
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of size, charge, and spin state on Hückel and Baird aromaticity in [N]annulenes†
Louis Van Nyvel a,
Mercedes Alonso*a and
Miquel Solà*b
a,
Mercedes Alonso*a and
Miquel Solà*b
aDepartment of General Chemistry (ALGC), Vrije Universiteit Brussel (VUB), Pleinlaan 2, 1050 Brussels, Belgium. E-mail: mercedes.alonso.giner@vub.be
bInstitut de Química Computacional i Catálisi (IQCC), Departament de Química, Universitat de Girona, C/ Maria Aurèlia Capmany, 69, 17003 Girona, Catalonia, Spain. E-mail: miquel.sola@udg.edu
First published on 21st February 2025
Abstract
The Hückel and Baird rules provide a framework to understand the aromaticity of monocyclic π-conjugated molecules in their singlet ground state and lowest-lying triplet state, respectively, particularly in the context of [N]annulenes. According to Hückel's rule, a molecule in the ground state is aromatic if it contains 4n+2 π-electrons, while Baird's rule states that a molecule in the lowest-lying triplet state is aromatic if it contains 4n π-electrons, where n = 0, 1, 2, and so on. A previous study (J. Am. Chem. Soc. 2021, 143, 2403) examined the changes in the aromaticity of singlet ground-state [N]annulenes as the ring size increased from N = 12 to 66. However, no systematic investigation has yet been conducted for the lowest-lying triplet state of [N]annulenes or charged [N]annulenes. In this work, we address this gap by performing DFT calculations across several aromaticity descriptors, including structural, electronic, magnetic, and energetic indicators of aromaticity, with a particular focus on aromatic stabilization energies (ASEs). Our findings reveal that both neutral and charged [N]annulenes adhere to the Hückel and Baird rules. Nevertheless, for larger ring sizes, these rules diminish in significance, and the distinction between ASEs (and other indices of aromaticity) of [N]annulenes with 4n and 4n+2 π-electrons becomes less and less pronounced.
Introduction
The Hückel rule describes the type of aromaticity that can be expected for π-conjugated monocycles in the singlet ground state.1–4 [N]Annulenes, CNHN, containing 4n+2 π-electrons are considered aromatic, while a structure containing 4n π-electrons is regarded as antiaromatic (n = 0, 1, 2, and so on).5 When considering the lowest-lying triplet (T1) state, the Baird rule should be applied, which is the counterpart of the Hückel rule.2,3,6 As a result, in the T1 state, aromatic cycles contain 4n π-electrons and antiaromatic ones 4n+2.All [N]annulenes, where N is even, up to N = 30 have been synthesised, with the exceptions of N = 26 and 28.7,8 The higher annulenes (N > 18) show significant bond alternation and conformational flexibility. In 1959, Longuet-Higgins and Salem5 theoretically predicted that [30]annulene would be nonaromatic, whereas, in 1964, Jackman and coworkers9 used NMR to conclude that [18]annulene is aromatic but [24]annulene is non-aromatic. Calculations by Dewar and Gleicher in 1965 found that [N]annulenes up to and including N = 22 are aromatic.10 Yoshizawa and coworkers11 using the MNDOC (C for correlation) method found that, for [18]annulene, the delocalised/aromatic structure is more stable, whereas the localized/Kekulé structure is more stable for [30]annulene. Choi and Kertesz conducted a study which demonstrated that annulenes become non-aromatic from 30π-electrons onwards.2,7 They attribute this transition from (anti)aromatic to nonaromatic to the pseudo-Jahn–Teller (PJT) effect, which is intensified by the reduction in the HOMO–LUMO gaps as the ring size increases.7 More recently, it has been stressed that Hückel's rule is not applicable to large annulenes, as it overestimates aromaticity and antiaromaticity due to the adopted geometry breaking the optimal overlap of the p orbitals, which is responsible for the π-conjugated system.2,12 Finally, Casademont-Reig et al.2 reported for a small set of [N]annulenes that Hückel and Baird rules disappear as the size, N, of the [N]annulene ring increases.2 This is particularly evident in the case of annulenes with 4n π-electrons,13 since molecules are more resilient to the loss of aromaticity than to the loss of antiaromaticity.2
Probably the most important characteristic of aromatic compounds is their increased stability as compared to their linear counterparts. Calculation of the aromatic stabilisation energy (ASE) is perhaps the most reliable method for determining the (anti)aromaticity of a given molecule.14–16 For the ASE calculation, a reference system is required, which can be either an acyclic polyene or a cyclic nonaromatic structure, allowing a comparison between a delocalised π-conjugated system and a localised one.15 However, it should be noted that the ASE result is highly dependent on the selected reaction scheme and chosen reference, each of which has its own advantages and disadvantages.15 Determining the ASE of annulenes can be achieved by utilising one of the two isomerisation stabilisation energy (ISE) methods proposed by Schleyer and colleagues.13,17,18 The methyl–methylene method (ISEI) involves the energy difference between a methylene (nonaromatic) and a methyl (aromatic) derivative of the annulene (Fig. 1). This method is suitable for use in constrained systems.17,19 The indene–isoindene method (ISEII) uses the energy difference between isomers in which a cyclopentadiene ring is fused to the annulene ring (Fig. 1).13 The benefit of the ISEII approach is that all carbon atoms in the ring remain sp2-hybridised in both isomers.13,19 It is important to note that both methods require correction to counteract the anti-syn mismatch.15 This is usually achieved by adding two extra reference structures in which a bond is saturated (Fig. 2). The convention in this paper is that aromatic structures have positive ASE values, while antiaromatic structures have negative ones.
