
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeep-blue phosphorescence from platinum(II) bis(acetylide) complexes with sulfur-bridged dipyridyl ligands†
Ka-Ming Tong ,
Jessica Toigo and
Michael O. Wolf*
,
Jessica Toigo and
Michael O. Wolf*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca
First published on 7th March 2025
Abstract
New approaches to prepare rarer emitters such as those that are deep-blue are needed to advance OLED technologies. Here, we demonstrate that a series of new platinum(II) bis(acetylide) complexes [Pt(N–N)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2] containing sulfur-bridged dipyridyl ligands (N–N) with various sulfur oxidation states: sulfide (S), sulfoxide (SO) and sulfone (SO2) give access to variable emission colors from green to deep-blue. Spectroscopic, electrochemical and computational studies show that mixed character excited states have energies which are significantly influenced by the oxidation state of sulfur and the presence of substituents. The sulfide and sulfoxide complexes are non-emissive in the solution state, while the sulfone complexes display 3MLCT/3LLCT excited-state yellow phosphorescence. In PMMA films the sulfide and sulfoxide complexes show intense deep-blue phosphorescence and green phosphorescence for the sulfone complexes, with photoluminescence quantum yields ranging from 0.35–0.91. Here we demonstrate the capability of changing the photophysical properties of these metal emitters by varying the oxidation state of sulfur to achieve intense deep-blue and green emitters.
CPh)2] containing sulfur-bridged dipyridyl ligands (N–N) with various sulfur oxidation states: sulfide (S), sulfoxide (SO) and sulfone (SO2) give access to variable emission colors from green to deep-blue. Spectroscopic, electrochemical and computational studies show that mixed character excited states have energies which are significantly influenced by the oxidation state of sulfur and the presence of substituents. The sulfide and sulfoxide complexes are non-emissive in the solution state, while the sulfone complexes display 3MLCT/3LLCT excited-state yellow phosphorescence. In PMMA films the sulfide and sulfoxide complexes show intense deep-blue phosphorescence and green phosphorescence for the sulfone complexes, with photoluminescence quantum yields ranging from 0.35–0.91. Here we demonstrate the capability of changing the photophysical properties of these metal emitters by varying the oxidation state of sulfur to achieve intense deep-blue and green emitters.
Introduction
Over the past few decades, ever growing demands for advanced lighting technologies and color displays for electronic devices have promoted extensive research into organic light-emitting diodes (OLEDs). Phosphorescent organometallic compounds are ideal for application in OLEDs as such triplet emitters can harvest both singlet and triplet excitons, and theoretically achieve 100% internal quantum efficiency.1 A large library of organometallic compounds, such as those based on iridium(III),2 platinum(II)3 and gold(III)4 have been explored for application as light-emitting materials in OLEDs. Despite substantial advances, the development of blue emitters for OLEDs remains challenging. According to the International Telecommunication Union (ITU), the standard for blue OLED displays (BT.2020) requires Commission Internationale de l'Éclairage (CIE) coordinates of CIEx,y = (0.131, 0.046).5 To achieve blue phosphorescence, compounds with high triplet excited-state energies are necessary, however such triplet states are often quenched by metal-centered (MC) states and deactivate through ligand substitution reactions that lead to decomposition.6 Blue emitters typically suffer from poor color purity, low emission efficiency and low photostability, hindering application in commercial devices.7 To overcome these issues, substantial work has been done towards advanced molecular design, especially incorporating strong σ-donor ligands such as N-heterocyclic carbenes (NHCs) and cyclometallating ligands. Various highly efficient platinum(II) and iridium(III) deep-blue emitters have been designed using this strategy, where the dark MC states are destabilized.8 Another approach to design blue emitters is replacing phosphorescent transition metal complexes with organic thermally activated delayed fluorescence (TADF) emitters. Rigid polycyclic aromatic TADF emitters with heteroatom dopants such as boron and nitrogen can exhibit short-range charge-transfer (SRCT) excited states, which can result in narrow-band blue TADF emission with high quantum efficiency.9 A downside of these strategies is that they typically require lengthy synthetic efforts for the construction of the ligands or TADF emitters, which limits practical industrial application.We have reported a simple approach to control the photophysical properties of metal complexes involving the installation of a sulfur bridge between the rings in π-conjugated aromatic ligands such as dipyridyl or dithiazolyl. The electronic properties of the ligand can be changed by simple oxidation of the sulfur bridge from sulfide (S) to sulfoxide (SO) and then sulfone (SO2), with the significant advantage that the geometry of the ligand is not affected as the addition of oxygens to the sulfur does not change the bent geometry at sulfur significantly. Examples of successfully incorporating these sulfur-bridged dipyridyl ligands into iridium(III)10 and copper(I)11 centers by our group has shown that the nature of the emissive excited state and the emission color change with different sulfur oxidation states, together with opening up thermochromic properties of the copper(I) complexes.12 We have also demonstrated that the solid-state phosphorescence quantum yields of rhenium(I) complexes are enhanced significantly when the oxidation state of the ligand is changed from sulfide or sulfoxide to sulfone, with a bathochromic shift of emission color from blue to yellow.13 However, none of the aforementioned complexes show deep-blue solid-state photoluminescence.
Inspired by our success in controlling the solid-state phosphorescence of rhenium(I) complexes with the oxidation state of the sulfur bridge, we now apply this strategy to a platinum(II) system by introducing sulfur-bridged dipyridyl ligands to platinum(II) bis(acetylide) complexes, [Pt(N–N)(CCPh)2]. The sulfur bridge not only can be used as a handle to tune the photophysical properties of the complexes via changes in oxidation state, but also reduces the π-conjugation in the dipyridyl ligand. Interrupting π-conjugation of the ligand by inserting sp3-carbons has been demonstrated as a useful strategy to increase the emission energies of platinum(II) complexes.14 We therefore anticipated that the sulfur bridges could also increase the emission energy and achieve blue phosphorescence. At the same time, the strong-field acetylide ligands are capable of destabilizing the MC state, which is expected to result in higher photoluminescence quantum yield.
