
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic CO2 reduction by a cobalt porphyrin mini-enzyme†
Alison A. Salamatiana,
Jose L. Alvarez-Hernandeza,
Karishma B. Ramesha,
Linda Leone b,
Angela Lombardib and
Kara L. Bren*a
b,
Angela Lombardib and
Kara L. Bren*a
aDepartment of Chemistry, University of Rochester, Rochester, NY 14627-0216, USA. E-mail: kara.bren@rochester.edu
bDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario Monte S. Angelo, Via Cintia, 80126 Naples, Italy
First published on 25th February 2025
Abstract
Cobalt-mimochrome VI*a (CoMC6*a), a synthetic mini-enzyme with a cobalt porphyrin active site, is developed as a biomolecular catalyst for electrocatalytic CO2 reduction in water. The catalytic turnover number reaches ∼14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for CO production with a selectivity of 86:5 over H2 production under the same conditions. Varying the applied potential and the pKa of the proton donor was used to gain insight into the basis for selectivity. The protected active site of CoMC6*a is proposed to enhance selectivity for CO2 reduction under conditions that typically favor H2 production by related catalysts. CoMC6*a activity and selectivity change only marginally under air, indicating excellent oxygen tolerance.
000 for CO production with a selectivity of 86:5 over H2 production under the same conditions. Varying the applied potential and the pKa of the proton donor was used to gain insight into the basis for selectivity. The protected active site of CoMC6*a is proposed to enhance selectivity for CO2 reduction under conditions that typically favor H2 production by related catalysts. CoMC6*a activity and selectivity change only marginally under air, indicating excellent oxygen tolerance.
Introduction
Electrochemical carbon dioxide (CO2) reduction is an appealing route to renewable fuel production.1,2 Achieving selectivity for CO2 reduction over proton reduction is an omnipresent challenge, since the reduction of CO2 to CO, or any stable product, requires protons (eqn (1) and (2)).3,4 Achieving selectivity in a protic solvent such as water is particularly challenging. However, there is significant interest in developing catalysis in water as an abundant source of protons and a desirable environmentally-friendly solvent.5–7 An additional challenge raised by use of water as a solvent is the poor solubility of CO2.8,9 Developing catalysts with microenvironments that sequester and activate CO2 in the presence of protons thus is of high interest.10–15| CO2 + 2H+ + 2e− → CO + H2O | (1) |
| 2H+ + 2e− → H2 | (2) |
Nature's enzymes achieve high selectivity and activity for reactions such as CO2 reduction by providing an active-site microenvironment to promote substrate binding and transformation and by controlling electron and proton delivery.16–19 Inspired by Nature's catalysts, artificial enzymes for CO2 reduction (see examples in Table S1†) have been prepared by incorporation of synthetic CO2 reduction catalysts, such as [Ni(cyclam)]2+,20 Ni(terpyridine),21 or cobalt porphyrins,22–24 into proteins including azurin,20 cytochrome b562,23 myoglobin,24 an artificial protein αRep,25 or an engineered photosensitizer protein.21 Some of these systems have been reported to achieve enhanced activity23 and selectivity20 relative to the synthetic catalyst outside of the protein environment. For example, improved selectivity for CO2 over proton reduction by [Ni(cyclam)]2+ bound to the protein azurin was attributed to the protein scaffold providing restricting conformational flexibility of the catalyst and an active site buried within a solvent-excluded hydrophobic patch.20
Inspired by the importance of proton transfer steps in enzymatic catalysis,17,26–29 roles for endogenous4,17,30–32 and exogenous7,22,30,33 proton donors in determining CO2 reduction selectivity and activity have been proposed. The use of relatively weak Brønsted acids as proton donors is proposed to slow metal-hydride formation and thus disfavor the competing H2 evolution pathway.1,34,35 Electrochemical studies on an iron–porphyrin electrocatalyst7 and a cobalt macrocyclic catalyst36 showed that using a higher-pKa buffer increases selectivity for CO over H2 production. Furthermore, in photochemical studies employing cobalt porphyrin catalysts, presence of a higher-pKa buffer (bicarbonate, as opposed to phosphate) was shown to increase selectivity for CO over H2 production.37,38 Other properties of buffers have also been implicated in determining selectivity. For [Ni(cyclam)]2+, buffer steric properties and charges were found to impact selectivity for CO over H2 production; cationic buffers were proposed to stabilize an activated Ni–CO2 species in a second-sphere interaction, favoring CO production.33
In a previous study, we reported CO2 reduction catalysis by a semisynthetic cobalt–porphyrin-containing mini-enzyme, CoMP11-Ac, consisting of a cobalt porphyrin with a covalently attached peptide donating an axial histidine ligand on the proximal side of the porphyrin (Fig. 1a). For CoMP11-Ac, selectivity for CO over H2 production in water is increased by using a higher-pKa buffer as an exogenous proton donor, which is proposed to disfavor the formation of a metal-hydride species that yields H2. Furthermore, catalysis at a more negative potential (−1.4 V vs. Ag/AgCl/KCl(1M)) lowers selectivity for CO over H2 production, while applying a less negative potential (−1.2 V) increases selectivity.22
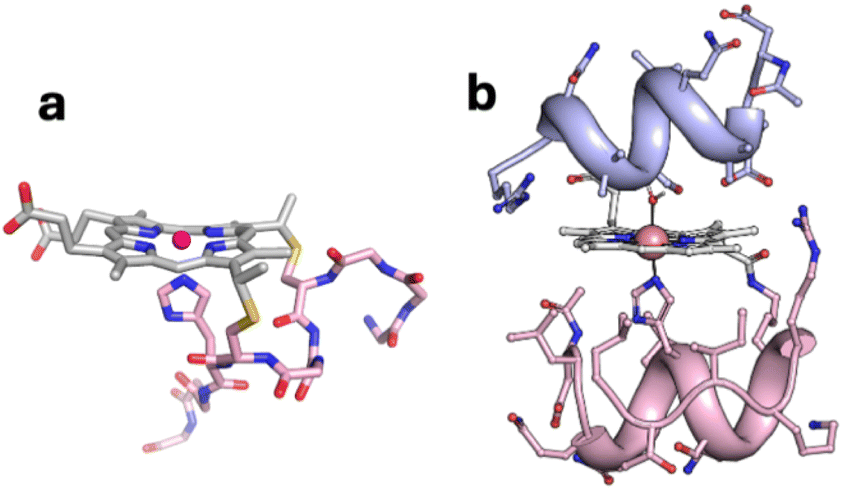 | ||
| Fig. 1 Models of (a) CoMP11-Ac; (b) CoMC6*a. | ||
We now investigate effects of biocatalyst structure on selectivity for CO2 vs. proton reduction. We have chosen a catalyst that, like CoMP11-Ac, has a cobalt porphyrin active site and axial His ligand, but that also has a peptide covering the distal side of the heme. This catalyst is a synthetic mini-enzyme, cobalt-mimochrome VI*a (CoMC6*a, Fig. 1b). Mimochromes are miniaturized porphyrin-based metalloproteins consisting of a deuteroporphyrin sandwiched between two peptide chains covalently bound to the porphyrin.39,40 MC6*a is a proven framework for catalysis, displaying peroxidase,41–43 peroxygenase42–44 or hydrogenase45,46 activities depending on conditions and the metal ion. Its scaffold consists of a distal decapeptide and a proximal tetradecapeptide that provides the axial His ligand to the metal ion. Helical secondary structure is favored by the inclusion of two 2-aminoisobutyric acid residues in the distal peptide.47
Previously, CoMC6*a was shown to act as an electrocatalyst for H2 evolution from water with a turnover number (TON) exceeding 230000 (ref. 45) as well as a catalyst in a system for photochemical H2 evolution.46 Subsequent studies of CoMC6*a catalysis of H2 evolution from water revealed that buffer acid species play a critical role in proton delivery to CoMC6*a during catalysis, with their structures and pKa values impacting catalytic rate, potential, and mechanism.48 In particular, proton-coupled electron transfer (PCET) was shown to be required for H2 production by CoMC6*a, with the catalytic potential shifting with the pKa of the buffer acid in a Nernstian fashion. Furthermore, catalytic rate was shown to depend on buffer sterics, an observation attributed to the impact of the distal peptide in hindering proton delivery by protonated buffer.48 Interestingly, the specific effects of buffer acid on H2 production catalysis differ from those observed for CoMP11-Ac, for which buffer pKa, but not buffer structure, plays a role in determining catalytic rate, likely as a result of the solvent-exposed active site of CoMP11-Ac.49
Having observed these impacts of catalyst structure on H2 evolution catalysis by CoMP11-Ac vs. CoMC6*a, we now turn to investigating the impact of structure on CO2 reduction by CoMC6*a. We hypothesized that the more hydrophobic and enclosed active site of CoMC6*a would favor CO2 reduction. Using conditions applied to CoMP11-Ac to facilitate comparison, the roles of both applied potential and exogenous proton donor pKa in determining CO2 vs. proton reduction selectivity and activity by CoMC6*a are investigated. Comparison to previous results on CoMP11-Ac indicates that the distal peptide plays a role in enhancing selectivity for CO2 reduction. Finally, we demonstrate that this catalyst exhibits excellent tolerance for oxygen, with minimal impact on CO2 reduction activity or selectivity.
