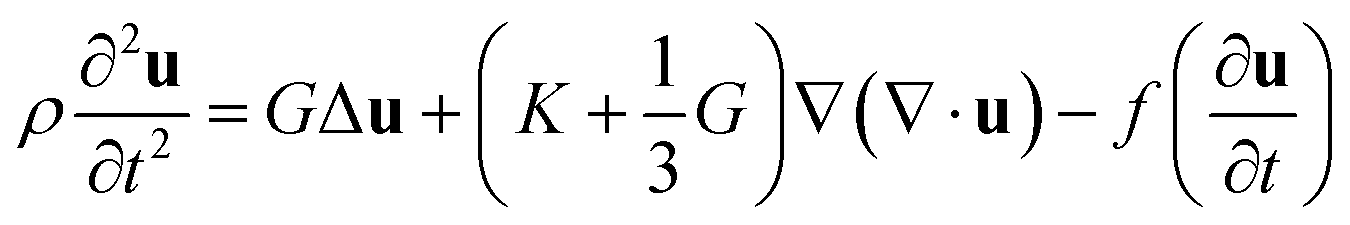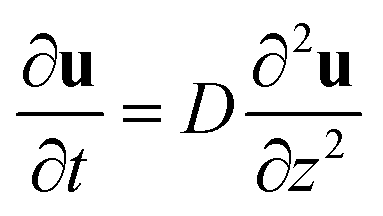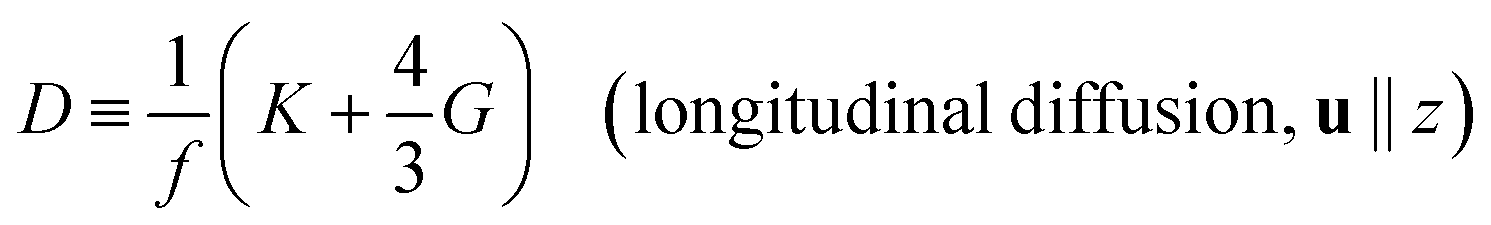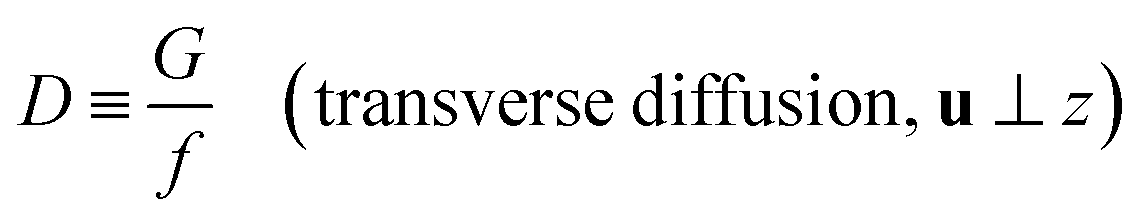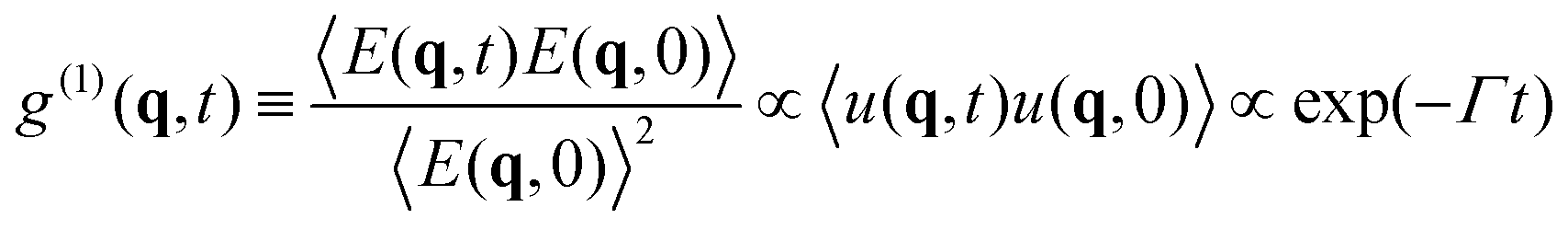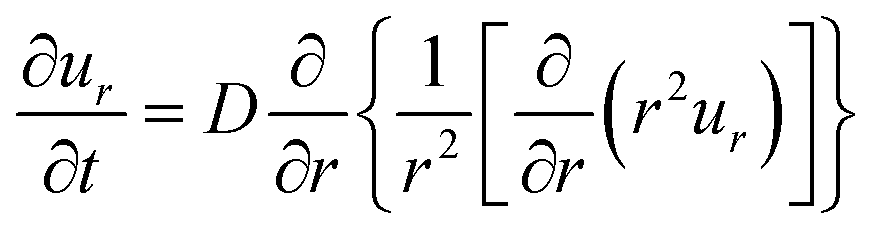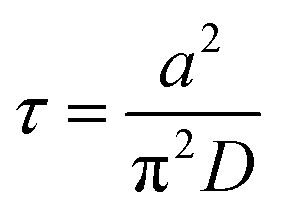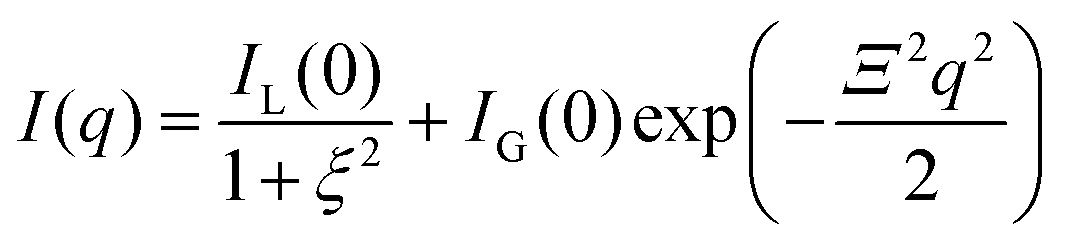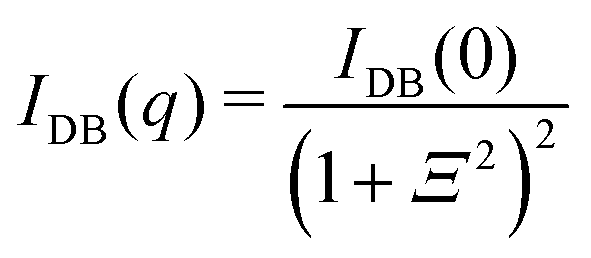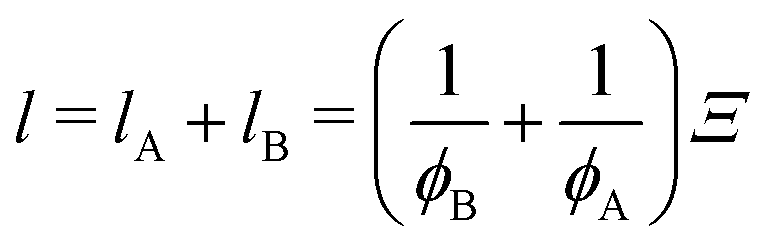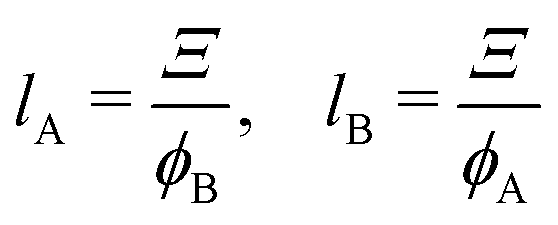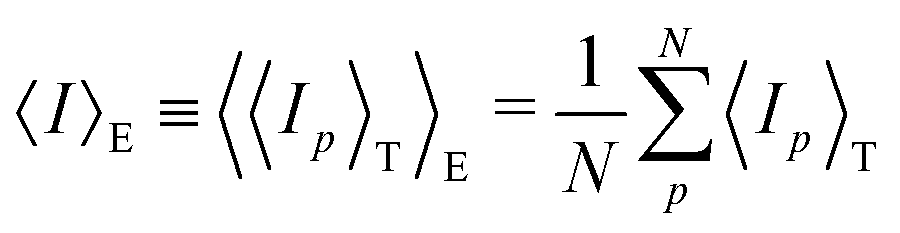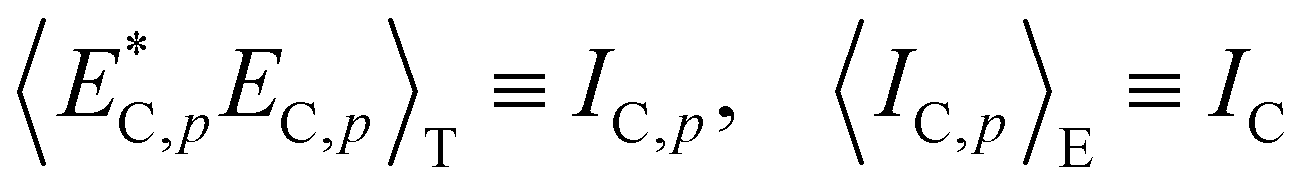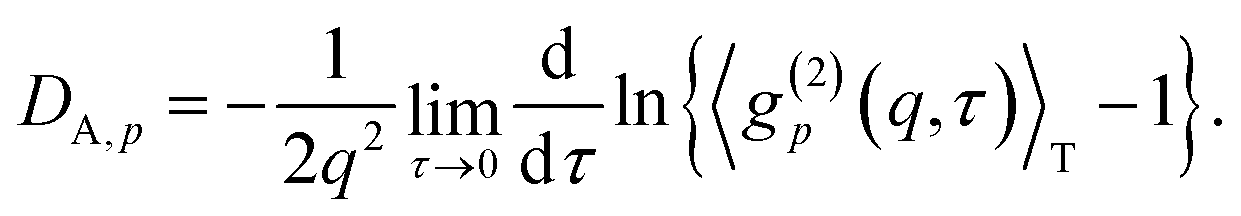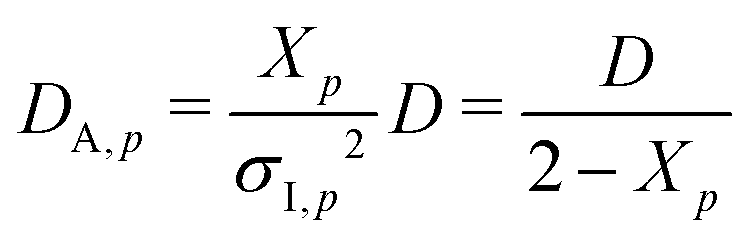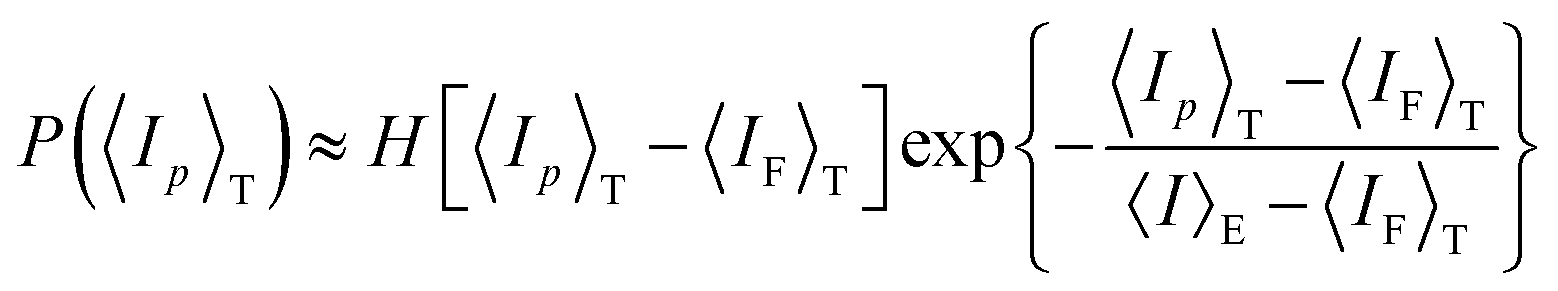DOI:
10.1039/D4SM01418A
(Review Article)
Soft Matter, 2025, Advance Article
Physics of polymer gels: Toyoichi Tanaka and after†
Received
29th November 2024
, Accepted 14th January 2025
First published on 3rd February 2025
Abstract
This review revisits the works of Toyoichi Tanaka on the physics of polymer gels and discusses their scientific significance with the keywords of volume phase transition, structure, dynamics, kinetics and inhomogeneities, followed by some recent topics including defect-free homogeneous gels. Then, the modern physics of polymer gels will be considered from the viewpoints of cross-linking, networking, and percolation, along with the scope of future directions of polymer gels and polymer networks.
1 Introduction
Polymer gels are ubiquitous and are found in many places and situations in daily life. They are interesting materials owing to their unique physical properties and various functions, such as swelling and shrinking, elastic and viscoelastic properties, and environmental responsiveness. Polymer gels have been intriguing in many fields, such as chemistry, physics, materials science, pharmacy, biology, food science, and so on, for more than half a century. They consist of cross-linked polymer chain networks and a large amount of solvent. In spite of this simple combination, polymer gels have been a difficult system to study in physics because of their topological nature, e.g., random cross-linking and imperfect networking.