Spectroscopic studies of the platinum(II) complexes in PMMA films shows that all the sulfide and sulfoxide complexes display intense deep-blue phosphorescence, with CIEx coordinates ranging from 0.15–0.16 and CIEy coordinates from 0.10–0.14, comparable to other blue phosphorescent or TADF emitters in PMMA films15 and devices;16 while the sulfone complexes emit in the green region, with CIEx,y coordinates = (0.34, 0.55) and (0.29, 0.53). Furthermore, these complexes show photoluminescence quantum yields as high as 0.91 in PMMA films with submicrosecond to microsecond lifetime. The relatively short emission lifetimes, which indicate fast radiative decay rates of the excited state, and high quantum yields of the emitters are crucial criteria for OLED applications, as these features should result in better device efficiency. Previous studies have demonstrated that platinum(II) complexes with similar properties can be used for OLED fabrications.17 The results demonstrate a simple strategy to achieve deep-blue phosphorescent platinum(II) emitters by installing a sulfur bridge to the dipyridyl ligand, where the emission color can be readily controlled by changing the oxidation state of the ligand with quantum yields that make these viable as candidates for application in OLEDs.
Results and discussion
Synthesis and characterization
Six platinum(II) complexes (1–6) containing sulfide- (S), sulfoxide- (SO) or sulfone- (SO2) bridged dipyridyl ligands were synthesized. Complexes 4–6 with methyl groups in the 4- and 4′-positions of the pyridyl rings were designed to study the substituent effect. The sulfide ligands were prepared using the literature methods from 2-bromopyridine or 2-chloro-4-methylpyridine with thiourea.10 The sulfoxide and sulfone ligands were synthesized by oxidizing the sulfide ligand using 30% H2O2 or m-chloroperoxybenzoic acid (m-CPBA). The reaction between the corresponding ligands with potassium tetrachloroplatinate(II), K2[PtCl4] gave the dichloro-platinum(II) complexes bearing the sulfur-bridged dipyridyl ligands. The platinum(II) acetylide complexes 1–6 were synthesized by coupling the dichloro-platinum(II) complexes with phenylacetylene in the presence of diisopropylamine and copper(I) iodide (Scheme 1). All complexes were characterized by 1H, 13C{1H} NMR spectroscopy, infrared spectroscopy (FT-IR) and high-resolution mass spectrometry (HR-ESI). The structures of the complexes 1, 2 and 4–6 were also confirmed by single-crystal X-ray diffraction. Similar to the previously reported rhenium(I) and iridium(III) complexes bearing sulfur-bridged dipyridyl ligands, the 1H and 13C{1H} NMR spectra show symmetrical signals for the pyridyl rings, suggesting the ligands coordinate to the metal via the N,N binding mode (Fig. S9–S20†). The IR spectra of complexes 1–6 show two bands from 2109–2121 cm−1 and 2125–2135 cm−1, corresponding to the symmetric and asymmetric CC stretches. The presence of two CC stretches indicates that the phenylacetylide ligands are in the cis- conformation.15c,18
 | ||
| Scheme 1 Synthesis of the platinum(II) complexes 1–6. | ||
Perspective drawings of the single crystal structures of complexes 1, 2 and 4–6 are depicted in Fig. 1, S21 and S22. The crystal structure determination data, and selected bond lengths and bond angles are summarized in Tables S1–S8.† The complexes adopt square-planar geometries with the N–Pt–N and C–Pt–C bite angles in the range of 88.03(5)–88.88(6)° and 88.42(6)–91.81(9)° respectively. The four-coordinate geometry indices19 τ4 are in the range of 0.02 to 0.05, indicating the complexes are close to perfectly square planar. The N–Pt–N bite angles are larger than in other platinum(II) diimine acetylide complexes, which generally have bite angles between 77 and 79°.18,20 The increase in bite angles is due to the presence of sulfur bridges that reduces the strain from the pyridyl rings to the platinum(II) center. On the other hand, the sulfur bridge also introduces non-planarity in the ligands such that the angles between ring planes are between 109.6 and 120.3°. The Pt–N bond lengths are in the range of 2.0828(14)–2.1048(18) Å, slightly longer than in the platinum(II) bipyridine acetylide complex in the literature.21 This can be attributed to the electron-withdrawing sulfur bridge reducing the σ-donating ability of the ligands compared to bipyridine. The Pt–C bond lengths of 1.947(2)–1.9699(17) Å are comparable to those in other platinum(II) diimine acetylide complexes.22
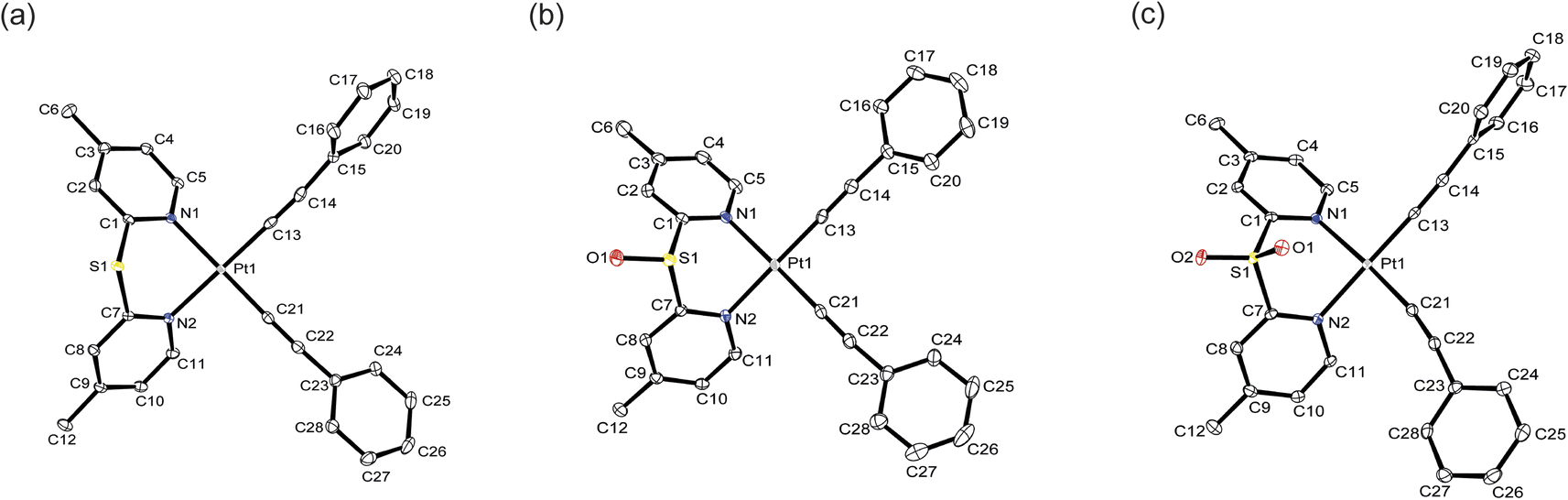 | ||
| Fig. 1 Crystal structures of complexes (a) 4, (b) 5 and (c) 6. Ellipsoids are plotted at the 50% probability level, and H atoms are removed for clarity. | ||
An examination of the crystal packing reveals that the complexes adopt an antiparallel stacking pattern, the shortest Pt⋯Pt distance ranges from 4.783 to 6.298 Å, indicating there are no metallophilic interactions between the molecules (Fig. S23†). Complexes 1, 2, 4 and 5 show the shortest intermolecular phenyl or pyridyl rings distances of 3.451–3.801 Å, suggesting that these complexes exhibit π⋯π interactions in the crystal structures.23 However, the shortest separation between the aromatic rings in complex 6 is 4.313 Å confirming π⋯π interactions are not present in the solid structure.