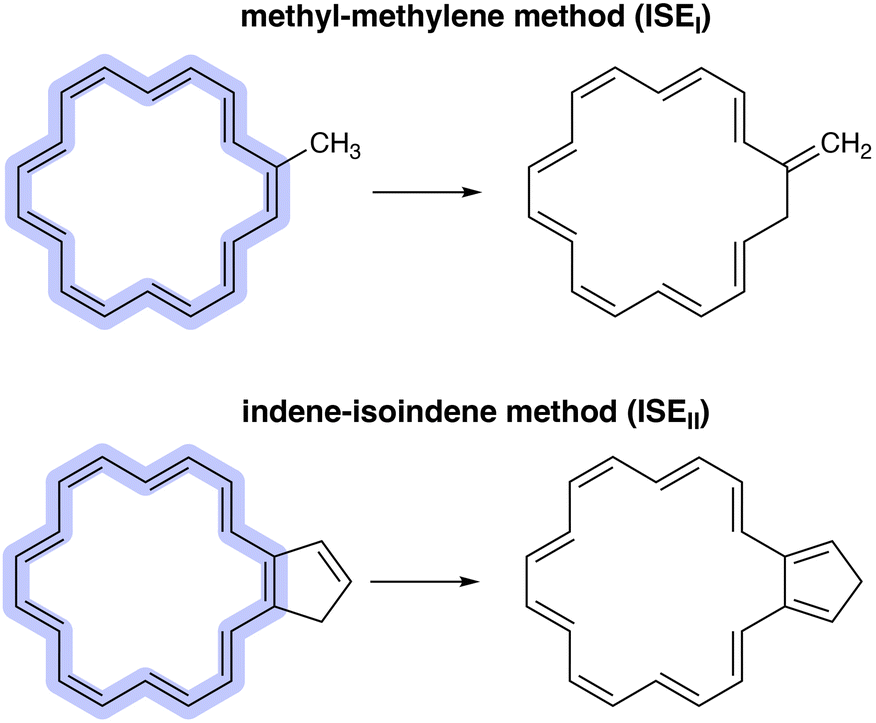 | ||
| Fig. 1 The methyl–methylene (ISEI) and indene–isoindene methods (ISEII) proposed by Schleyer and colleagues.13,17 | ||
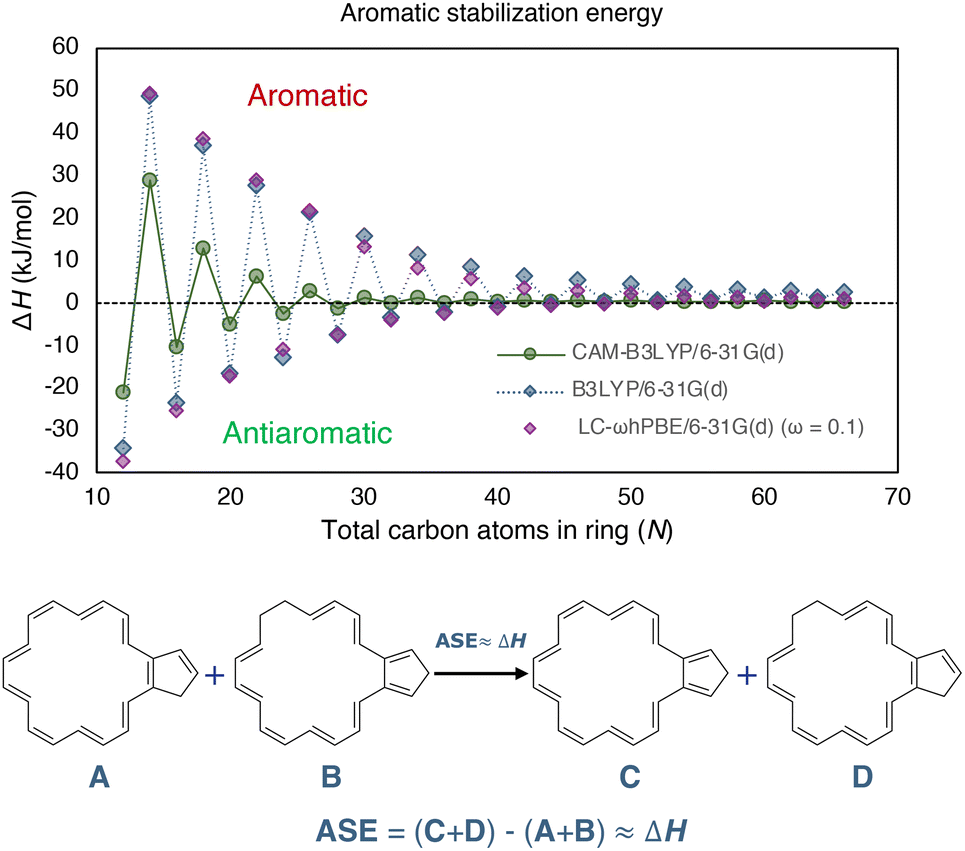 | ||
| Fig. 2 The closed-shell [N]annulene's (N = 12–66) ASE determined using the hyperhomodesmotic equation at the bottom. All structures were constrained to Cs symmetry during optimization. The B3LYP and LC-ωhPBE data were taken from ref. 20. | ||
Jirásek and coworkers20 and Wannere et al.13 investigated the ASE of annulenes in their neutral closed-shell singlet state by employing the ISEII approach. According to the Hückel rule, annulenes with 4n+2 π-electrons were expected to have positive values, while those with 4n were anticipated to have negative ones. Fig. 2 illustrates the obtained results and confirms the anticipated outcomes. Additionally, the ASE was found to be inversely proportional to the size of the rings even approaching zero for very large annulenes. This finding aligns with Casademont-Reig et al.‘s observations that the Hückel rule disappears as the annulene ring size increases.2
Stawski and colleagues recently presented a paper investigating the aromaticity of charged [18]annulenes.21 The dianion and tetraanion were expected to be antiaromatic (20π e−) and aromatic (22π e−), respectively, based on the Hückel rule.21 The predictions were confirmed by 1H-NMR, showing that this rule is not restricted to neutral compounds.21
The objective of the present investigation is to ascertain the impact of size, charge, and multiplicity on the aromaticity of even [N]annulenes. By applying Baird's rule, we anticipate that the ASE values of the neutral triplet annulenes will be the mirror image of those illustrated in Fig. 2 and, therefore, exhibit a similar trend. The overall number of π-electrons changes when two electrons are removed from the structure, thereby influencing the predicted aromaticity according to the Hückel and Baird rules. Thus, it is anticipated that the ASE trend of the charged structures will, once again, be the mirror image of the neutral molecules, as well as the trend between the triplet and closed-shell dications themselves.
Besides the energetic criterium, a range of structural, electronic, and magnetic aromaticity indices will be employed to further analyse the aromatic behaviour of annulenes. We anticipate these indices to possess an alternating pattern between aromaticity and antiaromaticity. In addition, their trend should indicate the disappearance of both rules when the size of the annulenes increases, as reported by Casademont-Reig and coworkers.2 Finally, the effectiveness of employing the ISEII method for determining the relative hardness (Δη) and predicting aromaticity, as previously demonstrated for the ISEI approach,22 will be evaluated.
Computational details
Scheme 1 depicts the structure of some of the [N]annulenes (N = 12–66) studied (for a complete representation, see Fig. S1 in the ESI†). For these annulenes, the indene (A) and isoindene (C) annulene structures employed in the ASE method were optimised utilising the (U)B3LYP/6-31G(d) level of theory and the Gaussian 16 software.20,23 In all cases, we started our geometry optimisations from the Cartesian coordinates provided by Jirásek and coworkers20 for the singlet neutral [N]annulenes. A subsequent frequency calculation at the same level of theory was conducted to obtain the zero-point vibrational energy (ZPVE) and to establish the absence of any imaginary frequency. These steps were carried out for the triplet as well as the dication with triplet and singlet multiplicity. The energies of the closed-shell B and D structures were extracted from the ESI† of ref. 20. These structures were optimised at the same level of theory but were constrained to the Cs symmetry (vide infra). We are aware of the fact that B3LYP tends to overestimate aromaticity and favour the delocalised structures.24–31 Nonetheless, several reasons justify the use of this approach in this work for the calculation of the ASE. First and foremost, the use of the B3LYP/6-31G(d) method allows for direct comparison with the findings of Jirásek et al.'s work20 which employed the same level of theory. Second, for trans-polyacetylene, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds calculated with B3LYP converge to 1.37 Å and 1.43 Å,7 almost the same as the experimentally determined values (1.36 and 1.44 Å).32 Third, ISE differences based on bond-equalized B3LYP and bond-localized Hartree–Fock (HF) optimised structures were found to be small.18 Fourth, owing to molecular orbital instabilities, DFT approaches with lower HF exchange, such as B3LYP, were found to be more robust for the calculation of benzenoid structures.33 