
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Colloidal organometallic synthesis of solution-processable barium titanate nanoparticles for nanoelectronic applications†
Lara Kim Linkea,
Katharina E. Dehma,
Kirill Gubanov b,
Rainer H. Finkb,
Bartłomiej M. Szyjac and
Ryan W. Crisp*a
b,
Rainer H. Finkb,
Bartłomiej M. Szyjac and
Ryan W. Crisp*a
aChemistry of Thin Film Materials, Department of Chemistry and Pharmacy, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstr. 3, 91058 Erlangen, Germany. E-mail: Ryan.Crisp@FAU.de
bDepartment of Chemistry and Pharmacy, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany
cInstitute of Advanced Materials, Faculty of Chemistry, Wrocław University of Science and Technology, Gdańska 7/9, 50-344 Wrocław, Poland
First published on 26th February 2025
Abstract
Perovskite oxides like barium titanate (BaTiO3) exhibit desirable properties: notably high dielectric constants, piezoelectricity, and ferroelectricity, thereby enabling more advanced electronic devices and actuators. There are numerous synthesis procedures for BaTiO3, among which, nanoparticle syntheses are versatile and well-studied. However, colloidal organometallic synthesis is less commonly employed for this material despite offering processing advantages like facile compositional control and customizable surface chemistry. Here, an organometallic synthesis route is explored to produce colloidally stable BaTiO3 nanoparticles with oleyl alkoxide ligands. Subsequently, we further develop ligand exchange procedures with X-type ligands using KOH and oxalic acid to produce colloidal inks applicable for solution-processed nanocrystalline films for dielectrics in devices for which there is still a need for better nanoscale control. The BaTiO3 nanoparticles and films were characterized using X-ray diffraction (XRD), scanning transmission electron microscopy (STEM), energy-dispersive X-ray spectroscopy (EDS), Fourier-transform infrared spectroscopy (FT-IR), atomic force microscopy (AFM), Kelvin probe force microscopy (KPFM), and density functional theory (DFT), to understand their properties and to develop processes for device applications.
Introduction
The advent of colloidal nanoparticles has significantly broadened the scope of material applications compared to their bulk counterparts. At the nanoscale, a higher surface-to-volume ratio is achieved, with a larger proportion of atoms residing at the surface. This larger surface area allows for greater manipulation of processing parameters such as dispersing media and crystal habit orientation, offering additional opportunities for tuning optoelectronic properties.1–4 Moreover, quantum effects become prominent at the nanoscale, leading to alterations in the electronic, optical, and magnetic characteristics of materials.5,6 Control over these properties has driven the rapid advance of nanotechnology into various sectors, including medicine, electronics, energy, and environmental science.7,8 Specifically, metal oxide perovskites exhibit properties like a high dielectric constant,9 piezoelectricity,10–12 ferroelectricity,13,14 dielectricity,15 and catalytic behaviour.16 Due to its dielectric and ferroelectric properties, BaTiO3 is a well-known representative of perovskites and is frequently used for applications ranging from general electronic devices17–19 to transducers20–22 and actuators,22–24 making it a focal point of research in materials science and engineering.Various methods have been explored to synthesize BaTiO3 nanoparticles, including sol–gel, hydrothermal, and sonochemical routes.25 Although organometallic synthesis methods have many advantages, they are less often employed to create BaTiO3 colloidal nanoparticles. These organometallic synthesis routes yield material with facile processing methods for assembling nanoparticles into thin films and other structured materials for device applications. Organic ligands are utilized to enhance dispersibility, for compatibility with substrates, and to introduce functional groups.26–28 Through steric stabilization, organic ligands help prevent agglomeration and ensure long-term stability in colloidal suspensions, which is problematic with other synthesis routes in aqueous environments or thermal decomposition methods.29,30
In this study, we fine-tune existing organometallic synthesis approaches with a focus on elucidating the reaction mechanisms to achieve an ink of BaTiO3 nanoparticles as a stable colloidal dispersion showing limited agglomeration and sedimentation over day-long timescales. We then develop both layer-by-layer (LbL) assembly and solution-phase ligand exchange (SLE) procedures with oxalic acid and KOH. Employing X-type ionic ligands with minimal steric hindrance — an approach not previously explored for BaTiO3 nanoparticles — facilitates deposition on substrates to ensure direct contact. With this approach, continuous crack-free dielectric coatings are possible which have applications and are needed in a variety of scenarios.31 The resulting films are evaluated for their physical, chemical, and dielectric properties. Finally, we explore the potential of these nanoparticles as inks for the fabrication of solution-processed piezoelectric films, thereby expanding their application scope.