The thermal stability of the complexes was assessed by thermogravimetric analysis (TGA), and the data are shown in Fig. S24.† The onset temperature for the thermal degradation of complexes 1, 2 and 3 are 200, 234 and 201 °C respectively, indicating the thermal stability is comparable to that of other platinum(II) acetylide complexes.15c,24
Electronic absorption spectroscopy
The electronic absorption spectra of complexes 1–6 in dichloromethane solutions at 298 K are shown in Fig. 2a, and the absorption data summarized in Table 1. In addition to high energy ligand-centered absorptions, the complexes also exhibit moderately intense low-energy absorption bands between 340 and 400 nm. According to previous studies on related platinum(II) complexes,22c,25 and computational studies (see below), these absorptions can be assigned to an admixture of MLCT [dπ(Pt) → π*(N–N)] and ligand-to-ligand charge-transfer (LLCT) [π(CCPh) → π*(N–N)] transitions. The lowest-energy absorptions are in the order of 1 (340 nm) > 2 (343 nm) > 3 (397 nm) and 4 (333 nm) > 5 (335 nm) > 6 (389 nm), which are in line with the electron-withdrawing ability of the sulfur bridge: sulfide (S) < sulfoxide (SO) < sulfone (SO2). It is worth noting that the absorption bands of sulfone complexes 3 and 6 tail beyond 450 nm while the bands of the sulfide and sulfoxide complexes do not. This is attributed to better stabilization of the π* orbital in the more electron-withdrawing sulfone ligand. The lowest-energy absorptions in complexes 4–6 are more blue-shifted than those of complexes 1–3, attributed to the electron-donating methyl groups destabilizing the π* orbital of the dipyridyl ligands. In comparison with [Pt(bpy)(CCPh)2] which shows the lowest-energy absorption shoulder at ca. 430 nm and tailing to 520 nm (Fig. S25†), the absorption shoulders of complexes 1–6 are more blue-shifted, suggesting the sulfur bridge between the pyridyl rings reduces the extent of conjugation of the ligand and results in a blue-shift in the absorption.
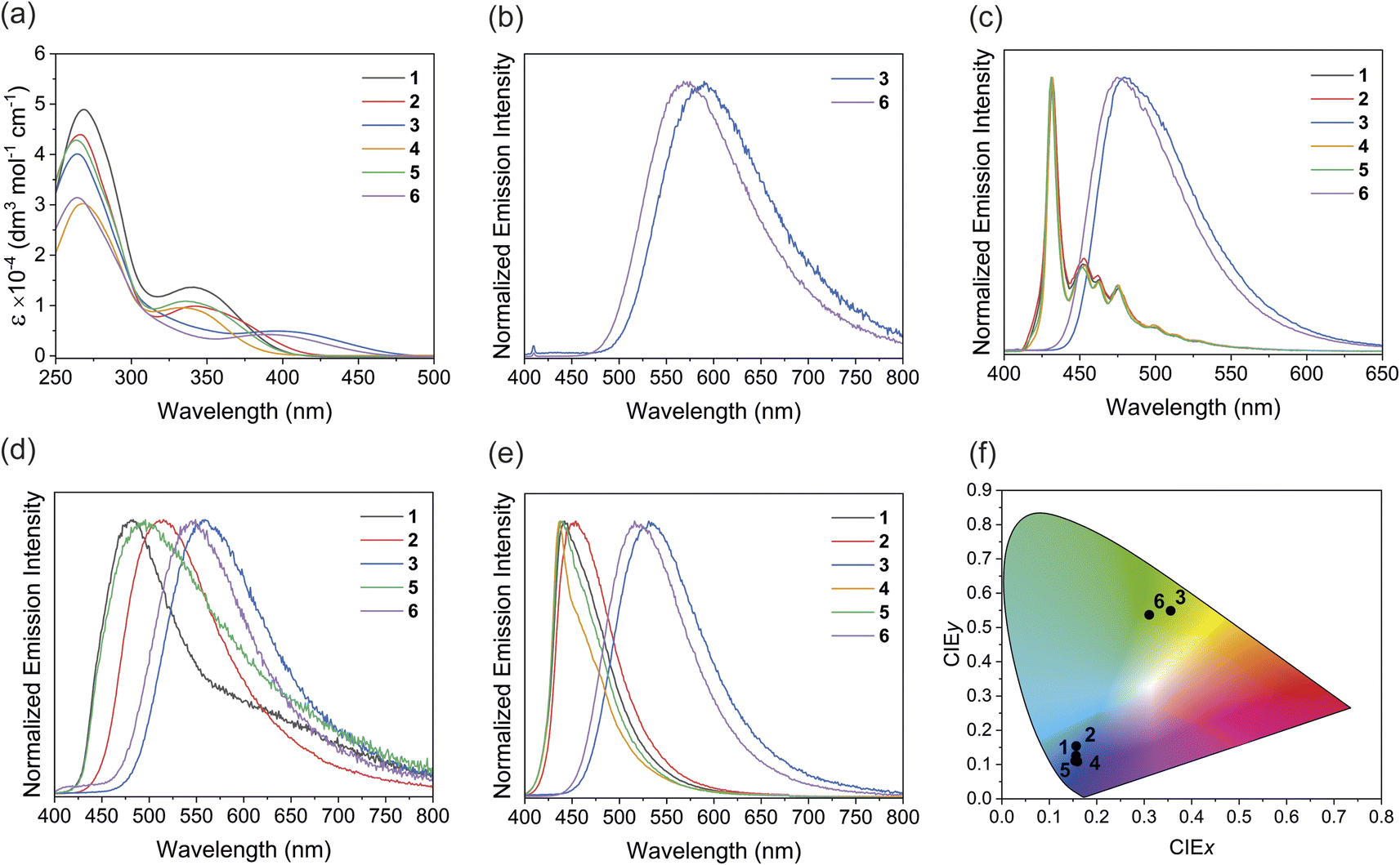 | ||
| Fig. 2 (a) Electronic absorption spectra of complexes 1–6 in dichloromethane. (b) Photoluminescence spectra of complexes 3 and 6 in dichloromethane at 298 K (λex = 365 nm). (c) Photoluminescence spectra of complexes 1–6 in 2-MeTHF glass at 77 K (λex = 365 nm). (d) Solid-state photoluminescence spectra of neat thin films of complexes 1–3, 5 and 6 drop-casting from dichloromethane (λex = 365 nm). (e) Photoluminescence spectra of complexes 1–6 in PMMA films with 2 wt% of Pt complexes (λex = 365 nm). (f) CIE 1931 chromaticity diagram for complexes 1–6 in PMMA thin films. | ||
| Complex | Absorption λabsa [nm] (ε dm3 mol−1 cm−1) |
|---|---|
| a Recorded in 2 × 10−5 mol dm−3 dichloromethane solution at 298 K. | |
| 1 | 269 (48900), 285 sh (38800), 340 (13700) |
| 2 | 266 (43900), 286 sh (29100), 343 (9870), 373 sh (6840) |
| 3 | 264 (40100), 288 sh (24000), 397 (4920) |
| 4 | 268 (30200), 285 sh (23600), 333 (9550) |
| 5 | 264 (42800), 283 sh (31300), 335 (10800) |
| 6 | 264 (31400), 219 sh (18200), 389 (423) |
Photoluminescence spectroscopy
The photoluminescence properties of complexes 1–6 in dichloromethane solution, solid-state neat thin films and PMMA films at 298 K, and 2-MeTHF glass at 77 K are summarized in Table 2. While the sulfide and sulfoxide complexes 1, 2, 4 and 5 are non-emissive in solution at 298 K, the sulfone complexes 3 and 6 display yellow photoluminescence with a structureless emission profile and submicrosecond lifetime in the solution state under argon (Fig. 2b). The excitation spectra for complexes 3 and 6 are shown in Fig. S26–27.