
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-stage photodegradation of indomethacin molecular nanocomposites under extreme confinement†
Cindy Yueli
Chen
 a,
Haonan
Wang
ab,
Ahmad
Arabi Shamsabadi
a and
Zahra
Fakhraai
*a
a,
Haonan
Wang
ab,
Ahmad
Arabi Shamsabadi
a and
Zahra
Fakhraai
*a
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: fakhraai@sas.upenn.edu
bCorning Research Center China, 201206 Shanghai, China
First published on 7th January 2025
Abstract
The incorporation of a glassy material into a self-assembled nanoparticle (NP) film can produce highly loaded nanocomposites. Reduction of the NP diameter can lead to extreme nanoconfinement of the glass, significantly affecting the thermal and physical properties of the nanocomposite material. Here, we investigate the photostability and photodegradation mechanisms of molecular nanocomposite films (MNCFs) produced from the infiltration of indomethacin (IMC) molecules into self-assembled films of silica NPs (11–100 nm in diameter). Upon UV irradiation in ambient conditions, IMC degrades in a two-stage process. We demonstrate that nanoconfinement only enhances the photostability of IMC during stage 1, which primarily involves decarboxylation and oxidation. These reactions are kinetically limited by the diffusion of CO2 and O2 and are thus affected by the increased glass transition temperature, Tg, and viscosity under confinement. In contrast, during prolonged UV exposure in ambient conditions, stage 2 of IMC degradation, which involves further reactions with water, is unaffected by confinement. This is attributed to the availability of locally adsorbed water in the nanocomposite under ambient conditions, which does not rely on transport through the confined matrix. Overall, unlike previous reports in inert environments, IMC photodegradation in ambient conditions cannot be improved by confinement. These findings highlight the significance of specific degradation pathways in determining whether a material can be stabilized through extreme nanoconfinement.
Introduction
There has been significant research interest in developing strategies to modulate the physicochemical properties of materials under confinement, which are in high demand for various applications—from medicine to energy to electronics.1–6 In principle, the introduction of nanomaterials into host materials is an effective way to induce nanoconfinement, and thus change their properties; these changes arise from the unique mechanical,7 barrier,8 electrical,9 and optical properties10 of the nanomaterials and their interfaces. Generally, modifying nanofillers (i.e., the materials introduced into hosts) in order to achieve a desired property in hybrid materials can be complex, especially at high concentrations, because it is difficult to independently control the various factors involved. The effect on the properties varies with the types of nanofillers and host materials, the dispersion of the nanofiller, the interfacial contact between nanofiller and host material, the method of preparation, and any potential catalytic effects generated by the nanofillers, among others.11–14The addition of nanofillers to improve material stability via confinement has been extensively studied. For example, it has been demonstrated that a moderate degree of loading (≤18 wt%) can reduce thermal degradation rates in the manufacturing processes of polymer/nanoparticle (NP) composites.12,13 The improved thermal stability, in this case, can be attributed to the barrier properties of the nanofillers, which have been reported to impede the movement and transportation of both polymers and their degradation products.12,15–17 On the other hand, at elevated concentrations, it is observed that nanofillers have a tendency to agglomerate, resulting in faster thermal degradation.13 Furthermore, the combination of NPs and a polymer matrix has been found to promote photocatalytic activity, thus decreasing photostability.18–21 Various natural biochemical systems also provide examples of nanofillers improving stability; these systems have catalytically active centers that are located in well-defined spaces, and they are significantly affected by nanoconfinement.22,23 In these systems, a specific reaction can be selectively accelerated by the confinement, producing high conversion yields even with moderate reaction conditions. Additionally, nanoporous materials, such as zeolites,24 metal–organic frameworks (MOFs),22 and covalent-organic frameworks (COFs),25,26 have ultra high surface areas and well-defined porous structures; these can play a crucial role in enhancing the efficiency of various catalytic reactions, facilitating chemical bond formation and breakage,23,25 modifying product selectivity,26 and optimizing processes related to mass transport,22 adsorption,24 and desorption equilibria.26 Given the diverse conditions of these systems and their dependence on specific chemical properties of the nanofillers, it is often difficult to distinguish the roles of thermodynamic and kinetic barriers in chemical reactions of confined systems. Control experiments can help investigate the interplay of kinetic factors and the local catalytic activity in order to predict how reactions would proceed with confinement.
Capillary rise infiltration (CaRI) is a facile and scalable method to produce highly loaded polymer nanocomposites.27 This technique involves heating a bilayer comprising glassy material (polymer) and a NP layer to above the glass transition temperature Tg. Capillary forces drive the glassy material into the interstices between the NPs, placing the polymer under nanoconfinement conditions that are more restrictive than in the aforementioned systems with added nanofillers. The extreme nanoconfinement can further stabilize the resulting nanocomposites. Polymer-infiltrated NP films (PINFs) produced using this method exhibit slower segmental dynamics as well as considerably higher Tg values than composites produced via other methods. This is because in these films, the nanoparticles are physically attached, and the polymer expansion does not change the average pore size; as such, the configurational entropy of the polymer or small molecules is significantly reduced.27–30 The Tg increase has been shown to originate primarily from limitations in segmental mobility under confinement, and it is thus primarily entropic in origin.29,30 We have recently shown that in polystyrene (PS)/SiO2 PINFs, the slow diffusion of oxygen and reaction products upon increased confinement significantly increases the barrier for PS degradation (by ∼50 kJ mol−1 in PINFs with 11-nm silica NPs).31 Furthermore, the slow rate of degradation allows heat dissipation, preventing the autocatalytic burning of PS, producing burn-resistant PS composites. Nanocomposites made using CaRI have been reported to exhibit exceptional properties, such as improved mechanical32,33 and optical properties.27
Another significant advantage of using CaRI to produce highly loaded nanocomposite systems is that it can be extended to produce a wide array of composites using molecular glasses, as opposed to polymers as the nanofiller.28,34 Similar to PINFs, molecular nanocomposite films (MNCFs) show higher Tg and slower relaxation dynamics upon confinement. In MNCFs made of SiO2 NP films infiltrated with N,N′-bis(3-methylphenyl)-N,N′-diphenylbenzidine (TPD), a Tg increase of ∼30 K is observed using 11-nm silica NPs, despite the weak molecule–NP interactions; this increase is similar in magnitude to the Tg increases in PINFs. Raman spectroscopy experiments indicate that the increased Tg originates from the reduced intra-molecular conformational degrees of freedom in TPD, imposed by the unusual geometry of the rigid self-assembled NP films,28 and is thus, of entropic origin. The properties of MNCFs can be tuned to a greater extent by controlling the interfacial interactions (Gibbs–Thomson effect) between the NPs and molecules.35,36 We have demonstrated that in indomethacin (IMC)/SiO2 MNCFs under inert environments, the Tg is also increased to a similar extent as in TPD (∼30 K in 11-nm silica NPs). The favorable interactions of IMC with silica, in addition to entropic effects, result in dramatically enhanced thermal stability (by up to ∼70 kJ mol−1) compared to unconfined films.34 In inert conditions, both the thermal stability and photostability of IMC depend on the kinetic transport of the reaction (degradation) products in the MNCF matrix, which is suppressed by the nanoconfinement.
The photostability of nanocomposites has not been widely studied. A few reports have shown enhanced photostability in nanocomposites that can inhibit the formation of photogenerated species.37,38 The improved photostability is only observed when a relatively low-energy light source is used (>300 nm).39,40 Since MNFCs have emerged as a unique platform to stabilize material properties, it is important to investigate the role of molecule–NP interactions and specific reaction pathways in these improvements. A fundamental understanding of key design rules for stability would allow for an informed design of stable molecular-glass nanocomposites for various applications, such as barrier coatings or multi-functional coatings for electronic augmented reality (AR) and virtual reality (VR) wearable devices,41,42 where photostability is imperative.
