
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise functionalization in nano-confinement: a bottom-up approach to the evolution of selective molecular receptors†
Ya-Mei Tana,
Lu-Mei Zhanga,
Qixia Baib,
Zhe Zhang b,
Pingshan Wangb and
Qi Zhang*a
b,
Pingshan Wangb and
Qi Zhang*a
aKey Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, China. E-mail: qi.zhang.ch@scu.edu.cn
bInstitute of Environmental Research at Greater Bay Area, Key Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Guangzhou University, Guangzhou 510006, China
First published on 20th January 2025
Abstract
Precise molecular recognition depends on the delicate interplay between a guest molecule and a host possessing complementary functional groups. The de novo design of selective artificial receptors remains a formidable challenge, given the complexity of predicting these interactions. We present herein a bottom-up approach to the evolution of selective molecular receptors through precise endo-functionalization of a supramolecular cage. Internal functional groups were introduced within the heteroleptic palladium coordination cage in a site-precise fashion. With just five different functional groups, we successfully created a library of 32 isoreticular nano-cages, each featuring a unique micro-environment, by varying the nature, location and combination of endo-functional groups. The nano-cage exhibited adaptive recognition ability towards guest molecules of distinct geometries and hydrogen bonding capabilities. Titration experiments demonstrated that the binding affinity for a specific guest can be finely tuned and optimized by changing the endo-functional groups. As a proof of principle, by strategically screening our nano-cage library, we identified a receptor with high affinity and specificity for the dihydrogen phosphate guest. X-ray analysis and DFT calculation highlighted the pivotal role of the synergistic interactions among distinct endo-functional groups in achieving high-fidelity molecular recognition. This study is expected to provide a versatile solution for the bottom-up construction of tailor-made molecular receptors.
Introduction
Molecular recognition is fundamental to a wide range of biological processes. The binding site of a natural enzyme is decorated by various amino acid residues. The exact spatial arrangement of these functional groups does more than just shape the cavity and filter the molecules that can dock within; it also facilitates a symphony of distinct non-covalent interactions between the receptor and the target molecule. These interactions collectively result in recognition with high affinity and specificity.Achieving an in-depth understanding of the operational mechanism of natural receptors has been a central objective, driving the creation of numerous artificial systems designed as simplified mimetics. The binding pocket of a natural receptor can be considered as a functionalized nanoscopic confinement. Therefore, a rational approach to mimic biomolecular receptors involves the introduction of functional groups within synthetic cavities. While synthetic receptors can achieve high-affinity binding for a specific guest molecule by integrating multiple recognition elements, differentiation between guest molecules that possess identical binding motifs using these artificial systems often poses a significant challenge.
Specific recognition depends on the intricate interactions between the guest and the host containing complementary functional groups. However, forecasting these interactions when designing a receptor structure is extremely challenging, despite advancements in computational chemistry. Screening potential guest molecules against an established host system may occasionally lead to the discovery of a selective receptor (Fig. 1a). However, this method is subject to serendipity and lacks predictability. A more rational approach involves the in situ synthesis of a host using the target guest molecule as a template.1 The guest molecule brings functionalized building blocks into close proximity through attractive interactions, and the linkage of these pre-organized building blocks can yield a selective receptor tailored to the template molecule (Fig. 1b).
 | ||
| Fig. 1 Strategies for identifying functionalized artificial hosts capable of selective molecular recognition. An optimal host–guest complex can be identified by screening potential guest molecules against an established host system (a). A selective host molecule can be synthesized using the target guest molecule as a template (b). In this study, a library of hosts was readily constructed through selective endo-functionalization of a supramolecular coordination cage. A receptor with both high affinity and specificity was identified by strategically screening the host library with the target guest molecule (c). | ||
Despite significant progress in the construction of synthetic receptors, two primary challenges persist. First, the cooperativity among various amino acid residues is essential for the precise molecular recognition that bio-receptors achieve. However, the majority of synthetic receptors incorporate a single type of endo-functional group. Systems with distinct endo-functional groups are rare.2 Second, modifying these endo-functional groups3 can alter the micro-environment within the cavity of synthetic receptors. This would offer the most direct and efficient means of fine-tuning and optimizing host–guest interactions. Yet, no synthetic systems have been developed that can effectively address both of these challenges simultaneously.
In this study, we report on a synthetic receptor capable of integrating distinct endo-functional groups and readily altering its internal micro-environment (Fig. 1c). A Pd2A2B2-type coordination cage was efficiently constructed via a one-pot, two-step synthesis. The heteroleptic nature of the Pd2A2B2 cage allows for the introduction of distinct endohedral groups. Facile modulation of the micro-environment within the coordination cages was achieved by varying the nature, location and combination of the internal functionalities. With only five different functional groups, a library of 32 coordination cages was readily established. Host–guest experiments demonstrated that the Pd2A2B2 cage can adapt to guest molecules of diverse geometries and hydrogen bonding capabilities by employing different operational endo-functional groups. Moreover, the binding affinity for a specific guest can be finely tuned and optimized by combining distinct endo-functional groups. A receptor for the dihydrogen phosphate guest with high affinity and specificity was obtained by strategic screening of the nano-cage library.
