DOI:
10.1039/D4NR04696J
(Paper)
Nanoscale, 2025,
17, 5732-5742
Mitochondria-targeting nanostructures from enzymatically degradable fluorescent amphiphilic polyesters†
Received
10th November 2024
, Accepted 7th January 2025
First published on 9th January 2025
Abstract
Water-soluble π-conjugated luminescent bioprobes have been broadly used in biomedical research but are limited by the nonbiodegradability associated with their rigid C–C backbones. In the present work, we introduced three naphthalene monoimide (NMI)-functionalized amphiphilic fluorescent polyesters (P1, P2, and P3) prepared by transesterification of functional diols with an activated diester monomer of adipic acid. These polyesters featured a side-chain NMI fluorophore, imparting the required hydrophobicity for self-assembly in water and endowing the polymeric nanoassemblies with green fluorescence. Two polymers (P1 and P2) were intrinsically cationic at physiological pH (7.4), while neutral P3 exhibited pH-triggered (pH ∼6.2) cationic features due to the protonation of the tertiary amine groups present in its backbone. These biocompatible polymers revealed around 85% cellular uptake after 1 hour of incubation. However, the initial uptake for the cationic polymers (P1 and P2) within 15 minutes was significantly greater than that of the neutral P3 because of their stronger electrostatic interactions with the negatively charged cell membranes. Notably, cationic P1 and P2 could specifically target mitochondria in cancerous HeLa cells by escaping the initial endosome/lysosome trap. In contrast, neutral P3 exhibited cell-selective mitochondria targeting in cancerous (HeLa) cells over non-cancerous (NKE) cells. This is attributed to P3's protonation-induced positive charge accumulation in the acidic environment of cancer cells, unlike in the non-acidic environment of non-cancerous cells. This possibly causes P3 nanoassemblies to behave similarly to P1 and P2 in HeLa cells despite P3 being intrinsically neutral. The insights gained from this work may be relevant for future development of cell-specific, mitochondria-targeted drug delivery systems from enzymatically degradable polyester backbones.
 Anindita Das | Anindita Das received her MSc in Chemistry from the University of Calcutta (2009) and her PhD from the Indian Association for the Cultivation of Science (IACS), India (2014), under the mentorship of Professor Suhrit Ghosh. She then pursued postdoctoral research as an Alexander von Humboldt Fellow with Professor Patrick Theato at the University of Hamburg, Germany, and later, worked with Professor E.W. Meijer at the Eindhoven University of Technology, Netherlands. In 2017, she joined IACS, where currently she holds a position of Associate Professor. Her research interests include precision (macro)molecular assemblies employing halogen bonding, supramolecular polymers, crystallization-driven polymer assemblies, and biodegradable polymers. |
Introduction
The last three decades have seen an upsurge in the application of amphiphilic polymeric nanoparticles (APNs) for drug delivery,1–3 intracellular targeting and bioimaging purposes.4–7 These applications require detailed understanding of the physiochemical mechanism by which such nanosystems enter the cells and their ultimate fate once they are internalized.8 With the advent of advanced synthetic and characterization techniques, a wide variety of structurally distinct APNs have been examined, focusing on their cellular internalization as a function of size, shape, and surface properties.9–11 Polymeric nanoparticles hold promise due to their multivalency, stability towards drug leakage, high encapsulation efficiency, potential for stimuli responsiveness12–16 and structural tunability, which can easily be integrated by appropriate functionalization of such polymers.1,17 In addition to the physiological stability and cell viability, which are the primary requisites for their specific accumulation at the desired site via the enhanced permeability and retention (EPR) effect,18–20 the cell-penetrating and intracellular targeting abilities of polymeric nanoparticles have also drawn simultaneous attention. Positively charged nanoparticles have been utilized to internalize molecules of interest inside the cell, as it is believed that the cationic groups aid in initiating favorable electrostatic interactions with negatively charged cell membranes for their rapid uptake.21 However, highly charged particles may also exhibit some cytotoxic effects due to the perturbation of the membrane potential.22,23 Based on numerous studies, endocytosis can be considered the most natural route to bring engineered polymeric nanoparticles inside the cell, and thus, many such endocytosis-promoting functionalities, like CPPs (cell penetrating peptides)10,24,25 and internalizable ligands like folates26,27 or biotins28 for cell-selective, receptor-mediated uptake for targeted delivery,29 have been extensively studied. Organelle-targeting polymeric nanoparticles with the ability to selectively image the subcellular structures provide critical information about the cellular microenvironment and are emerging systems for theranostic applications.30–32 Mitochondria, often described as the “powerhouse” of cells, are vital intracellular organelles that participate in crucial biological processes, such as cell differentiation and growth, cell apoptosis, and cell signalling. Mitochondrial dysfunction can contribute to a variety of diseases, including neural degeneration, various types of cancer, and metabolic diseases.33–35 Mitochondria feature a unique bilayer structure with a highly negative membrane potential on the inner hydrophobic membrane.36 Thus, the mitochondria-targeting strategy necessitates the utilization of lipophilic cations that are attracted to the negatively charged mitochondrial inner membrane and thereby accumulate inside. Lipophilic cations such as triphenylphosphonium (TPP)37–40 or cationic peptides,41,42 due to their combined electrostatic and hydrophobic interactions with negatively charged mitochondrial membranes, have been extensively explored for the generation of mitochondria-targeting nanocarriers.43–46 Moreover, TPP is frequently connected with self-assembling building blocks and auxiliary fluorescent probes to diagnose their intracellular mitochondrial localization.47,48 This often requires tedious and complex synthetic procedures or multicomponent self-assembly approaches for designing nanoparticles with simultaneous targeting and bioimaging abilities. Mitochondria-targeting polymeric nanoparticles have gained significant attention in the recent past;48–53 however, very few are reported with biodegradable polymer backbones.40,52–58 Degradable polymers that can undergo intracellular degradation in the presence of appropriate biological stimuli offer the