrificial reagents. This is ascribed to its sufficient valence band (VB) and conduction band (CB) to drive water oxidation and proton reduction, respectively. Thus, W-rich Sr2CoWO6 DP oxide can be used as a bifunctional oxide-based photocatalyst for photocatalytic water splitting.
:1, 3:2, and 1:4) were also synthesized by the SSR method.38 It is found that the Sr2CoWO6 (Co/W = 1:1, 3:2) samples could only drive water oxidation under visible light irradiation, whereas the W-rich Sr2CoWO6 (Co/W = 1:4) samples exhibited both photocatalytic OER and HER activities with the presence of sacrificial reagents. This is ascribed to its sufficient valence band (VB) and conduction band (CB) to drive water oxidation and proton reduction, respectively. Thus, W-rich Sr2CoWO6 DP oxide can be used as a bifunctional oxide-based photocatalyst for photocatalytic water splitting.
Although the SRR method can produce final products with particles at the nanometer scale, the control of morphology and crystallite size of the final products becomes challenging, especially during calcination at high temperatures.39 In general, high temperature annealing during the synthesis of perovskite oxides results in the formation of a pure phase. However, this also leads to a decrease in surface area.40 The ionic conductivity of perovskite oxides is influenced by the calcination temperature, as it affects the densification of atomic packing and leads to the formation of different crystal phases (e.g., rhombohedral, tetragonal, and cubic). These varying crystal structures in turn impact the material's ionic conductivity.41 Polycrystalline La2CuMnO6 (LCMO) DP ceramics was prepared by the SSR method.42 Rietveld refinement of X-ray diffraction confirmed their orthorhombic symmetry with the P21/n space group. The semiconducting behaviour was observed in LCMO ceramics. Raman spectra suggested the possibilities of strong spin–phonon coupling. Additionally, particle sizes have an impact on the catalytic behaviour during electrochemical reactions. Achieving a trade-off between specific surface areas, phase structure purity, and low-temperature calcination poses a significant challenge, as these three factors are closely interconnected.43
3.2. Molten salt synthesis (MSS) method
The MSS method is one of the simplest methods, by which DP oxides can be synthesized with the simplest salt flux reaction medium. The high mobility of the species in the molten salt flux reduces the diffusion distance of oxide mixtures and results in complete reaction in a relatively shorter time. This method has the advantages of reliability, simplicity, scalability, sustainability, low cost, ease of removal and a wide temperature window.44 La2MnCoO6 (LMCO) DP nanoparticles with an average size of 65 nm were synthesized by a molten salt method and used as promising catalysts for electrochemical hydrogen evolution reactions (HERs).45 LMCO nanoparticles exhibited promising catalytic performances for electrochemical HERs with a low Tafel slope value of 190 mV dec−1. The onset over-potential for HERs was found to be ∼120 mV, which matched well with the reported onset over-potentials of other electrocatalysts for the HER. The stability measurements demonstrated that the LMCO electrode materials exhibited a good cycling efficiency as promising HER electrocatalysts. Nano-sized (∼55 nm) La2Co0.5Fe0.5MnO6−δ DP particles were also synthesized by the MSS method,46 which exhibited an enhanced electrocatalytic activity for the OER and ORR in basic solution as compared with the counterpart La2CoMnO6−δ. The improved electrocatalytic activities are attributed to the enhanced surface active sites, modulated electronic structures, and fast charge transfer resistance.
3.3. Combustion method
The combustion method is a highly attractive route for the synthesis of nanomaterials due to its self-propagating high-temperature synthesis process. It has the advantages of flexibility and cost-effectiveness.47 La2CoMnO6 DP oxides were successfully synthesized by a modified combustion route.48 XRD analysis with Rietveld refinement revealed the formation of a monoclinic crystal structure with the space group P21/n. The Raman active modes of the MnO6 octahedra correspond to the Jahn–Teller stretching mode (antisymmetric stretching vibrations) and symmetric stretching vibrations, while the breathing mode represents the symmetric stretching vibrations. The La2CoMnO6 electrode material exhibited good electrochemical performance with 84% capacitance retention after 500 cycles.48 PrBaCo2O5+δ DP oxides, synthesized by the combustion method, crystalized in a tetragonal structure with a crystallite size of 48 nm.49 Their electrochemical performance demonstrated that the charge transfer during the ORR was the rate-limiting step. Thus, nanostructured PrBaCo2O5+δ can be considered as a potential oxide for the cathode for IT-OFC applications.
3.4. High-pressure (HP) synthesis
HP synthesis is an effective method to synthesize materials, which is remarkably effective to expand the chemical ranges of the final products beyond their limits set by the regular SSR method. Although this method is originally used for synthesizing hard materials and high-Tc superconductors, the development of DP oxides with strongly correlated electrons by this method has received much attention in recent years.50 Solid-state DP oxides containing a 5d element have recently attracted much attention owing to 5d elements exhibiting larger valence orbitals and more intense spin–orbit coupling than those of 3d and 4d elements.51 To date, a variety of chemical compositions of 5d (4d) and 3d hybrid DP oxides has been synthesized by the HP synthesis method.52 For example, osmium-based DP oxides such as A2MOsO6 (A = Ca, Sr, and Ba) and Ln2MOsO6 exhibit extraordinary magnetic properties as M is occupied by a 3d element. Sr2CrOsO6 underwent a magnetic transition at Tc = 725 K;53 unusual competing spin structures were discovered in Sr2FeOsO6;54 high-temperature ferrimagnetism was induced by a lattice distortion in Ca2FeOsO6;55 and independent ordering of two magnetic sublattices was observed in Sr2CoOsO6.56
3.5. Sol–gel method
As a simple, fast, and reasonable method for the synthesis of nanosized particles, the sol–gel method has the advantages of low processing temperatures, good stoichiometric control, and the formation of a homogenous multicomponent system.57 Typically, metal salts are used as precursors, and the most commonly used chelating agents are citric acid, ethylenediaminetetraacetic acid, propionic acid, ethylene glycol, glycine, and glacial acetic acid. These reactants are firstly mixed uniformly in the solvent to obtain a homogeneous sol. Then, the sol system will convert to gel by losing the fluid solvent. Finally, calcination is performed to obtain the final product.58 This method was utilized to prepare Sr2CoMoO6−δ (SCMO) DP nanoparticles, which exhibited a desirable rock salt structure.59 The presence of both Co and Mo elements in SCMO DP nanoparticles enhanced their redox capability, leading to enhanced oxygen mobility. It has a high oxygen vacancy content, which enhances the oxygen anion diffusion rate (2.03 × 10−11 cm2 s−1), and a remarkable capacitance of 747 F g−1 at 1 A g−1 as well as 125% retention after 5000 galvanostatic charge–discharge (GCD) cycles at 10 A g−1. La2B(II)MnO6 (B = Cu, Co, Ni) was synthesized as an electrode material using the sol–gel method and calcined at a moderate temperature.47 It was found that La2CuMnO6 (LCMO) exhibited a larger particle size with a specific surface area and remarkable capacitance of 781 F g−1 at a current density of 3.12 A g−1. Pr2CrMnO6 particles with mean size ∼870 nm and chain-like spherical morphology were synthesized by the sol–gel method, which exhibited a specific capacitance of 177.4 F g−1 at a high current density of 2 A g−1 and an excellent cyclic stability.60 Similarly, La2NiCrO6 (LNCO) DP nanoparticles with interconnected spherical morphology were also synthesized by the sol–gel method followed by solid-state reaction at different concentrations of citric acid.61 The interconnected spherical granular-structured LNCO had a high oxygen vacancy concentration, exhibiting ultra-high capacitance due to oxygen–anion intercalations. In a three-electrode system, LNCO@Ni-foam displayed a specific capacity of 529 C g−1 (147 mA h g−1) at a current density of 1 A g−1. La2MnNiO6 (LMNO) DP spherical nanoparticles with a diameter of 30 nm were synthesized via a modified sol–gel method followed by a firing process, which were used as promising electrode materials for OERs.62 These LMNO nanoparticles exhibited highly efficient electrocatalytic activity for OERs with a low onset over-potential (approximately 65 mV) and low Tafel slope value (120 mV dec−1) in alkaline media. The over-potential of LMNO nanoparticles at a current density of 10 mA cm−2 matches with the reported over-potential (η10) of double perovskites, commercial Pt/C and IrO2 electro-catalysts for promoting OERs. LMNO nanoparticles also display approximately 70% specific capacitance retention after 100 cycles.
3.6. Hydrothermal method
The hydrothermal method is a prominent and well-established approach for synthesizing DP oxides on the nanoscale. It offers advantages like lower working temperatures in comparison with the melting point of the reactants, a variety of autoclave options, and adjustable reaction parameters. Microspheres of Y2CoMnO6 (YCMO) DP oxides were synthesized via the hydrothermal process.63 The microspherical shape enhances the charge movement at the nanoscale, providing a larger surface area for the electrochemical reactions. The specific capacitance value was approximately 148.0 F g−1 at a current density of 0.5 A g−1, with a retention rate of ∼85% after 10000 cycles.63 Y2NiMnO6 (YNMO) DP oxide nanowires were used as an active material for the positive electrode in electrochemical supercapacitors, which were prepared by a simple and low-temperature hydrothermal method. YNMO nanowire-based electrodes exhibited a specific capacitance of 77.76 F g−1 (@ 30 mA g−1), energy density of 0.98 W h kg−1 (@ 30 mA g−1), power density of 19.27 W kg−1 (@ 150 mA g−1) and retention of 70.17% efficiency after 1800 cycles.64 Yttrium-based DP oxide nanorods such as YCMO and YNMO with average diameters of 300 nm were also prepared by a two-step hydrothermal method.65 It is found that when used as catalysts for the oxygen evolution reaction, YCMO nanorods displayed a much greater activity in comparison with YNMO ones. This behavior can be attributed to the differences in electronic configuration, wherein the existence of high-spin Co2+ species allows for vibronic super-exchange with Mn4+, thereby forming a catalytically active layer at the surface. Nanocrystallite assembled LNCO nanorods were synthesized by a facile solvothermal route followed by calcination, which exhibited a specific capacitance of 635.5 F g−1 at a current density of 1 A g−1.66 Furthermore, ∼75% specific capacitance retention at 5 A g−1 after 5000 cycles was obtained via chronopotentiometry. La2NiMnO6 (LNMO) nanocubes with average sizes of ∼200 nm were synthesized by a simple, surfactant-assisted hydrothermal method.67 The use of glycine was crucial for the formation of nanocubes. Moreover, the sizes of these nanocubes could be manipulated by controlling the pH value of the reaction medium. In addition, LNMO nanorods with average diameters of ∼50 nm were synthesized by using a surfactant-less hydrothermal method. Cobalt-free nanostructured LCMO is also synthesized via a facile hydrothermal process.68 The pseudocapacitive nature of the cobalt free LCMO was revealed by cyclic voltammetry and chronopotentiometry measurements. The specific capacitance was 205.5 F g−1 at 0.25 A g−1 with ∼78% specific capacitance retention after 1000 cycles at 1.50 A g−1. Mesoporous spheres of La2CrMnO6 DP oxides were synthesized by a hydrothermal process, which exhibited a high surface area of 57.07 m2 g−1, offering more active sites for electrochemical charge storage.69 The electrochemical performance of La2CrMnO6 as an electrode material displayed an excellent specific capacitance of 1416 F g−1 at 1 A g−1 in a three-electrode setup. A- and B-site ordered LaCaCoCrO6 DP oxides were synthesized using a one-step hydrothermal process, which crystallized in an orthorhombic crystal structure with spherical morphology.70 The LaCaCoCrO6 oxides exhibited a specific capacitance of ∼511 F g−1 at 2 A g−1, energy density of 13.75 W h kg−1 (@ 2 A g−1), power density of 439.84 W kg−1 (@ 2 A g−1), and ∼75% specific capacitance retention after 3000 cycles at 5.0 A g−1.
3.7. Co-precipitation methods
The precipitation method is considered as a convenient method to develop metal oxide nanomaterials. This method does not require any complicated process, hard precipitation conditions, or any special instruments. Particle size and shape can be easily controlled by this method. The precipitation method mixes different contents in solutions. The addition of precipitants leads to the formation of precursors. After filtering this precursor calcination is required to obtain the metal oxide. Faik et al. synthesized Sr2MWO6 DP oxides by the co-precipitation method at 947 °C in nitrogen environment.71 X-ray diffraction patterns confirmed that at room-temperature Sr2MWO6 oxides crystallized in a monoclinic crystal structure with a space group of P21/n. This compound presents the following temperature induced phase-transition sequence: P21/n → I4/m → Fmm. Polycrystalline Nd-doped Sr2FeMoO6 (SFMO) DP oxides were synthesized by a citrate co-precipitation method,72 which crystallized in the tetragonal symmetry in the range of 10–400 K and converted to cubic symmetry above 450 K. The unit cell volume increased with increasing Nd3+ concentration, which is an electronic effect in order to change the valence state of the B-site cations. Anti-site defects at the Fe–Mo sublattice increased with the Nd3+-doping content. The Curie temperature was increased from 430 K for Sr2FeMoO6 to 443 K for Sr1.6Fe0.4MoO6. The magnetic moment of the Fe-site decreased while the Mo-site moment increased with electron doping. Thus, antiferromagnetic (AFM) behavior caused the system to show a net ferrimagnetic moment. SFMO and Sr2CoMoO6 (SCMO) DP oxides used as cathode materials for the ORR in alkaline medium are reported,73 where the electrocatalysts (SFMO/C, SCMO/C) consisting of the DP oxides and carbon (Vulcan XC-72) are mixed and spread out into a thin layer on a glassy carbon substrate. At RT, a significant electrocatalytic activity was observed for both electrocatalysts. Compared to SFMO/C, the SCMO/C electrocatalyst exhibited a relatively high electrocatalytic activity for oxygen reduction due to its larger specific area.
3.8. Electrostatic spinning
Electrostatic spinning is used for the synthesis of DP oxides as electrocatalysts and supercapacitors. La2CoNiO6 (LCNO) inorganic nanofibers were successfully synthesized via an electrostatic spinning method, which were connected through rhombohedral LCNO nanoparticles forming a linear spatial network structure.74 These nanofibers were used as supercapacitor electrode materials, and the supercapacitors exhibited an enhanced electrochemical performance. The LCNO inorganic nanofibers had a specific capacitance of 335 F g−1 at 0.25 A g−1. La2CoMnO6 nanofibers were also synthesized by an electrostatic spinning method, which were used as electrode materials.75 They exhibited a specific capacitance of 109.7 F g−1 at 0.5 A g−1 and 90.9% specific capacitance retention after 1000 cycles at 1 A g−1, illustrating a good pseudo-capacitor performance and good cycle stability.
Table S1† summarizes the advantages and disadvantages of each method described above.
4. Characterization of the electrochemical performance of DP oxides
4.1. Evaluation of the performance of electrocatalysts based on DP oxides
DP oxide-based electrocatalysts have different electrochemical properties due to their different particle sizes. The OER process is significantly affected by many parameters, which are described below. The first electrochemical studies consist of recording cyclic voltammogram (CV) curves. The capacitance of the material can be evaluated from the double layer potential region of the CV. The electrocatalytic activities of electrocatalysts based on DP oxides can be evaluated by using linear sweep voltammogram (LSV) measurements at low scan rates of 1 mV s−1. LSV curves provide some of the best visual evidence to evaluate OER behavior, offering information about the onset potential (Eonset) and overpotential (η). The Eonset is one frequently used parameter to compare the catalytic activity of electrocatalysts. The potential, at which a sharp increase in current is observed, is known as onset potential. However, it is difficult to identify the exact value of Eonset. Therefore, the value of the potential at 10 mA cm−2 is considered as a parameter that is commonly used. The accurate Eonset can be measured by using a rotating ring disk electrode for the OER.76 Overpotential η, as one of the most important parameters to evaluate the performance of OER electrocatalysts, is usually referred to as the potential difference between the applied potential (E) and potential under equilibrium condition (Eeq.). Under an ideal condition, the applied potential E, required to carry out a specific reaction, should be equal to the equilibrium potential, Eeq. but actually a much higher applied potential than the equilibrium potential is required to overcome the energy barrier. Usually, the overpotential, η, is measured by using eqn (2).15
Particularly, the η needed to yield a current density of 10 mA cm−2 is widely utilized to evaluate the performance of different electrocatalysts.48 A high-quality electrocatalyst with lower Eonset and η will facilitate the OER process. Tafel plots can be also obtained from the LSV curves. Their analysis helps to compare the OER catalytic activity of different electrocatalysts and reaction kinetics, and is also used to reveal the OER mechanisms. From the Tafel slope, the dependence of steady-state current density on the anodic or cathodic overpotential can be obtained for water splitting. The overpotential, η, is generally logarithmically related to the current density (j) and its linear portion is given as the Tafel equation77
| |
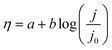 | (3) |
where
η is the overpotential,
a is a constant,
b denotes the Tafel slope,
j is the current density and
j0 is the exchange current density. The Tafel slope describes how fast the current increases against the overpotential. The
j0 can be obtained from the Tafel equation as
η is equal to zero. It represents the intrinsic electrocatalytic activity of the materials at the reversible potential and indicates the electron transfer rate between the electrode and electrolyte. Higher exchange current density indicates a better electrocatalytic ability. The Tafel slope indicates the current density changes with increasing overpotential. Thus, the best OER electrocatalyst based on DP oxides should have a low Tafel slope and a large exchange current density.