Results and discussion
Synthesis of the Pd2A2B2 nano-cage
Regulating the steric hindrance around the donor site4 of ligands has been demonstrated to be an effective strategy to control the outcome of assembly reactions. Utilizing this strategy, we previously reported the selective formation of a Pd2L2-type ring assembly instead of the regular Pd2L4-type cage structure.2e The steric hindrance at the donor site on ligand A1 impedes the coordination of additional A1 ligands on the ring complex, thereby ensuring its exceptional stability. In this study, it was found that the [Pd2(A1)2] ring was selectively transformed into a single species at room temperature upon treatment of 2 eq. of ligand B1 that lacks steric hindrance (Fig. 2a–d). Single crystals for X-ray analysis were obtained by slow diffusion of diethyl ether vapor into the reaction solution in acetonitrile. The product was identified to be the [Pd2(trans-A1)2(B1)2] cage2a,e,5 (Fig. 2e). The trans-cage likely occurred with a [Pd2(A1)2(B1)] species as an intermediate, which could be observed alongside the [Pd2(A1)2(B1)2] cage and the [Pd2(A1)2] ring when the ring precursor was treated with 1 eq. of ligand B1 (ESI-Fig. 17†). The Hiraoka group reported a similar ring-to-cage transformation.5r The Clever group constructed self-penetrating trans-Pd2A2B2 cages from a pair of ligands of complementary bite angles.5i,6 The [Pd2(A1)2(B1)2] cage is a meta-stable species. It underwent partial decomposition upon refluxing in acetonitrile (ESI-Chapter 6†). | ||
| Fig. 2 Selective synthesis of the [Pd2(A1)2(B1)2] cage. The reaction between ring assembly [Pd2(A1)2] and 2 eq. of ligand B1 yielded the cage [Pd2(A1)2(B1)2] (a). Partial 1H NMR spectra of ring assembly [Pd2(A1)2] (b), cage [Pd2(A1)2(B1)2] (c) and ligand B1 (d) in acetonitrile. Full spectra can be found in the ESI.† The crystal structure of cage [Pd2(A1)2(B1)2] in side view (e) and top view (f). Anions and solvent molecules are omitted for clarity. | ||
As shown by the crystal structure of the [Pd2(A1)2(B1)2] cage,7 each phenyl ring at the equator of the cage structure has a hydrogen atom directed into the cage cavity (Fig. 2e). Substituting these hydrogen atoms with functional groups would lead to endo-functionalized supramolecular cages. The distance between the functionalization sites on A-ligands differs slightly from that of B-ligands. This discrepancy can be attributed to the differing levels of distortion in the two types of ligands: the A-ligands lie almost flat without any distortion, whereas the B-ligands are twisted into opposite directions (Fig. 2f). Furthermore, the two functionalization sites on A-ligands point towards the center of the cage cavity, while those on B-ligands are directed away from the cavity center. These observations suggest that the functionalization sites on the A-ligand and B-ligand are non-equivalent. Therefore, three distinct arrangements of endo-functional groups can be achieved by utilizing the (1) functionalized A-ligand, (2) functionalized B-ligand or (3) a combination of A- and B-ligands both with functional groups (Fig. 3a).
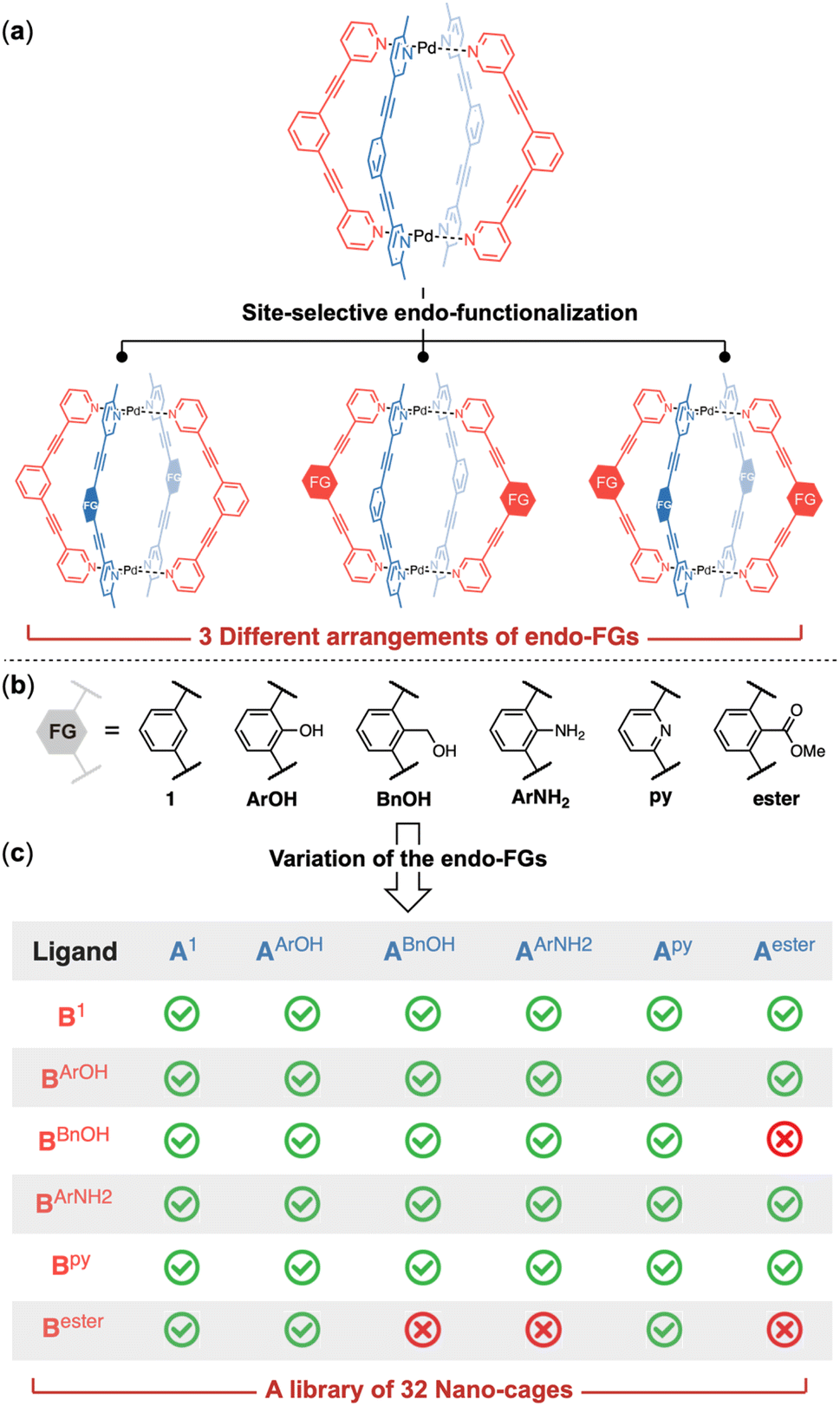 | ||
| Fig. 3 Construction of a library of Pd2A2B2 cages through endo-functionalization. Three different arrangements of the internal groups can be achieved through site-selective endo-functionalization (a). Phenol (ArOH), benzyl alcohol (BnOH), aniline (ArNH2), pyridine (py) and ester were selected as the endo-functional groups due to their hydrogen bonding capability (b). 32 Pd2A2B2 cages with different internal functionalities were obtained through variation of the nature, location and combination of the endo-functional groups (c). | ||
Modulation of the internal micro-environment through endo-functionalization