Fifth, 4n+2 triplets barely exhibit differences between B3LYP and other functionals.2 In addition, a benchmark was conducted to assess if the trends for the ASE obtained with B3LYP/6-31G(d) were comparable to those derived from CAM-B3LYP/6-31G(d) calculations for the neutral singlet ground state annulenes. Although the ASE values for B3LYP and LC-ωhPBE (ω = 0.1) are very similar, these ASEs are significantly reduced when going from B3LYP to CAM-B3LYP, and we achieved nonaromaticity, according to ASEs, earlier with this long-range corrected functional. From Fig. 2, we see that already from N = 32, the differences between 4n and 4n+2 are lower than 1 kJ mol−1. For B3LYP and LC-ωhPBE, this happens later, from N = 54. For the triplet and charged annulenes, the trends in the ASE obtained with (U)CAM-B3LYP/6-31G(d) were similar but less clear (see Fig. 4C and S7) and this is why we prefer to discuss here the B3LYP-computed ASE values. As said before, B3LYP overestimates aromaticity and the long-range corrected CAM-B3LYP functional is recommended to mitigate the impact of the delocalization error on the aromaticity descriptors.26,27 For this reason, the aromaticity descriptors have been computed with the CAM-B3LYP functional. The results of these descriptors obtained at the (U)B3LYP/6-31G(d) level of theory are displayed in the ESI (Fig. S21 and S22).† Finally, correlations plots showing the influence of the level of theory on the trends in aromaticity can be found in Fig. S20 and S39 of the ESI.†
C and C–C bonds calculated with B3LYP converge to 1.37 Å and 1.43 Å,7 almost the same as the experimentally determined values (1.36 and 1.44 Å).32 Third, ISE differences based on bond-equalized B3LYP and bond-localized Hartree–Fock (HF) optimised structures were found to be small.18 Fourth, owing to molecular orbital instabilities, DFT approaches with lower HF exchange, such as B3LYP, were found to be more robust for the calculation of benzenoid structures.33 Fifth, 4n+2 triplets barely exhibit differences between B3LYP and other functionals.2 In addition, a benchmark was conducted to assess if the trends for the ASE obtained with B3LYP/6-31G(d) were comparable to those derived from CAM-B3LYP/6-31G(d) calculations for the neutral singlet ground state annulenes. Although the ASE values for B3LYP and LC-ωhPBE (ω = 0.1) are very similar, these ASEs are significantly reduced when going from B3LYP to CAM-B3LYP, and we achieved nonaromaticity, according to ASEs, earlier with this long-range corrected functional. From Fig. 2, we see that already from N = 32, the differences between 4n and 4n+2 are lower than 1 kJ mol−1. For B3LYP and LC-ωhPBE, this happens later, from N = 54. For the triplet and charged annulenes, the trends in the ASE obtained with (U)CAM-B3LYP/6-31G(d) were similar but less clear (see Fig. 4C and S7) and this is why we prefer to discuss here the B3LYP-computed ASE values. As said before, B3LYP overestimates aromaticity and the long-range corrected CAM-B3LYP functional is recommended to mitigate the impact of the delocalization error on the aromaticity descriptors.26,27 For this reason, the aromaticity descriptors have been computed with the CAM-B3LYP functional. The results of these descriptors obtained at the (U)B3LYP/6-31G(d) level of theory are displayed in the ESI (Fig. S21 and S22).† Finally, correlations plots showing the influence of the level of theory on the trends in aromaticity can be found in Fig. S20 and S39 of the ESI.†
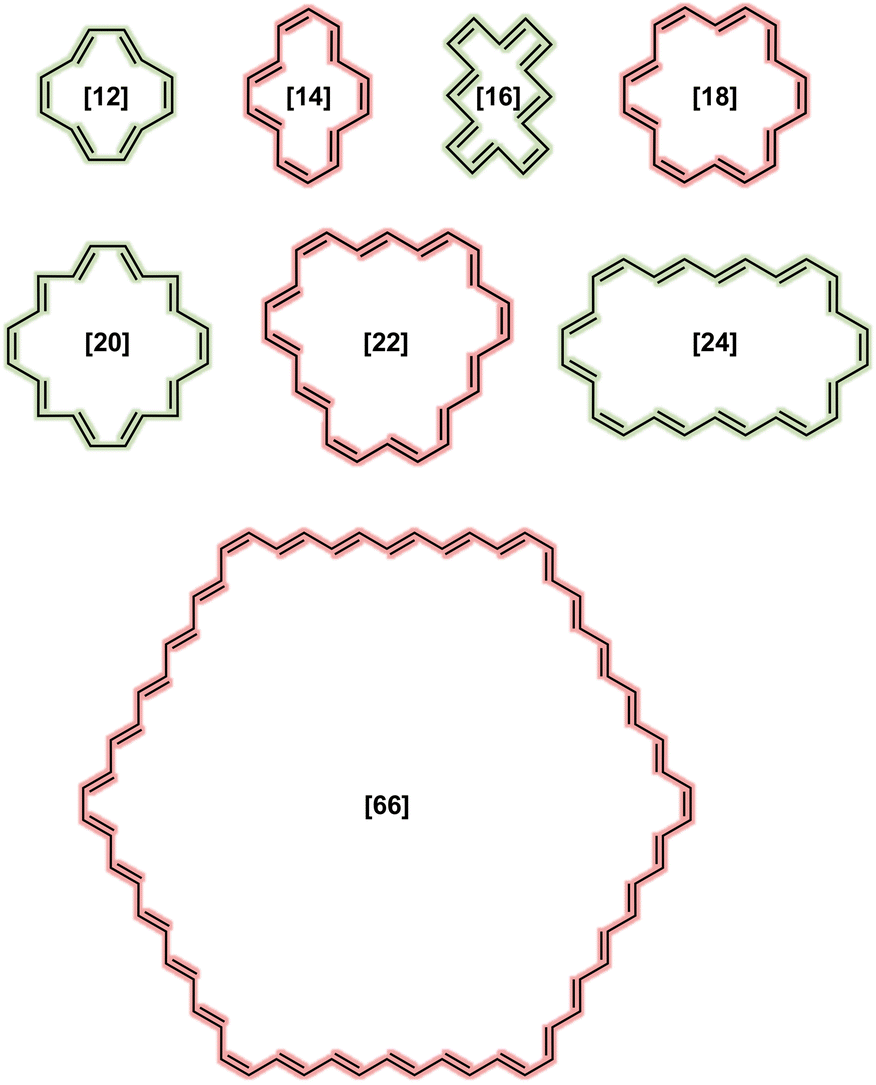 | ||
| Scheme 1 Representative structures of neutral singlet [N]annulenes, coloured based on the expected aromaticity according to the Hückel's rule. | ||
In the study by Jirásek and colleagues,20 the geometry of the different indene–isoindene isomers was imposed to be planar with Cs symmetry to observe the ‘true’ (anti)aromaticity of the annulene rings. However, there is a debate in the literature regarding whether or not small annulene rings (N < 22), especially [14]- and [18]annulenes, are planar due to the steric hindrance experienced by the hydrogen atoms located inside the ring.21,33–35 We decided not to enforce planarity in our research for three reasons. First, for smaller annulenes (N = 12–20), the steric hindrance from the hydrogen atoms within the ring sometimes prevents the formation of such a flat conformation. Second, this approach allows us to determine the real aromatic stabilisation energy of the selected annulenes. And third, we prefer to work with structures that are true minima with positive frequencies. Nevertheless, for small N values, this choice could lead to unexpected results in the calculated aromaticity indices, which may not perfectly align with the predictions of the Hückel and Baird rules.