The late MIT physics professor Toyoichi Tanaka was a leading authority on the physics of polymer gels. Tanaka brought about a revolution in the physics of polymer gels. He sublimated chaotic gels, with disparate molecular weight distributions of the strands between the cross-links and with the mesh of the polymer network being full of defects, into beautiful physical objects that can be described by mathematical equations. In particular, his discovery of the “volume phase transition (VPT)”1 shed new light on polymer science: analogous to the liquid–vapor transition of water, a VPT is a dramatic change in the volume of a gel in response to an infinitesimal change in one of the intensive properties, such as temperature (T), solvent composition, salt concentration and pH, as well as light, pressure (P), and magnetic field. Since a VPT is a macroscopic manifestation of molecular-level changes in polymer conformation, one can study various phenomena occurring at the molecular level by simply observing a size and/or shape change of the gel. Here, the cross-links connecting polymer chains play a major role. The discovery and the concepts of the VPT strongly influenced not only polymer science, but also disciplines such as soft-matter physics, biophysics, and bioengineering, as well as various applications, e.g., chemo-mechanical actuators, sensors, water absorbents, and other engineering and industrial applications.
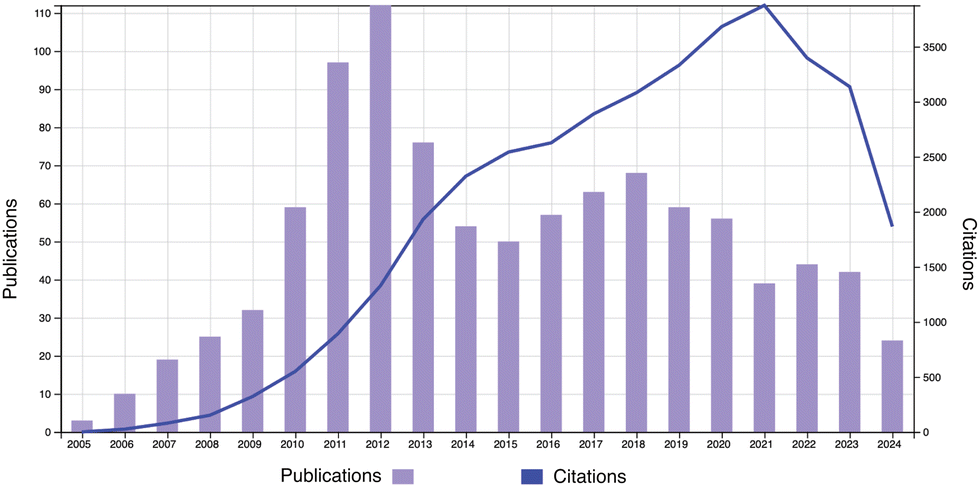
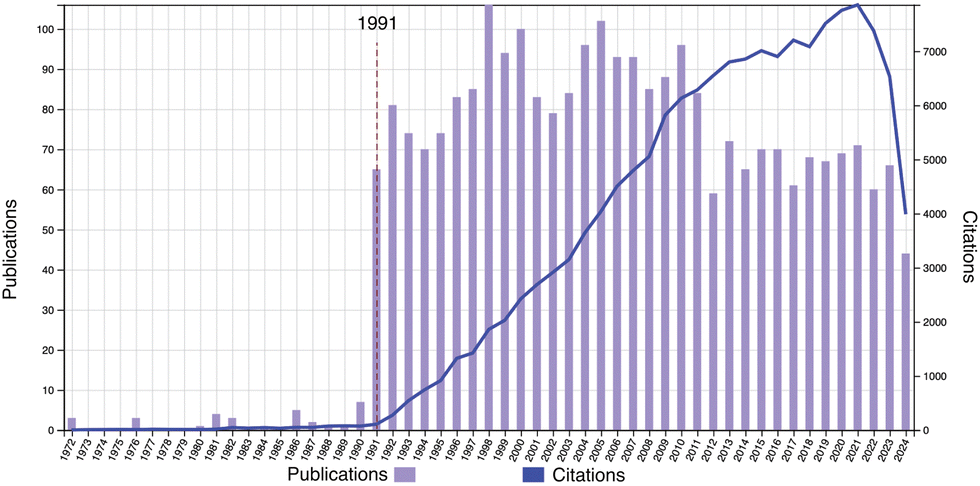
On the occasion of the 20th anniversary of Soft Matter, a review on the physics of polymer gels is presented. Fig. 1 shows the publication and citation statistics of Soft Matter in the period of 2005–2024 with the keyword of topic = polymer gel (Web of Science, as of Aug. 24, 2024). There are 987 publications with an H-index of 87. Typical subjects include double network hydrogels,2 stimuli-responsive polymer gels,3 friction and lubrication of hydrogels,4 and so on. Note that research on polymer gels was already at a blossoming stage and developing rapidly at the time of the first issue of Soft Matter. Fig. 2 shows the publication and citation statistics of Macromolecules (an ACS journal) in the period of 1972–2024 with the same keyword. It is quite surprising to notice that there is a discrete jump in the publications together with a steep increase in citations starting at the year of 1991. More surprisingly, the same trend is also found in the statistics of Physical Review Letters (PRL; an APS journal), Journal of Chemical Physics (an AIP journal), and Polymer (Elsevier) exactly in the same year (see Fig. S1–S3 in the ESI†)! We may now have a naive question: why did such a discrete jump in publications happen in 1991?
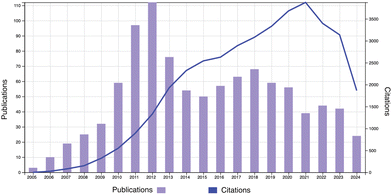 |
| | Fig. 1 Publication and citation statistics of Soft Matter with the topic of “polymer gel” (2005–2024). (Web of Science, as of Aug. 24, 2024.) | |
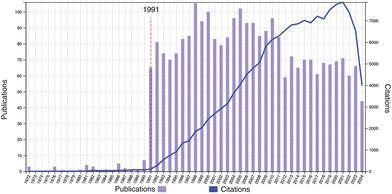 |
| | Fig. 2 Publication and citation statistics of Macromolecules with the topic of “polymer gel” (1972–2024). (Web of Science, as of Aug. 24, 2024.) | |
This review consists of the following: in Section 2, we overview the representative achievements of Toyoichi Tanaka, emphasizing their physical significance and influence in sciences. Here, we try to answer the above question. In Section 3, macroscopic as well as microscopic views of the VPT are reviewed. In Section 4, “Gel inhomogeneities”, which had not been explicitly reported in the works of Tanaka, are demonstrated based on shrinking kinetics, structure, and dynamics. Here, the frozen inhomogeneities are characterized via small-angle neutron scattering (SANS), and a decomposition method using dynamic light scattering (DLS) to separate the two types of concentration fluctuations, i.e., dynamic and static (frozen) concentration fluctuations, is reviewed. In Section 5, recent advances in polymer gels, i.e., quantitative understanding of polymer networks and gels consisting of defect-free ideal networks, the so-called “Tetra-PEG gels”, are reviewed as “Toward realization of ideal polymer networks”. Finally, future directions of the physics of polymer gels are considered in Section 6.
2 Physics driven by T. Tanaka
2.1 VPT: from theoretical prediction to experimental verification
Dušek and Patterson wrote a paper on solvent release from polymer gels due to poor solvation or high crosslink density and discussed the possibility that the composition dependence of the chemical potential of the solvent might exhibit minima and maxima that would predict phase separation.5 This was the first theoretical paper predicting the VPT, and they also indicated that “it would be difficult to attain the conditions necessary for the transition in the free-swelling case, but that it should be possible for a gel under tension”. As a matter of fact, the experimental verification of this theoretical prediction had to wait until 1978.1
In 1977, Tanaka and colleagues at the Massachusetts Institute of Technology (MIT) found in light scattering experiments that acrylamide hydrogel has a critical point at −17 °C and that it undergoes phase separation.6 Note that this is due not to water freezing but to phase separation. In 1978, as part of their cataract research, Tanaka et al. were experimenting with acrylamide gel particles in acetone–water mixtures to study cloudiness (phase separation or critical phenomena). One day, when they left the gels in various solvent compositions overnight, they found that cloudiness did not occur, and instead, the gels split into two types, swollen and shrunken, depending on the solvent composition. This was a historical discovery of the volume phase transition (VPT) of gels.1 However, reproducibility could not be achieved. After much trial and error, he realized that the gels that underwent the volumetric phase transition were aged gels that had been stored in a refrigerator and had been partially hydrolyzed, and that charged groups were necessary for the VPT.7 That is, in order to realize a VPT, it is necessary to introduce a strong repulsive term in the free energy of the gel, such as electrostatic interaction and the Donnan potential, so as to compete with the attractive interaction favoring gel shrinking.1
2.2 Representative achievements of Toyoichi Tanaka
Toyoichi Tanaka studied biophysics at the University of Tokyo under the auspices of Prof. A. Wada, where he conducted experimental and theoretical works on coil-globule transition.8,9 He was also involved in instrumentation and experimental works with dynamic light scattering (DLS).10 DLS is a technique that is commonly used to determine the size and size distribution of small particles in suspension or polymers in solution by taking the time correlation of the scattered intensity.11 He moved to MIT and began his gel research in the Benedek Lab in the Department of Physics. One of the themes was the study of cataracts, which is where the lens (intraocular lens) becomes cloudy, using the DLS technique, a specialty of Professor Benedek's. In the same year, he discovered that gels scatter light and fluctuate with time, leading to the development of a theory of gel fluctuation and its experimental demonstration. This was the birth of the cooperative diffusion theory of gels.12
Not only theoretical works but also experimental works on polymer gels were energetically conducted by Tanaka and his group until his tragic passing in 2001. T. Tanaka wrote 10 papers in Nature, 7 in Proceedings of the National Academy of Sciences (PNAS), 3 in Science, 16 in PRL, and many in other journals and books, of which representative works are listed in Table 1: the cooperative diffusion theory (the so-called THB theory; 1973),12 critical phenomena in gels (divergence of light scattering intensity at the critical temperature; 1977),6 the volume phase transition (VPT) of gels (1978,1 19817), kinetics of gel swelling (the Tanaka-Fillmore (TF) theory; the first theoretical prediction of the relaxation time of gel swelling being the inverse square of the gel size; 1979),13 collapse of gels in an electric field (1982),14 the VPT of poly(N-isopropylacrylamide) (PNIPAM) gel (1984),15 critical kinetics near the VPT (1985),16 instability patterns in gels (mechanical instability of gels at the phase transition; 1987),17 the universality class of gels (1989),18 visible-light-induced VPTs (1990),19 friction of gels (reversible decrease of gel-solvent friction at the VPT temperature; 1991),20 hydrogen-bond-induced VPTs (1991),21 saccharide-sensitive VPTs (1991),22 small-angle neutron scattering (SANS) studies on critical phenomena (1992)23 and microphase separation (1992),24 multiphases (existence of not only swollen and shrunken phases but also many intermediate phases; 1992),25 patterns in shrinking gels (1992),26 super absorbency (1992),27 a review of the VPT (1993),28 protein sequences (thermodynamic procedure to synthesize heteropolymers that can renature to recognize a given target molecule; 1994),29 first-order phase transitions and evidence for frustrations in polyampholytic gels (1999),30 reversible molecular adsorption (1999),31 gel catalysts (2000),32 and molecular imprinting (2000).33 They cover biophysics, physics, and physical chemistry. These papers, at least those published before 1991, inspired the subsequent rush in research, as was evidenced by the publication statistics (Fig. 2 and Fig. S1–S3, ESI†). In particular, the THB theory (1973),12 the volume phase transition (VPT) (1978),1 and the VPT of PNIPAM gel (1984)15 were extremely influential.
Table 1 Representative achievements of Toyoichi Tanaka
| Year |
Item |
Ref. |
| 1973 |
Cooperative diffusion theory and DLS (THB theory)12 |
J. Chem. Phys. |
| 1977 |
Critical phenomena6 |
PRL |
| 1978 |
Volume phase transition (VPT)1 |
PRL |
| 1979 |
Kinetics of gel swelling (TF theory)13 |
PRL |
| 1981 |
Volume phase transition7 |
Sci. Am. |
| 1982 |
Gel in an electric field14 |
Science |
| 1984 |
VPT of PNIPAM gel15 |
Nature |
| 1985 |
Critical kinetics of VPT16 |
PRL |
| 1987 |
Instability pattern17 |
Nature |
| 1989 |
Universality class18 |
J. Chem. Phys. |
| 1990 |
Light-induced VPT19 |
Nature |
| 1991 |
Friction of gel20 |
Science |
| 1991 |
VPT induced by hydrogen bonds21 |
Nature |
| 1991 |
Saccharide-sensitive VPT22 |
Nature |
| 1992 |
Neutron scattering studies of gels23,24 |
J. Chem. Phys. |
| 1992 |
Multiphases25 |
Nature |
| 1992 |
Patterns in shrinking gels26 |
Nature |
| 1992 |
Super absorbency27 |
Nature |
| 1993 |
Responsive gels: volume transitions28 |
Adv. Polym. Sci. |
| 1994 |
Nonrandomness in proteins29 |
PNAS |
| 1999 |
First-order VPT in polyampholytic gels30 |
PRL |
| 1999 |
Reversible molecular adsorption31 |
Science |
| 2000 |
Gel catalyst32 |
PNAS |
| 2000 |
Molecular imprinting33 |
PRL |
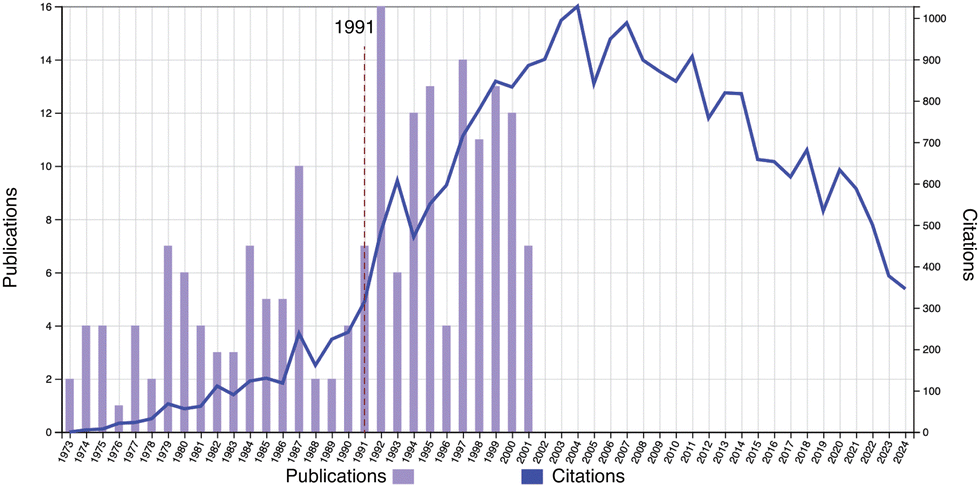
Fig. 3 shows the publications (1973–2001) and citations (1973–2024) of Toyoichi Tanaka, which started the work of the THB theory in 1973. It clearly shows a sharp increase in the citations around the year 1991, shown by a dashed line, and a marked correlation between the statistics in Fig. 2 and in Fig. 3. This eloquently answers the question posed in Fig. 2. Further evidence can be found in Fig. S4 (ESI†) (publication and citation statistics with the keywords of “volume phase transition AND polymer gels”).