We decided to incorporate functional groups capable of acting as hydrogen bonding donors or acceptors, given that hydrogen bonding interactions are pivotal for molecular recognition by natural enzymes. Pyridine and ester groups were selected for their ability to serve as hydrogen bonding acceptors, while phenol, aniline and benzyl alcohol were chosen for their dual role as both hydrogen bonding donors and acceptors, each with distinct capabilities. Additionally, the methylene linkage in the benzyl alcohol allows a different spatial arrangement of the hydroxy group compared to that in phenol. For this purpose, we synthesized ligands with pyridine (Apy/Bpy), aniline (AArNH2/BArNH2), phenol (AArOH/BArOH), benzyl alcohol (ABnOH/BBnOH) and ester (Aester/Bester) groups (Fig. 3b). We first investigated the reaction between the [Pd2(A1)2] ring and functionalized B-ligands. Selective cage formation was observed in all cases (ESI-Chapter 4.2†). Subsequently, we tried to incorporate endo-functional groups on A-ligands. We successfully obtained the functionalized ring intermediates (ESI-Chapter 3†). Subsequent reaction with ligand B1 produced the corresponding [Pd2(AX)2(B1)2] cages (ESI-Fig. 29/41/51/61/73†). Crystal structures were obtained for all di-endo-functionalized coordination cages (Fig. 4e–l and ESI-Fig. 307†)8 except for the [Pd2(A1)2(BBnOH)2] cage. We next employed functionalized A and B ligands to generate tetra-endo-functionalized hetero-cages, which not only possess additional hydrogen bonding recognition motifs but also enhance the diversity of the internal nanoscopic environment. The functionalized Pd2A2-ring assemblies smoothly reacted with B-ligand containing functional groups to yield tetra-functionalized hetero-cages (ESI-Chapter 4.3–4.7†) except for three cases ([Pd2(ABnOH)2(Bester)2], [Pd2(AArNH2)2(Bester)2] and [Pd2(Aester)2(BBnOH)2]). The assembly reactions halted at the stage of the Pd2A2B-bowl complexes likely due to the steric hindrance within the cavity of the complexes when these specific functional group combinations were employed. This method provides a facile and reliable two-step synthesis for the endo-functionalized Pd2A2B2-cage. By utilizing just five different functional groups, we successfully constructed a library of 32 nano-cages with different internal functionalities (Fig. 3c). | ||
| Fig. 4 Modulation of the micro-environment within the Pd2A2B2-cage through endo-functionalization. The incorporation of distinct endo-functional groups leads to varying degrees of axial contraction (Pd–Pd distance) and skeleton distortion of the nano-cages. Consequently, this results in diverse distances and orientations (θ angle) of the endo-functional groups. Selected crystal structures of the unfunctionalized [Pd2(A1)2(B1)2] (a), Pd2A2B2-cages with functionalized A-ligands (e–h), functionalized B-ligands (i–l) and a combination of A- and B-ligands, both with functional groups (b–d). Anions and solvent molecules are omitted for clarity. | ||
The crystal structures of the endo-functionalized hetero-cages were analyzed in detail. The introduction of endo-functional groups has a subtle influence on the geometry of the Pd2A2B2 cages. In the solid state, a counter ion was observed occupying the central cavity of the hetero-cage as a guest molecule. To accommodate the endohedral groups, the functionalized cages were widened at the equator, accompanied by slight contraction along the Pd–Pd axis. The presence of specific functional groups led to varying degrees of axial contraction (Fig. 4 and ESI-Table 4†). Despite featuring the same type of functional group, the cages exhibited different structural characteristics, which were influenced by the location of the endo-functional groups. The cages [Pd2(A1)2(BX)2] and [Pd2(AX)2(B1)2] demonstrated varying levels of axial contraction and skeleton distortion, resulting in different orientations of the functional groups (Fig. 4: (e) vs. (i); (f) vs. (j); (g) vs. (k) and (h) vs. (l)). We also obtained crystal structures for three tetra-endo-functionalized hetero-cages ([Pd2(AArOH)2(BArNH2)2],9 [Pd2(AArOH)2(Bpy)2]10 and [Pd2(Apy)2(BArNH2)2]11). Compared to their counterparts with two endo-functional groups, cages [Pd2(AArOH)2(Bpy)2] (Fig. 4c) and [Pd2(Apy)2(BArNH2)2] (Fig. 4d) experienced further contraction along the Pd–Pd axis, while the distortion of [Pd2(AArOH)2(BArNH2)2] (Fig. 4b) was comparable to that of the di-endo-functionalized parent ([Pd2(AArOH)2(B1)2], Fig. 4f).