78 Normally, a low Tafel slope value of 30–60 mV dec
−2 indicates the formation of surface-adsorbed species at the early stage of the OER catalytic cycle, suggesting the formation of numerous active sites at the surface. In contrast, a relatively higher value of Tafel slope over 120 mV dec
−2 implies the formation of surface adsorbed species just before the rate-determining-step sets in. This means a relatively low availability of surface-active sites.
79 Faradaic efficiency (faradaic yield, coulombic efficiency, or current efficiency) describes the capability of an electrochemical system to facilitate an electrochemical reaction
via electron transfer.
80 In the OER, it is the electron conversion efficiency to generate O
2 molecules and it can be evaluated by calculating the ratio of the experimentally produced O
2 amount (
nO2) to the theoretical produced O
2 amount
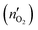
. The Faradaic efficiency can be calculated as follows,
| |
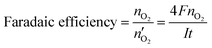 | (4) |
The OER activity of electrocatalysts can also be described in terms of turnover frequency (TOF). It is described as the number of O2 molecules formed per active metal site per second or the reactant required to form the desired product by using a catalyst per catalytic site per second. The TOF value can be evaluated according to eqn (5):
| |
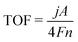 | (5) |
where
j (mA cm
−2) is the current density at a given overpotential,
A is the area of the working electrode,
F is the Faraday constant and
n is the number of active sites.
81 Voltammetry techniques or inductively coupled plasma techniques are used to determine the active species such as metal content. The intrinsic activity of the catalysts can also be represented by using TOF. Thus, a catalyst material with a high TOF value is regarded as a good OER catalyst.
76 To examine the kinetic mechanism of the OER, electrochemical impedance spectroscopy (EIS) is used. The measurement can be carried out in a three-electrode cell at different potentials over the capacitive region or the OER region. The experimental results can be fitted by an equivalent circuit model to extract the values of the non-faradaic double-layer capacitance (
Cdl), the charge transfer resistance (
Rct), and the electrolyte resistance (
Rs).
82 The Nyquist plot and Bode plot are dependent upon the potential applied for the measurement, the catalyst composition and the catalyst layer structure. The stability of the material towards the OER and long-term performance can be determined by measuring CV under different cycles. Chronoamperometry and chronopotentiometry are also widely utilized to test the stability in many references, which are schematically shown in
Fig. 4.
83 Table 4 presents the possible parameters that could be used in combination with the Tafel slope to evaluate the catalytic activity of an OER catalyst material.
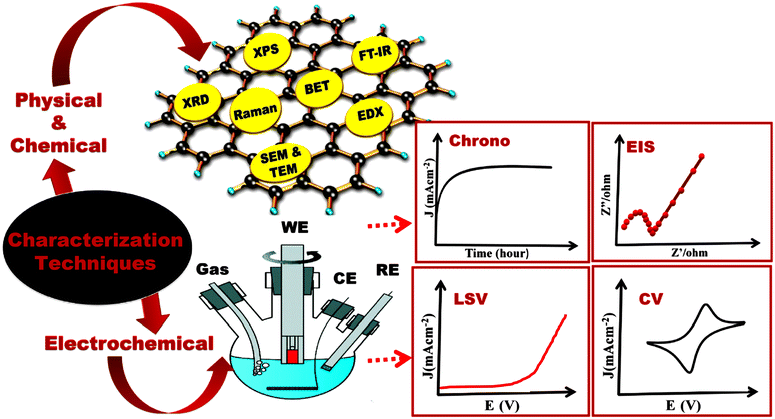 |
| | Fig. 4 Multiple techniques used for characterization of an OER catalyst.83 Copyright 2021, Elsevier Ltd. | |
Table 4 Kinetic parameters in conjunction with the Tafel slope to describe the OER catalytic activity of DP oxide-based electrocatalysts
| Parameter |
Symbol |
| Exchange current density |
j0 |
| Potential at a defined current density |
η(j), e.g. at 10 A gmetal−1 |
| Current density at a defined potential j(ϕ) |
j(η) |
4.2. Evaluation of the pseudocapacitive performance of DP oxides used for supercapacitors
Supercapacitors are often named as ultracapacitors or electrochemical capacitors, which use thin electrolytic dielectrics and high surface area electrode materials to produce capacitances that are many orders of magnitude higher than those of regular capacitors. Thus, supercapacitors can preserve the typical high-power density found in conventional capacitors while also achieving higher energy densities. Based on the energy storage mechanism, supercapacitors are mainly classified into three types: (i) electric double-layer capacitors (EDLCs), (ii) pseudocapacitors, and (iii) hybrid supercapacitors. EDLCs use charge separation to store energy in a manner like that of a conventional capacitor, while pseudocapacitors utilize a faradaic reaction to store energy, in which the transfer of charge between an electrode and an electrolyte is stored electrostatically. The term “pseudocapacity” was first coined by Grahame in 1941 to indicate surplus capacity not linked to the development of the electrical double-layer, where the word “pseudocapacitance” comes from Grahame.84 In a pseudocapacitor, the electrode material undergoes both reduction and oxidation when a voltage is applied. It entails the movement of charge through the double layer, which causes faradaic current to flow through the electrode material of the supercapacitor. In comparison with EDLCs, the faradaic technique used in pseudocapacitors accelerates the electrochemical reactions, leading to higher specific capacitance and energy densities. According to faradaic mechanisms, there have primarily been three types of pseudocapacitance. The first one is adsorption pseudocapacitance or underpotential deposition, in which cations in the electrolyte produce a monolayer that is adsorbed and formed on the surface of a metal electrode with greater redox potential. The second one is redox pseudocapacitance, which is more common. With an associated faradaic charge transfer between the ions in the liquid electrolyte and the solid electrode, it typically occurs on the electrode surface or subsurface. The third type is intercalation pseudocapacitance. It happens when ions are intercalated into the layers or tunnels of a redox-active material, and a quick faradaic charge transfer occurs alongside it without changing the crystallographic phase. As a result, during an electrochemical reaction, it maintains an ultra-stable structure.
The phenomenon known as pseudocapacity can result from a variety of causes, which is dependent on the response to (i) voltage sweep (as seen in cyclic voltammetry, CV); (ii) constant current (as seen in galvanostatic cycling); and (iii) alternating current, as seen in impedance spectroscopy. The sweep rate (v, mV s−1) for the experiment of CV is what determines how long the experiment will last. This parameter also establishes the chronology of the experiment. Based on whether the redox reaction is controlled by surface reactions or diffusion processes, the current approach to a given sweep rate will vary (capacitive). The capacitive and diffusive contribution can be quantified using different techniques. A power law given in eqn (6) relates the peak current to the scan rate (v) and is useful to quantify the capacitive (surface process) contribution and diffusive (bulk process) contribution.85
where
i is the peak current,
v is the scan rate and
a and
b are adjustable constants. Based on the value of
b, the type of charge storage kinetics can be determined. For diffusion limited, faradaic, bulk redox reactions, the peak current varies as a function of the square root of scan rate. For kinetically controlled surface processes like EDLC, the current varies as a linear function of scan rate. Hence,
b = 0.5 for diffusive processes (batteries) and
b = 1.0 for capacitive processes (EDLCs). For pseudocapacitors, particularly intercalation pseudocapacitors, there is a combination of both capacitive and diffusive processes. The related
b value lies between 0.5 and 1.0, revealing more information about the kinetics of the pseudocapacitive materials, which can be obtained from Dunn's method by measuring CV at different scan rates.
86 The logarithmic interpretation of
eqn (7) can be given as follows:
Hence, by plotting the log of the peak current
versus the log of the scan-rate (
v), the value of
b can be obtained from the slope, which is useful in identifying the dominating charge transfer process. The capacitive and diffusive current contributions can also be calculated from another interpretation of the power law as given in
eqn (8). As we know, the total current (
i) is the sum of capacitive and diffusion current,
| | |
i = icap + idiff = k1v + k2v1/2
| (8) |
| |
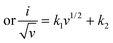 | (9) |
Eqn (9) is in the form of a line between 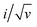 and
and 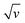 , where k1 and k2 are the slope and intercept, respectively. By measuring CV at different scan rates and by plotting
, where k1 and k2 are the slope and intercept, respectively. By measuring CV at different scan rates and by plotting 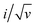 vs.
vs. 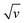 (the square root of the scan rate), the values of k1 and k2 can be obtained from the slope and intercept, respectively. By substituting k1 and k2 into eqn (8) for a particular scan rate, the current contribution can be determined for the diverse potentials in CV. This method can also be used to understand how the kinetics changes at different scan rates, where in general, for slower scan rates the diffusion takes a control, while at higher scan rates, the capacitive contribution dominates the current kinetically.
(the square root of the scan rate), the values of k1 and k2 can be obtained from the slope and intercept, respectively. By substituting k1 and k2 into eqn (8) for a particular scan rate, the current contribution can be determined for the diverse potentials in CV. This method can also be used to understand how the kinetics changes at different scan rates, where in general, for slower scan rates the diffusion takes a control, while at higher scan rates, the capacitive contribution dominates the current kinetically.
The specific capacitance (unit: F g−1) of a pseudocapacitor can be calculated by using the following formula,87
| |
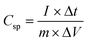 | (10) |
where
Csp is the specific capacitance of the electrode material,
I (unit: A) is the applied current, Δ
t (unit: s) is the discharge time,
m (unit: g) is the total mass of the electrode materials at the positive and negative poles, and Δ
V is the potential window (unit: V). At lower current densities, more interstitial sites are accessible by the ions for redox reactions to take place, leading to higher
Cs, while at higher values of current densities, fewer sites were utilized for ion adsorption, providing lower
Cs.
To evaluate the charge storage performance of an electrode material, two important parameters namely energy density (Ed, unit: W h kg−1) and power density (Pd, unit: W kg−1) were determined by using the following equations from the GCD curve:88
| |
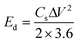 | (11) |
| |
 | (12) |
where
Cs, Δ
V, and Δ
t have the same meanings as described above.
5. Structural characterization of DP oxides used for OER electrocatalysts and supercapacitors
The structural characterization of DP oxides used for electrocatalysts and supercapacitors covers the qualitative and quantitative analyses of crystal structures, lattice defects, morphologies, crystallinity, and chemical compositions, providing the basis for elucidating the relationship between the crystal structures and their electrochemical performance. The structural characterization techniques are operated on the interactions between the incident X-ray (or high-energy electron beam) and the investigated samples. As one of the primary nondestructive techniques, powder X-ray diffraction (XRD) provides the structural information of crystal structures, coexistence of multiple phases, lattice doping, atom/ion vacancies, lattice strain, crystallite size, texture, crystal orientations and structural defects. Particularly, Rietveld refinement on XRD data can reveal atomic-scale structural changes that essentially govern the electrocatalytic behavior whereas the expansive/compressive lattice strains can be extracted from the Williamson–Hall equation.89–91 While XRD patterns provide global information from the sample, electron microscopy offers a localized structural crystal data. The lattice imperfections such as atom vacancies, dislocations, grain boundaries, and clusters are only visualized by high-resolution transmission electron microscopy HR(TEM) images. The atomic-scale details reveal the changes in interplanar spacing (d-spacing) of the crystallographic lattice planes due to atom/ion doping, induced lattice strains, secondary phase formation and dislocations. One of the effective methods is to deconvolute the d-spacing of individual reflections by a fast Fourier transform (FFT) mask filter and estimate the d-spacings at different regions after averaging multiple consecutive lattice planes from each region of the sample. In addition, in reciprocal space, the selected area electron diffraction (SAED) pattern is an extremely useful tool for local nanoscale structural characterization due to its high sensitivity to weak features of reciprocal space, particularly to the structured diffuse intensity distributions contributed by the local compositional ordering and the associated displaced structural relaxation. Techniques such as low energy electron diffraction (LEED) and low energy ion scattering (LEIS) are also promising for determining the surface structure especially the qualitative assessment of the symmetry and quantitative evaluation of atomic positions at the surface. For revealing the surface structure, more precisely the surface roughness, atomic force microscopy (AFM) is an effective tool. Few other metrological and advanced measurements like skewness (measure of symmetry of surface sites) and kurtosis (measure of the distribution of surface symmetry plots) can also be useful to evaluate the surface structure.
One of the primary reasons behind the lattice defects is elemental heterogeneity. Elemental mapping in the high-angle annular dark-field scanning TEM (HAADF-STEM) mode can unravel the extent of homogeneity of a sample.92 Depth profiling by energy dispersive X-ray (EDX) spectral analysis by scanning electron microscopy (SEM) is another method to reveal the compositional difference between the surface and the core of the sample, as well as to test the homogeneity across individual particles.93 X-ray photoelectron spectroscopy (XPS) is a valuable tool to detect elemental composition on the surface and the chemical states as well as electronic structures of materials. In XPS spectra, the sample is etched layer-by-layer by Ar+ ion sputtering to reveal the compositional heterogeneity especially for samples with a dissimilar surface composition and for core–shell structures.94 The surface composition can also be evaluated by Auger electron spectroscopy (AES), which is beneficial for depicting the chemical states for those elements in which the binding energies of two consecutive oxidation states are very similar such as CuO and Cu+. The cation and anion vacancies are commonly elucidated by temperature dependent electron paramagnetic resonance (EPR) spectroscopy. In particular, the lattice oxygen vacancies lead to oxygen defects which are mostly studied by EPR spectroscopy, duly supported by the qualitative and quantitative data from XPS, Raman spectroscopy and Rietveld refinement of the XRD patterns.95 Moreover, operando Raman and infrared (IR) spectroscopy can provide the fingerprint information about different intermediates adsorbed at the catalyst surface during the electrocatalytic reactions. In fact, this field has progressed at a rapid pace in establishing links between the catalytic pathways and the atomic scale defect modulated electronic structure along with the nature of dynamic active sites. In situ/operando techniques have become indispensable to provide a holistic view of the electrode dynamics. The operando techniques probe into the catalyst's interaction with its immediate environments such as the reaction intermediates and products, the ligands and the electrolyte. Electrochemical scanning tunneling microscopy (EC-STM) is such a well-established tool, in which the electrode–electrolyte interface can be probed in situ during electrochemical scanning.96 X-ray absorption spectroscopy (XAS) is one such tool coupled with advanced modeling methods to reveal the electronic structures and the local geometries of the atoms in materials.97 XAS is composed of X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). XANES spectra offer information about the valence state of the absorbing atom, while EXAFS spectra probe the local geometric structure (such as the number of neighboring atoms, distances, and disorder) of the absorbing atom. Oxygen defects or any type cation or anion defects will obviously change the oxidation states, which can be easily probed by XANES. The change in radial distance and coordination number can be detected from Fourier transform or wavelet transform EXAFS spectra. From the intensity ratios corresponding to different bonds, the amount of defects can be quantified easily. To understand the operational changes of a particular catalytic site, correlative microscopy with high surface sensitivity and spatial resolution is a useful approach.98 Scanning photoemission electron microscopy coupled to atom probe tomography can probe an identical location on a sample surface to unravel the surface oxidation states correlated to the atomic-scale composition and intrinsic chemical heterogeneities at the grain boundaries and defect sites. While the classical macroscopic electrochemical techniques give information about the electrochemical response over a large number of surface sites, correlative electrochemical multi-microscopy can target the nanoscopic single entities to probe the electrochemical interfaces.99 Scanning electrochemical cell microscopy can provide such information from hundreds of nanometers down to the atomic level such as the catalytic sites at the single-atom step defects or mapping the local electrical double-layer. In this section, we present a short review on the recent microstructural characterization of DP oxides used for electrocatalysts and supercapacitors.