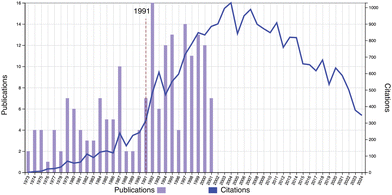 |
| | Fig. 3 Publication and citation statistics with the topic of “Toyoichi Tanaka AND MIT AND 1973–2001 ” (1973–2024). (Web of Science, as of Jan. 7, 2025.) | |
Tanaka's achievements can be categorized into the following three fields: (1) research of the origin of the helix–coil transition (–1973), (2) the physics of gels – discovery of the VPT and related phenomena (1973–1993), and (3) the physics of proteins – basic principles of biological functions (1994–2000). Although (1) and (3) are, respectively, the starting point of Dr Tanaka's research and the very science and life science he was aiming for, this review exclusively focuses on (2), i.e., the physics of polymer gels. Comprehensive reviews on the VPT are found in monographs.34,35
2.3 The THB theory and TF theory
The comprehensive theory titled “Spectrum of light scattered from viscoelastic gels”12 proposed by Tanaka–Hocker–Benedek (the THB theory) changed the perception of gels from phenomenological objects to theoretically predictable materials. This theory is based on the theory of elasticity.36 They described the equation of motion for gel networks with the displacement vector, u(r, t), which represents the displacement of a point r ≡ (x, y, z) in the network from its equilibrium location at time t, and derived the differential equation,| |
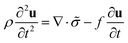 | (1) |
Here, ρ is the mass density of the small volume element, ![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) is the stress tensor (≡σik) and f is the friction factor. By conducting the operation on the divergence, they obtained the starting equation for the motion of gels:
is the stress tensor (≡σik) and f is the friction factor. By conducting the operation on the divergence, they obtained the starting equation for the motion of gels:| |
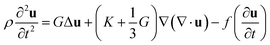 | (2) |
Here, K is the bulk modulus and G is the shear modulus.
The derived equation is
| |
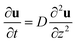 | (3) |
which is formally the same as the diffusion equation of molecules, and
D is the diffusion coefficient, defined by
| |
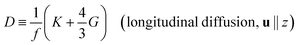 | (4) |
and
| |
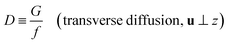 | (5) |
Eqn (3) ensures that the gel dynamics can be described with the conventional diffusion equation, although the phenomena are quite different.
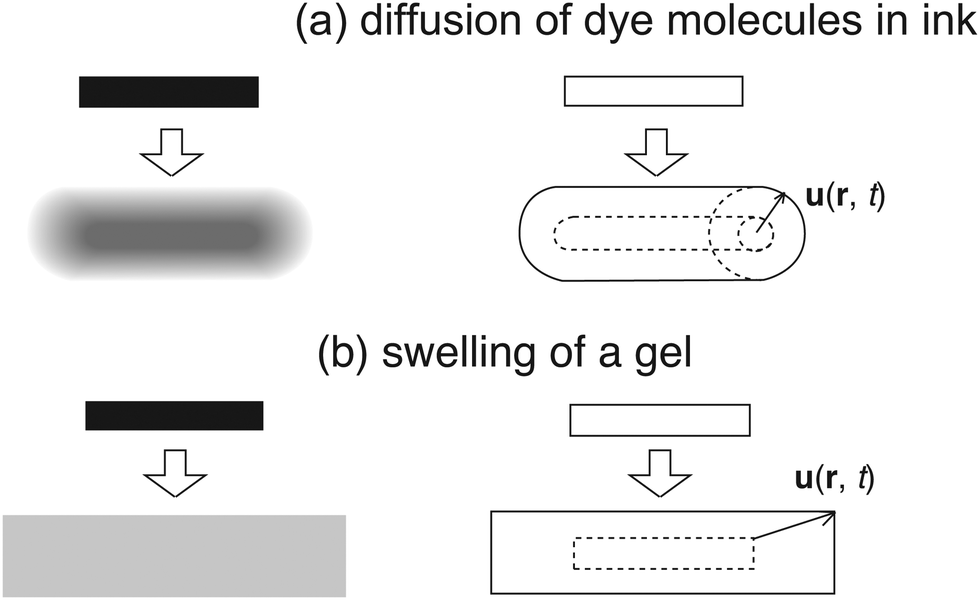
Fig. 4 schematically shows the difference in (a) diffusion of ink dye molecules on a filter paper and (b) swelling of a piece of gel. Note that in the case of (a) each dye molecule diffuses independently, while in (b) the gel is allowed to swell only while keeping its shape. This is the essential difference between the two, which originates from the absence/presence of connectivity, respectively, for ink dye and gel.
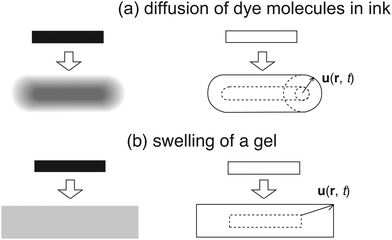 |
| | Fig. 4 Comparison of (a) diffusion of ink dye molecules on a wet filter paper and (b) swelling of a piece of gel. u(r, t) is the displacement vector. | |
2.3.1 DLS. DLS is a powerful tool for studying concentration fluctuations in soft matter.11 The time correlation function for the scattered electric field g(1)(q, t) is derived from eqn (3), and is proportional to the Fourier transform u(q, t) of the displacement u(r, t), and is given by| |
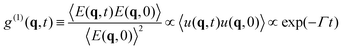 | (6) |
Here, q is the scattering vector and Γ is the relaxation rate defined by Γ = Dq2.The time correlation function of the scattered intensity is given by
| | |
g(2)(q, t) − 1 = |g(1)(q, t)|2 = exp(−2Γt)
| (7) |
Tanaka
et al. obtained the diffusion coefficient (
eqn (4)) of polyacrylamide hydrogels
via two methods; one using DLS with
eqn (7),
D =
DDLS, and the other
via a macroscopic static measurement of the frictional and elastic constants,
D =
Dmacro. Both were in good agreement with each other,
DDLS ≈
Dmacro,
12 which proved the validity of the THB theory beyond doubt.
2.3.2 Swelling kinetics: the TF theory. Since the driving force of the displacement of polymer chains is Brownian motion, the solution of the equation of motion (eqn (2)) is the diffusion equation (eqn (3)). On the other hand, in the case of the swelling kinetics of gels, the driving force is the osmotic pressure difference between the inside and outside of the gel and the effect of chain connectivity needs to be taken into account (as was schematically shown in Fig. 4).By solving the equation of motion of gels with the initial and boundary conditions of spherical gels in the spherical coordinate,13 the swelling equation was derived:
| |
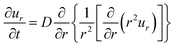 | (8) |
Here,
D is the diffusion coefficient defined in
eqn (4). The size change of a gel in the swelling process is given by:
| |
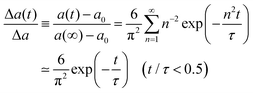 | (9) |
where
a0 and
a(
t = ∞) are the gel radius before swelling (
i.e.,
a(
t = 0) =
a0) and the final radius at swelling equilibrium,
a(
t = ∞) =
a∞ ≡
a, respectively, and
τ is the characteristic relaxation time of swelling given by
| |
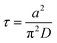 | (10) |
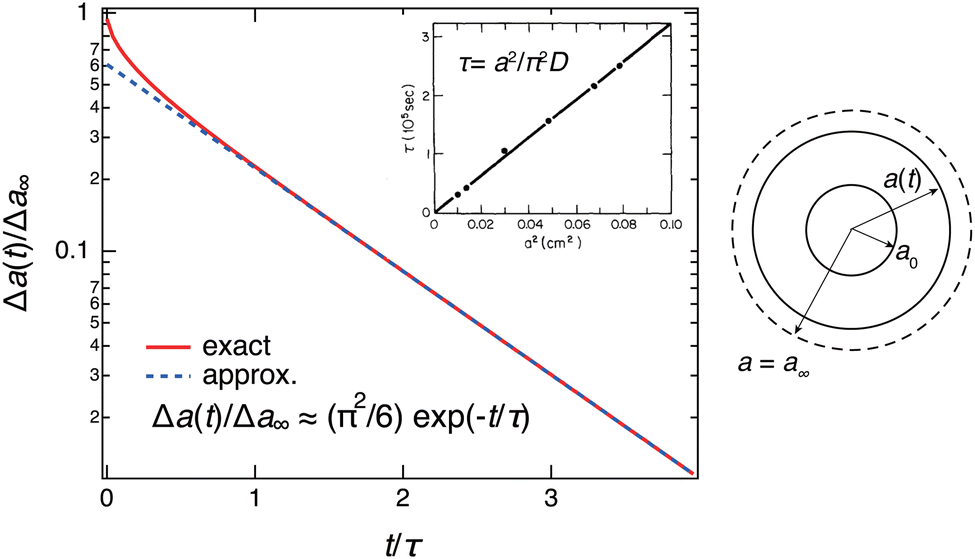
The time course of swelling for acrylamide hydrogels
13 is shown in
Fig. 5. The experimental results
13 agreed well with the theoretical prediction, and the characteristic relaxation time
τ and the diffusion coefficient of gel swelling
D(=
Dsw) were evaluated. It was confirmed that
τ is proportional to the square of the gel size
a2 (the inset of
Fig. 5) as predicted by the theory (
eqn (10)). In other words, the swelling rate 1/
τ becomes slower with the square of the gel size. This was a serious fundamental problem in the application of gels to gel actuators, which require rapid response. This problem has been solved by the novel design of gels,
i.e., comb-type grafted hydrogels.
37
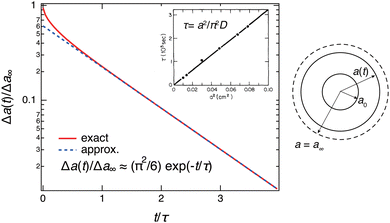 |
| | Fig. 5 Theoretical prediction of gel swelling kinetics (red-solid line, exact; blue-dashed line, predicted) and experimental results for acrylamide hydrogels with different final radii, a ≡ a∞. The definition of the symbols is shown on the right. The inset shows the experimental results on polyacrylamide hydrogels. (Replotted and modified from Fig. 4 and 6 in ref. 13.) | |
2.4 PNIPAM gels and responsive gels
Hirokawa and Tanaka discovered a VPT in nonionic gels, i.e., PNIPAM hydrogels.15 Here, the VPT is simply controlled by one of the easiest intensive variables, i.e., temperature, and the transition temperature is close to human body temperature. This sensational report on PNIPAM gels developed explosively in many fields both in fundamental science and also in applied science/engineering, namely in smart gels responsive not only to temperature, pH, and salt, but also to light and other external stimuli. Research on PNIPAM was reviewed by Schild38 and recently by Halperin.39
2.5 Thermodynamics of the VPT
Tanaka explained the VPT of polymer gels in terms of mean field theory based on the extension of Flory's theory.40 Here, the free energy of a gel consists of mixing enthalpy, mixing entropy, and elasticity terms. He derived the osmotic pressure of the gel and discussed its Taylor expansion with respect to the polymer fraction ϕ. The second virial coefficient becomes negative, corresponding to the discrete transition. This theory effectively reproduced the VPT.1,28 Hirotsu et al. studied the VPT of weakly charged poly(NIPAM-co-acrylic acid) copolymer gels P(NIPAM/AAc) and explained the VPT with the mean field theory.41 However, it should be noted here that there are some criticisms related to the mean-field assumption, namely the nonrandomness of the polymer–solvent interaction and hydrophobic interactions, the use of unrealistic values of the interaction parameter χ, and so on, in particular in the case of temperature induced VPTs (e.g., PNIPAM hydrogels).38,39 Hence, this issue has not been fully solved.42,43
2.6 Smart gels inspired by the VPT
The discovery of the VPT for temperature-responsive PNIPAM gels and its applications in smart gels seem to be other reasons why there was such a jump-wise increase in the publications. The smart gels44 referred to here are gel actuators (chemo-mechanical, electro-mechanical, photo-induced, etc.), gel sensors (pH, enzyme and antigen-responsive hydrogels,45 etc.), stimuli-responsive buckling gel films,46 shape-morphing materials,47 etc. Since there are a number of reviews in this regard, we focus on the structure and dynamics of the VPT in this review.