Methods and materials
Materials
Titanium(IV) isopropoxide (TTIP, 98+%) was purchased from Acros Organics. Metallic barium(0) (Ba) (99.99%), anhydrous benzyl alcohol (99.8%), toluene (≥99.7%), oleylamine (OLA) (≥98%), oleyl alcohol (≥85%) and dimethyl sulfoxide-d6 (DMSO-d6) were purchased from Sigma-Aldrich. Oxalic acid dihydrate was obtained from VWR Chemicals. Potassium hydroxide (KOH) (≥85%) was obtained from Honeywell Fluka. Hexane (∼95%) and methanol (MeOH) (≥99.9%) were purchased from Fisher Chemical. Silicon substrates with a native SiO2 layer were purchased from Silicon Materials, microscope glass slides were purchased from Epredia, indium tin oxide-coated (ITO) glass slides with a resistivity of 10 Ω sq−1 were purchased from Techinstro and were all cleaned by sonication in Hellmanex III (2% in water), acetone, isopropanol, and ultrapure water for 5 min each. The slides were dried under N2 stream. Formvar-supported 200 mesh copper grids were purchased from Plano. All chemicals were used without further purification unless stated.Devices
The nuclear magnetic resonance spectroscopy (NMR) data were obtained in DMSO-d6 and filtered over a PTFE syringe filter with a pore size of 45 μm to remove solids. The 1H NMR spectra were measured on a Bruker AVANCE 400WB 400 MHz spectrometer. The spectra were recorded at room temperature, without air exposure, and referenced to the solvent residual signal. The FT-IR measurements were performed using a Shimadzu IR Prestige 21 at room temperature and in air. UV-Vis measurements were performed with an Ocean Optics USB4000-UV-VIS in a 1 mm quartz cuvette with hexane as the solvent. The STEM images were acquired with a JSM-F100 (JEOL Ltd) with a field emission gun. Atomic composition analysis was performed with an attached EDS detector. The samples were prepared on carbon-coated copper grids or silicon wafers via drop-casting.XRD patterns were obtained using a D8 Advance instrument from Bruker equipped with a Lynxeye XE-T detector and Cu Kα source. The samples were prepared on glass slides via drop-casting.
For centrifugation of the BaTiO3 particles, a Multifuge X1R by Thermo Scientific was used at a speed of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and a relative centrifugal force (RCF) of 16747g for 10 min or 20 min.
000 rpm and a relative centrifugal force (RCF) of 16747g for 10 min or 20 min.
For centrifugation after the SLE, a VWR MiniStar Silverline with a maximum speed of 6000 rpm was used for 4 min.
The AFM/KPFM measurements were carried out using a JPK NanoWizard 4 system in non-contact mode, utilizing the ElectriMulti75-G silicon probes with an overall platinum coating and resonance frequency of 75 kHz. Surface potential was measured with KPFM by recording a contact potential difference (CPD) between a scanning probe (tip) and the sample surface. The surface morphology was simultaneously observed via AFM with 512 × 512 pixels resolution for film morphology and with 512 × 170 pixels for line scans; with a set point of 15 nm and line rate of 0.5 Hz. All samples were electrically grounded to avoid surface charging. The micrographs were analysed with JPK NanoWizard SPM Data Processing software.
Barium and titanium precursor preparation
For the precursor preparation, a procedure from literature was adapted from Z. Chen et al.32 Metallic Ba (0.5 mmol, 0.07 g) is dissolved in oleyl alcohol (OLOH) (2.5 mL) on a hot plate, which is set to 220 °C in a glovebox. The formation of gas bubbles is observed, and the mixture is stirred until a transparent solution is formed and the gas formation stops. After cooling to room temperature, TTIP (0.5 mmol, 0.15 mL) is added, and stirred overnight. A slightly yellow transparent solution is formed and contains two important components: the Ba and Ti species of Fig. S1† and the excess alcohol.An alternative synthetic approach with benzyl alcohol (BzOH) following a procedure from Z. Chen et al. was compared.32 Metallic Ba (0.5 mmol, 0.07 g) is dissolved in anhydrous BzOH (5.0 mL) at 100 °C and then stirred overnight at 60 °C after TTIP (0.5 mmol, 0.15 mL) is added. The precursor synthesis with BzOH formed the white precursor precipitate, as mentioned in ref. 32 but did not yield BaTiO3 nanoparticles in later steps and was not further explored.
For comparison to existing literature,32 the precursor synthesis was simplified to a one-step procedure (instead of two) by simultaneously adding the TTIP (0.5 mmol, 0.15 mL) and Ba (0.5 mmol, 0.07 g) to OLOH (2.5 mL) and heating the mixture on a hot plate set to 200 °C until the Ba and TTIP dissolve. The reaction proceeds similarly with our 1-step approach in half a day instead of overnight. This one-step approach was also done using BzOH (5.0 mL) at 80 °C. In both cases the precursor synthesis took place in under 4 h. Furthermore, the stability and scalability of the OLOH-based precursor was tested by scaling-up without issues by 5×. The stability of the precursor was tested by measuring the absorbance spectra before and after aging in a glovebox and in air. After 10 days, the precursor stored in an inert atmosphere showed no changes while the precursor stored in air showed a shift in the spectra that we attribute to oxidation (see Fig. S4†). This shift begins to occur after ca. 1 hour of air exposure.
BaTiO3 nanoparticle heat-up synthesis
For a heat-up synthesis, the procedure from Z. Chen et al.32 was adapted. The prepared precursor is transferred out of the glove-box in a syringe and injected immediately through a septum into a 50 mL three-neck flask filled with OLA (11.3 mL), equipped with a reflux condenser, a magnetic stir bar, and a thermocouple, sealed with septa and under N2-atmosphere. The OLA and precursor were then heated to 320 °C via a heating ramp of approximately 1.5 °C min−1 over 3 h starting at 20 °C while stirring and then maintained for 48 h. For isolating the as-synthesized particles, acetone (3:1) (vol:vol) was added as a polar anti-solvent to flocculate the particles, which were collected via centrifugation at 12000 rpm (RCF 16747g) for 10 min, at 15 °C. After centrifugation, the supernatant was discarded, and the nanoparticle pellet redispersed in toluene. This procedure was repeated a total of three times after which the nanoparticles were dispersed in hexane and stored under inert atmosphere. One aspect to note, the magnetic stir bars typically used in nanoparticle synthesis consist of neodymium or samarium magnets encased in PTFE (Polytetrafluoroethylene) and have been observed to swell during our syntheses though we find no leaching of the magnet's elements into the product when used only once. According to PTFE compatibility charts, there is no documented incompatibility with alkaline earth metals, amines, or any other reactants used in this reaction. However, DuPont reports a melting point of 327 °C for PTFE Teflon which is close to the reaction temperature of 320 °C. The prolonged exposure to these high temperatures could explain the observed swelling.33 Reusing stir bars leads to inconsistent results. Glass-coated stir bars are not suitable as the reaction temperature is above the supplier's (Sigma Aldrich) recommended maximum temperature of 274 °C.