Adaptive molecular recognition
Overall, all hetero-cages with endo-functional groups displayed unique structural features compared to the unfunctionalized parent cage ([Pd2(A1)2(B1)2]). These observations indicated that three key parameters – the nature, location, and combination of the endo-functional groups – can be utilized to manipulate the geometry of the Pd2A2B2 cages. The resulting distortion of the cage framework will subsequently influence the arrangement of the functional groups within the cavity. Together with the diverse combinations of functional groups, this approach could potentially yield a series of nano-cages with similar dimensions but distinct micro-environments. These distinct micro-environments could, in principle, accommodate different guest molecules. To test this hypothesis, we selected three guest molecules with different geometries and hydrogen bonding capabilities: trigonal planar nitrate (NO3−), tetrahedral perchlorate (ClO4−) and dihydrogen phosphate (H2PO4−). NO3− and ClO4− can only act as hydrogen bonding acceptors, whereas H2PO4− can function as both hydrogen bonding donors and acceptors. The eight inner pyridyl C–H bonds act as the primary binding sites for anionic guests through hydrogen bonding interactions and concomitant electrostatic attraction to the palladium cation, as demonstrated by the crystal structure of TfO−@[Pd2(A1)2(B1)2] (ESI-Fig. 300†). The bound triflate anion is also close to the functionalization sites at the equator of the cage host. Functional groups introduced at these positions could serve as a modifiable secondary binding site to affect the host–guest association (Fig. 5a). | ||
| Fig. 5 Adaptive host–guest association via the modulation of the secondary binding site. The functionalization sites within the Pd2A2B2-cage act as a tunable secondary binding site for the guest molecule that is bound to the inner pyridyl protons, which serve as the primary binding site (a). The impact of modifying the secondary binding site for guest recognition was examined through titration experiments performed in MeCN. The trigonal planar nitrate guest shows preference for the Pd2A2B2-cage with ester groups on A-ligands (b), whereas the tetrahedral perchlorate guest exhibits the strongest affinity for the nano-cage with aniline groups on B-ligands (c). | ||
Crystal structure analysis reveals that one counter ion of the coordination cages resides within the cavity as a primitive guest. To eliminate its interference, the original counter ion was replaced with the bulky BArF− anions12 (ESI-Chapter 5†), which are too large to fit within the cavity, prior to the host–guest studies. The host–guest association for NO3− and ClO4− conforms to the fast-exchange kinetics in acetonitrile, as evidenced by the observation of a single set of signals for the host (ESI-Fig. 198/232†). The binding constants for these two anions were determined by fitting the titration data to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding mode using BindFit.13 To elucidate the effect of the endo-functional groups on host–guest association, the binding constants with the endo-functionalized coordination cages were compared to those of the parent cage devoid of internal functional groups (Fig. 5 and ESI-Table 1/2†). The variation in association constants is sensitive to the nature and the location of the endo-functional groups. The incorporation of phenol (Fig. 5b, entry 2/3) and aniline (Fig. 5b, entry 6/7) led to a pronounced reduction in the binding affinity for the nitrate anion. A decline in the binding constant was also observed when the benzyl alcohol functionality was integrated into the B-ligand of the Pd2A2B2 cage (Fig. 5b, entry 5), while a comparable binding constant was measured with the [Pd2(ABnOH)2(B1)2] cage (Fig. 5b, entry 4). The Pd2A2B2 cages featuring two internal pyridine groups exhibited a slight to moderate enhancement in their affinity for NO3− (Fig. 5b, entry 8/9). The most substantial increase in the binding constant was observed with the incorporation of endohedral ester groups, which resulted in 4.9-fold (Fig. 5b, entry 11) and 8.1-fold (Fig. 5b, entry 10) increases with the [Pd2(A1)2(Bester)2] and [Pd2(Aester)2(B1)2] cages, respectively.
:1 binding mode using BindFit.13 To elucidate the effect of the endo-functional groups on host–guest association, the binding constants with the endo-functionalized coordination cages were compared to those of the parent cage devoid of internal functional groups (Fig. 5 and ESI-Table 1/2†). The variation in association constants is sensitive to the nature and the location of the endo-functional groups. The incorporation of phenol (Fig. 5b, entry 2/3) and aniline (Fig. 5b, entry 6/7) led to a pronounced reduction in the binding affinity for the nitrate anion. A decline in the binding constant was also observed when the benzyl alcohol functionality was integrated into the B-ligand of the Pd2A2B2 cage (Fig. 5b, entry 5), while a comparable binding constant was measured with the [Pd2(ABnOH)2(B1)2] cage (Fig. 5b, entry 4). The Pd2A2B2 cages featuring two internal pyridine groups exhibited a slight to moderate enhancement in their affinity for NO3− (Fig. 5b, entry 8/9). The most substantial increase in the binding constant was observed with the incorporation of endohedral ester groups, which resulted in 4.9-fold (Fig. 5b, entry 11) and 8.1-fold (Fig. 5b, entry 10) increases with the [Pd2(A1)2(Bester)2] and [Pd2(Aester)2(B1)2] cages, respectively.