Results and discussion
CoMC6*a was prepared and characterized as described in the ESI (Fig. S1 and S2)† as well as previous publications.45,47 Cyclic voltammetry (CV) of 1 μM CoMC6*a was carried out using a hanging mercury drop electrode, used in previous related work.22,45,48 As was observed for CoMP11-Ac,22 dip-and-stir experiments50 indicate that CoMC6*a adsorbs to the electrode, acting as an immobilized catalyst (Fig. S3 and S4†).Effects of applied potential
CV of 1 μM CoMC6*a at pH 6 in 50 mM 3-morpholinopropane-1-sulfonic acid (MOPS, pKa 7.2) under N2 (Fig. 2) shows faradaic current beginning at an onset potential of ∼ −1.2 V vs. Ag/AgCl/KCl(1M) (all potentials reported herein are reported against this reference). The rise in current forming a single peak is attributed to CoMC6*a electrocatalytic H2 evolution activity via protonated buffer consumption, which was previously reported under similar conditions.45,48 When the solution is saturated with CO2 and placed under 1 atm CO2, two peaks are observed at ∼ −1.2 V and ∼ −1.5 V (Fig. 2). The resulting increase in current at ∼ −1.2 V may indicate selective CO2 reduction over proton reduction at this potential. Furthermore, the anodic shift of the catalytic onset potential may be due to CO2 coordination and reduction or a coupled EC/CE reaction.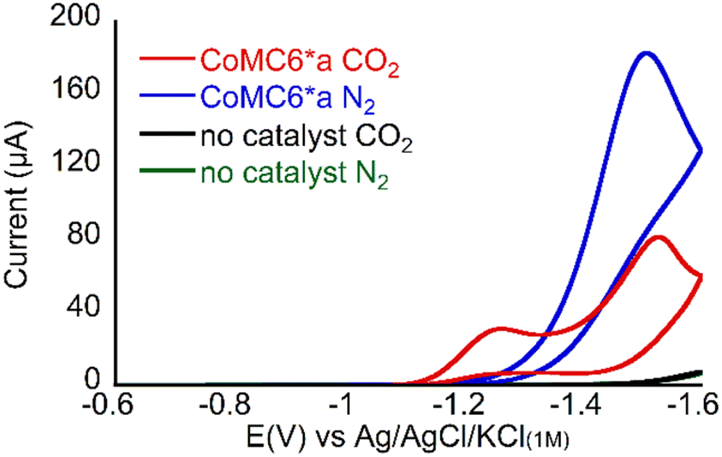 | ||
| Fig. 2 Cyclic voltammograms of 1 μM CoMC6*a pH 5.9 in 50 mM MOPS, 0.1 M KCl, at 100 mV s−1, scan 2, under 1 atm of the indicated gas. | ||
To characterize product formation, controlled potential electrolysis (CPE) experiments were run on 1 μM CoMC6*a in the presence of MOPS for two hours, after which the headspace gas was sampled and analyzed by gas chromatography (GC). Experiments were run at −1.2 and −1.4 V to aid comparison to published results on CoMP11-Ac at these conditions (Table S2†).22 At −1.4 V under N2 with no CO2 present, H2 is produced with nearly quantitative faradaic efficiency (FEH2 96 ± 4%), consistent with previous results.45,48 When a CO2-saturated solution of CoMC6*a under one atmosphere of CO2 is subjected to CPE, the major product is CO (Tables 1, S3 and Fig. S5†). However, selectivity for CO formation over H2 under these conditions changes with applied potential, with higher selectivity (85:6 FECO:FEH2) at −1.2 V compared to 68:24 at −1.4 V (Tables 1, S3 and Fig. S5†). The turnover number (TON) for CO production also is dependent on potential, with double the value (2200 ± 300) at the less cathodic potential of −1.2 V. In comparison with results on CoMP11-Ac under the same conditions (Table S2†), FECO (85 ± 2%) and FEH2 (8 ± 2%) are nearly the same as the values for CoMC6*a at −1.2 V. However, at −1.4 V (Table 2), CoMP11-Ac favors H2 production, with FECO of 21 ± 5% and FEH2 of 63 ± 13%. Thus, under these conditions at −1.4 V, CoMC6*a shows significantly greater selectivity for CO2 over proton reduction compared to CoMP11-Ac, supporting the hypothesis that protection of the CoMC6*a active site by the distal peptide enhances selectivity.