Layer-by-layer ligand exchange of the BaTiO3 nanoparticles via dip coating
Solution-phase transfer ligand exchange of the BaTiO3 nanoparticles
:MeOH 1:2, vol:vol), shaken, and left to separate following a similar recipe as reported in ref. 34. After the phase transfer, the colourless hexane phase is on top and the white MeOH phase at the bottom, which contains the agglomerated BaTiO3 nanoparticles. This can be seen in Fig. 2. The hexane phase was carefully pipetted off, the vial centrifuged, and the sedimented precipitate was redispersed in MeOH (0.4 ml) and used as-is for further characterization.:MeOH 1:2, vol:vol), shaken, and left to stand until the phase separation occurred. The colourless hexane phase on top was separated from the slightly turbid MeOH phase at the bottom, which contains agglomerated nanoparticles, which were again isolated from the MeOH with centrifugation and dispersed in fresh MeOH (0.4 ml).Annealing the BaTiO3 nanoparticles
The BaTiO3 particles dispersed in hexane were drop-cast on a glass slide and subsequently annealed in a N2 glovebox to 250 °C and 350 °C for 45 min.DFT modelling
The structure of the BaTiO3 unit cell was taken from Materials Project database35 (mp-2998) from database version v2023.11.1. The unit cell had a cubic structure with the lengths a = b = c = 4.01 Å and angles α = β = γ = 90°. Based on the unit cell, three slabs corresponding to (100), (110), and (111) Miller indices were built to investigate the electronic structures of the surfaces. Each slab contained a stoichiometric number of Ba, Ti, and O atoms (Ba48, Ti48, O144) – in total 240 atoms.The structure with (100) Miller indices was built with the supercell of 4 × 4 × 3 unit cells, consisted of 6 layers of atoms, and its dimensions were a = 16.031 Å, b = 16.031 Å, c = 30.000 Å, with all angles equal to α = β = γ = 90°. The structure corresponding to (110) Miller indices contained 8 layers of atoms, had the dimensions of a = 16.028 Å, b = 17.003 Å, c = 30.000 Å, and all angles equal to α = β = γ = 90°. The (111) structure contained 12 layers of atoms, had the dimensions of a = 11.335 Å, b = 22.670 Å, c = 30.000 Å, with the angles of α = β = 90°, γ = 120°.
All calculations were carried out with VASP (ver. 5.4.4)36,37 within the framework of the DFT. The exchange–correlation energy was used in the Perdew–Burke–Ernzerhof form.38 The energy cutoff was set to 500 eV in all calculations. The electron–ion interactions were described by the projector-augmented wave method.37,39 Spin-polarized calculations have been performed to account for possible magnetic effects. The Brillouin zone was sampled with Γ point only due to sufficiently large system size. To account for the strong on-site Coulomb interactions, we used the Hubbard U correction scheme, with U = 10.0 eV for Ti atoms.40
Results and discussion
The formation mechanism of the metallic precursor involves two steps and can be seen in Fig. S1.† In the first step, metallic Ba reacts with the employed alcohol, either OLOH or BzOH, to form the barium alkoxide, releasing hydrogen gas (see Fig. S1†). The hydrogen release can be observed through the formation of bubbles on the surface of the metallic barium. The second step, as proposed by M. Niederberger et al., involves the formation of a Ti complex. In this step, a β-carbon atom from the isopropoxide nucleophilically attacks the methyl group of the alcohol.41 This process is activated by the interaction between the alcohol's hydroxyl group (–OH) and Ti. The outcome is a Ti complex with the coordinated alcohol and hydroxyl groups, which then forms a Ti–O–Ti species through the elimination of the alcohol. This proposed reaction mechanism suggests that the white precursor precipitate is not a bimetallic precursor, as often described,32,41,42 but rather a mixture of two metallic precursor species with excess alcohol. To gain further insights into the precursor and its reaction mechanism, NMR spectra, FT-IR spectra, and EDS analysis were used. For analysis via FT-IR spectroscopy and NMR spectroscopy, the precursor synthesized with BzOH was chosen over the precursor synthesized with OLOH. This decision was based on the higher boiling point and more complex structure of OLOH, due to its long carbon chain, which makes it harder to remove excess organic residues and complicates the assignment of the NMR signals. The precursor preparation, which in the beginning required overnight heating, was turned into a one-step reaction. When Ba and TTIP are added together and react simultaneously, the released hydrogen from the reaction of Ba with the alcohol can catalyse the Ti–O–Ti species forming reaction. With this one-step approach, the precursor forms faster and no longer requires overnight heating.The NMR spectrum of the barium complex in Fig. S2a† shows the aromatic signals between 7.29 ppm and 7.20 ppm and the methylene group as a singlet at 4.49 ppm. A signal of the OH-group is not detected. The missing chemical shift of the hydroxyl group, supports the formation of the barium alkoxide, seen in Fig. S1a.†
The aromatic signals between 7.31 ppm and 7.22 ppm and the methylene group 4.48 ppm–4.50 ppm are also detected in the NMR spectrum of the dried white precipitate (Fig. S2b†). However, the methylene peak presents as a doublet and an additional triplet at 5.17 ppm–5.14 ppm appeared. This peak can be explained by the hydroxyl group of residual BzOH. The remaining singlet signal at 3.32 ppm could potentially be attributed to the Ti–O–Ti species with the two anticipated signals of the isopropyl group overlapping, although they do not appear as the expected septet and doublet.