advantages of controlled drug release and low toxicity,3 which are often the limitations of in vivo studies using nondegradable polymer backbones for their poor renal clearance. Thus, biodegradable polymers have emerged as a promising class of biomaterials59–62 for drug and gene delivery,63–65 tissue engineering,66,67 and a host of other applications.68,69 Aliphatic polyesters70 such as poly(lactic acid) (PLA), poly(ε-caprolactone) (PCL), poly(glycolic acid) (PGA), poly(lactic-co-glycolic acid) (PLGA), or poly(hydroxyalkanoates) (PHA) have attracted great attention in biomedical applications61,71,72 due to their high biocompatibility and known lysosomal enzymatic cleavage of their ester linkages.73 However, these conventional aliphatic polyesters are semi-crystalline, hydrophobic, and devoid of any side chain functionality. Optimal hydrophobicity/hydrophilicity balance with pronounced polar characteristics are essential features for the biocompatibility and biodegradability of polyesters. While these are crucial parameters in water-dispersible polymeric nanoparticle designs,73,74 for expanding their scope in biological applications, additional needs for the conjugation of prodrugs, bioimaging probes or targeting ligands become essential. Thus, multifunctional amphiphilic polyesters75,76 capable of generating cell-viable, biodegradable nanoparticles that display rapid cellular internalization, efficient bioimaging capabilities, and cell-specific affinity towards mitochondria are in great demand for targeted therapeutic applications.40
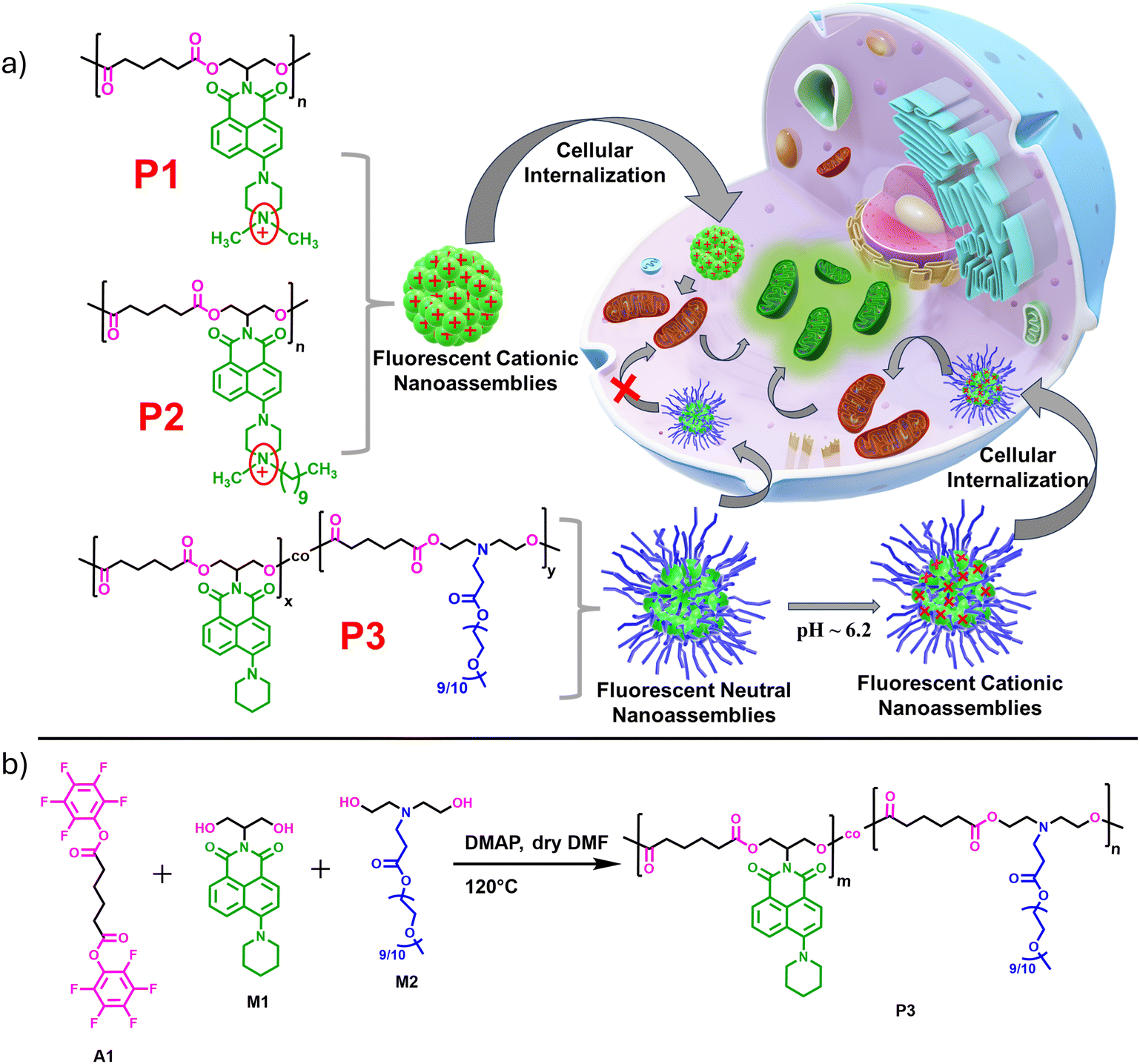
Recently, we reported the broad spectrum antibacterial properties of two enzymatically degradable amphiphilic polyesters, P1 and P2 (Scheme 1a), which possessed a green-emitting cationic naphthalene monoimide (NMI) derivative as side chain pendants.77 These polyesters were prepared under mild conditions by step-growth polymerization following our own established transesterification-based methodology employing activated ester chemistry.78–80P1 and P2 had identical chemical structure but differed only in the hydrophobic group attached to the quaternary ammonium unit, where the former possessed a methyl group and the latter was equipped with a decyl chain. This structural variation led to contrasting differences in their minimum inhibitory concentration (MIC) values in water. Much-enhanced antibacterial properties were observed for P2, owing to the higher positive charge density and balanced hydrophobicity of its polymeric nanoaggregates. Herein, we examined the potential of these amphiphilic polyesters (P1 and P2) for dual mitochondria targeting and imaging due to their combined cationic and luminescent characteristics, an aspect that has not been previously investigated. Furthermore, to rationalize the significance of the cationic NMI pendants on mitochondria targeting, we compared the cellular uptake and mitochondria-tracking behavior of P1 and P2 with a newly designed neutral NMI-functionalized polyester P3 (Scheme 1a), which could also produce water-dispersible fluorescent nanoassemblies of comparable size to P1 and P2. Additionally, P3 possessed a tertiary amine group in its repeating unit that could be protonated under mild acidic conditions (pH ∼6.2) in the tumor microenvironment81,82 or in more acidic (pH = 5–5.5) intracellular organelles like endosomes and lysosomes.83 The pH-triggered84–87 positive charge accumulation in P3 possibly enables its nanoaggregates to preferentially locate in the mitochondria of cervical cancer (HeLa) cells without imparting any notable cytotoxicity at the tested concentrations. In contrast, in non-cancerous normal kidney epithelial (NKE) cells, self-assembled P3 gets trapped in other cytosolic compartments, likely due to differences in mitochondrial membrane potential or the lack of sufficient positive charge to target mitochondria effectively. Notably, the intrinsically cationic P1 and P2 nanoassemblies, although rapidly internalized and accumulated into the mitochondria of both cancerous and non-cancerous cells, failed to exhibit any cell selectivity. This work presents a new avenue to rationally design and prepare pH-responsive nanoassemblies for cell-selective mitochondria targeting from a water-dispersible chromophore-appended polyester with an enzymatically degradable backbone, which is rare in the field.
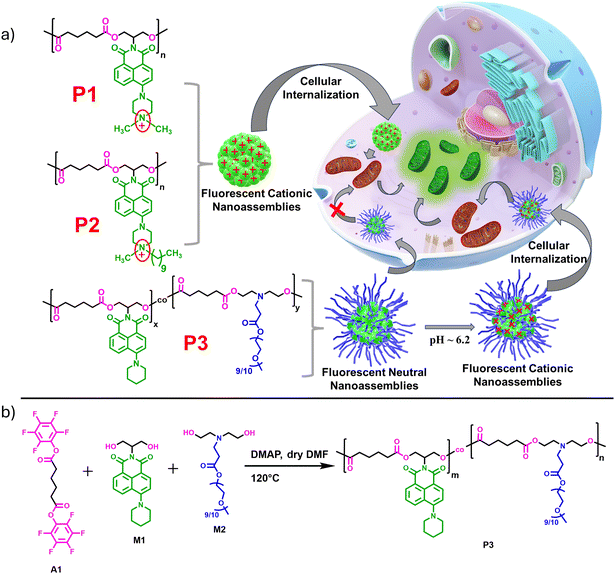 |
| | Scheme 1 (a) Structure of amphiphilic polyesters P1, P2, and P3 (left) and the schematic representation of the mitochondria-targeting property of their fluorescent nanoassemblies in water (right). (b) Synthetic scheme of the polymer P3. | |
Results and discussion
Synthesis, characterization and self-assembly
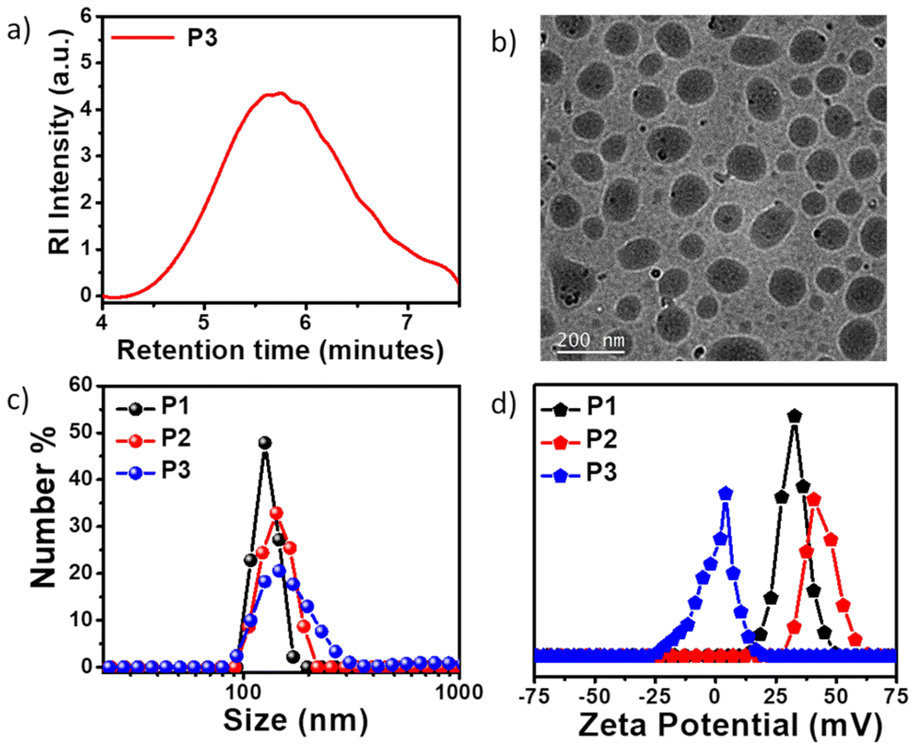
Detailed synthesis and characterization of the P3 polymer and its respective monomers have been discussed in the ESI.† For the synthesis of copolymer P3 (Scheme 1b), pentafluorophenyl adipate (A1) was reacted with a mixture of a neutral NMI-functionalized diol M1 and a newly synthesized hydrophilic diol M2 (Scheme S1†) via activated ester-mediated step-growth polymerization in the presence of 4-dimethylaminopyridine (DMAP) as an organocatalyst in DMF at 120 °C (Scheme 1b). From the 1H NMR studies (Fig. S1†), the molar ratio of the attached NMI to PEG in the side chains was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The size exclusion chromatography (SEC) curve (Fig. 1a) of P3 revealed a monomodal peak with a number average molecular weight (Mn) of ∼9000 g mol−1 and dispersity (Đ) of 1.73. Due to the presence of the polar cationic or polyethylene glycol (PEG) moiety in the side chains, all three polymers exhibit efficient self-assembly behavior in aqueous medium and intrinsic fluorescent properties that originate from the attached hydrophobic π-scaffold. Spherical nanoaggregates with comparable sizes ranging between 100–140 nm were obtained for the polymers P1, P2 and P3 in water (C = 100 μg mL−1), as can be seen from the cryo-transmission electron microscopy (Cryo-TEM) images (Fig. S2a, b† and Fig. 1b), which corroborated the DLS results (Fig. 1c). The critical aggregation concentrations (CAC) of P1 (56 μg mL−1), P2 (46 μg mL−1) and P3 (36 μg mL−1) were determined using pyrene as a hydrophobic probe (Fig. S3†).88,89 As anticipated, nanoparticles of P1 and P2 possessed high zeta potential (ζ) values of +33 and +41 mV (Fig. 1d), respectively, at the neutral physiological pH (pH ∼7.4). In contrast, P3 exhibited negligible positive charge (ζ ∼+4 mV) at the same physiological pH due to the absence of the quaternary ammonium group in the NMI side chains, unlike in P1 and P2. Absorption normalized emission spectra of P1, P2, and P3 in water showed comparable emission intensity for P1 and P2 but significantly weaker emission for P3 at the equivalent