| Gas | Buffer | Eb (V) | FE(H2) % | FE(CO) % | TON(H2) | TON(CO) | QT (C) |
|---|---|---|---|---|---|---|---|
| a Two-hour CPE experiments conducted on 1 μM catalyst in 0.5 M buffer with 1 M KCl. Data shown corresponds to the average of at least three individual runs, the error corresponds to the difference between the average and the replicate with the greatest difference from the average; ESI shows detailed results. The pH of all MOPS, CHES, and CAPS solutions after purging with CO2 was 6.5 ± 0.2; and 7.2 ± 0.2 when purged with N2.b Potentials reported vs. Ag/AgCl/KCl(1M).c Activity is not reported if it did not exceed three times background in more than one replicate. | |||||||
| CO2 | CAPS (pKa 10.4) | −1.4 | 4 ± 1 | 76 ± 10 | 110 ± 20 | 2100 ± 600 | 2.6 ± 0.4 |
| −1.2 | 4 ± 4 | 73 ± 5 | 11 ± 10 | 230 ± 10 | 0.3 ± 0.1 | ||
| CO2 | CHES (pKa 9.3) | −1.4 | 14 ± 1 | 67 ± 12 | 280 ± 10 | 1300 ± 400 | 1.9 ± 0.1 |
| −1.2 | 11 ± 1 | 86 ± 11 | 100 ± 20 | 800 ± 200 | 0.9 ± 0.1 | ||
| CO2 | MOPS (pKa 7.2) | −1.4 | 24 ± 4 | 68 ± 8 | 390 ± 120 | 1100 ± 200 | 1.6 ± 0.5 |
| −1.2 | 6 ± 1 | 85 ± 11 | 160 ± 40 | 2200 ± 300 | 2.5 ± 0.2 | ||
| N2 | CAPS (pKa 10.4) | −1.4 | 88 ± 10 | ∼0 | 1100 ± 400 | ∼0 | 1.2 ± 0.3 |
| −1.2 | No above-background activityc | ||||||
| N2 | CHES (pKa 9.3) | −1.4 | 97 ± 14 | ∼0 | 1800 ± 200 | ∼0 | 1.8 ± 0.1 |
| −1.2 | 78 ± 14 | ∼0 | 130 ± 30 | ∼0 | 0.2 ± 0.1 | ||
| N2 | MOPS (pKa 7.2) | −1.4 | 96 ± 4 | 1.0 ± 0.3 | 3900 ± 1500 | 45 ± 12 | 3.9 ± 1.4 |
| −1.2 | No above–background activity | ||||||
| Buffer | Catalyst | FE(H2) % | FE(CO) % |
|---|---|---|---|
| a Data on CoMP-11 from ref. 22 Data collected under 1 atm CO2, 0.5 M buffer, pH 6.5. Full table of comparative results in ESI. | |||
| CAPS (pKa 10.4) | CoMP11-Ac | 29 ± 6 | 48 ± 10 |
| CoMC6*a | 4 ± 1 | 76 ± 10 | |
| CHES (pKa 9.3) | CoMP11-Ac | 43 ± 9 | 57 ± 4 |
| CoMC6*a | 14 ± 1 | 67 ± 12 | |
| MOPS (pKa 7.2) | CoMP11-Ac | 63 ± 13 | 21 ± 5 |
| CoMC6*a | 24 ± 4 | 68 ± 8 | |
Effects of proton donor pKa
An important tool for addressing product selectivity and gaining mechanistic insights in CO2 reduction electrocatalysis is tuning proton donor properties.31,51 For a number of catalysts in water, protonated buffers have been shown to be the primary proton donors in proton-requiring catalysis (except at low pH values)52 for H2 production48–50 and CO2 reduction,7,33,49 with buffer properties impacting catalytic rate, mechanism, and selectivity.36–38 For CoMC6*a, properties of buffer acids have been shown to impact electrocatalytic H2 evolution efficiency, activity, and mechanism: lower-pKa buffers result in an anodic shift in the catalytic wave, which has been attributed to their role in PCET,48 and less bulky buffers increase catalytic current, a phenomenon attributed to distal CoMC6*a peptide hindering proton donor access to the active site.48 To determine the effect of proton donor on CO2 reduction selectivity by CoMC6*a, we chose three structurally related buffers: MOPS, used above (pKa = 7.2), N-cyclohexyl-2-aminoethanesulfonic acid (CHES, pKa = 9.3) and 3-(cyclohexylamino)-1-ethanesulfonic acid (CAPS pKa = 10.4; structures are shown in Fig. 3).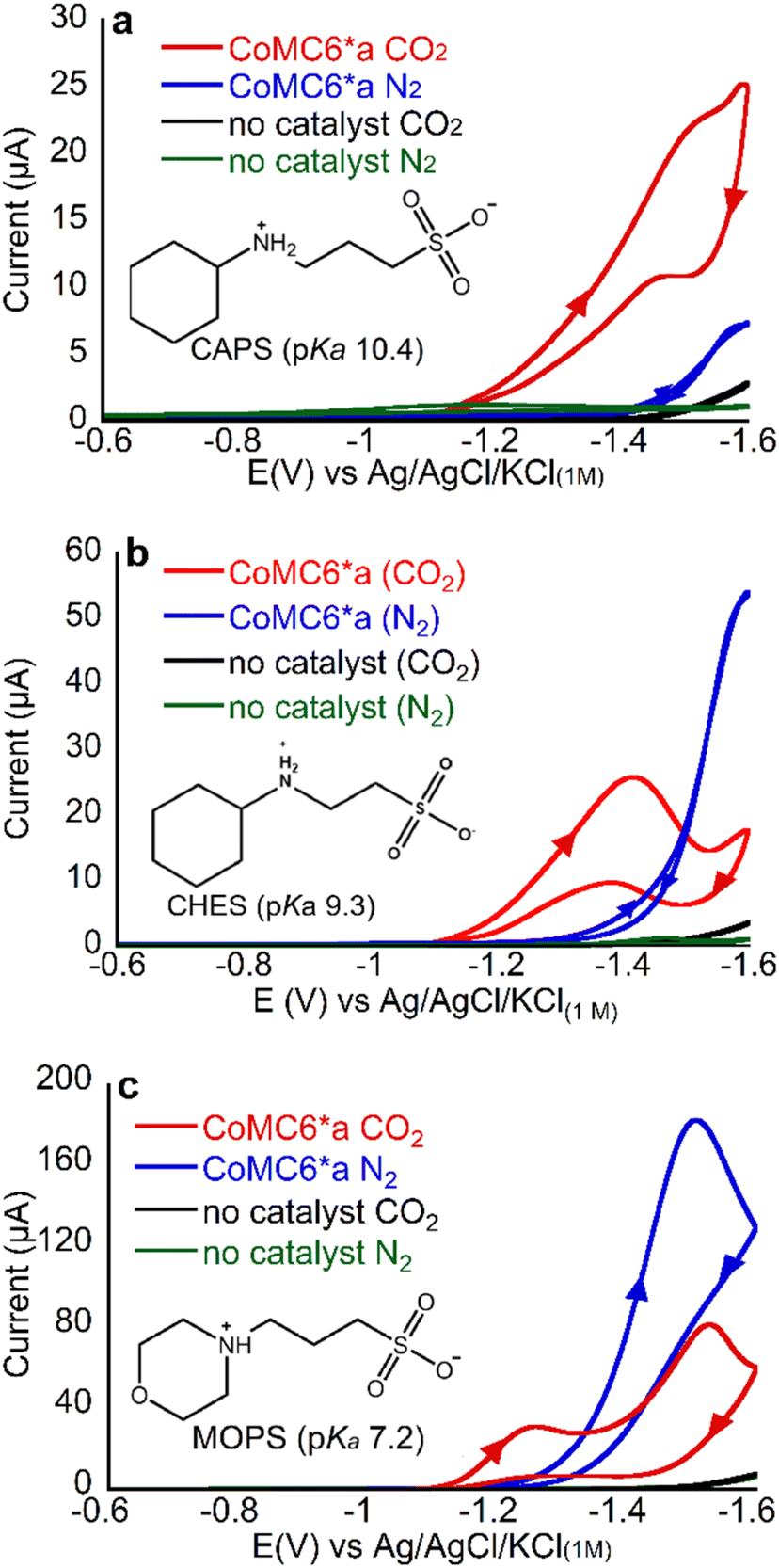 | ||
| Fig. 3 CVs of 1 μM CoMC6*a in 50 mM (a) CAPS, (b) CHES, (c) MOPS. For all CVs, pH = 5.9, [KCl] = 0.1 M and scan rate = 100 mV s−1. Arrows in the CV traces indicate the scanning direction. | ||
First, we collected CVs of CoMC6*a under N2 or CO2, with the solution saturated with the respective gas. Under N2, there is only one feature, which is between −1.4 and −1.6 V, and was previously shown to be associated with catalytic H2 evolution.45,48 The peak current of this low-potential feature decreases with increasing buffer pKa, consistent with lower H2 production activity with less acidic proton donors (Fig. 3).
Under CO2, the CV changes dramatically but in a manner dependent on the buffer present. With all three buffers, an increase in current under CO2 relative to that under N2 is seen at ∼ −1.2 V, a potential at which CPE experiments show (vide infra) there is minimal H2 production (Fig. 3 and Table 1). This result suggests that there may be enhanced CO2 reduction ∼ −1.2 V.
