
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mercapto-NSAIDs generate a non-steroidal anti-inflammatory drug (NSAID) and hydrogen sulfide†
Simran M.
Gupta
 ,
Pratiksha S.
Mohite
and
Harinath
Chakrapani
*
,
Pratiksha S.
Mohite
and
Harinath
Chakrapani
*
Department of Chemistry, Indian Institute of Science Education and Research Pune, Pune 411 008, Maharashtra, India. E-mail: harinath@iiserpune.ac.in
First published on 5th February 2025
Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the frontline treatments for inflammation and pain. Hydrogen sulfide (H2S) and related persulfide (RS-SH) are important mediators of antioxidant response and protect cells from oxidative stress. Hybrids of these pharmacological agents have shown promise in clinical trials and are superior to the parent NSAID. Here, we report a new class of NSAID–H2S hybrids, where a strategic placement of a sulfhydryl group adjacent to a carbonyl of a NSAID facilitates the enzymatic generation of H2S. We show that α-mercapto-nabumetone, a derivative of the clinical drug nabumetone, is a substrate for 3-mercaptopyruvate sulfurtransferase (3-MST), an enzyme involved in H2S biosynthesis. The key step of 3-MST catalysis is the cleavage of a C–S bond adjacent to a carbonyl group, which generates an enolate and 3-MST persulfide, which in turn is cleaved under reducing conditions to generate H2S. Guided by a molecular docking study with 3-MST, we prepared two mercapto-nabumetone derivatives, protected as their thioacetates. In the presence of 3-MST, both mercapto-nabumetone derivatives generated H2S and the NSAID in a nearly quantitative yield, produced glutathione persulfide (GS-SH), an important mediator of cellular antioxidant response, and permeated cells to generate H2S. Lastly, to gain insights into the scope of this strategy, we prepared mercapto-NSAID derivatives containing a carboxylic acid. We found that the propensity to generate H2S depended on the nature of the enol that is produced during the transformation of the mercapto-NSAID into the parent NSAID. This offers new insights into 3-MST catalysis and how reaction outcomes can be modulated by the keto–enol equilibrium. Taken together, the atom economical transformation of a clinical NSAID with one strategically placed sulfhydryl group to generate H2S presents new opportunities to enhance the properties of NSAIDs through participation in endogenous H2S biosynthesis.
Introduction
Hydrogen sulfide (H2S) and related sulfane sulfur species are components of a cell's antioxidant machinery, and therapeutic strategies to use such species to promote antioxidant response to stress are in development.1–8 Exogenous sources of H2S, known as H2S donors, are small molecules that dissociate to generate H2S.4–9 From inorganic sources to compounds that require enzymatic activation, many classes of H2S donors are known, and some have promising antioxidant properties.8–12 Non-steroidal anti-inflammatory drugs (NSAIDs) are the mainstay treatment of pain and inflammation.13–15 These drugs have some limitations that have been shown to be mitigated by H2S.16–18 Hence, NSAID–H2S hybrids (Fig. 1a) are in development, and these hybrids have distinct pharmacological effects and appear to synergize to improve the overall efficacy of the NSAID. For example, the thioamide ATB-346 is a promising candidate for the treatment of post-operative pain, inflammation and osteoarthritis.19 This compound undergoes metabolism to generate H2S and the NSAID and has been found to reduce incidences of ulcers in patients when compared with the parent NSAID. A number of other hybrids have been similarly developed, with combinations of NSAIDs with varying degrees of success, and typically involve the addition of bulky groups and linkers.19–24 Some generate H2S spontaneously, while others are activated by thiols, enzymes, or reactive oxygen species (ROS).25–31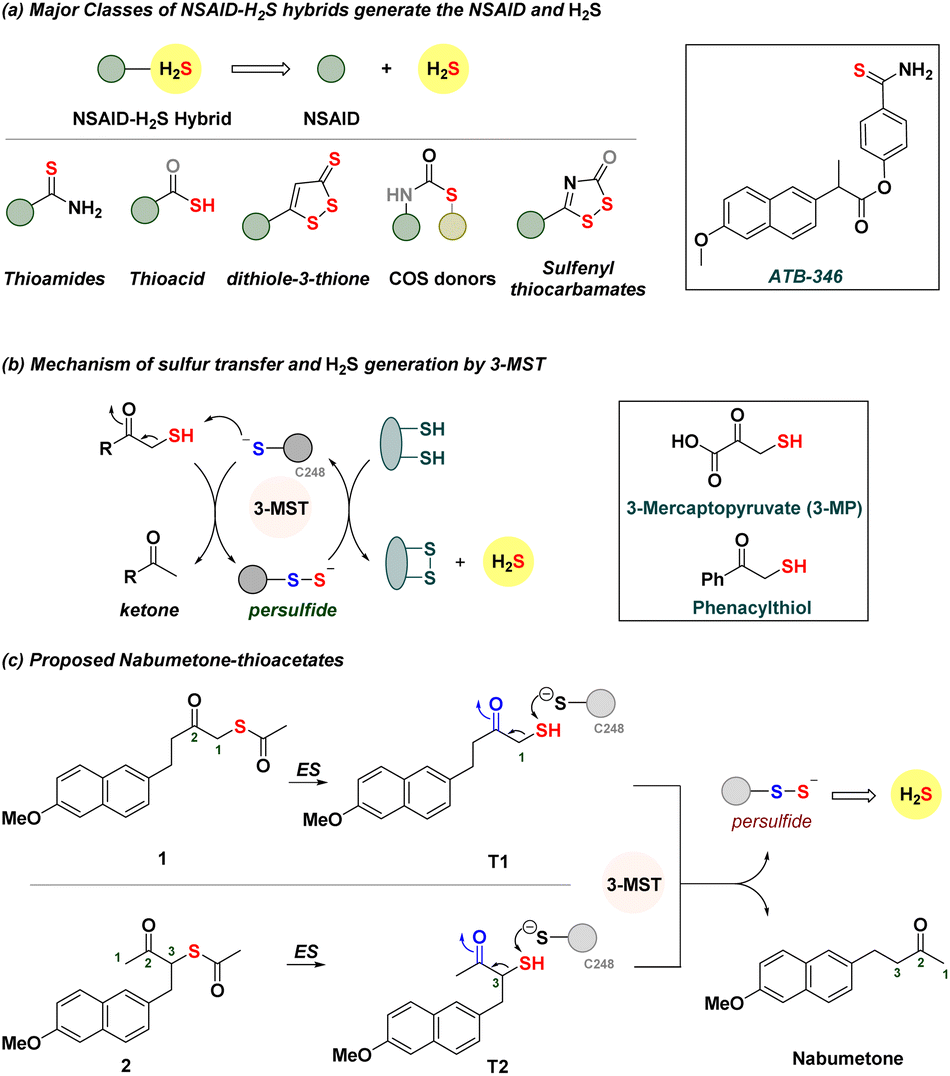 | ||