dye concentrations (Fig. S4†). Unlike the self-assembled nanoassemblies of P1 and P2, where the cationic NMI pendants are exposed to the bulk water for facilitating their colloidal stability (Scheme 1a), the P3 polymer exhibits a different stacking arrangement of the aromatic dyes. In P3, the hydrophobic NMI moieties are shielded from the surrounding aqueous environment and form the core of the nanoparticles, while the polar PEG chains constitute the stabilizing corona. This possibly makes the hydrophobically-assisted π-stacking within the NMI core more rigid in P3, leading to its substantially reduced emission compared to P1 and P2. Variable temperature 1H NMR studies (Fig. S5†) with P3 in D2O showed shielding effects of the aromatic protons at lower temperatures, further establishing the involvement of the attached NMI chromophores in π–π stacking within the hydrophobic core. Also, such collapsed self-assembled nanostructures were supported by a 17 nm red shift in the UV-vis spectrum of the P3 polymer in water compared to that in chloroform, where it remains in its molecularly dissolved state (Fig. S6†).
:1. The size exclusion chromatography (SEC) curve (Fig. 1a) of P3 revealed a monomodal peak with a number average molecular weight (Mn) of ∼9000 g mol−1 and dispersity (Đ) of 1.73. Due to the presence of the polar cationic or polyethylene glycol (PEG) moiety in the side chains, all three polymers exhibit efficient self-assembly behavior in aqueous medium and intrinsic fluorescent properties that originate from the attached hydrophobic π-scaffold. Spherical nanoaggregates with comparable sizes ranging between 100–140 nm were obtained for the polymers P1, P2 and P3 in water (C = 100 μg mL−1), as can be seen from the cryo-transmission electron microscopy (Cryo-TEM) images (Fig. S2a, b† and Fig. 1b), which corroborated the DLS results (Fig. 1c). The critical aggregation concentrations (CAC) of P1 (56 μg mL−1), P2 (46 μg mL−1) and P3 (36 μg mL−1) were determined using pyrene as a hydrophobic probe (Fig. S3†).88,89 As anticipated, nanoparticles of P1 and P2 possessed high zeta potential (ζ) values of +33 and +41 mV (Fig. 1d), respectively, at the neutral physiological pH (pH ∼7.4). In contrast, P3 exhibited negligible positive charge (ζ ∼+4 mV) at the same physiological pH due to the absence of the quaternary ammonium group in the NMI side chains, unlike in P1 and P2. Absorption normalized emission spectra of P1, P2, and P3 in water showed comparable emission intensity for P1 and P2 but significantly weaker emission for P3 at the equivalent dye concentrations (Fig. S4†). Unlike the self-assembled nanoassemblies of P1 and P2, where the cationic NMI pendants are exposed to the bulk water for facilitating their colloidal stability (Scheme 1a), the P3 polymer exhibits a different stacking arrangement of the aromatic dyes. In P3, the hydrophobic NMI moieties are shielded from the surrounding aqueous environment and form the core of the nanoparticles, while the polar PEG chains constitute the stabilizing corona. This possibly makes the hydrophobically-assisted π-stacking within the NMI core more rigid in P3, leading to its substantially reduced emission compared to P1 and P2. Variable temperature 1H NMR studies (Fig. S5†) with P3 in D2O showed shielding effects of the aromatic protons at lower temperatures, further establishing the involvement of the attached NMI chromophores in π–π stacking within the hydrophobic core. Also, such collapsed self-assembled nanostructures were supported by a 17 nm red shift in the UV-vis spectrum of the P3 polymer in water compared to that in chloroform, where it remains in its molecularly dissolved state (Fig. S6†).
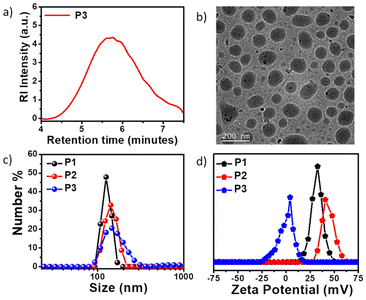 |
| | Fig. 1 (a) SEC plot of P3 with DMF as the eluent. (b) Cryo-TEM image of the polymer P3 nanoassemblies in water (C = 100 μg mL−1). (c) DLS plot of P1, P2, and P3 in water (C = 100 μg mL−1). (d) Zeta potential plots of P1, P2, and P3 polymers at pH = 7.4 (C = 100 μg mL−1). | |
Degradability and cytotoxicity assay
All three polymers P1, P2 and P3 possessed degradable ester linkages in their backbones, which are susceptible to degradation by enzymatic hydrolysis.90 As a representative example, the degradation of P3 was studied by its incubation with the Lipase B enzyme from Pseudomonas cepacia for 18 hours. The FT-IR spectra of P3 showed the absence of the characteristic ester peak at around 1733 cm−1, suggesting the breakdown of the polyester chain (Fig. 2a). The broad peak centered at 1667 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the peptide bonds in Lipase B. The polymer degradation of P3 was further validated by its SEC profile (Fig. S7†), which revealed an increment in the elution time following enzyme treatment. The degradation of P2 and P3 was also assessed in the presence of esterase enzyme from porcine liver.91 Time-dependent FT-IR studies showed partial disappearance of the ester carbonyl peaks of P2 and P3 after 4 hours and 18 hours and complete disappearance after 24 hours, suggesting the enzymatic degradability of the polyester backbone with increasing time (Fig. S8†).77 Furthermore, at an acidic pH of 4, the polymer P3 was found to be partially degraded as can be observed from the increment in retention time in the SEC trace (Fig. S8c†) compared to that at the physiological pH of 7.4, suggesting plausible degradation in the acidic lysosomal cavity. Enzymatic degradation was further validated by the stability test of the polymeric nanoassemblies in complete DMEM media, where their average size was retained over the tested period of 24 hours (Fig. S9a, b and c†). This was corroborated by the retention of the ester peak of the polymers in complete DMEM media after 24 hours in the absence of the enzyme, as observed from the FT-IR spectra (Fig. S9d, e and f†). The nanoparticles of the three polymers were also found to retain their stability up to the tested experimental temperature of 55 °C, as can be observed in the DLS plots (Fig. S9g, h and i†). The cytotoxicity of P1, P2, and P3 was estimated by MTT {3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide} assay using HeLa cells, following the standard protocol (Fig. 2b).92,93 All three polymer-treated cells were incubated for 24 hours at different concentrations. P3 demonstrated outstanding biocompatibility as it was found to be nontoxic up to a concentration of 400 μg mL−1, which is about 11 times its CAC. Considering the cationic nature of P1 and P2, their cell viability exceeding 80% even at 7–8 times higher CAC is noteworthy. Our previous study77 reveals that even the cationic monomers for P1 and P2 exhibit appreciable cell viability at similar concentration ranges, indicating that the degradation products have no discernible toxicity, which is a prerequisite for biodegradable polymers.