Transitioning from the trigonal planar NO3− to the tetrahedral ClO4−, the endo-functional group had a notably distinct impact. The ester functionality, which was highly effective for the recognition of NO3−, led to the weakest binding for ClO4− (Fig. 5c, entry 21/22). Furthermore, coordination cages decorated with pyridine groups demonstrated a reduced affinity for ClO4− (Fig. 5c, entry 19/20) relative to their unfunctionalized counterparts. In contrast, cages modified with functional groups capable of acting as hydrogen bond donors exhibited enhanced binding constants, with the effect being more pronounced when these groups were integrated into the B-ligands. Aniline emerged as the most effective binding motif for ClO4−, resulting in a 5.9-fold increase in the association constant with [Pd2(A1)2(BArNH2)2] (Fig. 5c, entry 18).
Evolution of a selective molecular receptor
The binding of dihydrogen phosphate exhibits a slow-exchange characteristic with the formation of 1:2 host–guest complexes (ESI-Fig. 268†). Preliminary titration experiments with H2PO4− yielded relatively close binding constants for the Pd2A2B2-cages functionalized with aniline, phenol and benzyl alcohol groups (ESI-Table 3†). To evaluate the influence of endo-functionalization more precisely, competitive binding experiments were employed (Fig. 6a and ESI-Chapter 8†). Upon introducing H2PO4− to an acetonitrile solution containing an equimolar mixture of two distinct Pd2A2B2-cages, the guest molecule exhibited a slow exchange rate between the two host–guest complexes formed, facilitating the quantification of each species involved. In contrast, NO3− and ClO4−, obeying a fast-exchange binding mechanism, precluded such analysis, as only one set of host signals could be observed for the two distinct competitive coordination cages. We first performed competition experiments between the unfunctionalized cage and its di-endo-functionalized counterparts (Fig. 6b, phase 1). The cages featuring phenol, benzyl alcohol and aniline functionalities on the B-ligands outperformed the parent cage. Encapsulation of H2PO4− was only observed with these functionalized cages (Fig. 6b/phase 1, lines 1–3 and ESI-Fig. 279–281†). In contrast, cages with ester and pyridine groups displayed reduced binding affinity for H2PO4− (Fig. 6b/phase 1, lines 4/5). Interestingly, the presence of ester groups altered the binding kinetics from slow to fast exchange (ESI-Fig. 277/283†). The pyridine-functionalized cage completely rejected the encapsulation of H2PO4− (ESI-Fig. 276/282†). Among the three endohedral groups that enhance the binding towards H2PO4−, aniline demonstrated superior recognition ability (Fig. 6b/phase 1, lines 6/7 and ESI-Fig. 284/285†).
 | ||
| Fig. 6 Competitive binding experiments with the dihydrogen phosphate guest. The impact of endo-functionalization on the recognition of H2PO4− was evaluated through competitive binding experiments (a). Cage [Pd2(ABnOH)2(BArNH2)2] was identified as the most effective receptor (b). X-ray crystal structures of the host–guest complexes (H2PO4−)@[Pd2(A1)2(BArNH2)2] (c) and (H2PO4−)@[Pd2(AArOH)2(BArNH2)2] (d). DFT model at the ωB97X-D/def2-SV(P) level of the host–guest complex (H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] refined using Wavefunction Spartan'24 (e). | ||
Similar observations were made with cages bearing functional groups on A-ligands. [Pd2(A1)2(BArNH2)2] showed a slightly stronger binding affinity towards H2PO4− than [Pd2(AArNH2)2(B1)2] in the competitive experiment (Fig. 6b/phase 1, line 8 and ESI-Fig. 286†).
Having established the [Pd2(A1)2(BArNH2)2] cage as a preliminary optimal receptor for H2PO4−, our subsequent investigation focused on the [Pd2(AX)2(BArNH2)2] series of cages, which incorporate additional functional groups on the A-ligands (Fig. 6b, phase 2). The recognition ability of the tetra-endo-functionalized cages is attributed to the synergistic interplay among the functional groups on both A- and B-ligands. The incorporation of ester and pyridine groups led to a decreased binding for H2PO4− (Fig. 6b/phase 2, line 5/6 and ESI-Fig. 287/288†) compared to [Pd2(A1)2(BArNH2)2]. The binding affinities for H2PO4− were further increased with [Pd2(AArNH2)2(BArNH2)2], [Pd2(AArOH)2(BArNH2)2] and [Pd2(ABnOH)2(BArNH2)2] (Fig. 6b/phase 2, lines 1/3/4 and ESI-Fig. 289/290/291†). Interestingly, the homoleptic [Pd2(BArNH2)4] (Fig. 6b/phase 2, line 2 and ESI-Fig. 292†), which also features four endohedral aniline groups as [Pd2(AArNH2)2(BArNH2)2] was outcompeted by [Pd2(A1)2(BArNH2)2]. Further series of competition experiments eventually unveiled the [Pd2(ABnOH)2(BArNH2)2] cage as the best receptor for H2PO4− among all candidates evaluated (Fig. 6b/phase 3 and ESI-Fig. 293/294†). Notably, the cage [Pd2(ABnOH)2(BArNH2)2] completely outperformed [Pd2(AArOH)2(BArNH2)2], which is also decorated with internal hydroxy and amino groups. These results nicely highlighted the delicate impact of the arrangement of the internal functionalities on molecular recognition. The association constant for (2H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] reached 1.21 × 108 M−2, which is approximately 3.8 times that of the unfunctionalized [Pd2(A1)2(B1)2].