Consistent with the NMR in Fig. S2a† no broad O–H band is detected in the FT-IR spectrum (Fig. S3†), which is expected in the range of 3000 cm−1–3500 cm−1.43 The absorption peaks between 1400 cm−1–1500 cm−1 can be associated with the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the benzyl alkoxide.43 The absorption peak at 3020 cm−1 is also associated with the aromatic ring, as it results from C–H stretching vibrations. At 2840 cm−1 and 2882 cm−1 peaks of CH3 vibrations are visible, which do not result from the barium alkoxide. These bands could be attributed to the Ti species. The bands at 752 cm−1 and 826 cm−1 correspond to those attributed to the Ti–O–Ti vibrations reported in another similar Ti complex (with bands attributed to the Ti–O–Ti vibrations at 763 cm−1 and 820 cm−1) likely indicating that a Ti–O–Ti species is also present in our case.44 Although the FT-IR spectrum cannot prove the structure of the literature-suggested Ti complex, the two bands in the fingerprint area can support the complex in Fig. S1b† step 4 and 5′.
C stretching vibrations of the benzyl alkoxide.43 The absorption peak at 3020 cm−1 is also associated with the aromatic ring, as it results from C–H stretching vibrations. At 2840 cm−1 and 2882 cm−1 peaks of CH3 vibrations are visible, which do not result from the barium alkoxide. These bands could be attributed to the Ti species. The bands at 752 cm−1 and 826 cm−1 correspond to those attributed to the Ti–O–Ti vibrations reported in another similar Ti complex (with bands attributed to the Ti–O–Ti vibrations at 763 cm−1 and 820 cm−1) likely indicating that a Ti–O–Ti species is also present in our case.44 Although the FT-IR spectrum cannot prove the structure of the literature-suggested Ti complex, the two bands in the fingerprint area can support the complex in Fig. S1b† step 4 and 5′.
Without excess alcohol that the white precipitate is dispersed in (isolated via centrifugation), the nanoparticle synthesis results in titanium dioxide (TiO2) and barium oxide (BaO) instead of the tertiary BaTiO3 particles (see Fig. S5†). Performing the synthesis without the supernatant of the metallic precursor, the reaction mixture turned black and cloudy instead of remaining a transparent yellow. This indicates oxygen deficiency and the formation of black metal oxides, which is well-documented in the literature and can be explained by oxygen vacancies.45–47 The supernatant, consisting of excess alcohol, therefore, must act as an oxygen source in the reaction driving it to the ternary compound as opposed to the binaries.
The XRD patterns for the syntheses using the precursor without the supernatant and using the precursor in BzOH show signals for the binary oxides BaO and TiO2 instead of the desired BaTiO3 (see Fig. S5†). The three reflections between 10° and 20° can be attributed to the polymerization of organic residue solvents, already well reported for unsaturated hydrocarbons.48
The BaTiO3 nanoparticles, resulting from the heat-up synthesis with OLOH as the employed alcohol, are confirmed by the XRD pattern (Fig. 1a). The reflections are broadened as a result of their small crystallite size that was approximated with the Scherrer equation, defined as:
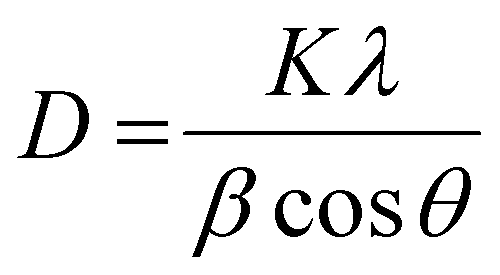 | (1) |
 | ||
| Fig. 1 Physical characterization of the as-synthesized BaTiO3 particles with (a) the XRD pattern and BaTiO3 reference patterns of the cubic and tetragonal phases. * indicates a reflection from the holder. (b) Smaller step-size diffraction pattern collected from 43° to 46.5° indicating the particles are of mixed cubic and tetragonal phases. The reference patterns are retrieved from the Materials Project database version v2023.11.1 and from the Crystallography Open Database (COD), accessed in June 2024. (c) Depicts the size distribution of the nearly spherical BaTiO3 nanoparticles, based on image analysis of 60 particles from STEM micrographs shown at two different scales in (d) and (e). (f) shows an EDS area analysis of a BaTiO3-coated Si/SiO2 wafer. The averaged Ba:Ti ratio over 4 different spots is 1:1.81. The signals of Si, C, and additional O can be attributed to the ligands and the silicon wafer. Three spots showed Al impurities with a maximum of 0.13 (at%). | ||
For the most dominant diffraction peak at 25°–35° the calculated crystallite size is 2.14 nm (see Table 1). For the diffraction peak at 52°–59° the calculated crystallite size is 4.4 nm, and for the diffraction peak at 60°–72° the calculated crystallite size is 3.31 nm. Since the particle shape looks nearly spherical in the STEM image (Fig. 1c), the different values for the crystallite size were averaged. The averaged calculated value is 3.3 nm with a standard error of 0.05 nm. This value was compared to the particle size distribution of the nearly spherical BaTiO3 particles based on the measured diameter of 60 particles. This resulted in a mean size of 3.7 nm with a standard deviation of 0.5 nm. The agreement between the particle sizes obtained from the STEM images and those calculated from the XRD data using the Scherrer formula suggests that each nanoparticle is a single crystal.