O stretching vibrations of the peptide bonds in Lipase B. The polymer degradation of P3 was further validated by its SEC profile (Fig. S7†), which revealed an increment in the elution time following enzyme treatment. The degradation of P2 and P3 was also assessed in the presence of esterase enzyme from porcine liver.91 Time-dependent FT-IR studies showed partial disappearance of the ester carbonyl peaks of P2 and P3 after 4 hours and 18 hours and complete disappearance after 24 hours, suggesting the enzymatic degradability of the polyester backbone with increasing time (Fig. S8†).77 Furthermore, at an acidic pH of 4, the polymer P3 was found to be partially degraded as can be observed from the increment in retention time in the SEC trace (Fig. S8c†) compared to that at the physiological pH of 7.4, suggesting plausible degradation in the acidic lysosomal cavity. Enzymatic degradation was further validated by the stability test of the polymeric nanoassemblies in complete DMEM media, where their average size was retained over the tested period of 24 hours (Fig. S9a, b and c†). This was corroborated by the retention of the ester peak of the polymers in complete DMEM media after 24 hours in the absence of the enzyme, as observed from the FT-IR spectra (Fig. S9d, e and f†). The nanoparticles of the three polymers were also found to retain their stability up to the tested experimental temperature of 55 °C, as can be observed in the DLS plots (Fig. S9g, h and i†). The cytotoxicity of P1, P2, and P3 was estimated by MTT {3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide} assay using HeLa cells, following the standard protocol (Fig. 2b).92,93 All three polymer-treated cells were incubated for 24 hours at different concentrations. P3 demonstrated outstanding biocompatibility as it was found to be nontoxic up to a concentration of 400 μg mL−1, which is about 11 times its CAC. Considering the cationic nature of P1 and P2, their cell viability exceeding 80% even at 7–8 times higher CAC is noteworthy. Our previous study77 reveals that even the cationic monomers for P1 and P2 exhibit appreciable cell viability at similar concentration ranges, indicating that the degradation products have no discernible toxicity, which is a prerequisite for biodegradable polymers.
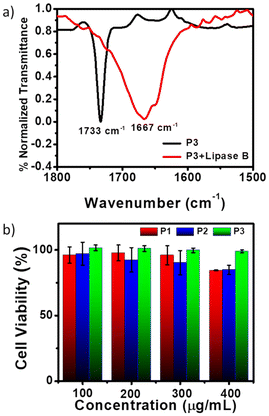 |
| | Fig. 2 (a) FT-IR spectra of the P3 polymer before and after degradation with the enzyme Lipase B from Pseudomonas cepacia. (b) Cytotoxicity of the HeLa cell line in the presence of different concentrations of P1, P2, and P3 after incubation for 24 h. Data are shown as the mean ± SD of three experimental replicates. | |
Cellular internalization and mitochondria tracking
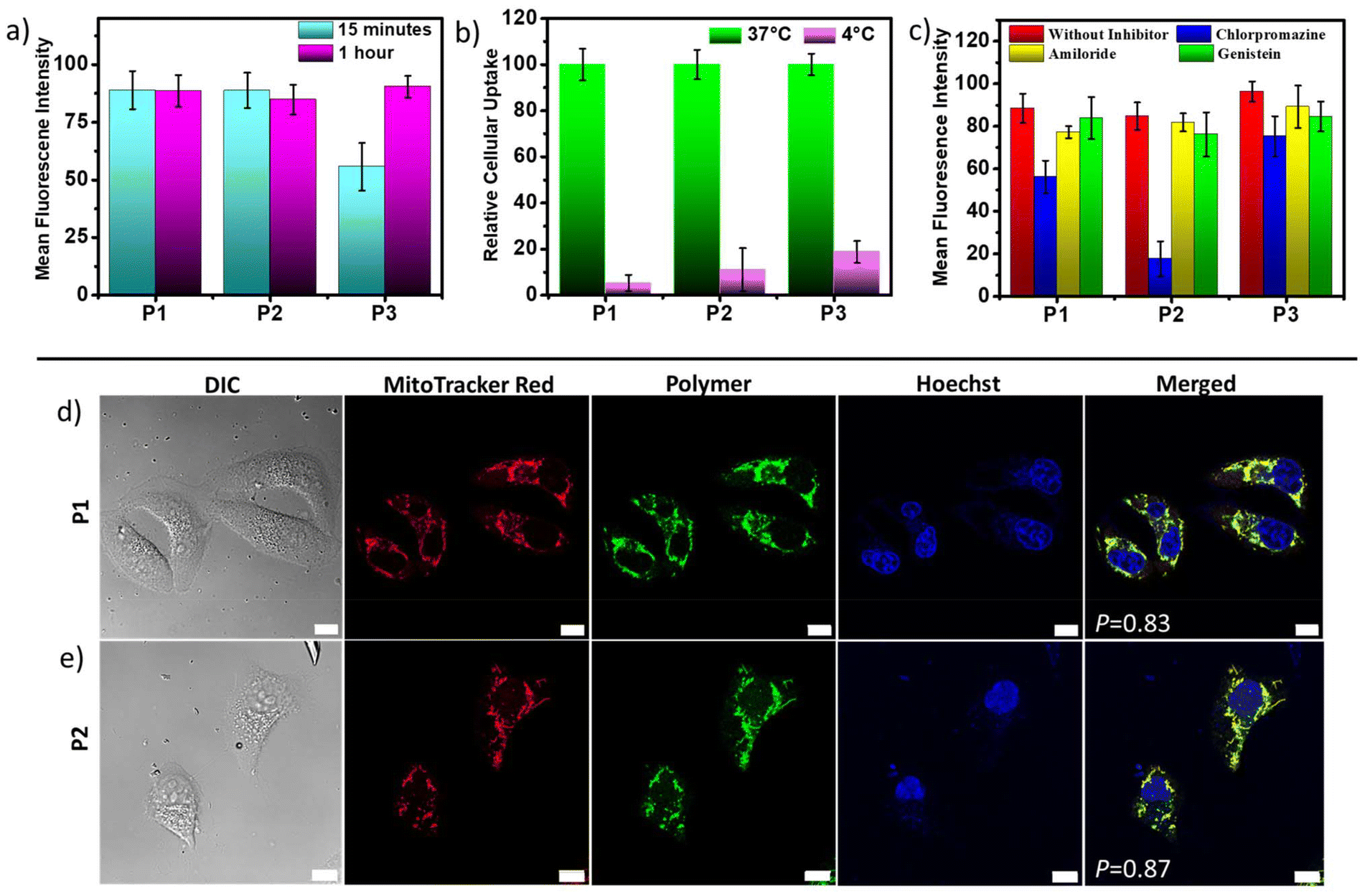
The aqueous nanoassemblies of P1, P2, and P3 apparently appear to be promising candidates for cellular uptake and transport studies due to their optimal size, excellent cell viability, fluorescence properties, and enzymatic degradability. To investigate this, we studied their cellular internalization at their absorbance normalized concentrations (C = 100 μg mL−1 for P1 and P2, 200 μg mL−1 for P3) in the HeLa cell line. The analysis was conducted through fluorescence-activated cell sorting (FACS), and the cellular uptake data were recorded after incubation of the polymers for 1 hour. A comparative study reveals that cellular internalization for all three polymers was close to 85% after 1 hour (Fig. 3a). However, the initial uptake rate in just 15 minutes is notably faster for P1 and P2, as compared to P3 (Fig. 3a). This is also complemented by the representative FACS data showing the relative fluorescence intensity variation with different incubation time (Fig. S10†). This difference likely stems from the fact that positively charged P1 and P2 nanoassemblies show higher affinity toward negatively charged cell membranes due to favorable electrostatic interactions, which promote their rapid internalization by the cells.21,81 Furthermore, all three polymer aggregates followed energy-dependent endocytosis, as evident from their drastically lower uptake values at 4 °C as compared to 37 °C (Fig. 3b and S11†). At lower temperatures, the receptors that are essential for the internalization process by the endocytosis pathway are said to be “frozen” for the entry of foreign particles,5,94 thereby reducing the endocytic uptake. To gain a better understanding of the cellular uptake mechanisms, the internalization of self-assembled P1, P2, and P3 in HeLa cells was investigated in the presence of three commonly used inhibitors, viz. amiloride hydrochloride, chlorpromazine, and genistein. These inhibitors are known to block micropinocytosis, clathrin-mediated endocytosis, and caveolae-mediated endocytosis, respectively (Fig. 3c).94 The results show that amiloride hydrochloride and genistein had negligible effects on the cellular uptake of all three polymers; however, chlorpromazine significantly inhibited the uptake of P1 and P2 to different extents, indicating that clathrin-mediated endocytosis plays an important role in their cellular internalization. P2 showed maximum inhibition in the presence of chlorpromazine when compared to P1, suggesting that P2 is predominantly internalized via clathrin-mediated endocytosis,94,95 while P1 might follow other pathways of internalization in addition to clathrin-mediated endocytosis. On the other hand, neutral P3 was anticipated to internalize through a mechanism that might be different from those of cationic P1 and P2, because none of the tested inhibitors significantly inhibited P3. A more detailed investigation is necessary to determine the actual endocytic pathway for P3.