3 Macroscopic and microscopic views of the VPT
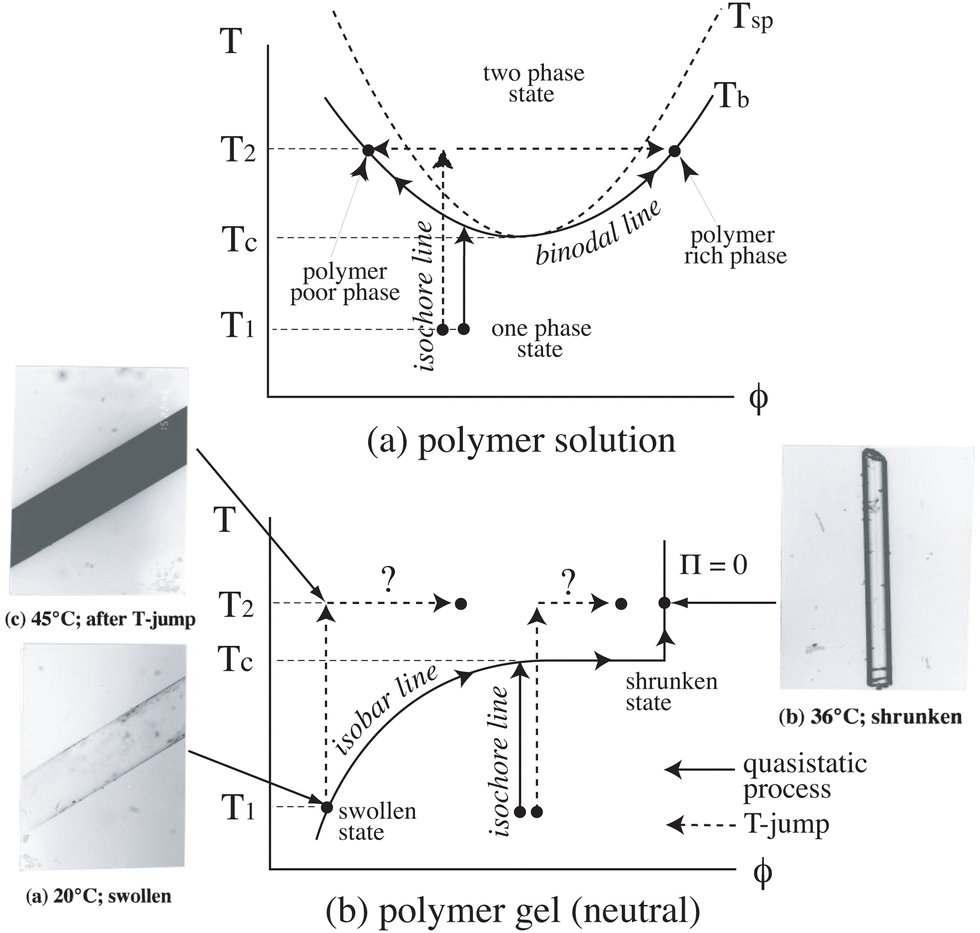
3.1 Phase diagrams: polymer gels vs. polymer solutions
Fig. 6 shows a comparison between phase diagrams of (a) polymer solutions and (b) polymer gels. Unlike polymer solutions, a polymer gel is an open system with its surrounding medium, e.g., water. This is why a gel can change its size so as to balance the chemical potentials in and out of the gel. This is realized by setting the osmotic pressure, Π, to 0. The photographs in Fig. 6b show the case of a cylindrical PNIPAM gel of submillimeter thickness. When the temperature is changed much slower than the relaxation rate of the gel, the gel can change its size along the isobar line and reaches a transparent shrunken state or a swollen state vice versa ((a) → (b) or (b) → (a)) without phase separation. However, when the temperature is changed much faster, the gel cannot reach a new equilibrium state, resulting in phase separation (turbid gel) ((a) → (c)). This is followed by slow structural relaxation and results in a transparent shrunken gel, as shown in the photographs ((c) → (b)).48 This example demonstrates a marked difference between polymer solutions and polymer gels. The VPT has been applied to many fields, such as drug delivery systems, sensors and actuators. For example, Miyata et al. demonstrated antigen-responsive hydrogels.45
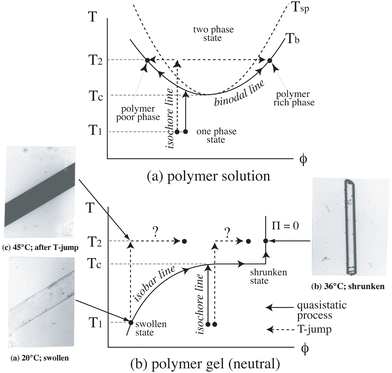 |
| | Fig. 6 Phase diagrams of (a) polymer solutions and (b) polymer gels (neutral/noncharged), and optical photographs of cylindrical gels. (Modified from parts of Fig. 1 in ref. 49). | |
3.2 Microscopic view of VPT: SANS
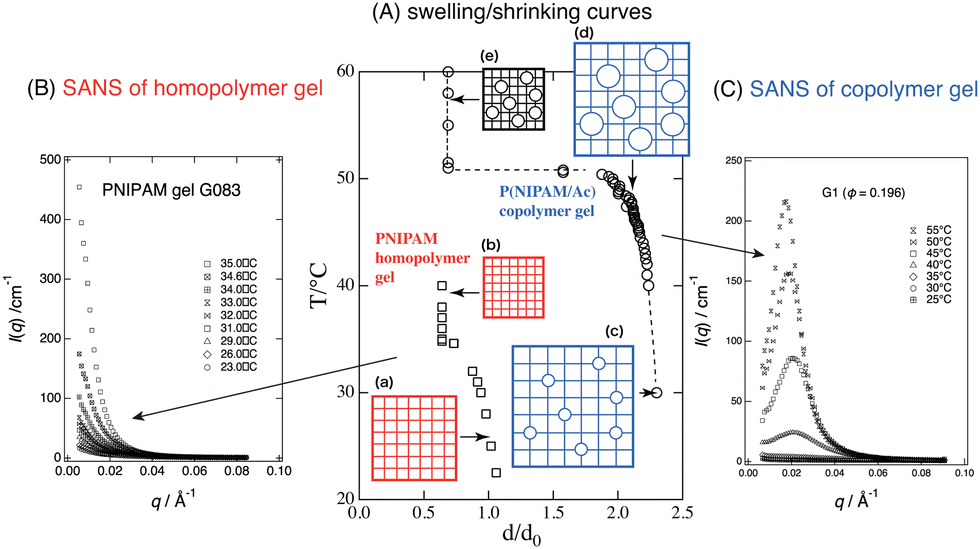
Small-angle neutron scattering (SANS) is a very powerful tool to study the structure of polymer gels because of various advantages: (i) large scattering contrast between hydrogenous polymer networks and deuterated solvent, (ii) a suitable target spatial range for gel mesh size, (iii) sensitivity to network inhomogeneities, and so on.50 The difference between continuous and discrete shrinking transitions of polymer gels was clearly elucidated via SANS experiments.
Fig. 7(A) shows the swelling/shrinking curves of PNIPAM (homopolymer) gel and P(NIPAM/AAc) copolymer gels, where d and d0 are the gel diameters at observation and in the reference state (i.e., at preparation; Tprep ≈ 22 °C), respectively, and d/d0 is the linear swelling ratio. When increasing the temperature, the homopolymer gel underwent a continuous or a slightly discrete VPT, as shown by the squares, while the copolymer gel underwent a discontinuous transition, shown by circles. The reason for the difference could not be elucidated via macroscopic observations, such as visual or optical microscopy.
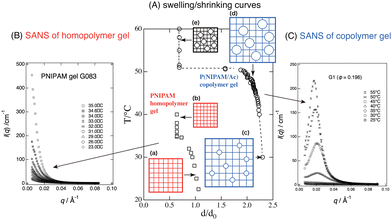 |
| | Fig. 7 Swelling/shrinking curves (T vs. d/d0) (A), and SANS profiles for PNIPAM (homopolymer) gel (B) and P(NIPAM/AAc) (copolymer) gel (C). Homopolymer gels undergo a continuous VPT, while copolymer gels undergo a discrete VPT. SANS of the former shows critical divergence at TPNIPAM ≈ 32 °C, while the latter shows a marked peak at T > TPNIPAM. The cartoons of square grids and circles show PNIPAM networks and ionized chain regions associated with AAc comonomers, respectively. The cartoons indicate that the size of the gel (grids) becomes smaller via hydrophobic contraction and the circles become larger via electrostatic repulsion, with changing T. (Modified from parts of Fig. 2 in ref. 49.) | |
Fig. 7(B) (homopolymer gel) shows divergence of the scattered intensity with increasing T, indicating critical fluctuations ((a) → (b)).23 On the other hand, Fig. 7(C) (copolymer gel) shows a first-order discrete transition accompanied by microphase separation ((c) → (d) → (e)).24 Here, the microphase separation was induced by antagonistic interactions: the hydrophobic interaction of the PNIPAM network chains (shown by the lattice; inward favored) vs. the Donnan potential originating from the charged acrylic acid groups (shown by circles; outward favored). This is the first demonstration and evidence of a microscopic view of a discrete volume phase transition.
4 Gel inhomogeneities
Tanaka did not explicitly discuss inhomogeneities of gels in his works, but he recognized them through experimental observations, such as patterns in shrinking gels.26 The existence of inhomogeneities in gels was already reported in the 1970s via static light scattering (SLS)51 and later via DLS, small-angle X-ray scattering (SAXS), and SANS.52–55 Geissler et al. proposed empirical equations for SANS and SAXS of gels in swollen poly(dimethylsiloxane) gels:55| |
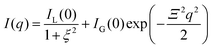 | (11) |
where ξ is the correlation length in the solution-like part of the gel, and Ξ is the size of solid-like domains, e.g., highly cross-linked parts in the network. IL(0) and IG(0) are the corresponding prefactors. Later, the inhomogeneities were theoretically explained by Panyukov and Rabin (hereafter we call this the PR theory) as frozen inhomogeneities56,57 formed during the gelation/cross-linking process.
4.1 Shrinking kinetics
Since gel inhomogeneities are introduced during the gelation process, the structure and properties strongly depend on how the gel is prepared, e.g., the temperature Tprep and concentration Cprep at gel preparation. Certainly, the gel structure also depends on these at observation, i.e., Tobs and Cobs. Hence, in order to describe the gel structure, two sets of parameters are required, i.e., (Tobs, Cprep) and (Tprep, Cprep), as discussed for the PR theory.56,57
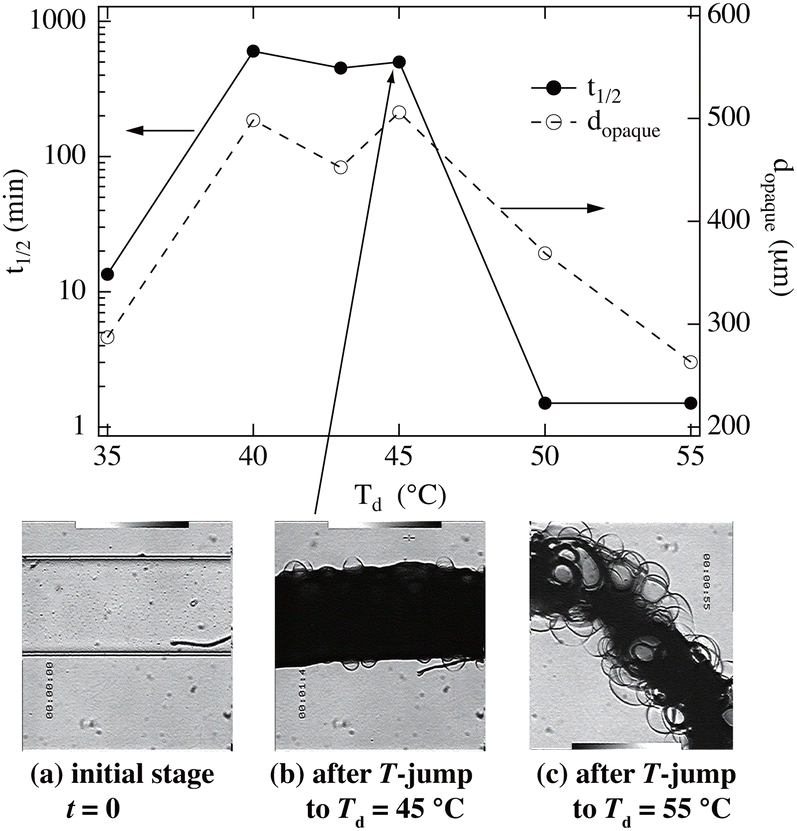
Fig. 8 demonstrates the shrinking kinetics and morphology of PNIPAM gels prepared at 20 °C and when jumping to different destination temperatures, Td.58 This figure shows that the shrinking kinetics strongly depend on Td. If Td ≈ TPNIPAM, the shrinking half time t1/2 is around 20 min. It dramatically increases to 600 min for 40 ≤ Td ≤ 45 °C, whereas it becomes much faster for Td ≥ 50 °C. Interestingly, this change is well-correlated with the gel diameter dopaque at which the gel becomes opaque (an indication of macroscopic phase separation). This means that the gel shrinking is slowed down (quenched) by macroscopic phase separation (the middle photograph). On the other hand, if the Td is much higher than TPNIPAM, the gel does not have time to macrophase-separate and shrinks to the desired shrunken state. Here, bubbles are formed, as shown in the right photograph (Fig. 8(c)). This photograph also indicates that gels are, in general, inhomogeneously prepared with water-rich and -poor domains.