5.1. DP oxides used for OER electrocatalysts
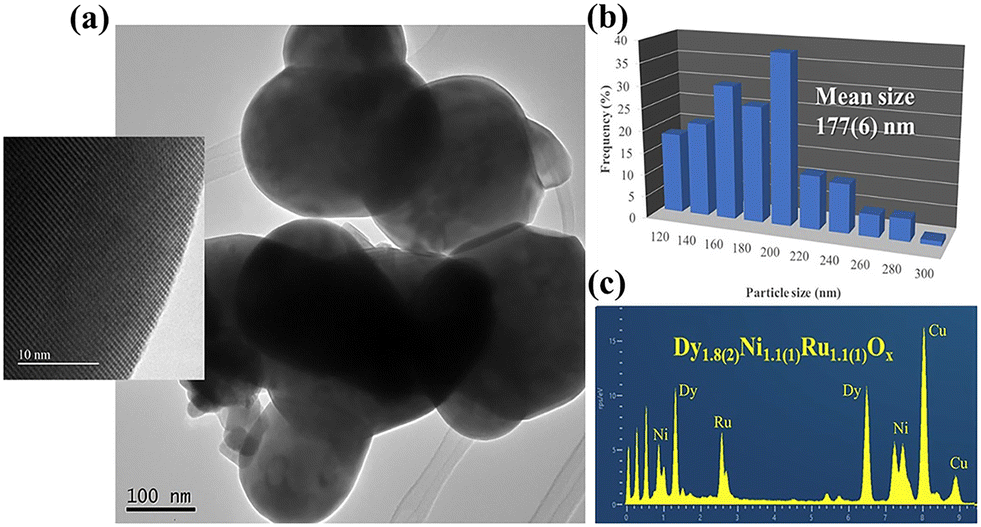
5.1.1. DP oxide nanoparticles. As a sub-class of perovskite materials, DP oxides denoted as A2BB′O6 (where the A-site is normally occupied by alkaline-earth metal ions while B-sites are occupied alternately by different B and B′ cations) exhibit intriguing physical and chemical properties due to enhanced coupling effects via intervening oxygen bridging every B′ and B atom pair.17–19 Profitting from ordered B-site ion arrangement and physical correlation effects are prominent in DP oxides, which offer novel modulation approaches for their electronic structures and electric transport properties, and pave a distinct avenue to tuning OER performance and investigating the OER mechanism of DP catalysts.100–102 In LNMO nanoparticles with ∼33 nm diameter, Tong et al. demonstrated a super-exchange (SE) effect, which redistributes the d electron configuration in B-site cations from a Mn4+–O–Ni2+ static SE effect to a Mn3+–O–Ni3+ vibronic SE effect.103 Such a vibronic SE synergistically optimized the eg electron filling state and led to the optimal eg electron filling states of Mn and Ni ions towards unity, increasing the formation of active species on the surface of catalysts. Thus, a superior OER catalytic performance with a higher current density and lower Tafel slope than its bulk counterpart was observed in the LNMO nanoparticles. Fig. 5 shows the systematic structural characterization of the LNMO nanoparticles. As shown in Fig. 5, the LNMO nanoparticles crystallized in a monoclinic (P21/n) crystal structure and no crystalline phase separation happened during the annealing process. As the particle size of the LNMO catalysts decreased from bulk to ∼33 nm, the SE interaction in LNMO transferred from static Mn4+–O–Ni2+ to vibronic SE of Mn3+–O–Ni3+ with the e1g electron filling state of both Mn and Ni ions. Furthermore, such vibronic SE interaction of Mn3+–O–Ni3+ induces strong Jahn–Teller distortions of MnO6 and NiO6 octahedra, elongating the metal–O bonds, which is beneficial for the formation of active species of Mn/Ni hydroxide/oxide at the surface of catalysts. This is the reason why LNMO nanoparticles exhibit superior OER catalytic performance with a higher current density and lower Tafel slope in comparison with their bulk counterpart. Rodríguez-García et al. also reported a family of Ru-based R2NiRuO6 DP oxides (R3+ = Pr3+, Nd3+, Tb3+, Dy3+, Y3+, Ho3+ and Er3+, and with Ni2+ and Ru4+ occupying the B and B′ sites), which combines both high activity and durability for the OER in acidic electrolyte.104 It is found that the OER activities of R2NiRuO6 mixed oxides are dependent upon the nature of R3+, where the Dy2NiRuO6 oxides exhibit the highest OER activity (1.507 V at 10 mA cm−2) and stability for more than 400 consecutive OER cycles measured at a slow scan rate. In comparison with the rest of the series, such higher performance of Dy2NiRuO6 can be ascribed to the shorter Ru–O bonds in the crystal structure, which offers more hybridization, a higher Ru oxidation state and suitable adsorption energies of the OER intermediates. The morphological, compositional and structural variations in Dy2NiRuO6 during the OER are examined using TEM and HRTEM images for the cycled Dy2NiRuO6 samples. As shown in Fig. 6(a) and (b), initial-Dy2NiRuO6 had a mean particle size of 177(6) nm. Their atomic composition obtained by EDX (Fig. 6(c)) was Dy1.8(2)Ni1.1(1)Ru1.1(1)Ox, approaching the nominal stoichiometric value. In addition, a few particles without nickel, ascribed to the presence of Dy2Ru2O7 pyrochlore, were observed. This phase was also identified by XRD refinement as a minor impurity (close to 4% wt). The representative TEM images of 100-Dy2NiRuO6 particles are shown in Fig. 7(a) and (b), which demonstrate that they remain stable, and the size and morphology are similar to those of initial-Dy2NiRuO6 particles. However, a careful examination of the TEM images of 100-Dy2NiRuO6 particles reveals the presence of a few small particles, probably indicative of an incipient degradation process. The presence of these small particles became more evident in 1000-Dy2NiRuO6 particles, where the larger particles also became more irregular with less smooth and rougher borders, as illustrated in Fig. 7(c). Patterns (ED, inset) for the fresh and used catalysts are shown in Fig. 8. The pristine Dy2NiRuO6 was a crystalline material with diffraction spots ascribed to the (110) and (002) planes of the perovskite structure (inset of Fig. 8(a)). In 100-Dy2NiRuO6, the crystallographic structure within the particles remained crystalline with well-defined diffraction spots corresponding to the initial perovskites, as shown in Fig. 8(b). However, the 100-Dy2NiRuO6 surface is slightly rougher than that in the pristine one, indicating a higher level of disorder of the atoms located at the periphery of the particles. In 1000-Dy2NiRuO6, it is impossible to detect crystallographic planes in the larger particles, indicating that the catalyst loses its crystallinity as a result of its severe degradation. However, by XRD it is still possible to detect perovskite reflections merged with a large amorphous component. The surface composition of initial-Dy2NiRuO6, and the environment and oxidation states of Dy, Ru and Ni cations were investigated by XPS. The Ru 3d and C 1s core-level regions of the XPS spectrum are shown in Fig. 9(a). The strong XPS peak at 284.6 eV is contributed to C 1s. In addition, a minor contribution of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species results in a broad peak at ∼289.0 eV. The presence of Ru is deduced from the Ru 3d5/2 peak at ∼281.9 eV (green peak in Fig. 9(a)). This binding energy (BE) is usually ascribed to oxidized Ru species, typically Ru4+, as the expected oxidation state of Ru cations in Dy2NiRuO6. The XPS spectrum of 100-Dy2NiRuO6 is shown in Fig. 9(b). A single Ru species with the Ru 3d5/2 peak at ∼281.9 eV is observed, suggesting that the catalyst remains stable during cycling, in agreement with the TEM and XRD analyses. Conversely, the spectra of 500-Dy2NiRuO6 and 1000-Dy2NiRuO6 display two Ru species, with Ru 3d5/2 peaks at ca. 281.6 and 282.7 eV. The first peak characterizes the original Ru4+ species, whereas the latter peak at 282.7 eV is usually ascribed in the literature to Ru3+ species.105 In view of these assignments, and taking into account the environment in which the OER cycles are recorded, the Ru peaks observed in the spectra of 500-Dy2NiRuO6 and 1000-Dy2NiRuO6 can be assigned to Ru3+ species, probably Ru hydroxides. Surface atomic ratios of the initial and cycled Dy2NiRuO6 catalysts can be determined by XPS spectra. First of all, a clear dissolution of Dy occurs at the beginning of the reaction but becomes more evident after 500 cycles. The atomic surface ratio for Ru/Ni evolves more slowly, as it increases from 1.9 in Dy2NiRuO6 to 4.1 in 100-Dy2NiRuO6, and remains roughly constant in 500- Dy2NiRuO6, with an atomic Ru/Ni ratio of 4.3. This ratio decreases with further cycling to a value of 2.3 in 1000-Dy2NiRuO6. This trend suggests that the increment of the initial activity is mainly related to the dissolution of Dy cations, together with a slight enrichment of Ru at the surface, which is still rich in both Ru and Ni during the OER. Surface reconstruction occurs in numerous OER catalysts in view of the harsh reaction conditions. In fact, Ru enrichment has been previously reported in acids for Ru-based catalysts such as pyrochlores.106,107 After 1000 cycles measured at 10 mV s−1 the perovskite phase loses its crystallinity and most Ni atoms are gone, segregated into small Ni-based particles, which evinces catalyst degradation. Oroumi et al. also reported spherical-shaped Y2CrMnO6 nanoparticles used as highly active electrocatalysts in hydrogen storage activity, which were synthesized via sol–gel auto-combustion using grape juice as a fuel and complexing agent.108 Fig. 10 shows FE-SEM images of the morphologies and size distributions of the as-obtained Y2CrMnO6 nanoparticles synthesized under different quantities of grape fruit juice. As shown in Fig. 10(a and b), a mixed interconnected microrods and sphere-like particles with large diameters can be formed by selecting 5.0 mL of grape fruit juice during the self-combustion synthesis of Y2CrMnO6 structures. As the fuel value is increased up to 10.0 mL, the final products show a highly agglomerated chunk structure covered with a number of spherical particles, as illustrated in Fig. 10(c and d). In spite of this, the FE-SEM micrographs in Fig. 10(e and f) depict puffy sponge-like clusters with a smooth surface that can be more readily accessed due to the large volume of 15.0 mL required for reducing and capping agents. The improved kinetic performance of Y2CrMnO6 as well as the rapid diffusion of ions during electrochemical energy storage demonstrates the structural stability of this material. As a result of the coexistence of sugars and organic acids in grape juice, the observation data are based upon the influence of strong steric hindrance on the fabrication of nanostructures from grape juice.109 Based on the above SEM images shown in Fig. 10, it can be concluded that the shielding ability of natural fuel in the synthesis reaction affects the isotropic crystal growth of particles.
O species results in a broad peak at ∼289.0 eV. The presence of Ru is deduced from the Ru 3d5/2 peak at ∼281.9 eV (green peak in Fig. 9(a)). This binding energy (BE) is usually ascribed to oxidized Ru species, typically Ru4+, as the expected oxidation state of Ru cations in Dy2NiRuO6. The XPS spectrum of 100-Dy2NiRuO6 is shown in Fig. 9(b). A single Ru species with the Ru 3d5/2 peak at ∼281.9 eV is observed, suggesting that the catalyst remains stable during cycling, in agreement with the TEM and XRD analyses. Conversely, the spectra of 500-Dy2NiRuO6 and 1000-Dy2NiRuO6 display two Ru species, with Ru 3d5/2 peaks at ca. 281.6 and 282.7 eV. The first peak characterizes the original Ru4+ species, whereas the latter peak at 282.7 eV is usually ascribed in the literature to Ru3+ species.105 In view of these assignments, and taking into account the environment in which the OER cycles are recorded, the Ru peaks observed in the spectra of 500-Dy2NiRuO6 and 1000-Dy2NiRuO6 can be assigned to Ru3+ species, probably Ru hydroxides. Surface atomic ratios of the initial and cycled Dy2NiRuO6 catalysts can be determined by XPS spectra. First of all, a clear dissolution of Dy occurs at the beginning of the reaction but becomes more evident after 500 cycles. The atomic surface ratio for Ru/Ni evolves more slowly, as it increases from 1.9 in Dy2NiRuO6 to 4.1 in 100-Dy2NiRuO6, and remains roughly constant in 500- Dy2NiRuO6, with an atomic Ru/Ni ratio of 4.3. This ratio decreases with further cycling to a value of 2.3 in 1000-Dy2NiRuO6. This trend suggests that the increment of the initial activity is mainly related to the dissolution of Dy cations, together with a slight enrichment of Ru at the surface, which is still rich in both Ru and Ni during the OER. Surface reconstruction occurs in numerous OER catalysts in view of the harsh reaction conditions. In fact, Ru enrichment has been previously reported in acids for Ru-based catalysts such as pyrochlores.106,107 After 1000 cycles measured at 10 mV s−1 the perovskite phase loses its crystallinity and most Ni atoms are gone, segregated into small Ni-based particles, which evinces catalyst degradation. Oroumi et al. also reported spherical-shaped Y2CrMnO6 nanoparticles used as highly active electrocatalysts in hydrogen storage activity, which were synthesized via sol–gel auto-combustion using grape juice as a fuel and complexing agent.108 Fig. 10 shows FE-SEM images of the morphologies and size distributions of the as-obtained Y2CrMnO6 nanoparticles synthesized under different quantities of grape fruit juice. As shown in Fig. 10(a and b), a mixed interconnected microrods and sphere-like particles with large diameters can be formed by selecting 5.0 mL of grape fruit juice during the self-combustion synthesis of Y2CrMnO6 structures. As the fuel value is increased up to 10.0 mL, the final products show a highly agglomerated chunk structure covered with a number of spherical particles, as illustrated in Fig. 10(c and d). In spite of this, the FE-SEM micrographs in Fig. 10(e and f) depict puffy sponge-like clusters with a smooth surface that can be more readily accessed due to the large volume of 15.0 mL required for reducing and capping agents. The improved kinetic performance of Y2CrMnO6 as well as the rapid diffusion of ions during electrochemical energy storage demonstrates the structural stability of this material. As a result of the coexistence of sugars and organic acids in grape juice, the observation data are based upon the influence of strong steric hindrance on the fabrication of nanostructures from grape juice.109 Based on the above SEM images shown in Fig. 10, it can be concluded that the shielding ability of natural fuel in the synthesis reaction affects the isotropic crystal growth of particles.
 |
| | Fig. 5 (a) Rietveld refined XRD patterns of LNMO-1 nanoparticles, where the experimental data are marked by red dots, calculated profile by cyan lines, allowed positions of Bragg reflection by green vertical bars, and difference curves by blue lines. (b) TEM image of LNMO-1 nanoparticles. (c) Particle size distribution of LNMO-1 nanoparticles. (d) XPS spectra of Mn 2p for LNMO samples. (e) Mn K-edge XANES spectra of LNMO samples. The inset is an enlarged spectrum of photon energy in the range of 6552–6556 eV. (f) Ni K-edge XANES spectra of LNMO samples. Samples LNMO-1, LNMO-2, and LNMO-3 refer to the LNMO precursors annealed at 700, 900, and 1300 °C, respectively.103 Copyright 2018, American Chemical Society. | |
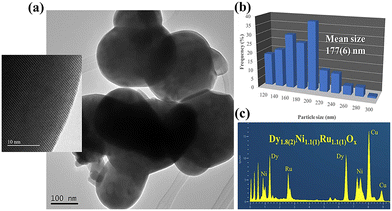 |
| | Fig. 6 (a) TEM images of initial Dy2NiRuO6 particles. Inset: HRTEM. (b) Distribution and mean particle size. (c) EDX corroborating the stoichiometry obtained in the Rietveld analysis of XRD.104 Copyright 2024, The Royal Society of Chemistry. | |
 |
| | Fig. 7 (a) TEM images of a region of 100-cycles Dy2NiRuO6 catalyst. (b) One of the isolated Ni-based particles formed during the OER. (c) TEM image of 1000-Dy2NiRuO6 particles, where the larger particles became more irregular with less smooth and rougher borders. The inset is an enlarged TEM image.104 Copyright 2024, The Royal Society of Chemistry. | |
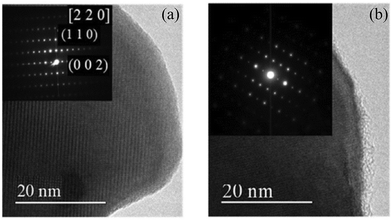 |
| | Fig. 8 HRTEM images of (a) initial-Dy2NiRuO6 and (b) 100-Dy2NiRuO6 particles. The insets are selected area electron diffraction (SAED) patterns.104 Copyright 2024, The Royal Society of Chemistry. | |
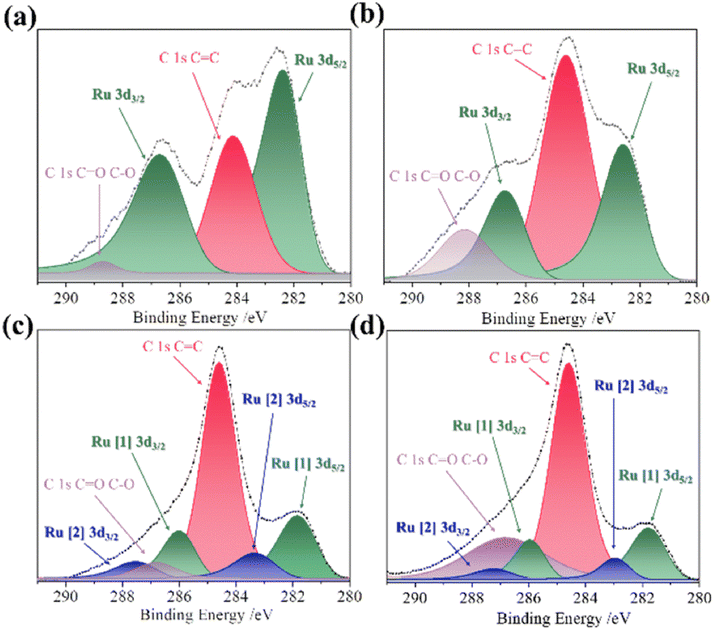 |
| | Fig. 9 XPS of the Ru 3d and C 1s core-level regions of (a) initial-Dy2NiRuO6, (b) 100-Dy2NiRuO6, (c) 500-Dy2NiRuO6 and (d) 1000-Dy2NiRuO6.104 Copyright 2024, The Royal Society of Chemistry. | |
 |
| | Fig. 10 FE-SEM micrographs of morphological evaluation for the YCMO nanoparticles synthesized via a green sol–gel approach by using different quantities of grape fruit juice. (a and b) YCMO-5 mL, (c and d) YCMO-10 mL, and (e and f) YCMO-15 mL.108 Copyright 2024, Elsevier Ltd. | |
A simple molten-salt method was used to synthesize nano-sized La2Co0.5Fe0.5MnO6−δ (LCFMO) DP nanoparticles with a high specific surface area and uniform morphology and particle size.46 They exhibited improved OER and ORR electrocatalytic activities, which were ascribed to the enhanced surface active sites, modulated electronic structure, and fast charge transfer resistance. Fig. 11(a) shows the SEM image of LCFMO DP nanoparticles, which clearly reveals uniform size distribution of the nanoparticles. Based on the histogram of particle size distribution (Fig. 11(b)), the average particle size of the LCFMO was determined to be about 54.1 ± 7.1 nm.