To determine products formed, two-hour CPE experiments on CoMC6*a in MOPS, CHES and CAPS buffers at pH 6 were performed at −1.2 and −1.4 V, with results in Tables 1, S3–S5 and Fig. S5–S7.† The UV-vis spectrum of the catalyst in bulk solution shows minimal change before and after CPE, indicating catalyst robustness (Fig. S8†). Under N2 at −1.2 V, no activity above background was observed in the presence of CAPS or MOPS, and very low activity was observed in CHES, indicating that minimal H2 production occurs at −1.2 V in the presence of all three buffer acids under these conditions, consistent with prior results on CoMC6*a.48 At −1.4 V under N2, the charge passed exceeds background for all three buffers, with H2 formation with FEH2 values from 88 to 97%. As we lower buffer pKa, we see an increase in TONH2, supporting the hypothesis that more acidic proton donors enhance H2 production activity, in line with prior results.48
When CPE of CoMC6*a is performed under CO2, CO becomes the major product under all conditions used here. At −1.2 V under CO2, FECO is approximately the same for experiments run with the three different buffer acids (ranging from 73 to 85%) and the FEH2 values are also similar (4–11%), indicating that the pKa of the buffer does not have a significant impact on selectivity at −1.2 V. In contrast, at −1.4 V under CO2, FEH2 increases from 4 ± 1% to 14 ± 1% to 24 ± 4% as buffer pKa decreases, showing that increased buffer acidity enhances H2 evolution under a CO2 atmosphere, possibly by promoting formation of a metal hydride or its protonation. FECO shows minimal change with buffer pKa at −1.4 V, (67–76%), indicating that the effect of increased buffer pKa on enhancing selectivity for CO production at −1.4 V results primarily from decreasing H2 production.
Comparison to results on CoMP11-Ac (Fig. 1) provides insight into how catalyst structure impacts selectivity. Similar to CoMC6*a, at −1.2 V, CO:H2 selectivity of CoMP11-Ac shows no dependence on buffer acid pKa (Table S2†). At −1.4 V, also like CoMC6*a, CoMP11-Ac shows an increase in selectivity for CO2 reduction over proton reduction as the pKa of the buffer acid is increased (Table 2 and S2†).22 CoMP11-Ac and CoMC6*a thus show similar trends in CO:H2 selectivity with buffer acid pKa, with no dependence at −1.2 V and an increased FECO:FEH2 with decreased buffer acidity at −1.4 V, dominated by an impact on FEH2. However, CoMC6*a has a higher CO:H2 selectivity under all conditions, always in favor of CO2 reduction. These results indicate that the CoMC6*a structure enhances CO2 reduction selectivity over proton reduction, an effect primarily seen at the more negative potential used herein.
For CoMP11-Ac, two mechanisms were proposed at the two different potentials.22 At −1.4 V, a mechanism invoking formal Co(I) formation was proposed, consistent with an estimated Co(II/I) reduction potential of −1.42 V.52 Cobalt hydride is proposed to yield H2 upon protonation, and this process accounts for the greater FEH2 at a more negative potential. This mechanism is in line with the observed selectivity dependency on the buffer acid pKa at −1.4 V, as a more acidic proton donor will favor Co(I) protonation,48 thus biasing the system toward H2 formation. At −1.2 V, a mechanism in which CO2 binding couples to electron transfer to form a formal Co(I)–CO2 adduct was invoked, which avoids directly forming a Co(I) species and accounts for the lack of dependence of selectivity on buffer pKa at this potential. This mechanism has a selectivity-determining step prior to any protonation step, which suggests that selectivity will not depend on proton donor pKa, in line with the experimental results at −1.2 V.
