
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comprehensive analysis from the basics to the application of V-cathodes in Zn–V static and flow batteries
Yufei Li†
,
Jie Chen†
 and
Guanjie He
*
and
Guanjie He
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk
First published on 11th April 2025
Abstract
Electrochemical energy storage devices using zinc anodes and aqueous solutions have the characteristics of low cost, easy manufacture, and intrinsic safety. As an important part of modern aqueous batteries, zinc batteries have attracted extensive attention in the academic community. Among them, vanadium-based materials have been widely gaining attention as cathode materials in static aqueous zinc-ion batteries owing to their multiple valence states and abundant resources. In addition, zinc–vanadium flow batteries using the vanadium electrolyte for energy storage have also been gradually developed, which further expanded the application of vanadium-based materials in aqueous zinc batteries. In this review, an overview of zinc–vanadium batteries (including static batteries and flow batteries) is briefly discussed, including their working mechanism, classification, structure, existing problems, and improvement strategies, for promoting further development of this field.
 Yufei Li | Yufei Li is currently an undergraduate student in the Department of Chemistry at University College London (UCL). She has done research on electrochemistry, macrocyclic compounds and synthesis of COF materials. Her research interests focus on electrocatalysis and battery materials in aqueous zinc-ion batteries. |
 Jie Chen | Jie Chen is currently a PhD student in the Department of Chemistry at University College London (UCL). He obtained his Bachelor's (2020) and Master's (2023) degrees in Physics from Lanzhou University and served as a visiting student at Fudan University. His research interests mainly focus on the interface between electrodes and electrolytes in zinc-ion batteries and interdisciplinary discussions between physics and electrochemistry. |
 Guanjie He | Guanjie He is an Associate Professor in the Department of Chemistry at University College London (UCL). He received his PhD degree from UCL in 2018 and visited Yale University during his PhD study. He has worked at Queen Mary University of London and the University of Lincoln. His research fields are mainly aqueous batteries, electrocatalytic materials and devices, advanced characterization, and simulation. He has received the EPSRC New Investigator Award, ERC Starting Grant and STFC Early Career Awards for his research on zinc-ion batteries. |
1. Introduction
With the increasing demand for energy, continuous consumption of non-renewable energy sources and increasing environmental pollution, the development and energy storage of renewable energy sources, such as solar, wind and tidal power, are attracting significant attention.1 Therefore, it is necessary to vigorously research and develop large-scale energy storage equipment. In recent years, traditional ion batteries, such as lithium-ion and sodium-ion batteries, have dominated the entire rechargeable battery market because of their mature large-scale manufacturing processes. Although lithium-ion and sodium-ion batteries are very successful in commercial sales, lithium-ion batteries have problems, such as safety and easy thermal runaway, and sodium-ion batteries offer relatively low energy density and poor cycling performance, which have limited the development of these two types of batteries and made them unsuitable for large-scale energy storage. On the contrary, the new zinc-ion battery is safe, cost-effective, and environmentally friendly, making it a strong competitor as a large-scale energy storage equipment.1The research on modern static water zinc-ion batteries can be traced back to 2012; Kang et al. constructed a secondary aqueous zinc-ion battery based on an alpha-MnO2 cathode and ZnSO4 aqueous electrolyte and revealed the intercalation/extraction mechanism of Zn2+ ions.2 Later in 2016, Nazar's team synthesized layered Zn0.25V2O5·nH2O for the first time and used it as a cathode material for aqueous zinc-ion batteries. Its specific capacity reached 300 mA h g−1, and the capacity retention rate was more than 80% after 1000 cycles, showing the huge application potential of vanadium oxide.1 Since then, manganese oxide and vanadium oxide have become the most typical cathode materials for static aqueous zinc-ion batteries.
In addition to zinc-ion static batteries, Zn flow batteries can also address the problem of large-scale energy storage, and their potential advantages are greater than those of static batteries, because the power and capacity of flow batteries can be configured independently; the power is determined by the number of stacks, and the capacity is determined by the volume of the electrolyte.3 This design makes them more flexible in large-scale and long-term energy storage scenarios. In the process of deep charging and discharging, the performance of flow batteries is more stable and suitable for long-term energy storage (more than 4 hours). At present, the commercial market circulates all-vanadium flow batteries and zinc–bromine flow batteries, but the development of these two flow batteries are limited owing to their low energy density and safety problems caused by electrolyte corrosion and growth of zinc dendrites. In 2012, the concept of Zn–V flow battery was first proposed, and V(IV)/V(V) and Zn2+/Zn redox couples were assembled into a flow battery, which broadened the application of vanadium-based materials in aqueous zinc batteries.3
In this review, zinc–vanadium batteries are separated into static batteries and flow batteries, as shown in Fig. 1. We first describe the different energy storage mechanisms of these two batteries, then introduce the existing problems of vanadium-based zinc-ion batteries and Zn–V flow batteries, and finally put forward some corresponding improvement strategies, and further prospect the future development.
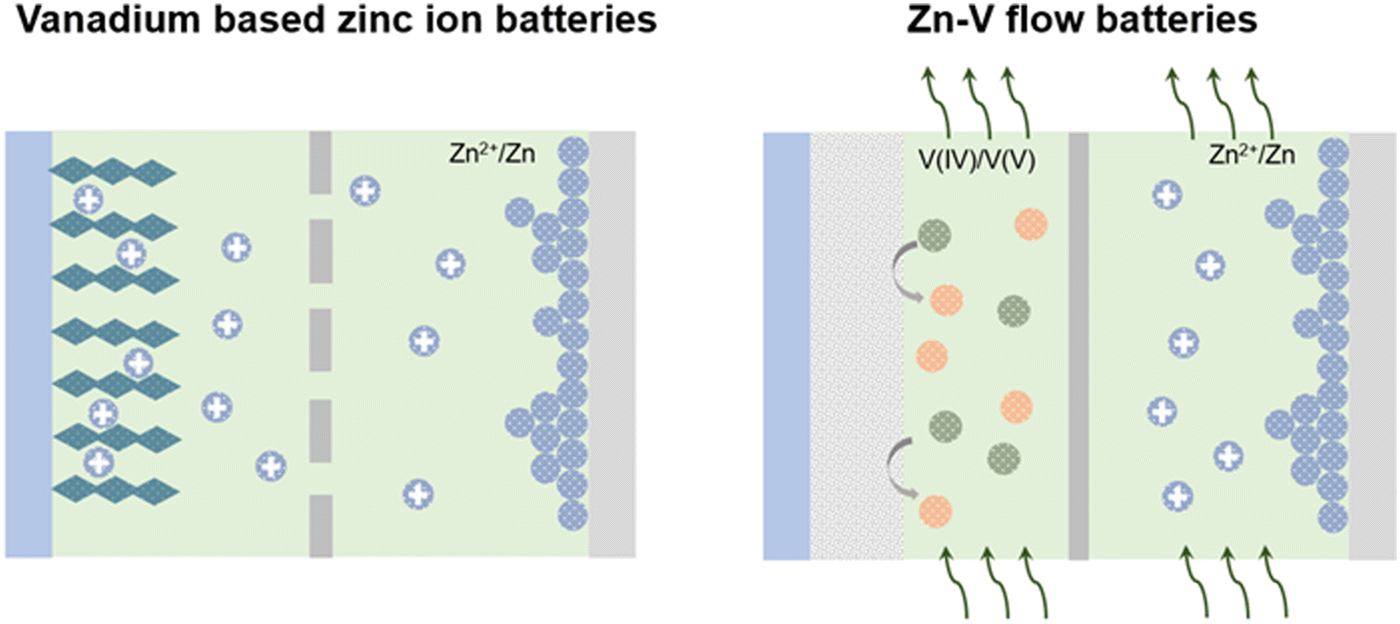 | ||
| Fig. 1 Schematic of the static Zn–V battery (vanadium-based zinc-ion batteries) and Zn–V flow battery. | ||
2. Zn–V static batteries
2.1. Mechanisms
 | ||
| Fig. 2 Schematic of (a)–(c) Structures of common vanadium oxides with the Zn2+ intercalation/extraction mechanism. (d) Zn2+ intercalation/extraction mechanism of V6O13.4 Copyright 2019, Wiley-VCH GmbH. (e) Zn2+ intercalation/extraction mechanism of V2O5.5 Copyright 2020, Elsevier. (f) General Zn2+ intercalation/extraction mechanism in static Zn–V batteries (vanadium-based zinc-ion batteries).6 Copyright 2021, Elsevier. | ||
In many studies, V2O5 has been used to explain the intercalation/extraction mechanism of Zn2+ ions because its layered structure and large laminar gap are conducive to the movement of Zn2+ ions, as shown in Fig. 2e, and it can attain a high theoretical capacity of 589 mA h g−1 for the whole charge–discharge process.2 The redox reactions of vanadium ions occur during the insertion and extraction of Zn2+. During discharging, Zn2+ ions embed between the layers of V2O5, and vanadium ions undergo reduction (V5+ to V4+ ions). As the concentration of intercalated Zn2+ ions increases, part of V4+ ions will continue to reduce to V3+ ions. During charging, an oxidation reaction occurs (V3+ to V4+ or V5+ ions), and the V2O5 structure detaches from the Zn2+ ions.3 The structural distortion of V2O5 due to the intercalation of Zn2+ ions will revert to the structure after the removal of Zn2+ ions, which is proven by the remained crystal structure and phase transition of the battery material.2 Therefore, the Zn2+ intercalation/extraction reaction is reversible, and the battery can continuously charge and discharge. In addition, the high structural stability of the cathode material makes the cycle life of the battery long.