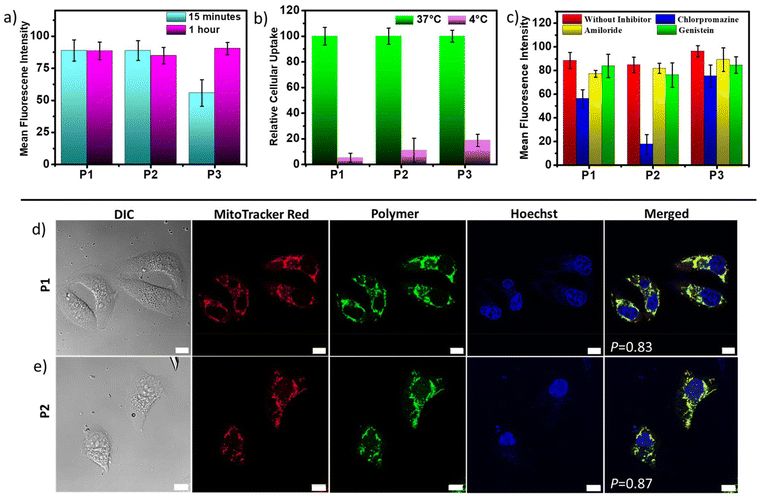 |
| | Fig. 3 Cellular uptake studies with HeLa cells treated with P1, P2, and P3 by FACS analysis. Comparative bar graph showing (a) 15 minutes and 1 hour incubation data, (b) 37 °C and 4 °C data (37 °C data normalized to 100) and (c) in the presence of different endocytosis inhibitors. The results are expressed with respect to the control experiments without the polymers. Data are shown as the mean ± SD of three experimental replicates. CLSM images of HeLa cells incubated with (d) P1 and (e) P2 for 30 minutes (C = 100 μg mL−1). Images from left to right are arranged as follows: differential interference contrast (DIC), red, green and blue channel emissions due to mitochondria-staining MitoTracker Red, intracellular polymer emission, and Hoechst 33342 staining of cell nuclei, respectively, and an overlay of these three images. P = Pearson's correlation coefficients. Scale bar = 10 μm. | |
Due to their inherent green emission, the fate of these self-assembled polyesters in HeLa cells after their cellular internalization could be traced by direct visualization using confocal laser scanning microscopy (CLSM). To further investigate their subcellular location, two commercially available organelle staining dyes, viz., MitoTracker Red and Hoechst (33342) Blue, were used for imaging the mitochondria and nucleus, respectively. Freshly grown HeLa cells were treated with cationic P1 and P2 for 24 hours, followed by incubation for 30 minutes at 37 °C, and then stained with these two biomarkers. The CLSM images (Fig. 3d and e) revealed a very high degree of colocalization between the red-emitting MitoTracker Red and the green-emitting polymers, with Pearson's correlation coefficients (P) of 0.83 for P1 and 0.87 for P2. Conversely, there was negligible overlap between the blue-emitting Hoechst dye and the green-emitting polymers, indicating that these cationic polyesters were mainly located in the cytosol rather than the nucleus. These findings indicate that both P1 and P2 exhibit a strong tendency to target mitochondria, which is not surprising considering their cationic nature. To further track the intracellular transport behavior of P1 and P2 and their dynamic localization in different cellular compartments, CLSM images were collected at different time intervals (Fig. 3d, e and S12†). For this study, we used the same red-emitting mitochondrial marker and switched to LysoTracker Blue for lysosome staining in place of Hoechst 33342 Blue, as the polymers did not show any tendency to target the nucleus. This enabled us to track the localization and dynamics of the nanoassemblies with respect to mitochondrial and lysosomal compartments. Even at a shorter incubation time of only 15 minutes, the P values for P1 and P2 reached ∼0.88 and ∼0.92, respectively (Fig. 4a and c), between the overlapping red and green fluorescent signals, indicating rapid cellular internalization and mitochondria staining in HeLa cells by both P1 and P2. Notably, after 4 hours of incubation, the polymers appeared to partly transport from the mitochondria to the endo-lysosomal cavity96 without causing any harm to the cells. The CLSM images revealed a substantial overlap of the green-emitting polymers with both MitoTracker Red and LysoTracker Blue, as indicated by the yellow and cyan emissions in the merged images, respectively. The P values for the correlation between blue and green signals significantly increased from 0.27 to 0.54 for P1 (Fig. 4a and b) and 0.29 to 0.78 for P2 (Fig. 4c and d), whereas the P values for the correlation between red and green signals decreased from 0.88 to 0.69 for P1 (Fig. 4a and b) and 0.92 to 0.66 for P2 (Fig. 4c and d), confirming gradual escape from the mitochondria to the lysosomal compartment over a period of 4 hours (Fig. 4b and d). The hydrophobic chain length difference between P1 and P2 did not show any significant difference in the cellular transport property during the observed time scale. The cellular internalization study using CLSM was complemented with the FACS data (Fig. 4e and f). For this, the HeLa cells were incubated with green-emitting P1 and P2 (C = 100 μg mL−1) for 15 minutes and their relative fluorescence intensities with respect to the unlabeled cells were monitored immediately and after 4 hours. To eliminate the effect of freely dispersed nanoparticles, if any, that were not internalized by the cells, the medium was aspirated, and the cell pellet was collected after centrifugation, to which fresh complete medium was added. The FACS data showed ∼85% uptake for both P1 and P2 within 15 minutes, as was also observed before (Fig. 3a). Furthermore, the fluorescence intensity peak shifted to lower values from 15 minutes to 4 hours (Fig. 4e and f), signifying that although the initial uptake is very high, after 4 hours, a gradual cellular excretion of these polymers possibly through the lysosomal compartment takes place. This validates the results obtained from the CLSM images.