| Fig. 1 (a) Major classes of NSAID–H2S hybrids. These compounds dissociate to produce the NSAID and H2S. (b) Mechanism of persulfide and H2S generation by 3-MST. In the box are 3-mercaptopyruvate and phenacylthiol. (c) Proposed NSAID hybrids should be cleaved by esterase (ES) to generate the thiol, which reacts with 3-MST to generate a persulfide and the NSAID nabumetone. | ||
We considered using 3-mercaptopyruvate sulfurtransferase (3-MST), an endogenous enzyme that generates persulfide and H2S (Fig. 1b).32–35 The natural substrate is 3-mercaptopyruvate (3-MP), which interacts with the active site cysteine (C248) of 3-MST.36 Transfer of the sulfhydryl group of 3-MP by the cleavage of a C–S bond to 3-MST produces a persulfide, which, under reductive conditions, generates H2S.37 We recently developed phenacylthiol as an artificial substrate for this enzyme and found that this α-mercaptoketone produced H2S in the presence of 3-MST (Fig. 1b).38,39 Taking a cue from this result, we considered the clinically used ketone nabumetone (Fig. 1c)40 as a test case for insertion of a sulfhydryl group adjacent to the ketone, at either the C-1 or the C-3 position (structures T1 and T2, Fig. 1c). Nabumetone is among the widely used NSAIDs for the treatment of pain and inflammation as well as conditions such as osteoarthritis and rheumatoid arthritis.41–43 If these mercapto-NSAIDs are substrates for 3-MST, they are expected to regenerate the NSAID, nabumetone, and produce H2S. Since thiols themselves are prone to oxidation44 and are difficult to store for extended durations, we designed two thioacetates 1 and 2; hydrolysis by esterase (ES) should generate T1 and T2, respectively.45 Here, we report the synthesis and evaluation of α-mercapto-nabumetone, a new class of NSAID–H2S hybrids.
Results and discussion
Molecular docking study
The active site of the enzyme 3-mercaptopyruvate sulfurtransferase (PDB ID: 4JGT) was identified based on previous reports.46,47 The residues that are involved in binding and catalysis of 3-MP are C248, R188, and R197 (Fig. 2). Using standard molecular docking protocols, the binding of thiols T1 and T2 was first analysed. The lowest energy binding conformation of T1 was found to be favorable (−5.5 kcal mol−1). An important parameter that facilitates turnover and product formation is the S⋯S distance between the cysteine sulfur and T1.38,46 This distance was found to be 4.6 Å, which from our previous studies supports a favorable binding interaction (Fig. 2A).38 The carbonyl group was also found to be proximal to the two arginine residues, and this interaction has been previously considered important for the stabilization of the incipient enolate.48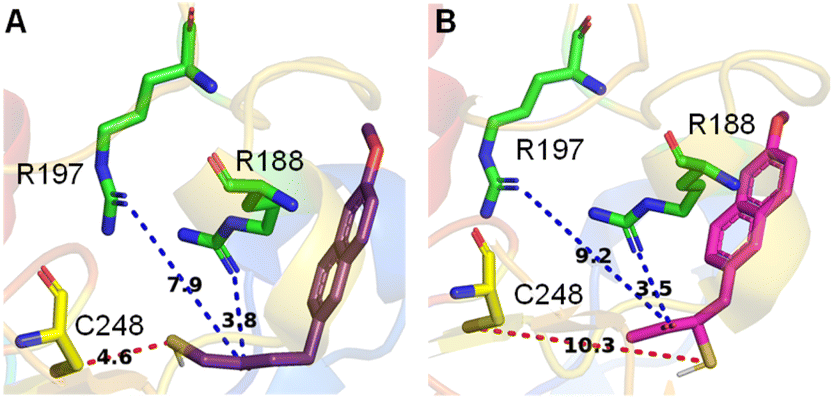 | ||
| Fig. 2 Lowest energy conformations found by molecular docking studies with human 3-MST of (A) T1 and (B) T2. Both T1 and T2 show comparable docking energy, i.e., −5.5 kcal mol−1 and −5.3 kcal mol−1, respectively. C is the cysteine residue and R represents arginine. The distance between the sulfur of T1 and the sulfur of active residue C248 is 4.6 Å, whereas for T2 it is 10.3 Å. | ||
Similarly, when T2 was analysed, although the binding energy was favorable (−5.3 kcal mol−1), the S⋯S distance between the cysteine and T2 was somewhat longer (10.3 Å), suggesting a less favorable interaction that eventually leads to persulfide formation (Fig. 2B). Hence, both thiols are predicted to be substrates for 3-MST, with T1 being more favourable for binding and positioning in the active site, as compared with T2 (Table S1, see the ESI†).