Due to the multiple manifestations of aromaticity, characterisation of aromaticity should be done with a set of descriptors rooted in distinct criteria.36–39 In the present work, additional structural, electronic, and magnetic descriptors were evaluated to characterise the aromaticity of the different [N]annulenes, including the harmonic oscillator model of aromaticity (HOMA),40 bond-length alternation (BLA),2,41 aromatic fluctuation index (FLU),42 bond-order alternation (BOA),2,27,43 AV1245 index,44 AVmin index,2,43 electron density of delocalized bonds (EDDB),45,46 as well as nucleus independent chemical shifts (NICS) and related indices (see ESI† for a more detail description of each index) at the (U)CAM-B3LYP/6-311G(d,p)//(U)CAM-B3LYP/6-31G(d) level of theory.47–50 The ESI-3D program41 was employed for the computation of the electronic and structural indices. To evaluate the atomic overlap matrices required for the ESI-3D code, the AIMAll software was employed,51 which relies on the quantum theory of atoms-in-molecules (QTAIM) atomic partitioning. The electron density of delocalised bonds was analysed using the RunEDDB code.52 The natural atomic orbitals used for the EDDB calculation were visualised using the Avogadro software in order to distinguish between the different symmetry components (σ, π, …).53 This distinction was necessary since it allowed the separation of the contribution of the delocalised π-electrons from the overall EDDBH(r) function. Finally, the π-EDDB function was visualised with the Avogadro software to assess the electron density of the π-delocalised electrons.53 The NICS-based indices were obtained from NMR calculations at the (U)CAM-B3LYP/6-311G(d,p)//(U)CAM-B3LYP/6-31G(d) level of theory using the GIAO approach54 and the Gaussian 16 software.23 The same method was used to evaluate the atomic orbital density matrices required by the GIMIC program55 to obtain the current densities (nA/T), with the magnetic field oriented along the z-axis perpendicular to the molecular plane.56,57 To quantify the strength of the ring current, a numerical integration was conducted utilising the two-dimensional Gauss–Lobatto algorithm58 on a plane situated halfway between two selected atoms and extending from the centre of the molecule to a region where the current density vanishes.56,57 The integration plane was subdivided into slices of 0.05 × 5 Å and the total current strength was obtained by summing up all the contributions from the slides.56
Finally, the relative hardness (Δη) was obtained by taking the difference between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) orbital energies (ε) of the A and C structures (eqn (1)) from the ISEII method (Fig. 2) computed with the (U)CAM-B3LYP/6-31G(d) method.22 The Δη of the triplet structures was determined by employing the HOMO orbital energies of the α electrons and the LUMO orbital energies of the β counterparts because, in our systems, the HOMOα–LUMOβ gap is the lowest.
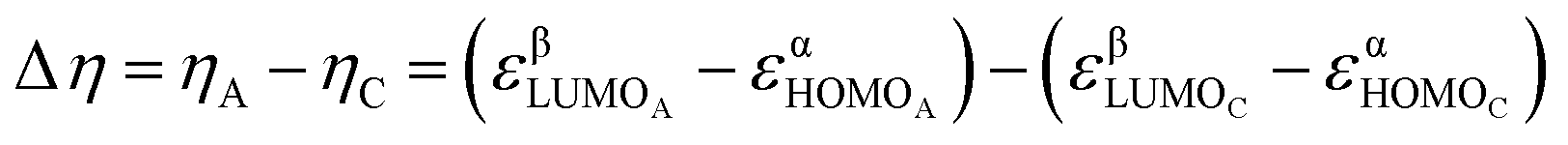 | (1) |
Results and discussion
To ascertain the presence of Hückel and Baird aromaticity in [N]annulenes and establish the influence of size and charge, aromaticity indices from five different criteria were employed. A diverse range of ring sizes, encompassing 12 to 66 atoms, were selected for this investigation. As previously mentioned, Jirásek et al.20 and Wannere et al.13 already conducted investigations into the ASE of closed-shell [N]annulenes from the reaction A + B → C + D in Fig. 2. A summary of our ASE results together with the previously published ASE data is illustrated in Fig. 3. Initially, our geometries were optimised without imposing any constraints (Fig. 3A) in contrast to the previously published results. The curves illustrate the anticipated trends with alternation occurring between the 4n and 4n+2 π-electron structures. However, the location of the curves of the neutral triplet and dication singlet annulenes is unexpected as they indicate that all the structures with N > 16 would be antiaromatic with negative ASE values. Nevertheless, the other aromaticity indices belonging to the electronic, structural, and magnetic criteria contradict this conclusion (Fig. 4). | ||
| Fig. 3 ASE results of the neutral closed-shell and triplet [N]annulenes as well as the triplet and singlet dicationic counterparts computed at the (U)B3LYP/6-31G(d) level of theory. Energies of A, B, C, and D are zero-point energy corrected electronic energies. (A) The structures to evaluate the ASE values were not subjected to any constraints. The geometries of A, B, C, and D do not contain any imaginary frequencies. (B) All the structures in the ASE calculations are enforced to be planar. Consequently, several geometries contain one or more imaginary frequencies. The neutral singlet molecules were taken from ref. 20. (C) The electronic energy and ZPVE of the B and D structures employed in the ISEII method were derived from ref. 20 and exhibit Cs symmetry. By contrast, the structure of A and C were optimised without any constraints. The fully optimized geometry of the indene isomer of the neutral triplet [12]annulene is also shown (A) together with its planar counterpart (B). | ||
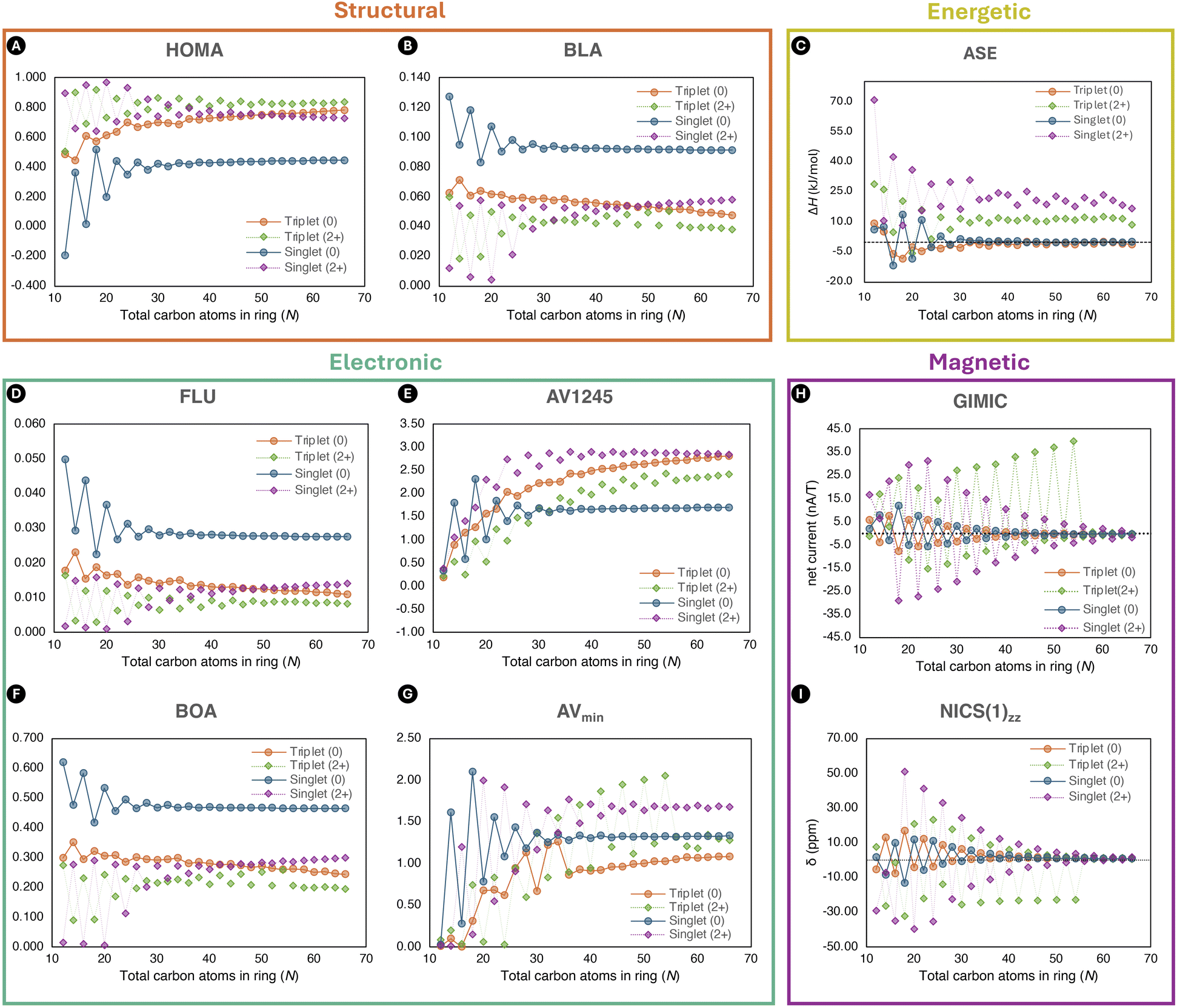 | ||
| Fig. 4 The evolution of selected aromaticity descriptors rooted in distinct criteria structural (A and B), energetic (C), electronic (D–G), and magnetic (H and I) of the neutral and charged [N]annulenes in the singlet and triplet states (N = 12–66). Except the ASE values, all the aromaticity descriptors are computed at the (U)CAM-B3LYP/6-311G(d,p)//(U)CAM-B3LYP/6-31G(d) level of theory. For the ASEs, we used the (U)CAM-B3LYP/6-31G(d) method. | ||