 | ||
| Fig. 3 Schematic of (a) Structure of H–VO2. (b) Possible sites in H–VO2 for H+/Zn2+ co-intercalation/extraction. (c) Migration pathways for H+ (left) and Zn2+ (right) ions in H–VO2.8 Copyright 2020, Elsevier. (d) H+/Zn2+ co-intercalation/extraction mechanism in static Zn–V batteries (vanadium-based zinc-ion batteries).6 Copyright 2021, Elsevier. | ||
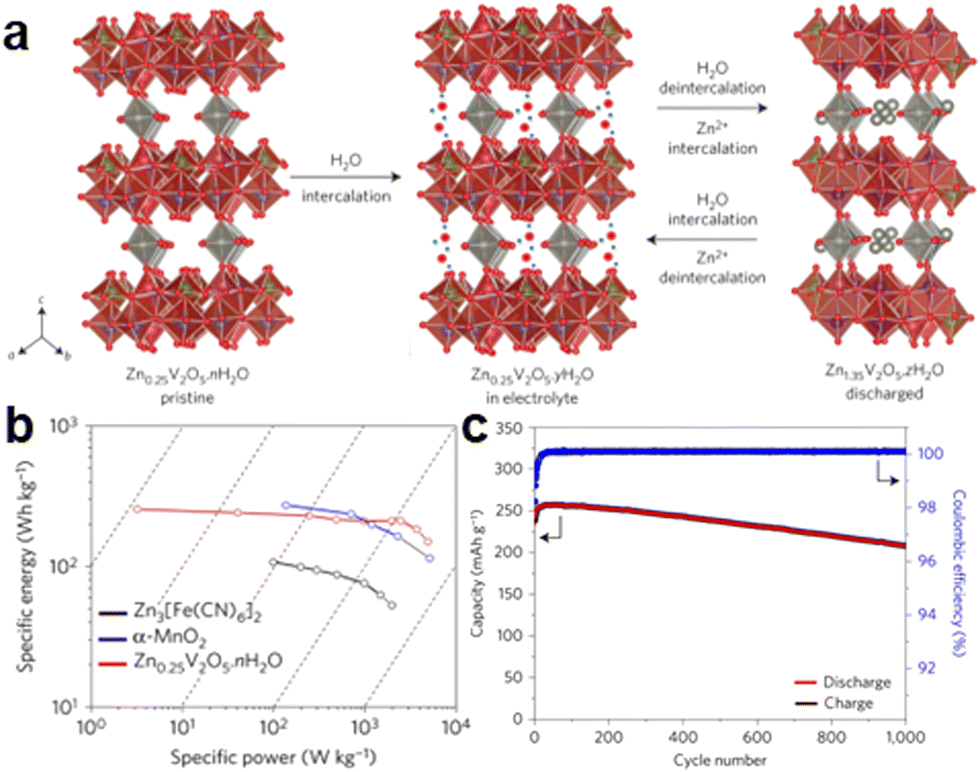 | ||
| Fig. 4 Schematic of (a) H2O/Zn2+ co-intercalation/extraction mechanism of Zn0.25V2O5·nH2O in static Zn–V batteries (vanadium-based zinc-ion batteries). (b) Energy density value of Zn0.25V2O5·nH2O compared with two other known materials for AZIBs. (c) Extended cycling performance and the corresponding coulombic efficiency at an 8C rate showing a capacity retention rate exceeding 80% over 1000 cycles.1 Copyright 2016, Nature Energy. | ||
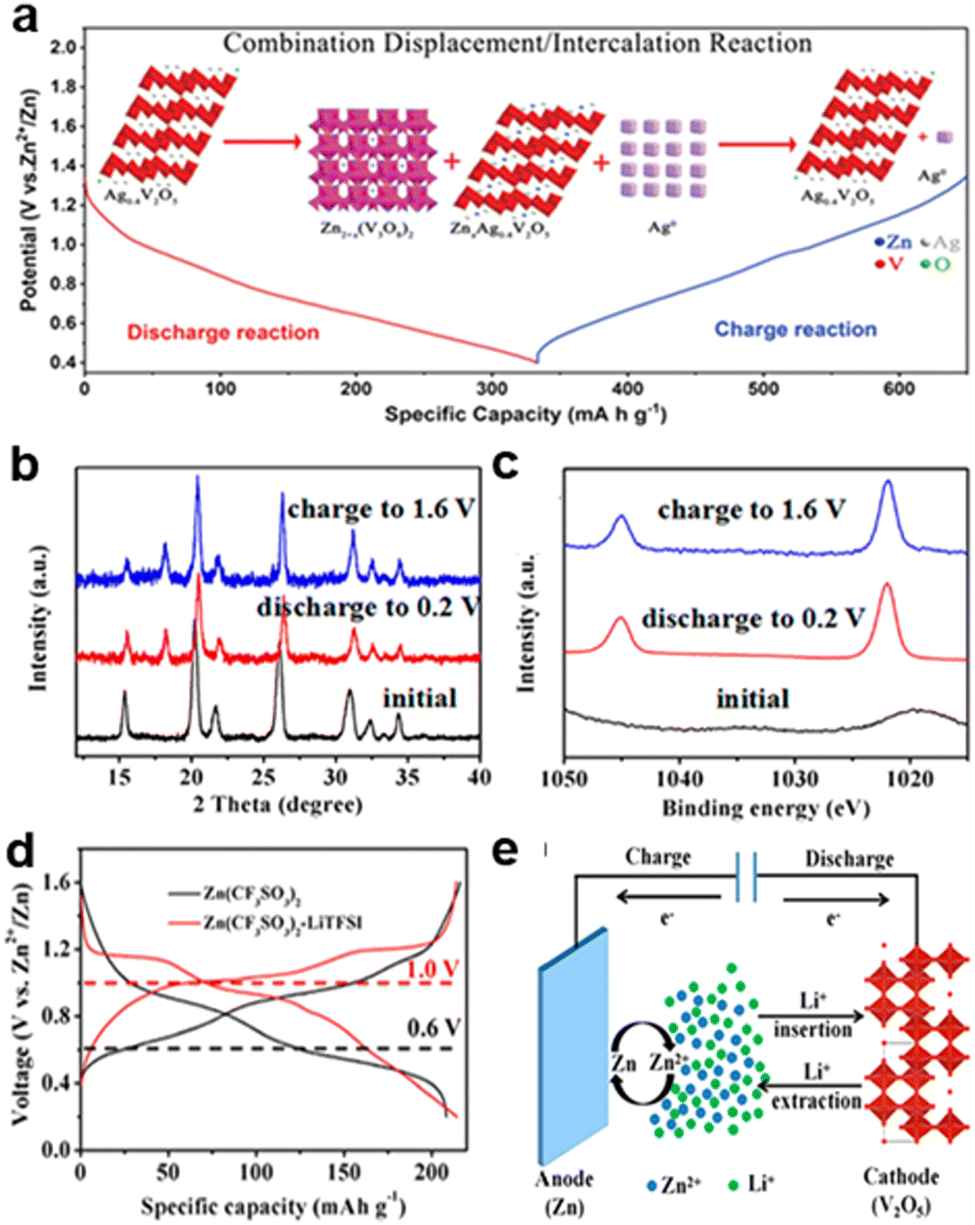 | ||
| Fig. 6 Schematic of (a) Combination substitution/interaction reaction mechanism of Ag0.4V2O5 during discharging/charging processes.19 Copyright 2020, Wiley-VCH GmbH. (b) Ex situ XRD patterns collected in the Zn (CF3SO3)2 electrolyte (black: initial state, red: discharged to 0.2 V; and blue: charged to 1.6 V) at 100 mA g−1. (c) Ex situ XPS collected in the Zn (CF3SO3)2–LiTFSI electrolyte at 100 mA g−1. (d) Higher voltage platform of Zn/V2O5 batteries at 100 mA g−1. (e) Hybrid metal ion mechanism.20 Copyright 2017, the American Chemical Society. | ||
The mixed metal ion mechanism involves the simultaneous participation of multiple cations, such as Zn2+ and Li+, in the charge storage process. In the Zn/V2O5 hybrid-ion battery system, the use of a “water-in-salt” electrolyte (Zn (CF3SO3)2–LiTFSI) enables both Zn2+ and Li+ to intercalate into the V2O5 cathode during discharging, as shown in Fig. 6e. Ex situ XRD and XPS analyses confirm that Zn2+ primarily intercalates into V2O5, while Li+ enhances structural stability by acting as an interlayer pillar, preventing structural collapse, and improving cycle life, as shown in Fig. 6b and c. This dual-ion interaction leads to a higher voltage platform, enhanced rate capability, and superior cycling stability compared to conventional single-ion Zn2+ intercalation, as shown in Fig. 6d and e. The hybrid system demonstrates high-capacity retention (80% after 2000 cycles at 2000 mA g−1), highlighting its potential for high-performance energy storage.20
2.2. Classification and structures
 | ||
| Fig. 7 Schematic of (a) and (f) Structures of V2O5 and V2O3.22 Copyright 2016, Nature. (b)–(e) Structures of VO2 (B, A, M1, and R).23 Copyright 2020, Wiley-VCH GmbH. | ||
To compare battery performance among different materials and before and after improvements in the same material, either diffusion coefficient or kinetics is discussed. VO2 exhibits a significantly higher diffusion coefficient (∼10−6.5 cm2 s−1) than most layered vanadium oxides such as MnVO and VOH (∼10−13 cm2 s−1), suggesting a lower migration energy barrier that facilitates faster charging/discharging and higher power density. This indicates that VO2 nanorods