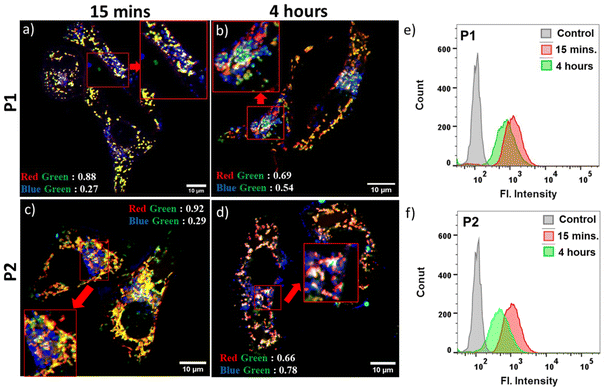 |
| | Fig. 4 CLSM images of HeLa cells incubated with (a and b) P1 and (c and d) P2 (C = 100 μg mL−1) for 15 minutes (a and c) and 4 hours (b and d). The data presented show an overlay of the images from the red, green and blue channel emissions due to the MitoTracker Red, internalized polymers, and LysoTracker Blue. Scale bar = 10 μm. (e and f) FACS analysis showing relative fluorescence intensity variation of polymers P1 and P2 (C = 100 μg mL−1) at different time points of incubation in HeLa cells. The left shift of the chromatogram indicates a decrease in intracellular polymer concentration after 4 hours. | |
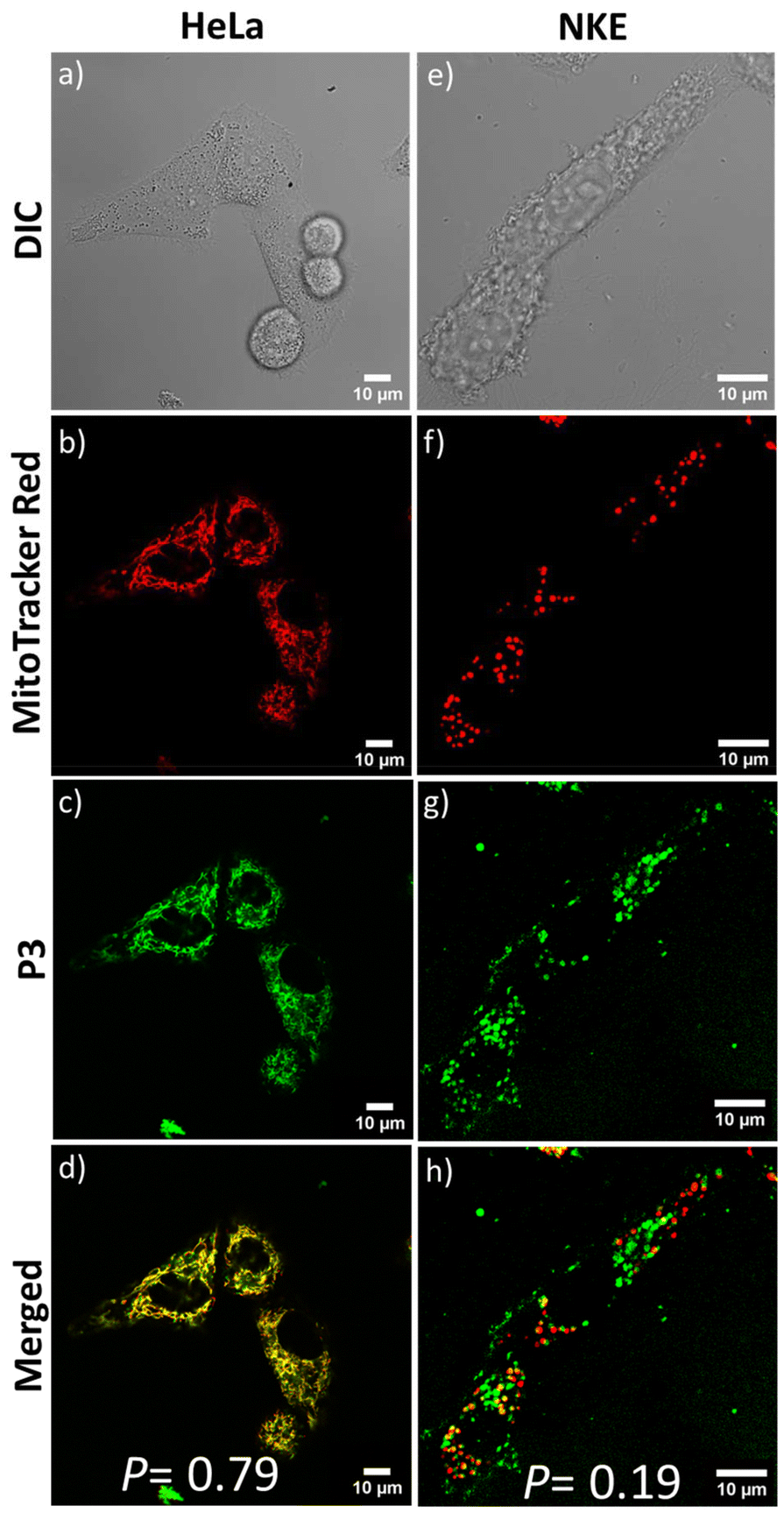
Next, to assess the impact of the positive charges, we examined the cellular uptake of P3. CLSM studies were performed to visualize the intracellular distribution of the neutral P3 inside HeLa cells. Similar to P1 and P2, the confocal images showed significant accumulation of P3 nanoassemblies in the mitochondria. This is evident from the merged image (Fig. 6d) showing yellow emission with significant fluorescence overlap between MitoTracker Red and the green-emitting P3 (P = 0.79). P3's ability to target mitochondria was unexpected as it did not possess the cationic NMI pendant. It is hypothesized that the acidic environment (pH ∼6.5) prevailing in the extracellular environment97 of the cancerous HeLa cells75,98,99 could protonate the repeating tertiary amine groups present in the P3 backbone. This can lead to the acquisition of sufficient positive charge by self-assembled P3, facilitating its interaction with the negatively charged mitochondrial membranes in a cancerous microenvironment, similar to cationic P1 and P2. The zeta potential value of +24 mV for P3 at an acidic pH ∼6.2 corroborates the above hypothesis (Fig. S13†). To further testify the significance of positive charge accumulation in deciding the fate of the polymer P3 in the cellular environment, we tested the mitochondrial targeting in a non-cancerous normal kidney epithelial (NKE) cell line. The intracellular pH of non-cancerous cell lines is known to be in the near-neutral range (pH ∼7.4), where P3 nanoaggregates have negligible positive charge (ζ ∼+4 mV) (Fig. 1d). After 1 hour of incubation, all three polymers showed more than 70% cellular internalization in NKE cells, as evident from the FACS data (Fig. 5a and b). However, the CLSM images of NKE cells incubated with self-assembled P3 (Fig. 6e–h) revealed negligible overlap between the red emission of MitoTracker Red and the green emission of P3 (P = 0.19), unlike their significant overlap observed in HeLa cells (P = 0.79). This further strengthens our hypothesis that the accumulation of positive charge by P3 nanoassemblies in the acidic environment of HeLa cells enhances their electrostatic interactions, enabling them to target cancer cell mitochondria, akin to P1 and P2, whereas in a normal cell line having neutral pH (∼7.4), the fate of distribution of P3 differs significantly. This is also facilitated by the fact that the mitochondrial membrane potential is more hyperpolarized in cancerous cells (−220 mV) than in normal cells (−140 mV).100 This significant difference may also facilitate preferential accumulation of P3 nanoaggregates in cancel cell mitochondria,100–102 despite acquiring a lower positive charge than P1 and P2. As anticipated, due to the inherent cationic nature of P2, it failed to distinguish between the mitochondria of cancerous and non-cancerous cells, which is evident from the appreciable overlap (P = 0.74) of its emission with that of MitoTracker Red in the normal NKE cell line also (Fig. S14†). Hence, P3 is able to target mitochondria in a cell-specific manner driven by its pH-dependent protonation under acidic conditions, which in turn decides its intracellular distribution, as evident from the contrasting results observed when P3 is internalized in cancerous compared to normal cells.