In this work, we employ in situ spectroscopic ellipsometry (SE) and ultra-performance liquid chromatography/mass spectrometry (UPLC/MS) to investigate the photostability of IMC/SiO2 MNCFs in ambient conditions, and analyze the reaction products. SE is a robust nondestructive technique used to measure optical properties,43 film thickness,44,45 surface coverage or homogeneity,31,34 material porosity,46,47 dielectric constants,48–50 and conductivity of materials50,51 in the visible and near-infrared spectral ranges. Degradation reactions due to heat, UV exposure, or environmental conditions can be probed by in situ SE through changes in the effective index of refraction28,34,51 or other properties, such as conductivity.50 IMC is chosen as the model system because of its multi-stage degradation. Upon UV irradiation in ambient conditions, the stage 1 degradation of IMC primarily involves decarboxylation and oxidation.34,52–54 Our results show that the photodegradation rate during this stage depends on the degree of confinement, and it slows down as the NP diameter is decreased. This is attributed to the slower molecular relaxation dynamics, and slower transport of CO2 and O2 products in confinement, due to increased Tg. We find that the stage 2 degradation, which also includes secondary reactions with H2O, is unaffected by the degree of confinement. This is attributed to the availability of locally adsorbed water in the porous silica network in ambient conditions, which does not depend on the kinetic transport of reactants or products. These findings are corroborated by UPLC/MS measurements. The results highlight the importance of the design of degradation mechanisms in achieving an optimum level of stability in MNCF systems.
Materials and methods
Preparation of wafers and reagents
One-side-polished silicon wafers (Virginia Semiconductor 〈100〉) were cut to 1.5 cm × 1.5 cm squares. Solutions of indomethacin (IMC) (MilliporeSigma, MW = 357.79 kg mol−1) in tetrahydrofuran (THF) were prepared at ∼0.02 wt% and filtered using 0.2-μm polytetrafluoroethylene (PTFE) syringe filters prior to film deposition. Suspensions of SiO2 NPs with various diameters, SiO2(11 nm) and SiO2(100 nm) from Nissan Chemical (30–31 wt% suspension in isopropanol (IPA)) and SiO2(25 nm) from Sigma-Aldrich (Ludox TM-50, 50 wt% suspension in water), were diluted with their respective solvents. Diluted NP suspensions were filtered using 0.45-μm PTFE syringe filters before use.Preparation of IMC and IMC/SiO2 nanocomposite films (MNCFs)
A pure IMC film (∼75 nm) was prepared by spin-coating (Laurell, WS-400BZ-6NPP/Lite spin-coater) the IMC solution onto a silicon wafer at ∼4000 rpm. The sample was then annealed using a vacuum oven at 348 K for ∼30 min to remove residual solvent. For the IMC/SiO2 nanocomposite films (the MNCFs), a layer with a thickness of either ∼200 nm or ∼400 nm (where specifically mentioned in the manuscript) of self-assembled SiO2 NPs (diameter, D = 11, 25, or 100 nm) was first spin-coated onto a silicon wafer. The NP layer was then sintered at 773 K for 30 min on a Linkam temperature controller (THMS600) to remove the residual solvent and stabilize the NP film. Once the NP layer was cooled to room temperature, a ∼75 nm (or ∼150 nm for 400-nm MNCFs) IMC layer was added by spin-coating an IMC solution over the NP layer, generating a bilayer film. The bilayer was then heated at 373–393 K using a vacuum oven to remove residual solvent and to allow IMC to further infiltrate into the pores of the NP layer. The resulting bilayer consists of a residual IMC layer (1–5 nm) on top of a fully infiltrated IMC/SiO2 nanocomposite layer, as shown in Table S1 of the ESI.†In situ spectroscopic ellipsometry (SE) measurements of photodegradation in ambient conditions
To perform SE measurements, a pure IMC film or an IMC/SiO2 MNCF was placed on the SE stage and illuminated by a UV lamp (UVP 95-0007-05 Model UVGL-58, 115 V, 6 W) at normal incidence at a distance of ∼30 mm from the film's surface, under ambient conditions (humidity 40–50%, RT = 298 ± 1 K). In situ SE measurements were performed at a sampling rate of 10 s with high-accuracy zone averaging at an incidence angle of 70°. The raw SE data, amplitude Ψ(λ) and phase difference Δ(λ) (λ: wavelength), were fitted to a model containing the optical properties of the silicon substrate, a 1-nm native oxide layer and a ∼200-nm transparent Cauchy layer to account for the MNCF properties (n(λ) = A + B/λ2, κ(λ) = 0). Here, n and κ are the real and imaginary parts of the index of refraction, and A and B are fitting parameters along with the film thickness, h.After the initial fitting, the thickness of the nanocomposite layer was held constant, as the sintered NP layers are rigid, and their thicknesses are not expected to change during these experiments. The change in the index of refraction of the nanocomposite layer was then monitored in situ as a function of UV irradiation time. For MNCFs with a top residual IMC layer, both one-layer and two-layer models were employed and compared. In the two-layer model, the thickness of the top IMC layer was fitted as an additional variable, with its refractive index held constant. In this report, the one-layer model was utilized because including the thin IMC layer (∼1–5 nm) did not impact the fitting quality and resulting refractive indices. More fitting details can be found in the ESI† (Table S1 and Fig. S1–S3) and our previous publications.31,34 The data were fitted within the wavelength range of 600 nm ≤ λ ≤ 1600 nm, where IMC is transparent (ESI,† Fig. S7b).
As degradation proceeded, graded optical properties were assumed for the nanocomposite layer to account for the inhomogeneity of the index of refraction at various depths from the substrate. The data were fit either to a linearly or an exponentially graded Cauchy model, which generally produced lower mean-square error (MSE) values than homogeneous models (more details can be found in the ESI,† Fig. S4, and our previous reports31,34). Linearly graded models were used to measure the rate of IMC mass loss during UV irradiation. The relative IMC mass loss (m/m0) in the nanocomposite layer was calculated based on measurements of the index of refraction of a porous NP film (nNP), the index of refraction of a fully infiltrated nanocomposite layer before UV irradiation (nnanocomposite), and the measured average index of refraction of the linearly graded nanocomposite layer (〈n〉) as:
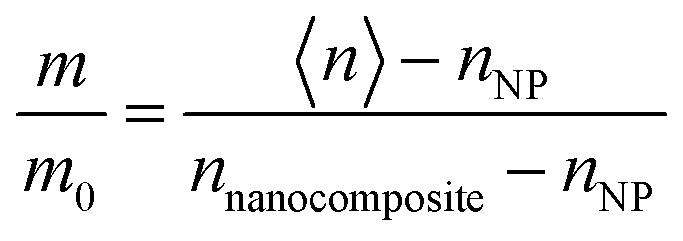 | (1) |
Exponentially graded models were utilized to gain a more thorough understanding of the degradation details within the films at various degradation times, as they generally provide better fits to the data compared to the linearly graded models but are more time-consuming to calculate. To perform the fitting for the exponentially graded model, the thick composite layer (h ∼ 200 nm) was divided vertically into 100 sublayers, each having its unique refractive index, while imposing physical boundary conditions through an exponential equation, as detailed in our previous reports.31,34 The refractive index of each slice was calculated at various time points during degradation, resulting in index profile curves consisting of 100 data points along the depth of the film at each given time point. As the degradation proceeds and the index of refraction of the film is decreased everywhere, the profile line appears to move downwards uniformly. When the surface layer degrades more rapidly than the middle of the film, a lower refractive index near the surface is expected, showing a curved index profile, similar to previous reports.31,34
Ultra-performance liquid chromatography/mass spectrometry (UPLC/MS) analysis of the degradation products
UPLC/MS analysis was performed on a Waters Acquity UPLC system equipped with a Waters TUV detector and a Waters SQD single-quadrupole mass analyzer with electrospray ionization. A degraded IMC or IMC/SiO2 film on Si substrate was placed into a vial to which 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/acetonitrile were added to a concentration of ∼0.8 mM. The solution was filtered using a 0.2-μm PTFE syringe filter. 6 μL of the solution was injected into a reverse-phase chromatography column (Acquity UPLC HSS C18, 1.7 μm, 2.1 × 50 mm). The mobile phase used for gradient elution consisted of (A) water and (B) acetonitrile with 0.1% v/v formic acid. The gradient was operated from 40% B for 1.3 min, followed by a 5-min gradient to 5% A/95% B, for 1 min. The flow rate was 0.75 mL min−1. Details (e.g., mass-to-charge ratio (m/z)) for the determination of IMC and the main photodegraded IMC products can be found in the ESI† (Fig. S9, S10 and Table S2). Quantitative analysis of the IMC and photodegraded byproducts was performed using MestReNova Software. The relative values of the area under each base peak (%) were obtained from UPLC–MS chromatograms and used to determine the relative fractions of IMC and the seven observed photodegradation products, P1–P7.