Structural analysis of the host–guest complexes provided compelling evidence for the involvement of endo-functional groups in the recognition of the guest molecules. Crystal structures were obtained for the inclusion complexes of [Pd2(A1)2(BArNH2)2] and [Pd2(AArOH)2(BArNH2)2] with H2PO4−. In the solid state, it was observed that only a single H2PO4− ion was encapsulated within the cages, which contrasts with the 1:2 host–guest stoichiometry observed in acetonitrile solution. Within the cavity of [Pd2(A1)2(BArNH2)2], two oxygen atoms of H2PO4− were engaged in hydrogen bonding with the endohedral amino functional groups14 (Fig. 6c). In the case of [Pd2(AArOH)2(BArNH2)2], which is a more potent receptor for H2PO4−, three distinct hydrogen bonding interactions were identified. An additional interaction was noted between an oxygen atom of the anion and one of the internal hydroxy groups15 (Fig. 6d), enhancing the binding affinity. For the best receptor [Pd2(ABnOH)2(BArNH2)2], attempts to grow single crystals have thus far been unsuccessful. Consequently, a density functional theory (DFT) geometry optimization (ωB97X-D/def2-SV(P)) was conducted to simulate the inclusion complex. The model suggested that, compared to the hydroxy groups present in the [Pd2(AArOH)2(BArNH2)2] cage, both benzyl alcohols of cage [Pd2(ABnOH)2(BArNH2)2] are positioned more closely to the bound guest molecule, enabling all four endo-functional groups to contribute to the stabilization of the bound H2PO4− ion (Fig. 6e).
The design of artificial receptors primarily emphasizes on identifying functional groups that amplify the receptor's recognition capabilities. Nevertheless, functionalities that can attenuate recognition are equally valuable. In our H2PO4− recognition experiments, the incorporation of pyridine groups was found to markedly reduce, or even entirely negate, the binding affinity for H2PO4−. By harnessing this property, we have successfully achieved selective host–guest paring using a mixture of distinct Pd2A2B2 cages and anions. Upon treating an equimolar mixture of [Pd2(A1)2(B1)2] and [Pd2(A1)2(Bpy)2] with a solution of H2PO4− and ClO4− in a 2:1 ratio, it was observed that [Pd2(A1)2(Bpy)2] selectively encapsulated ClO4−, effectively excluding the binding of H2PO4−. In contrast, [Pd2(A1)2(B1)2] exhibited no selectivity in binding between H2PO4− and ClO4− (ESI-Fig. 295†). When [Pd2(A1)2(B1)2] was substituted with [Pd2(A1)2(BArNH2)2], there was significant enhancement in selectivity for H2PO4−. Only a minor amount of the complex ClO4−@[Pd2(A1)2(BArNH2)2] was detected (ESI-Fig. 296†). Utilizing the optimal receptor [Pd2(ABnOH)2(BArNH2)2], the exclusive formation of host–guest complexes (2H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] and (ClO4−)@[Pd2(A1)2(Bpy)2] was observed (Fig. 7). A similar pairing behavior ((2H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] and (NO3−)@[Pd2(A1)2(Bpy)2]) was also observed with a mixture of H2PO4− and NO3− (ESI-Fig. 297–299†). While host–guest complexation driven by the electrostatic interaction is robust, it typically lacks specificity. These experiments demonstrated that the introduction and modification of secondary binding sites within a cationic host could confer specific binding for an anionic guest, meanwhile preserving strong affinity.
 | ||
| Fig. 7 Selective host–guest pairing through strategical selection of operational endo-functional groups. The host–guest complexes (2H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] and (ClO4−)@[Pd2(A1)2(Bpy)2] were selectively formed from a mixture containing [Pd2(ABnOH)2(BArNH2)2], [Pd2(A1)2(Bpy)2], H2PO4− and ClO4− in acetonitrile (a). Partial 1H NMR spectra of the host–guest complex (ClO4−)@[Pd2(A1)2(Bpy)2] (b), the mixture containing [Pd2(A1)2(Bpy)2], [Pd2(ABnOH)2(BArNH2)2], ClO4− and H2PO4− (c), and the host–guest complex (2H2PO4−)@[Pd2(ABnOH)2(BArNH2)2] (d). | ||
Conclusions
In summary, we present the selective synthesis of heteroleptic trans-Pd2A2B2 coordination cages from readily available bispyridyl ligands, utilizing a Pd2A2 ring assembly as a key intermediate. The Pd2A2B2 cage features two pairs of distinct sites for endo-functionalization, which provide a versatile platform for incorporating a variety of functional groups as endohedral moieties. By altering the internal functional groups, we have achieved facile derivatization of the cage cavity. Through the variation of the nature, location, and combination of endo-functional groups, a library of 32 coordination cages, each with its own unique micro-environment, was readily constructed using only five different functional groups. The library members displayed adaptive recognition capability towards guest molecules with different geometries and hydrogen bonding capabilities. Host–guest experiments demonstrated that the binding affinity, the binding mechanism and even the very occurrence of binding events could be regulated through the selection of the operational endohedral groups. The evolution of a receptor for H2PO4− that exhibits both high affinity and selectivity highlights the critical role of the synergistic action of distinct endo-functional groups in achieving precise molecular recognition. This was confirmed by X-ray analysis and density functional theory (DFT) calculations. Furthermore, by strategically selecting endo-functional groups, we have accomplished the high-fidelity pairing of host and guest from a mixture of Pd2A2B2-cages and anions. The modification of the host's micro-environment presents a broadly applicable methodology for the identification of an optimal receptor for a specific guest molecule. The heteroleptic Pd2A2B2 cage serves as a reliable platform for implementing this strategy. We are currently broadening the application of this methodology to other biologically relevant anions and neutral molecules. We anticipate that our work will offer a new avenue for the development of tailor-made artificial receptors.