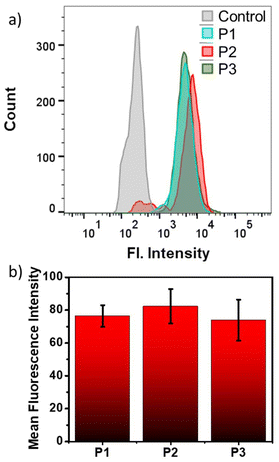 |
| | Fig. 5 (a) FACS analysis showing relative fluorescence intensity variation of polymers P1, P2 and P3 after 1 hour of incubation at 37 °C in the NKE cell line. (b) Comparative bar diagram showing cellular uptake data in the NKE cell line upon 1 hour of incubation at 37 °C. Data are shown as the mean ± SD of three experimental replicates. | |
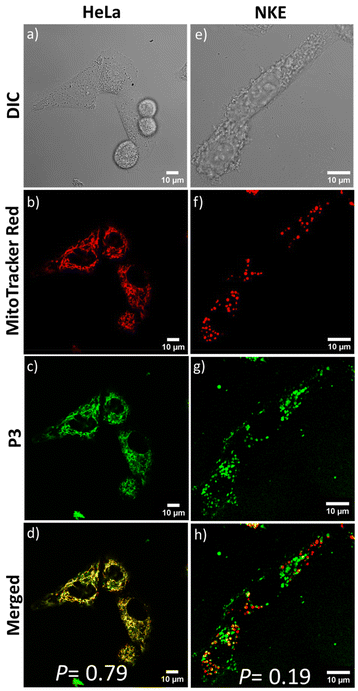 |
| | Fig. 6 CLSM images of HeLa and NKE cells incubated with P3 (C = 100 μg mL−1) for 1 hour. From top to bottom: images from differential interference contrast (DIC) (a and e), red (b and f) and green (c and g) channel emissions due to MitoTracker Red, and the P3 polymer, respectively, and their overlay (d and h). Scale bar = 10 μm. | |
Conclusions
In summary, we present three naphthalene monoimide (NMI)-functionalized amphiphilic polyesters, P1 and P2 (cationic), and P3 (neutral), and investigated their cellular uptake and mitochondria-targeting ability with the help of their inherent fluorescence properties. All three water-dispersible polymers form nanoassemblies of comparable size in water that exhibit high biocompatibility and efficient cellular uptake in HeLa cells via receptor-mediated endocytic pathways. While self-assembled P1 and P2 demonstrate rapid mitochondrial-targeting ability in HeLa cells originating from their cationic nature, time-dependent CLSM images validated by FACS data suggest their slow lysosomal excretion over time. Contrarily, nanoassemblies of polyester P3 target mitochondria in a cell-specific manner, which stems from their pH-triggered cationic properties. In acidic environments, such as those found in a tumor microenvironment, protonation of the tertiary amine groups in P3 increases its surface positive charge significantly, thereby enabling P3 nanoassemblies to selectively stain the mitochondria of cancerous cells (HeLa) over non-cancerous cells (NKE), also facilitated by their higher negative membrane potential. Typically, biocompatible polyesters such as polylactides or polycaprolactone that have great biomedical implications are hydrophobic, and they necessitate the incorporation of suitable hydrophilic segments (majorly non-biodegradable) for their water dispersibility. In the present study, we demonstrated an efficient molecular design for mitochondria-targeting amphiphilic polymers from cleavable polyester backbones that exhibit complete enzymatic breakdown under mild physiological conditions. Notably, the dual cationic and fluorescent properties of P1 and P2 provide a more straightforward approach for mitochondrial-targeting nanocarrier designs, eliminating the need for additional incorporation of mitochondrial localizing moieties, such as TPP (triphenylphosphonium), as previously reported.39,103 Additionally, judicial incorporation of a pH-dependent functional group enables cell-selective mitochondria targeting in P3. The findings of the present work appear to be highly promising for the future development of cell-specific, organelle-targeting drug delivery systems from structurally diverse polyester scaffolds, which can be prepared following our straightforward activated ester-mediated step-growth polymerization methodology.
Author contributions
S.B. and P.R. contributed equally to the design, experimentation, and analysis of the work. All the authors approved the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. B., and A. B. thank IACS for the research fellowship and infrastructural facilities. P. R. thanks Technical Research Centre (TRC) at IACS for the research fellowship. A. D. thanks the SERB India (Grant No. CRG/2022/003069) and TRC for funding and instrumental facilities. All the authors thank Professor S. Ghosh from IACS for providing the laboratory facilities for the cell culture work. All the authors thank Mr. Soumyadip Show for assisting in the graphical illustration.
References
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef PubMed.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef PubMed.
- I. Altinbasak, Y. Alp, R. Sanyal and A. Sanyal, Nanoscale, 2024, 16, 14033–14056 RSC.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef PubMed.
- K. Haupt, P. X. M. Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- P. Rajdev, P. Dey, I. Ghosh, R. Khamrui, J. Kar, S. S. Jana and S. Ghosh, ACS Macro Lett., 2021, 10, 1467–1473 CrossRef CAS.
- W. Zhang, R. Taheri-Ledari, F. Ganjali, S. S. Mirmohammadi, F. S. Qazi, M. Saeidirad, A. KashtiAray, S. Zarei-Shokat, Y. Tian and A. Maleki, RSC Adv., 2023, 13, 80–114 RSC.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- Y. Li, J. Liu, J. He, A. Dey, V. D. Bui and J. H. Park, Chem. Mater., 2024, 36, 4054–4077 CrossRef CAS.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- A. Gibalova, L. Kortekaas, J. Simke and B. J. Ravoo, Chem. – Eur. J., 2023, 29, e202302215 CrossRef CAS PubMed.
- S. R. Mane, A. Sathyan and R. Shunmugam, ACS Appl. Nano Mater., 2020, 3, 2104–2117 CrossRef CAS.
- S. Liu, R. Maheshwari and K. L. Kiick, Macromolecules, 2009, 42, 3–13 CrossRef CAS PubMed.
- E. A. Azzopardi, E. L. Ferguson and D. W. Thomas, J. Antimicrob. Chemother., 2013, 68, 257–274 CrossRef CAS PubMed.
- A. Nel, E. Ruoslahti and H. Meng, ACS Nano, 2017, 11, 9567–9569 CrossRef PubMed.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef PubMed.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2004, 56, 1023–1050 CrossRef PubMed.
- A. M. Weiss, M. A. Lopez, B. W. Rawe, S. Manna, Q. Chen, E. J. Mulder, S. J. Rowan and A. P. Esser-Kahn, Macromolecules, 2023, 56, 7286–7299 CrossRef PubMed.
- D. Tang, M. Cui, B. Wang, G. Liang, H. Zhang and H. Xiao, Nat. Commun., 2024, 15, 6026 CrossRef PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef PubMed.
- P. Ebrahimnejad, A. S. Taleghani, K. Asare-Addo and A. Nokhodchi, Drug Discovery Today, 2022, 27, 471–489 CrossRef PubMed.
- H. Kim, A. Jo, S. Baek, D. Lim, S.-Y. Park, S. K. Cho, J. W. Chung and J. Yoon, Sci. Rep., 2017, 7, 41090 CrossRef PubMed.
- S. Bag, M. P. Gadpayle, D. Ghosh, S. Maiti and P. De, Biomacromolecules, 2024, 25, 4233–4245 CrossRef PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef PubMed.
- C. Qian, Y. Chen, P. Feng, X. Xiao, M. Dong, J. Yu, Q. Hu, Q. Shen and Z. Gu, Acta Pharmacol. Sin., 2017, 38, 764–781 CrossRef PubMed.
- D. Wei, Y. Sun, H. Zhu and Q. Fu, ACS Nano, 2023, 17, 23223–23261 CrossRef CAS PubMed.
- A. Sharma, P. Verwilst, M. Li, D. Ma, N. Singh, J. Yoo, Y. Kim, Y. Yang, J.-H. Zhu and H. Huang, Chem. Rev., 2024, 124, 2699–2804 CrossRef CAS.
- L. Galluzzi, E. Morselli, O. Kepp, I. Vitale, A. Rigoni, E. Vacchelli, M. Michaud, H. Zischka, M. Castedo and G. Kroemer, Mol. Aspects Med., 2010, 31, 1–20 CrossRef CAS.
- J. S. Modica-Napolitano and K. K. Singh, Mitochondrion, 2004, 4, 755–762 CrossRef CAS.
- M. Z. Rasheed, H. Tabassum and S. Parvez, Protoplasma, 2017, 254, 33–42 CrossRef CAS.
- W. Kühlbrandt, BMC Biol., 2015, 13, 1–11 CrossRef.
- Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- J. Wang, J. Li, Y. Xiao, B. Fu and Z. Qin, RSC Med. Chem., 2020, 11, 858–875 RSC.
- A. Ashokan, M. Birnhak, B. Surnar, F. Nguyen, U. Basu, S. Guin and S. Dhar, Nanoscale, 2025, 17, 1260–1269 RSC.
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed.
- S. Kim, H. Y. Nam, J. Lee and J. Seo, Biochemistry, 2019, 59, 270–284 CrossRef PubMed.