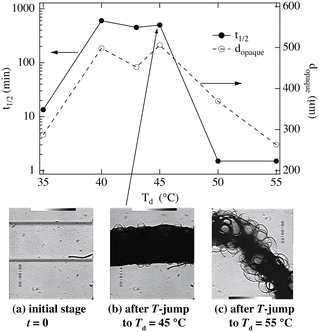 |
| | Fig. 8 Shrinking kinetics of PNIPAM gels with T-jumps to different temperatures, Td.58 (Upper) t1/2 (left) and dopaque (right) vs. Td. (Lower) Optical photographs of the gel (a) in the initial state, and after T-jumping to (b) Td = 45 °C and (c) to Td = 55 °C. (Modified from parts of Fig. 3 in ref. 49.) | |
Note that skin formation is believed to be one of the reasons that delay gel shrinking.59 However, this is a competition between thermal diffusion vs. structural relaxation of the gel network. As discussed for eqn (10), the latter is strongly dependent on the size of the gel. In the case of the gel shrinking kinetics treated in this work, the gel diameter is submillimeter, where the thermal diffusion (T-jump) is much faster than the time for skin formation.
The molecular picture of the distinct difference between the fast and slow response of gel shrinking relates to the connectivity. This leads to cooperative motion as discussed in Fig. 4. That is, due to chain connectivity, it takes a certain period of time, τequib to reach equilibrium, but if the temperature change is much faster than τequib, the structure is frozen. In comparison with polymer gels, PNIPAM solutions (not gels) instantaneously undergo phase separation or precipitation at TPNIPAM irrespective of the rate of temperature change. This instant response is due to the absence of cross-linkers and each polymer chain can behave independently (not collectively as in the case of polymer gels).
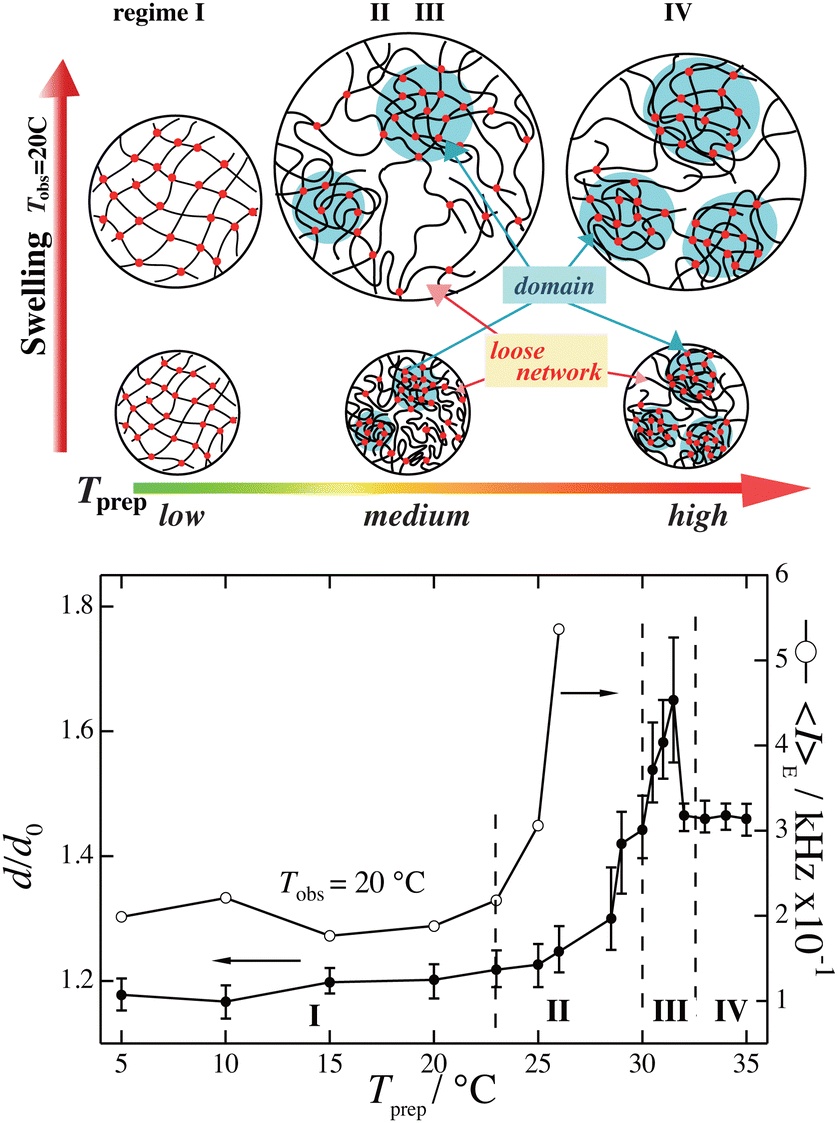
Fig. 9 shows the preparation temperature, Tprep, dependence of the linear swelling ratio, d/d0, of PNIPAM gels observed at Tobs = 20 °C.60 As shown in the figure, d/d0 strongly depends on Tprep and dramatically increases when approaching TPNIPAM ≈ 32 °C. The right axis shows the light scattering intensity, 〈I〉E, which also increases with Tprep, where 〈I〉E is the ensemble average scattering intensity as defined below. The changes in the gel network structure are schematically shown in the top of Fig. 9, indicating that gels prepared near TPNIPAM are more inhomogeneous than those prepared at lower temperatures. This is an example of the case of PNIPAM hydrogels, but such inhomogeneities are commonly observed in polymer gels.
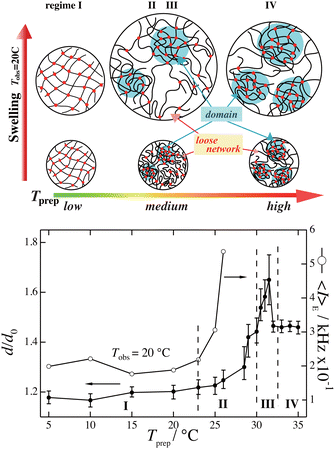 |
| | Fig. 9 (Top) Gel inhomogeneity model for PNIPAM gels prepared at different temperatures Tprep and observed at Tobs = 20 °C. (Bottom) Linear swelling ratio d/d0 vs. Tprep (left; filled circles) and light scattering intensity 〈I〉E vs. Tprep (right; open circles).60 (Modified from parts of Fig. 4 in ref. 49.) | |
4.2 Structure and dynamics
In the late 1970s, de Gennes anticipated the importance of the two new techniques in polymer physics, “neutron diffraction” and “light scattering”, in the preface of his book “Scaling Concepts in Polymer Physics.”61 These refer to SANS and DLS, respectively. Certainly, developments of DLS studies of polymer gels are owed to Tanaka. Later, the concept of inhomogeneities were taken into account in DLS analyses both theoretically and experimentally.62–64 On the other hand, SANS of gels developed rather phenomenologically until the PR theory was proposed.56,57 In this section, both SANS and DLS studies on polymer gels are overviewed.
4.2.1 Gel research with SANS. SANS became one of the most powerful tools for investigations of the structure of polymer gels because of the following reasons:50 (1) it is not necessary to deuterate the polymer component, but the major component, i.e., the solvent, can be simply substituted with a deuterated solvent in most cases to enhance the scattering contrast, as well as to reduce the strong contribution of incoherent scattering from hydrogenous components.65 (2) A variety of labeling methods,50 such as labeled networks, labeled paths, labeled chains, and labeled cross-links, can be chosen depending on the purpose of the study. (3) Reference samples, such as non-cross-linked polymer solutions, are easily available to study the effects of cross-links. (4) Versatile sample environments, such as gel deformation, shearing, rheometry and high pressure, are available because of the strong neutron penetration power.48As a result of many experimental works and discussions, the following SANS function is commonly used to analyze the scattering functions of polymer gels:66
| | |
I(q) = Isoln + Iexcess ≈ IOZ(q) + IDB(q)
| (12) |
| |
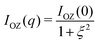 | (13) |
| |
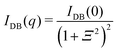 | (14) |
where
IOZ(
q) is the scattering function of polymer solutions and
ξ is the correlation length, meaning the screening length of a semidilute solution (the blob length). This equation is the Ornstein–Zernike equation.
67 On the other hand,
IDB(
q) is the excess scattering arising from gel inhomogeneities. This equation is the so-called Debye–Bueche equation, representing two-phase structures with the characteristic inhomogeneity length
Ξ.
68 Ξ is related to the chord lengths of the A domain (
e.g., the cross-link-rich domain)
lA and of the B matrix (
e.g., the cross-link-poor matrix)
lB (or
vice versa), respectively, as follows:
69| |
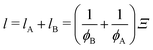 | (15) |
| |
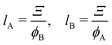 | (16) |
where
ϕA and
ϕB are the volume fraction of the cross-link-rich domain and cross-link-poor matrix, respectively. The validity of
eqn (12) is supported by the theory of Panyukov and Rabin.
56,57 A review of SANS works for polymer gels can be found in ref.
50 and
66, where other topics, such as deformation and contrast variation, are also discussed.
4.2.2 Gel research with DLS. Starting from the THB theory, dynamic light scattering (DLS) studies have also been a common tool to study the structure of polymer gels. In the case of polymer solutions, the scattering intensity is independent of the sampling position (ergodic system). In the case of polymer gels, on the other hand, the scattering intensity strongly depends on the sampling position.62,64 This is because gels are nonergodic systems in which the time average is not equal to the ensemble average:Hence, in order to describe polymer gels, two types of average scattering intensities have to be defined, i.e., the time average scattering intensity 〈Ip〉T, and ensemble average scattering intensity 〈I〉E. Here, 〈Ip〉T is the scattering intensity of a long-term average at a sample position p, and 〈I〉E is an ensemble average of 〈Ip〉T as given by| |
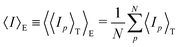 | (18) |
Here, p is the sampling position and N is the number of sampling positions.The scattering intensity 〈Ip〉T is given by
| | |
〈Ip〉T = 〈IF〉T + IC,p
| (19) |
where the subscript F means the fluctuating (dynamic) component.
The scattering field now consists of a dynamic component EF(t) and a static component EC,p, and the latter is sample-position (p)-dependent, as is written by
| | |
Ep(t) = EF(t) + EC,p
| (20) |
Here, the time correlation functions of these two fluctuations correspond to the respective intensities,
IF and
IC:
| |
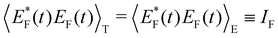 | (21) |
| |
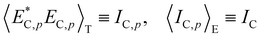 | (22) |
The time correlation function for the dynamic fluctuations is the same as that given by Tanaka, i.e., eqn (6) and (7). On the other hand, that for nonergodic systems is now given by64
| | |
〈g(2)p(q, t)〉T − 1 = Xp2|g(1)(q, t)|2 + 2Xp(1 − Xp)|g(1)(q, t)|
| (23) |
where
| |
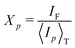 | (24) |
DLS analyses can be done by simply fitting the correlation function, 〈g(2)p(q,τ)〉T, by using eqn (7). However, it is more convenient to use the following equation:
| | |
〈g(2)p(q,τ)〉T − 1 = σI,p2exp(−2DA,pq2τ)
| (25) |
where
DA,p (
D ≤
DA,p ≤ 2
D) is the apparent diffusion coefficient at position
p. This is because the difference between
D and 2
D is not easy to distinguish in the fitting process.
σI,p2 is the initial amplitude of the correlation function given by
| | |
σI,p2 = 〈g(2)p(τ = 0)〉T − 1 = Xp(2 − Xp)
| (26) |
By taking the
τ = 0 limit of the equation, one obtains
DA,p:
| |
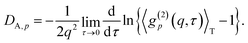 | (27) |
Here, the relationship among
DA,p,
Xp, and
σI,p is given by
| |
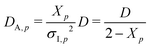 | (28) |
On the other hand, 〈
Ip〉
T has its lower bound 〈
IF〉
T. Since 〈
Ip〉
T is the scattering intensity corresponding to the thermal fluctuations, its lower bound is proportional to the absolute temperature
T.
The distribution of 〈Ip〉T is given by
| |
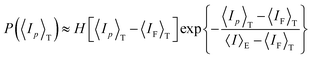 | (29) |
Here,
H(
x) is the Heaviside step function;
H(
x) = 1, (
x ≥ 0),
H(
x) = 0, (
x < 0).
From eqn (24) and (28), one obtains
| |
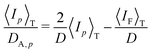 | (30) |
This equation is useful to decompose the scattering intensity (and/or the concentration fluctuations) to the static and dynamic components and to evaluate the diffusion coefficient
D (=
DDLS).
70,71
Fig. 10 shows an example of the decomposition plot of polymer gels: (a) the scattering intensity variations with sample position, i.e., speckle patterns, typical for polymer gels; (b) two correlation functions for solution-like and solid-like parts in a gel; (c) the distribution function of the apparent diffusion coefficient DA, which is a decreasing function of 〈Ip〉T with a lower cutoff 〈IF〉T; and (d) the decomposition plot from which D(= DDLS), 〈IF〉T, and 〈IE〉T can be evaluated.71
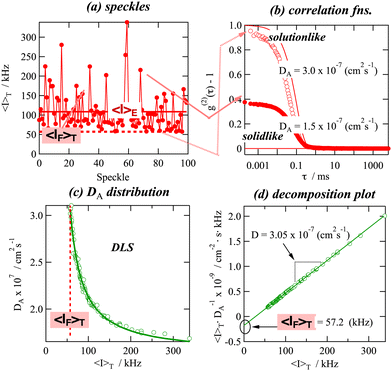 |
| | Fig. 10 DLS decomposition plot. (a) Speckles, (b) correlation functions, (c) DA distribution, and (d) decomposition plot. (Reprinted with permission from ref. 71. Copyright 2006 Chemical Society of Japan.) | |
DLS has made long strides in incorporating the concept of inhomogeneities, and provides various pieces of information: the mesh size, the sol–gel transition point,71–73 the separation of static/dynamic concentration fluctuations,70,71 probe diffusion,74,75 etc.