† Based on studies on related complexes,25 the emission origin is ascribed to phosphorescence from a mixture of 3MLCT [dπ(Pt) → π*(N–N)] and 3LLCT [π(CCPh) → π*(N–N)] excited states. Moreover, computational studies also show that the first triplet excited state (T1) involves MLCT and LLCT transitions, consistent with the aforementioned argument (see below). Complex 6 shows a blue-shift in emission energy of ca. 20 nm, presumably due to the methyl groups increasing the energy of the emitting T1 state. The non-emissive nature of the sulfide and sulfoxide complexes in the solution state can be explained by their higher non-radiative decay rates (knr) and the lower spin–orbit coupling (SOC) from the respective T1 and ground states (see the computational studies for details). Therefore, the oxidation state of the sulfur bridge can serve as a tool to turn on the photoluminescence properties of the complexes in the solution state. The photoluminescence quantum yield of complex 6 (Φem = 0.06) is higher than that of complex 3 (Φem = 0.03), possibly due to the stronger σ-donating methyl-substituted ligand destabilizing the non-emissive MC excited states, reducing non-radiative decay.
| Complex | Medium (T/K) | Emission λem (nm) | τem (μs) | Φem |
|---|---|---|---|---|
| a In 2-MeTHF.b Non-emissive under the experimental conditions.c Quantum yield too low to be accurately measured (Φem < 0.01). | ||||
| 1 | CH2Cl2 (298) | —b | — | — |
| Solid (298) | 482 | 0.11 (38%), 0.02 (32%), 0.71 (30%) | —c | |
| PMMA (298) | 442 | 2.13 (38%), 8.02 (6%), 0.39 (56%) | 0.40 | |
| Glass (77)a | 432 | 13.3 (66%), 28.0 (34%) | — | |
| 2 | CH2Cl2 (298) | —b | — | — |
| Solid (298) | 514 | 0.16 (42%), 0.74 (41%), 0.03 (17%) | 0.14 | |
| PMMA (298) | 453 | 2.90 (48%), 0.96 (45%), 7.84 (7%) | 0.82 | |
| Glass (77)a | 432 | 10.8 (65%), 25.5 (35%) | — | |
| 3 | CH2Cl2 (298) | 590 | 0.09 (81%), 0.14 (19%) | 0.03 |
| Solid (298) | 562 | 0.03 (56%), 0.01 (24%), 0.12 (20%) | 0.02 | |
| PMMA (298) | 532 | 0.38 (73%), 0.97 (27%) | 0.49 | |
| Glass (77)a | 482 | 3.29 (62%), 5.16 (38%) | — | |
| 4 | CH2Cl2 (298) | —b | — | — |
| Solid (298) | —b | — | — | |
| PMMA (298) | 436 | 14.1 (55%), 4.03 (41%), 0.47 (4%) | 0.35 | |
| Glass (77)a | 432 | 16.8 (60%), 21.6 (40%) | — | |
| 5 | CH2Cl2 (298) | —b | — | — |
| Solid (298) | 497 | 0.11 (38%), 0.63 (31%), 0.02 (31%) | —c | |
| PMMA (298) | 439 | 3.47 (44%), 0.95 (44%), 9.7 (11%) | 0.71 | |
| Glassa (77) | 432 | 13.9 (64%), 28.5 (36%) | — | |
| 6 | CH2Cl2 (298) | 570 | 0.09 (95%), 0.22 (5%) | 0.06 |
| Solid (298) | 547 | 0.14 (54%), 0.36 (36%), 0.03 (10%) | 0.09 | |
| PMMA (298) | 519 | 0.44 (68%), 1.05 (32%) | 0.91 | |
| Glassa (77) | 476 | 3.09 (55%), 4.76 (45%) | — | |
All the complexes display strong photoluminescence in 2-MeTHF glass at 77 K (Fig. 2c). Interestingly, the sulfide and sulfoxide complexes 1, 2, 4 and 5 show drastically different emission profiles from the sulfone complexes 3 and 6. The sulfide and sulfoxide complexes show almost identical emission spectra with maxima at 432 nm and vibronic structures between 440–550 nm. Previous studies of platinum(II) phenylacetylide complexes,26 and the insensitivity of the energies to the dipyridyl ligands, suggest that the emission originates from the ligand-centered (LC) [π → π*(CCPh)] excited state. On the other hand, the sulfone complexes 3 and 6 show more structureless emission bands at 482 and 476 nm respectively, suggesting that the emission predominately arises from the 3MLCT and 3LLCT excited states. These results reveal that changing the sulfur oxidation state not only can manipulate the emission energies but can also influence the origin of emission of the complexes at low temperatures.