To consider this model for CoMC6*a, we measured the formal Co(II/I) reduction potential. This was accomplished under N2 at high pH and with a rapid scan rate, conditions at which H2 evolution is suppressed. From quasi-reversible CVs at pH 10–12, a midpoint potential of ∼ −1.58 V was measured (Fig. S9†). Thus, under the conditions used here for catalysis, direct formation of Co(I) is not possible. For CO2 reduction, reaching this formal oxidation state will require CO2 binding before or coupled with reduction. For proton reduction, PCET is required, as was previously demonstrated.48 These observations lead to the proposed mechanism in Fig. 4, which has its basis in published mechanisms for CO2 reduction and proton reduction by cobalt porphyrins.53 However, the low potential of Co(II/I)MC6*a precludes direct formation of a Co(I) species under these conditions, a process typically invoked in related systems.22,37,53 To provide additional data to test this model, effects of CO2 concentration on catalysis were measured.
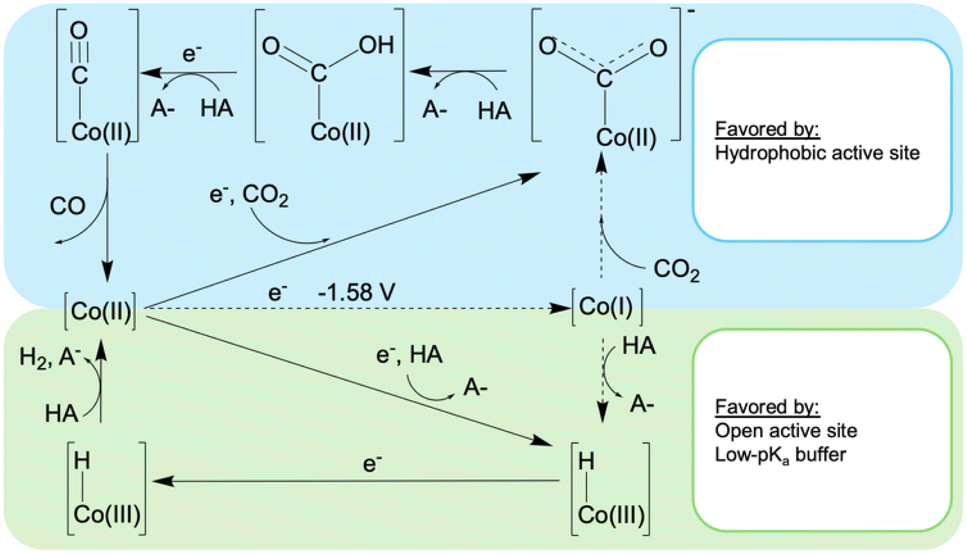 | ||
| Fig. 4 Proposed mechanisms for H2 and CO formation catalyzed by CoMC6*a. The dotted lines indicate processes not observed or expected under the conditions used herein. | ||
Effects of CO2 partial pressure
Prior experiments examined the effect of proton donor (buffer) concentration on catalysis. Next, we examined effects of CO2 by collecting voltammograms as a function of CO2 partial pressure (PCO2).22 In the presence of increasing partial pressures of CO2 (Fig. 5), a CV wave develops on the anodic side of the voltammogram, consistent with a process that is dependent on the concentration of CO2. The proposed mechanism, invoking coupled CO2 binding and reduction, should be dependent on the following equation under equilibrium conditions. Note that Eh refers to the half-wave potential:| M + e− + CO2 ⇄ [M–CO2]− | (3) |
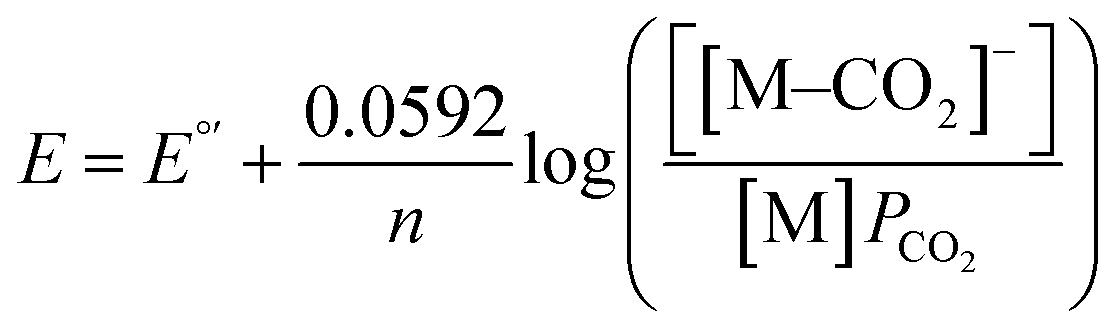 | (4) |
|
Eh = E°′− 0.0592log(PCO2)
| (5) |
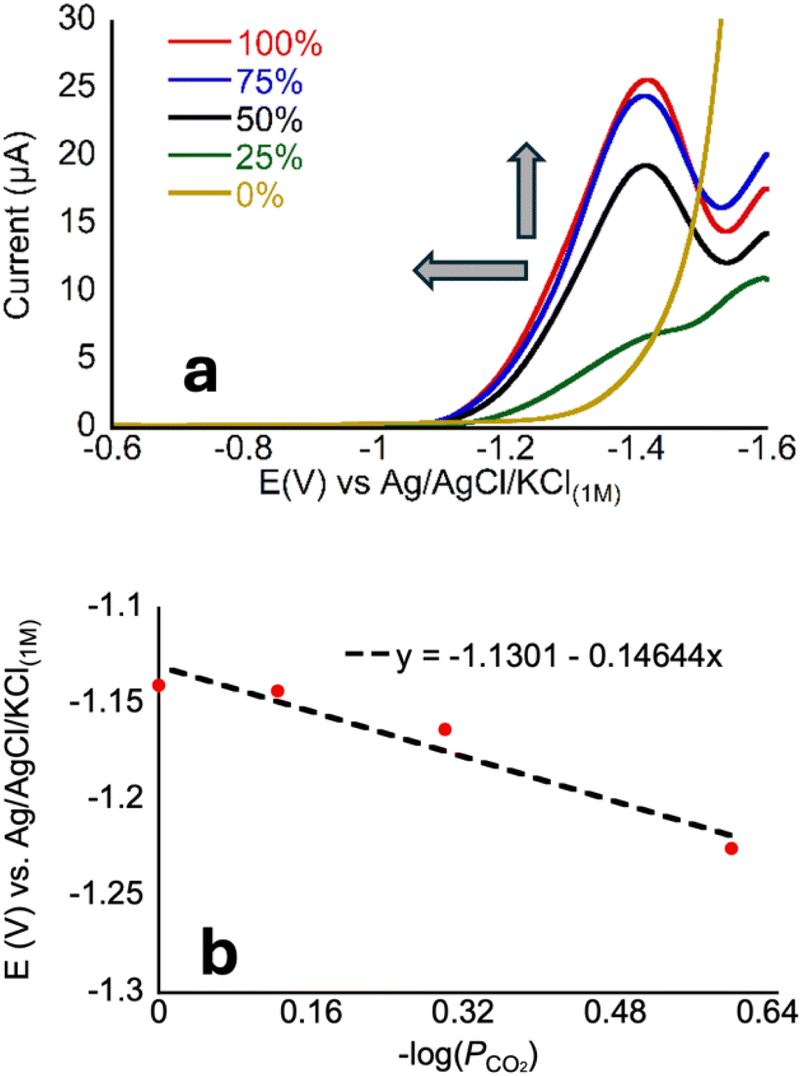 | ||
| Fig. 5 (a) Linear sweep voltammograms of 1 μM CoMC6*a in 50 mM CHES, 0.1 M KCl, pH 5.9 ± 0.1 at 100 mV s−1 under different PCO2, the arrows indicate the direction of increasing PCO2. (b) Plot of Ei vs. −log(PCO2) showing a slope of ∼150 mV per decade. R2 = 0.94. | ||
To analyze these data, we chose a current near the foot of the wave (1.5 μA) to reflect the CO2-dependent process that occurs at less cathodic potentials than H2 production because a distinct peak is not always present in the voltammograms of CoMC6*a. We then define Ei as the potential at which this current is reached; we have used this approach when Eh (eqn (5)) cannot be readily defined (Fig. 5).22
|
Ei = −0.0592log(PCO2) + E°′
| (6) |
The negative non-zero slope seen in Fig. 5 reflects the increasing current with increasing PCO2, consistent with a relationship between CO2 concentration and electron transfer, which supports our proposed mechanism. However, because a clear peak is not present reflecting primarily CO2 reduction, defining a quantitative relationship is not possible from these data.
Examination of Fig. 5a reveals that the voltammogram is nearly the same under 75% and 100% CO2, which contrasts with the clear changes from 0 to 75%. This change in dependence suggests that, above 75%, substrate (CO2) availability is no longer a limiting factor in catalysis. Notably, this observation differs from what is seen for CoMP11-Ac, for which the anodic shift continues for all PCO2 values in the same range. To determine whether the proton donor becomes limiting under these conditions, we measured CVs for CoMC6*a under a CO2 atmosphere under varied concentrations of CHES buffer (the buffer used in Fig. 5). In contrast with the increase in catalytic current seen as a function of [CHES] (and all buffers)48 under N2, the CVs under CO2 are nearly invariant as a function of [CHES] (Fig. S10 and S11†). These observations for CoMC6*a indicate that, in the presence of CO2, a process other than CO2 or proton delivery limits catalysis. This may be a conformational rearrangement of the catalyst, i.e., of the distal peptide to facilitate substrate access, or a later step in catalysis such as C–O bond breakage.