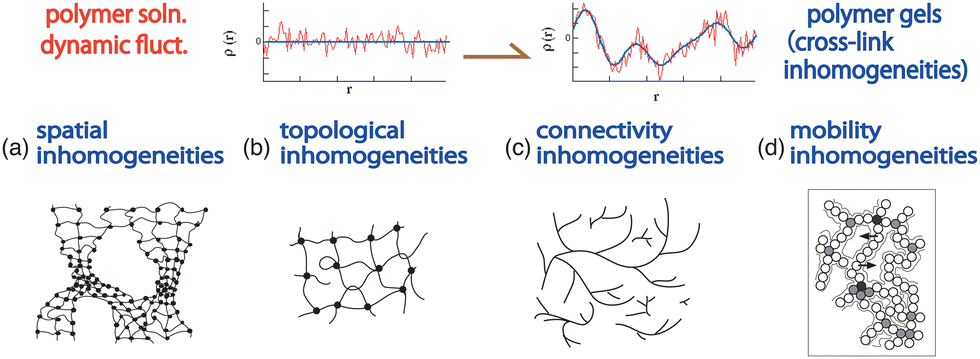
4.3 Classification of gel inhomogeneities
Polymer gels inherently contain various types of inhomogeneities that are introduced during gel preparation stages, such as the polymerization/cross-linking stage, or kinetically trapped in the sol–gel transition stage. Shibayama discussed gel inhomogeneities76 and classified gel inhomogeneities into spatial, topological, connectivity, and mobility inhomogeneities.71 Fig. 11 schematically shows gel inhomogeneities.71 The upper row shows the concentration fluctuations of polymer solutions (left) and polymer gels (right), where frozen inhomogeneities (blue line) are superimposed on dynamic fluctuations (red line). The lower row shows the types of inhomogeneities, i.e., spatial, topological, connectivity, and mobility inhomogeneities (from left to right). These various inhomogeneities can be investigated via SANS, swelling/shrinking experiments, DLS, and light scattering intensity speckles, respectively. A recent review by Seiffert followed up the literature of SANS as well as small-angle X-ray scattering (SAXS) and static and dynamic light scattering.77
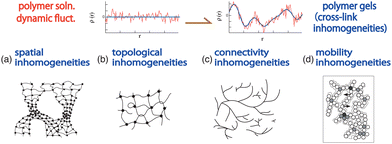 |
| | Fig. 11 Various types of inhomogeneities. (Upper row) Schematics of concentration fluctuations: (left) polymer solutions and (right) polymer gels where frozen inhomogeneities (blue) are introduced by cross-linking, superimposed onto thermal fluctuations (red). (Lower row; from left to right) (a) Spatial, (b) topological, (c) connectivity, and (d) mobility inhomogeneities. | |
5 Toward realization of ideal polymer networks
5.1 Tetra-PEG gels
Gels are believed to be inherently inhomogeneous because cross-linking is an inevitable process of gel preparation. Many researchers have tried to prepare defect-free homogeneous gels, but have failed. In 2008, Sakai et al. succeeded in fabrication of near-ideal polymer gels with monodisperse mesh sizes, called tetra-PEG gels.78 The tetra-PEG gels were prepared by cross-end-coupling of N-hydroxysuccinimidyl (NHS)-terminated tetra-PEG macromers and amine-terminated tetra-PEG macromers. The typical molecular weights Mw of commercially available PEG macromers are 5 kDa to 40 kDa. These tetra-PEG macromers were separately dissolved in neutral buffered water. By simply mixing the two aqueous solutions with vigorous stirring, gelation occurred.
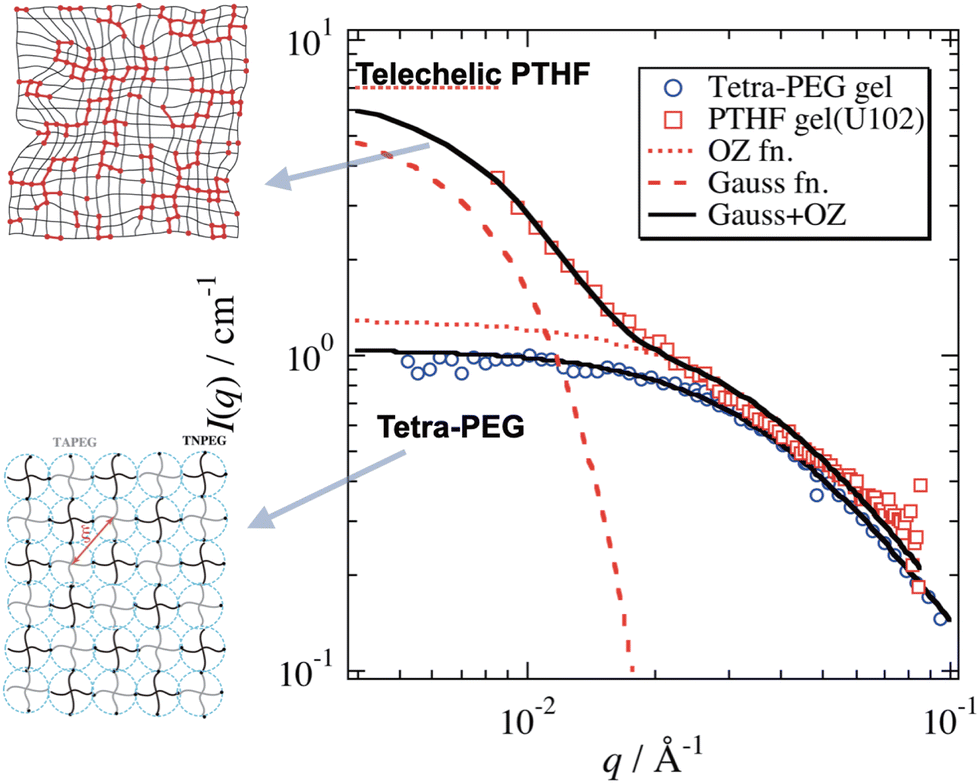
Fig. 12 shows the SANS functions of a poly(tetrahydrofuran) (PTHF) network gel and tetra-PEG gel. The former consisted of end-linked PTHF networks having a fairly narrow molecular weight distribution, with a polydispersity index of 1.20.79 Even though it was regarded as a unimodal model polymer network, its SANS function had a strong forward scattering, which was fitted with eqn (11). On the other hand, the SANS function of tetra-PEG did not show such a forward scattering80,81 and was simply fitted with the Ornstein–Zernike function IOZ(q) in eqn (13). In the review paper of the structure–mechanical properties of tough hydrogels,66 the difference in SANS between conventional gels (PTHF gel) and tetra-PEG gels is discussed. The main conclusion of the difference was the presence/absence of low q-scattering, respectively, for a conventional gel and tetra-PEG gel.
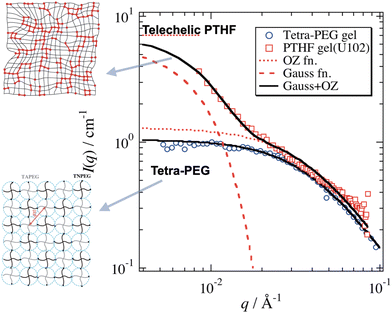 |
| | Fig. 12 SANS functions of a conventional PTHF gel and tetra-PEG gel. Solid lines are fitted with eqn (11) with/without a Gaussian component for the PTHF gel and tetra-PEG, respectively. The cartoons on the right schematically show the network structures. (Modified from Fig. 3 of ref. 66.) | |
Though the combination of NHS-terminated and amine-terminated tetra-PEG macromers was very successful,78 it had a drawback. Because the NHS-terminated tetra-PEG macromers gradually hydrolyzed spontaneously in water, the activity of the NHS end-group deteriorated with time.82 For preparation of defect-free gels, a combination of maleimide-terminated and thiol-terminated tetra-PEG macromers is more suitable for preparation of precisely controlled tetra-PEG gels, because there is negligible byproduct in this coupling reaction.83,84
5.2 Physics driven by tetra-PEG gels
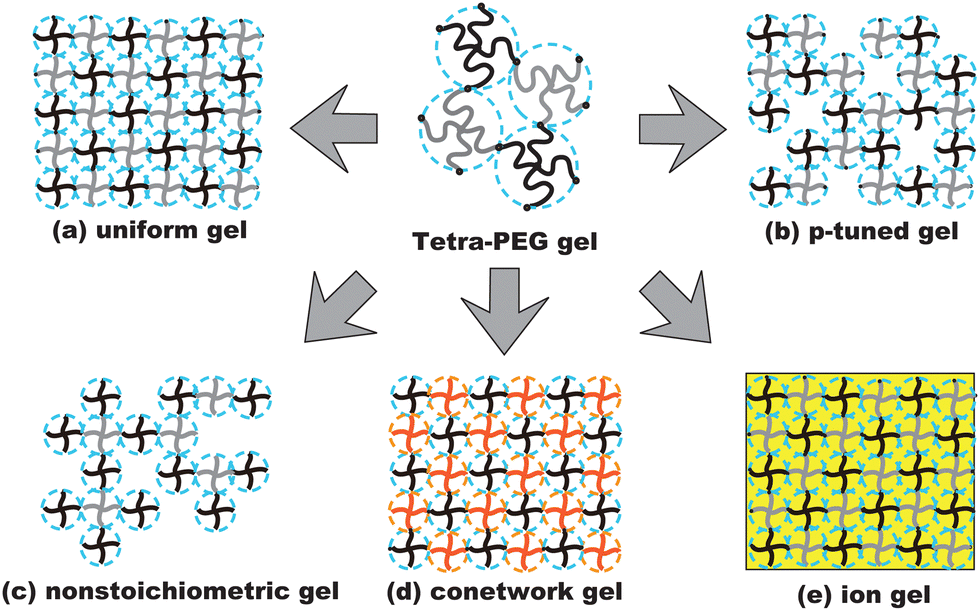
Because of their uniform network structure, tetra-PEG gels have been used for various purposes and applications.85 Fig. 13 illustrates the physics developed because of the discovery of tetra-PEG gels. The uniform network structure of tetra-PEG gels (a) allowed investigations of the deformation mechanism,86 a model network system for examination of rubber elasticity,87,88 molecular sieves,82 microscopic dynamics with probe diffusion,75 and the kinetics of tetra-PEG gels.89,90 The tunability (p-tuning) of the network structure (b) is used for fabrication of controlled-defect networks (p-tuned gels) and for percolation studies, where p (0 < p ≤ 1) is the bond probability of the network.88,91 Non-stoichiometric gelation of tetra-PEG gels (nonstoichiometry) (c) is used for studying critical gels.92 Conetwork gels (d) obtained by copolymerization with environment-sensitive tetra-macromers have also opened a new direction for various applications, including thermosensitive gels,93,94 solvent-sensitive gels,95 and model polyelectrolyte gels.96,97 Regarding (d), nonswellable gels that do not swell in the body have been fabricated. In medical applications, nonswellability is crucial to avoid damaging tissue. Such a nonswellable gel was made by fabricating a conetwork in which the components have opposite temperature dependence.94 Fabrication of tetra-PEG gels in ionic liquids (ion gels) (e) opened a new door for realization of non-volatile gels.98–100
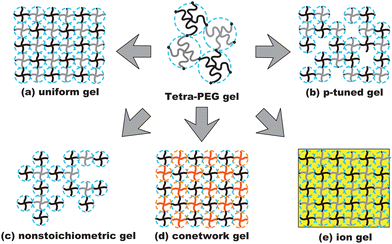 |
| | Fig. 13 Schematics of the derivatives of tetra-PEG gels: (a) uniform gels, (b) p-tuned gels, (c) nonstoichiometric gels, (d) conetworks, and (e) ion gels. Black and grey lines show a set of NHS-terminated tetra-PEG and amine-terminated tetra-PEG macromers, or a set of maleimide-terminated and thiol-terminated tetra-PEG macromers. Red lines denote environment-sensitive macromers, such as thermosensitive macromers. The yellow box shows an ionic liquid environment instead of an aqueous environment. | |
Here, percolation theory is a theory that deals with how target substances are connected within a system and how their characteristics are reflected in the properties and dynamics of the system.101,102 There are two basic types of percolation: bond percolation and site percolation. Let us consider a lattice model, which can be a two dimensional or three dimensional model or something else. In the case of bond percolation, all sites are pre-occupied by monomers and the connectivity is statistically controlled by the bond probability, pb. When any finite clusters interconnected via bonds reach the system size, a bond percolation is attained. In the case of site percolation, on the other hand, monomers are randomly placed in sites of the lattice. If there is a neighbor, they spontaneously bond to each other and form clusters. The fraction of occupancy is defined as the site probability, ps. When any of the finite clusters reach both ends of the system, it is called percolation. Another type of model called site-bond percolation, proposed by Coniglio et al.,103 is more suitable for description of polymer gels because the site corresponds to a monomer and the bond corresponds to a cross-linker.71,104
In the following, two examples, i.e., re-exploration of rubber elasticity and of diffusion coefficients, are given.