represent a more promising cathode material for achieving superior battery performance.24,25 Additionally, material modifications such as Mn ion intercalation and the creation of oxygen vacancies significantly enhance the battery performance by improving reaction kinetics and energy density. Specifically, Mn ion intercalation reduces charge transfer resistance, as evidenced by V2O5·nH2O's CV b-values shifting from 0.65, 0.76, 0.81, and 0.73 to 0.65, 0.86, 0.81, and 0.84, respectively.26 In parallel, the introduction of oxygen vacancies lowers diffusion barriers and is applied to modify HVO. The modified HVO (T-HVO) shows improved b-values—approximately 0.90 and 0.89 compared to the 0.74 and 0.83 for unmodified HVO—indicating a transition to surface-controlled processes.27 These changes lead to enhanced rate performance and faster charge/discharge capabilities.
 | ||
| Fig. 8 Schematic structure of (a) VS2 and (b) VS4. The purple balls are V atoms, and the yellow balls are S atoms.29 Copyright 2013, the American Chemical Society. (c) Charge and discharge curves of VS2 at a current density from 0.05 to 2.0 A g−1. (d) Long-term cycling properties of VS2 at a current density of 0.5 A g−1.30 Copyright 2017, Wiley-VCH GmbH. | ||
In recent studies, He et al. reported that VS2 nanosheets achieve high capacity (190.3 mA h g−1 at 0.05 A g−1) and demonstrate stable cycling performance, as shown in Fig. 8c and d.30 In comparison to VS2, the VS4 @rGO cathode shows impressive capacity of 450 mA h g−1 at 0.5 A g−1, attributed to the unique crystal structure of VS4 and excellent conductivity of rGO, with capacity retention of 82% over 3500 cycles.31
 | ||
| Fig. 9 Schematic of (a)–(c) Structures of vanadium phosphates in α1, α2 and β phases.32 Copyright 2013, the Royal Society of Chemistry. (d) Reversible capacity of the VOPO4 and KVOPO4 electrodes at 1 A g−1. (e) Zn-storage mechanism of the KVOPO4 electrode during the cycling process.33 Copyright 2024, Elsevier. | ||
As cathodes in aqueous zinc-ion batteries, vanadium phosphates can support multi-electron redox reactions, which significantly enhance their energy storage capabilities. The layered structure of VOPO4 facilitates the intercalation of Zn2+ ions, enabling high specific capacity. For example, a new cathode material, potassium vanadyl phosphate (KVOPO4), has been created by incorporating K+ ions into the layers of VOPO4, as shown in Fig. 9e. This approach not only maintains the stability of the crystal structure but also improves the diffusion kinetics of Zn2+ ions, leading to enhanced cycling stability and rate performance. The KVOPO4 cathode exhibits significant reversible capacity of 153.2 mA h g−1 after 400 cycles at a current rate of 1 A g−1 and exhibits excellent rate performance with 119.4 mA h g−1 at 5.0 A g−1, as shown in Fig. 9d.33 This indicates that vanadium phosphates, due to their distinct structural characteristics and adjustable properties, can serve effectively as cathodes in aqueous zinc-ion batteries (AZIBs), offering high capacity, stability, and rate performance.