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Q. Z. conceived and supervised the project; Y.-M. T. performed the synthesis, characterization (except the HRMS) of all metal–ligand assemblies and the corresponding host–guest studies with L.-M. Z.; Q. B. measured the HRMS of all metal–ligand assemblies and analysed the data with Z. Z. and P. W.; Y.-M. T. and Q. Z. compiled the first draft of the manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171192), the “1000-Youth Talents Program” and the Fundamental Research Funds for the Central Universities. We would like to thank Dai-Bing Luo (Analytical & Testing Center, Sichuan University) for X-ray crystal diffraction, Dong-Yan Deng (the College of Chemistry, Sichuan University) and Xiao-Yan Wang (Analytical & Testing Center, Sichuan University) for NMR measurements, and Jing Li (the College of Chemistry, Sichuan University) for HR-ESI-MS measurements. Yang Shao (Analytical & Testing Center, Sichuan University) is acknowledged for graphic design.Notes and references
- (a) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed; (b) B. Brisig, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2003, 42, 1270–1273 CrossRef CAS PubMed; (c) R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS PubMed; (d) I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751–756 CrossRef CAS PubMed; (e) M. J. Langton and P. D. Beer, Chem. Commun., 2014, 50, 8124–8127 RSC; (f) M. Matache, E. Bogdan and N. D. Hădade, Eur. J. Chem., 2014, 20, 2106–2131 CrossRef CAS PubMed; (g) V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J.-F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555–1559 CrossRef CAS PubMed; (h) A. Galán, E. C. Escudero-Adán and P. Ballester, Chem. Sci., 2017, 8, 7746–7750 RSC; (i) O. Shyshov, R.-C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed; (j) R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed; (k) Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef CAS PubMed; (l) G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef PubMed; (m) L. Jing, E. Deplazes, J. K. Clegg and X. Wu, Nat. Chem., 2024, 16, 335–342 CrossRef CAS PubMed.
- (a) D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef PubMed; (b) M. Otte, M. Lutz and R. J. M. Klein Gebbink, Eur. J. Org Chem., 2017, 2017, 1657–1661 CrossRef CAS; (c) S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed; (d) S. C. Bete and M. Otte, Angew. Chem., Int. Ed., 2021, 60, 18582–18586 CrossRef CAS PubMed; (e) S. C. Bete, L. K. May, P. Woite, M. Roemelt and M. Otte, Angew. Chem., Int. Ed., 2022, 61, e202206120 CrossRef CAS PubMed; (f) Y. Liu, S.-H. Liao, W.-T. Dai, Q. Bai, S. Lu, H. Wang, X. Li, Z. Zhang, P. Wang, W. Lu and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217215 CrossRef CAS PubMed; (g) K. G. Andrews, T. K. Piskorz, P. N. Horton and S. J. Coles, J. Am. Chem. Soc., 2024, 146, 17887–17897 CrossRef CAS PubMed.
- (a) M. Tominaga, K. Suzuki, T. Murase and M. Fujita, J. Am. Chem. Soc., 2005, 127, 11950–11951 CrossRef CAS PubMed; (b) S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS; (c) K. Suzuki, M. Kawano, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 10652–10653 CrossRef CAS PubMed; (d) K. Suzuki, J. Iida, S. Sato, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 5780–5782 CrossRef CAS PubMed; (e) K. Harris, Q.-F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed; (f) M. C. Young, A. M. Johnson, A. S. Gamboa and R. J. Hooley, Chem. Commun., 2013, 49, 1627–1629 RSC; (g) R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS PubMed; (h) S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC; (i) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed; (j) Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed; (k) L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed; (l) P. M. Bogie, L. R. Holloway, C. Ngai, T. F. Miller, D. K. Grewal and R. J. Hooley, Eur. J. Chem., 2019, 25, 10232–10238 CrossRef CAS PubMed; (m) X. Yan, P. Wei, Y. Liu, M. Wang, C. Chen, J. Zhao, G. Li, M. L. Saha, Z. Zhou, Z. An, X. Li and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 9673–9679 CrossRef CAS PubMed; (n) L.-P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 CrossRef CAS PubMed; (o) K. Xu, B. Li, S. Yao, Z. Li, Y. Lu, M. Dong, J. Qiu, L. Luo and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202203016 CrossRef CAS PubMed; (p) C. M. Brown, D. J. Lundberg, J. R. Lamb, I. Kevlishvili, D. Kleinschmidt, Y. S. Alfaraj, H. J. Kulik, M. F. Ottaviani, N. J. Oldenhuis and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 13276–13284 CrossRef CAS PubMed; (q) A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS PubMed.
- (a) M. Yoshizawa, M. Nagao, K. Umemoto, K. Biradha, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2003, 1808–1809 RSC; (b) M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383–5388 CrossRef CAS.