To identify the underlying cause of the unexpected results, the spin density of the different indene–isoindene isomers was evaluated (spin density maps of the annulenes alone can be found in Fig. S25 and S26 of the ESI†). The spin density plots of the indene-isoindene isomers revealed that the distribution of the neutral triplet in the B geometries is notably concentrated in a specific region of the molecule, in contrast to the A, C, and D structures, where the spin density is more evenly distributed throughout the molecule (Fig. S23†). This uneven spin excess distribution for the B structure was observed consistently for both aromatic and antiaromatic annulenes. Remarkably, the spin density of the charged triplet structures was likewise distributed throughout the entire A, B, C, and D molecules (Fig. S24†), suggesting a potential issue with the B geometries of the neutral triplet annulenes. Our hypothesis is that the localization of the spin density in B destabilises this reference molecule and, as a result, the ASE becomes artificially more negative in the triplet species.
When quantifying the ASE, an anti-syn correction is required to correct the mismatch between the number of syn- and anti-diene configurations at both sides of the ISEI and ISEII reactions (Fig. 1). For this reason, the dihydro derivatives B and D needs to be included as illustrated in Fig. 2. To avoid the destabilization observed in the triplet state of B, we implemented the closed-shell anti-syn correction by Jirasek et al.20 Thus, the sum of electronic energy (EE) and the ZPVE of B and D were taken from the ESI of ref. 20 in which all geometries exhibit Cs symmetry ensuring planarity. This approach effectively shifted the curve, ensuring alignment with the results from the remaining aromaticity indices (Fig. 3C). The same correction was applied to the triplet dications to ensure consistency with the results of the singlet counterparts. From these plots, firstly, it is evident that there is alternation between the aromatic and antiaromatic regions. Secondly, the anticipated mirror images were established, as predicted by both rules. Thirdly, the convergence of ASE towards zero is faster for the neutral triplet species than the singlet ones, although when normalized the two series converge at similar rate (see Fig. S8†). Fourthly, annulenes in their T1 states are less (anti)aromatic than in the S0 state. Fifthly, the difference in ASE between the aromatic and antiaromatic structures disappears when the annulene rings became larger. This latter result demonstrates that the Hückel and Baird rules are no longer applicable for large annulene rings, in accordance with the findings of Casademont-Reig et al.2 Finally, it appears that a residual aromaticity of about 10 kJ mol−1 is present for the charged structures of both multiplicities. We attribute this residual aromaticity to the resonance structures that can be formed by the positive charges present in the system (see Fig. S40†). Hole delocalization results in electronic delocalization, and therefore we consider the extra stability of large dicationic annulenes as a residual aromaticity.
Upon initial examination, the proposed correction appears somewhat unconventional. To demonstrate that this approach leads the anticipated outcomes, the ASE results with all structures possessing Cs symmetry were additionally calculated (Fig. 3B). Notably, the position of the curves in Fig. 3A and B is nearly identical and inherently different from the ASE values obtained by using the proposed correction. The rationale behind the decision not to impose planarity do not provide an explanation for the observed behaviour. A paper by Zhu, An, and Schleyer demonstrated that the conformation of the anti-butadiene in the triplet state is less stable than in the singlet state.59 For the systems investigated in this paper, no issues were encountered when applying the anti-syn correction. However, their correction was applied using an acyclic compound, which was not the case in our research. This could explain why the initial results did not meet the expectations set by Baird's rule. Regarding the cations, the charge might play an additional role in shifting the curve for both the singlet and triplet states (Fig. 3C). The application of the correction yielded the anticipated outcomes; however, it remains uncertain whether this approach can be generalised to other hyperhomodesmotic treatments.
As found in previous studies, ASE of [N]annulenes is large for small rings and decreases with the size of the ring. This trend is the same for neutral and charged singlet and triplet [N]annulenes. For large N values, the ASE becomes similar for aromatic and antiaromatic annulenes. Considering that energetic stabilization is one of the key aspects of aromaticity, we conclude that neutral singlet and triplet [N]annulenes with N > 30 should not be considered aromatic or antiaromatic. Convergence of ASE values for charged species occurs at higher N values.
An inspection of the other selected indices, depicted in Fig. 4, revealed the anticipated alternation between 4n and 4n+2 annulenes, in accordance with the predictions of the Hückel and Baird rules. Furthermore, we notice the difference between the aromatic and antiaromatic structures disappearing with the increase in ring size, in accordance with the results by Casademont-Reig et al. based on electronic descriptors.2 The observed convergence of the aromaticity descriptor to zero for the neutral structures is only met by the energetic, magnetic, and reactivity (Δη) criteria (Fig. 4 and 5). The other indices only satisfied this trend at the beginning of the curve, eventually converging to different values. Additionally, the presence of residual aromaticity in the charged annulenes is supported by the majority of structural, electronic, and magnetic indices, as the cationic closed-shell and triplet structures consistently exhibited the highest or lowest values, respectively, depending on the index. Moreover, the trend of the neutral and charged triplet and singlet annulenes should be each other mirror image, as well as the trends between the neutral and charged structures bearing the same multiplicity. In the case of the dications, the closed-shell and triplet results of all indices are each other's mirror image across most of the curve. However, unexpectedly, the aromatic triplet dication systems do not show the expected convergence with the size of the annulene.