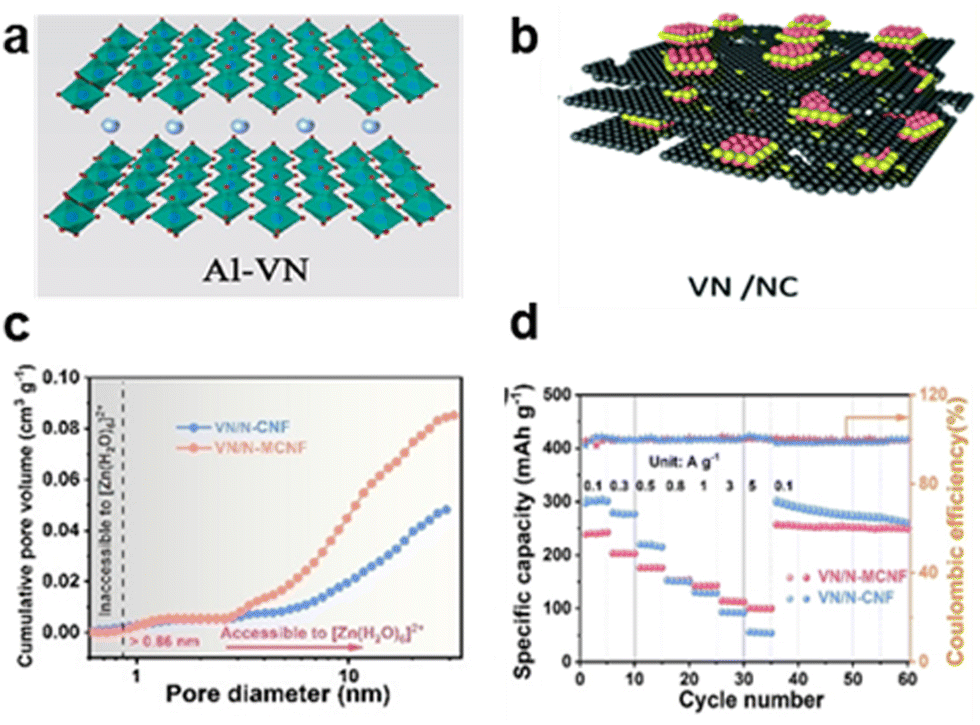 | ||
| Fig. 10 Schematic of (a) Al-VN structure.34 Copyright 2025, Elsevier. (b) Structure of VN/NC hybrid nanosheets.35 Copyright 2022, the Royal Society of Chemistry. (c) Corresponding pore size distributions of VN/N-CNF compared with VN/N-MCNF showing the mesoporous structure. (d) Specific capacity of VN/N-CNF (242 mA h g−1 at 0.1 A g−1) shown by rate performance.36 Copyright 2025, European Chemical Societies. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) due to its expanded interlayer spacing (0.29 nm) and strong hydrogen-bonding network, as shown in Fig. 11a and b.39 Sodium vanadate (NaV3O8·nH2O) adopts a tunnel structure composed of VO6 octahedra and VO5 trigonal bipyramids, offering rapid ion diffusion kinetics of 410 mA h g−1 at 0.1 A g−1, as shown in Fig. 11c and d.7
000 cycles) due to its expanded interlayer spacing (0.29 nm) and strong hydrogen-bonding network, as shown in Fig. 11a and b.39 Sodium vanadate (NaV3O8·nH2O) adopts a tunnel structure composed of VO6 octahedra and VO5 trigonal bipyramids, offering rapid ion diffusion kinetics of 410 mA h g−1 at 0.1 A g−1, as shown in Fig. 11c and d.7
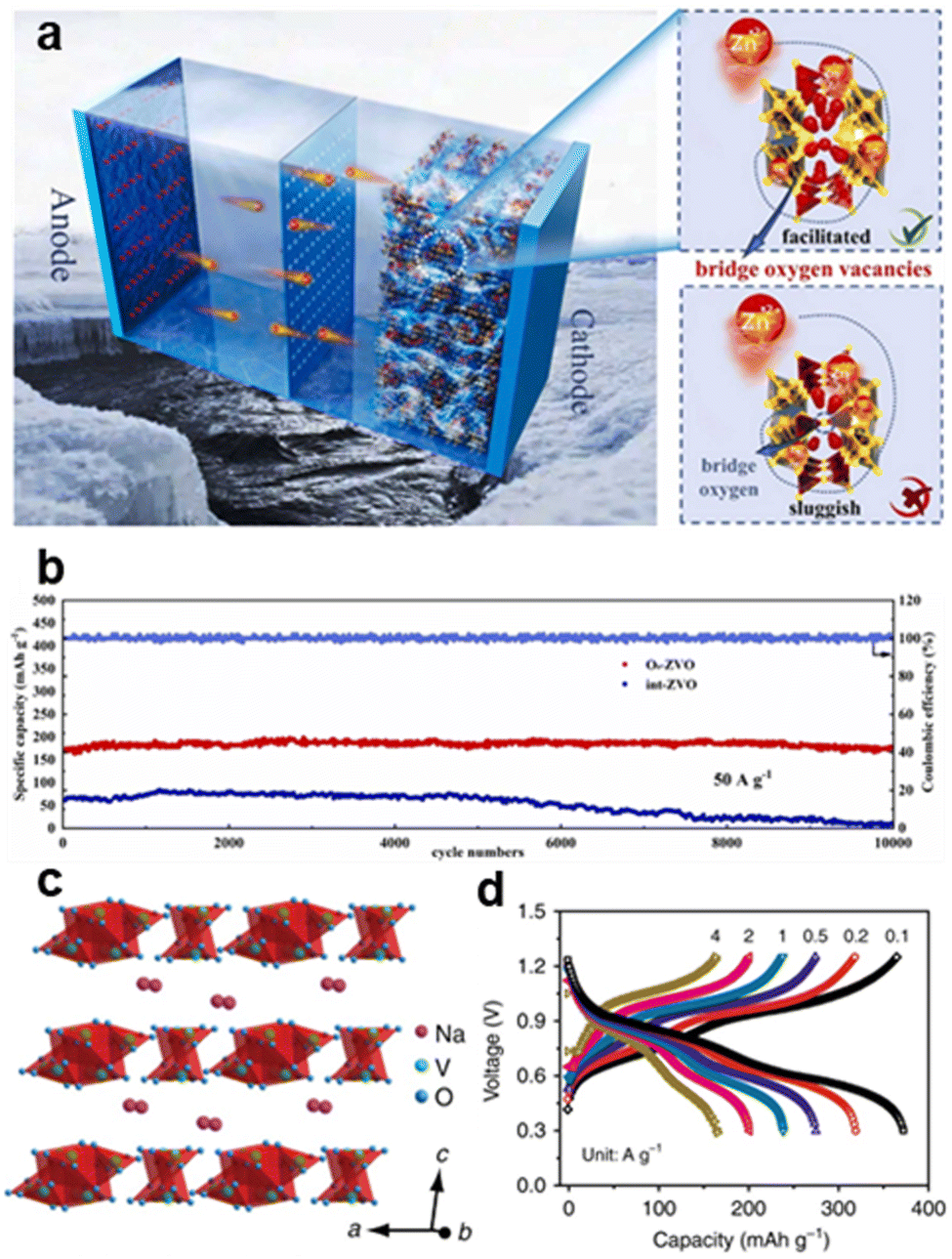 | ||
| Fig. 11 Schematic diagram of (a) ZVO applied in a zinc-ion battery. (b) High cycling stability (88% retention over 10000 cycles) compared with long cycle stability of O8–ZVO and int–ZVO at 50 A g−1.40 Copyright 2023, Elsevier. (c) Crystal structure of NaV3O8·nH2O; Na+ exists in the form of hydrated ions. (d) Rate performance of NVO electrodes in ZnSO4/Na2SO4 electrolyte showing 410 mA h g−1 at 0.1 A g−1.7 Copyright 2018, Nature Communications. | ||
Vanadates are stable in alkaline and neutral environments but can undergo reduction in acidic conditions. In low pH solutions, vanadium can reduce to lower oxidation states. Vanadates exhibit excellent electrochemical properties due to the multiple oxidation states of vanadium (V5+/V4+). This allows them to undergo reversible redox reactions, making them suitable for energy storage applications.38 Vanadates are extensively researched as cathode materials for AZIBs because of their high theoretical capacity, excellent cycling stability, and outstanding electrochemical characteristics. Materials like NaVO3 and CaVO3 have demonstrated encouraging outcomes regarding energy density and power density.41,42
2.3. Problems
The structural instability would degrade the ion diffusion rate. The insertion and extraction of Zn2+ ions can cause structural changes and even collapse in the vanadium cathode material during repeated charge/discharge cycles.48 In some cases, the coexistence of distinct phases (e.g. VO2 (M) and VO2 (R)) can significantly retard the ion diffusion rate. This phenomenon, known as super-susceptibility, can lead to an order of magnitude decrease in effective diffusivity when the material undergoes a first-order phase transition.49
2.4. Modification strategies
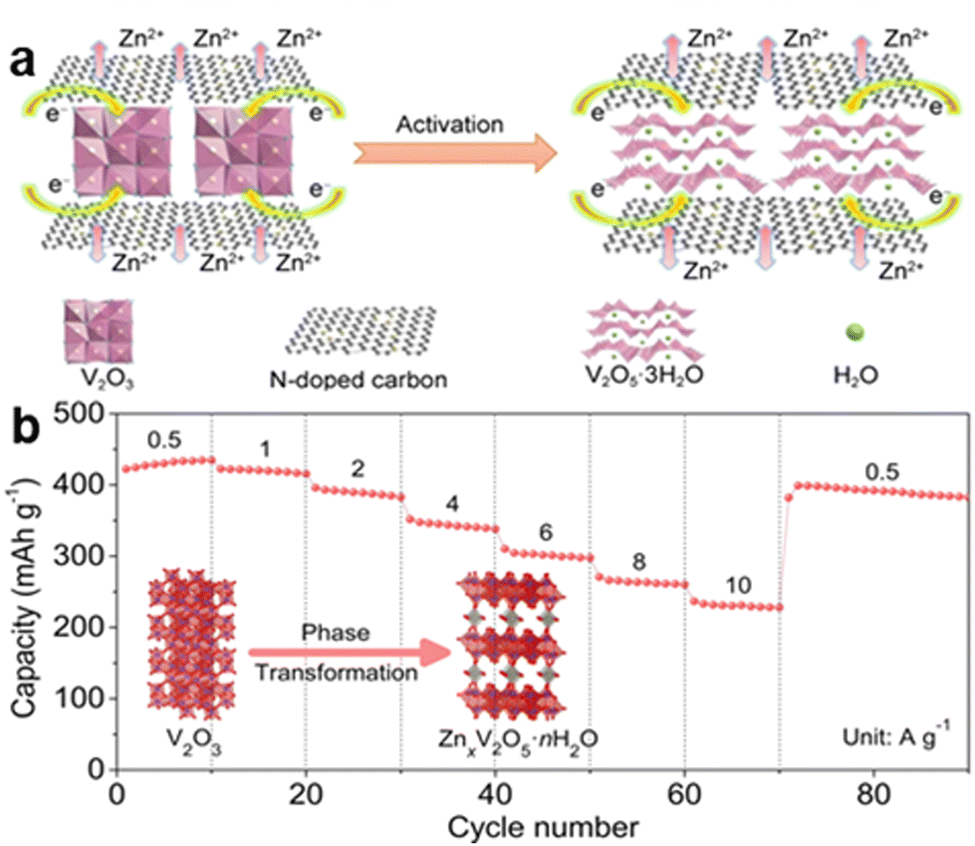
More examples and studies of guest ion pre-intercalation are shown. Studies have shown that the pre-intercalation of metal cations like Fe2+, Co2+, Ni2+, Mn2+, Zn2+, and Cu2+ into V2O5 can significantly increase the Zn2+ transport rate and cycle stability. Additionally, the introduction of conductive polymers as guest species can improve electron/ion transport rates, resulting in better rate and cycle performances.18
More examples referring to the self-anchoring effects and synergistic effects have been reported. In self-anchoring, part of the Zn2+ ions may anchor in the vanadium vacancies, enhancing the stability of the structure. This self-anchoring effect can prevent the complete extraction of Zn2+, maintaining the structural integrity.54 Sulphur doping and heterojunction formation are two well-known examples of synergistic effects that can limit the production of inactive byproducts and improve the kinetics and reaction reversibility of the Zn2+ and H+. Heterostructures form by integrating materials with distinct physical and chemical properties, creating synergistic effects and addressing the key challenges of poor charge transfer and limited active surface area. They reduce ion transport barriers, creating low-resistance channels for rapid Zn2+ movement.55 These modifications not only increase the specific capacity and rate performance but also contribute to better cycle stability, making vanadium-based cathodes more viable for high-performance aqueous zinc-ion batteries.