:1 water/acetonitrile were added to a concentration of ∼0.8 mM. The solution was filtered using a 0.2-μm PTFE syringe filter. 6 μL of the solution was injected into a reverse-phase chromatography column (Acquity UPLC HSS C18, 1.7 μm, 2.1 × 50 mm). The mobile phase used for gradient elution consisted of (A) water and (B) acetonitrile with 0.1% v/v formic acid. The gradient was operated from 40% B for 1.3 min, followed by a 5-min gradient to 5% A/95% B, for 1 min. The flow rate was 0.75 mL min−1. Details (e.g., mass-to-charge ratio (m/z)) for the determination of IMC and the main photodegraded IMC products can be found in the ESI† (Fig. S9, S10 and Table S2). Quantitative analysis of the IMC and photodegraded byproducts was performed using MestReNova Software. The relative values of the area under each base peak (%) were obtained from UPLC–MS chromatograms and used to determine the relative fractions of IMC and the seven observed photodegradation products, P1–P7.
Scanning electron microscopy (SEM) characterization of photodegraded nanocomposite films
A FEI-600 Quanta ESEM microscope was used to observe the morphology of photodegraded composite films. A sputter-coater (Q150T S/E/ES, Quorum Technologies) was used to apply a 4-nm iridium layer to each sample prior to imaging. SEM images were captured at an accelerating voltage of 30 kV, a spot size of 3.0, and a working distance of 10 mm.Results and discussion
Photodegradation of IMC films
Upon UV irradiation in ambient conditions, IMC is prone to decarboxylation and oxidation, resulting in the production of CO2, which can evaporate away, and H2O, which can either evaporate or participate in additional degradation reactions.53,55,56 Based on the byproducts detected by ULPC/MS, three major degradation pathways are involved in the photodegradation of IMC under UV irradiation at 254 and 365 nm (Fig. 1). In this first stage of photodegradation (stage 1), these pathways result in decarboxylation (I), oxidation (II), or hydrolysis (III). During pathway I, IMC undergoes a mesolytic cleavage at C–COOH, which is a bond-breaking reaction yielding a neutral radical,55 followed by a hydrogen shift. This reaction produces P1 (Fig. 1) as well as other intermediates, shown as P4 and P5 in the ESI† (Fig. S9 and S10, detailed in Table S2).52,53,57 In pathway II, P1 is formed as in pathway I and then further oxidized to produce an aldehyde (P2) and alcohol (P3). In pathway III, P1 is formed as in other pathways, but it then reacts with water to produce P3.54,55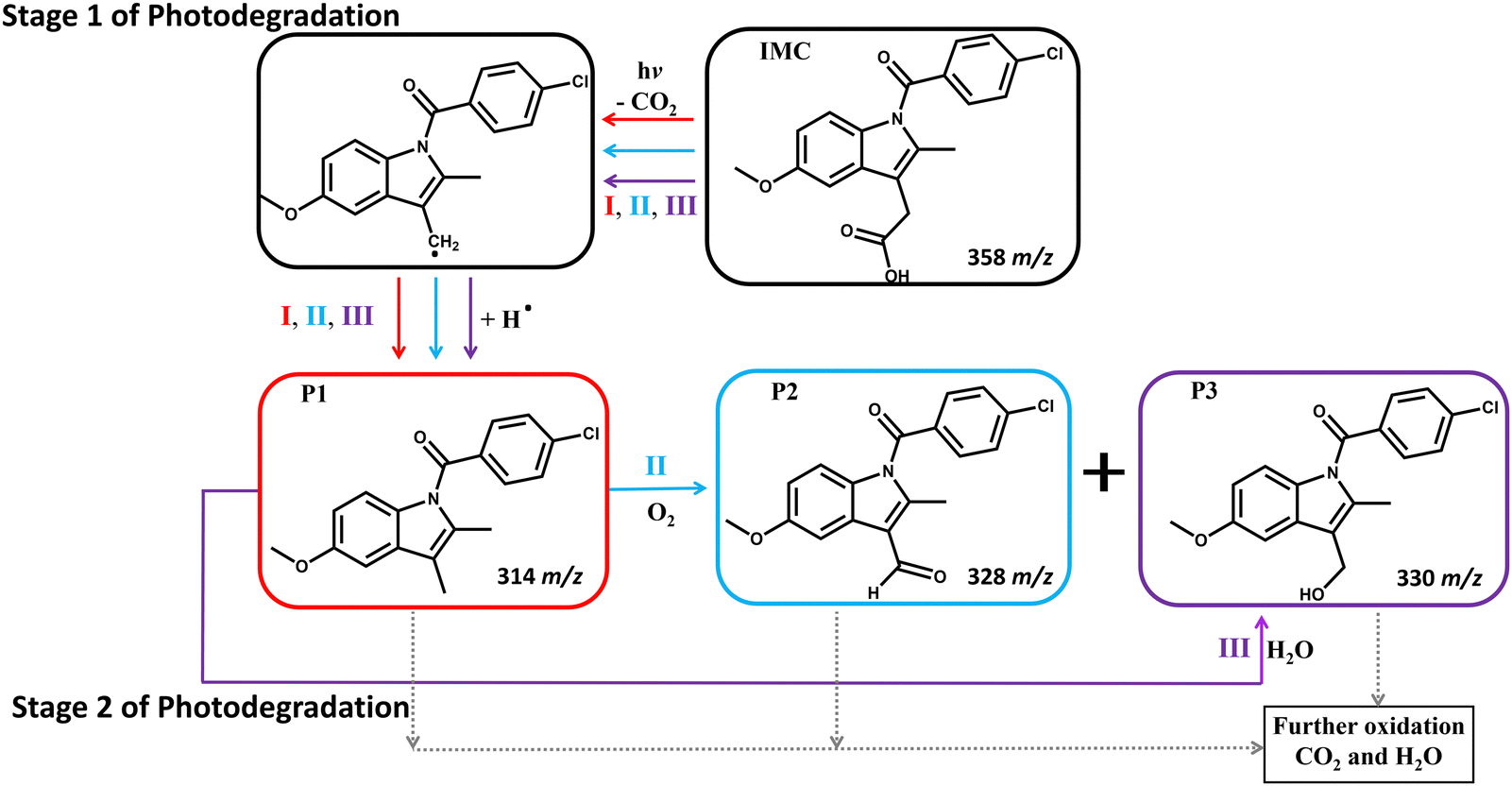 | ||
| Fig. 1 Proposed reaction pathways for the main products of the first stage of IMC photodegradation at either 254- or 365-nm UV exposure under ambient conditions. Pathway I involves decarboxylation to generate P1. Pathway II begins similarly but ends with the decarboxylated compound further oxidizing to P2 and P3. Pathway III proceeds similarly to P1, which then reacts with water to yield P3. Note that P3 can be generated via pathway II or III. The provided m/z values were determined by MS measurements. | ||
As seen in Fig. 2 and Fig. S5 (ESI†), the photodegradation of IMC shows a two-stage decay pattern. Since pathway I (decarboxylation) is needed for the subsequent pathways to occur, stage 1 primarily involves pathway I, with smaller contributions from oxidation and H2O generation (see Fig. 6 and related discussions for more details). Since CO2 and water can evaporate, the IMC degradation during stage 1 leads to a reduction in IMC mass and the reduction of the film thickness in pure IMC films.