The solid-state photoluminescence spectra of the neat thin films prepared by drop-casting a dichloromethane solution of each complex onto a quartz plate were recorded (Fig. 2d). All complexes, except for complex 4, show photoluminescence between 482 and 562 nm in the solid state. The non-emissive nature of complex 4 is probably due to the methyl groups enhancing vibrational relaxation facilitating non-radiative decay.13 The emission energies of the complexes follow the trend 1 (482 nm) > 2 (514 nm) > 3 (562 nm) and 5 (497 nm) > 6 (547 nm), and complexes 5 and 6 with methyl groups possess higher emission energy than their unsubstituted analogs. The energy is dependent on the electron-withdrawing ability of the dipyridyl ligands, further supporting the assignment of the emission origin to the mixture of 3MLCT and 3LLCT excited states. Complexes 3 and 6 show a blue-shift in energies compared to their solution-state emission. The blue-shift can be explained by the rigidochromic behavior of the complexes, which is typically observed in photoluminescence involving charge-transfer character.27 Interestingly, the platinum(II) complexes show broad emission profiles in the neat thin films, with the sulfide and sulfoxide complexes showing a tail extending to the low-energy region. According to previous studies on phosphorescent platinum(II) materials, Pt⋯Pt interactions are possible in the powder form due to aggregation, even these are not observed in the single crystal lattice.28 Therefore, the low energy emission feature could potentially originate from a metal-metal-to-ligand charge-transfer (MMLCT) excited state arising from the Pt⋯Pt interaction in the neat film.
To further investigate the luminescence behavior of the complexes in the solid state, the emission of 1–6 in PMMA films containing 2 wt% of the complexes were measured (Fig. 2e, S28 and S29†). In sharp contrast to the neat thin films, the sulfide and sulfoxide complexes display intense blue phosphorescence with significantly narrower emission bands in the PMMA films. The sulfone complexes also show slight narrowing and a hypsochromic shift in the emission bands. Similar to in the neat thin films, the sulfone complexes show higher emission energies in PMMA films than in solution, this can also be attributed to rigidochromic effects in the PMMA films. The absence of a low-energy emission tail supports the hypothesis that Pt⋯Pt interactions are present in the powder form due to the aggregation. In the PMMA film containing a 2 wt% loading of the complex, aggregation is significantly suppressed, so Pt⋯Pt interactions and, therefore, the MMLCT transitions are less viable, leading to the disappearance of the low-energy emission feature. Moreover, complex 4, which is non-emissive in the neat thin film, becomes brightly emissive at 436 nm in PMMA. This can be attributed to the suppression of aggregation and self-quenching due to the low doping concentration. The CIE 1931 chromaticity diagram was used to analyze the luminescence color of the PMMA films (Fig. 2f). The sulfide and sulfoxide complexes 1, 2, 4 and 5 emit in the deep-blue region, as characterized by the CIE coordinates CIEy < 0.15 and (CIEx + CIEy) < 0.30 (Table S9†)29 with photoluminescence quantum yields between 0.35 and 0.82, which is generally higher than in other recently reported platinum(II) deep-blue emitters (Table S10†).15c,30 As compared to the bipyridyl analogue, [Pt(bpy)(CCPh)2], which emits at 543 nm in the PMMA film with CIE coordinates of (0.35, 0.57) (Fig. S30 and Table S9†), the simple addition of a sulfur bridge between the pyridyl rings can result in deep-blue emission, a desirable feature for fabricating blue-emitting devices. On the other hand, the sulfone complexes 3 (Φem = 0.49) and 6 (Φem = 0.91) emit in the green region, demonstrating the influence of sulfur oxidation state on the solid-state emission color.
Photoluminescence lifetimes of complexes 3 and 6 in deaerated dichloromethane solutions were measured; both complexes exhibit submicrosecond lifetimes with biexponential decay, the lifetimes are comparable to other related platinum(II) diimine acetylide systems.22a,31 The biexponential decay could arise from different emitting states due to the presence of aggregated species in solution,32 or from different sublevels in the lowest triplet state (T1).33 Biexponential decay behavior has also been observed in solutions of other platinum(II) complexes in previous studies.34 All the complexes show submicrosecond to microsecond decay lifetime with two or three components in the solid-state neat thin film and PMMA films. Multiexponential decay of platinum(II) complexes is typical in the solid state, which might arise from the difference in structural environments.32 The lifetimes in PMMA films are generally longer than in neat films, due to reduced self-quenching in PMMA. The short emission lifetime suggests fast radiative decay rate (kr) is observed in these complexes, which is crucial for OLED applications as that could surpass the non-radiative decay rate and increase the quantum efficiency of the device.35 When the complexes are cooled down to 77 K in 2-MeTHF, the sulfide and sulfoxide complexes 1, 2, 4 and 5 show biexponential decays with both components longer than 10 μs, while for the sulfone complexes 3 and 6, the two components are shorter than 10 μs. This result is due to the difference in emission origin with the sulfide and sulfoxide complexes emitting from the 3LC excited state and the sulfone complexes emitting from the LLCT/MLCT excited state. The biexponential decay at 77 K can be attributed to the individual and aggregated molecules in the glass medium, which is commonly observed in platinum(II) complexes.36
Electrochemical studies