Synthesis
Nabumetone (3) was first converted to 1-bromo-nabumetone using copper bromide (CuBr2),49 and the yield was found to be 45% (Scheme 1). The bromide was subsequently converted to 1 using potassium thioacetate with a yield of 87% (Scheme 1). Similarly, to synthesize 2, 3-bromo-nabumetone was prepared using phenyltrimethyl ammonium tribromide (PTT)50 as the brominating agent in 72% yield (Scheme 1). The reaction of this bromide with potassium thioacetate gave 2 in 67% yield. The oxygen versions 4 and 5 were synthesized as negative controls in 68% and 62% yields, respectively (Schemes 1 and S1, see the ESI†).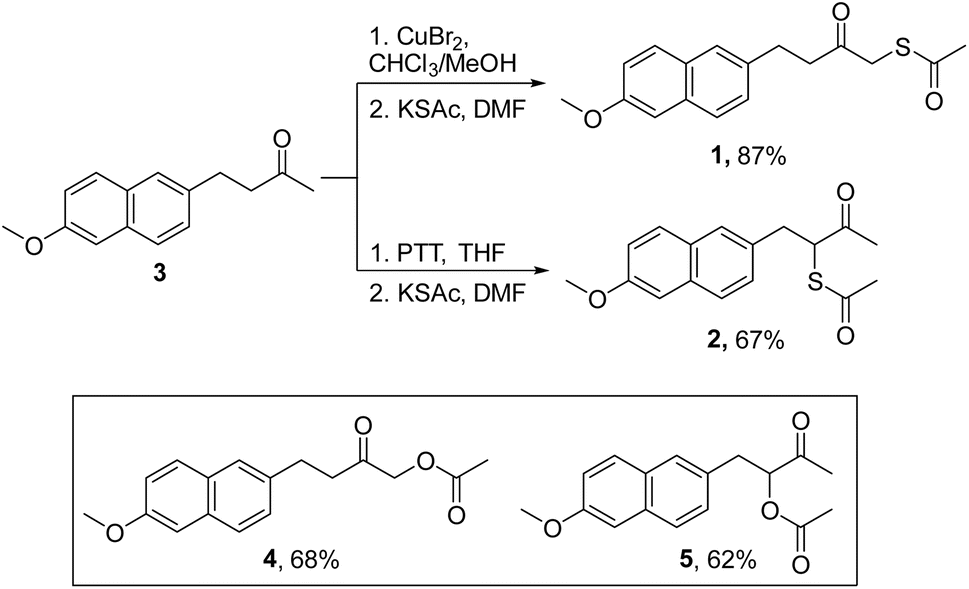 | ||
| Scheme 1 Synthesis of mercapto-nabumetone derivatives 1 and 2; inset: structures of negative controls 4 and 5 (Schemes S1, see the ESI†). | ||
Stability studies
First, to determine whether 1 was stable in pH 7.4 buffer, HPLC analysis was carried out. The thioacetate was found to be stable over 90 min (Fig. S1A, see the ESI†). In the presence of esterase, as expected, the thioacetate produced T1 (Fig. S2A, see the ESI†) along with a disulfide, due to aerial oxidation. Independently, T1 was trapped by mono-bromobimane (mBBr) as a bimane adduct (MALDI analysis, Scheme S2, Fig. S3, see the ESI†). In the presence of ES and dithiothreitol (DTT), T1 was produced and remained stable (Fig. S4A, see the ESI†). In the case of 2, under similar conditions, only 54% of the thioacetate remained in 90 min in pH 7.4 buffer (Fig. S1, see the ESI†). The formation of T2 as the major product was seen, indicating that the thioester of 2 was cleaved faster than 1. The origin of this increased lability is not clear but the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency for the thioacetate for 2 was higher than for 1 (2, 1695 cm−1vs.1, 1689 cm−1) suggesting an increased reactivity. No further reaction of the thiol with DTT was seen, suggesting that both T1 and T2, once formed, largely remained intact under reducing conditions (Fig. S4B, see the ESI†). Derivatives 1 and 2 were also found to be stable at acidic pH for 60 min (pH 2 and 6.8, Fig. S5 and S6, see the ESI†). Hence, the thioacetate can be converted to the thiol in the presence of biothiols as well as ES; the thiols T1 and T2 were themselves quite stable at acidic and neutral pH as well as in the presence of biothiols at pH 7.4.
O stretching frequency for the thioacetate for 2 was higher than for 1 (2, 1695 cm−1vs.1, 1689 cm−1) suggesting an increased reactivity. No further reaction of the thiol with DTT was seen, suggesting that both T1 and T2, once formed, largely remained intact under reducing conditions (Fig. S4B, see the ESI†). Derivatives 1 and 2 were also found to be stable at acidic pH for 60 min (pH 2 and 6.8, Fig. S5 and S6, see the ESI†). Hence, the thioacetate can be converted to the thiol in the presence of biothiols as well as ES; the thiols T1 and T2 were themselves quite stable at acidic and neutral pH as well as in the presence of biothiols at pH 7.4.
Catalysis by 3-MST to generate nabumetone
Next, to determine whether the thiol could generate persulfide through sulfur transfer, a reaction of 1 in the presence of ES, 3-MST, and DTT was performed (Scheme S3 and Fig. S7A, see the ESI†). Under these conditions, it is expected that 1 is converted to T1, which in turn would be turned over by 3-MST to produce 3-MST persulfide and nabumetone (Fig. 3A). DTT is expected to cleave 3-MST persulfide to regenerate the active catalyst. In the presence of ES, 3-MST, and DTT, a nearly complete disappearance of 1 was seen in 30 min. The thiol T1 was formed and was gradually converted to nabumetone over several hours (Fig. 3C). The observed rate constant for the disappearance of T1 (kT1) was 0.38 h−1 and for the formation of nabumetone (k3) was 0.57 h−1. The yield of nabumetone was nearly quantitative, supporting a clean and efficient conversion of 1 to produce the NSAID (Fig. S7A, see the ESI†). The hybrid 2 also produced T2, which disappeared (kT2 = 0.4 h−1) to produce nabumetone as the exclusive product, again in a nearly quantitative yield at a rate of k3 = 0.6 h−1 (Fig. 3B, D, and S7B, see the ESI†). No significant difference in the rates of nabumetone formation in the aforementioned reactions was seen. The disappearance of thiols also occurred at a comparable rate. Both thiols are equally good substrates for 3-MST, and it appears that the barrier for conversion of the most favorable but high energy conformation of T2 to a less favorable productive conformation is low. | ||