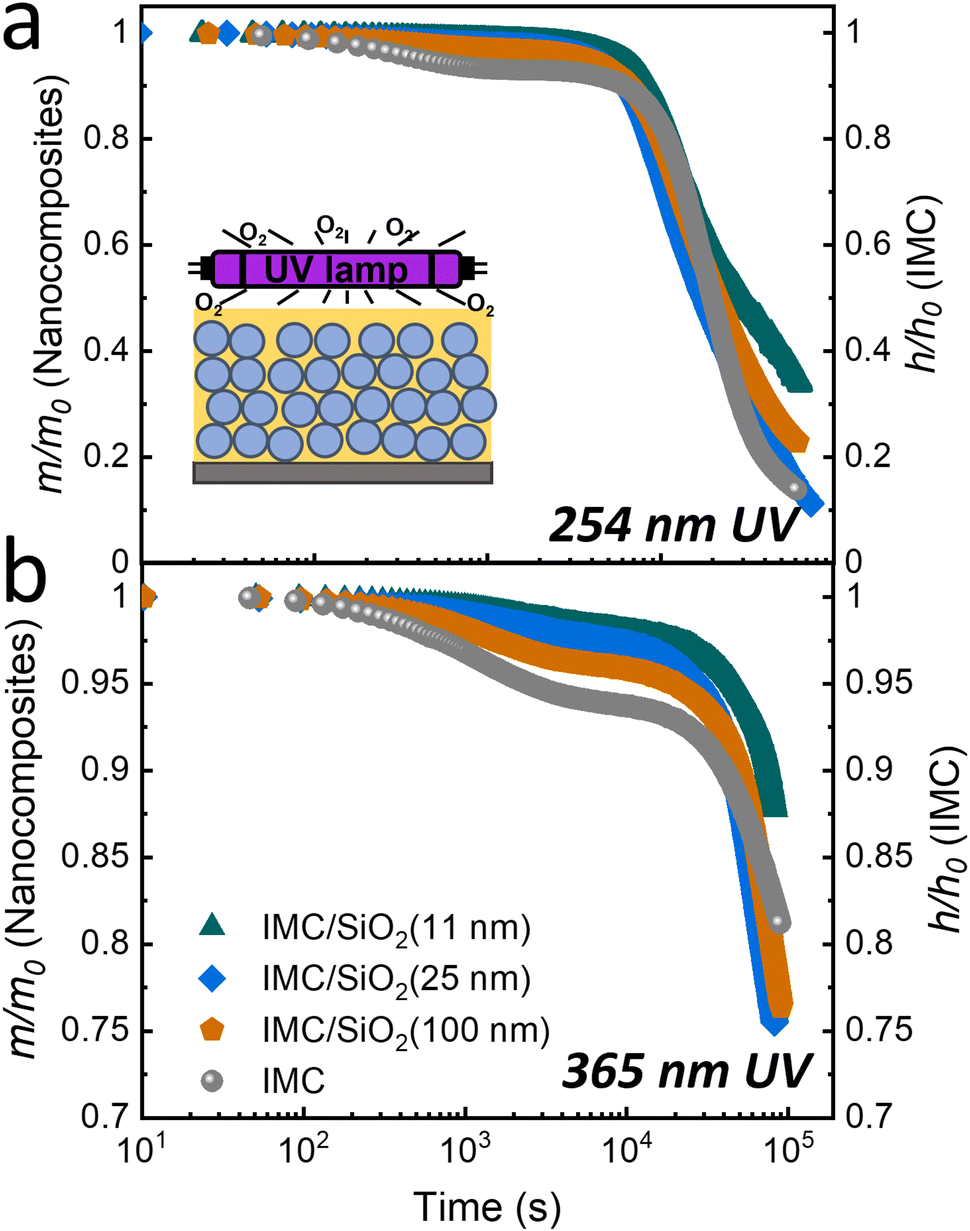 | ||
| Fig. 2 Photodegradation under ambient conditions due to (a) 254- and (b) 365-nm UV irradiation. Degradation of a pure IMC film (∼75 nm) was measured through changes in relative film thickness with time (gray circles, right axes); and that of IMC/SiO2 MNCFs (∼200 nm) was measured through changes in the relative IMC mass in the nanocomposite (left axes). The IMC/SiO2 MNCFs were prepared from IMC and various diameters of SiO2 NPs: IMC/SiO2(11 nm) (green triangles), IMC/SiO2(25 nm) (blue diamonds), and IMC/SiO2(100 nm) (orange pentagons). The average refractive index of the film, fitted by linearly graded models (not shown), was used for the mass-loss calculations. The inset schematically shows the set-up of the nanocomposite film and UV lamp. Two-stage photodegradation is observed both for IMC and the IMC/SiO2 MNCF after prolonged UV irradiation under ambient conditions. | ||
Prolonged UV irradiation at either 254 or 365 nm results in a second stage of photodegradation (stage 2), where additional byproducts with smaller molecular weights such as P6 and P7 are also observed (ESI,† Fig. S9, S10 and Table S2), further reducing the thicknesses of pure IMC films or refractive indices of nanocomposite layers. Eventually, with continuous irradiation, IMC and its byproducts completely degrade/evaporate to CO2 and H2O, consistent with previous observations.53,56 Photodegraded IMC products that arise from the C(O)–N bond cleavage via O2 attack were expected, based on previous reports,52,53 but such species were not detected by UPLC/MS. Future experiments, such as those using total correlation spectroscopy, may be able to identify products with smaller yields;58,59 however, this is beyond the scope of the current study.
Role of confinement in determining photodegradation rate
In confinement, the IMC Tg is increased, slowing down the relaxation dynamics in the glass matrix.34 As such, the viscosity is expected to increase, and the diffusion rate for gases, such as O2 and CO2, are expected to decrease. We have previously shown that under inert conditions, where only one degradation pathway is expected (one similar to pathway I of Fig. 1, ending in decarboxylation), the photodegradation rate can be significantly reduced by confinement.34 In contrast, under ambient conditions, multiple reaction pathways can compete during UV irradiation, some of which do not depend so much on gas diffusion (for example, pathway III in Fig. 1).To explore the role of nanoconfinement in determining the overall photodegradation rate under ambient conditions, the degree of confinement in IMC MNCFs was varied by using SiO2 NPs of different diameters, ranging from 11 to 100 nm; this produced average pore sizes from 3 to 33 nm.34,46,47,60 We have previously reported that the Tg increases from Tg(IMC) = 315 ± 2 K to Tg(IMC/SiO2(100 nm)) = 324 ± 3 K, Tg(IMC/SiO2(25 nm)) = 339 ± 3 K, and Tg(IMC/SiO2(11 nm)) = 344 ± 3 K in 100 nm, 25 nm, and 11 nm SiO2 NP films, respectively.34 This increase in Tg is also expected to increase the viscosity by at least 1.5 decades, similar to unentangled polymers.30 The degree of photodegradation and the resulting relative mass loss (m/m0) in ∼200-nm-thick IMC/SiO2 MNCFs were evaluated based on changes in the nanocomposites’ index of refraction as a function of irradiation time (as detailed in the Materials and methods section). This relative mass loss was compared with that observed for pure IMC films (∼75 nm in thickness, similar initial IMC mass) by considering changes in their relative film thickness (h/h0).34
Fig. 2 summarizes this data, which were obtained at two irradiation wavelengths, 254 and 365 nm. Additional details of the refractive index and thickness changes vs. degradation time for the films are in the ESI† (Fig. S5). In the ∼200-nm-thick MNCFs, approximately 40% of the 254-nm light and 10% of the 365-nm light are absorbed by the IMC layer (see ESI,† Fig. S7). As described in the Materials and Methods section, the mass loss in the nanocomposite layers was calculated using linearly graded models. The data of Fig. 2 indicate a two-stage decay in the IMC mass for both pure IMC films and the IMC/SiO2 MNCFs during photodegradation.
Fig. 3 shows only a portion of what is shown in Fig. 2, but with more detail; only the first stage (stage 1) of the degradation is shown (up to ∼1 × 103–2 × 103 s for 254-nm UV irradiation and ∼6 × 103–8 × 103 s for 365-nm irradiation). In stage 1 of photodegradation, the mass loss of MNCFs is shown to be dependent on the degree of confinement; the overall mass loss decreases as the degree of confinement increases. With 254-nm irradiation, the mass loss of IMC/SiO2(11 nm) is ∼1.7%, while it is ∼7% in pure IMC film (Fig. 3(a), green triangles vs. gray circles). The same trend is found at the longer wavelength (365 nm): a mass loss of ∼1.7% in IMC/SiO2(11 nm) and ∼6% in pure IMC film (Fig. 3(b)). In addition, the rate of degradation is slower. The reduced mass loss and slower degradation can be attributed to the increased Tg in confined IMC films (by ∼30 K in IMC/SiO2(11 nm)34) which results in slower IMC molecular relaxation dynamics as well as slower diffusion of CO2 and O2 products (from pathways I and II, Fig. 1), reducing the overall reaction rate, similar to our previous report of thermal and UV degradation under vacuum conditions.34 The combined effects lead to higher photostability, minimizing stage 1 degradation. With continued irradiation, the rate of mass loss gradually slows and eventually reaches a relatively steady-state plateau (ending stage 1) at ∼2 × 103 s for 254-nm irradiation and ∼6 × 103 s for 365-nm irradiation. With prolonged UV irradiation, a second stage of photodegradation (stage 2) begins; the degradation rate becomes independent of the degree of confinement, and the rate becomes similar to that of pure IMC films (Fig. 2).