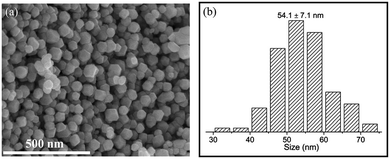 |
| | Fig. 11 (a) SEM image of the La2Co0.5Fe0.5MnO6−δ (LCFMO) nanoparticles, and (b) histogram of particle size distribution of LCFMO.46 Copyright 2023, Elsevier B.V. | |
La2MnCoO6 (LMCO) DP nanoparticles were also synthesized by a molten salt method, which were utilized as promising catalysts for electrochemical hydrogen evolution reactions (HERs).45 The average particle size was determined to be ∼65 nm from the high- magnification FE-SEM images.
5.1.2. One-dimensional DP oxides. Recently, to design high-performance electrocatalysts, well-regulated DP oxide nanostructures (e.g., nanorods, nanofibers, nanotubes) have been developed. McGuire et al. reported on yttrium-based DP nanorods of Y2CoMnO6 (YCMO) and Y2NiMnO6 (YNMO), which can be used not only as supports for methanol and ethanol oxidation reactions (MOR/EOR) but also as catalysts for the OER.65 The YCMO and YNMO nanorods with ∼300 nm average diameter were synthesized by a two-step hydrothermal procedure, wherein (i) a metal hydroxide composite intermediate is formed by a hydrothermal procedure followed by (ii) annealing at high temperature to yield the desired double perovskites. Fig. 12(a) shows the XRD patterns of the as-prepared YCMO and YNMO nanorods, and their SEM images are displayed in Fig. 12(b). The average diameters of YNMO and YCMO nanorods were determined to be 324 ± 58 and 353 ± 46 nm, respectively, along with apparent lengths in microns. Moreover, EDS element mappings for YNMO and YCMO nanorods are shown in Fig. 13, which indicated that all of the individual (Y, Co, Mn, and O) elements are evenly and uniformly distributed throughout the isolated nanorods while maintaining overall chemical compositions approaching the expected stoichiometries. The individual effects of synthetic parameters (e.g., pH value, annealing temperature, and precursor ratios) on both the composition and morphology were systematically investigated. Specifically, the optimal hydrothermal synthesis conditions for YCMO nanorods were a Y:Mn:Co precursor ratio of 1.9:1:1 and a pH of 12.3, followed by annealing at 1000 °C for an interval of 4 h, whereas for fabricating YNMO nanorods, the relevant reaction conditions were associated with a Y:Mn:Ni precursor ratio of 1.6:1:1 and a pH of 13.4, coupled with annealing at 1000 °C for a period of 12 h. Zhao et al. reported on the PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) DP oxide nanofibers as a highly efficient and robust catalyst for the OER,101 which was attributed to both co-doping of Sr and Fe into PrBaCo2O5+δ (PBC) DP oxide and nanostructure engineering. A PBSCF nanofiber with ∼20 nm diameter exhibited ∼20 times enhancement in mass activity at an overpotential of 0.37 V as compared with the PBC powders. The nanofiber morphology enhanced its mass activity remarkably as the diameter was reduced to ∼20 nm. This was ascribed to the increased surface area and an unexpected intrinsic activity enhancement possibly due to a favorable eg electron filling associated with partial surface reduction, as clarified from chemical titration and electron energy-loss spectroscopy. Fig. 14 show shows the structural and morphological characterizations of PBC powders and PBSCF nanofibers. The XRD patterns shown in Fig. 14(b) suggest that pure DP phases (ICDD04-015-0633) are obtained. The observed splitting of the (100)/(002) and (110)/(102) diffraction peaks in PBC powders is characteristic of the DP phase. These two pairs of peaks were overlapped for PBSCF, suggesting that the lattice parameter c is very close to 2ap (the cell parameter of the ideal cubic perovskite), which is consistent with the literature.110 The morphologies of these powders were similar with particle diameters in the range of ∼0.4–2.5 μm. The fast Fourier transform pattern along the [110] zone axis (Fig. 14(c)) shows the presence of superlattice reflections along the c axis, confirming the successful formation of double perovskite structure for PBSCF powders. However, the superlattice reflection dots became weak in some selected regions, suggesting that the double perovskite structure is somewhat disordered mainly because the ionic radius of Sr2+ is comparable to that of Pr3+. Zhang et al. prepared the cubic SmBa0.5Sr0.5Co2O6-δ (SBSC) DP oxide nanofibers by using an electrospinning method via altering the starting raw materials and calcination temperatures, where the morphological structure, phase structure, and electronic structure were simultaneously modulated.111 As shown in Fig. 15(a), SBSC nanofibers were successfully synthesized by using the electrospinning method followed by calcination at 800 °C (SBSC-E(800)), 900 °C (SBSC-E(900)), and 1100 °C (SBSC-E(1100)). A schematic diagram of the crystal structure of SBSC is presented in Fig. 15(b). Based on Rietveld refinement results of the XRD patterns shown in Fig. 15(c), the sample calcined at 800 °C had a cubic structure with a space group of Pmm (a = b = c = 3.849 Å). To further extract the structural information, the high-resolution transmission electron microscopy (HRTEM) images and the corresponding fast Fourier transformation (FFT) patterns of the sample (inset) are shown in Fig. 15(d). The HRTEM image confirmed the phase structure of SBSC-E(800), with a spacing of d = 0.271 nm for the (110) plane. Fig. 15(e) shows the SEM-image of SBSC-E(800) and it maintained the nanofiber morphology structure with an average diameter of ∼80 nm. In addition, Fig. 15(f) presents the EDS elemental mapping images of Sm, Ba, Sr, Co and O elements, which were homogeneously distributed in SBSC-E(800) nanofibers, as revealed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images. Fig. 15(g) and (h) demonstrate the HRTEM images of SBSC-E(950) and SBSC-E(1100) nanofibers, respectively. The HRTEM image in Fig. 15(g) displays lattice fringes with a resolved d-spacing of 0.270 nm, corresponding to the (110) plane of SBSC-E(950), and lattice fringes with a d-spacing of 0.372 nm, corresponding to the (002) plane of SBSC-E (1100), are clearly resolved in Fig. 15(h). A cubic phase is formed in the 950 °C calcined nanofiber while a tetragonal phase was formed in the 1100 °C calcined one (SBSC-E(1100)) with doubling of the c axis. The nanofiber morphology with a large specific area enhanced the electrochemical surface area, enabling more active sites to be exposed, resulting in improved OER mass activity and kinetics. Benefiting from the synergistic modulation, the as-obtained cubic SBSC DP nanofibers exhibited excellent catalytic activity with a low overpotential of 370 mV to reach 10 mA cm−2. Zhu et al. also reported the PBSCF nanotubes (NTs) with diameters of 80–200 nm prepared by electrospinning followed by a calcination process.112 It was found that the OER activity of PBSCF NTs was degraded after thermal annealing in H2 atmosphere whereas significantly improved OER performance was observed in the ones subjected to electrochemical reduction. The distinct different OER activities between the PBSCF NTs after thermal annealing and electrochemical reduction are ascribed to the fact that after thermal annealing the PBSCF NTs were broken into short pieces and agglomerated together (seen in Fig. 16) whereas the PBSCF NTs treated by electrochemical reduction maintained well the hollow nanostructure of the pristine NTs, as demonstrated in Fig. 17. This is the main reason why the PBSCF NTs exhibited degraded performance after thermal annealing while the ones subjected to electrochemical reduction exhibited significantly improved OER performance.
 |
| | Fig. 12 (A) XRD patterns of the as-prepared YCMO and YNMO nanorods, and SEM images of (B) YNMO and (C) YCMO nanorods.65 Copyright 2022, American Chemical Society. | |
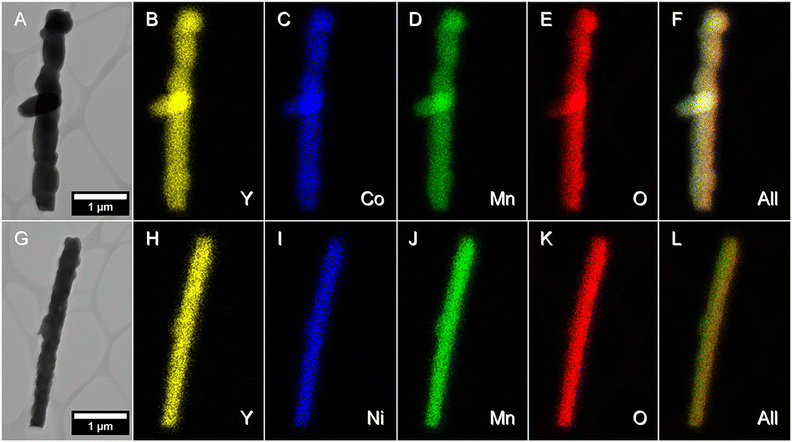 |
| | Fig. 13 (A and G) STEM images and (B–F, H–L) EDS mapping data, associated with (A–F) YCMO and (G–L) YNMO nanorods.65 Copyright 2022, American Chemical Society. | |
 |
| | Fig. 14 Structural and morphological characterization of PrBaCo2O5+δ (PBC) and PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) DP oxides. (a) Schematic of the PBC/PBSCF DP crystal structure, (b) XRD patterns of PBC and PBSCF powders. (c) Fast Fourier transform (FFT) pattern (obtained from the region of ∼23 nm × 23 nm)101 copyright 2017, Springer Nature. | |
 |
| | Fig. 15 (a) XRD patterns for SmBa0.5Sr0.5Co2O6−δ (SBSC) nanofibers at calcination temperatures of 800, 950, and 1000 °C. (b) Crystal structure representations of SBSC-E(800) perovskite. (c) Rietveld refinement XRD patterns of SBSC-E(800). (d) HRTEM image of SBSC-E(800). The inset is fast Fourier transform (FFT) patterns of the SBSC-E(800) (e) SEM images of SBSC-E(800). (f) EDX mappings of the Sm, Ma, Sr, Co, and O element distributions in SBSC-E(800). (g) HRTEM image of SBSC-E(950), and (h) HRTEM image of SBSC-E(1100). The insets are selected area electron diffraction (SAED) patterns.111 Copyright 2019, The Royal Society of Chemistry. | |
 |
| | Fig. 16 SEM and TEM images, EDX line-scanning of (a) the PBSCF NTs-pristine, (b) NT-H-6 h and (c) NT-V-20 min NT samples. The insets are magnified views of the SEM spectra.112 Copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA. | |
 |
| | Fig. 17 TEM image of the (a and d) PBSCF NTs-pristine, (b and e) NT-H-6 h and (c and f)) NT-V-20 min NT samples.112 Copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA. | |
5.1.3. DP oxide thin films. DP oxide thin films can combine atomic phase control and catalytic activity close to those of large-surface-area samples, thus providing an ideal model to study the details of the oxide–electrolyte interface during the OER. Gunkel et al. have demonstrated that epitaxial praseodymium barium cobalt (PBCO) oxide thin film is an efficient catalyst for the OER during electrochemical water splitting, with an activity that is mainly ascribed to PBCO's inherent atomic structure and band alignment.113 They grew epitaxial PBCO oxide thin films by RHEED-controlled pulsed laser deposition (PLD) on (100) SrTiO3 (STO) single-crystal substrates, which display electrocatalytic properties with current densities of up to 10 mA cm−2 at 1.8 V vs. RHE. Dense PBCO thin films can be synthesized in a disordered perovskite phase second CoO2−δ atomic plane along the out-of-plane direction. Fig. 18(a) shows the HR-STEM image of a PBCO thin film grown at 650 °C (here only 20 nm thick), revealing a few unit cell thick coherent layer formed at the interface to the STO substrate, consistent with the observation of RHEED-intensity oscillations. At larger thickness, a disordered grain-like microstructure was formed, similar to that reported in the literature,114,115 while a general c axis orientation was maintained. When the growth temperature was increased to 850 °C (Fig. 18(b)), a more defined microstructure was formed throughout the entire film thickness. In some regions, a fringe contrast was observed at 850 °C, indicating double-spaced atomic ordering. This was confirmed by the appearance of half-order peaks after Fourier transformation (top right). Further increasing the growth temperature to 950 °C, the films demonstrated coherent ordering within the entire thin films, as was evident in the overview image as well as in the magnified inset (red arrows indicate ordered low-intensity CoO2−δ planes). White arrows indicate Ruddlesden–Popper-like stacking faults. The intensity line scan displayed in the bottom panel reveals decreased intensity in every second B-site plane and enlarged interatomic distances between every second A-site plane (line scan taken between the black lines indicated in the top panel). The fringe contrast observed for high-temperature-grown samples is characterized by a lowered relative intensity in every second CoO2−δ atomic plane, indicating that oxygen vacancies are formed mainly in the CoO2−δ atomic planes, rather than in AO planes. The intensity profile (bottom panel of Fig. 18(c)) taken perpendicular to the ordering direction (indicated by the black arrow) revealed not only the lowered intensity of every other B-site atomic plane (red arrows) but also an enlarged interplanar spacing between the A-site cations in the vicinity of the oxygen deficient CoO2−δ planes (green bars), yielding the lowered symmetry of the unit cell responsible for the superlattice peaks observed in XRD. Based on the above structural analyses, a systematic transition from fully disordered to an oxygen vacancy ordered crystal phase can be solely controlled by the growth temperature of the thin films. At low temperature, no ordering was observed. At intermediate growth temperature, a mixed phase involving ordered and disordered phases was established. At the highest growth temperature, a fully oxygen vacancy ordered (double) perovskite PBCO thin film was obtained, in which oxygen vacancies were formed within the CoO2−δ plane, while A-site cation ordering was not evident.
 |
| | Fig. 18 HR-STEM images of the PBCO thin films grown at (a) 650 °C, (b) 850 °C, and (c) 950 °C. The enlarged image in (a) illustrates the disordered perovskite phase obtained at low growth temperature. (b) In some regions, fringe contrast is observed at 850 °C, indicating double-spaced atomic ordering, confirmed by the appearance of half-order peaks after Fourier transformation (top right). (c) Films grown at 950 °C show coherent ordering within the entire thin films, as is evident in the overview image as well as in the magnified inset (red arrows indicate ordered low-intensity CoO2−δ planes). White arrows indicate Ruddlesden–Popper-like stacking faults. The intensity line scan displayed in the bottom panel reveals decreased intensity in every second B-site plane and enlarged interatomic distances between every second A-site plane (line scan taken between the black lines indicated in the top panel).113 Copyright 2017, American Chemical Society. | |
5.2. DP oxides used as electrode materials for supercapacitors
Supercapacitors are electrochemical devices where the processes are carried out at the electrode/electrolyte interface, so the load transport is electronic/ionic. The electrode is the key part of the supercapacitors, thus, the material of the electrodes is the most important factor to determine the properties of supercapacitors, while the operating voltage window is usually dependent on the stability of the electrolyte. The capacitance of these devices depends to a large extent on the characteristics of the electrode material; the most important are a high specific surface, high capacity mainly due to the double electric layer with a pseudocapacitive contribution, good electrical conductivity, and being easily processable to assemble electrodes, using them alone or mixed with plasticizers.116 In the past several years, extensive research has been performed on DP oxides that are used as electrode materials for supercapacitors due to their relatively low-cost and high chemical and thermal stability, high oxygen surface exchange kinetics, fast oxygen ion diffusion rates, and excellent mixed ionic and electronic conductivity at high temperatures. The electrochemical performance of DP oxides can be enhanced by proper tuning of morphology and crystal structure. It is reported that the morphologies of the electrode materials have high influence on the supercapacitor performances; nanosized DP oxide particles can enhance the charge kinetics and this will provide more active sites for redox reaction mechanisms, improving the supercapacitor performances.