Cyclic voltammetry data of complexes 1–6 are summarized in Table 3 and the cyclic voltammograms are shown in Fig. S31 and S32.† All the complexes show an irreversible oxidation wave between +0.93 and +1.12 V vs. Fc/Fc+ with a small anodic shift when the sulfur oxidation state is increased. Based on electrochemical studies of related compounds, the oxidation wave is tentatively assigned to the PtII/III metal-centered oxidation and acetylide-based oxidation.25,28a Complexes 1, 2 and 5 display an irreversible reduction wave in the range −2.03 to −2.22 V vs. Fc/Fc+, while complexes 3 and 6 show an irreversible reduction couple at −1.68 and −1.83 V vs. Fc/Fc+ respectively. These reduction potentials are sensitive to the oxidation state of the sulfur with 1 (−2.22 V) < 2 (−2.03 V) < 3 (−1.68 V) and 5 (−2.19 V) < 6 (−1.83 V), suggesting these reductions are pyridyl ligand-centered. The second reduction peak at −2.13 V in complex 3 and –2.26 V in complex 6 can be assigned to the reduction of the sulfone group in the ligand.10 No reduction wave to ligand-centered reduction is observed in complex 4 within the potential window studied. This is likely due to the higher energy of the LUMO in complex 4 as the methyl groups destabilize the π* orbital of the pyridyl ligand. Cathodic shifts are also observed when comparing complex 2 with 5 and complex 3 with 6.| Complex | Oxidation,b Epa vs. Fc/Fc+ [V] | Reduction,c Epc vs. Fc/Fc+ [V] |
|---|---|---|
| a Scan rate: 100 mV s−1.b Irreversible oxidation; anodic peak potential (Epa vs. Fc/Fc+).c Irreversible reduction; cathodic peak potential (Epa vs. Fc/Fc+).d Not observed. | ||
| 1 | +0.96 | −2.22 |
| 2 | +1.07 | −2.03 |
| 3 | +1.12 | −1.68, −2.13 |
| 4 | +0.93 | —d |
| 5 | +1.01 | −2.19 |
| 6 | +1.05 | −1.83, −2.26 |
Computational studies
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed with the PBE0/def2-TZVP(-f) and ZORA scalar relativistic corrections level of theory on the optimized geometries of complexes 1–6 to gain more insight into their electronic and excited-state properties. The ground-state geometries (Tables S11 and S12†) showed excellent agreement with the X-ray crystal structures, with average errors for the bond length of 0.70–0.92% and bond angles of 0.73–1.72% on average across the series. The optimized geometry for the first triplet state (T1) was also obtained (Tables S11 and S12†), where a decrease in the Pt–C and Pt–N bond lengths and an increase in the CC bond length were observed compared to the ground-state geometry. This aligns with the expected LLCT/MLCT excited state for these complexes. The electronic density contours for the frontier orbitals of the complexes are depicted in Fig. S33 and S34,† and the energies are tabulated in Table S13.† The HOMO and HOMO–1 are mainly localized on the platinum(II) center and the phenylacetylide ligands, with minor contributions on the dipyridyl ligand. The energies of the HOMO are in the range of −5.91 to −6.07 eV, only slightly affected by the oxidation state of the sulfur due to the limited contribution from the dipyridyl ligand. Nevertheless, the HOMOs are more stabilized with increasing sulfur oxidation state, which is in line with the trend of the oxidation potentials in electrochemical studies. The LUMOs are mainly localized on the dipyridyl ligand, and the energies are significantly influenced by the sulfur oxidation state with sulfide (−1.93 eV) > sulfoxide (−2.15 eV) > sulfone (−2.58 eV) in the non-methylated series. The stabilization of the LUMO arises from the increasing number of electron-withdrawing oxygen atoms in the ligand that lowers the energy of the π* orbital. On the other hand, installing methyl groups on the pyridyl rings destabilizes the π* orbital and results in higher LUMO energies in complexes 4–6. The DFT calculations are in agreement with the results of the photophysical and electrochemical studies.
Based on the optimized structures, the electronic transition energies and simulated electronic absorption spectra of complexes 1–6 were calculated by TD-DFT and SOC-TD-DFT. Selected singlet and triplet excited states are listed in Tables S14–S16,† and the spectra with the electronic difference plots showing charge accumulation (blue) and depletion (red) are shown in Fig. 3 and S35–S39.† The simulated electronic absorption spectra closely resemble the experimental spectra. The first singlet excited states (S1) in all complexes consist of more than 90% of HOMO → LUMO transition, while the S2 state corresponds to either the HOMO–1 → LUMO transition or a combination of HOMO–1 → LUMO and HOMO → LUMO+1 transitions. Similarly, the first triplet excited state (T1) is composed primarily of the HOMO → LUMO transition, with an increase in the contribution of this transition as the oxidation state of the sulfur is increased. State T2 is energetically accessible through the S1 excited state only for complexes 1, 2, 4 and 5, and is composed primarily of HOMO–1 → LUMO (61–80%). These transitions are composed of predominantly MLCT [dπ(Pt) → π*(N–N)] and LLCT [π(CCPh) → π*(N–N)] transitions.
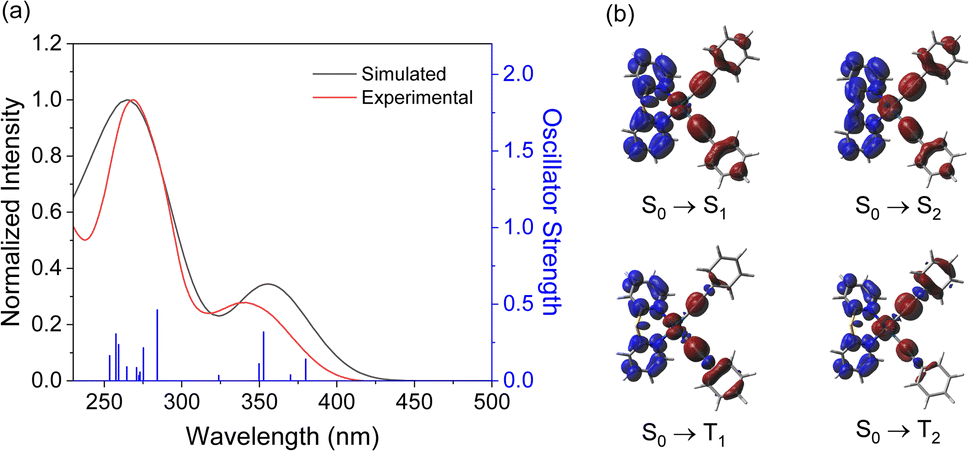 | ||
| Fig. 3 (a) Simulated (black trace) and experimental (red trace) electronic absorption spectra for complex 1. Transition energies and oscillator strengths calculated from TD-DFT calculations are shown by blue vertical lines, and the simulated spectrum is deconvoluted with Gaussian functions. (b) Electronic difference plots with the accumulation (blue) and depletion (red) of charge for the lowest-energy transitions and the triplet states. | ||
Spin–Orbit Coupling Matrix Elements (SOCMEs) between the S1 and triplet states and between the ground and triplet states were calculated at the ground-state geometry (Table S17†). Intersystem crossing (ISC) primarily occurs from the S1 state and is influenced by the magnitude of the SOCME between S1 and the lower-energy triplet states. Complexes 1, 2, 4, and 5 possess high SOCME values, ranging from 584 to 714 cm−1 mainly due to the high SOCME between T2 and S1 states. This may open additional decay channels that are not energetically accessible in complexes 3 and 6. In contrast, complexes 3 and 6 show higher SOCME values between the T1 and ground states compared to 1, 2, 4, and 5, leading to an increased radiative decay rate (kr). These results align with the shorter lifetime observed in these complexes, resulting in a higher Φem.37 Additionally, the adiabatic and vertical transition energy difference between T1 and the ground state in vacuum were calculated and compared to the experimental emission energies in the solid-state PMMA films (Table S18†). The theoretical values showed good agreement with the maximum wavelength of emission. This agrees with experimental data, where oxidizing the ligands from sulfide to sulfoxide and sulfone results in a red-shifted emission, and the addition of the methyl groups results in a small blue-shift in the emission.