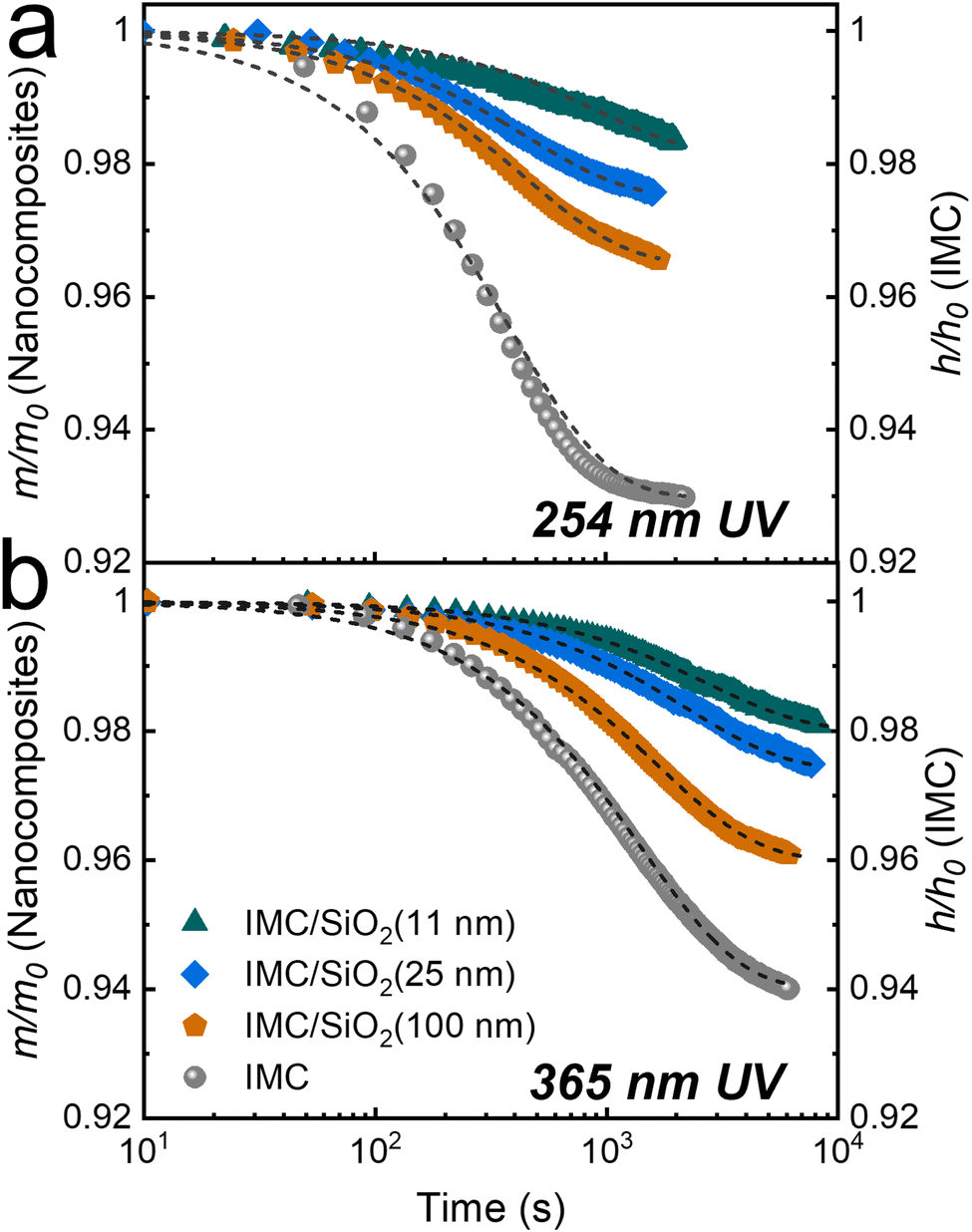 | ||
| Fig. 3 Stage 1 of photodegradation under ambient conditions due to (a) 254-nm (time <2 × 103 s) and (b) 365-nm UV irradiation (time <7 × 103 s). Degradation of a pure IMC film (∼75 nm) was measured through changes in relative film thickness with time (gray circles, right axes); and that of IMC/SiO2 MNCFs (∼200 nm) was measured through changes in the relative IMC mass in the nanocomposite (left axes). The IMC/SiO2 MNCFs are the same as those in Fig. 2: IMC/SiO2(11 nm) (green triangles), IMC/SiO2(25 nm) (blue diamonds), and IMC/SiO2(100 nm) (orange pentagons). The average refractive index of the film, fitted by the linearly graded models (not shown), was used for mass-loss calculations. Dashed lines are exponential decay fits to the data. | ||
Evolution of morphology during stage 1 and stage 2 photodegradation
To gain additional details about the two stages of photodegradation, each time point of the SE data was fit to the exponentially graded refractive index (ESI,† Fig. S4 and S6). Fig. 4 shows typical results of the refractive index as a function of the distance from the surface for an IMC/SiO2(25 nm) MNCF (data for other MNCFs are available in the ESI† (Fig. S6)). Two patterns are observed in the data, and they can be associated with the aforementioned two stages of photodegradation, stage 1 and stage 2. In stage 1 (during the first ∼2 × 103 s), the overall refractive index throughout the nanocomposite layer (the flat region of the profile at each given time) drops only slightly. The index of refraction at the film's surface region (indicated by the red arrow in Fig. 4) also decreases, but only slightly faster than the film center (see early curves of Fig. 4). this non-uniform degradation, also measured with the inhomogeneity index in linearly-graded model remains nearly constant during stage 1 (ESI,† Fig. S2), indicating a uniform degradation process throughout the entire nanocomposite layer.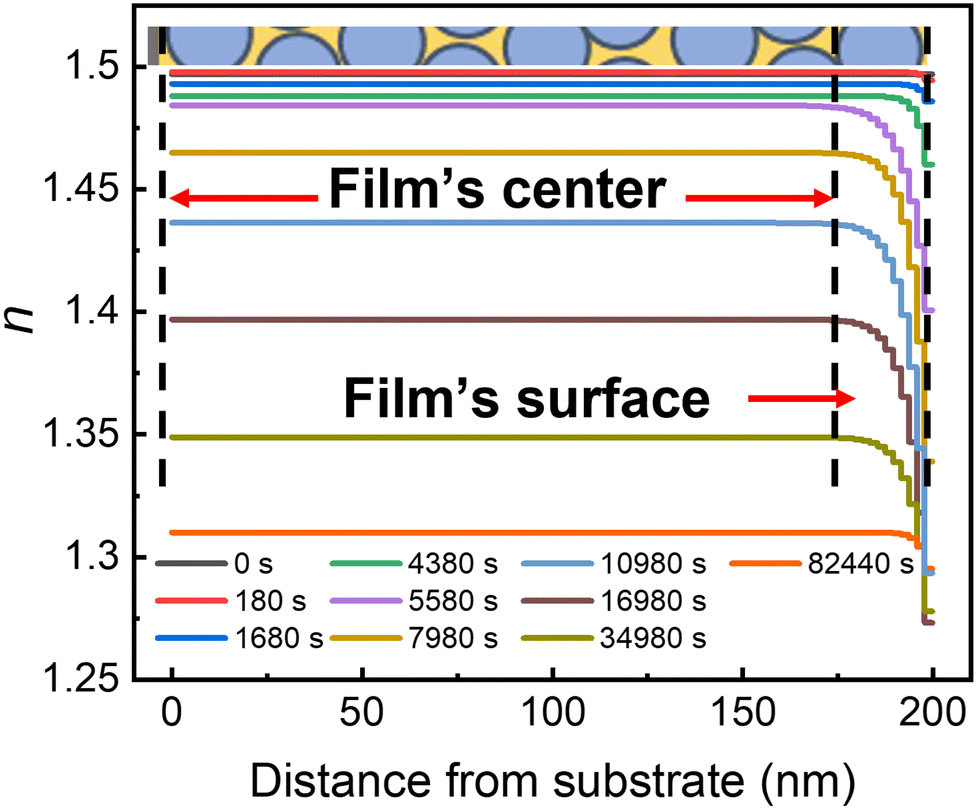 | ||
| Fig. 4 Index of refraction as a function of the distance from the substrate in an IMC/SiO2(25 nm) MNCF at various UV exposure times (legend at bottom inset indicates times; λ = 254 nm). Each profile is extracted based on the index of refraction profile obtained from the exponentially-graded model fitting. The schematic on top shows a slice of the composite film. Vertical dashed lines indicate from left to right: the location of the substrate, the end of the central portion of the film coinciding with the beginning of its surface region with a thickness of ∼25 nm (NP diameter), and the free surface. | ||
In stage 2 (after ∼2 × 103 s), the material in the nanocomposite begins to evaporate more rapidly, producing a greater change in the index values both at the film center and at the film surface, which is seen as a rapid change in the baseline values of the index of refraction (the flat regions in Fig. 4) and increased curvature near the surface in each profile, respectively. The initial evaporation rate at this stage is faster at the free surface (see data between 4 × 103 and 4 × 104 s in Fig. 4), also resulting in increased inhomogeneity in the index of refraction (ESI,† Fig. S2). Eventually, material loss throughout the film decreases the index of refraction of the film center, and the inhomogeneity also decreases (see data for >4 × 104 s and the ESI,† Fig. S2). This observation is partially consistent with our previous report:31,34 when thermal degradation of IMC or polystyrene (PS) occurred in a confined system, the near-surface nanocomposite layer degraded first, followed by the film's center. However, in stage 2 of photodegradation (as opposed to thermal degradation), confinement does not protect IMC from degradation under ambient conditions. In contrast, under vacuum, stage 2 is fully suppressed in the absence of water and oxygen. It is likely that as IMC byproducts in the nanocomposite layer start to evaporate the resulting porous network allowing the condensation of water, facilitating the degradation without the need for products or water to diffuse through the glass network. Water absorption and dissolution in IMC and its products can also potentially alter molecular mobility and dynamics by reducing the viscosity.61–63
To further corroborate the SE results, SEM images were obtained for IMC/SiO2(100 nm) MNCFs irradiated with 254-nm UV light (Fig. 5). Samples were analyzed after different amounts of IMC mass loss: ∼10% (after ∼1.5 × 103 s of irradiation time, stage 1), ∼30% (∼1 × 104 s, stage 2), ∼50% (∼2 × 104 s, stage 2), and ∼70% (∼4 × 104 s, stage 2). This data also supports two-stage photodegradation and is consistent with SE measurements. During stage 1, no pronounced morphology change is observed (Fig. 5(a) and (e)). During stage 2, we first observe the surface layer degradation/evaporation (Fig. 5(b) and (f)); the film's center then degrades, and gaseous products diffuse out (Fig. 5(c), (g), (d) and (h)).