| Fig. 3 (A and B) Representative HPLC traces of 1 or 2 (100 μM) in the presence of ES (1 U mL−1), 3-MST (1 μM), and DTT (1 mM), showing the formation of nabumetone 3 from t = 0 to t = 540 min. T1 and T2 are traces where 1 or 2 has been treated with ES and DTT; (C and D): curve fitting gave an apparent rate constant for the decomposition of T1 and T2 with a kT1 of 0.38 h−1 and a kT2 of 0.4 h−1, and the formation of nabumetone with 0.57 h−1 and 0.6 h−1, respectively. All data are represented as mean ± SD (n = 2). SD is the standard deviation. | ||
Sulfur transfer and sulfane sulfur generation
Next, two independent assays to assess sulfur transfer and persulfide generation were carried out (Fig. 4A). An assay based on the persulfide-sensitive fluorogenic probe SSP-2 was next used to detect 3-MST persulfide during catalysis (Scheme S4, see the ESI†).51 As expected, AS in the presence of ES and 3-MST gave a signal in this assay (Fig. 4B). In the absence of 3-MST, no significant signal was observed, supporting the need for catalysis by 3-MST to generate persulfide. Under similar conditions, 1 or 2 alone showed no detectable increase in the fluorescence signal (Fig. S8, see the ESI†). In the presence of ES and 3-MST, a significant enhancement of fluorescence, comparable with AS, was recorded. When this reaction mixture was co-treated with DTT, a nearly complete abrogation of fluorescence that is consistent with the cleavage of the 3-MST persulfide was observed (Fig. S8, see the ESI†). The oxygen versions 4 and 5 did not show any fluorescent signal (Fig. 4B).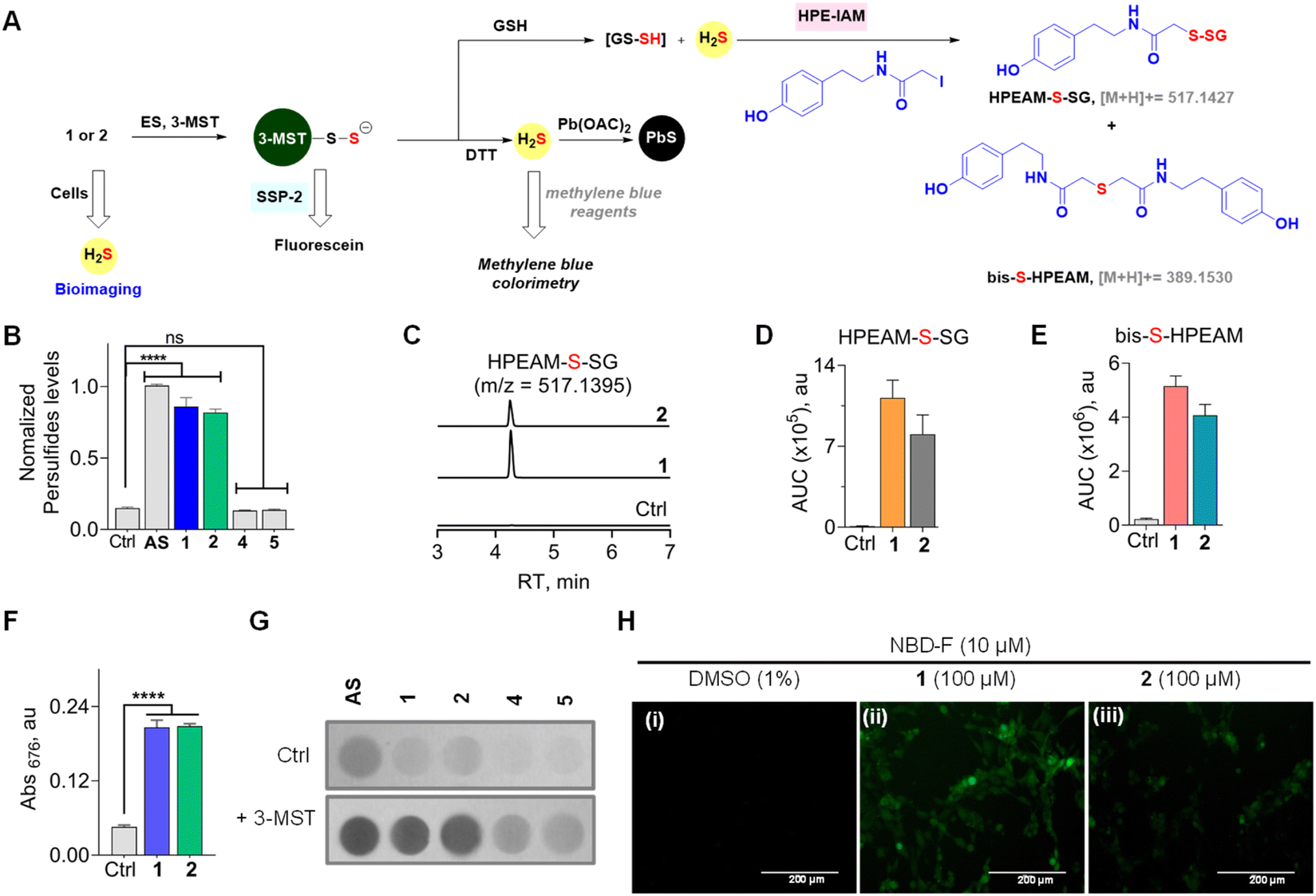 | ||
| Fig. 4 (A) Compounds 1 or 2 in the presence of ES and 3-MST should produce 3-MST persulfide, and when glutathione is added, the formation of glutathione persulfide (GS-SH) and H2S is expected. The intermediates of the above are independently detected by various assays. (B) An SSP-2-based fluorescence assay for the detection of persulfide: compounds (100 μM) were incubated with 3-MST (15 μM), ES (1 U mL−1), and SSP-2 (50 μM). Ctrl represents 3-MST only. λex = 482 nm and λem = 518 nm. (C) LC-MS/MS analyses for persulfide and H2S: extracted ion chromatogram of the HPEAM-S-SG adduct (expected m/z = 517.1427; observed m/z = 517.1395) formed upon incubation of 1 or 2 with 3-MST (25 μM) in the presence of ES (1 U mL−1), followed by sequential addition of the thiol acceptor (GSH; 400 μM) and electrophilic trapping reagent (HPE-IAM; 1 mM). The formation of HPEAM-S-SG (GSSH formation) and bis-S-HPEAM (H2S formation) was monitored, and these data are available in (D) and (E). Ctrl represents only 3-MST under standard conditions. (D) The area under the curve (AUC) for the peak corresponds to HPEAM-S-SG. (E) The area under the curve (AUC) for the peak corresponds to the bis-S-HPEAM adduct (expected m/z = 389.1530; observed m/z = 389.1516). (F) H2S detection using methylene blue assay: 1 or 2 (100 μM) was incubated with 3-MST (1 μM), ES (1 U mL−1), and DTT (10 mM). Absorbance at 676 nm is a measure of H2S generated. Incubation time is 120 min. See ESI Fig. S9† for time course and kinetics analysis. (G) H2S detection using lead acetate assay: 1 or 2 (100 μM) was incubated in the presence of 3-MST (1 μM), ES (1 U mL−1), and DTT (10 mM). Ctrl represents the absence of 3-MST. (H) Representative images of MEF cells. The H2S-sensitive fluorogenic probe NBD-fluorescein (10 μM) was cotreated with (i) DMSO (1%); (ii) 1, 100 μM; (iii) 2, 100 μM for 1 h, followed by imaging using a green fluorescent protein filter (GFP). The scale bar is 200 μm. All data are represented as mean ± SD (n = 3). Statistical analysis carried out in (B) and (F): Student's two-tailed unpaired parametric t-test was used to determine significance: ****p < 0.0001 and ns = non-significant with respect to 3-MST only. | ||
Since 3-MST has been reported to persulfidate low molecular weight thiols, which could contribute to antioxidant response, we next tested if GS-SH, the persulfide of glutathione (GSH), could be formed under standard reaction conditions.37,52–54 Compound 1 or 2 was treated with β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) (Scheme S5, see the ESI†); this electrophile reacts with H2S and persulfides to produce distinct adducts, which can be analysed by LC-MS/MS (Fig. 4A).55,56 The thioesters were independently treated with ES and 3-MST, and the reaction mixtures were incubated with GSH followed by HPE-IAM treatment. Independently, a control experiment for 3-MST alone was carried out with GSH, and no significant persulfide generation was observed. We found that both mercapto-nabumetone derivatives were capable of generating GS-SH (Fig. 4C and D). AS was also used independently for the generation of persulfides, and a distinct HPEAM-S-SG signal was observed (Fig. S9†). In addition, bis-S-HPEAM, which is consistent with the formation of H2S, was also detected (Fig. 4E and S9, see the ESI†). In the absence of 3-MST, the mercapto-nabumetone adducts were unable to generate GS-SH and H2S adducts, suggesting the importance of 3-MST catalysis.