One approach is the use of elemental doping and in situ activation. For instance, cadmium doping combined with in situ activation has shown to introduce abundant oxygen vacancies and interlayered water, which significantly enhance the electronic and ionic conductivity of vanadium oxides.56 This results in improved capacity and cycling stability. Another strategy is the pre-intercalation of molecules or ions, such as NH4+, which can stabilize the structure via hydrogen bonding and facilitate ion diffusion.57 Structural compositing, such as incorporating conductive polymers or creating oxygen vacancies, is also effective in improving the overall performance of the cathode material. These macro structure design mechanisms collectively contribute to higher capacity, better rate capability, and enhanced cycling stability, making vanadium-based cathodes more suitable for advanced Zn–V static batteries. Additionally, materials such as graphene and other conductive carbon compounds have been utilized to boost the overall conductivity and structural stability of vanadium-based cathodes.58 The advancement of high-performance vanadium cathode materials for aqueous zinc-ion batteries is significantly supported by these macrostructure design approaches.
To solve those problems described above, either direct electrolyte control strategies, such as optimizing the operating potential, controlling the electrolyte pH and using the established pH- and potential-species relationships, or electrolyte modification such as using novel electrolyte additives (e.g. Al2(SO4)3 or polyethylene glycol combined with a 1 m Zn(TFSI)2 salt (and compared with Zn(OTf)2)) or capping agents (e.g. Citric Acid (CA), hexadecyltrimethylammonium bromide (CTAB) and polyvinyl pyrrolidone (PVP)) could be applied.60–62 The electrolyte additive, Al2(SO4)3, can suppress the H2O-involved hydrogen evolution, consume the OH− ions through hydrolysis, and act as a robust pillar.60 The advanced molecular crowding electrolyte, using polyethylene glycol (PEG) combined with a 1 m Zn (TFSI)2 salt (and compared with Zn (OTf)2), confines water molecules, which reduces water-induced side reactions and leads to an improved electrochemical stability window with an increase of 0.3 V, and enhances ion transport.61 Capping agents work to form a robust dynamic cathode electrolyte interface, which would isolate free water molecules from the electrodes and prevent the intercalation of hydrated Zn2+ ions. The three capping agents undergo different mechanisms to improve the battery performance. Both CA and PVP work through the selective absorption mechanism, differing in targeting different zinc crystal planes, while CTAB works through the surface interaction mechanism.62
Both strategies have improved battery performance, where they enhanced the capacity retention (0.2/10 A g−1 after 100/4000 cycles for the electrolyte additive, and 91% after over 500 cycles for capping agents). Additionally, Al2(SO4)3 has also given batteries an ultralong cycle life (97.6%/98.7%), and the molecular crowding electrolyte has achieved high specific energy (∼190 W h kg−1 based on the cathode).60–62
3. Zn–V redox flow batteries
3.1. Mechanisms
 | ||
| Fig. 12 Schematic of the reversible redox reaction of Zn and V in the Zn–V flow battery.63 Copyright 2023, the American Chemical Society. | ||
3.2. Problems
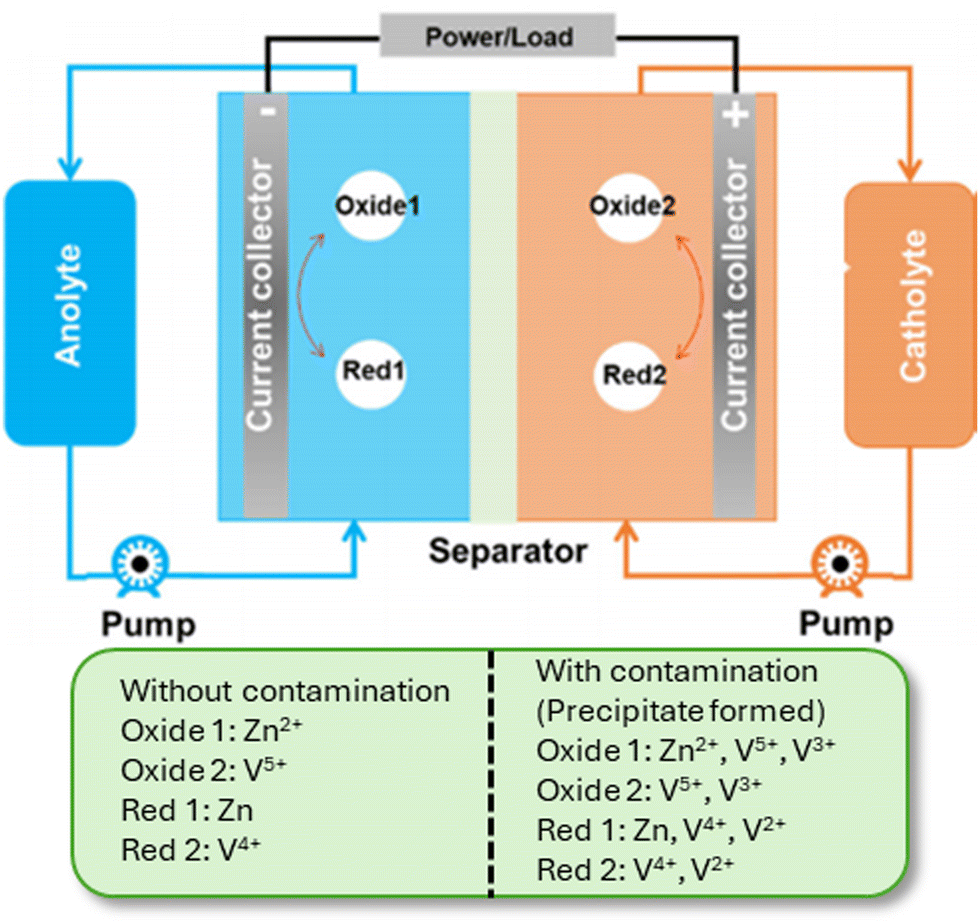 | ||
| Fig. 13 Schematic of the cross contamination of vanadium ions in the Zn–V flow battery.64 Reproduced with permission: Copyright 2023, Wiley-VCH GmbH. | ||
3.3. Modification strategies
 | ||
| Fig. 14 Schematic of (a) V precipitation without MSA addition at −5 °C and 40 °C at 0 h and 24 h. V precipitation after the stability test of (b) V(II) electrolyte with MSA addition and (c) V(V) electrolyte with MSA addition.65 Copyright 2021, Elsevier. (d)–(f) Cyclic voltammograms of a stationary Pt electrode in V(V)/V(IV)-MSA solutions with different concentrations of V(V)/V(IV) and scan rates from 1 to 100 mV s−1.3 Copyright 2012, Elsevier. | ||