The first report on DP oxides used as electrodes in supercapacitors appeared in 2015, where LCNO nanofibers prepared by an electrostatic spinning method were used as electrode materials in supercapacitors.74 The LCNO nanofibers were connected through rhombohedral LCNO nanoparticles resulting in a linear spatial network structure, as demonstrated in Fig. 19. The diameter of the nanofibers carbonized at 700 °C was 700 nm with a large specific area of 10.12 m2 g−1. The LCNO nanofiber electrode material has good capacitor performance. For the three-electrode system the electrode achieved a respectable specific capacitance of 335.0 F g−1 at 0.25 A g−1. For the symmetrical two-electrode system the electrode achieved a specific capacitance of 129.1 F g−1 at the same current density. In 2016, Alam et al. synthesized YNMO nanowires (NWs) by a hydrothermal method.64 Their detailed structural characterization through XRD, HRTEM, EFTEM, EDAX and FESEM is illustrated in Fig. 20, which reveals the successful formation of YNMO NWs. They display superior electrochemical properties with a specific capacitance of 77.76 F g−1, energy density of 0.98 W h kg−1, power density of 19.27 W kg−1, and retention of 70.17% efficiency after 1800 cycles. To improve the energy density of supercapacitors, much effort has been made to develop new kinds of DP oxides used as electrode materials in supercapacitors, such as reduced PrBaMn2O6−δ (r-PBMO),117 PrBaCo2O5+δ (PBCO) (A-site cation-ordered DP),118 La2CoMnO6 hollow spheres,75 B-site cation ordered Ba2Bi0.1Sc0.2Co1.7O6−δ,119 Ba2FeCoO6−δ (BFCO) polycrystalline powders,120 La2FeCoO6 nano-crystallites,121 nanostructured cobalt-free La2CuMnO6,68 La2NiMnO6 nanostructures,122 Pr2CrMnO6 chain-like spherical particles,60 Gd2NiMnO6 powders,123 and Dy2CoMnO6 powders.124 Recent progress in the development of nanostructured DP oxide electrodes has improved their energy density, power density, and cycling stability. However, the nanostructure may lead to a low pack density of the electrode, leading to poor volumetric energy density of the corresponding supercapacitor. Importantly, in many portable electronics applications, the volumetric energy density is a more important characteristic than the gravimetric energy density. It is still a considerable challenge to develop energy materials with high power densities, high volumetric energy densities, and good cycling stability at low cost. Thus, a long journey lies ahead to enhance the electrochemical performance of DP oxide materials for future supercapacitor applications.
 |
| | Fig. 19 SEM images of La2CoNiO6 nanofibers carbonized at different temperatures for 3 hours. (a1 and a2) 500 °C; (b1 and b2) 600 °C; (c1 and c2) 700 °C; (d1 and d2) 800 °C.74 Copyright 2015, Acta Physico-Chimica Sinica, CCME, PKU. | |
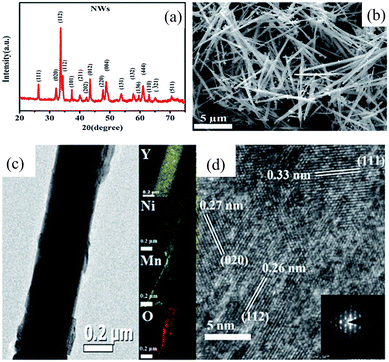 |
| | Fig. 20 (a) XRD pattern of Y2NiMnO6 NWs; (b) FE-SEM image of Y2NiMnO6 NWs; (c) TEM image of the NWs and elemental mapping of Y (in yellow), Ni (in blue), Mn (in green), and O (in red); (d) HRTEM image and SAED pattern (inset) of the Y2NiMnO6 NWs.64 Copyright 2016, The Royal Society of Chemistry. | |
6. Applications of DP oxides in OER electrocatalysts and supercapacitors
6.1. DP oxides used as OER electrocatalysts
As a sub-class of perovskite oxides, DP oxides have the formula of AA′BB′O6, where the A or B cations are substituted by different cations, and B and B′ sites are considered as a prime location for implementing a distinct method to regulate electrocatalytic OER performance.100,125 Thus, DP oxides are key candidates as promising OER electrocatalysts. The different approaches for generating DP oxides via cation substitution (or doping), surface treatment, and nanostructure engineering play an important role in enhancing the catalytic activity and durability of the resultant DP oxides.30 For example, partially substituted B-site ordered DP oxides of Sr2Fe0.8Co0.2Mo0.6Co0.4O6−δ (SFC0.2MC0.4) exhibited a better OER activity in alkaline media with an overpotential of 345 mV to reach a 10 mA cm−2 current density and a Tafel slope of 60 mV dec−1,126 in comparison with Sr2FeMoO6−δ for the OER.127 The excellent OER activity of SFC0.2MC0.4 demonstrated that the introduction of OER active elements simultaneously into the B-site and B′-site of the ordered DP oxides is an effective strategy for enhancing the OER catalytic activity.126 To enhance the activity of DP oxide-based OER electrocatalysts, it is necessary to understand the correlation between the material properties and the OER activity. Based on the conventional OER mechanism for transitional-metal oxides in alkaline solution, all of the intermediates interact with the surface of transitional metal oxides through an oxygen atom. Thus, the M–O bonding interactions (M is a catalytically active metal center) within the intermediates (e.g., MOH, MO, and MOOH) are crucial for the overall OER activity.15,128–131 More specifically, the stronger the oxygen atoms interact with the surface, the easier the adsorption of OH− and the more difficult the desorption of O2 becomes. Therefore, the interaction between the adsorbate and electrocatalysts should be neither too strong nor too weak.132,133 As a result, the M–O bonding strength is considered to be a universal descriptor for the catalytic activity of the OER. In addition, molecular orbital principles also demonstrate that the OER activity is strongly correlated with the eg occupation level of transitional metal cations because the electrons in eg orbits can interact effectively with oxygen-containing adsorbates. So, a good OER catalyst should have a surface cation eg occupancy approaching unity, to facilitate the charge transfer between the transition metal cations and the reaction adsorbates (M–O, M–OH, M–OOH), accelerating the water splitting kinetics.58,128,134–137 Therefore, developing effective strategies to modify the electronic structures (or optimize the eg electron occupation) and tune the interactions between the adsorbates and active sites is highly desirable. It is well known that oxygen vacancies exist widely in transitional metal oxides due to their low formation energy. The formation energy of oxygen vacancies increases with the energy difference between the upper edge of the O 2p band and the Fermi level. Thus, only a certain amount of oxygen vacancies can be created in oxides and they have a direct influence on the M–O bonds near the oxygen vacancies.138–140 Several studies have shown that a suitable amount of oxygen vacancies indeed plays a positive role in the OER activity of transition oxides by significantly changing the bulk (such as energy level, conductivity) and surface (such as molecular adsorption) properties of materials.141–143 For example, the layered PrBaMn2O5+δ DP oxide electrocatalyst demonstrates the presence of additional oxygen vacancies and an optimized eg filling for the Mn ions leads to an enhanced OER activity.144 In spite of oxygen-deficient DP oxides showing high catalytic activity for the OER, large oxygen vacancies in the lattice would reduce the performance of the OER.145 In addition, how to generate oxygen vacancies with uniform distribution and controllable concentration remains a great challenge. Advanced methods to quantify oxygen vacancies accurately and confirm their real location are also highly desirable. Rational control of the surface reconstruction process is considered as an effective strategy to develop high-performance OER electrocatalysts.146 During the electrocatalytic OER, the DP oxide surface allows changes on the surface via A- or B-site leaching and surface reconstruction. For example, LNMO nanoparticles show the reconstruction of the surface depending upon the number of OER cycles as compared with the pristine unreacted sample.103 As shown in Fig. 21(a), the pristine LNMO-1 catalyst exhibited a clean surface. However, after 50 charge–discharge tests, a thin active amorphous phase with a thickness of about 1.0 nm appeared on the surface of the catalyst (Fig. 21(b)). When prolonging the CV tests, the thickness of the electroactive species further increased and gradually reached a steady state, as demonstrated in Fig. 21(c) and (d). However, the active phases on the surface of LNMO-2 and LNMO-3 catalysts after 500 charge–discharge tests were obviously thinner than that of LNMO-1, as shown in Fig. 21(e)–(h), which demonstrated the active role of Mn3+and Ni3+ in forming active species during the OER process. The above results reveal that the appearance of Mn3+and Ni3+ could facilitate the formation of an active phase on the surface of the LNMO sample, further promoting the catalytic activity. A similar scenario appeared in the YCMO nanorods, where the thickness of the isolated amorphous layer was increased after 5, 20, and 60 minute intervals as compared with the pristine sample.
 |
| | Fig. 21 HRTEM images of (a) pristine LNMO-1 sample (without activated) and after (b) 50, (c) 100, and (d) 500 cycles. (e) and (f) HRTEM images of the LNMO-2 catalyst after 100 and 500 cycles, respectively. (g) and (h) HRTEM images of the LNMO-3 catalyst after 100 and 500 cycles, respectively.103 Copyright 2018, American Chemical Society. | |
Recently, well-regulated nanostructures (e.g., nanofibers, nanotubes, and nanoarrays) have been developed to construct high-performance electrocatalysts.147,148 PBC and PBSCF DP oxide nanofibers were synthesized by an electrospinning method followed by the calcination process, which were used as highly efficient and stable electrocatalysts for the OER.101 As shown in Fig. 22, (i) PBC/PBSCF has an ideal layered structure. A synergistic effect of Fe and Sr in PBSCF creates pore channels which enhances oxygen ion diffusion and surface oxygen exchange. The PBSCF nanofibers with three different diameters (PBSCF-I, PBSCF-II, and PBSCF-III) were prepared by varying the calcination temperature. From the TEM images, it is clear that PBSCF-I nanofibers have discontinuous pores inside and PBSCF-II nanofibers have a hollow structure with continuous mesopores. In the case of PBSCF-III, some adjoined nanofibers were fused with no observable pores (Fig. 22(ii)). Fig. 22(iii) shows the polarization curves of the synthesized materials along with the commercial IrO2. From this figure, it was clear that ultrafine PBSCF-III exhibited superior OER activity as compared to the other synthesized materials. Porous structures such as microporous, mesoporous, and macroporous structures with a rich porous architecture generally provide large surface areas. The rich pores can offer free pathways for electrolyte diffusion, enabling good interaction between the active sites on the surface and the reactants. The nano-size effect was also demonstrated by synthesizing hierarchically porous PBSCF via an inorganic salt-template strategy.149 The hierarchically porous structure facilitated the mass transfer and provided a high surface area (148 m2 g−1), thus exposing more surface-active sites. Particularly, 3D ordered meso- and macro-porous morphologies have allowed new electrocatalysts to be designed. The periodic pore arrangement further enhances certain catalytic properties, such as the operational durability. 3D ordered macroporous (3DOM) DP oxides La2NiMO6 (M = Cu, Co, Fe, Mn) were synthesized via the colloidal crystal template approach.150 The unique 3DOM structure improves the interactions between the soot and the catalyst. Meanwhile, the periodic arrangement of MO6 and NiO6 octahedra in the DP structure induced the generation of coordination unsaturated B-site ions. However, the synthesis of DP oxide-based ordered porous structures and their applications to electrocatalysis still remain limited. Besides nanostructural engineering, Zhu et al. evaluated the effect of the crystal orientation on proton-coupled electron transfer processes and the OER activity.151 The PBSCF thin films with different orientations were deposited on single crystal substrates by the PLD method. Fig. 23(a) illustrates the crystal structure of PBSCF with different surface orientations of (100), (110), and (111). The PBSCF thin films exhibit OER activity in the order of (100) > (110) > and (111).101 The proton diffusion path at the (100) surface displayed energy-saving and fast reaction kinetics characteristics (Fig. 23(b) and (c)). With the rational selection of the crystal orientation of the A-site ordered DP, the OER activity can be effectively improved.
 |
| | Fig. 22 (i) Schematic diagram of PrBaCo2O5+δ (PBC)/PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) DP oxide crystal structure; (ii) TEM images of PBSCF I, PBSCF II, and PBSCF III nanofibers; (iii) iR corrected polarization curves of IrO2, PBC-0, PBSCF-0, PBSCF-I, PBSCF-II, and PBSCF-III.101 Copyright 2017, Springer Nature. | |
 |
| | Fig. 23 (a) Crystal structure of PBSCF showing different surface orientations of (100), (110), and (111). (b) Energy profiles for proton diffusion in bulk PBSCF. (c) Geometrical demonstration of oxygen and proton bulk diffusion in PBSCF along the (100) direction.152 Copyright 2020, Springer Nature. | |
6.2. DP oxides as electrode materials for supercapacitors
As the outcomes of single perovskite oxides, DP oxides with the general formula of AA′BB′O6 were first discovered by Steward and Rooks in 1951. Their fast oxygen ion diffusion rates and good ionic as well as electronic conductivity make them attractive for various applications. Also, due to their more ordered arrangement, these materials have shown less lattice distortion, thereby improving the cyclic stability of supercapacitor devices. Bavio et al. prepared LNMO DP oxides by the citrate precursor route and used them as electrode materials for supercapacitors.47 The electrochemical performance of the devices was measured in both alkaline and acid solutions. The results demonstrated that the LNMO DP oxides performed better in alkaline medium than in acidic media. The GCD measurements demonstrated that in alkaline medium the LNMO DP oxides had a specific capacitance of 1681 F g−1 at 3 A g−1, and the power density and energy density were 2903 W kg−1 and 378 W h kg−1, respectively. While in acidic medium, these values were 492 F g−1, 3225 W kg−1, and 137 W h kg−1, respectively. Liu et al.152 developed Ni-doped Sr2CoMo1−xNixO6−δ (x = 0.15, 0.25, 0.35) (SCMNx-S1) DP oxides via the sol–gel method to improve the specific capacitance of the material. The SCMNx-S3 sample showed a specific capacitance of 930 F g−1 at a current density of 1 A g−1, and exhibited a capacitance retention of 93.1% over 5000 charge–discharge cycles. The enhanced electrochemical performance could be due to the improvement in the oxygen vacancies of the nanomaterial and surface faradaic redox reactions. PrBaCo2O5+δ, an anion inserted pseudopotential DP oxide was synthesized via sol–gel methods.118 It was found that the presence of oxygen vacancies at Pr3+ by removing oxygen partially or entirely helped to improve the electrochemical performance of the material. Thus, the material exhibited a high specific capacity of 428.2 C g−1 at 1 mV s−1. Moreover, the material had notable stability of 93% over 2000 charge–discharge cycles. The enhanced specific capacitance and stability of the material were ascribed to the improvement in electrical conductivity and oxygen vacancy concentration of the A-site cation ordered DP oxide. Besides A site-ordered DP oxides, B-site cation-ordered DP oxides were also synthesized for supercapacitors.117 Ba2Bi0.1Sc0.2Co1.7O6−δ (BBSC) electrode materials, with large oxygen vacancy concentrations and high oxygen diffusion rates, exhibited a high specific capacitance of 1050 F g−1 and maintained up to 780 F g−1 at 1 A g−1 even after 3000 charge–discharge cycles. This indicated that the BBSC materials had high capacitance retention. PBMO and PBCO DP oxides exhibit high conductivity and oxygen surface exchange coefficient, good chemical stability and low-cost. Their electrochemical performance was further improved by doping new elements at B-site cations. Zhong et al.153 synthesized PBCO DP oxide with partial substitution of Co by Ni at the B-site. The Ni-doped PrBaCo2−xNixO5+δ (PBCN) is an oxygen ion-embedded electrode material for supercapacitors with a maximum specific capacity of 513 C g−1. The asymmetric capacitor of PBCN//activated carbon (AC) displayed an energy density of 48.3 W h kg−1 with a maximum power density of 20011.8 W kg−1. Developing electrode materials with hollow microstructures exhibits great advantages in storage applications. The hollow structure allows for fast movement of the electrolyte ions and shortens the diffusion length, such that the volume expansion of the materials during charge storage is considerably reduced. Singh et al. synthesized mesoporous La2CrMnO6 DP powders as a positive electrode.69 The material showed an outstanding specific capacitance of 1416 F g−1 at 1 A g−1 and a capacitance retention of ∼42% over 5000 charge–discharge cycles. Details are shown in Fig. 24. The La2CrMnO6 electrode material also demonstrated a high energy density of 24.1 W h kg−1 and a power density of 350 W kg−1 (@1 A g−1), indicating that mesoporous La2CrMnO6 DP oxides can be promising electrode materials for supercapacitor applications. The microsphere-like structure of Y2CoMnO6 DP oxides prepared by a hydrothermal method is also used for supercapacitors.63 Their electrochemical performance revealed a specific capacitance value of about 148 F g−1 at 0.5 A g−1 and a capacitance retention of ∼85% over 10000 charge–discharge cycles. Recently, well-regulated nanostructures such as nanofibers have been developed to be used as electrode materials for supercapacitors. The YNMO DP nanowire electrode material demonstrated a specific capacitance value of 77.76 F g−1 at 30 mA g−1, energy density of 0.89 W h kg−1 @ 30 mA g−1, power density of 19.27 W kg−1 @ 150 mA g−1, and capacitance retention of ∼70% over 1800 charge–discharge cycles.64
 |
| | Fig. 24 (a) Galvanostatic charge–discharge curve with varying current densities, (b) specific capacitance and specific capacity at current densities of 1, 2, 4, 6, 8, and 10 A g−1, (c) Ragone plot showing the relationship between energy and power density, and (d) retention rate after completing 5000 cycles.69 Copyright 2023, Wiley-VCH GmbH. | |
7. Theoretical studies of DP oxides used for OER electrocatalysts and supercapacitors
In parallel with experimental investigations on DP oxides used for OER electrocatalysts and supercapacitors, theoretical computations (e.g., density functional theory (DFT), Monte Carlo simulation, molecular dynamics simulations, and machine learning (ML)) have been widely utilized to better understand the reaction mechanisms of the OER based on DP oxides and the electrochemical storage mechanisms of the supercapacitors based on DP oxides. Grimaud et al.154 reported that Ln0.5Ba0.5CoO3−δ (LnBaCo2O5+δ′ with δ′ = 1–2δ and Ln = Pr, Sm, Gd and Ho) DP oxides exhibited high activity and stability, which could be explained by their O p-band center neither too close nor too far from the Fermi level. The O p-band centers were calculated by the DFT approach, as shown in Fig. 25, indicating the computed oxygen p-band center can be used as a descriptor to screen the OER activity and stability of oxides. Moving the computed O p-band center closer to the Fermi level can increase OER activities, but having the computed O p-band center too close to the Fermi level would decrease oxide stability during the OER. The DP oxides with the O p-band center much close to the Fermi level not only have the highest OER activities in alkaline solution but also exhibit the highest activities for surface oxygen exchange kinetics upon oxygen reduction at elevated temperatures,155 which highlights the importance of the oxide electronic structure on oxygen electrocatalysis. To understand the atomic and electronic level mechanism of the effect of Sr, Fe co-doping in PBC on the OER activity, DFT calculations for the surface OER reactions of PBC and PBSCF nanofibers were carried out.101 The results are shown in Fig. 26. As shown in Fig. 26(a), a widely accepted OER mechanism in an alkaline medium involves a four-step, four-electron OER,132 where the OER reaction started on the catalyst adsorbed with *OH unlike some other reported OER mechanisms (inverse ORR process) including a step with a bare catalyst surface.156,157 Fig. 26(b) shows the calculated Gibbs free energy of reactions in the whole system for the OER as a function of the reaction coordinate, where the OER energy profiles for both PBC and PBSCF nanofibers are very similar with step 1 from *OH to *O and step 4 from *OO to *OH + O2 (uphill/endothermic), indicating an external driving force (for example, electrical potential) necessary to drive the OER. It has been recently demonstrated that among the family of DP oxides the composition of PrBaCo2O5+δ with the O p-band center much close to the Fermi level can exhibit very high oxygen evolution activity.154 The above theoretical calculation clearly demonstrates that Fe/Sr co-doping in PBC enhances the OER activity on the Fe-site, which is consistent with the electrochemical measurement results. As compared to the experimental results, DFT calculations have significantly improved the efficiency of catalyst screening, but they require substantial computational resources. Moreover, due to the inherent limitations of DFT, bulk rapid screening of efficient catalysts is very costly. Therefore, it is crucial to find low-cost and high-speed methods for screening high-performance catalysts. In recent years, ML has been gradually adopted to quickly discover and screen candidate materials and assist in unlocking the reaction mechanisms. ML can predict the performance of new catalysts based on feature engineering, greatly reducing the cost of screening catalysts and improving efficiency. Additionally, ML can analyze feature importance, providing deep insights into the impact of different features on catalytic performance and enhancing theoretical understanding of catalysis. Thus, the simulation-based, ML-accelerated prediction strategy offers quantitative guidance for rational screening the DP oxides to fabricate OER electrocatalysts with high electrocatalyst activity and at a significantly reduced cost.158 In 2020, Li et al. developed an adaptive learning strategy to explore high-performance AA′B2O6 cubic perovskites for catalyzing the OER.159 Through mapping the correlations between a large amount of available informatics and the adsorption energies (i.e., *O and *OH), the probabilistic Gaussian processes quickly estimated the adsorption energies of reaction intermediates and the corresponding uncertainties of a rich material space. This adaptive learning strategy gradually improves the robustness of the model by verifying promising samples, albeit with large uncertainties. After iteratively validating/refining the candidates with theoretical overpotentials below 0.5 V, an excellent ML model with RMSE (root mean squared error) less than 0.5 eV was attained. This model rapidly predicted nearly 4000 AA′B2O6 DP compounds and proposed nine stable cubic perovskite candidates with the optimal OER performance. Furthermore, they also revealed the potential relationship between the electronic structure descriptors and the OER activity of the perovskites, indicating that the orbital electronic structure characteristics of the B-site ion might be latent factors governing the OER activity. This work demonstrates that adaptive learning is a cost-effective strategy that can reduce the uncertainty of model predictions in high-dimensional feature spaces with the least computational cost.