Effect of air on catalysis
Since practical sources of CO2 such as flue gas tend to have impurities such as oxygen (O2), which has been shown to negatively affect many CO2 reduction catalysts, developing catalysts that can facilitate CO2 reduction in the presence of oxygen is a priority.54 To test whether O2 impacts CO2 reduction catalysis by CoMC6*a, a CV of a CoMC6*a solution saturated with CO2 was collected under room air (Fig. 6). The CV of CoMC6*a was not significantly impacted by the presence of air, overlaying closely with CVs under CO2 and nitrogen, suggesting the possibility of air-tolerant CO2 reduction. Results were similar for CVs of CoMC6*a solutions saturated with CO2 whether under 1 atmosphere of CO2, N2, or air. Next, two-hour CPEs were run to determine the impact of air on product formation. The resulting CPEs (Fig. 6 and Table 3) showed no significant difference in selectivity. The overall charge passed and TON values decreased when CO2 was removed from the headspace. This observation is consistent with lower activity with a decrease in available substrate and demonstrates an effect of changing the headspace on the two-hour CPE experiment. These results indicate that CoMC6*a maintains CO2 reduction activity and selectivity in the presence of O2. Note that air tolerance for H2 evolution by CoMC6*a was previously demonstrated.45 | ||
| Fig. 6 (a) CVs of 1 μM CoMC6*a in 50 mM MOPS, pH 5.9 ± 0.1. For all CVs, [KCl] = 0.1 M and scan rate = 100 mV s−1. Arrows in the CV traces indicate the scanning direction. (b) CPE experiments run in 0.5 M MOPS, 1 M KCl, the concentration of catalyst was 1 μM when present. The pH of all MOPS after purging with CO2 was 6.5 ± 0.1; and 7.2 ± 0.2 when purged with N2. Potentials reported vs. Ag/AgCl/KCl(1M). H: headspace S: solution. | ||
| GASHeadspace | GASSolution | Eb (V) | FE(H2) % | FE(CO) % | TON(H2) | TON(CO) | QT (C) |
|---|---|---|---|---|---|---|---|
| a Two-hour CPE experiments conducted on 1 μM catalyst in 0.5 M MOPS with 1 M KCl. Results correspond to the average of at least three individual runs, the error corresponds to the difference between the average and the replicate with the greatest difference from the average. The pH of all solutions was adjusted to 6 for experiments. CPEs under air were purged with CO2 before the headspace was replaced with air ∼99% of the CO2 was replaced.b Potentials reported vs. Ag/AgCl/KCl(1M).c Activity is not reported if it did not exceed three times background in more than one replicate. | |||||||
| CO2 | CO2 | −1.2 | 6 ± 1 | 85 ± 11 | 160 ± 40 | 2200 ± 300 | 2.5 ± 0.2 |
| Air | CO2 | −1.2 | 4 ± 1 | 86 ± 7 | 67 ± 30 | 1500 ± 500 | 1.7 ± 0.6 |
| N2 | CO2 | −1.2 | 5 ± 4 | 90 ± 10 | 80 ± 60 | 1500 ± 200 | 1.6 ± 0.1 |
| N2 | N2 | −1.2 | No above-background activityc | ||||
While more investigations are needed to understand the basis for this air tolerance, there are a few reported examples that provide context. One is a cobalt phthalocyanine catalyst anchored to carbon nanotubes for CO2 reduction. In this system, FECO drops from 93% to 0% in the presence of 5% O2. However, protecting the cobalt phthalocyanine with a bioinspired polymer of intrinsic microporosity increased FECO in the presence of 5% O2 to 75.9%. At levels of O2 in air of 22%, however, FECO decreased to 49.7%.55 Another oxygen-tolerant transition-metal catalyst for CO2 reduction is an iron–porphyrin catalyst with four ferrocenes in its distal site that displays a 500-fold faster rate of CO2 binding compared to O2 binding, giving the catalyst high FEco of 84% in the presence of 25% O2.56 Its O2 tolerance is also attributed to its favorable 4-electron reduction of O2 to H2O that avoids the formation of destructive reactive oxygen species, as well as rapid CO2 binding.56
Insights into effects of catalyst structure on activity
Nature's enzymes have enviable properties, typically rapid catalysis, high substrate and product specificity, and great efficiency (i.e. low overpotential). These properties are attributed to the active-site microenvironment provided by the polypeptide matrix.16,57 However, Nature's metalloenzymes can be challenging to isolate in significant quantities and often are large structures with a low density of active sites. Furthermore, many enzymes that make H2 and that reduce CO2 are sensitive to oxygen. Thus, there has been interest in developing biomolecular catalysts that are relatively easy to prepare and work with, but retain the advantage of having polypeptide matrix that can be tuned to engineer the active site environment.6,58 However, despite the progress made to date, there are few examples in which structure–function relationships have been demonstrated in engineered biomolecular catalysts,20,23,45,59,60 especially for systems that exhibit high activity and robustness (i.e., high TON values).Prior investigation of the mechanism of electrochemical proton reduction by CoMC6*a revealed that proton delivery to CoMC6*a is slow relative to CoMP11-Ac and is impacted by steric hindrance of the proton donor.48 The data are consistent with the requirement of a conformational rearrangement of CoMC6*a to facilitate proton delivery, i.e., to expose the distal side of the porphyrin, which is protected by a helix in the folded mini-protein (Fig. 1). In contrast, CoMP11-Ac reacts with proton donors in a diffusion-controlled manner, provided the proton donor has a pKa below ∼7.5.49 Those results revealed the impact of the distal helix on H2 evolution reactivity of CoMC6*a: it slows proton delivery, changes mechanism, and increases catalyst robustness, as reflected by TONH2 values nearly 10-fold higher (230000) than what is seen for CoMP11-Ac (25000).45,61
Given the more hydrophobic nature of the CoMC6*a active site relative to CoMP11-Ac, we hypothesized that it may display greater CO2 reduction activity and/or selectivity compared to CoMP11-Ac. This prediction is consistent with reports that hydrophobic microenvironments can improve activity and selectivity for CO2 reduction in MOF- and materials-based catalytic systems.14,62–64 and also for catalysts within protein environments.20,23
For electrocatalytic CO2 reduction at −1.2 V, CoMP11-Ac22 and CoMC6*a (Table 1) yield similar and high selectivities for CO production (Table S2† compares results on these catalysts). For CoMP11-Ac at −1.2 V in the presence of MOPS, CHES, or CAPS buffers, values of FECO range from 81 to 88%, and FEH2 ranges from 5 to 8%, similar to the respective ranges for CoMC6*a (73–86% and 4–11%). The measure that does change when comparing these catalysts under these conditions is TON measured in 2-hour experiments; CoMP11-Ac generally has higher TON values for both H2 and CO production at −1.2 V, by a factor of four- to six-fold for CO production and two- to seven-fold for H2 production, suggesting that the more solvent-accessible active site of CoMP11-Ac facilitates reaction turnover at −1.2 V. However, when CPE is run at −1.2 V for 24 hours (Fig. S12†), the gap in TON values for CO production between these catalysts closes, with a TONCO of 14000 for CoMC6*a compared to 32000 for CoMP11-Ac (Table S6†). This result is attributed to a loss of overall activity for CoMP11-Ac in this longer experiment, in which it yields FECO of 61% compared to 86% for CoMC6*a. We propose that the more protected nature of the CoMC6*a active site maintains catalyst integrity and activity in this longer experiment. Its total value of FEH2 + FECO is 91%, but this value is only 70% for CoMP11-Ac. We propose that catalyst degradation, which is significant for CoMP11-Ac, accounts for the balance of FE, consistent with the observation that CoMP11-Ac undergoes deactivation and degradation in longer CPE experiments.61 These results illustrate how supermolecular structure confers advantages for CoMC6*a catalysis that translate to it maintaining high activity and selectivity for CO production in longer (24-hour) experiments.
These differences in selectivity between these catalysts change substantially for reactions run at more negative potential. At −1.4 V in the three different buffers, CoMC6*a has FECO values that vary little (67–76%), while FECO is lower and more variable (21–48%) for CoMP11-Ac. FEH2 values differ significantly between these two catalysts at −1.4 V, ranging from 4 to 24% for CoMC6*a and 29–63% for CoMP11-Ac in the three buffers. Overall, for both catalysts, a decreased buffer acid pKa is correlated with a higher FEH2. We also see that the TONCO value for CoMC6*a at −1.4 V is highest with the least acidic proton donor (CAPS), but for CoMP11-Ac, TONCO at −1.4 V with CAPS is its lowest value among the three buffers. While the basis for this difference is speculative, we propose that these observations support the proposal that the protected and hydrophobic active site of CoMC6*a facilitates CO2 binding and inhibits proton delivery to both enhance CO production and inhibit H2 evolution, especially at lower potentials that enhance H2 evolution activity. However, in CoMP11-Ac, with its solvent-exposed distal site, the pKa of the proton donor is the key factor determining overall catalytic activity, such that CO production activity (TON) increases with a more acidic proton donor even as FECO decreases.
Conclusions
CoMC6*a is a synthetic mini-enzyme that electrochemically catalyzes CO2 reduction to CO in water. We provide evidence that its selectivity for CO2 over proton reduction is enhanced relative to CoMP11-Ac, particularly at more negative potentials, which we attribute to protection of its active site and its lower Co(II/I) potential. The catalytic mechanism for CO formation requires CO2 binding before or coupled with Co(II) reduction for CO formation. CoMC6*a displays an outstanding TONCO of 14000 over 24 hours and excellent selectivity of 86:5 CO:H2 products in the same 24-hour experiment, demonstrating that a small artificial biocatalyst can be active, robust, and selective for CO2 reduction in water. Furthermore, the activity of CoMC6*a is minimally impacted by air, an unusual and desirable property for a CO2 reduction catalyst.