| Reflection | Unannealed (nm) | 250 °C (nm) | 350 °C (nm) |
|---|---|---|---|
| 25–35° | 2.14 ± 0.027 | 2.16 ± 0.034 | 2.31 ± 0.012 |
| 52–59° | 4.40 ± 0.085 | 4.68 ± 0.058 | 4.86 ± 0.053 |
| 60–72° | 3.31 ± 0.051 | 3.50 ± 0.027 | 3.57 ± 0.062 |
BaTiO3 exists in five different crystal structures: rhombohedral, orthorhombic, cubic, tetragonal, and hexagonal.49 Dependent on their crystal structure the particles exhibit different properties. While the tetragonal phase exhibits ferroelectric, piezoelectric, and thermoelectric properties, the cubic phase shows paraelectric properties and high-temperature stability.50 To take advantage of the piezoelectric properties of BaTiO3, a tetragonal crystal structure is preferred. In Fig. 1b the diffraction peak at 45° is magnified. The cubic reference pattern shows no (200) peak splitting, while the tetragonal reference pattern has a peak splitting of the (200) and (002) planes.51,52 When the step size was increased, it became visible that the broad signal entails at least two reflections. Due to their positioning and distance, they do not match the tetragonal nor the cubic structure accurately but instead it is likely that the particles are a mixture of the two crystal structures with the (200) plane signal of the tetragonal structure being overlapped by the (200) plane of the cubic crystal structure.
Fig. 2a shows the changes observed during the SLE process. Initially, the vial shows a clear, colourless hexane phase with the dispersed BaTiO3 particles at the top. After the oxalic acid in MeOH is added the particles transfer to the bottom MeOH phase, indicating a ligand exchange process. To confirm, FT-IR measurements further detail this ligand exchange. In Fig. 2b, overlapping absorbance signals between 2957 cm−1–2851 cm−1 for the oleyl alkoxide ligands of the as-synthesized particles are clearly visible. After the SLE with 1 M oxalic acid, the signals were no longer detectable thereby indicating the removal of the oleyl alkoxide, and subsequently a complete ligand exchange. After the LbL ligand exchange via dipcoating with 0.1 M oxalic acid, the oleyl alcohol signals decreased significantly, indicating successful ligand exchange.
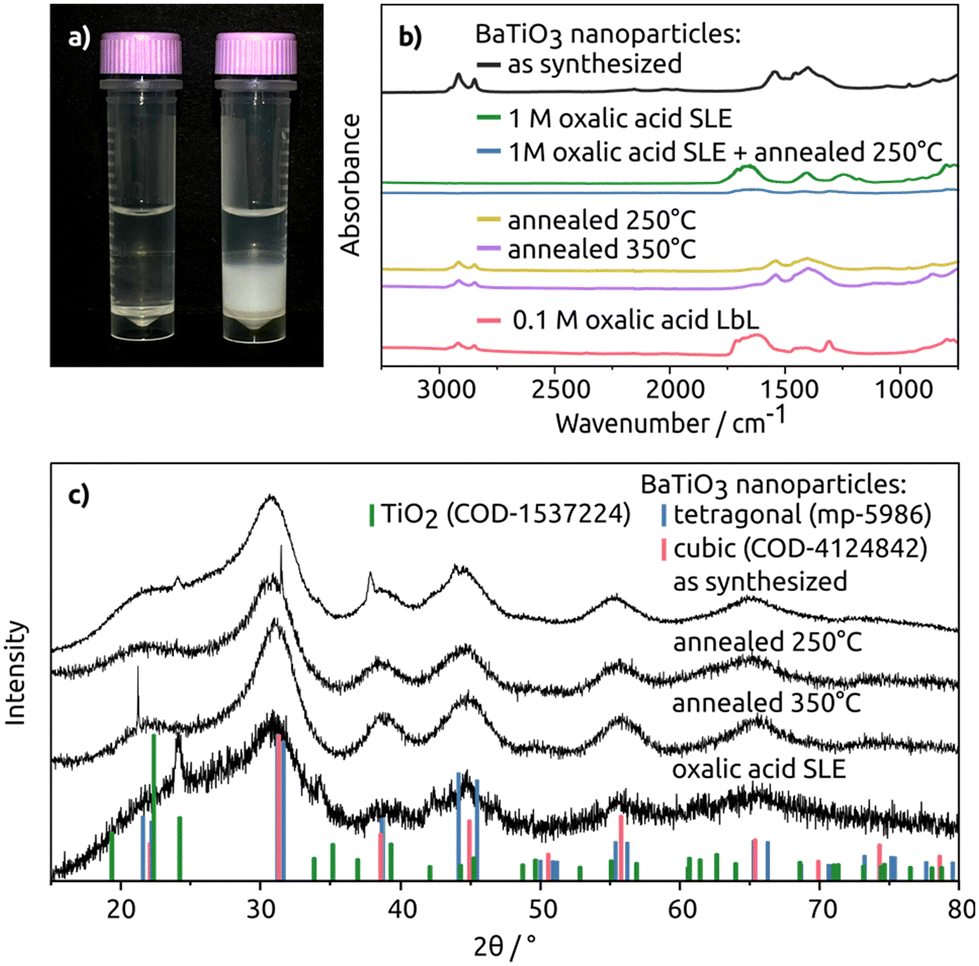 | ||
| Fig. 2 In (a) on the left side is a vial with the colourless hexane phase with the well-dispersed BaTiO3 particles on top, while the phase on the bottom is neat MeOH. On the right side is the vial after oxalic acid addition to the MeOH for the SLE whereby the solution turns white as the particles agglomerate due to the bidentate oxalic acid ligands. (b) Shows FT-IR spectra of the BaTiO3 without and with the ligand exchange ((SLE) and (LbL)) and with different annealing temperatures (250 °C and 350 °C). The as-synthesized BaTiO3 and LbL ligand exchange samples were measured on ITO, the SLE and SLE annealed sample were measured on Au, and the annealed samples were measured on glass. Note an offset for clarity. (c) Depicts the XRD pattern of BaTiO3 at room temperature, annealed BaTiO3 at 250 °C and 350 °C, and BaTiO3 after a SLE with oxalic acid. The BaTiO3 reference patterns are retrieved from the Materials Project, from database version v2023.11.1 and from the Crystallography Open Database (COD), accessed in June 2024. | ||
XRD analysis of the ligand exchanged BaTiO3 particles with oxalic acid as ligands revealed an additional reflection at 24°. This reflection matches the reflection of the hexagonal TiO2 reference pattern (see Fig. S5†). This suggests the partial degradation of the particles, indicating that prolonged exposure to the acidic environment should be avoided as was also observed in ref. 53 where the Ba was selectively etched out of BaTiO3 powders. This is the reason for using MeOH for the SLE, however, water in solution from the hydrated salt and air still leads to slight etching of the particles.