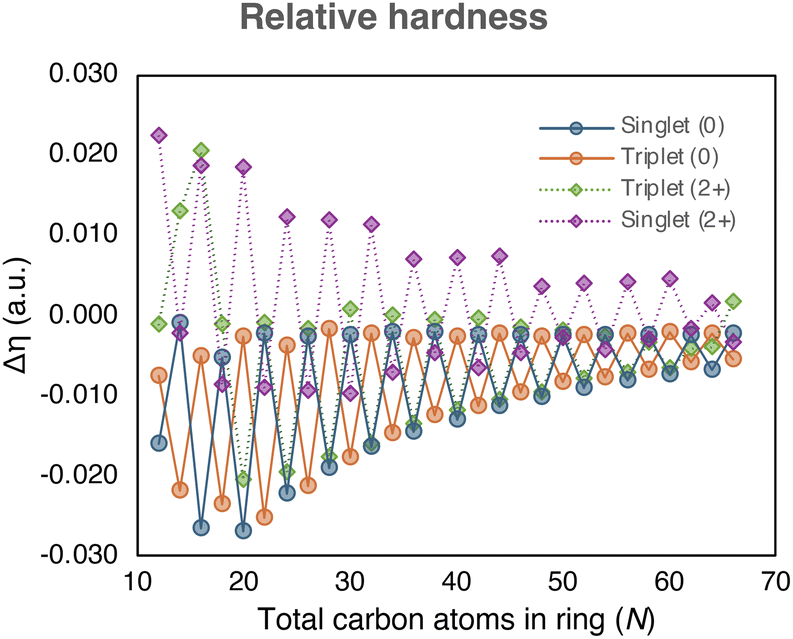 | ||
| Fig. 5 The relative hardness of the neutral and charged [N]annulenes (N = 12–66) computed with the (U)CAM-B3LYP/6-31G(d) level of theory. | ||
The presence of the Hückel and Baird rules can be further demonstrated by visualising the electron density of the π-electrons with the EDDB method (Fig. S28–S31†), which provided a clear distinction between (de)localised electron densities in aromatic and antiaromatic [N]annulenes. In the case of smaller rings, an apparent difference is noticed between the aromatic (delocalised) and antiaromatic (localised) structures. As the rings became larger, this distinction become less pronounced and eventually disappears. In addition, we observe the influence of the multiplicity and the positive charge as the density transitions between localised and delocalised states, and vice versa. These results provided further evidence of the presence of Hückel and Baird (anti)aromaticity in neutral and charged, singlet and triplet annulenes.
The efficacy of indene–isoindene derivatives in determining relative hardness (Δη) was evaluated. The effective use of the methyl–methylene isomers for determining Δη in monocycles was previously reported by De Proft and Geerlings and later extended to macrocycles.22 The evolution of Δη with the annulene size (Fig. 5) show that the indene-isoindene isomers in the ISEII method can be similarly applied to evaluate the Δη of closed-shell and triplet annulenes. In contrast to ASE, this reactivity index does not require the anti-syn corrections. The alternating and converging trends adhered to the Hückel and Baird rules in line with the rest of aromaticity indices. However, the potential residual aromaticity, thought to be present in the dications, is not observed with this reactivity index.
Among the different indices employed to assess the (anti)aromaticity of [N]annulenes, there are several findings that should be highlighted. First, the ASE shows a convergence to zero for the singlet and triplet species and a convergence to ca. 10 kJ mol−1 for the charged singlet and triplet states, implying that large [N]annulenes are non-aromatic. This result is largely supported by the reactivity Δη index and the magnetic indices (GIMIC and NICS(1)zz), with the exception of the aromatic triplet dication systems in the case of the magnetic indices. Both, NICS(1)zz and the ring current strength, increases when going from large to small annulenes. However, for the smallest annulenes (N ≤ 20), the ring current intensity is reduced because of the distortion present in these annulene structures that quenches the π-delocalisation, decreasing the intensity of the ring current and the absolute value of the NICS(1)zz. As for the ASE values, for the magnetic descriptors, there is not a clear convergence for the charged species, showing a distinct behaviour than the neutral ones. For the structural and electronic criteria, we can see the convergence to a given value as the size of the annulene increases. However, in all cases, the converged value does not correspond to that of a non-aromatic species. Accordingly, these indices suggest that large [N]annulenes keep some aromatic character, even those classified as antiaromatic ones based on the Hückel or Baird rule. Moreover, most indices indicate that the charged species are more aromatic than the neutral ones. Our interpretation is that in large [N]annulenes electron delocalization and bond length equalisation are still significant, but this electron delocalization does not have an impact in the energetic stabilization of the systems. For this reason, we conclude based on the energetic criterion that large [N]annulenes are non-aromatic. Our results clearly indicate that energetic (de)stabilisation is only important for small π-conjugated rings, in line with the findings of Jirásek et al.20 For large rings, the energetic (de)stabilisation by aromaticity is minor.
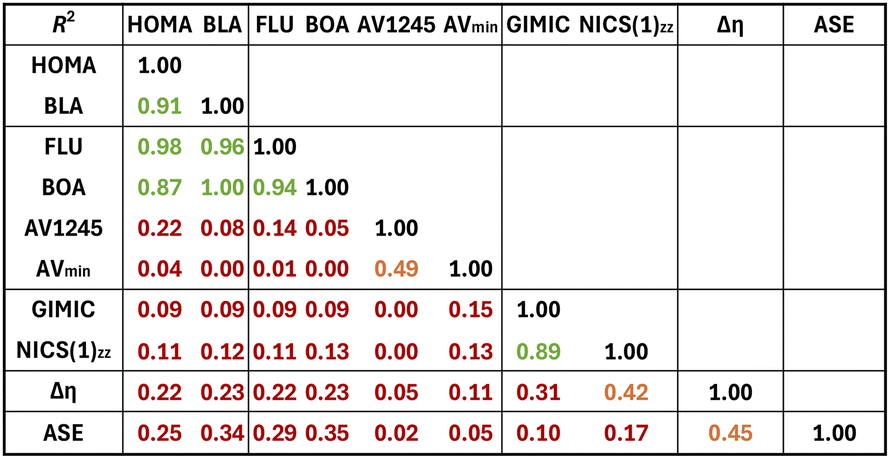
Finally, the correlation between the different aromaticity indices was examined using the R2 values (Table 1). The magnetic indices exhibit excellent correlation with an R2 value of 0.89. For the electronic criterion, the indices could be divided into two groups depending on the correlation between them. The first group comprised FLU and BOA, whilst the following group consists of AV1245 and AVmin. When R2 values between indices rooted in different criteria are considered, HOMA and BLA exhibit strong correlation with FLU and BOA. Interestingly, when separated into four different groups, namely, Hückel species (both charged and uncharged), Baird species (both charged and uncharged), charged species (both Hückel and Baird), and uncharged (both Hückel and Baird), correlations, in general but not always, improve somewhat but still in many cases correlations are poor (see Fig. S37 and S38†). As it is well known, in general, different indicators of aromaticity may not always produce results that are consistent with one another. For example, predictions based on energetic grounds frequently differ from those based on magnetic or structural criteria of aromaticity.60 Addressing the so-called multidimensional feature of aromaticity frequently resolves the apparent inconsistencies between descriptors of aromaticity.61–65 In this sense, since one compound may be more aromatic than another in one dimension and less aromatic in another dimension, it can be argued that it makes sense that different indices provide diverging orderings. This is why many authors recommend characterizing aromatic compounds using a set of indices based on various physical properties.36,66
 |
Conclusions
The objective of this paper was to assess the influence of size and charge on Hückel and Baird aromaticity in 4n and 4n+2 annulenes. Aromaticity indices from five different criteria were employed to investigate the existence of Hückel and Baird aromaticity in neutral and positively charged closed-shell and triplet annulenes. Furthermore, the disappearance of the Hückel and Baird rules with an increase in ring size was also explored. Applying these aromaticity rules to the selected macrocycles led to the hypothesis that the trend of the neutral and charged triplet and singlet annulenes should be each other's mirror image, as well as between the trends of neutral and charged structures bearing the same multiplicity.Initially, our findings indicated that the selected indices were consistent with the Hückel and Baird aromaticity hypothesis, thereby demonstrating the influence of charge on the aromaticity. Subsequently, our results indicated that both rules disappeared as the size of the annulenes increased, which was consistent with the findings published by Casademont-Reig and colleagues.2 We also found that [N]annulenes in their T1 states are less (anti)aromatic than in the S0 state, according to the ASE values. Additionally, it was also shown that the dication structures of both multiplicities potentially exhibited residual aromaticity, which could be attributed to the resonance structures resulting from the delocalisation of the positive charge. Moreover, our ASE values prove that energetic (de)stabilisation is only important for small π-conjugated rings. For large rings, the energetic (de)stabilisation by aromaticity is minor. Finally, we demonstrated that the indene-isoindene isomers (A and C) of the ISEII method can be employed to determine the relative hardness and, consequently, aromaticity, in a similar manner analogous to previously observed for the methyl–methylene isomers of the ISEI approach by De Proft, Geerlings, and Alonso.22
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. V. N. performed the calculations, analysed the data, and wrote the initial draft of the manuscript. M. A. supervised the work and reviewed and edited the final manuscript. M. S. conceived the idea, supervised the work, reviewed and edited the final manuscript, and provided financial support.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. is grateful for the financial support from the Ministerio de Ciencia e Innovación (MCIN/AEI/10.13039/50110001103, Projects PID2023-147424NB-I00 and PID2020-113711GB-I00) and the Generalitat de Catalunya (Project 2021-SGR-623 and ICREA Academia prize 2024). L. V. N. thanks the Erasmus + program. M. A. thanks the VUB for the Strategic Research Program awarded to the ALGC research group. The resources and services used in this work were provided by the VSC (Flemish Super-computer Center), funded by the Research Foundation—Flanders (FWO) and the Flemish Government.References
- (a) E. Hückel and I. Quantentheoretische beiträge zum benzolproblem, Die Elektronenkonfiguration des Benzols und werwandter Verbindungen, Z. Phys., 1931, 70, 204–286 CrossRef; (b) E. Hückel, Quanstentheoretische beiträge zum benzolproblem: II. quantentheorie der induzierten polaritäten, Z. Phys., 1931, 72, 310–337 CrossRef; (c) E. Hückel, Quantentheoretische Beiträge zum Problem der aromatischen und ungesättigten Verbindungen. III, Z. Phys., 1932, 76, 628–648 CrossRef.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, How do the Hückel and Baird rules fade away in annulenes?, Molecules, 2020, 25, 711 CrossRef CAS PubMed.