To better understand the reaction of 1 in the presence of ES, GSH, and 3-MST, HPLC analysis of this reaction mixture was carried out (Scheme S3, see the ESI†). Under these conditions, the transformation of 1 or 2 to nabumetone was observed, albeit at a lower efficiency compared to DTT (Fig. S10, see the ESI†). Previous reports suggest that GSH is not a good sulfur acceptor to regenerate the 3-MST enzyme in its active state.35,46 This lack of efficiency could contribute to diminished yields of nabumetone under these conditions. In the cellular milieu, enzymes such as thioredoxin reductase predominantly convert 3-MST persulfides to 3-MST and H2S.46 DTT is a mimic of thioredoxin reductase, and in our hands, mercapto-nabumetone derivatives are transformed to the NSAID in excellent yield.
To further analyse the production of H2S under reducing conditions, we used two independent assays. First, a methylene blue assay, where the aforementioned adduct of H2S has a distinct absorbance at 676 nm, was used (Scheme S6, see the ESI†).29,46 Under the standard reaction conditions, both 1 and 2 were found to generate H2S (Fig. 4F). A time course of H2S generation was recorded and curve fitting gave observed rate constants of 0.68 h−1 and 0.62 h−1, for 1 and 2, respectively (Fig. S11, see the ESI†).
These values are comparable with the observed rate constants of conversion of thiols, T1 or T2, to nabumetone (0.57 h−1 and 0.6 h−1). Hence, there appear to be no other byproducts during the transformation of these thiols into the parent NSAID.
To further validate H2S generation from α-mercapto-nabumetone derivatives 1 and 2, a lead acetate assay was carried out.57 A lead acetate solution-soaked paper was used to trap lead sulfide (black coloration), which is formed by the reaction of lead acetate and H2S (Scheme S7, see the ESI†). Both derivatives 1 and 2 gave an intense black coloration in this assay (Fig. 4G). Under similar conditions, no significant signal for H2S was observed with 4 or 5 (Fig. S12, see the ESI†).
Cellular H2S generation and effect on cell viability
Having established that mercapto-nabumetone derivatives were able to generate persulfide, H2S, and the parent NSAID, we next tested if these derivatives were able to permeate cells to generate H2S. Mouse Embryonic Fibroblast (MEF) cells58 were co-treated with mercapto-nabumetone derivatives 1 or 2 as well as the H2S-sensitive fluorogenic NBD-fluorescein dye.59 The cells were imaged after 1 h of incubation using a green fluorescent protein (GFP) filter. A distinct fluorescent signal for 1 and 2 was observed, supporting the ability of these derivatives to enhance intracellular H2S in MEF cells (Fig. 4H). Derivative 2 has somewhat lower fluorescence intensity when compared with 1. This may be due to differences in permeability but could also be due to differences in the reactivity of the substrate with 3-MST in cellulo. The oxygen derivative 4 did not show any detectable fluorescence signal in this experiment (Fig. S13, see the ESI†).Making modifications to a clinical drug can result in adverse effects on pharmacological properties. To understand the impact of an additional sulfhydryl group on cell viability in MEF cells, 1 and 2 were independently tested and compared with nabumetone. We found that much like nabumetone, neither derivative had any adverse effects on cell viability (Fig. S14, see the ESI†).
Applicability of the strategy to other mercapto-NSAIDs
Previous strategies to produce NSAID and H2S have relied on making large-scale modifications to the NSAID. For example, the clinical agent ATB-346 has a thiobenzamide ring attached to the NSAID, which adds a number of carbons to the parent drug itself.19,60 The most atom-economical approach is to make the thioacid of the NSAID; such thioacids are hydrolyzed to generate H2S.20 The acidity of thioacids is significantly higher than that of carboxylic acids, and this may alter permeability and these compounds are likely to generate H2S outside as well as inside cells. Our approach is also similarly minimalistic, and the major modification is to add a thioacetate group and, in principle, a sulfhydryl group to generate H2S along with the NSAID. Since nabumetone is consumed orally, we compared the NSAID, T1 and T2 using SwissADME software (see the ESI†). We found that the physicochemical properties of the thiols are similar to those of nabumetone (Tables S2 and S3 and Fig. S15, see the ESI† for detailed physicochemical properties). Several NSAID derivatives have a free carboxylic acid,13–15 and we considered mercapto-6-MNA.61 6-MNA is a product of metabolism of nabumetone and has excellent anti-inflammatory activity. We next prepared a series of 6-MNA ester derivatives with a thioacetate at the α-position (Schemes 2 and S8, see the ESI†).62,63 The carboxylic acid esters are expected to undergo hydrolysis to produce 6-MNA.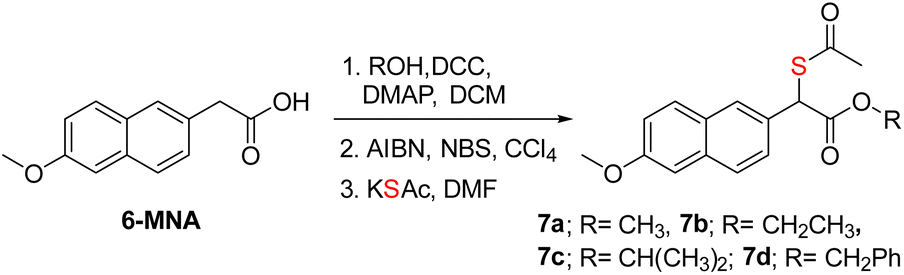 | ||
| Scheme 2 Synthesis of mercapto-6-MNA with different esters. Overall yields were 79–85%. | ||
Compounds in series 7 should be cleaved by ES to produce T3, which, in the presence of 3-MST, should generate persulfide (Fig. 5A). First, 7a was reacted with ES and 3-MST, and the formation of persulfides was analysed by SSP-2 assay. This assay when conducted in the absence of ES, and a significant increase in persulfides for 7a was observed (Fig. 5B). A comparable result was observed with lead acetate as well (Fig. S16, see the ESI†). Although 7a produces persulfides and H2S, the yield was significantly lower than that of 2. Similar results were recorded for 7b–7d with both SSP-2 and lead acetate assays (Fig. S17 and S18, see the ESI†). These data suggest that the methyl ester was a better persulfide generator when compared with carboxylic acids and other ester groups. To further understand the ester group effect on binding with 3-MST, molecular docking was carried out with thiols of 7a–7d (Table S1, see the ESI†). Molecular docking analysis supports favorable binding of thiol T3 (in the carboxylic acid form) with the active site of 3-MST, similar to 2 (Fig. 5C and Table S1, see the ESI†). Hence, the diminished rate might be due to reduced catalysis.