Conclusions
In conclusion, a series of platinum(II) bis(acetylide) complexes with sulfur-bridged dipyridyl ligands in various oxidation states have been investigated. These complexes have been characterized by 1H and 13C{1H} NMR spectroscopy, infrared spectroscopy and high-resolution mass spectrometry. Complexes 1, 2, and 4–6 were also structurally confirmed by single-crystal X-ray diffraction. Spectroscopic studies reveal that the absorption energy of the complexes is sensitive to the oxidation state of the sulfur bridge and the substituents present on the pyridyl rings. The increase in sulfur oxidation state leads to a red-shift of absorption energy while the introduction of methyl groups results in a blue-shift in energy. In solution, only the sulfone complexes (3 and 6) display yellow 3MLCT [dπ(Pt) → π*(N–N)] and 3LLCT [π(CCPh) → π*(N–N)] excited-state phosphorescence. The solid-state neat thin films of the complexes, except 4 which was found to be non-emissive, show broad emission bands with a tail extending into the low-energy region in the photoluminescence spectra, suggesting the possibility of Pt⋯Pt interactions due to the aggregation of molecules in the neat films. All solid-state PMMA films with 2 wt% of the sulfide and sulfoxide complexes display intense deep-blue phosphorescence while the sulfone complexes show green phosphorescence with a short decay lifetime. We have illustrated a simple approach to prepare deep-blue phosphorescent platinum(II) complexes via incorporating a sulfur bridge to the dipyridyl ligand and demonstrated the capability of controlling the solution- and solid-state photophysics by changing the sulfur oxidation state. This approach enables the fabrication of light-emitting devices with a wide range of colors by simple structural modification of the platinum(II) complexes.
Data availability
All experimental procedures, NMR spectra, crystal and structure determination data, additional photophysical, electrochemical and computational data can be found in the article or in the ESI.†Author contributions
K.-M. Tong performed the experiments. J. Toigo performed DFT calculations. M. O. Wolf contributed to project design and supervision. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC). We would like to acknowledge the Laboratory for Advanced Spectroscopy and Imaging Research (LASIR) for facility access and thank Dr Saeid Kamal for assistance with spectroscopic studies. We thank Dr Daniel B. Leznoff and Leanna Karn for assistance with solid-state photoluminescence quantum yield measurement.Notes and references
- E. Longhi and L. De Cola, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons Inc., Chichester, 2017, pp. 205–274 Search PubMed.
- (a) W.-Y. Lai, J. W. Levell, A. C. Jackson, S.-C. Lo, P. V. Bernhardt, I. D. W. Samuel and P. L. Burn, Macromolecules, 2010, 43, 6986–6994 CrossRef CAS; (b) M. T. Sajjad, N. Sharma, A. K. Pal, K. Hasan, G. Xie, L. S. Kölln, G. S. Hanan, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 8939–8946 RSC; (c) C. F. R. Mackenzie, S.-Y. Kwak, S. Kimb and E. Zysman-Colman, Dalton Trans., 2023, 52, 4112–4121 RSC.
- (a) P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS; (b) T. Riesebeck and T. Strassner, Chem. Eur. J., 2024, 30, e202304263 CrossRef CAS; (c) A. K. Gupta, R. Gupta, H. C. Potter, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel, E. Zysman-Colman and A. K. Pal, Inorg. Chem., 2024, 63, 14811–14815 CrossRef CAS PubMed.
- (a) M.-C. Tang, M.-Y. Leung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 13115–13124 CrossRef CAS PubMed; (b) M.-C. Tang, W.-K. Kwok, S.-L. Lai, W.-L. Cheung, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2019, 10, 594–605 RSC; (c) H. Beucher, S. Kumar, E. Merino, W.-H. Hu, G. Stemmler, S. Cuesta-Galisteo, J. A. González, J. Jagielski, C.-J. Shih and C. Nevado, Chem. Mater., 2020, 32, 1605–1611 CrossRef CAS.
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- (a) A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed; (b) M. A. Kinzhalov, E. V. Grachova and K. V. Luzyanin, Inorg. Chem. Front., 2022, 9, 417–439 RSC.
- (a) S. Lee and W.-S. Han, Inorg. Chem. Front., 2020, 7, 2396–2422 RSC; (b) L. M. Cañada, K. Kölling, Z. Wen, J. I.-C. Wu and T. S. Teets, Inorg. Chem., 2021, 60, 6391–6402 CrossRef.
- (a) X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753–6756 CrossRef CAS PubMed; (b) H. J. Park, J.-H. Jang, J.-W. Lee and D.-H. Hwang, ACS Appl. Mater. Interfaces, 2022, 14, 34901–34908 CrossRef CAS; (c) J. Yan, Y. Pan, I.-C. Peng, W.-Y. Hung, B. Hu, G. Ni, S.-M. Yiu, Y. Chi and K. C. Lau, Inorg. Chem. Front., 2024, 11, 2413–2426 RSC.
- (a) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS; (b) C.-Y. Chan, S. M. Suresh, Y.-T. Lee, Y. Tsuchiya, T. Matulaitis, D. Hall, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, C. Adachi and E. Zysman-Colman, Chem. Commun., 2022, 58, 9377–9380 RSC; (c) S. M. Suresh, L. Zhang, T. Matulaitis, D. Hall, C. Si, G. Ricci, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2023, 35, 2300997 CrossRef CAS.
- C. M. Brown, M. Kitt, Z. Xu, D. Hean, M. Ezhova and M. O. Wolf, Inorg. Chem., 2017, 56, 15110–15118 CrossRef CAS.
- C. M. Brown, C. Li, V. Carta, W. Li, Z. Xu, P. H. F. Stroppa, I. D. W. Samuel, E. Zysman-Colman and M. O. Wolf, Inorg. Chem., 2019, 58, 7156–7168 CrossRef CAS.
- C. M. Brown, V. Carta and M. O. Wolf, Chem. Mater., 2018, 30, 5786–5795 CrossRef CAS.
- K.-M. Tong, J. Toigo, B. O. Patrick and M. O. Wolf, Inorg. Chem., 2023, 62, 13662–13671 CrossRef CAS PubMed.