The FT-IR data (see Fig. S6†) of the ligand exchange conducted with KOH also indicated a ligand exchange through the decreasing oleyl alcohol signal. However, exposure to KOH over longer time periods etched the material, leaving behind only traces of TiO2, meaning that KOH-based ligand exchanges lead to more significant degradation of BaTiO3 particles than the oxalic acid-based ligand exchange.
The particles were sintered by heating them to 250 °C and 350 °C in an approach to further immobilize them. Three different reflections (see Table 1) were analysed using the Scherrer equation (see eqn (1)). A trend of increasing particle size for increasing temperature is seen. This is expected as the particles fuse together during the heating process. To compare the particle sizes of the different annealing temperatures, the most prominent reflection of the BaTiO3 reflection patterns in Fig. 2(c) at 31° was used. The calculated average size of the unheated particles is 2.14 nm after heating to 250 °C, the average particle size increased to 2.16 nm, which amounts to an increase of ∼1% which is negligible within the error range. After further increasing the temperature to 350 °C the particle size further increased to 2.31 nm, which increases the particle size by a statistically significant ∼7% (see Table 1). For the other two reflections around 55° and 65° after heating to 350 °C, the size increases by 10% and 8%, respectively. This indicates fusing and ripening of the particles, a desired effect for a continuous coating or layer to be formed.
In order to test the electrical properties, LbL assembly and drop-cast films were prepared on ITO for KPFM measurements. The LbL films using oxalic acid have a thickness of 110 nm and a root-mean-square (RMS) roughness of 3.5 nm determined by AFM at a step-edge. Similarly, the resulting film from LbL with KOH was 95 nm thick with an RMS roughness of 4.2 nm. These films are 20× less rough than the as-synthesized drop-cast film (with an average thickness of 150 nm and an RMS roughness of 82 nm) as seen in the AFM maps in Fig. 3.
 | ||
| Fig. 3 AFM topography maps (a, c and e) and surface potential difference maps (b, d and f) for the BaTiO3 particles with the indicated ligands. | ||
Furthermore, the contact potential difference maps from KPFM show a uniform potential across the LbL films indicating a homogeneous energy landscape. After exchanging the ligands, the surface potential difference offset was compared to ITO by scanning over a step-edge and it changes from 30 mV with oleyl alkoxide, to 35 mV for OH-, and to 40 mV with oxalate-capped particles. This indicates the Fermi level of the particles with each of the ligand treatments moves further from the ITO levels. For creating electronic actuators or other devices, the potential difference between the contacts and the material are important values needed to design the highest performance devices. We then sought to further understand the nature of the electronic states responsible for these differences and carried out DFT calculations to uncover their origin.
Due to the size of the nanoparticles making them unfeasible for direct calculation, we used the approach of multiple Miller index slabs simulated separately to approximate the overall behaviour of spherical nanoparticles. Fig. S7† shows the projected density of states (DOS) plots for the slabs with (100), (110), and (111) Miller indices. Importantly, the slab with a (100) surface exposed shows a calculated bandgap of approximately 2.7 eV, which is less than the experimental value of 3.2 eV but typical for the lower values expected from DFT calculations.54,55 This discrepancy is the result of the presence of surface states that do not exist in the bulk. Despite the narrowing, the gap is clearly visible, which is consistent with the high stability of this surface reported in the literature.56 As expected, the valence band contains mostly oxygen states, due to their anionic character. On the other hand, the states of Ti which forms a reducible oxide, are visible in the conduction band.
Different observations have been made for the (110) and (111) terminated slabs. There is no well-defined bandgap in these systems because states arise and fill the gap at energies between where the bulk values of the conduction and valence bands. Accordingly, these systems should exhibit conductive properties.57 This is again a result of surface states appearing in the gap, mostly belonging to Ti atoms. This observation is consistent with the significantly lesser stability of these surfaces.56 In addition, the surface spin polarization has been observed, with the total magnetic moment of the system equal to 43.4μB and 15.1μB for (110) and (111) surfaces, respectively. This effect is typically associated with vacancies or other defects in the structure but has also been reported for ultra-thin films based on computational analysis.57 These calculations allow us to speculate that the majority of the surface for these particles would be the more stable (100) facet.