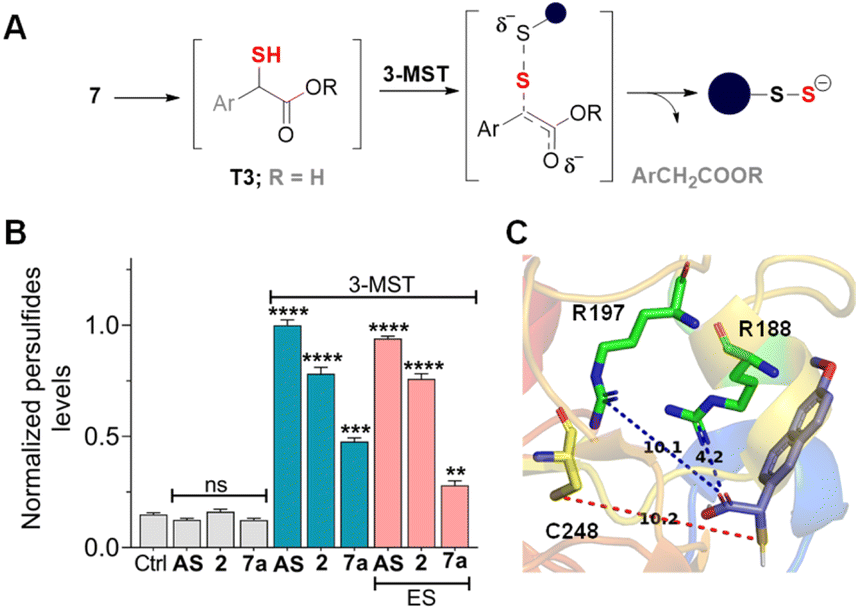 | ||
| Fig. 5 (A) Compounds 7a–7d are expected to produce the corresponding thiol (Ar = 6-methoxynaphthyl and R = various groups), which reacts with 3-MST to generate persulfide. When ES is added, the thiol T3 (Ar = 6-methoxynaphthyl) is the likely intermediate. (B) Sulfane sulfur detection in the presence of 3-MST (15 μM) and SSP-2 (50 μM). The assay was conducted both in the absence of ES and in the presence of ES. Persulfide generation is significantly higher in the absence of ES. Results are presented as mean ± SD (n = 3). Student's two-tailed unpaired parametric t-test was used to determine significance: ns = nonsignificant, ****p < 0.0001, ***p < 0.001, and **p < 0.01 with respect to 3-MST only. Ctrl represents 3-MST only. (C) Lowest energy conformations found through molecular docking studies with human 3-MST and T3. Thiols T1, T2, and T3 show comparable docking energy, i.e., −5.4 kcal mol−1, −5.5 kcal mol−1, and −5.3 kcal mol−1, respectively. The distance between the sulfur of T3 and the sulfur of active residue C248 is 10.2 Å. | ||
The key step of C–S bond cleavage depends on the formation of an enol(ate), and in the case of 1 or 2, the enolate of a ketone is produced, while in the case of 6-MNA derivatives, an enolate of an ester is the likely intermediate (Fig. 5A). The keto–enol equilibria of ketones and esters suggest that their equilibrium constants (K) are in favor of the keto form but are quite different. The values of K for ketones range from 10−7 to 10−8. Although the position of the keto–enol equilibrium for nabumetone has not been experimentally determined, the closest structure, acetone, has a K of 10−8.64 The K for acetophenone, the product of the 3-MST catalysed reaction of phenacylthiol, is 10−7, which supports a relatively stable enol.38 Previously, we showed that the presence of an electron withdrawing group on the aromatic ring of phenacylthiol induced moderate acceleration of H2S generation through 3-MST catalysis.38 This is presumably due to the additional stability of the intermediary enolate that is formed during C–S bond cleavage (Fig. 5A).
The keto–enol K for an ester is estimated to be 10−19.64–67 These values are reflective of a less stable enol in the case of an ester, and hence, the transition state leading to the formation of the ester enolate (Fig. 5A) is likely to have a higher barrier. Yet, 3-MST is able to catalyse, in some cases, the transformation of a mercapto-ester to a persulfide. This result underscores the versatility of this enzyme and provides insights into the binding and catalysis by 3-MST (Table S1, see the ESI†). However, when the ester is hydrolysed to the carboxylate of T3, the reaction does not proceed well. 3-MP is also expected to be in the carboxylate form, but 3-MST catalysis proceeds efficiently. We infer that, in the case of T3, the anionic form (carboxylate) reduces the stability of the enolate, leading to reduced catalysis rates. Hence, enolate stability or lack therefor drives the reaction outcome. Further studies on the structure–enzyme activity relationship are necessary to better understand these aspects of 3-MST catalysis. Nevertheless, this strategy can be applicable to other NSAIDs that have a ketone functional group68,69 and perhaps an amide.70,71 Lastly, the mercapto-NSAID can be functionalized with suitable protective groups that can help with site-specific activation by enzymes or ROS to facilitate release of the NSAID as well as H2S.