Data availability
Data supporting this article have been published as ESI.†Author contributions
Conceptualization: JLA-H, KLB, AL; funding acquisition: AAS, KLB, AL; investigation: AAS, JLA-H, LL, KBR; supervision: KLB, AL; writing – original draft: AAS, JLA-H; writing – review & editing: KLB, AL, LL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Marco Chino for participating in fruitful discussions. The electrocatalytic experiments and analysis of products was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0002106. The design and preparation of CoMC6*a samples was supported by the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.3, theme 2.a “Green Energies for the Future”, funded by the European Union – NextGenerationEU – Project Title “NEST – Network 4 Energy Sustainable Transition” – CUP E63C22002160007. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. (DGE-1939268). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.References
- C. Costentin, M. Robert and J.-M. Savéant, Catalysis of the electrochemical reduction of carbon dioxide, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- J. Schneider, H. F. Jia, J. T. Muckerman and E. Fujita, Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- P. Saha, S. Amanullah and A. Dey, Selectivity in Electrochemical CO2 Reduction, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- K. K. Häckl, W., Some aspects of green solvents, C. R. Chim., 2018, 21, 572–580 CrossRef.
- J. M. Le and K. L. Bren, Engineered Enzymes and Bioinspired Catalysts for Energy Conversion, ACS Energy Lett., 2019, 4, 2168–2180 CrossRef CAS.
- C. Costentin, M. Robert, J.-M. Saveant and A. Tatin, Efficient and selective molecular catalyst for the CO2-to-CO electrochemical conversion in water, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 6882–6886 CrossRef CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, Advances and challenges in electrochemical CO2 reduction processes: an engineering and design perspective looking beyond new catalyst materials, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization, Angew Chem. Int. Ed. Engl., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- J.-J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z.-J. Wang, H. Jin, X. Wang and S. Wang, Microenvironment Engineering for the Electrocatalytic CO2 Reduction Reaction, Angew Chem. Int. Ed. Engl., 2022, 61, e202207252 CrossRef CAS PubMed.
- E. Fujita, D. C. Grills, G. F. Manbeck and D. E. Polyansky, Understanding the Role of Inter- and Intramolecular Promoters in Electro- and Photochemical CO2 Reduction Using Mn, Re, and Ru Catalysts, Acc. Chem. Res., 2022, 55, 616–628 CrossRef CAS PubMed.
- E. S. Wiedner, A. M. Appel, S. Raugei, W. J. Shaw and R. M. Bullock, Molecular Catalysts with Diphosphine Ligands Containing Pendant Amines, Chem. Rev., 2022, 122(14), 12427–12474 CrossRef CAS PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Towards molecular understanding of local chemical environment effects in electro- and photocatalytic CO2 reduction, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- T. L. Soucy, W. S. Dean, J. K. Zhou, K. E. R. Cruz and C. C. L. McCrory, Considering the Influence of Polymer-Catalyst Interactions on the Chemical Microenvironment of Electrocatalysts for the CO2 Reduction Reaction, Acc. Chem. Res., 2022, 55, 252–261 CrossRef CAS PubMed.
- S. Amanullah, P. Saha and A. Dey, Recent developments in the synthesis of bio-inspired iron porphyrins for small molecule activation, Chem. Commun., 2022, 58, 5808–5828 RSC.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- J. Y. Yang, T. A. Kerr, X. S. Wang and J. M. Barlow, Reducing CO2 to HCO2− at Mild Potentials: Lessons from Formate Dehydrogenase, J. Am. Chem. Soc., 2020, 142, 19438–19445 CrossRef CAS PubMed.
- L. B. Maia, I. Moura and J. J. G. Moura, Molybdenum and tungsten-containing formate dehydrogenases: Aiming to inspire a catalyst for carbon dioxide utilization, Inorg. Chim. Acta, 2017, 455, 350–363 CrossRef CAS.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimküshler, S. Hirota, Y. L. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Second and Outer Coordination Sphere Effects in Nitrogenase, Hydrogenase, Formate Dehydrogenase, and CO Dehydrogenase, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- C. R. Schneider and H. S. Shafaat, An internal electron reservoir enhances catalytic CO2 reduction by a semisynthetic enzyme, Chem. Commun., 2016, 52, 9889–9892 RSC.
- X. H. Liu, F. Y. Kang, C. Hu, L. Wang, Z. Xu, D. D. Zheng, W. M. Gong, Y. Lu, Y. H. Ma and J. Y. Wang, A genetically encoded photosensitizer protein facilitates the rational design of a miniature photocatalytic CO2-reducing enzyme, Nat. Chem., 2018, 10, 1201–1206 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, A. A. Salamatian, J. W. Han and K. L. Bren, Potential- and Buffer-Dependent Selectivity for the Conversion of CO2 to CO by a Cobalt Porphyrin-Peptide Electrocatalyst in Water, ACS Catal., 2022, 12, 14689–14697 CrossRef CAS PubMed.
- R. Alcala-Torano, N. Halloran, N. Gwerder, D. J. Sommer and G. Ghirlanda, Light-Driven CO2 Reduction by Co-Cytochrome b562, Front. Mol. Biosci., 2021, 8, 609654 CrossRef CAS PubMed.
- Y. Deng, S. Dwaraknath, W. O. Ouyang, C. J. Matsumoto, S. Ouchida and Y. Lu, Engineering an Oxygen-Binding Protein for Photocatalytic CO2 Reductions in Water, Angew Chem. Int. Ed. Engl., 2023, 62, e202215719 CrossRef CAS PubMed.
- G. A. O. Udry, L. Tiessler-Sala, E. Pugliese, A. Urvoas, Z. Halime, J.-D. Maréchal, J.-P. Mahy and R. Ricoux, Photocatalytic Hydrogen Production and Carbon Dioxide Reduction Catalyzed by an Artificial Cobalt Hemoprotein, Int. J. Mol. Sci., 2022, 23, 14640 CrossRef CAS.
- A. A. Salamatian and K. L. Bren, Bioinspired and biomolecular catalysts for energy conversion and storage, FEBS Lett., 2023, 597, 174–190 Search PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Biomimetic assembly and activation of FeFe-hydrogenases, Nature, 2013, 499, 66–70 CrossRef CAS PubMed.
- S. B. Carr, R. M. Evans, E. J. Brooke, S. A. M. Wehlin, E. Nomerotskaia, F. Sargent, F. A. Armstrong and S. E. V. Phillips, Hydrogen activation by NiFe -hydrogenases, Biochem. Soc. Trans., 2016, 44, 863–868 CrossRef CAS PubMed.
- H. L. Tai, S. Hirota and S. T. Stripp, Proton Transfer Mechanisms in Bimetallic Hydrogenases, Acc. Chem. Res., 2021, 54, 232–241 CrossRef CAS PubMed.
- Y. S. Liu and C. C. L. McCrory, Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Saveant, A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- S. Lense, K. A. Grice, K. Gillette, L. M. Wolf, G. Robertson, D. McKeon, C. Saucedo, P. J. Carroll and M. Gau, Effects of Tuning Intramolecular Proton Acidity on CO2 Reduction by Mn Bipyridyl Species, Organometallics, 2020, 39, 2425–2437 CrossRef CAS.
- C. R. Schneider, L. C. Lewis and H. S. Shafaat, The good, the neutral, and the positive: buffer identity impacts CO2 reduction activity by nickel(ii) cyclam, Dalton Trans., 2019, 48, 15810–15821 RSC.