- M. Solà, Aromaticity rules, Nat. Chem., 2022, 14, 585–590 CrossRef PubMed.
- E. Von, W. Doering and L. Knox, The cycloheptatrienylium (tropylium) ion, J. Am. Chem. Soc., 1954, 76, 3203–3206 CrossRef.
- H. C. Longuet-Higgins and L. Salem, The alternation of bond lengths in long conjugated chain molecules, Proc. R. Soc. London, Ser. A, 1959, 251, 172–185 CAS.
- N. C. Baird, Quantum organic photochemistry. II. Resonance and aromaticity in the lowest 3ππ* state of cyclic hydrocarbons, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- C. H. Choi and M. Kertesz, Bond length alternation and aromaticity in large annulenes, J. Chem. Phys., 1998, 108, 6681–6688 CrossRef CAS.
- E. L. Spitler, C. A. Johnson and M. M. Haley, Renaissance of annulene chemistry, Chem. Rev., 2006, 106, 5344–5386 CrossRef CAS PubMed.
- L. Jackman, F. Sondheimer, Y. Amiel, D. Ben-Efraim, Y. Gaoni and R. Wolovsky, et al., The nuclear magnetic resonance spectroscopy of a series of annulenes and dehydro-annulenes, J. Am. Chem. Soc., 1962, 84, 4307–4312 CrossRef CAS.
- M. J. Dewar and G. J. Gleicher, Ground states of conjugated molecules. II. Allowance for molecular geometry, J. Am. Chem. Soc., 1965, 87, 685–692 CrossRef CAS.
- K. Yoshizawa, T. Kato and T. Yamabe, Electron correlation effects and possible D6h structures in large cyclic polyenes, J. Phys. Chem., 1996, 100, 5697–5701 CrossRef CAS.
- C. Gellini and P. R. Salvi, Structures of annulenes and model annulene systems in the ground and lowest excited states, Symmetry, 2010, 2, 1846–1924 CrossRef CAS.
- C. S. Wannere, D. Moran, N. L. Allinger, B. A. Hess, L. J. Schaad and P. v. R. Schleyer, On the stability of large [4n] annulenes, Org. Lett., 2003, 5, 2983–2986 CrossRef CAS PubMed.
- M. Dewar and H. Schmeising, A re-evaluation of conjugation and hyperconjugation: The effects of changes in hybridisation on carbon bonds, Tetrahedron, 1959, 5, 166–178 CrossRef CAS.
- M. Alonso, and I. Fernández, Quantifying aromaticity according to the energetic criterion, Elsevier, Aromaticity, 2021, pp. 195–235 Search PubMed.
- M. K. Cyrański, Energetic aspects of cyclic pi-electron delocalization: evaluation of the methods of estimating aromatic stabilization energies, Chem. Rev., 2005, 105, 3773–3811 CrossRef PubMed.
- P. v. R. Schleyer and F. Pühlhofer, Recommendations for the evaluation of aromatic stabilization energies, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS PubMed.
- C. S. Wannere and P. v. R. Schleyer, How aromatic are large (4n+2) π annulenes?, Org. Lett., 2003, 5, 865–868 CrossRef CAS PubMed.
- K. An and J. Zhu, Evaluation of triplet aromaticity by the indene-isoindene isomerization stabilization energy method, Eur. J. Org Chem., 2014, 2764–2769 CrossRef CAS.
- M. Jirásek, M. Rickhaus, L. Tejerina and H. L. Anderson, Experimental and theoretical evidence for aromatic stabilization energy in large macrocycles, J. Am. Chem. Soc., 2021, 143, 2403–2412 CrossRef PubMed.
- W. Stawski, Y. Zhu, I. Rončević, Z. Wei, M. A. Petrukhina and H. L. Anderson, The anti-aromatic dianion and aromatic tetraanion of [18]annulene, Nat. Chem., 2024, 16, 998–1002 CrossRef CAS PubMed.
- (a) F. De Proft and P. Geerlings, Relative hardness as a measure of aromaticity, Phys. Chem. Chem. Phys., 2004, 6, 242–248 RSC; (b) M. Alonso, P. Geerlings and F. De Proft, Viability of Möbius Topologies in [26]- and [28]Hexaphyrins, Chem.–Eur. J., 2012, 18, 10916–10928 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji; X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi; J. Goings; B. Peng; A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega; G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven; K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. A. King, T. D. Crawford, J. F. Stanton and H. F. Schaefer, Conformations of [10] annulene: more bad news for density functional theory and second-order perturbation theory, J. Am. Chem. Soc., 1999, 121, 10788–10793 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Insights into current limitations of density functional theory, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
- D. W. Szczepanik, M. Solà, M. Andrzejak, B. Pawełek, J. Dominikowska and M. Kukułka, et al., The role of the long-range exchange corrections in the description of electron delocalization in aromatic species, J. Comput. Chem., 2017, 38, 1640–1654 CrossRef CAS PubMed.
- I. Casademont-Reig, T. Woller, J. Contreras-García, M. Alonso, M. Torrent-Sucarrat and E. Matito, New electron delocalization tools to describe the aromaticity in porphyrinoids, Phys. Chem. Chem. Phys., 2018, 20, 2787–2796 RSC.
- M. Torrent-Sucarrat, S. Navarro, F. P. Cossío, J. M. Anglada and J. M. Luis, Relevance of the DFT method to study expanded porphyrins with different topologies, J. Comput. Chem., 2017, 38, 2819–2828 CrossRef CAS PubMed.