 |
| | Fig. 25 Computed oxygen p-band center for oxygen evolution. (a) Schematic representation of the O p-band for transition metal oxides and (b) evolution of the iR-corrected potential at 0.5 mA cm−2 oxide versus the O p-band center relative to EF (eV) of Ln0.5Ba0.5CoO3−δ with Ln = Pr, Sm, Gd and Ho, for LaCoO3 (LCO), La0.4Sr0.6CoO3−δ (LSC46), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF82), Ba0.5Sr0.5Co0.4Fe0.6O3−δ (BSCF46) and SrCo0.8Fe0.2O3−δ (SCF82). The O p-band center relative to the Fermi level was computed by DFT for the fully oxidized and relaxed structure. DFT calculations with the Hubbard U (Ueff = 3.3 eV) correction for the Co 3d electrons were performed with the Vienna ab initio simulation package using the projector-augmented plane-wave method. Error bars represent s.d. from at least four independent measurements.154 Copyright 2013, Macmillan Publishers Limited. | |
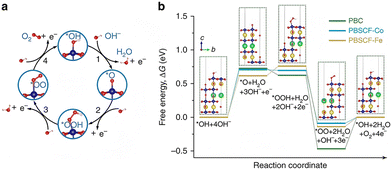 |
| | Fig. 26 DFT calculation for the OER mechanism. (a) Illustration of the four electron-proton step mechanism of the OER in alkaline electrolyte. (b) The computed Gibbs free energy changes for the whole system for the OER at η = 0 V/298.15 K on a PrBaCo2O5+δ (PBC)/PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) surface; the B-sites in PBC (Co) and PBSCF (Co/Fe) are treated as the catalytically active centers, all adsorbates are bonded to these redox centers; insets are atomic structures of PBC slabs with adsorbates. In all schematics, each deep blue ball is Co or Fe, fluorescent yellow ball is Pr, green ball is Ba or Sr, red ball is O and light pink ball is H.101 Copyright 2017, Springer Nature. | |
DP oxides are useful electrode materials in supercapacitors. However, adopting their special structures to improve the electrochemical performances of supercapacitor electrodes is still challenging. In recent years, the relationship between the structure and properties of DP oxides has been illuminated by implementing a number of new computational methods that incorporate data. Recently, Dimitrovska et al. predicted the length of unit cell edge (a) of DP oxides with the general formula A2BB′O6 by two methodologies: multiple linear regression (MLR) and artificial neural networks (ANN).160 The unit cell edge length was expressed as a function of six independent variables: the effective ionic radii of the constituents (A, B and B′), the electronegativities of B and B′, and the oxidation state of B. In this analysis, 147 perovskites of the A2BB′O6 type, having the cubic structure and belonging to the Fm3m space group, are included. They are divided into two sets; 98 compounds are used in the calibration set and 49 are used in the test set. Both models give consistent results and could be successfully used to predict the lattice cell parameter of new members of this series. Although the ANNs are capable of modeling possible nonlinearities among independent and dependent variables, in this case there is no statistically significant difference between the predictions of the two models. ML classification models based on random forests were also built to predict formability and thermodynamic stability of DP oxides.161 The ML models were trained to maximize accuracy while minimizing variance. To start with, a large number of elemental features were considered by using domain knowledge, intuition, and prior work by researchers, to screen for novel stable DP oxides. This study identifies that 437828 DP compounds that are likely to be stable and 891188 DP compounds that are likely to be formable. From the intersection of this large chemical space of formable and stable oxide perovskites, 414 compositions are identified as the most promising candidates for future experimental synthesis of novel oxide perovskites. The developed ML models could be generalized and expanded into other families of compounds. It is realized that ML has great potential and advantages in discovering novel DP oxides and revealing their relationships between the structural, compositional, and technological descriptors and physio-chemical performance based on known material information. Despite some successful research achieved in the applications of ML in perovskite materials, the application of ML in materials research is still in its infancy and much work needs to be further deepened in the future.
8. Summary and perspectives
We have reviewed the structural and compositional flexibilities, synthesis methods and structural characterization techniques of DP oxides and their promising applications in the fields of electrochemical OER and supercapacitors. The relationships between the unique structural and compositional flexibilities and the remarkable electrochemical performance and charge storage performance are especially emphasized. The evaluation methods of OER electrocatalysts and supercapacitors are also described. Though significant progress has been made in the past decade, more is anticipated since there are still a large number of DP oxides that are theoretically predicted to exist, but have not been explored experimentally. The main findings of the recently reported state-of-the-art electrocatalysts and supercapacitors based on DP oxides have been summarized in Tables 5 and 6 for comparison.117,162–166 Despite these inspiring achievements, developing high-performance and low-cost OER electrocatalysts and high energy density supercapacitors remains a great challenge. Below, we discuss the opportunities and perspectives for future studies.
Table 5 Summary of recently reported OER electrocatalysts based on DP oxides
| Catalysts |
Synthesis method |
Applications (electrolyte) |
Onset@0.3 mA cm−2 (mV vs. NHE) |
η (@10 mA cm−2 (mV) |
Tafel slope (dec mV−1) |
Main findings |
Ref. |
| La2Co0.5Fe0.5MnO6−δ |
Molten-salt method |
OER (1 M KOH) |
N/A |
400 |
|
Remarkable electrocatalytic activity towards OER46 |
46 |
| La2MnNiO6 (nanoparticles) |
Sol–gel method |
OER (0.5 M KOH) |
65 |
440 |
120 |
Retain approximately 70% of their catalytic efficiency after62 100 cycles |
62 |
| La2MnNiO6 (nanoparticles annealed at 700 °C) |
Sol–gel method |
OER (1 M KOH) |
N/A |
370 |
58 |
La2NiMnO6 nanoparticles exhibit superior OER catalytic performance with higher current density and lower Tafel slope than its bulk counterpart103 |
103 |
| La2MnNiO6 (nanoparticles annealed at 900 °C) |
430 |
73 |
| La2MnNiO6 (nanoparticles annealed at 1300 °C) |
470 |
91 |
| BaPrCo2O6 |
Glycerol nitrate method |
OER (1 M KOH) |
296 |
480 |
96 |
Highly active Co-based DP oxide125 |
125 |
| BaPrCo1.4Fe0.6O6 |
OER (1 M KOH) |
254 |
430 |
92 |
| BaGdCo1.8Fe0.2O6 |
OER (1 M KOH) |
373 |
477 |
60.1 |
| Sr2Fe0.8Co0.2Mo0.6Co0.4O6−δ |
Sol–gel method |
OER (0.1 M KOH) |
283 |
345 |
132 |
Significantly enhanced OER activity in alkaline media compared with the host material126 |
126 |
| PrBaCo2O6−δ |
Sol–gel method |
OER (1 M KOH) |
|
360 |
70 |
Role in the oxygen vacancy content on the OER activity162 |
162 |
Table 6 Summary of recently reported electrochemical performance of supercapacitors based on different DP oxides
| Electrode material |
Synthesis method |
Specific capacitance (F g−1) /electrolyte |
Energy density (W h kg−1) |
Power density (W kg−1) |
Cycling stability |
Ref. |
| Pr0.95Ba1.05Co2O6−δ |
Solid-state reaction method |
235.66 @ 25 mV s−1, 6 M KOH |
N/A |
N/A |
N/A |
37 |
| Pr0.90Ba1.10Co2O6−δ |
598.40 @ 25 mV s−1, 6 M KOH |
N/A |
N/A |
| Pr0.80Ba1.20Co2O6−δ |
408.37 @ 25 mV s−1, 6 M KOH |
N/A |
N/A |
| La2CuMnO6 |
Sol–gel method |
718.25 @ 3.12 A g−1, 0.5 M KOH |
72.6 |
113.4 |
N/A |
38 |
| La2CoMnO6 |
Combustion method |
0.013 @ 0.44 mA g−1, 1 M Na2SO4 |
0.0018 |
0.2238 |
84% cyclic stability after 500 cycles |
50 |
| Sr2CoMoO6-δ |
Sol–gel method |
747 @ 1 A g−1, 6 M KOH |
64 |
855 |
125% retention after 5000 GCD cycles at 10 A g−1 |
59 |
| Pr2CrMnO6 |
Sol–gel method |
177.4 @ 2 A g−1, 2 M KOH |
∼23.3 |
∼224.9 |
∼87% retention after 5000 GCD cycles at 100 mV s−1 scan rate |
60 |
| Y2CoMnO6 (microspheres) |
Hydrothermal process |
148.0 @ 0.50 A g−1, 6 M KOH |
N/A |
N/A |
85% retention after 10000 GCD cycles at 10 A g−1 |
63 |
| Y2NiMnO6 (nanowires) |
Hydrothermal process |
77.76 @ 30 mA g−1, 0.5 M KOH |
0.8 |
19.27 @ 150 mA g−1 |
70.17% retention after 1800 GCD cycles at 150 mA g−1 |
64 |
| La2NiCrO6 (nanorods) |
Solvothermal synthesis |
635.5 @ 1 A g−1, 0.5 M KOH |
17.8 |
229.8 |
∼75% retention after 5000 GCD cycles at 5 A g−1 |
66 |
| La2CuMnO6 (nanostructure) |
Hydrothermal process |
205.5 @ 0.25 A g−1, 2 M KOH |
N/A |
N/A |
∼78% retention after 1000 GCD at 1.50 A g−1 |
68 |
| La2CrMnO6 (mesoporous spheres) |
Hydrothermal process |
1416 @ 1 A g−1 |
241.1 |
350 |
∼42% retention after 5000 cycles at 12 A g−1 |
69 |
| LaCaCoCrO6 |
Hydrothermal process |
511 @ 1 A g−1, 6 M KOH |
3.21 @ 2 A g−1 |
449.99 @ 2 A g−1 |
∼75% retention after 3000 cycles at 12 A g−1 |
70 |
| La2CoNiO6 (nanofibers) |
Electrostatic spinning process |
335 @ 0.25 A g−1, 0.5 M KOH |
N/A |
N/A |
∼70% retention after 1000 GCD cycles at 1 A g−1 |
74 |
| La2CoMnO6 (nanofiber) |
Electrostatic spinning process |
109.7 @ 0.5 A g−1, 6 M KOH |
N/A |
N/A |
90.9% retention after 1000 GCD cycles at 1 A g−1 |
75 |
| Ba2Bi0.1Sc0.2Co1.7O6−δ |
EDTA–citric acid method |
1050 @ 1 A g−1, 6 M KOH |
58.8 |
362.8 |
92.9% retention after 5000 GCD cycles at 1 A g−1 |
119 |
| Ba2FeCoO6−δ |
Wet chemical method |
820 @ 3 A g−1, 2 M KOH |
23.06 |
677.92 |
N/A |
120 |
| La2FeCoO6 |
Sol–gel method |
831.1 @ 1 A g−1, 2 M KOH |
∼23.3 |
∼224.9 |
∼88% retention after 1000 GCD cycles at 6 A g−1 |
121 |
| Gd2NiMnO6 |
Wet chemical method |
400.46 @ 1 A g−1, 4 M KOH |
20.23 |
421.75 |
N/A |
123 |
| Sr2CoMo1−xNixO6−δ |
EDTA–citric acid combined with sol–gel |
930 @ 1 A g−1, 6 M KOH |
N/A |
N/A |
74.28% retention after 3000 GCD cycles at 1 A g−1 |
152 |
| Y2CoNiO6 |
Sol–gel method |
711 @ 1 A g−1 |
19.97 |
1324 |
∼98.5% retention after 1200 cycles at 6 A g−1 |
163 |
| La2CoMnO6 (hollow spherical porous) |
Template impregnation method |
376 @ 1 A g−1 |
65.8 |
1000 |
89.2% retention after 3000 cycles at 3 A g−1 |
164 |
| PrBaMn2O6−δ |
DTA–CA) complexing approach |
1034.8 @1 A g−1, 6 M KOH |
N/A |
N/A |
92.84% retention after 5000 GCD at 1 A g−1 |
117 |
| La2ZnMnO6 (nanoflakes) |
Hydrothermal process |
515.5@ 0.5 A/g, 2 M KOH |
N/A |
N/A |
86% retention after 1000 GCD at 2.5 A g−1 |
165 |
| La2CoMnO6 |
Template impregnation method |
376 @ 1 A g−1, 1 M Na2SO4 |
65.8 |
1000 |
98.2% retention after 3000 GCD cycles at 5 A g−1 |
164 |
| Y2CuMnO6 |
Wet chemical method |
15.6 @ 0.2 A g−1, 1 M KOH |
0.43 |
∼56.2 |
75% retention after 1000 GCD at 0.5 A g−1 |
166 |
8.1. Exploiting new DP oxides with high OER activity and long-term stability
The exploration of new materials based on DP structure with enhanced electrocatalytic performance is in strong demand. To improve the OER activity, several strategies such as A, B and B′-site cation doping or defect creation, and creation of oxygen vacancies have been proposed and investigated by researchers. The presence of oxygen vacancies has been proved to enhance the electronic conductivity of DP oxides via both experimental and theoretical routes, and promote charge-transfer, thus improving OER activity.134,137 Many methods (e.g., thermal treatment, cation/anion doping, reduction processing, plasma, flame, and laser technology) have been successfully adopted to generate oxygen vacancies, but how to generate oxygen vacancies in a controllable manner remains a great challenge. Recently considerable advancement on DP oxides as OER electrocatalysts has been achieved by researchers, even though the as-reported DP oxides are a long way off from commercial application. The low electronic conductivity and long-term stability are critical concerns for DP oxides. Therefore, more efforts should be made for improving their electronic conductivity or preparing hybrids between DP oxides and other highly conductive materials. A solution for minimizing the stability issues is the construction of DP structure with special morphology, introduction of dopants, etc. For the efficient transport of electrons and ions and also for the accommodation of the reactants and the reaction intermediates on the surface of the perovskite oxide, the development of 3D structures and nanoarrays with considerable interconnected channels is required. The stability of the OER electrocatalysts based on DP oxides is essential for practical applications of energy devices. Although previous studies have demonstrated that oxygen vacancies can effectively enhance the OER activity, the stability of oxygen vacancies themselves is elusive in many cases. Based on previous research, oxygen was intercalated into oxygen vacancies of some DP oxides in alkaline solution.