Conclusions
BaTiO3 nanoparticles, between 2 nm to 5 nm in diameter, were successfully synthesized with oleyl alkoxide as the initial capping ligands. Characterizing the reaction mechanisms with 1H NMR confirmed the single-step formation of a Ba- and Ti-containing organometallic precursor also supported by EDS and FT-IR analyses. The synthesized particles were characterized using XRD, EDS analysis, and STEM imaging which indicated titanium-rich materials with mixed cubic and tetragonal crystal phases. Solution-phase and solid-state layer-by-layer ligand exchange strategies resulted in films with minimal organic residue remaining that were mapped with AFM and KPFM. AFM topology maps showed smooth films with an RMS roughness on the same order as the particle diameter. The contact potential difference measured with KPFM showed a homogeneous distribution across the film surface with a potential difference offset of 30 mV to 40 mV between the particles and the ITO-glass substrate. This fabrication of films of BaTiO3 in an ink-based approach provides a path to creating dielectric and piezoelectric films without harsh processing conditions. DFT results suggest that these properties stem from the presence of surface states on the less stable facets of the nanoparticles.Author contributions
L. K. L. synthesized the precursors and BaTiO3 nanoparticles, performed ligand exchanges, XRD, and EDS measurements, prepared NMR samples and FT-IR films, planned experiments, analysed data, prepared figures, and drafted the manuscript; K. E. D. performed STEM imaging, XRD, and FT-IR measurements, assisted with laboratory work, and contributed to figure conceptualization; R. W. C. initiated and managed the project, developed the experimental plan, supervised the research activities of L. K. L. and K. E. D., analysed and aided in performing AFM and KPFM measurements, aided with the ligand exchanges, performed crystallite size analysis using the Scherrer equation, and provided critical review, commentary, and editing of the manuscript draft; K. G. performed and analysed AFM and KPFM measurements with supervision from R. H. F.; and B. M. S. conducted the DFT calculations and edited the manuscript. This manuscript was written with contributions from all authors, and all authors approve the final version of the manuscript.Data availability
Data for this article, including XRD patterns, STEM images, FTIR spectra, AFM maps, KPFM maps, EDS spectra, and NMR spectra are available from Zenodo.org at https://doi.org/10.5281/zenodo.14277513.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Julien Bachmann and Markus Halik for lab space and equipment access; Achim Zahl for NMR measurements; Selina Kern, Vincent Mauritz, and Lisa Ngo for laboratory assistance. Funding from the Deutsche Forschungsgemeinschaft (DFG – German Research Foundation) for project number 542141531 is acknowledged. KED acknowledges scholarship funding from the Deutsche Bundesstiftung Umwelt (DBU – German Federal Environmental Foundation). KG and RF are grateful for the financial support by the Federal Ministry of Education and Research (BMBF, contract 05K22WE2), and “Solar Technologies Go Hybrid” – (SolTech) initiative by the State of Bavaria. This research was carried out with the support of the Interdisciplinary Centre for Mathematical and Computational Modelling University of Warsaw (ICM UW) under computational allocation no G100-2264.References
- C. Giansante, Acc. Chem. Res., 2020, 53, 1458–1467 CrossRef CAS PubMed.
- F. Hernandez, M. Yang, N. Nagelj, A. Y. Lee, H. Noh, K. P. Hur, X. Fu, C. J. Savoie, A. M. Schwartzberg and J. H. Olshansky, Nanoscale, 2024, 16, 5624–5633 RSC.
- N. Kirkwood, J. O. Monchen, R. W. Crisp, G. Grimaldi, H. A. Bergstein, I. Du Fossé, W. Van Der Stam, I. Infante and A. J. Houtepen, J. Am. Chem. Soc., 2018, 140, 15712–15723 CrossRef CAS PubMed.
- T. Singletary, G. Drazer, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi and C. E. Colosqui, Nanoscale, 2024, 16, 5374–5382 Search PubMed.
- V. Lesnyak, J. Phys. Chem. Lett., 2021, 12, 12310–12322 Search PubMed.
- C. Ge, T. Masalehdan, M. Shojaei Baghini, V. Duran Toro, L. Signorelli, H. Thomson, D. Gregurec and H. Heidari, Adv. Sci., 2024, 2404254 CrossRef CAS PubMed.
- F. Montanarella and M. V. Kovalenko, ACS Nano, 2022, 16, 5085–5102 CrossRef CAS PubMed.
- A. Eigen, V. Schmidt, M. Sarcletti, S. Freygang, A. Hartmann-Bausewein, V. Schneider, A. Zehetmeier, V. Mauritz, L. Müller and H. Gaß, et al., Nano Sel., 2024, 5, 2300130 CrossRef CAS.
- D. Damjanovic, Rep. Prog. Phys., 1998, 61, 1267 Search PubMed.
- M. Stachiotti, Appl. Phys. Lett., 2004, 84, 251–253 Search PubMed.
- J. Varghese, R. W. Whatmore and J. D. Holmes, J. Mater. Chem. C, 2013, 1, 2618–2638 Search PubMed.
- W. S. Yun, J. J. Urban, Q. Gu and H. Park, Nano Lett., 2002, 2, 447–450 Search PubMed.
- D. Berlincourt and H. Jaffe, Phys. Rev., 1958, 111, 143–148 Search PubMed.
- S. Wada, K. Yamato, P. Pulpan, N. Kumada, B.-Y. Lee, T. Iijima, C. Moriyoshi and Y. Kuroiwa, J. Appl. Phys., 2010, 108, 094114 CrossRef.
- A. Haroon, P. Rai and I. Uddin, Int. J. Nanosci., 2020, 19, 1950001 CrossRef CAS.
- C. Srilakshmi, G. M. Rao and R. Saraf, RSC Adv., 2015, 5, 45965–45973 Search PubMed.
- P. Lin, Z. Liu and B. W. Wessels, J. Opt. A: Pure Appl. Opt., 2009, 11, 075005 CrossRef.
- T. Takenaka, H. Nagata, Y. Hiruma, Y. Yoshii and K. Matumoto, J. Electroceram., 2007, 19, 259–265 CrossRef CAS.
- R. Maranganti and P. Sharma, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 054109 CrossRef.