- L. J. Karas, S. Jalife, R. V. Viesser, J. V. Soares, M. M. Haley and J. I. Wu, Tetra-tert-butyl-s-indacene is a bond-localized C2h structure and a challenge for computational chemistry, Angew. Chem., Int. Ed., 2023, 62, e202307379 CrossRef CAS PubMed.
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, Benchmarking magnetizabilities with recent density functionals, J. Chem. Theory Comput., 2021, 17(3), 1457–1468 CrossRef CAS PubMed.
- M. Orozco-Ic, M. Dimitrova, J. Barroso, D. Sundholm and G. Merino, Magnetically induced ring-current strengths of planar and nonplanar molecules: New insights from the pseudo-π model, J. Phys. Chem. A, 2021, 125(26), 5753–5764 CrossRef CAS PubMed.
- C. Yannoni and T. Clarke, Molecular geometry of cis-and trans-polyacetylene by nutation NMR spectroscopy, Phys. Rev. Lett., 1983, 51, 1191 CrossRef CAS.
- R. A. King, P. R. Schreiner and T. D. Crawford, Structure of [18] annulene revisited: challenges for computing benzenoid systems, J. Phys. Chem. A, 2024, 128, 1098–1108 CrossRef CAS PubMed.
- C. S. Wannere, K. W. Sattelmeyer, H. F. Schaefer III and P. v. R. Schleyer, Aromaticity: The alternating C–C bond length structures of [14]-, [18]-, and [22]annulene, Angew. Chem., Int. Ed., 2004, 43, 4200–4206 CrossRef CAS PubMed.
- K. Mislow, Aromaticity of conjugated monocyclic hydrocarbons, J. Chem. Phys., 1952, 20, 1489–1490 CrossRef CAS.
- J. Poater, I. García-Cruz, F. Illas and M. Solà, Discrepancy between common local aromaticity measures in a series of carbazole derivatives, Phys. Chem. Chem. Phys., 2004, 6, 314–318 RSC.
- J. I. Wu, I. Fernández and P. v. R. Schleyer, Description of aromaticity in porphyrinoids, J. Am. Chem. Soc., 2013, 135, 315–321 CrossRef CAS PubMed.
- M. Solà, Why aromaticity is a suspicious concept? Why?, Front. Chem., 2017, 5, 22 Search PubMed.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Photochemistry driven by excited-state aromaticity gain or antiaromaticity relief, Chem.–Eur. J., 2023, 29, e202203748 CrossRef CAS PubMed.
- J. Kruszewski and T. Krygowski, Definition of aromaticity basing on the harmonic oscillator model, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- E. Matito, 3D: electron sharing indices program for 3D molecular space partitioning, Institute of Computational Chemistry and Catalysis (IQCC), 2006 Search PubMed , https://ematito.dipc.org/programs.html.
- E. Matito, M. Duran and M. Solà, Erratum: The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization, 2005, 122, 014109, J. Chem. Phys., 2006, 125, 059901 CrossRef.
- I. C. Reig, Computational study of aromaticity in porphyrinoid systems and photosensitizers from chemical bonding descriptors PhD thesis, Universidad del País Vasco-Euskal Herriko Unibertsitatea, 2021 Search PubMed.
- E. Matito, An electronic aromaticity index for large rings, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- D. W. Szczepanik, M. Andrzejak, K. Dyduch, E. Zak, M. Makowski, G. Mazur and J. Mrozek, A uniform approach to the description of multicenter bonding, Phys. Chem. Chem. Phys., 2014, 16, 20514–20523 RSC.
- D. W. Szczepanik and M. Solà, The electron density of delocalized bonds (EDDBs) as a measure of local and global aromaticity, Elsevier, Aromaticity, 2021, pp. 259–284 Search PubMed.
- A. Stanger, NICS–past and present, Eur. J. Org Chem., 2020, 3120–3127 CrossRef CAS.
- R. Gershoni-Poranne and A. Stanger, Magnetic criteria of aromaticity, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism: I. A gauge-invariant LCAO method for NMR chemical shifts, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- C. S. López, and O. N. Faza, Overview of the computational methods to assess aromaticity. Aromaticity, Elsevier, Dordretch, The Netherlands, 2021, pp. 41–71 Search PubMed.
- T. Keith, AIMAll (version 19.10.12), TK Gristmill Software, Overland Park KS, 2019, http://aim.tkgristmill.com/ Search PubMed.
- D. W. Szczepanik, RunEDDB, https://aromaticity.eu/eddb/, accessed: April 19, 2024.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- P. Pulay, J. Hinton, and K. Wolinski, Efficient implementation of the GIAO method for magnetic properties: theory and application, in Nuclear magnetic shieldings and molecular structure, ed. J. A. Tossell, Springer, Dordretch, 1993, pp. 243–262 Search PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, The gauge including magnetically induced current method, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- L. Leyva-Parra, R. Pino-Rios, D. Inostroza, M. Solà, M. Alonso and W. Tiznado, Aromaticity and magnetic behavior in benzenoids: unraveling ring current combinations, Chem.–Eur. J., 2024, 30, e202302415 CrossRef CAS PubMed.
- L. Leyva-Parra, I. Casademont-Reig, R. Pino-Rios, L. Ruiz, M. Alonso and W. Tiznado, New perspectives on delocalization pathways in aromatic molecular chameleons, ChemPhysChem, 2024, 25, e202400271 CrossRef CAS PubMed.
- Handbook of mathematical functions with formulas, graphs, and mathematical Tables, ed. M. Abramowitz, and I. A. Stegun, Dover, New York, 1965 Search PubMed.
- J. Zhu, K. An and P. v. R. Schleyer, Evaluation of triplet aromaticity by the isomerization stabilization energy, Org. Lett., 2013, 15, 2442–2445 CrossRef CAS PubMed.
- A. R. Katritzky, P. Barczynski, G. Musumarra, D. Pisano and M. Szafran, Aromaticity as a quantitative concept. 1. A statistical demonstration of the orthogonality of classical and magnetic aromaticity in five-and six-membered heterocycles, J. Am. Chem. Soc., 1989, 111, 7–15 CrossRef CAS.
- K. Jug and A. M. Köster, Aromaticity as a multi-dimensional phenomenon, J. Phys. Org. Chem., 1991, 4, 163–169 CrossRef CAS.
- A. R. Katritzky, K. Jug and D. C. Oniciu, Quantitative measures of aromaticity for mono-, bi-, and tricyclic penta-and hexaatomic heteroaromatic ring systems and their interrelationships, Chem. Rev., 2001, 101, 1421–1450 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyrański, Structural aspects of aromaticity, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed.
- A. R. Katritzky, M. Karelson, S. Sild, T. M. Krygowski and K. Jug, Aromaticity as a quantitative concept. 7. Aromaticity reaffirmed as a multidimensional characteristic, J. Org. Chem., 1998, 63, 5228–5231 CrossRef CAS.
- M. K. Cyranski, T. M. Krygowski, A. R. Katritzky and P. V. R. Schleyer, To what extent can aromaticity be defined uniquely?, J. Org. Chem., 2002, 67, 1333–1338 CrossRef CAS PubMed.
- M. Alonso, P. Geerlings and F. De Proft, Topology switching in [32] heptaphyrins controlled by solvent, protonation, and meso substituents, Chem.–Eur. J., 2013, 19, 1617–1628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Theoretical background of the aromaticity indices; benchmark study; spin density plots and aromaticity descriptors of neutral and charged [N]annulenes in singlet and triplet states. See DOI: https://doi.org/10.1039/d4sc08225g |
| This journal is © The Royal Society of Chemistry 2025 |