Therefore, it is necessary to understand how oxygen vacancy concentration changes over time under the OER process and how does it impact the activity. To accomplish this goal, accurate and advanced in situ technologies along with comprehensive theoretical calculation should be developed for attaining more information regarding the active sites and intermediates, which are beneficial to understanding the mechanism of the OER and guiding the rational design of efficient OER electrocatalysts. In the past years, significant advances in in situ TEM and spectroscopic techniques (e.g., in situ Fourier-transform infrared spectroscopy and in situ Raman spectroscopy, and in situ XAS) as well as time-resolved measurements have shown promise in providing in-depth insight into electrocatalytic behavior. These in situ characterization techniques could offer real-time information on potential structural changes, chemical states, and reaction mechanisms under varying electrochemical conditions.167–171 The results will be helpful to improve the performance and stability of electrocatalysts and binding trends along with parameters for databases. Ongoing efforts to gather data from numerous experiments have enhanced theoretical calculations too.172 Thus, combinations of advanced in situ/operando techniques and theoretical calculations can greatly promote the design of more promising OER electrocatalysts based on DP oxides.
8.2. Exploiting new high energy density supercapacitors
Since the past decades, DP oxides have gained much attention in the applications of high energy density supercapacitors due to their pseudocapacitive behavior. To improve the electrochemical performance of DP oxides for the forthcoming applications, there is still much work to be done. Investigating novel DP oxides is crucial for improving the energy storage performance metrics, including power conversion efficiency, specific capacity, cycle life, and energy/power densities. The multi-dimensional (0D, 1D, 2D, and 3D) DP oxides are the subsets of perovskite materials that have demonstrated significant potential for energy storage and harvesting devices.173 However, currently the available DP oxides are not enough for commercial level applications. The electrochemical performance of DP oxides can be improved by tuning their morphologies and particle sizes of the electrode materials. In addition, A-site and B-site engineering and substitution with new elements can also enhance the electrochemical performance. They could reveal rich active sites, modulate the charging kinetics and enhance supercapacitor performances. In supercapacitors, it is very complicated to investigate their reaction kinetics, especially when several types of interactions are involved. Over the years, extensive research has been performed on DP oxides to explore the high oxygen surface exchange kinetics, fast oxygen ion diffusion rates, and excellent mixed ionic and electronic conductivity at high temperatures. However, the accurate mechanisms of electrochemical storage in DP oxide-based supercapacitors are still ambiguous. Thus, this is one of the issues that need to be addressed in future research. The recently developed advanced characterization techniques such as Cs-corrected TEM and in situ spectroscopic techniques now can ascertain the specific atomic structural changes of metal oxide electrodes, especially in the research of catalysts and batteries. These advanced structural characterization techniques can be applied to in situ investigate the strains generated in DP oxides during charging and discharging processes, which induces capacitance attenuation. Nevertheless, it has seldom been reported, due to the limitations of advanced characterization. Finally, the research area of employing DP oxides as pseudocapacitive materials has to be more deeply investigated as it displays very promising results for future ultra-energy supercapacitors.
Data availability
No primary research results, software or code has been included and no new data were generated or analyzed as part of this review.
Author contributions
L. D. Chen: investigation, conceptualization, draft writing, review, and editing. J. Ding: investigation, and draft review. X. H. Zhu: conceptualization, supervision, review, editing, and funding acquisition.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (granted no. 11974170) for the present work.
References
- S. Höysniemi, Energy Res. Soc. Sci., 2022, 93, 102840 CrossRef.
- S. Li and Y. C. Chang, Energy Strat. Rev., 2021, 38, 100700 CrossRef.
- J. Song, C. Wei, Z. Huang, C. Liu, L. Zeng, X. Wang and Z. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- J. O. G. Posada, A. J. R. Rennie, S. P. Villar, V. L. Martins, J. Marinaccio, A. Barnes, C. F. Glover, D. A. Worsley and P. J. Hall, Renewable Sustainable Energy Rev., 2017, 68, 1174–1182 CrossRef CAS.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- E. Zhu, X. Yan, S. Wang, M. Xu, C. Wang, H. Liu, J. Huang, W. Xue, J. Cai, H. Heinz, Y. Li and Y. Huang, Nano Lett., 2019, 19, 3730–3736 CrossRef CAS PubMed.
- Y. Heo, S. Choi, J. Bak, H.-S. Kim, H. B. Bae and S.-Y. Chung, Adv. Energy Mater., 2018, 8, 1802481 CrossRef.
- R. Zhang, L. Wang, Y.-H. Ma, L. Pan, R. Gao, K. Li, X. Zhang and J.-J. Zou, J. Mater. Chem. A, 2019, 7, 10010–10018 RSC.
- S. Mondal, S. K. De, R. Jana, A. Roy, M. Mukherjee, A. Datta, B. Satpati and D. Senapati, ACS Appl. Energy Mater., 2021, 4, 3017–3032 CrossRef CAS.
- X. Liu, L. Zhang, M. Li and X. Hu, Chem. Commun., 2018, 54, 2502–2505 RSC.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J. Xie, Y. Gao, G. Chen, Y. Wang, J. Yu, F. Ciucci, D. Chen and Z. Shao, Small, 2022, 18, 2204109 CrossRef CAS PubMed.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- K. I. Kobayashi, T. Kimura, H. Sawada, K. Terakura and Y. Tokura, Nature, 1998, 395, 677–680 CrossRef CAS.
- M. García-Hernández, J. L. Martínez, M. J. Martínez-Lope, M. T. Casais and J. A. Alonso, Phys. Rev. Lett., 2001, 86, 2443–2446 CrossRef PubMed.
- H. Sun, Z. Hu, X. Xu, J. He, J. Dai, H.-J. Lin, T.-S. Chan, C.-T. Chen, L. H. Tjeng, W. Zhou and Z. Shao, Chem. Mater., 2019, 31, 5919–5926 CrossRef CAS.
- A. Banerjee, M. K. Awasthi, P. Maji, M. Pal, S. T. Aziz, G. K. Lahiri and A. Dutta, ChemElectroChem, 2023, 10, e202201098 CrossRef CAS.
- A. K. Tomar, A. Joshi, G. Singh and R. K. Sharma, Coord. Chem. Rev., 2021, 431, 213680 CrossRef CAS.
- A. Chroneos, R. V. Vovk, I. L. Goulatis and L. I. Goulatis, J. Alloys Compd., 2010, 494, 190–195 CrossRef CAS.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- X. Xu, Y. Zhong and Z. Shao, Trends Chem., 2019, 1, 410–424 CrossRef CAS.
- H.-S. Nan, X.-Y. Hu and H.-W. Tian, Mater. Sci. Semicond. Process., 2019, 94, 35–50 CrossRef CAS.
- Y. Liu, S. P. Jiang and Z. Shao, Mater. Today Adv., 2020, 7, 100072 CrossRef.
- M. Mohan, N. P. Shetti and T. M. Aminabhavi, J. Power Sources, 2023, 574, 233166 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- M. M. Omran, A. I. Abdel-Salam, D. Aman and S. G. Mohamed, in Pseudocapacitors: Fundamentals to High Performance Energy Storage Devices, ed. R. K. Gupta, Springer Nature Switzerland, Cham, 2024, pp. 133–155 Search PubMed.
- M. T. Anderson, K. B. Greenwood, G. A. Taylor and K. R. Poeppelmeier, Prog. Solid State Chem., 1993, 22, 197–233 CrossRef CAS.
- M. W. Lufaso, P. W. Barnes and P. M. Woodward, Acta Crystallogr., Sect. B:Struct. Sci., 2006, 62, 397–410 CrossRef PubMed.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B:Struct. Sci., 2001, 57, 725–738 CrossRef CAS PubMed.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- W. Pies and A. Weiss, in Crystal Structure Data of Inorganic Compounds, Numerical Data and Functional Relationships in Science and Technology Group III: Crystal and Solid State Physics, ed. K.-H. Hellwege and A. M. Hellwege, Springer-Verlag, Berlin-Heidelberg, New York, 1976, vol. 7 Search PubMed.
- A. S. Bangwal, M. Chauhan and P. Singh, Int. J. Hydrogen Energy, 2022, 47, 12582–12591 CrossRef CAS.
- A. M. Idris, T. Liu, J. H. Shah, X. Zhang, C. Ma, A. S. Malik, A. Jin, S. Rasheed, Y. Sun, C. Li and H. Han, Sol. RRL, 2020, 4, 1900456 CrossRef CAS.
- Q. Tang and X. Zhu, J. Mater. Chem. C, 2022, 10, 15301–15338 RSC.
- C. Li, Z. Yu, H. Liu and K. Chen, J. Phys. Chem. Solids, 2018, 113, 151–156 CrossRef CAS.
- L. C. C. B. Oliveira, R. Venâncio, P. V. F. de Azevedo, C. G. Anchieta, T. C. M. Nepel, C. B. Rodella, H. Zanin and G. Doubek, J. Energy Chem., 2023, 81, 1–19 CrossRef.
- D. N. Singh, T. P. Sinha and D. K. Mahato, Mater. Today: Proc., 2017, 4, 5640–5646 Search PubMed.
- D. Liu, M. Chen, X. Du, H. Ai, K. H. Lo, S. Wang, S. Chen, G. Xing, X. Wang and H. Pan, Adv. Funct. Mater., 2021, 31, 2008983 CrossRef CAS.
- S. K. Gupta and Y. Mao, Prog. Mater. Sci., 2021, 117, 100734 CrossRef CAS.
- J. Ahmed, T. Ahamad, R. M. Alotaibi, N. Alhokbany and S. M. Alshehri, Ceram. Int., 2023, 49, 18818–18824 CrossRef CAS.
- T. Li, W. Guo and Q. Shi, Int. J. Electrochem. Sci., 2023, 18, 100103 CrossRef CAS.
- M. A. Bavio, J. E. Tasca, G. G. Acosta, M. F. Ponce, R. O. Fuentes and A. Visintin, J. Solid State Electrochem., 2020, 24, 699–710 CrossRef CAS.
- A. Shereef, P. A. Aleena, J. Kunjumon, A. K. Jose, S. A. Thomas, M. Tomy, T. S. Xavier, S. Hussain and D. Sajan, Mater. Sci. Eng., B, 2023, 289, 116262 CrossRef CAS.
- A. P. Khandale, R. V. Kumar and S. S. Bhoga, Bull. Mater. Sci., 2023, 46, 119 CrossRef CAS.
- K. Yamaura, J. Solid State Chem., 2016, 236, 45–54 CrossRef CAS.
- X.-Y. Wang, C. Avendaño and K. R. Dunbar, Chem. Soc. Rev., 2011, 40, 3213–3238 RSC.
- J. Chen, H. L. Feng and K. Yamaura, Mater. Today Phys., 2024, 40, 101302 CrossRef CAS.
- Y. Krockenberger, K. Mogare, M. Reehuis, M. Tovar, M. Jansen, G. Vaitheeswaran, V. Kanchana, F. Bultmark, A. Delin, F. Wilhelm, A. Rogalev, A. Winkler and L. Alff, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 020404 CrossRef.
- A. K. Paul, M. Reehuis, V. Ksenofontov, B. Yan, A. Hoser, D. M. Többens, P. M. Abdala, P. Adler, M. Jansen and C. Felser, Phys. Rev. Lett., 2013, 111, 167205 CrossRef PubMed.
- H. L. Feng, M. Arai, Y. Matsushita, Y. Tsujimoto, Y. Guo, C. I. Sathish, X. Wang, Y.-H. Yuan, M. Tanaka and K. Yamaura, J. Am. Chem. Soc., 2014, 136, 3326–3329 CrossRef CAS PubMed.
- R. Morrow, R. Mishra, O. D. Restrepo, M. R. Ball, W. Windl, S. Wurmehl, U. Stockert, B. Büchner and P. M. Woodward, J. Am. Chem. Soc., 2013, 135, 18824–18830 CrossRef CAS PubMed.
- D.-H. Chen and X.-R. He, Mater. Res. Bull., 2001, 36, 1369–1377 CrossRef CAS.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- A. K. Tomar, A. Joshi, S. Atri, G. Singh and R. K. Sharma, ACS Appl. Mater. Interfaces, 2020, 12, 15128–15137 CrossRef CAS PubMed.
- R. K. Muddelwar, J. Pani, A. B. Lad, K. U. Kumar, V. M. Gaikwad and H. Borkar, Mater. Chem. Phys., 2023, 302, 127726 CrossRef CAS.
- I. Ajin, R. Balamurugan and A. Chandra Bose, ACS Appl. Energy Mater., 2023, 6, 9764–9777 CrossRef CAS.
- J. Ahmed, N. Alhokbany, M. Ubaidullah, S. Mutehri, M. A. M. Khan and S. M. Alshehri, Ceram. Int., 2020, 46, 20038–20044 CrossRef CAS.
- M. Devi, D. Nagpal, A. Vasishth, A. Kumar and A. Kumar, Phys. Status Solidi A, 2023, 220, 2200444 CrossRef CAS.
- M. Alam, K. Karmakar, M. Pal and K. Mandal, RSC Adv., 2016, 6, 114722–114726 RSC.
- S. C. McGuire, W. Wesley, K. Sasaki, X. Tong and S. S. Wong, ACS Appl. Mater. Interfaces, 2022, 14, 30914–30926 CrossRef CAS PubMed.
- J. Singh and A. Kumar, J. Nanopart. Res., 2021, 23, 208 CrossRef CAS.
- S. C. McGuire, C. Koenigsmann, C. C. Chou, X. Tong and S. S. Wong, Catal. Sci. Technol., 2022, 12, 613–629 RSC.
- J. Singh, U. K. Goutam and A. Kumar, Solid State Sci., 2019, 95, 105927 CrossRef CAS.
- A. Singh, A. Vasishth and A. Kumar, Phys. Status Solidi A, 2023, 220, 2300198 CrossRef CAS.
- D. Nagpal, A. Singh, A. Vasishth, R. Singh and A. Kumar, Phys. Status Solidi A, 2023, 220, 2300495 CrossRef CAS.
- A. Faik, J. M. Igartua, G. J. Cuello, F. Jiménez-Villacorta, G. R. Castro and L. Lezama, J. Mol. Struct., 2009, 933, 53–62 CrossRef CAS.
- A. K. Azad, S. G. Eriksson, A. Khan, A. Eriksson and M. Tseggai, J. Solid State Chem., 2006, 179, 1303–1311 CrossRef CAS.
- M. Cheriti and A. Kahoul, Mater. Res. Bull., 2012, 47, 135–141 CrossRef CAS.
- Y.-B. Wu, J. Bi and B.-B. Wei, Acta Phys.-Chim. Sin., 2015, 31, 315–321 CAS.
- J. Fu, H.-Y. Zhao, J.-R. Wang, Y. Shen and M. Liu, Int. J. Miner., Metall. Mater., 2018, 25, 950–956 CrossRef CAS.
- Z.-F. Huang, J. Wang, Y. Peng, C.-Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- P. K. Shen, C. Y. Wang, S. P. Jiang, X. Sun and J. Zhang, Electrochemical Energy: Advanced Materials and Technologies, CRC Press, Taylor & Francis Group, Boca Raton, 1st edn, 2016 Search PubMed.
- K. Fominykh, J. M. Feckl, J. Sicklinger, M. Döblinger, S. Böcklein, J. Ziegler, L. Peter, J. Rathousky, E.-W. Scheidt, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2014, 24, 3123–3129 CrossRef CAS.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- A. Vazhayil, L. Vazhayal, J. Thomas, S. Ashok C and N. Thomas, Appl. Surf. Sci. Adv., 2021, 6, 100184 CrossRef.
- D. C. Grahame, J. Am. Chem. Soc., 1941, 63, 1207–1215 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- H. Shao, Z. Lin, K. Xu, P.-L. Taberna and P. Simon, Energy Storage Mater., 2019, 18, 456–461 CrossRef.
- A. K. Tomar, R. B. Marichi, G. Singh and R. K. Sharma, Electrochim. Acta, 2019, 296, 120–129 CrossRef CAS.
- A. Das, M. Ojha, P. Subramanyam and M. Deepa, Nanoscale Adv., 2020, 2, 2925–2942 RSC.
- R. Majee, Q. A. Islam, S. Mondal and S. Bhattacharyya, Chem. Sci., 2020, 11, 10180–10189 RSC.
- Q. A. Islam, R. Majee and S. Bhattacharyya, J. Mater. Chem. A, 2019, 7, 19453–19464 RSC.
- A. Mandal, A. Ghosh, D. Ghosh and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2021, 13, 43104–43114 CrossRef CAS PubMed.
- A. Ghosh, D. K. Chaudhary, A. Mandal, S. Prodhan, K. K. Chauhan, S. Vihari, G. Gupta, P. K. Datta and S. Bhattacharyya, J. Phys. Chem. Lett., 2020, 11, 591–600 CrossRef CAS PubMed.