- G. H. Haertling, J. Am. Ceram. Soc., 1999, 82, 797–818 Search PubMed.
- T. Hueter and E. Dozois, J. Acoust. Soc. Am., 1952, 24, 85–86 CrossRef.
- R. J. Meyer, R. E. Newnham, A. Amin and B. M. Kulwicki, J. Am. Ceram. Soc., 2003, 86, 934–938 CrossRef CAS.
- A. Dent, C. Bowen, R. Stevens, M. Cain and M. Stewart, J. Eur. Ceram. Soc., 2007, 27, 3739–3743 CrossRef CAS.
- C. M. Landis and R. M. McMeeking, Ferroelectrics, 2001, 255, 13–34 CrossRef CAS.
- B. Jiang, J. Iocozzia, L. Zhao, H. Zhang, Y.-W. Harn, Y. Chen and Z. Lin, Chem. Soc. Rev., 2019, 48, 1194–1228 RSC.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss and D. J. Milliron, et al., ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- S. B. Kim, C. Cai, J. Kim, S. Sun and D. A. Sweigart, Organometallics, 2009, 28, 5341–5348 CrossRef CAS.
- V. Mauritz and R. W. Crisp, J. Mater. Chem. C, 2024, 12, 11319–11334 RSC.
- H. H. Liu, S. Surawanvijit, R. Rallo, G. Orkoulas and Y. Cohen, Environ. Sci. Technol., 2011, 45, 9284–9292 Search PubMed.
- C. Grote, T. Cheema and G. Garnweitner, Langmuir, 2012, 28, 14395–14404 CrossRef CAS PubMed.
- H. Pan, A. Kursumovic, Y.-H. Lin, C.-W. Nan and J. L. MacManus-Driscoll, Nanoscale, 2020, 12, 19582–19591 RSC.
- Z. Chen, L. Huang, J. He, Y. Zhu and S. O'Brien, J. Mater. Res., 2006, 21, 3187–3195 CrossRef CAS.
- C. P. Laboratory, Teflon (PTFE) Chemical Compatibility Chart, 2024, https://www.calpaclab.com/teflon-ptfe-compatibility/, Accessed: 2024-06-11.
- R. W. Crisp, R. Callahan, O. G. Reid, D. S. Dolzhnikov, D. V. Ta-lapin, G. Rumbles, J. M. Luther and N. Kopidakis, J. Phys. Chem. Lett., 2015, 6, 4815–4821 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al., APL Mater., 2013, 1, 011002 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. Podsiadły-Paszkowska, I. Tranca and B. M. Szyja, J. Phys. Chem. C, 2019, 123, 5401–5410 CrossRef.
- M. Niederberger, G. Garnweitner, N. Pinna and M. Antonietti, J. Am. Chem. Soc., 2004, 126, 9120–9126 CrossRef CAS PubMed.
- S. O'Brien, L. Brus and C. B. Murray, J. Am. Chem. Soc., 2001, 123, 12085–12086 CrossRef PubMed.
- Sigma-Aldrich, Infrared Spectrum Table, 2024, https://www.sigmaaldrich.com/DE/de/technical-documents/technical-article/analytical-chemistry/photometry-and-reflectometry/ir-spectrum-table?report=reader, Accessed: 2024-05-28.
- V. A. Zeitler and C. A. Brown, J. Phys. Chem., 1957, 61, 1174–1177 CrossRef CAS.
- G. Zhuang, Y. Chen, Z. Zhuang, Y. Yu and J. Yu, Sci. China Mater., 2020, 63, 2089–2118 Search PubMed.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513 CrossRef CAS.
- W. Lipinska, K. Grochowska, J. Ryl, J. Karczewski, M. Sawczak, E. Coy, V. Mauritz, R. W. Crisp and K. Siuzdak, J. Mater. Sci., 2024, 1–16 Search PubMed.
- E. Dhaene, J. Billet, E. Bennett, I. Van Driessche and J. De Roo, Nano Lett., 2019, 19, 7411–7417 CrossRef CAS PubMed.
- A. F. Suzana, S. Liu, J. Diao, L. Wu, T. A. Assefa, M. Abeykoon, R. Harder, W. Cha, E. S. Bozin and I. K. Robinson, Adv. Funct. Mater., 2023, 33, 2208012 Search PubMed.
- Y. V. Kolenko, K. A. Kovnir, I. S. Neira, T. Taniguchi, T. Ishigaki, T. Watanabe, N. Sakamoto and M. Yoshimura, J. Phys. Chem. C, 2007, 111, 7306–7318 Search PubMed.
- A. Thanki and R. Goyal, Mater. Chem. Phys., 2016, 183, 447–456 CrossRef CAS.
- D.-H. Yoon, J. Ceram. Process. Res., 2006, 7, 343–354 Search PubMed.
- A. Neubrand, R. Lindner and P. Hoffmann, J. Am. Ceram. Soc., 2000, 83, 860–864 CrossRef CAS.
- M. G. Elmahgary, A. M. Mahran, M. Ganoub and S. O. Abdellatif, Sci. Rep., 2023, 13, 4761 Search PubMed.
- D. Bagayoko, AIP Adv., 2014, 4, 127104 CrossRef.
- K. Wang, V. Fung, Z. Wu and D.-E. Jiang, J. Phys. Chem. C, 2020, 124, 18557–18563 CrossRef CAS.
- H. Gao, Z. Yue, Y. Liu, J. Hu and X. Li, Nanomaterials, 2019, 9, 269 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction mechanism schemes and unit cell geometry along with supporting data from NMR, FTIR, XRD, and EDS. See DOI: https://doi.org/10.1039/d4nr05133e |
| This journal is © The Royal Society of Chemistry 2025 |