- T. A. Tabish and M. R. Hamblin, Biomater. Biosyst., 2021, 3, 100023 Search PubMed.
- H. A. Prag, D. Kula-Alwar, L. Pala, S. T. Caldwell, T. E. Beach, A. M. James, K. Saeb-Parsy, T. Krieg, R. C. Hartley and M. P. Murphy, Mol. Pharm., 2020, 17, 3526–3540 CrossRef PubMed.
- H. Zhou, Q. Qian, Q. Chen, T. Chen, C. Wu, L. Chen, Z. Zhang, O. Wu, Y. Jin and X. Wang, Small, 2024, 20, 2308167 Search PubMed.
- Q. Chen, Q. Qian, H. Xu, H. Zhou, L. Chen, N. Shao, K. Zhang, T. Chen, H. Tian and Z. Zhang, ACS Nano, 2024, 18, 8885–8905 CrossRef PubMed.
- S. Sivagnanam, K. Das, I. Pan, A. Stewart, A. Barik, B. Maity and P. Das, RSC Chem. Biol., 2024, 5, 236–248 RSC.
- J. Wang, Q. Zhou, X. Li, D. Dutta and Z. Ge, ACS Macro Lett., 2022, 11, 543–548 CrossRef PubMed.
- A. Sevilla-Pym, W. L. Primrose, B. T. Luppi, K. Bergmann and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2024, 16, 46133–46144 CrossRef.
- J. Jin, P. Yuan, W. Yu, J. Lin, A. Xu, X. Xu, J. Lou, T. Yu, C. Qian, B. Liu, J. Song, L. Li, Y. Piao, T. Xie, Y. Shen, H. Tao and J. Tang, ACS Nano, 2022, 16, 10327–10340 CrossRef PubMed.
- Y. Geng, Y. Zhong, Q. Zhou, S. Chen, Y. Piao, W. Yin, H. Lu and Y. Shen, Chem. Commun., 2019, 55, 10015–10018 RSC.
- J. Gao, K. Dutta, J. Zhuang and S. Thayumanavan, Angew. Chem., Int. Ed., 2020, 59, 23466–23470 CrossRef.
- S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16288–16293 CrossRef.
- M. Twomey, E. Mendez, R. K. Manian, S. Lee and J. H. Moon, Chem. Commun., 2016, 52, 4910–4913 RSC.
- Z. Khatun, Y. S. Choi, Y. G. Kim, K. Yoon, M. Nurunnabi, L. Li, E. Lee, H. C. Kang and K. M. Huh, Biomacromolecules, 2017, 18, 1074–1085 CrossRef CAS.
- Kenry and B. Liu, Biomacromolecules, 2018, 19, 1783–1803 CrossRef CAS PubMed.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., 2019, 131, 7739–7743 CrossRef.
- M. Redrado, Z. Xiao, K. Upitak, B. Doan, C. M. Thomas and G. Gasser, Adv. Funct. Mater., 2024, 2401950 CrossRef CAS.
-
E. Piskin, J. Biomater. Sci., Polym. Ed, 1995, 6, 775–795 Search PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- Z. Y. Ong, C. Yang, W. Cheng, Z. X. Voo, W. Chin, J. L. Hedrick and Y. Y. Yang, Acta Biomater., 2017, 54, 201–211 CrossRef CAS PubMed.
- Y. Li, D. Maciel, J. Rodrigues, X. Shi and H. Tomás, Chem. Rev., 2015, 115, 8564–8608 CrossRef CAS.
- C.-S. Ha and J. A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef CAS.
- B. Jia, H. Huang, Z. Dong, X. Ren, Y. Lu, W. Wang, S. Zhou, X. Zhao and B. Guo, Chem. Soc. Rev, 2024, 53, 4086–4153 RSC.
- H. Ye, K. Zhang, D. Kai, Z. Li and X. J. Loh, Chem. Soc. Rev., 2018, 47, 4545–4580 RSC.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef PubMed.
- A. Kirillova, T. R. Yeazel, D. Asheghali, S. R. Petersen, S. Dort, K. Gall and M. L. Becker, Chem. Rev., 2021, 121, 11238–11304 CrossRef.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L.-W. Majumder and G. W. Huber, Chem. Rev., 2023, 123, 9915–9939 CrossRef.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- V. Fakhri, C.-H. Su, M. T. Dare, M. Bazmi, A. Jafari and V. Pirouzfar, J. Mater. Chem. B, 2023, 11, 9597–9629 RSC.
- R. N. Darie-Niţă, M. Râpă and S. Frąckowiak, Polymers, 2022, 14, 951 CrossRef.
- A. K. Pearce and R. K. O'Reilly, Biomacromolecules, 2021, 22, 4459–4469 CrossRef PubMed.
- S. Kashyap, N. Singh, B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 384–398 CrossRef PubMed.
- S. Saxena and M. Jayakannan, Biomacromolecules, 2017, 18, 2594–2609 CrossRef PubMed.
- S. Biswas, R. Barman, M. Biswas, A. Banerjee and A. Das, Polym. Chem., 2024, 15, 2753–2762 RSC.
- S. Biswas and A. Das, Chem. – Eur. J., 2023, 29, e202203849 CrossRef PubMed.
- A. Das, S. Lin and P. Theato, ACS Macro Lett., 2017, 6, 50–55 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS.
- X. Hu, X. Guan, J. Li, Q. Pei, M. Liu, Z. Xie and X. Jing, Chem. Commun., 2014, 50, 9188–9191 RSC.
- S. Santra, S. Das, S. Dey, A. Sengupta, B. Giri and M. R. Molla, Biomacromolecules, 2024, 25, 1724–1737 CrossRef CAS.
- M. Y. Chin, A. R. Patwardhan, K.-H. Ang, A. L. Wang, C. Alquezar, M. Welch, P. T. Nguyen, M. Grabe, A. V. Molofsky, M. R. Arkin and A. W. Kao, ACS Sens., 2021, 6, 2168–2180 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Z. Wang, T. Luo, R. Sheng, H. Li, J. Sun and A. Cao, Biomacromolecules, 2016, 17, 98–110 CrossRef CAS.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS.
- M. A. Beach, S. L. Y. Teo, M. Z. Chen, S. A. Smith, C. W. Pouton, A. P. R. Johnston and G. K. Such, ACS Appl. Mater. Interfaces, 2021, 14, 3653–3661 CrossRef PubMed.
- Y. Xia, H. Yao, Z. Miao, Y. Ma, M. Cui, L. Yan, H. Ling and Z. Qi, RSC Adv., 2015, 5, 50955–50961 RSC.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- M. T. Zumstein, D. Rechsteiner, N. Roduner, V. Perz, D. Ribitsch, G. M. Guebitz, H.-P. E. Kohler, K. McNeill and M. Sander, Environ. Sci. Technol., 2017, 51, 7476–7485 CrossRef CAS.
- M. Okada, K. Tsunoda, K. Tachikawa and K. Aoi, J. Appl. Polym. Sci., 2000, 77, 338–346 CrossRef.
- M. C. Arno, J. D. Simpson, L. D. Blackman, R. P. Brannigan, K. J. Thurecht and A. P. Dove, Biomater. Sci., 2023, 11, 908–915 RSC.
- L. Chambre, A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2018, 29, 1885–1896 CrossRef PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef PubMed.
- X. Liu, F. Wu, Y. Tian, M. Wu, Q. Zhou, S. Jiang and Z. Niu, Sci. Rep., 2016, 6, 24567 CrossRef PubMed.
- N. A. Bright, L. J. Davis and J. P. Luzio, Curr. Biol., 2016, 26, 2233–2245 CrossRef PubMed.
- S. Kolay, M. Das, A. Mondal, A. Sengupta, S. Bag, P. De and M. R. Molla, Biomacromolecules, 2024, 25, 5068–5080 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- H. Hou, Y. Zhao, C. Li, M. Wang, X. Xu and Y. Jin, Sci. Rep., 2017, 7, 1759 CrossRef PubMed.
- M. D. Forrest, BioRxiv, 2015, 25197 Search PubMed.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef PubMed.
- M. Huang, C. R. Myers, Y. Wang and M. You, Cancer Prev. Res., 2021, 14, 285–306 CrossRef PubMed.
- L. Jiang, S. Zhou, X. Zhang, C. Li, S. Ji, H. Mao and X. Jiang, Nat. Commun., 2021, 12, 2390 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*