 | ||
| Fig. 5 Top-view (top) and cross-sectional (bottom) SEM images of ∼400-nm-thick IMC/SiO2(100 nm) MNCFs during the two-stage photodegradation under 254-nm irradiation and ambient conditions: stage 1 (red) and stage 2 (orange). All scale bars are 200 nm. Percentage of IMC mass loss is ∼10% for (a) and (e), ∼30% for (b) and (f), ∼50% for (c) and (g), and ∼70% for (d) and (h). | ||
UPLC/MS determination of main byproducts during photodegradation of IMC
To understand the differences in the reaction kinetics during stage 1 and stage 2, UPLC/MS can be utilized to identify the byproducts of photodegradation. More details, such as byproduct information and associated degradation processes during the two stages of IMC photodegradation, are summarized in the ESI† (Fig. S9, S10 and Table S2). Fig. 6(a) shows the proportions of the remaining IMC and the degradation products of IMC films and MNCFs after 103 s of 254-nm UV irradiation; this corresponds to stage 1 degradation. As the degree of confinement is increased, the amount of remaining IMC is higher, and smaller amounts of degradation products are produced. In the most confined system, IMC/SiO2(11 nm) MNCF, which has an average pore size of 3 nm, ∼15% more IMC is detected than in pure IMC film. This is consistent with data shown in Fig. 2, where the relative mass loss is smaller in the IMC/SiO2(11 nm) MNCF than in pure IMC films. We note that the mass loss obtained from SE experiments corresponds to gaseous products (likely CO2), which may evaporate away; meanwhile, the UPLC/MS data can reveal the remaining ratios of high-molecular-weight products. Given that decarboxylated IMC is the main degradation product during stage 1 (20–30% of total mass), we can conclude that this degradation stage predominantly proceeds through pathway I (Fig. 1), which is limited by the diffusion of CO2 products. With confinement and the subsequent Tg increase, gas diffusion is expected to slow down,34 thus explaining the decrease in the reaction (i.e., degradation) rate for confined systems in stage 1. Limited amounts of other products are also detected at this point in stage 1, indicating parallel reactions/degradation through alternative pathways. These degradation processes can also be slowed down by the reduced diffusion rates of O2 reactants in confinement.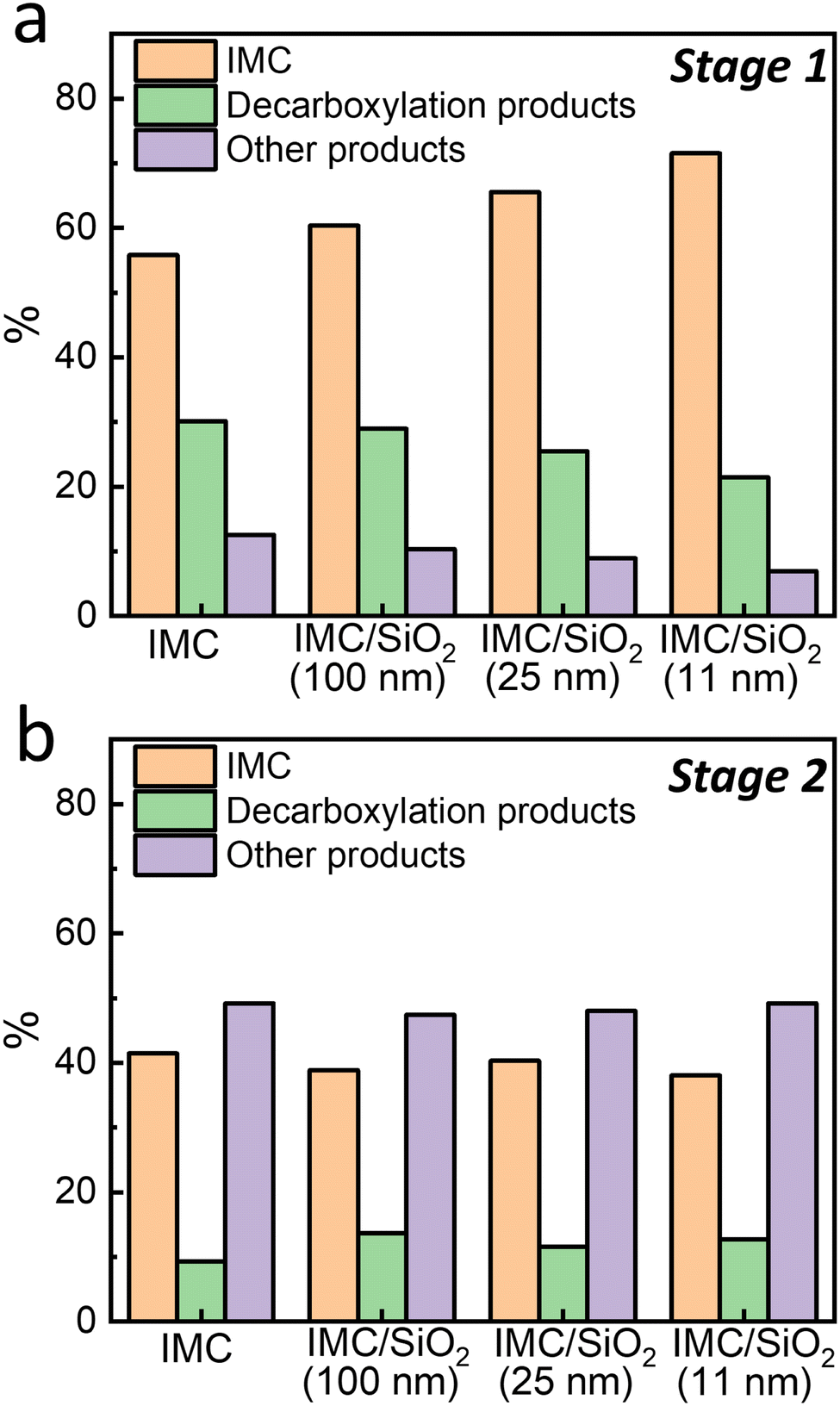 | ||
| Fig. 6 Fractions of IMC and degradation products detected by UPLC/MS measurements after UV irradiation at 254 nm under ambient conditions: (a) after 103 s (the end of stage 1 photodegradation), and (b) 104 s (during stage 2, ∼20% of IMC has degraded) of UV irradiation. Degradation products have been categorized as decarboxylation products (decarboxylated IMC (P1)) and other products. | ||
After prolonged UV irradiation (104 s, during stage 2 of photodegradation), the fractions of remaining IMC (∼40%) and degradation products are similar for MNCFs and IMC films (Fig. 6(b)). The UPLC/MS data also indicate that the primary products are degradation products other than decarboxylated IMC (P1), suggesting the prominent role played by locally available water in UV degradation. In stage 2, confinement and diffusion are no longer key factors in degradation kinetics, as the mass loss also generates a more porous environment (Fig. 5), allowing for the products to evaporate. These observations are consistent with SE measurements (Fig. 2), where confinement effects are only observed during stage 1 of photodegradation.