- A. Sadhu and S. Bhattacharyya, J. Phys. Chem. C, 2013, 117, 26351–26360 CrossRef CAS.
- N. S. Gajbhiye and S. Bhattacharyya, Nanotechnology, 2005, 16, 2012 CrossRef CAS PubMed.
- B. Debnath, S. Parvin, H. Dixit and S. Bhattacharyya, ChemSusChem, 2020, 13, 3875–3886 CrossRef CAS PubMed.
- J. H. K. Pfisterer, Y. Liang, O. Schneider and A. S. Bandarenka, Nature, 2017, 549, 74–77 CrossRef CAS PubMed.
- C. H. M. van Oversteeg, H. Q. Doan, F. M. F. de Groot and T. Cuk, Chem. Soc. Rev., 2017, 46, 102–125 RSC.
- K. Schweinar, R. L. Nicholls, C. R. Rajamathi, P. Zeller, M. Amati, L. Gregoratti, D. Raabe, M. Greiner, B. Gault and O. Kasian, J. Mater. Chem. A, 2020, 8, 388–400 RSC.
- C. L. Bentley, M. Kang and P. R. Unwin, J. Am. Chem. Soc., 2019, 141, 2179–2193 CrossRef CAS PubMed.
- H. Sun, G. Chen, J. Sunarso, J. Dai, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 16939–16942 CrossRef CAS PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- S. Tao and J. T. S. Irvine, Nat. Mater., 2003, 2, 320–323 CrossRef CAS PubMed.
- Y. Tong, J. Wu, P. Chen, H. Liu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2018, 140, 11165–11169 CrossRef CAS PubMed.
- I. Rodríguez-García, J. L. Gómez de la Fuente, D. Galyamin, Á. Tolosana-Moranchel, P. Kayser, M. A. Salam, J. A. Alonso, F. Calle-Vallejo, S. Rojas and M. Retuerto, J. Mater. Chem. A, 2024, 12, 16854–16862 RSC.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- D. Galyamin, J. Torrero, I. Rodríguez, M. J. Kolb, P. Ferrer, L. Pascual, M. A. Salam, D. Gianolio, V. Celorrio, M. Mokhtar, D. Garcia Sanchez, A. S. Gago, K. A. Friedrich, M. A. Peña, J. A. Alonso, F. Calle-Vallejo, M. Retuerto and S. Rojas, Nat. Commun., 2023, 14, 2010 CrossRef CAS PubMed.
- M. A. Hubert, A. M. Patel, A. Gallo, Y. Liu, E. Valle, M. Ben-Naim, J. Sanchez, D. Sokaras, R. Sinclair, J. K. Nørskov, L. A. King, M. Bajdich and T. F. Jaramillo, ACS Catal., 2020, 10, 12182–12196 CrossRef CAS.
- G. Oroumi, R. Monsef, E. A. Dawi, A. M. Aljeboree, M. H. S. Alubiady, A. M. Al-Ani and M. Salavati-Niasari, J. Energy Storage, 2024, 85, 111161 CrossRef.
- B. Y. Hussein and A. M. Mohammed, Mater. Today: Proc., 2021, 42, A18–A26 CAS.
- S. Choi, S. Yoo, J. Kim, S. Park, A. Jun, S. Sengodan, J. Kim, J. Shin, H. Y. Jeong, Y. Choi, G. Kim and M. Liu, Sci. Rep., 2013, 3, 2426 CrossRef PubMed.
- H. Zhang, D. Guan, X. Gao, J. Yu, G. Chen, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 19228–19233 RSC.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- F. Gunkel, L. Jin, D. N. Mueller, C. Hausner, D. S. Bick, C.-L. Jia, T. Schneller, I. Valov, R. Waser and R. Dittmann, ACS Catal., 2017, 7, 7029–7037 CrossRef CAS.
- Z. Yuan, J. Liu, C. L. Chen, C. H. Wang, X. G. Luo, X. H. Chen, G. T. Kim, D. X. Huang, S. S. Wang, A. J. Jacobson and W. Donner, Appl. Phys. Lett., 2007, 90, 212111 CrossRef.
- M. Liu, J. Liu, G. Collins, C. R. Ma, C. L. Chen, J. He, J. C. Jiang, E. I. Meletis, A. J. Jacobson and Q. Y. Zhang, Appl. Phys. Lett., 2010, 96, 132106 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- Y. Liu, Z. Wang, J.-P. M. Veder, Z. Xu, Y. Zhong, W. Zhou, M. O. Tade, S. Wang and Z. Shao, Adv. Energy Mater., 2018, 8, 1702604 CrossRef.
- Z. Wang, Y. Liu, Y. Chen, L. Yang, Y. Wang and M. Wei, J. Alloys Compd., 2019, 810, 151830 CrossRef CAS.
- Z. Xu, Y. Liu, W. Zhou, M. O. Tade and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 9415–9423 CrossRef CAS PubMed.
- A. Kumar and A. Kumar, Ceram. Int., 2019, 45, 14105–14110 CrossRef CAS.
- J. Singh and A. Kumar, Mater. Sci. Semicond. Process., 2019, 99, 8–13 CrossRef CAS.
- M. A. Bavio, J. E. Tasca, G. G. Acosta and A. E. Lavat, Rev. Mater., 2018, 23, 12132 Search PubMed.
- A. Kumar, A. Kumar and A. Kumar, Solid State Sci., 2020, 105, 106252 CrossRef CAS.
- R. K. Muddelwar, J. Pani, A. B. Lad, H. Borkar and V. M. Gaikwad, J. Solid State Chem., 2024, 329, 124455 CrossRef CAS.
- H. Andersen, K. Xu, D. Malyshkin, R. Strandbakke and A. Chatzitakis, Chem. Commun., 2020, 56, 1030–1033 RSC.
- H. Sun, X. Xu, G. Chen, Y. Zhou, H.-J. Lin, C.-T. Chen, R. Ran, W. Zhou and Z. Shao, ChemSusChem, 2019, 12, 5111–5116 CrossRef CAS PubMed.
- H. Sun, X. Xu, Z. Hu, L. H. Tjeng, J. Zhao, Q. Zhang, H.-J. Lin, C.-T. Chen, T.-S. Chan, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 9924–9932 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- M. García-Mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116, 21077–21082 CrossRef.
- K. Zhu, T. Wu, M. Li, R. Lu, X. Zhu and W. Yang, J. Mater. Chem. A, 2017, 5, 19836–19845 RSC.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- S. Lee, G. Nam, J. Sun, J.-S. Lee, H.-W. Lee, W. Chen, J. Cho and Y. Cui, Angew. Chem., Int. Ed., 2016, 55, 8599–8604 CrossRef CAS PubMed.
- Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- Y. Liu, W. Wang, X. Xu, J.-P. Marcel Veder and Z. Shao, J. Mater. Chem. A, 2019, 7, 7280–7300 RSC.
- A. Sarkar and G. G. Khan, Nanoscale, 2019, 11, 3414–3444 RSC.
- J. Kim, X. Yin, K.-C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef.
- D. Chen, J. Wang, Z. Zhang, Z. Shao and F. Ciucci, Chem. Commun., 2016, 52, 10739–10742 RSC.
- H. A. Tahini, X. Tan, U. Schwingenschlögl and S. C. Smith, ACS Catal., 2016, 6, 5565–5570 CrossRef CAS.
- H. Sun, Y. Zhu and W. Jung, Molecules, 2021, 26, 5476 CrossRef CAS PubMed.
- H. Sun, J. Dai, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 10547–10567 CrossRef CAS.
- X. Huang, G. Zhao, G. Wang and J. T. S. Irvine, Chem. Sci., 2018, 9, 3623–3637 RSC.
- Y. Li, F. Chu, Y. Bu, Y. Kong, Y. Tao, X. Zhou, H. Yu, J. Yu, L. Tang and Y. Qin, Chem. Commun., 2019, 55, 7828–7831 RSC.
- C. Zhang, P. Zhao, S. Liu and K. Yu, Chin. J. Catal., 2019, 40, 1324–1338 CrossRef CAS.
- X. Mei, J. Xiong, Y. Wei, Y. Zhang, P. Zhang, Q. Yu, Z. Zhao and J. Liu, Appl. Catal., B, 2020, 275, 119108 CrossRef CAS.
- Y. Zhu, Z. He, Y. Choi, H. Chen, X. Li, B. Zhao, Y. Yu, H. Zhang, K. A. Stoerzinger, Z. Feng, Y. Chen and M. Liu, Nat. Commun., 2020, 11, 4299 CrossRef CAS PubMed.
- Y. Liu, Z. Wang, Y. Zhong, X. Xu, J.-P. M. Veder, M. R. Rowles, M. Saunders, R. Ran and Z. Shao, Chem. Eng. J., 2020, 390, 124645 CrossRef CAS.
- S. Zhong, Y. Chen, L. Yang, Y. Liu, X. Chen and C. Wang, Mater. Lett., 2021, 297, 130013 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- A. Tarancón, M. Burriel, J. Santiso, S. J. Skinner and J. A. Kilner, J. Mater. Chem., 2010, 20, 3799–3813 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- Q. Tao, P. Xu, M. Li and W. Lu, npj Comput. Mater., 2021, 7, 23 CrossRef.
- Z. Li, L. E. K. Achenie and H. Xin, ACS Catal., 2020, 10, 4377–4384 CrossRef CAS.
- S. Dimitrovska, S. Aleksovska and I. Kuzmanovski, Cent. Eur. J. Chem., 2005, 3, 198–215 Search PubMed.
- A. Talapatra, B. P. Uberuaga, C. R. Stanek and G. Pilania, Chem. Mater., 2021, 33, 845–858 CrossRef CAS.
- X. Miao, L. Wu, Y. Lin, X. Yuan, J. Zhao, W. Yan, S. Zhou and L. Shi, Chem. Commun., 2019, 55, 1442–1445 RSC.
- M. Ickler, M. Devi, I. Rogge, J. Singh and A. Kumar, J. Mater. Sci.: Mater. Electron., 2020, 31, 6977–6987 CrossRef CAS.
- Z. Meng, J. Xu, P. Yu, X. Hu, Y. Wu, Q. Zhang, Y. Li, L. Qiao, Y. Zeng and H. Tian, Chem. Eng. J., 2020, 400, 125966 CrossRef CAS.
- J. Singh, A. Kumar, U. K. Goutam and A. Kumar, Appl. Phys. A: Mater. Sci. Process., 2019, 126, 11 CrossRef.
- F. N. Mansoorie, J. Singh and A. Kumar, Mater. Sci. Semicond. Process., 2020, 107, 104826 CrossRef CAS.
- Z. Fan, L. Zhang, D. Baumann, L. Mei, Y. Yao, X. Duan, Y. Shi, J. Huang, Y. Huang and X. Duan, Adv. Mater., 2019, 31, 1900608 CrossRef PubMed.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, Nano-Micro Lett., 2019, 11, 47 CrossRef CAS PubMed.
- Y. Yang, Y. Wang, Y. Xiong, X. Huang, L. Shen, R. Huang, H. Wang, J. P. Pastore, S.-H. Yu, L. Xiao, J. D. Brock, L. Zhuang and H. D. Abruña, J. Am. Chem. Soc., 2019, 141, 1463–1466 CrossRef CAS PubMed.
- Y. Zhu, T.-R. Kuo, Y.-H. Li, M.-Y. Qi, G. Chen, J. Wang, Y.-J. Xu and H. M. Chen, Energy Environ. Sci., 2021, 14, 1928–1958 RSC.
- H. Sun and W. Zhou, Energy Fuels, 2021, 35, 5716–5737 CrossRef CAS.
- L. Wu, T. Guo and T. Li, iScience, 2021, 24, 102398 CrossRef CAS PubMed.
- P. Goel, S. Sundriyal, V. Shrivastav, S. Mishra, D. P. Dubal, K.-H. Kim and A. Deep, Nano Energy, 2021, 80, 105552 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*




 ap ×
ap ×  ap ×
ap ×  ap
ap ap ×
ap ×  ap ×
ap ×  ap
ap

 . The Faradaic efficiency can be calculated as follows,
. The Faradaic efficiency can be calculated as follows,

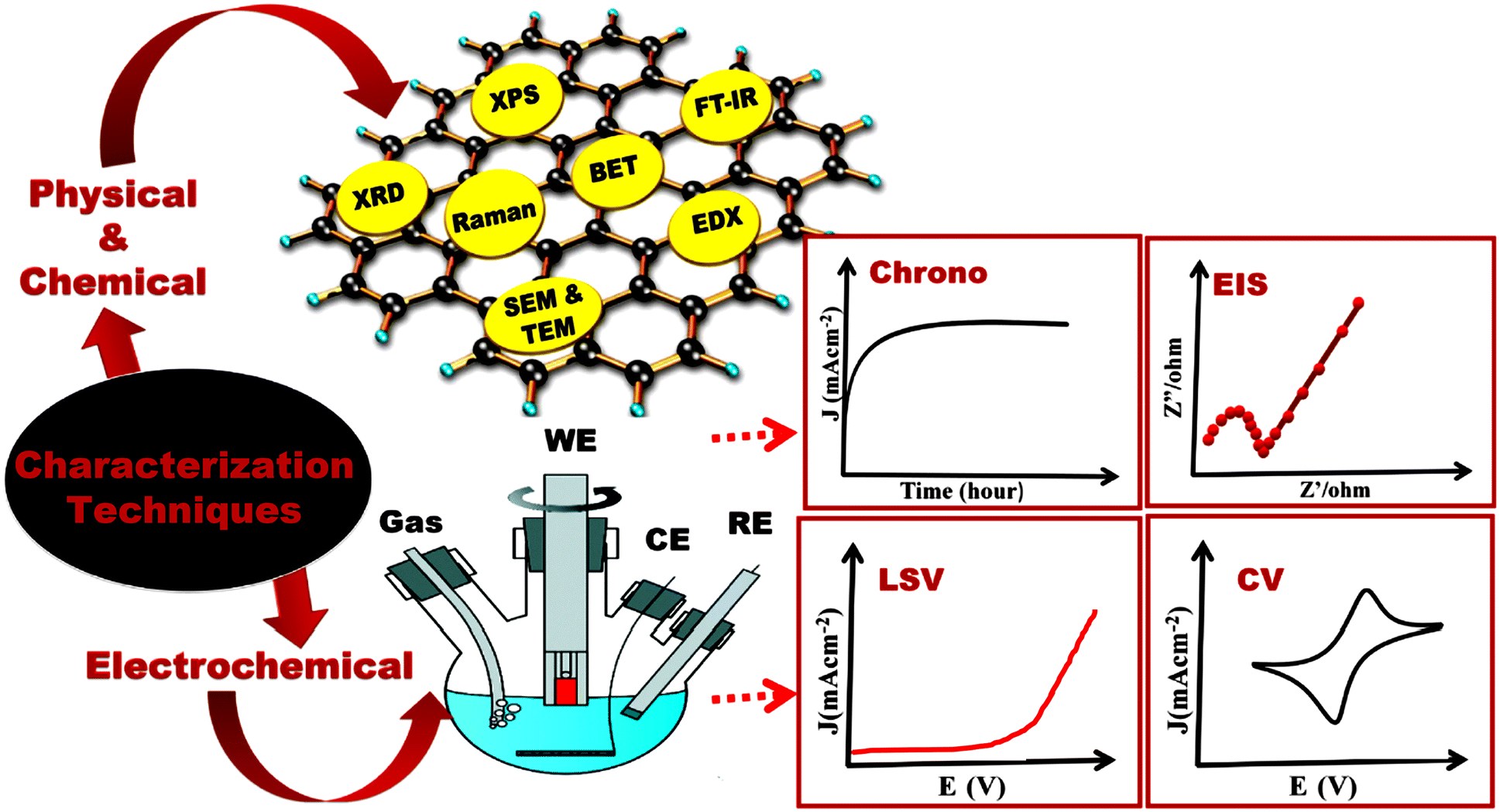
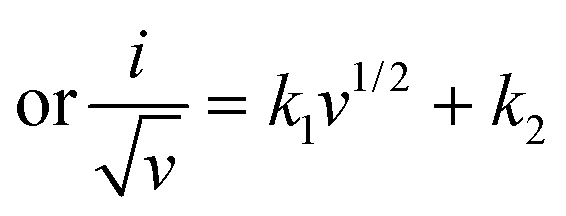
 and
and  , where k1 and k2 are the slope and intercept, respectively. By measuring CV at different scan rates and by plotting
, where k1 and k2 are the slope and intercept, respectively. By measuring CV at different scan rates and by plotting  vs.
vs.  (the square root of the scan rate), the values of k1 and k2 can be obtained from the slope and intercept, respectively. By substituting k1 and k2 into eqn (8) for a particular scan rate, the current contribution can be determined for the diverse potentials in CV. This method can also be used to understand how the kinetics changes at different scan rates, where in general, for slower scan rates the diffusion takes a control, while at higher scan rates, the capacitive contribution dominates the current kinetically.
(the square root of the scan rate), the values of k1 and k2 can be obtained from the slope and intercept, respectively. By substituting k1 and k2 into eqn (8) for a particular scan rate, the current contribution can be determined for the diverse potentials in CV. This method can also be used to understand how the kinetics changes at different scan rates, where in general, for slower scan rates the diffusion takes a control, while at higher scan rates, the capacitive contribution dominates the current kinetically.