MSA offers higher solubility of vanadium ions, better stability, lower viscosity, lower corrosivity, and better electrochemical performance, all of which contribute to improved energy efficiency and longer operational life of the battery, as shown in Fig. 14d–f. These advantages make MSA a promising candidate for enhancing the performance of vanadium redox flow batteries.3 Electrolyte modification is a common strategy for aqueous zinc-ion batteries, which is expected to bring novel improvement strategies to Zn–V flow batteries in the future.66
 | ||
| Fig. 15 GCD profiles of (a) Zn–V system and (b) Zn–V flow cell with a Daramic microporous separator and Nafion-117 membrane. Cyclic life of the Zn–V metal hybrid system (c) with a Nafion-117 membrane and (d) with a micro porous separator.67 Copyright 2019, the American Chemical Society. | ||
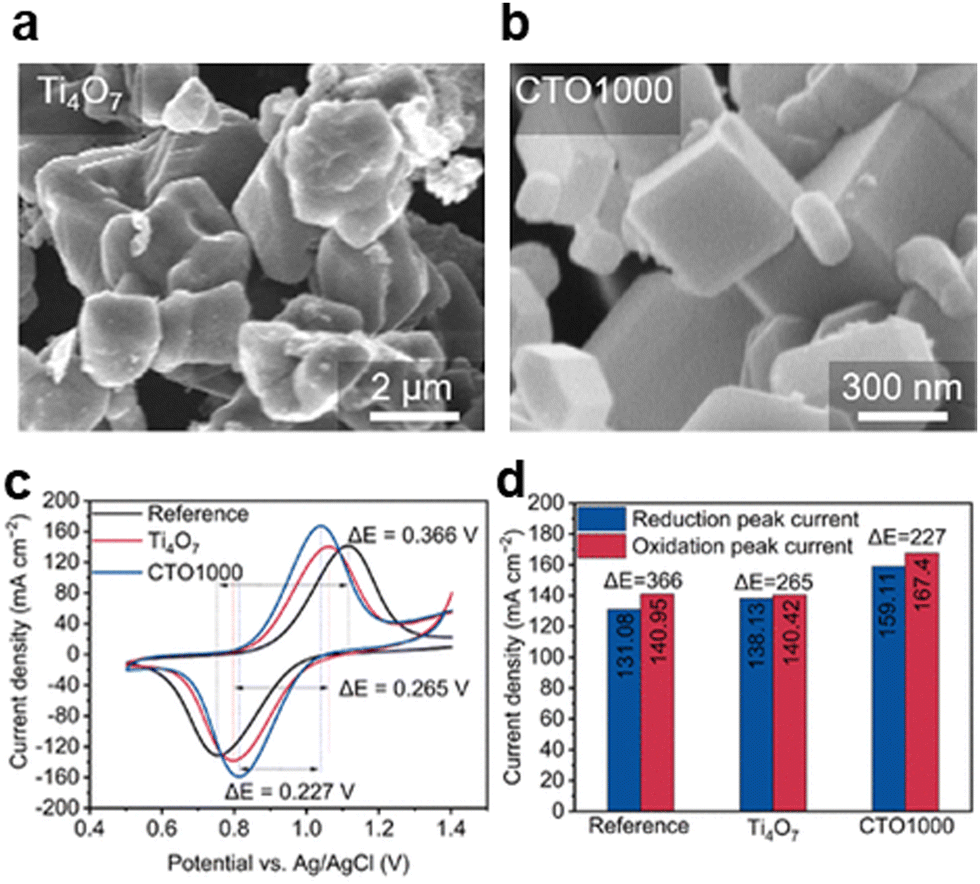 | ||
| Fig. 16 SEM images of (a) Ti4O7 and (b) CTO1000 electrocatalyst. (c) Cyclic voltammograms of pristine CF, Ti4O7, and CTO1000 electrodes. (d) Reduction and oxidation peak currents and potential difference of pristine CF, Ti4O7, and CTO1000 electrodes.63 Copyright 2023, the American Chemical Society. | ||
In 2023, Choi et al. have reported a binary cerium titanium oxide for cathode optimization, and the inversely proportional relationship between the values of oxidation states and the number of formations of oxygen vacancies which function as active sites, which is found by researchers, as shown in Fig. 16a and d. The crystal structure of CTO1000 may affect its catalytic activity through structural defects (e.g. oxygen vacancies), so researchers adjust the crystal structure by controlling the temperature during the synthesis process to enhance the battery performance, as shown in Fig. 16b.63
3.4. Advantages of the Zn–V redox flow battery system
Zinc-vanadium redox flow batteries (Zn–V RFBs) exhibit distinct advantages over Zn–Br2 and all-vanadium (All V) systems, particularly in terms of voltage window, power density, and environmental compatibility. While Zn–Br batteries are able to achieve a high open-circuit voltage (2.1 V), their practical current density is significantly limited (<100 mA cm−2) by sluggish Br−/Br2 redox kinetics and safety concerns due to toxic bromine volatility, which results in low energy efficiency (∼63%) and high maintenance costs. On the other hand, Zn–V RFBs combine competitive voltage (1.96 V) with high power density (840 mW cm−2), enabled by the rapid multi-electron reactions of vanadium species (V5+/V4+) and optimized electrolyte flow rates. Comparing the Zn–V system with all V systems, the anode reaction of the Zn–V system is the reaction of Zn2+/Zn, which has lower potential, leading to a larger voltage window. This is the advantage that all vanadium redox flow batteries do not have. Additionally, Zn–V RFBs leverage zinc's abundance and low toxicity, avoiding the environmental risks of bromine leakage and offering scalable, cost-effective energy storage solutions compared to resource-intensive all V systems. However, the low energy density (16 W h L−1) and short lifespan of Zn–V redox flow batteries need to be overcome to unleash more application potential. These quantitative advantages underline the potential of Zn–V flow batteries as a cost-effective, high-performance alternative for large-scale energy storage, thereby facilitating further development in this field.644. Conclusion
Zn–V battery systems, encompassing static and flow configurations, have emerged as promising solutions for large-scale energy storage, renewable energy integration, and grid stability, leveraging vanadium's multivalent redox chemistry, natural abundance, and environmental compatibility. Vanadium-based cathodes and electrolytes endow these systems with high theoretical capacity, energy density, and safety, driving significant advancement in recent years.For Zn–V static batteries, progress has been marked by innovations in vanadium-based cathode materials (e.g., oxides, nitrides, and vanadates) with tailored nanostructures, defect engineering, and composite designs to enhance Zn2+ storage kinetics and cycling stability. However, challenges persist, including irreversible phase transitions during cycling, vanadium dissolution, and ambiguous energy storage mechanisms—such as debates over Zn2+ intercalation versus conversion reactions, active phase identification, and byproduct formation—which hinder long-term performance and consensus within the scientific community.