The confinement-insensitivity of stage 2 photodegradation in ambient conditions is in contrast with our previous studies of thermal degradation31,34 and with the photodegradation of IMC under inert environments.34 The thermal degradation of IMC and of the polymer PS in similarly confined environments are rate-dependent after the removal of the surface/top layer.31,34 Above the Tg, these systems were in the supercooled-liquid state. The material in larger pores had higher mobility, lower viscosity, and higher diffusion and degraded faster than those in smaller and more confined pores. As the degradation proceeded, the remaining material moved toward smaller pores within the system due to capillary forces, contributing to slower degradation rates. During photodegradation, where the material is below the Tg, these systems are in the glassy (solid) state. As reaction products are removed, the material is not driven toward smaller pores. Under inert conditions where only pathway I was present, the degradation was found to be fully suppressed due to Le Chatelier's principle, as the system was practically under constant volume constraints.34 In contrast, under ambient conditions, it is possible for water to condense on the surface, replacing the evaporated material, reducing the constant volume constraints imposed by the rigid NP walls. Locally absorbed water is also present in the system from the beginning. In addition, other reaction pathways (pathways II and III) are not kinetically tied to the relaxation dynamics of the IMC as transport of the products is not needed for them to occur. In this case, a two-part degradation process is observed. While a similar protection is observed during stage I as in vacuum conditions, which is primarily limited by the relaxation dynamics of the glass matrix and gas diffusion kinetics, after the removal of the top layer, the material can locally degrade. Here, the material in larger pores still degrades before those at smaller pores, but the degradation products can readily evaporate, through the porous network, without further rearrangements of the remaining material towards smaller pores, which is limited by the glass viscosity. As such in this case, we attribute the lack of a straightforward rate-dependence in stage 2 photodegradation to the exposure to ambient conditions; IMC and its degradation products are able to adsorb water to varying degrees,61,62 leading to unpredictable amounts of vapor condensation driven by NP capillary forces,64,65 which is independent of molecule rearrangements and the system viscosity. In addition, the adsorbed water can also reduce the local viscosity of the system, further facilitating the degradation. Investigating this latter possibility requires a detailed characterization of the water content and dynamics, which can be the focus of future studies.
In summary, we studied the photodegradation under ambient conditions of IMC molecular nanocomposite films (MNCFs) in extreme confinement, which was induced by the CaRI technique. Consistent with our previous studies, the degradation process begins with the degradation and removal of material from the film surface, followed by more uniform degradation of the film's center. We observed a two-stage IMC photodegradation in the system. During stage 1, the photostability of IMC increased due to confinement; it is mainly a diffusion-limited process that correlates with the degree of confinement. In stage 2, photodegradation involves local reactions not dependent on diffusion and thus confinement. These reactions are facilitated by locally available water, and their products can readily evaporate through the porous network created by degradation. The adsorbed water can also mix with IMC to enhance molecule mobility and reduce viscosity. Our study highlights that while MNCFs can be used as an effective approach to stabilize glass coatings, the details of stability highly depend on the degradation pathways, where reactions involving gas diffusion are strongly suppressed by extreme nanoconfinement, while more diffusion-independent reactions are unaffected. This method also provides a new approach to changing the balance of various reactions to investigate chemical reactivity in thin glass films and to understand the role of physical and chemical properties in material degradation. Materials with highly confined structures have many potential technological applications, including energy conversion, environmental remediation, and pharmaceutical synthesis. Our results highlight the need to carefully optimize the properties of component NPs in these materials, and they emphasize the importance of understanding changes in the thermodynamic and kinetic properties of a reaction. Both are crucial for realizing applications for materials with extreme confinement.
Data availability
All data supporting the findings in this study are available via Figshare at https://doi.org/10.6084/m9.figshare.26932447.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding provided by the University of Wisconsin Materials Research Science and Engineering Center (MRSEC) grant (DMR-2309000). C. Y. C. was supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) (DGE-1845298). A. A. S. was supported with a postdoctoral fellowship from the Vagelos Institute of Energy Science and Technology (VIEST). SEM measurements were performed at the Singh Center for Nanotechnology with support from the University of Pennsylvania MRSEC (DMR-1720530). We thank Dr. Jamie Ford for technical support for SEM measurements. We thank Dr. Charles W. Ross III, the director of the Automated Synthesis and Characterization Center of Penn Chemistry, and Mr. Ryan Kubanoff, BCRC Lab Manager, for providing chromatographic and mass spectral method development, training, analyses, and data interpretation. We acknowledge Dr. Lianne M. C. Beltran for professional scientific editing and graphical assistance.References
- S. Dragojevic, J. S. Ryu and D. Raucher, Molecules, 2015, 20, 21750–21769 CrossRef CAS
.
- S. Karaosmanoglu, M. Zhou, B. Shi, X. Zhang, G. R. Williams and X. Chen, J. Controlled Release, 2021, 329, 805–832 CrossRef CAS
- V. S. Saji, J. Electrochem. Soc., 2020, 167, 121505 CrossRef CAS
- T. AlAbdulaal, M. AlShadidi, M. Hussien, V. Ganesh, A.-F. Bouzidi, S. Rafique, H. Algarni, H. Zahran, M. Abdel-wahab and I. Yahia, Environ. Sci. Pollut. Res. Int., 2022, 29, 19109–19131 CrossRef CAS PubMed
- C. K. Jeong, C. Baek, A. I. Kingon, K.-I. Park and S.-H. Kim, Small, 2018, 14, 1704022 CrossRef
- H. Liu, R. Jian, H. Chen, X. Tian, C. Sun, J. Zhu, Z. Yang, J. Sun and C. Wang, Nanomaterials, 2019, 9, 950 CrossRef CAS PubMed
- S. Sen, J. D. Thomin, S. K. Kumar and P. Keblinski, Macromolecules, 2007, 40, 4059–4067 CrossRef CAS
- S. Qin, M. G. Pour, S. Lazar, O. Köklükaya, J. Gerringer, Y. Song, L. WÅgberg and J. C. Grunlan, Adv. Mater. Interfaces, 2019, 6, 1970009 Search PubMed
- P. Bonnet, D. Sireude, B. Garnier and O. Chauvet, Appl. Phys. Lett., 2007, 91, 201910 CrossRef