Conclusions
A new approach to modifying an NSAID to generate hydrogen sulfide has been developed. The introduction of a sulfhydryl group at the α-position of a ketone or an ester was found to be a viable strategy to generate H2S along with the parent carbonyl compound. The yield of the NSAID in the case of ketone was excellent (>90%), and the hybrids produced key cellular antioxidant response molecules, GS-SH and H2S. Since persulfides have superior antioxidant properties, these compounds are expected to have translational value. The α-mercapto-ester produced persulfide and H2S under these conditions but was less efficient than the ketone. Hence, enolate stability is a major contributor to sulfur transfer by 3-MST and will help with future design of artificial substrates for this enzyme. Taken together, we provide evidence for an atom economical approach to co-generating H2S and an NSAID, and further studies are underway to evaluate the therapeutic potential of these compounds.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All experiments and computational studies were carried out by SMG and PSM, under the supervision of HC. The manuscript was written with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial assistance for this project was provided by IISER Pune, Science and Engineering Research Board (HC, CRG/2023/003892). SMG acknowledges a fellowship from the University Grant Commission (UGC). The authors thank the laboratory of Dr Amrita B Hazra, IISER Pune, for their help with cloning and purification of 3-MST.Notes and references
- V. S. Khodade, S. C. Aggarwal, A. Eremiev, E. Bao, S. Porche and J. P. Toscano, Antioxid. Redox Signaling, 2022, 36, 309–326 CrossRef CAS PubMed.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- C. Munteanu, M. A. Turnea and M. Rotariu, Antioxidants, 2023, 12, 1737 CrossRef CAS PubMed.
- M. Whiteman, S. Le Trionnaire, M. Chopra, B. Fox and J. Whatmore, Clin. Sci., 2011, 121, 459–488 CrossRef CAS PubMed.
- Q. Hu and J. C. Lukesh, Antioxidants, 2023, 12, 650 CrossRef CAS PubMed.
- S. Xu, M. Shieh, B. D. Paul and M. Xian, Br. J. Pharmacol., 2023, 1–13 Search PubMed.
- Y. Zheng, X. Ji, K. Ji and B. Wang, Acta Pharm. Sin. B, 2015, 5, 367–377 CrossRef PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Antioxid. Redox Signaling, 2020, 32, 96–109 CrossRef CAS PubMed.
- E. Magli, E. Perissutti, V. Santagada, G. Caliendo, A. Corvino, G. Esposito, G. Esposito, F. Fiorino, M. Migliaccio, A. Scognamiglio, B. Severino, R. Sparaco and F. Frecentese, Biomolecules, 2021, 11, 1899 CrossRef CAS PubMed.
- E. Zaorska, L. Tomasova, D. Koszelewski, R. Ostaszewski and M. Ufnal, Biomolecules, 2020, 10, 323 CrossRef CAS PubMed.
- B. Yu, X. Yang, Z. Yuan and B. Wang, Curr. Opin. Chem. Biol., 2023, 75, 102329 CrossRef CAS PubMed.
- B. G. Katzung, Basic and Clinical Pharmacology, McGraw-Hill Education, 14th edn, 2018 Search PubMed.
- L. L. Brunton, R. Hilal-Dandan, B. C. Knollmann, Goodman & Gilman's: The Pharmacological Basis of Therapeutics, McGraw-Hill Education, 13th edn, 2018 Search PubMed.
- W. J. Phillips and B. L. Currier, J. Am. Acad. Orthop. Surg., 2004, 12, 221–233 CrossRef PubMed.
- J. L. Wallace, Can. J. Gastroenterol., 1996, 10, 451–459 CrossRef CAS PubMed.
- C. Sostres, C. J. Gargallo, M. T. Arroyo and A. Lanas, Best Pract. Res., Clin. Gastroenterol., 2010, 24, 121–132 CrossRef CAS PubMed.
- J. L. Wallace, World J. Gastroenterol., 2013, 19, 1861 CrossRef PubMed.
- J. L. Wallace, P. Nagy, T. D. Feener, T. Allain, T. Ditrói, D. J. Vaughan, M. N. Muscara, G. de Nucci and A. G. Buret, Br. J. Pharmacol., 2020, 177, 769–777 CrossRef CAS PubMed.
- W. Peng, L. Qin, T. Wang, Y. Sun, Z. Li, D. J. Lefer, C. Luo, F. Ye, B. Wang, W. Guo and Y. Zheng, J. Med. Chem., 2024, 67, 17350–17362 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef CAS PubMed.
- M. Chattopadhyay, R. Kodela, N. Nath, C. R. Street, C. A. Velázquez-Martínez, D. Boring and K. Kashfi, Biochem. Pharmacol., 2012, 83, 733–740 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef CAS PubMed.
- J. Kang, Z. Li, C. L. Organ, C. M. Park, C. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef CAS PubMed.
- Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef CAS PubMed.
- A. Sufian, N. Parihar, M. Badirujjaman, P. Barman, R. Kesarwani, D. B. Pemmaraju and K. P. Bhabak, J. Mater. Chem. B, 2024, 12, 4248–4261 RSC.
- X. Ni, X. Li, T. L. Shen, W. J. Qian and M. Xian, J. Am. Chem. Soc., 2021, 143, 13325–13332 CrossRef CAS PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef CAS PubMed.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef CAS PubMed.
- Y. Zhao, H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2017, 139, 16365–16376 CrossRef CAS PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, J. Am. Chem. Soc., 2019, 141, 13610–13618 CrossRef CAS PubMed.
- Y. Kimura, S. Koike, N. Shibuya, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2017, 7, 10459 CrossRef PubMed.
- B. Pedre, D. Talwar, U. Barayeu, D. Schilling, M. Luzarowski, M. Sokolowski, S. Glatt and T. P. Dick, Nat. Chem. Biol., 2023, 19, 507–517 CrossRef CAS PubMed.
- N. Nagahara, Br. J. Pharmacol., 2018, 175, 577–589 CrossRef CAS PubMed.
- Y. Kimura, S. Koike, N. Shibuya, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2017, 7, 10459 CrossRef PubMed.
- N. Nagahara and T. Nishino, J. Biol. Chem., 1996, 271, 27395–27401 CrossRef CAS PubMed.
- M. Libiad, P. K. Yadav, V. Vitvitsky, M. Martinov and R. Banerjee, J. Biol. Chem., 2014, 289, 30901–30910 CrossRef CAS PubMed.