In contrast, Zn–V flow batteries, though still in early-stage research, offer inherent scalability, and adjustable reaction electrode surface area and electrolyte content of storage tanks, attracting growing attention for grid applications. Yet, their development faces hurdles like electrolyte optimization, membrane durability, and cost-effective system integration. The existing knowledge of V cathodes in Zn–V static batteries could be helpful for improvements of Zn–V flow batteries. For example, the electrolyte modification strategy that effectively mitigates vanadium dissolution in the Zn–V static battery system can also be directly used in the flow battery system to solve the problem of ion cross-contamination. In addition, the idea of membrane optimization in Zn–V flow battery systems provides a direction for further battery improvement in static battery systems to weaken the vanadium dissolution and shuttle of Zn–V static batteries (Fig. 17).
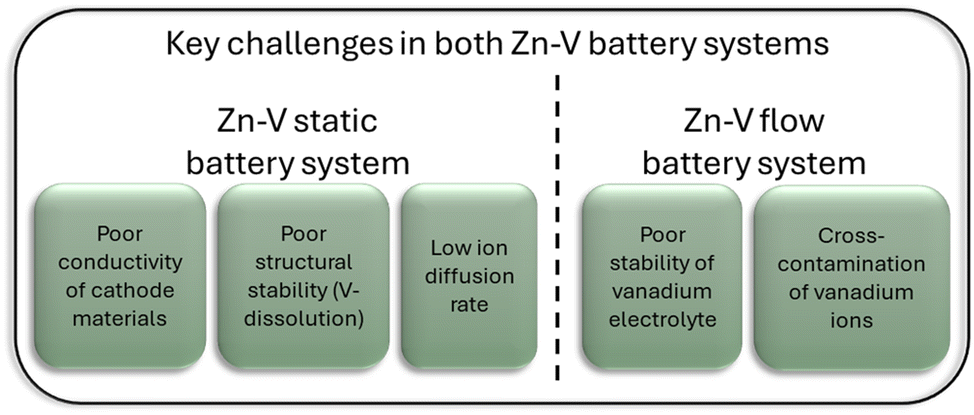 | ||
| Fig. 17 Schematic of the key challenges of both types of Zn–V batteries. | ||
Moving forward, resolving mechanistic ambiguities through advanced in situ/operando characterization, stabilizing vanadium cathodes via structural engineering (e.g., pre-intercalation, doping, or protective coatings), and accelerating flow battery innovation through hybrid electrolyte designs and scalable prototypes are critical. Bridging fundamental research with practical engineering will be essential to unlock the full potential of Zn–V batteries, ensuring their viability in the global transition to sustainable energy storage (Table 1).
| Category | Zn–V static system | Zn–V flow system |
|---|---|---|
| Mechanism | Zn2+ intercalation/extraction | Reversible redox reactions of Zn and V, including Zn2+/Zn and the V5+/V4+ redox couples |
| H+/Zn2+ co-intercalation | ||
| H2O/Zn2+ co-intercalation | ||
| Chemical conversion reactions | ||
| Other types (substitution/interaction reaction, mixed metal ion mechanism) | ||
| Structures of vanadium cathode materials | Vanadium oxide | A redox-active vanadium electrolyte (VO2+/VO2+ species) |
| Vanadium sulphide | ||
| Vanadium phosphate | ||
| Vanadium nitride | ||
| Vanadate | ||
| Major challenges | Poor conductivity | Poor electrolyte stability |
| Poor structural stability (V-dissolution) | Cross-contamination of vanadium ions | |
| Low ion diffusion rate | ||
| Modification strategies | Guest ion pre-intercalation | Electrolyte modification |
| Defect engineering | Membrane optimization | |
| Macrostructure design | Cathode optimization | |
| Electrolyte modification |
Author contributions
Y. F. L. wrote and modified the manuscript. J. C. modified the manuscript and gave suggestions and guidance on manuscript improvements. G. J. H. supervised the project and secured the funding. All authors have discussed and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Jie Chen thanks the funding support from University College London and UK Research and Innovation (UKRI) for the CDT scholarship. This work was financially supported by the Cooperation project of UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding (101077226; EP/Y008707/1).References
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2011, 51, 933–935 CrossRef PubMed.
- C. Tang and D. Zhou, Electrochim. Acta, 2012, 65, 179–184 CrossRef CAS.
- L. Shan, J. Zhou, W. Zhang, C. Xia, S. Guo, X. Ma, G. Fang, X. Wu and S. Liang, Energy Technol., 2019, 7, 1900022 CrossRef.
- M. Sufyan Javed, H. Lei, Z. Wang, B. Liu, X. Cai and W. Mai, Nano Energy, 2020, 70, 104573 CrossRef.
- N. Liu, B. Li, Z. He, L. Dai, H. Wang and L. Wang, J. Energy Chem., 2021, 59, 134–159 CrossRef CAS.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- K. Zhu, T. Wu, S. Sun, W. van den Bergh, M. Stefik and K. Huang, Energy Storage Mater., 2020, 29, 60–70 CrossRef.
- J. Ding, H. Gao, D. Ji, K. Zhao, S. Wang and F. Cheng, J. Mater. Chem. A, 2021, 9, 5258–5275 RSC.
- E. Roex, F. Boschini, V. Delaval, A. Schrijnemakers, R. Cloots and A. Mahmoud, J. Electroanal. Chem., 2023, 929, 117133 CrossRef CAS.
- T. Lv, Y. Peng, G. Zhang, S. Jiang, Z. Yang, S. Yang and H. Pang, Adv. Sci., 2023, 10, 2206907 CrossRef CAS PubMed.
- Y. Ding, Y. Peng, S. Chen, X. Zhang, Z. Li, L. Zhu, L.-E. Mo and L. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 44109–44117 CrossRef CAS PubMed.
- K. Hu, D. Jin, Y. Zhang, L. Ke, H. Shang, Y. Yan, H. Lin, K. Rui and J. Zhu, J. Energy Chem., 2021, 61, 594–601 CrossRef CAS.
- G. Fang, S. Liang, Z. Chen, P. Cui, X. Zheng, B. Lu and X. Lu, Adv. Funct. Mater., 2019, 29, 1905267 CrossRef CAS.
- L. Wang, K.-W. Huang, J. Chen and J. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef CAS PubMed.
- M. Chen, S.-C. Zhang, Z.-G. Zou, S.-L. Zhong, W.-Q. Ling, J. Geng, F.-A. Liang, X.-X. Peng, Y. Gao and F.-G. Yu, Rare Met., 2023, 42, 2868–2905 CrossRef CAS.
- L. Shan, Y. Wang, S. Liang, B. Tang, Y. Yang, Z. Wang, B. Lu and J. Zhou, InfoMat, 2021, 3, 1028–1036 CrossRef CAS.
- T. Zhou and G. Gao, Nano Energy, 2024, 127, 109691 CrossRef CAS.
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Adv. Energy Mater., 2020, 10, 2000477 CrossRef CAS.
- P. Hu, M. Yan, T. Zhu, X. Wang, X. Wei, J. Li, L. Zhou, Z. Li, L. Chen and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 42717–42722 CrossRef CAS PubMed.
- D. Chen, X. Rui, Q. Zhang, H. Geng, L. Gan, W. Zhang, C. Li, S. Huang and Y. Yu, Nano Energy, 2019, 60, 171–178 CrossRef CAS.
- S. Lee, I. N. Ivanov, J. K. Keum and H. N. Lee, Sci. Rep., 2016, 6, 19621 CrossRef CAS PubMed.
- X. Xu, F. Xiong, J. Meng, C. Wang, C. Niu, Q. An and L. Mai, Adv. Funct. Mater., 2020, 30, 1904398 CrossRef CAS.
- L. Chen, Y. Ruan, G. Zhang, Q. Wei, Y. Jiang, T. Xiong, P. He, W. Yang, M. Yan, Q. An and L. Mai, Chem. Mater., 2019, 31, 699–706 CrossRef CAS.
- H. Chen, J. Huang, S. Tian, L. Liu, T. Qin, L. Song, Y. Liu, Y. Zhang, X. Wu, S. Lei and S. Peng, Adv. Sci., 2021, 8, 2004924 CrossRef CAS PubMed.
- C. Liu, Z. G. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- J. Chen, Y. Liu, B. Xiao, J. Huang, H. Chen, K. Zhu, J. Zhang, G. Cao, G. He, J. Ma and S. Peng, Angew. Chem., Int. Ed., 2024, 63, e202408667 CrossRef CAS PubMed.