- For related heteroleptic assemblies, reviews: (a) S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed; (b) D. Bardhan and D. K. Chand, Eur. J. Chem., 2019, 25, 12241–12269 CrossRef CAS PubMed; (c) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC; (d) C.-B. Tian and Q.-F. Sun, Eur. J. Chem., 2023, 29, e202300195 CrossRef CAS PubMed ; Selected examples: ; (e) A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed; (f) Q.-F. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 13510–13513 CrossRef CAS PubMed; (g) D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed; (h) W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed; (i) W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285–8289 CrossRef CAS PubMed; (j) S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 880 CrossRef CAS PubMed; (k) S. Sudan, R.-J. Li, S. M. Jansze, A. Platzek, R. Rudolf, G. H. Clever, F. Fadaei-Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2021, 143, 1773–1778 CrossRef CAS PubMed; (l) R.-J. Li, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Eur. J. Chem., 2021, 27, 9439–9445 CrossRef CAS PubMed; (m) K. Wu, B. Zhang, C. Drechsler, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2021, 60, 6403–6407 CrossRef CAS PubMed; (n) J. Tessarolo, H. Lee, E. Sakuda, K. Umakoshi and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 6339–6344 CrossRef CAS PubMed; (o) S.-C. Li, L.-X. Cai, M. Hong, Q. Chen and Q.-F. Sun, Angew. Chem., Int. Ed., 2022, 61, e202204732 CrossRef CAS PubMed; (p) J. E. M. Lewis, Angew. Chem., Int. Ed., 2022, 61, e202212392 CrossRef CAS PubMed; (q) D. Preston and J. D. Evans, Angew. Chem., Int. Ed., 2023, 62, e202314378 CrossRef CAS PubMed; (r) T. Abe, N. Sanada, K. Takeuchi, A. Okazawa and S. Hiraoka, J. Am. Chem. Soc., 2023, 145, 28061–28074 CrossRef CAS PubMed; (s) C. F. Espinosa, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2023, 145, 9965–9969 CrossRef CAS PubMed; (t) P. Molinska, A. Tarzia, L. Male, K. E. Jelfs and J. E. M. Lewis, Angew. Chem., Int. Ed., 2023, 62, e202315451 CrossRef CAS PubMed; (u) K. Wu, E. Benchimol, A. Baksi and G. H. Clever, Nat. Chem., 2024, 16, 584–591 CrossRef CAS PubMed; (v) R. G. DiNardi, S. Rasheed, S. S. Capomolla, M. H. Chak, I. A. Middleton, L. K. Macreadie, J. P. Violi, W. A. Donald, P. J. Lusby and J. E. Beves, J. Am. Chem. Soc., 2024, 146, 21196–21202 CrossRef PubMed; (w) M. Parbin, V. Sivalingam and D. K. Chand, Angew. Chem., Int. Ed., 2024, 63, e202410219 CrossRef CAS PubMed; (x) B. Zhang, H. Lee, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2024, 63, e202404682 CrossRef CAS PubMed; (y) E. Benchimol, I. Regeni, B. Zhang, M. Kabiri, J. J. Holstein and G. H. Clever, J. Am. Chem. Soc., 2024, 146, 6905–6911 CrossRef CAS PubMed; (z) W.-T. Dai, T.-T. Liu, Q. Bai, Z. Zhang, P. Wang, W. Lu and Q. Zhang, Sci. China:Chem., 2024, 67, 4110–4115 CrossRef CAS.
- I. Regeni, R. Chowdhury, K. Terlinden, S. Horiuchi, J. J. Holstein, S. Feldmann and G. H. Clever, Angew. Chem., Int. Ed., 2023, 62, e202308288 CrossRef CAS PubMed.
- Deposition Number 2382614 (cage [Pd2(A1)2(B1)2](OTf)4) contains the supplementary crystallographic data for this paper.†.
- Deposition Numbers 2382615 (cage [Pd2(A1)2(BArNH2)2](OTf)4), 2382616 (cage [Pd2(A1)2(BArOH)2](OTf)4), 2382617 (cage [Pd2(A1)2(Bester)2](OTf)4), 2382618 (cage [Pd2(A1)2(Bpy)2](OTf)4), 2382619 (cage [Pd2(AArNH2)2(B1)2](OTf)4), 2382620 (cage [Pd2(AArOH)2(B1)2](OTf)4), 2382621 (cage [Pd2(ABnOH)2(B1)2](OTf)4), 2382622 (cage [Pd2(Aester)2(B1)2](OTf)4) and 2382623 (cage [Pd2(Apy)2(B1)2](OTf)4) contain the supplementary crystallographic data for this paper.†.
- Deposition Number 2382626 (cage [Pd2(AArOH)2(BArNH2)2](OTf)4) contains the supplementary crystallographic data for this paper.†.
- Deposition Number 2382624 (cage [Pd2(AArOH)2(Bpy)2](OTf)4) contains the supplementary crystallographic data for this paper.†.
- Deposition Number 2382625 (cage [Pd2(Apy)2(BArNH2)2](OTf)4) contains the supplementary crystallographic data for this paper.†.
- D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed.
- http://app.supramolecular.org/bindfit/.
- Deposition Number 2382627 (host–guest complex (H2PO4−)@[Pd2(A1)2(BArNH2)2]4+ contains the supplementary crystallographic data for this paper.†.
- Deposition Number 2382628 (host–guest complex (H2PO4−)@[Pd2(AArOH)2(BArNH2)2]4+ contains the supplementary crystallographic data for this paper.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2382614, 2382615, 2382616, 2382617, 2382618, 2382619, 2382620, 2382621, 2382622, 2382623, 2382624, 2382625, 2382626, 2382627 and 2382628. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08176e |
| This journal is © The Royal Society of Chemistry 2025 |