- P. Bora, S. Manna, M. A. Nair, R. R. M. Sathe, S. Singh, V. S. Sreyas Adury, K. Gupta, A. Mukherjee, D. K. Saini, S. S. Kamat, A. B. Hazra and H. Chakrapani, Chem. Sci., 2021, 12, 12939–12949 RSC.
- S. Manna, R. Agrawal, T. Yadav, T. A. Kumar, P. Kumari, A. Dalai, S. Kanade, N. Balasubramanian, A. Singh and H. Chakrapani, Angew. Chem., Int. Ed., 2024, 63, e202411133 CrossRef CAS PubMed.
- Drug Approval Package: Nabumetone NDA #75-179.
- T. Hedner, O. Samulesson, P. Währborg, H. Wadenvik, K. A. Ung and A. Ekbom, Drugs, 2004, 64, 2315–2343 CrossRef CAS PubMed.
- B. J. Lister, M. Poland and R. E. DeLapp, Am. J. Med., 1993, 95, S2–S9 CrossRef PubMed.
- R. D. Brobyn, Am. J. Med., 1987, 83, 50–54 CrossRef CAS PubMed.
- D. S. Tarbell, Organic Sulfur Compounds, Elsevier, 1961, pp. 97–102 Search PubMed.
- Carboxylesterases are known to cleave thioesters: D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Acta Pharm. Sin. B, 2018, 8, 699–712 CrossRef PubMed.
- P. K. Yadav, K. Yamada, T. Chiku, M. Koutmos and R. Banerjee, J. Biol. Chem., 2013, 288, 20002–20013 CrossRef CAS PubMed.
- G. T. Huang and J. S. K. Yu, J. Phys. Chem. B, 2016, 120, 4608–4615 CrossRef CAS PubMed.
- J. C. Lec, S. Boutserin, H. Mazon, G. Mulliert, S. Boschi-Muller and F. Talfournier, ACS Catal., 2018, 8, 2049–2059 CrossRef CAS.
- Y. Zai, Y. Feng, X. Zeng, X. Tang, Y. Sun and L. Lin, RSC Adv., 2019, 9, 10091–10093 RSC.
- M. Nobilis, C. Chamseddin, T. Jira and R. Ku, J. Pharm. Biomed. Anal., 2013, 80, 164–172 CrossRef CAS PubMed.
- W. Chen, C. Liu, B. Peng, Y. Zhao, A. Pacheco and M. Xian, Chem. Sci., 2013, 4, 2892 RSC.
- K. Aquilano, S. Baldelli and M. R. Ciriolo, Front. Pharmacol, 2014, 5, 196 Search PubMed.
- J. V. Lana, A. Rios, R. Takeyama, N. Santos, L. Pires, G. S. Santos, I. J. Rodrigues, M. Jeyaraman, J. Purita and J. F. Lana, Nutrients, 2024, 16, 2476 CrossRef CAS PubMed.
- U. Barayeu, T. Sawa, M. Nishida, F. Wei, H. Motohashi and T. Akaike, Br. J. Pharmacol., 2023, 1–14, DOI:10.1111/bph.16246.
- H. A. Hamid, A. Tanaka, T. Ida, A. Nishimura, T. Matsunaga, S. Fujii, M. Morita, T. Sawa, J. M. Fukuto, P. Nagy, R. Tsutsumi, H. Motohashi, H. Ihara and T. Akaike, Redox Biol., 2019, 21, 101096 CrossRef PubMed.
- T. Takata, M. Jung, T. Matsunaga, T. Ida, M. Morita, H. Motohashi, X. Shen, C. G. Kevil, J. M. Fukuto and T. Akaike, Nitric Oxide, 2021, 116, 47–64 CrossRef CAS PubMed.
- R. A. M. Williams, S. M. Kelly, J. C. Mottram and G. H. Coombs, J. Biol. Chem., 2003, 278, 1480–1486 CrossRef CAS PubMed.
- M. Tomita, N. Nagahara and T. Ito, Molecules, 2016, 21, 1707 CrossRef PubMed.
- C. Wei, Q. Zhu, W. Liu, W. Chen, Z. Xi and L. Yi, Org. Biomol. Chem., 2014, 12, 479–485 RSC.
- B. Gemici, W. Elsheikh, K. B. Feitosa, S. K. P. Costa, M. N. Muscara and J. L. Wallace, Nitric Oxide, 2015, 46, 25–31 CrossRef CAS PubMed.
- K. Matsumoto, T. Hasegawa, K. Ohara, C. Takei, T. Kamei, J. Koyanagi, T. Takahashi and M. Akimoto, Xenobiotica, 2020, 50, 783–792 CrossRef CAS PubMed.
- A. Fortenberry, S. A. Mohammad, T. A. Werfel and A. E. Smith, Macromol. Rapid Commun., 2022, 43 Search PubMed.
- C. M. D. Swarbrick, J. D. Nanson, E. I. Patterson and J. K. Forwood, Prog. Lipid Res., 2020, 79, 101036 CrossRef CAS PubMed.
- J. R. Keefe, A. J. Kresge and N. P. Schepp, J. Am. Chem. Soc., 1990, 112, 4862 CrossRef.
- E. A. Jefferson, J. R. Keefe and A. J. Kresge, J. Chem. Soc., Perkin Trans. 2, 1995, 2041 RSC.
- R. P. Bell and P. W. Smith, J. Chem. Soc. B, 1966, 241–243 RSC.
- J. P. Richard, G. Williams, A. M. C. O'Donoghue and T. L. Amyes, J. Am. Chem. Soc., 2002, 124, 2957 CrossRef CAS PubMed.
- R. G. Child, A. C. Osterberg, A. E. Sloboda and A. S. Tomcufcik, J. Pharm. Sci., 1977, 66, 466–476 CrossRef CAS PubMed.
- R. N. Brogden, R. C. Speight and G. S. Avery, Drugs, 1981, 21, 1–22 CrossRef CAS PubMed.
- D. A. Gamache, G. Graff, M. T. Brady, J. M. Spellman and J. M. Yanni, Inflammation, 2000, 24, 357–370 CrossRef CAS PubMed.
- A. Bezatis, I. Georgou, J. Dedes, P. Theodossiadis and I. Chatziralli, Clin. Exp. Optom., 2022, 105, 263–267 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08525f |
| This journal is © The Royal Society of Chemistry 2025 |