- Y. Zhang, A. Chen and J. Sun, J. Energy Chem., 2021, 54, 655–667 CrossRef CAS.
- C. S. Rout, B.-H. Kim, X. Xu, J. Yang, H. Y. Jeong, D. Odkhuu, N. Park, J. Cho and H. S. Shin, J. Am. Chem. Soc., 2013, 135, 8720–8725 CrossRef CAS PubMed.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- K. Chen, X. Li, J. Zang, Z. Zhang, Y. Wang, Q. Lou, Y. Bai, J. Fu, C. Zhuang, Y. Zhang, L. Zhang, S. Dai and C. Shan, Nanoscale, 2021, 13, 12370–12378 RSC.
- R. Gautier, R. Gautier, O. Hernandez, N. Audebrand, T. Bataille, C. Roiland, E. Elkaïm, L. Le Pollès, E. Furet and E. L. Fur, Dalton Trans., 2013, 42, 8124 RSC.
- Y. Li, W. Li, H. Chen, Z. Liu, X. Li and D. Zhou, J. Electroanal. Chem., 2024, 974, 118755 CrossRef CAS.
- J. Chen, K. Guo, T. Ren, G. Feng, W. Guo and F. Bao, J. Power Sources, 2024, 626, 235751 CrossRef.
- Y. Niu, W. Xu, Y. Ma, Y. Gao, X. Li, L. Li and L. Zhi, Nanoscale, 2022, 14, 7607–7612 RSC.
- H. Wang, W. Hou, X. Wang, X. Xie, H. Peng, G. Ma, L. Zhu and Y. Xu, Chem. - Eur. J., 2025, 31, e202403903 CrossRef CAS PubMed.
- H. Chen, Z. Yang, J. Wu, Y. Rong and L. Deng, J. Power Sources, 2021, 507, 230286 CrossRef CAS.
- X.-F. Ma, H.-Y. Li, W. Ren, D. Gao, F. Chen, J. Diao, B. Xie, G. Huang, J. Wang and F. Pan, J. Mater. Sci. Technol., 2023, 153, 56–74 CrossRef CAS.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2017, 30, 1705580 CrossRef PubMed.
- M. Wang, G. Zhao, X. Yu, X. Bai, A. Chen, C. Zhao, P. Lyu and N. Zhang, Nano Energy, 2023, 110, 108336 CrossRef CAS.
- L. Chen, H. Wu, H. Wang, L. Chen, X. Pu and Z. Chen, Electrochim. Acta, 2019, 307, 224–231 CrossRef CAS.
- C. R. Tang, G. Singh, L. M. Housel, S. J. Kim, C. D. Quilty, Y. Zhu, L. Wang, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, Phys. Chem. Chem. Phys., 2021, 23, 8607–8617 RSC.
- X. Chen, H. Zhang, J.-H. Liu, Y. Gao, X. Cao, C. Zhan, Y. Wang, S. Wang, S.-L. Chou, S.-X. Dou and D. Cao, Energy Storage Mater., 2022, 50, 21–46 CrossRef CAS.
- X. Ding, J. Le, Y. Yang, L. Liu, Y. Shao, Y. Xiao, Y. Li and L. Han, Energy Storage Mater., 2025, 76, 104098 CrossRef.
- X. Ren, M. Sun, Z. Gan, Y. Sun, N. Wang, L. Hu, Z. Yan, C. Jia and Z. Li, Chem. Eng. J., 2024, 494, 152994 CrossRef CAS.
- Y. D. Luna, Z. Mohamed, A. Dawoud and N. Bensalah, RSC Adv., 2024, 14, 39193–39203 RSC.
- D. Sun, M. Zhang, W. Wan, Y. Cao and H. Chai, J. Energy Storage, 2023, 73, 109236 CrossRef.
- S. Bai, Z. Wu, X. Zhang, J. Qiu, J. Chen, Z. Liu and Y. Zhang, Chem. Eng. J., 2024, 500, 157281 CrossRef CAS.
- Y. Cai, Z. Wang, J. Wan, J. Li, R. Guo, J. W. Ager, A. Javey, H. Zheng, J. Jiang and J. Wu, Nat. Commun., 2024, 15, 5814 CrossRef CAS PubMed.
- B. Hu, X. Yang, D. Li, L. Luo, J. Jiang, T. Du, H. Pu, G. Ma, B. Xiang and Z. Li, J. Alloys Compd., 2024, 1008, 176801 CrossRef CAS.
- T. Zhou and G. Gao, J. Energy Storage, 2024, 84, 110808 CrossRef.
- Z. Chen, H. Liu, S. Fan, Q. Zhang, C. Yuan, W. Peng, Y. Li and X. Fan, Adv. Energy Mater., 2024, 14, 2400977 CrossRef CAS.
- J. Gao, C. Cheng, L. Ding, G. Liu, T. Yan and L. Zhang, Chem. Eng. J., 2022, 450, 138367 CrossRef CAS.
- K. Zhu, S. Wei, H. Shou, F. Shen, S. Chen, P. Zhang, C. Wang, Y. Cao, X. Guo, M. Luo, H. Zhang, B. Ye, X. Wu, L. He and S. Li, Nat. Commun., 2021, 12, 6878 CrossRef CAS PubMed.
- S. Hou, X. Chen, G. He, X. Peng, J. Wang, C. Huang, H. Liu, T. Liu, X. Wang, S. Hou and L. Zhao, J. Mater. Chem. A, 2025, 13, 1240–1248 RSC.
- T. Chen, Q. Wu, L. Lang, Z. Chen, G. Luo, L. Hu, G. Liu and W. Chen, Chem. Eng. J., 2024, 499, 156295 CrossRef CAS.
- K. Fang, H. Zhang, P. Chen, H.-Y. Zhang, Z. Wei, L. Ding, X.-A. Ye, J. Liu, Y.-L. Liu, G.-G. Wang and H. Y. Yang, Chem. Eng. J., 2024, 496, 153736 CrossRef CAS.
- T.-H. Wu, T.-K. Li and L.-J. Guo, J. Power Sources, 2024, 623, 235503 CrossRef CAS.
- Y. Li, H. Ba, Z. Wang, S. Wu, Y. Shang, S. Huang and H. Y. Yang, Mater. Today Energy, 2023, 39, 101460 CrossRef.
- D. Liu, Z. Zhang, Y. Zhang, M. Ye, S. Huang, S. You, Z. Du, J. He, Z. Wen, Y. Tang, X. Liu and C. C. Li, Angew. Chem., 2023, 135, e202215385 CrossRef.
- D. E. Ciurduc, C. de la Cruz, N. Patil, A. Mavrandonakis and R. Marcilla, Mater. Today Energy, 2023, 36, 101339 CrossRef CAS.
- R. Chen, Y. Zhong, P. Jiang, H. Tang, F. Guo, Y. Dai, J. Chen, J. Wang, J. Liu, S. Wei, W. Zhang, W. Zong, F. Zhao, J. Zhang, Z. Guo, X. Wang and G. He, Adv. Mater., 2025, 2412790, DOI:10.1002/adma.202412790.
- J. Choi, J. Park, J. Park, M. Kim, S. Lee, C. R. Cho, J. H. Lee, Y. Park, M. G. Kim, J. Choi, J.-W. Park and M. Park, ACS Appl. Mater. Interfaces, 2023, 15, 55692–55702 CrossRef CAS PubMed.
- Q. Zhao, G. Yin, Y. Liu, R. Tang, X. Wu and X. Zeng, Carbon Neutralization, 2023, 2, 90–114 CrossRef CAS.
- G. Kim, Y. Kim, T. Yim and K. Kwon, J. Ind. Eng. Chem., 2021, 99, 326–333 CrossRef CAS.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef PubMed.
- M. Ulaganathan, S. Suresh, K. Mariyappan, P. Periasamy and R. Pitchai, ACS Sustainable Chem. Eng., 2019, 7, 6053–6060 CrossRef CAS.
Footnote |
| † Yufei Li and Jie Chen contribute to the work equally. |
| This journal is © The Royal Society of Chemistry 2025 |