- K. M. O. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- I. L. Bhugun, D. Lexa and J.-M. Savéant, Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins: Synergystic effect of weak Brönsted acids, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- J. W. Wang, K. Yamauchi, H. H. Huang, J. K. Sun, Z. M. Luo, D. C. Zhong, T. B. Lu and K. Sakai, A Molecular Cobalt Hydrogen Evolution Catalyst Showing High Activity and Outstanding Tolerance to CO and O2, Angew Chem. Int. Ed. Engl., 2019, 58, 10923–10927 CrossRef CAS PubMed.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst, ACS Catal., 2019, 9, 4867–4874 Search PubMed.
- X. Zhang, K. Yamauchi and K. Sakai, Earth-Abundant Photocatalytic CO2 Reduction by Multielectron Chargeable Cobalt Porphyrin Catalysts: High CO/H2 Selectivity in Water Based on Phase Mismatch in Frontier MO Association, ACS Catal., 2021, 11, 10436–10449 CrossRef CAS.
- L. Leone, M. Chino, F. Nastri, O. Maglio, V. Pavone and A. Lombardi, Mimochrome, a metalloporphyrin-based catalytic Swiss knife, Biotechnol. Appl. Biochem., 2020, 67, 495–515 CrossRef CAS PubMed.
- L. Leone, M. De Fenza, A. Esposito, O. Maglio, F. Nastri and A. Lombardi, Peptides and metal ions: A successful marriage for developing artificial metalloproteins, J. Pept. Sci., 2024, 30, e3606 Search PubMed.
- M. Chino, S. La Gatta, L. Leone, M. De Fenza, A. Lombardi, V. Pavone and O. Maglio, Dye Decolorization by a Miniaturized Peroxidase Fe-MimochromeVI*a, Int. J. Mol. Sci., 2023, 24, 11070 Search PubMed.
- G. Zambrano, A. Sekretareva, D. D'Alonzo, L. Leone, V. Pavone, A. Lombardi and F. Nastri, Oxidative dehalogenation of trichlorophenol catalyzed by a promiscuous artificial heme-enzyme, RSC Adv., 2022, 12, 12947–12956 Search PubMed.
- L. Leone, A. B. Muñoz-García, D. D’Alonzo, V. Pavone, F. Nastri and A. Lombardi, Peptide-based metalloporphyrin catalysts: unveiling the role of the metal ion in indole oxidation, J. Inorg. Biochem., 2023, 246, 112298 Search PubMed.
- L. Leone, D. D'Alonzo, O. Maglio, V. Pavone, F. Nastri and A. Lombardi, Highly Selective Indole Oxidation Catalyzed by a Mn-Containing Artificial Mini-Enzyme, ACS Catal., 2021, 11, 9407–9417 Search PubMed.
- V. Firpo, J. M. Le, V. Pavone, A. Lombardi and K. L. Bren, Hydrogen evolution from water catalyzed by cobalt-mimochrome VI*a, a synthetic mini-protein, Chem. Sci., 2018, 9, 8582–8589 Search PubMed.
- E. H. Edwards, J. Le, A. Salamatian, N. L. Peluso, L. Leone, A. Lombardi and K. L. Bren, A Cobalt Mimochrome for Photochemical Hydrogen Evolution from Neutral Water, J. Inorg. Biochem., 2022, 230, 11753 CrossRef PubMed.
- G. Caserta, M. Chino, V. Firpo, G. Zambrano, L. Leone, D. D'Alonzo, F. Nastri, O. Maglio, V. Pavone and A. Lombardi, Enhancement of peroxidase activity in artificial catalysts through rational design, ChemBioChem, 2018, 19, 1823–1826 CrossRef CAS PubMed.
- J. M. Le, G. Alachouzos, M. Chino, A. J. Frontier, A. Lombardi and K. L. Bren, Tuning Mechanism through Buffer Dependence of Hydrogen Evolution Catalyzed by a Cobalt Mini-enzyme, Biochemistry, 2020, 59, 1289–1297 Search PubMed.
- J. L. Alvarez-Hernandez, A. E. Sopchak and K. L. Bren, Buffer pK(a) Impacts the Mechanism of Hydrogen Evolution Catalyzed by a Cobalt Porphyrin-Peptide, Inorg. Chem., 2020, 59, 8061–8069 Search PubMed.
- J. L. Alvarez-Hernandez, J. W. Han, A. E. Sopchak, Y. X. Guo and K. L. Bren, Linear Free Energy Relationships in Hydrogen Evolution Catalysis by a Cobalt Tripeptide in Water, ACS Energy Lett., 2021, 6, 2256–2261 Search PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Saveant, Pendant Acid-Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, A. A. Salamatian, A. E. Sopchak and K. L. Bren, Hydrogen evolution catalysis by a cobalt porphyrin peptide: A proposed role for porphyrin propionic acid groups, J. Inorg. Biochem., 2023, 249, 112390 CrossRef CAS PubMed.
- J. Shen, M. J. Kolb, A. J. Göttle and M. T. M. Koper, DFT Study on the Mechanism of the Electrochemical Reduction of CO2 Catalyzed by Cobalt Porphyrins, J. Phys. Chem. C, 2016, 120, 15714–15721 CrossRef CAS.
- N. J. Harmon and H. Wang, Electrochemical CO2 Reduction in the Presence of Impurities: Influences and Mitigation Strategies, Angew Chem. Int. Ed. Engl., 2022, 61, e202213782 Search PubMed.
- X. Lu, Z. Jiang, X. Yuan, Y. Wu, R. Malpass-Evans, Y. Zhong, Y. Liang, N. B. McKeown and H. Wang, A bio-inspired O2-tolerant catalytic CO2 reduction electrode, Sci. Bull., 2019, 64, 1890–1895 CrossRef CAS PubMed.
- B. Mondal, P. Sen, A. Rana, D. Saha, P. Das and A. Dey, Reduction of CO2 to CO by an Iron Porphyrin Catalyst in the Presence of Oxygen, ACS Catal., 2019, 9, 3895–3899 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- R. E. Treviño and H. S. Shafaat, Protein-based models offer mechanistic insight into complex nickel metalloenzymes Regina E. Trevino and Hannah S. Shafaat, Curr. Opin. Chem. Biol., 2022, 67, 102110 CrossRef PubMed.
- A. E. Wertz, P. Teptarakulkarn, R. E. Stein, P. J. Moore and H. S. Shafaat, Rubredoxin Protein Scaffolds Sourced from Diverse Environmental Niches as an Artificial Hydrogenase Platform, Biochemistry, 2023, 62, 2622–2631 CrossRef CAS PubMed.
- M. Bacchi, G. Berggren, J. Niklas, E. Veinberg, M. W. Mara, M. L. Shelby, O. G. Poluektov, L. X. Chen, D. M. Tiede, C. Cavazza, M. J. Field, M. Fontecave and V. Artero, Cobaloxime-Based Artificial Hydrogenases, Inorg. Chem., 2014, 53, 8071–8082 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, Hydrogen Evolution from Neutral Water under Aerobic Conditions Catalyzed by Cobalt Microperoxidase-11, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- X. Yang, Q.-X. Li, S.-Y. Chi, H.-F. Li, Y.-B. Huang and R. Cao, Hydrophobic perfluoroalkane modified metal-organic frameworks for the enhanced electrocatalytic reduction of CO2, SmartMat, 2022, 3, 163–172 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Enhancing carbon dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst microenvironment, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- C. D. Sahm, A. Ciotti, E. Mates-Torres, V. Badiani, K. Sokołowski, G. Neri, A. J. Cowan, M. García-Melchor and E. Reisner, Tuning the local chemical environment of ZnSe quantum dots with dithiols towards photocatalytic CO2 reduction, Chem. Sci., 2022, 13, 5988–5998 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07026g |
| This journal is © The Royal Society of Chemistry 2025 |