- G. Li, K. Klimes, T. Fleetham, Z.-Q. Zhu and J. Li, Appl. Phys. Lett., 2017, 110, 113301 CrossRef.
- (a) H. Na, L. M. Cañada, Z. Wen, J. I.-C. Wu and T. S. Teets, Chem. Sci., 2019, 10, 6254–6260 RSC; (b) R. Hojo, D. M. Mayder and Z. M. Hudson, J. Mater. Chem. C, 2021, 9, 14342–14350 RSC; (c) Y. H. Nguyen, V. Q. Dang, J. V. Soares, J. I. Wu and T. S. Teets, Chem. Sci., 2023, 14, 4857–4862 RSC.
- (a) C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS; (b) C. F. R. Mackenzie, L. Zhang, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Adv. Optical Mater., 2023, 11, 2201495 CrossRef CAS; (c) E. Tankelevičiūtė, I. D. W. Samuel and E. Zysman-Colman, J. Phys. Chem. Lett., 2024, 15, 1034–1047 CrossRef.
- (a) G. Li, S. Liu, Y. Sun, W. Lou, Y.-F. Yang and Y. She, J. Mater. Chem. C, 2022, 10, 210–218 RSC; (b) Y. H. Nguyen, L. T. M. Dang, M. Cornu, C. Jiang, A. C. Darrow, V. Q. Dang and T. S. Teets, ACS Appl. Opt. Mater., 2024, 2, 2019–2030 CrossRef CAS.
- Y. Kang, J. Lee, D. Song and S. Wang, Dalton Trans., 2003, 3493–3499 RSC.
- L. Yang, L. D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. Hissler, W. B. Connick, D. K. Geiger, J. . E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447–457 CrossRef CAS.
- H. Lang, A. del Villar and G. Rheinwald, J. Organomet. Chem., 1999, 587, 284–289 CrossRef CAS.
- (a) T. J. Wadas, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2003, 42, 3772–3778 CrossRef CAS; (b) J. K.-W. Lee, C.-C. Ko, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Organometallics, 2007, 26, 12–15 CrossRef CAS; (c) J. Ni, Y.-G. Wang, H.-H. Wang, L. Xu, Y.-Q. Zhao, Y.-Z. Pan and J.-J. Zhang, Dalton Trans., 2014, 43, 352–360 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- J. Kang, X. Zhang, H. Zhou, X. Gai, T. Jia, L. Xu, J. Zhang, Y. Li and J. Ni, Inorg. Chem., 2016, 55, 10208–10217 CrossRef CAS.
- E. C.-H. Kwok, M.-Y. Chan, K. M.-C. Wong, W. H. Lam and V. W.-W. Yam, Chem.–Eur. J., 2010, 16, 12244–12254 Search PubMed.
- (a) J. Benito, J. R. Berenguer, J. Forniés, B. Gil, J. Gómeza and E. Lalinde, Dalton Trans., 2003, 4331–4339 RSC; (b) Y. Wu, Z. Wen, J. I.-C. Wu and T. S. Teets, Chem.–Eur. J., 2020, 26, 16028–16035 CrossRef CAS PubMed.
- (a) T. G. Kotch, A. J. Lees, S. J. Fuerniss, K. I. Papathomas and R. W. Snyder, Inorg. Chem., 1993, 32, 2570–2575 CrossRef CAS; (b) A. J. Howarth, M. B. Majewski, C. M. Brown, F. Lelj, M. O. Wolf and B. O. Patrick, Dalton Trans., 2015, 44, 16272–16279 RSC; (c) K.-C. Chan, K.-M. Tong, S.-C. Cheng, C.-O. Ng, S.-M. Yiu and C.-C. Ko, Inorg. Chem., 2018, 57, 13963–13972 CrossRef CAS PubMed.
- (a) M. Bachmann, D. Suter, O. Blacque and K. Venkatesan, Inorg. Chem., 2016, 55, 4733–4745 CrossRef CAS PubMed; (b) C.-H. Tseng, M. A. Fox, J.-L. Liao, C.-H. Ku, Z.-T. Sie, C.-H. Chang, J.-Y. Wang, Z.-N. Chen, G.-H. Lee and Y. Chi, J. Mater. Chem. C, 2017, 5, 1420–1435 RSC.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- (a) Y. Wu, Z. Wen, J. I.-C. Wu and T. S. Teets, Chem.–Eur. J., 2020, 26, 16028–16035 CrossRef CAS PubMed; (b) J. D. Bullock, Z. Xu, S. Valandro, M. Younus, J. Xue and K. S. Schanze, ACS Appl. Electron. Mater., 2020, 2, 1026–1034 CrossRef CAS.
- H. Na, M. Song and T. S. Teets, Chem.–Eur. J., 2019, 25, 4833–4842 CrossRef CAS PubMed.
- D. Poveda, Á. Vivancos, D. Bautista and P. González-Herrero, Inorg. Chem., 2023, 62, 20987–21002 CrossRef CAS PubMed.
- (a) H. Yersin and J. Strasser, Coord. Chem. Rev., 2000, 208, 331–364 CrossRef CAS; (b) H. Na and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 6353–6360 CrossRef CAS PubMed.
- (a) M. Krause, J. Friedel, S. Buss, D. Brünink, A. Berger, C. A. Strassert, N. L. Doltsinis and A. Klein, Dalton Trans., 2022, 51, 16181–16194 RSC; (b) S. A. Fitzgerald, X. Xiao, J. Zhao, P. N. Horton, S. J. Coles, R. C. Knighton, B. D. Ward and S. J. A. Pope, Chem.–Eur. J., 2023, 29, e202203241 CrossRef CAS PubMed; (c) O. Bysewski, N. Klosterhalfen, R. Jordan, L. Kletsch, A. Winter, A. Klein, B. Dietzek-Ivanšić and U. S. Schubert, Eur. J. Inorg. Chem., 2024, 27, e202300620 CrossRef CAS.
- P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, Chem. Sci., 2021, 12, 6172–6180 RSC.
- S. A. Archer, T. Keane, M. Delor, E. Bevon, A. J. Auty, D. Chekulaev, I. V. Sazanovich, M. Towrie, A. J. H. M. Meijer and J. A. Weinstein, Chem.–Eur. J., 2017, 23, 18239–18251 CrossRef CAS PubMed.
- H. Yersin, R. Czerwieniec, U. Monkowius, R. Ramazanov, R. Valiev, M. Z. Shafikov, W.-M. Kwok and C. Ma, Coord. Chem. Rev., 2023, 478, 214975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2393601–2393605. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08205b |
| This journal is © The Royal Society of Chemistry 2025 |