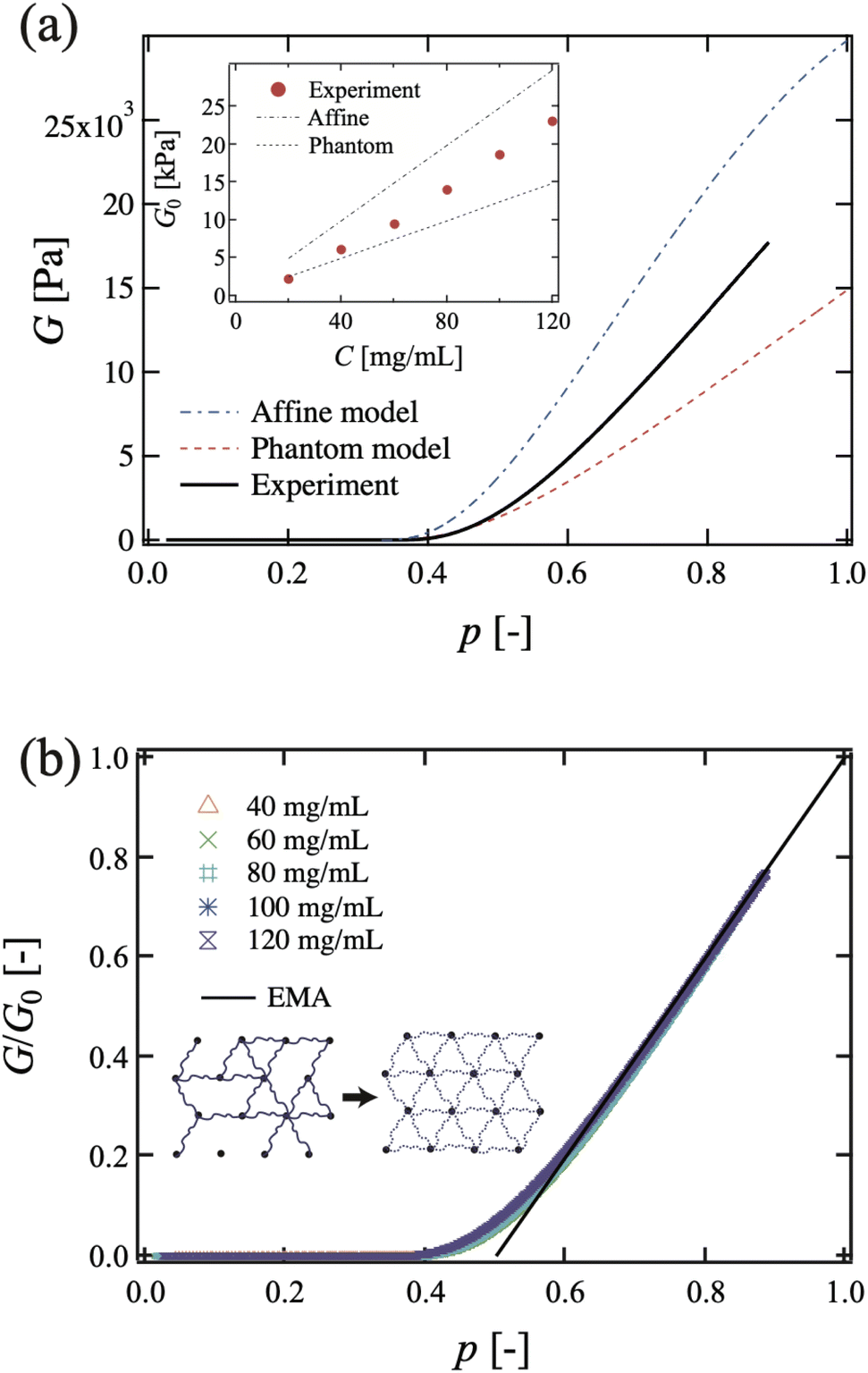
5.2.1 Rubber elasticity. Nishi et al. examined rubber elasticity by using entanglement-free model networks.106 They prepared well-defined model polymer networks with maleimine- and thiol-terminated tetra-PEGs, and measured the elastic modulus G for a broad range of polymer concentrations and connectivity probabilities, p.106 They first confirmed that the percolation threshold is pc ≈ 0.39, which is analogous to the conductivity of a resistor network107 and not to the classical model (0.33 for a tetra-arm Bethe lattice). They also observed two distinct features different from those believed for classical Gaussian chain behaviors: (1) the critical exponent t on the shear modulus G near the gelation threshold, G ∝ |p − pc|t, where p and pc are the fraction of bonds that connect neighbor sites and the percolation threshold, respectively, and (2) deviation from affine and phantom chain models. Regarding (1), the classical theories predict t = 3,61 whereas de Gennes and Daoud predict t = 1.9107 and 2.6,108 respectively. Nishi et al. observed t = 1.95 ± 0.05 for a wide range of initial gel concentrations, 40 mg mL−1 ≤ C0 ≤ 120 mg mL−1 and 0.03 ≤ p − pc. They confirmed the validity of the effective medium theory (EMA) of percolation on a central-force elastic network.88Regarding (2), Fig. 14 shows (a) an experimental G–p plot and theoretical predictions for the affine model and phantom model (C0 = 120 mg mL−1), and (b) the p dependence of the reduced elastic modulus at various C0 values. Note that the G/G0–p curves fall onto a single master curve, where G0 is that of p = 1 and the solid line corresponds to the theoretical prediction from the effective medium approximation (EMA). In the inset, g0 is the elastic constant of a single chain and gm is the normalized elastic constant of the uniform effective medium.105,106
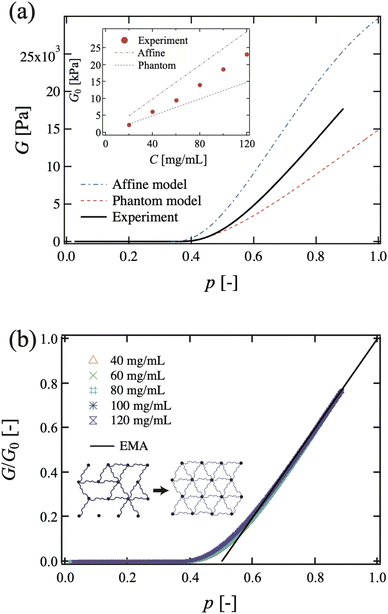 |
| | Fig. 14 (a) Experimental G–p plot and theoretical predictions for the affine model and phantom model (C0 = 120 mg mL−1). Inset: Concentration dependence of the elastic modulus at p = 1, as evaluated from the fitting result. The theoretical predictions of the affine and phantom network model are also shown. (b) The p dependence of the reduced elastic modulus at various C0 values. The solid line corresponds to the theoretical prediction from the effective medium approximation (EMA). Inset: Schematic illustration of the EMA. The network on the left represents the original system in which neighboring sites are randomly connected with chains of elastic constant g0. The network on the right represents the EMA with a nondisordered structure in which all neighboring sites are connected with chains of elastic constant gm. (Reprinted from ref. 106 with permission. Copyright 2017 American Physical Society.) | |
5.2.2 Diffusion coefficients. Sakai et al. revisited some of the essential problems of the physics of polymer gels, such as solvent diffusion, the shear modulus and the friction factor, using near-ideal polymer network gels.109 Their studies include the permeation of water through hydrogels with a controlled network structure,83 comparison of the three diffusion coefficients (Dsw, via swelling experiments; DDLS, via DLS; and Dw, via water permeation),110 mixing and elastic contributions to the diffusion coefficient of polymer networks,111 the shear modulus dependence of the diffusion coefficient of a polymer network,112 and structure–property relationships of a model network containing solvent.113 Here, the scaling theory on the friction factor and the blob size is experimentally derived,83 and the validity of the THB and TF theories is discussed.110–112 These works could only be achieved with monodisperse-strand model networks free of entanglements and defects.
5.3 Flexible and highly ordered three-dimensional networks: super-homogeneous gels
Although tetra-PEG gels are very homogeneous with a low level of defects and are regarded as “near-ideal polymer networks”, there still remain noticeable speckles and forward scattering. Recently, Li et al. employed an innovative strategy and succeeded in the fabrication of “super-homogeneous gels.”114 This is based on the “bond percolation” model, which is one of the classical percolation models,103 as discussed above. The bond percolation model assumes that the space is uniformly prepacked with mutually exclusive units in the beginning, and the percolation occurs as a consequence of cross-linking between the nearby units, i.e., a reaction-controlled process.115 Because the units are mutually exclusive, the space is always uniformly filled by the units regardless of the extent of cross-linking and network formation, leading to a highly ordered, ideal network structure after the reaction completes.
The following is the protocol of the preparation of the bond-percolation gels: (i) it starts with monodisperse star polymers, e.g., four-armed poly(ethylene glycol), as space filling units (Fig. 15A) because polymers with multiple arms show a strong excluded volume effect that prevents other polymers from coming into the pervaded volume.116 Then, (ii) dissolving the star polymers in a good solvent at a concentration well above their chain overlapping concentration ensures that the star polymers uniformly and tightly fill the space (Fig. 15B) as the bond percolation model assumes. (iii) To prevent the segregation of polymer chains during cross-linking,117 dehydrated acetonitrile is chosen as the solvent because it shows excellent affinity to the star polymers used in this study. Moreover, (iv) to remove dust and nanobubbles118 from the solution as much as possible, the dissolved polymer solutions are filtered through ultrafine syringe filters. Although all the aforementioned issues have already been pointed out separately in past studies, they have never been resolved altogether in a single system.
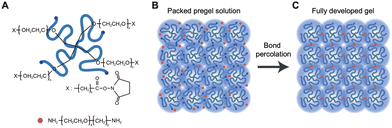 |
| | Fig. 15 Schematic of the gel preparation via bond percolation. (A) Star polymer: tetrafunctional poly(ethylene glycol) (PEG) with active ester end groups; bifunctional cross-linker: 1,14-diamino-3,6,9,12-tetraoxatetradecane (amino-PEG4-amine). (B) Stoichiometric mixture of the star polymer and the cross-linker in a good solvent. The system is uniformly prepacked with the star polymers. (C) Polymer gel formed by end-linking of the star polymers with the small cross-linkers via bond percolation. 2D schematics are shown instead of the real 3D polymer network for the sake of legibility. (Reprinted from a part of Fig. 1 of ref. 114 with permission. Copyright 2019 American Association for the Advancement of Science.) | |
The thus-obtained gel (Fig. 15C) was examined using SLS and SAXS over a wide q range and it was confirmed that the scattering function of the gel is almost identical to that of the pregel solution and is well represented by the OZ equation. This indicates that (1) gelation of the star polymers proceeded via bond percolation and (2) the spatial correlation between the polymer chains did not change with cross-linking. Furthermore, speckle patterns, which are an indication of spatial defects and the sol–gel transition, were not observed at all throughout the whole gelation process.
The success of the development of “super-homogeneous gels” has opened a new door in gel science for further refinement of the theories of rubber elasticity, polymer gels, polymer networks, and polymer solutions, but also for advanced applications in filtration,119 sensing,120 drug release,121 electronics,122 and so on. The realization of super-homogeneous gels would be a key theoretical and technological advance in polymer science and technology because of their defect-free and entanglement-free three-dimensional network structure.
6 Future directions
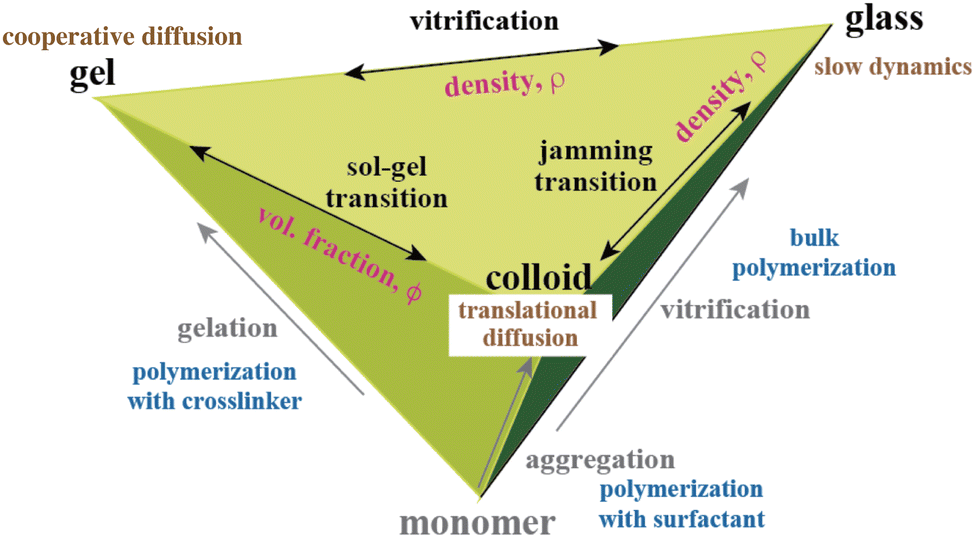
The history of the physics of polymer gels has been considered along with the footprints of Toyoichi Tanaka. One of the future directions of gel physics would be deeper and unified understanding of connectivity- or percolation-related phenomena, i.e., sol–gel transitions, vitrification, and jamming transitions. The physics of polymer gels, polymer glasses, and colloids has been developed rather independently, but could be generalized via the key word of “connectivity” or “percolation”.
Fig. 16 shows the relationship among polymer gels, polymer glasses, and polymer colloids. Gels are made by polymerization in the presence of cross-linkers. Polymer colloids are made from monomers in solvent in the presence of a surfactant. Glasses are formed by bulk polymerization of monomers. Here, the significant phenomena in physics are sol–gel transitions, vitrification, and jamming transitions. As shown in the figure, these transitions also exist between gels and colloids, between gels and glasses, and between colloids and glasses by tuning the “density” (ρ = monomer concentration, cross-linker concentration, surfactant concentration, etc.). Here, translational diffusion, cooperative diffusion, and slow dynamics have been hot topics in soft-matter physics. Scattering and spectroscopic methods, including DLS and SANS, have been used for unveiling the physics connecting these different materials, namely, vitrification vs. gelation123 and jamming transitions,124 which will be further advanced in the future by using modern experimental methods.