- A. Chandra, L.-S. Turng, P. Gopalan, R. M. Rowell and S. Gong, Compos. Sci. Technol., 2008, 68, 768–776 CrossRef CAS
- N. Jouault, D. Zhao and S. K. Kumar, Macromolecules, 2014, 47, 5246–5255 CrossRef CAS
- A. Laachachi, E. Leroy, M. Cochez, M. Ferriol and J. L. Cuesta, Polym. Degrad. Stab., 2005, 89, 344–352 CrossRef CAS
- O. Bera, B. Pilić, J. Pavličević, M. Jovičić, B. Holló, K. M. Szécsényi and M. Špirkova, Thermochim. Acta, 2011, 515, 1–5 CrossRef CAS
- J. Moll and S. K. Kumar, Macromolecules, 2012, 45, 1131–1135 CrossRef CAS
- K. Chrissafis and D. N. Bikiaris, Thermochim. Acta, 2011, 523, 25–45 CrossRef
- S. Bourbigot, J. Gilman, C. Wilkie, S. Bourbigot, J. W. Gilman and C. A. Wilkie, Polym. Degrad. Stab., 2004, 84, 483–492 CrossRef CAS
- H. S. Vaziri, I. A. Omaraei, M. Abadyan, M. Mortezaei and N. Yousefi, Mater. Des., 2011, 32, 4537–4542 CrossRef CAS
- S. Lu, R. M. Rodrigues, S. Huang, D. A. Estabrook, J. O. Chapman, X. Guan, E. M. Sletten and C. Liu, Chem. Catal., 2021, 1, 704–720 CrossRef CAS PubMed
- O. Weizman, J. Mead, H. Dodiuk, A. Ophir and S. Kenig, Polym. Degrad. Stab., 2022, 195, 109811 CrossRef CAS
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS
- H. Zhao and S. L. Simon, Polymer, 2011, 52, 4093–4098 CrossRef CAS
- T. Man, C. Xu, X.-Y. Liu, D. Li, C.-K. Tsung, H. Pei, Y. Wan and L. Li, Nat. Commun., 2022, 13, 305 CrossRef CAS PubMed
- J. Young Maeng, J. Hyun Yang, H. Ji Jang, M. Hee Joo, Y. Jun Kim, C. Kyun Rhee and Y. Sohn, Appl. Surf. Sci., 2023, 609, 155349 CrossRef CAS
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54, 2894–2904 CrossRef CAS
- Y. Zhai, G. Liu, F. Jin, Y. Zhang, X. Gong, Z. Miao, J. Li, M. Zhang, Y. Cui, L. Zhang, Y. Liu, H. Zhang, Y. Zhao and Y. Zeng, Angew. Chem., Int. Ed., 2019, 58, 17679–17683 CrossRef CAS
- C. Guo, F. Duan, S. Zhang, L. He, M. Wang, J. Chen, J. Zhang, Q. Jia, Z. Zhang and M. Du, J. Mater. Chem. A, 2022, 10, 475–507 RSC
- J. L. Hor, Y. Jiang, D. J. Ring, R. A. Riggleman, K. T. Turner and D. Lee, ACS Nano, 2017, 11, 3229–3236 CrossRef CAS
- H. Wang, K. L. Kearns, A. Zhang, A. A. Shamsabadi, Y. Jin, A. Bond, S. M. Hurney, C. Morillo and Z. Fakhraai, Nano Lett., 2021, 21, 1778–1784 CrossRef CAS PubMed
- H. Wang, J. L. Hor, Y. Zhang, T. Liu, D. Lee and Z. Fakhraai, ACS Nano, 2018, 12, 5580–5587 CrossRef CAS PubMed
- J. L. Hor, H. Wang, Z. Fakhraai and D. Lee, Soft Matter, 2018, 14, 2438–2446 RSC
- H. Wang, Y. Qiang, A. A. Shamsabadi, P. Mazumder, K. T. Turner, D. Lee and Z. Fakhraai, ACS Macro Lett., 2019, 8, 1413–1418 CrossRef CAS PubMed
- Y. Jiang, J. L. Hor, D. Lee and K. T. Turner, ACS Appl. Mater. Interfaces, 2018, 10, 44011–44017 CrossRef CAS
- Y. Qiang, S. S. Pande, D. Lee and K. T. Turner, ACS Nano, 2022, 16, 6372–6381 CrossRef CAS PubMed
- C. Y. Chen, H. Wang, A. A. Shamsabadi and Z. Fakhraai, J. Phys. Chem. B, 2024, 128, 12798–12807 Search PubMed
- D. Jin and J. Zhong, J. Phys. Chem. B, 2023, 127, 5295–5307 CrossRef CAS PubMed
- G. Kaptay, J. Nanosci. Nanotechnol., 2012, 12, 2625–2633 CrossRef CAS PubMed
- N. Pradal, G. Chadeyron, S. Thérias, A. Potdevin, C. V. Santilli and R. Mahiou, Dalton Trans., 2014, 43, 1072–1081 RSC
- V. Viswanath, G. Subodh and C. Muneera, Opt. Laser Technol., 2020, 127, 106168 CrossRef CAS
- S. A. Kumar, J. S. Shankar, B. K. Periyasamy and S. K. Nayak, Phys. Chem. Chem. Phys., 2021, 23, 22804–22816 RSC
- W. Sun, Y. Li, W. Shi, X. Zhao and P. Fang, J. Mater. Chem., 2011, 21, 9263–9270 RSC
- J. Xiong, E.-L. Hsiang, Z. He, T. Zhan and S.-T. Wu, Light: Sci. Appl., 2021, 10, 1–30 CrossRef
- L. Godet, R. Thijssen, J. Fu, R. Hourani, S. Doshay and R. J. Visser, 51-2: Invited Paper: Developing a Platform for Creating Waveguide Combiners for AR Headsets and Metasurface-based Optics, SID Symposium Digest of Technical Papers, 2023, pp. 731–733.
- W. Li, X. Wang, Z. Liu, X. Zou, Z. Shen, D. Liu, L. Li, Y. Guo and F. Yan, Nat. Mater., 2024, 23, 688–694 CrossRef
- A. Zhang, A. R. Moore, H. Zhao, S. Govind, S. E. Wolf, Y. Jin, P. J. Walsh, R. A. Riggleman and Z. Fakhraai, J. Chem. Phys., 2022, 156, 244703 CrossRef CAS PubMed
- Y. Jin, A. Zhang, S. E. Wolf, S. Govind, A. R. Moore, M. Zhernenkov, G. Freychet, A. Arabi Shamsabadi and Z. Fakhraai, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2100738118 CrossRef CAS
- B. M. Baklanov, K. Mogilnikov, V. Polovinkin and F. Dultsev, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2000, 18, 1385–1391 Search PubMed
- B. Q. Kim, M. Füredi, R. B. Venkatesh, S. Guldin and D. Lee, Small, 2023, 19, 2302676 CrossRef CAS
- B. Song, F. Liu, H. Wang, J. Miao, Y. Chen, P. Kumar, H. Zhang, X. Liu, H. Gu, E. A. Stach, X. Liang, S. Liu, Z. Fakhraai and D. Jariwala, ACS Photonics, 2020, 7, 2896–2905 CrossRef CAS
- J. Lynch, E. Smith, A. Alfieri, B. Song, M. Klein, C. E. Stevens, C. Y. Chen, C. F. K. Lawrence, C. R. Kagan, H. Gu, S. Liu, L.-M. Peng, S. Vangala, J. R. Hendrickson and D. Jariwala, Nat. Photonics, 2024, 18, 1176–1184 CrossRef CAS
- A. A. Shamsabadi, H. Fang, D. Zhang, A. Thakur, C. Y. Chen, A. Zhang, H. Wang, B. Anasori, M. Soroush, Y. Gogotsi and Z. Fakhraai, Small methods, 2023, 7, 2300568 CrossRef CAS PubMed
- S. G. Tarasov, V. Gaponenko, O. M. Howard, Y. Chen, J. J. Oppenheim, M. A. Dyba, S. Subramaniam, Y. Lee, C. Michejda and N. I. Tarasova, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2400084121 CrossRef
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao, W. Lv and G. Liu, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS
- Q. Zhang, P. Chen, M. Zhuo, F. Wang, Y. Su, T. Chen, K. Yao, Z. Cai, W. Lv and G. Liu, Appl. Catal., B, 2018, 221, 129–139 CrossRef CAS
- P.-Y. Lin, F.-A. Chen, A.-B. Wu, S.-H. Chao, Y.-T. Peng, V. M.-H. Lee and C.-Y. Chen, J. Iran. Chem. Soc., 2016, 13, 1777–1783 CrossRef CAS
- F. Temussi, F. Cermola, M. DellaGreca, M. R. Iesce, M. Passananti, L. Previtera and A. Zarrelli, J. Pharm. Biomed. Anal., 2011, 56, 678–683 CrossRef CAS PubMed
- J. J. Jiménez, M. I. Sánchez, R. Pardo and B. E. Muñoz, J. Environ. Sci., 2017, 51, 13–20 CrossRef
- Y. Qiu, S. S. Dalal and M. D. Ediger, Soft Matter, 2018, 14, 2827–2834 RSC
- F. Zhang and R. Brüschweiler, Angew. Chem., Int. Ed., 2007, 46, 2639–2642 CrossRef CAS
- J.-N. Dumez, Chem. Commun., 2022, 58, 13855–13872 RSC
- A. Bertei, B. Nucci and C. Nicolella, Mech. Chem. Eng. Trans., 2013, 32, 1531–1536 Search PubMed
- V. Andronis, M. Yoshioka and G. Zografi, J. Pharm. Sci., 1997, 86, 346–351 CrossRef CAS PubMed
- K. J. Dawson, K. L. Kearns, M. D. Ediger, M. J. Sacchetti and G. D. Zografi, J. Phys. Chem. B, 2009, 113, 2422–2427 CrossRef CAS
- T. Pérez-Castañeda, C. Rodríguez-Tinoco, J. Rodríguez-Viejo and M. A. Ramos, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11275–11280 CrossRef
- S. Gruener, T. Hofmann, D. Wallacher, A. V. Kityk and P. Huber, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 067301 CrossRef PubMed
- K. Yamashita and H. Daiguji, J. Phys. Chem. C, 2015, 119, 3012–3023 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional SE and UPLC/MS data. See DOI: https://doi.org/10.1039/d4sm01266f |
| This journal is © The Royal Society of Chemistry 2025 |