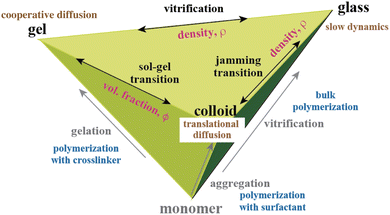 |
| | Fig. 16 Gel–glass–colloid diagram. Polymer gels, polymer glasses, and polymer colloids have a common origin, monomers, but are formed in different environments/conditions. There are at least interconversions via connectivity transitions, such as sol–gel transitions (chemical and/or physical bond; polymer volume fraction ϕ), jamming transitions (space filling; colloid density ρ), and vitrification (mobility freezing; gel density ρ). | |
Another and more alluring direction of gel physics is the understanding of life and biological functions, in which Tanaka himself was deeply involved in his last decade. As is well known, life is defined by its capacities for homeostasis, organization, metabolism, growth, adaptation, response to stimuli, and reproduction. We have been learning and imitating these biological functions. Responsive gels are typical examples and have been developed since the 1990s.34,35 Today, synthetic gels with sophisticated biological functions are designed and realized, such as self-healing gels, self-growing gels,125,126 etc. This direction will be a great step forward.
7 Conclusions
The history of the physics of polymer gels over about half a century, starting with the work of Toyoichi Tanaka, has been reviewed. In particular, (1) the Tanaka–Hocker–Benedek theory, and the discoveries of (2) the volume phase transition of polymer gels and (3) the temperature-induced volume phase transition have influenced not only polymer science but also other fields.
The understanding of gel inhomogeneities led not only to the developments of various gel characterization methods, such as neutron scattering, X-ray scattering, and pulsed NMR, but also to providing novel ideals to circumvent various problems that conventional gels have and to the search for defect-free homogeneous ideal polymer gels.
The fabrication of ideal polymer networks has great physical significance that can be compared with the success of the preparation of monodisperse polymers by Szwarc in 1956.127 He developed an anionic living polymerization method for the preparation of narrow-molecular-weight dispersed polystyrene. After the acquisition of this “standard polymer”, the theories of polymer solutions128 and rubber elasticity,129 polymer rheology,130 and the scaling theory of polymer physics61 have made great advances. Now, tetra-PEG gels are a model polymer network system for the refinement of rubber elasticity, percolation theory,101,102 and so on.
Lastly, this review closes with repeating a part of the preface of a book Tanaka edited:122 “Polymers are considered among the most important materials in science and technology for the 21th century. The uses of polymers in our everyday life are being extended and diversified day by day. The chemical, medical, and agricultural industries as well as many others are heavily dependent on a wide variety of polymers. Moreover, polymers are the materials that nature chose as the vehicle for life that appeared on this earth.” Here, Tanaka would have substituted the term “polymers” by “polymer gels”.
Data availability
Since this is a review paper, in principle, no new data are provided in this work and the data can be obtained in the references. If not, the data that support the findings of this study are available from the corresponding author, MS, upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- T. Tanaka, Phys. Rev. Lett., 1978, 40, 820–823 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- S. K. Ahn, R. M. Kasi, S. C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- J. P. Gong, Soft Matter, 2006, 2, 544–552 RSC.
- K. Dušek and D. Patterson, J. Polym. Sci., 1968, 6, 1209–1216 CrossRef.
- T. Tanaka, S. Ishiwata and C. Ishimoto, Phys. Rev. Lett., 1977, 38, 771–774 CrossRef CAS.
- T. Tanaka, Sci. Am., 1981, 244, 124–136 CrossRef CAS PubMed.
- T. Tanaka, A. Wada and M. Suzuki, J. Chem. Phys., 1973, 59, 3799–3810 CrossRef CAS.
- T. Tanaka, K. Soda and A. Wada, J. Chem. Phys., 1973, 58, 5707–5715 CrossRef CAS.
- A. Wada, K. Soda, T. Tanaka and N. Suda, Rev. Sci. Instrum., 1970, 41, 845–853 CrossRef CAS.
- B. Chu, Laser Light Scattering, Academic Press, 2nd edn, 1991 Search PubMed.
- T. Tanaka, L. O. Hocker and G. B. Benedek, J. Chem. Phys., 1973, 59, 5151–5159 CrossRef CAS.
- T. Tanaka and D. J. Fillmore, J. Chem. Phys., 1979, 70, 1214 CrossRef CAS.
- T. Tanaka, I. Nishio, S.-T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CrossRef CAS PubMed.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379–6380 CrossRef.
- T. Tanaka, E. Sato, Y. Hirokawa, S. Hirotsu and J. Peetermans, Phys. Rev. Lett., 1985, 55, 2455–2458 CrossRef CAS PubMed.
- T. Tanaka, S. T. Sun, Y. Hirokawa, S. Katayama, J. Kucera, Y. Hirose and T. Amiya, Nature, 1987, 325, 796 CrossRef CAS.
- Y. Li and T. Tanaka, J. Chem. Phys., 1989, 90, 5161–5166 CrossRef CAS.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- M. Tokita and T. Tanaka, Science, 1991, 253, 1121–1123 CrossRef CAS PubMed.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef CAS.
- E. Kokufuta, Y. Q. Zhang and T. Tanaka, Nature, 1991, 351, 302–304 CrossRef.
- M. Shibayama, T. Tanaka and C. C. Han, J. Chem. Phys., 1992, 97, 6829–6841 CrossRef CAS.
- M. Shibayama, T. Tanaka and C. C. Han, J. Chem. Phys., 1992, 97, 6842–6854 CrossRef CAS.
- M. Annaka and T. Tanaka, Nature, 1992, 355, 430–432 CrossRef CAS.
- E. Sato-Matsuo and T. Tanaka, Nature, 1992, 358, 482 CrossRef.
- Y. Q. Zhang, T. Tanaka and M. Shibayama, Nature, 1992, 360, 142–144 CrossRef CAS.
- M. Shibayama and T. Tanaka, in Responsive Gels: Volume Transitions I, Advances in Polymer Science, 1993, vol. 109, pp. 1–62 Search PubMed.
- V. J. Pande, A. Y. Grosberg and T. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12972–12975 CrossRef CAS PubMed.
- Y. Takeoka, A. N. Berker, R. Du, T. Enoki, A. Y. Grosberg, M. Kardar, T. Oya, K. Tanaka, G. Wang, X. Yu and T. Tanaka, Phys. Rev. Lett., 1999, 82, 4863–4865 CrossRef CAS.
- T. Oya, T. Enoki, A. Y. Grosberg, S. Masamune, T. Sakiyama, Y. Takeoka, K. Tanaka, G. Wang, Y. Yilmaz, M. S. Feld, R. Dasari and T. Tanaka, Science, 1999, 286, 1543–1545 CrossRef CAS PubMed.
- G. Q. Wang, K. Kuroda, T. Enoki, A. Grosberg, S. Masamune, T. Oya, Y. Takeoka and T. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9861–9864 CrossRef CAS PubMed.
- T. Enoki, K. Tanaka, T. Watanabe, T. Oya, T. Sakiyama, Y. Takeoka, K. Ito, G. Wang, M. Annaka, H. Hara, R. Du, J. Chuang, K. Wasserman, A. Y. Grosberg, S. Masamune and T. Tanaka, Phys. Rev. Lett., 2000, 85, 5000–5003 CrossRef CAS PubMed.
- K. Dusek, Responsive Gels: Volume Transitions I, Springer-Verlag, Berlin, 1993, vol. 109 Search PubMed.
- K. Dusek, Responsive Gels: Volume Transitions II, Springer-Verlag, Berlin, 1993, vol. 110 Search PubMed.
- L. D. Landau and E. M. Lifshitz, Theory of Elasticity, Nauka, SSSR, 1965 Search PubMed.
- R. Yoshida, K. Uchida, Y. Kaneko, K. Sakai, A. Kikuchi, Y. Sakurai and T. Okano, Nature, 1995, 374, 240 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Halperin, M. Kroger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell Univ, Ithaca, 1953 Search PubMed.
- S. Hirotsu, Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1987, 87, 1392–1395 CrossRef CAS.
- A. Y. Grosberg and D. V. Kuznetsov, Macromolecules, 1992, 25, 1970–1979 CrossRef CAS.
- F. Tanaka and K. Nishinari, Macromolecules, 1996, 29, 3625 CrossRef CAS.
- M. News, Toyoichi Tanaka, MIT physicist who discovered “smart” gels, dies at 54, 2000, https://news.mit.edu/2000/tanaka.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- D. Chen, J. Yoon, D. Chandra, A. J. Crosby and R. C. Hayward, J, Polym. Sci., Part B: Polym. Phys., 2014, 52, 1441–1461 CrossRef CAS.
- S. J. Jeon, A. W. Hauser and R. C. Hayward, Acc. Chem. Res., 2017, 50, 161–169 CrossRef CAS PubMed.
- M. Shibayama, Polym. J., 2011, 43, 18–34 CrossRef CAS.
- M. Shibayama, Kobunshi High Polym., 2005, 54, 462–465 CrossRef CAS.
- M. Shibayama, in Small-angle Neutron Scattering on Gels, ed. P. Pecora and R. Borsali, Springer-Verlag, New York, 2008, vol. 2, book section 14, pp. 783–832 Search PubMed.
- E. Pines and W. Prins, J. Polym. Sci., Polym. Phys. Ed., 1972, B10, 719–724 Search PubMed.
- V. K. Soni and R. S. Stein, Macromolecules, 1990, 23, 5257–5265 CrossRef CAS.
- S. Mallam, F. Horkay, A. M. Hecht and E. Geissler, Macromolecules, 1989, 22, 3356–3361 CrossRef CAS.
- S. Mallam, A. M. Hecht and E. Geissler, J. Chem. Phys., 1989, 91, 6447 CrossRef CAS.
- S. Mallam, F. Horkay, A. M. Hecht, A. R. Rennie and E. Geissler, Macromolecules, 1991, 24, 543–548 CrossRef CAS.
- S. Panyukov and Y. Rabin, Phys. Rep., 1996, 269, 1–132 CrossRef CAS.
- S. Panyukov and Y. Rabin, Macromolecules, 1996, 29, 7960–7975 CrossRef CAS.
- M. Shibayama and K. Nagai, Macromolecules, 1999, 32, 7461–7468 CrossRef CAS.
- T. G. Park and A. S. Hoffman, J. Appl. Polym. Sci., 1994, 52, 85–89 CrossRef CAS.
- S. Takata, K. Suzuki, T. Norisuye and M. Shibayama, Polymer, 2002, 43, 3101–3107 CrossRef CAS.
- P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University, Ithaca, 1979 Search PubMed.
- P. N. Pusey and W. van Megen, Phys. A, 1989, 157, 705–741 CrossRef CAS.
- J. G. H. Joosten, E. T. F. Gelade and P. N. Pusey, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 2161–2175 CrossRef CAS PubMed.
- J. G. H. Joosten, J. L. McCarthy and P. N. Pusey, Macromolecules, 1991, 24, 6690–6699 CrossRef CAS.
- M. Shibayama, M. Nagao, S. Okabe and T. Karino, J. Phys. Soc. Jpn., 2005, 74, 2728–2736 CrossRef CAS.
- M. Shibayama, Soft Matter, 2012, 8, 8030–8038 RSC.
- L. Ornstein and F. Zernike, Proc. Acad. Sci., 1914, 17, 793–806 Search PubMed.
- P. Debye and A. M. Bueche, J. Appl. Phys., 1949, 20, 518–525 CrossRef CAS.
- P. Debye, H. R. Anderson and H. Brumberger, J. Appl. Phys., 1957, 28, 679–683 CrossRef CAS.
- M. Shibayama, Y. Fujikawa and S. Nomura, Macromolecules, 1996, 29, 6535–6540 CrossRef CAS.
- M. Shibayama and T. Norisuye, Bull. Chem. Soc. Jpn., 2002, 75, 641–659 CrossRef CAS.
- T. Norisuye, M. Shibayama and S. Nomura, Polymer, 1998, 39, 2769–2775 CrossRef CAS.
- T. Matsunaga and M. Shibayama, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 030401 CrossRef PubMed.
- M. Shibayama, Y. Isaka and Y. Shiwa, Macromolecules, 1999, 32, 7086–7092 CrossRef CAS.
- X. Li, N. Watanabe, T. Sakai and M. Shibayama, Macromolecules, 2017, 50, 2916–2922 CrossRef CAS.
- M. Shibayama, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS.
- S. Seiffert, Prog. Polym. Sci., 2017, 66, 1–21 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- M. Shibayama, H. Takahashi and S. Nomura, Macromolecules, 1995, 28, 6860–6864 CrossRef CAS.
- T. Matsunaga, T. Sakai, Y. Akagi, U. Chung and M. Shibayama, Macromolecules, 2009, 42, 1344–1351 CrossRef CAS.
- T. Matsunaga, T. Sakai, Y. Akagi, U. Chung and M. Shibayama, Macromolecules, 2009, 42, 6245–6252 CrossRef CAS.
- M. Kurakazu, T. Katashima, M. Chijiishi, K. Nishi, Y. Akagi, T. Matsunaga, M. Shibayama, U. Chung and T. Sakai, Macromolecules, 2010, 43, 3935–3940 CrossRef CAS.
- T. Fujiyabu, X. Li, M. Shibayama, U.-I. Chung and T. Sakai, Macromolecules, 2017, 50, 9411–9416 CrossRef CAS.
- K. Hayashi, F. Okamoto, S. Hoshi, T. Katashima, D. C. Zujur, X. Li, M. Shibayama, E. P. Gilbert, U. Chung, S. Ohba, T. Oshika and T. Sakai, Nat. Biomed. Eng., 2017, 1, 0044 CrossRef CAS.
- M. Shibayama, X. Li and T. Sakai, Colloid Polym. Sci., 2019, 297, 1–12 CrossRef CAS.
- T. Matsunaga, H. Asai, Y. Akagi, T. Sakai, U. Chung and M. Shibayama, Macromolecules, 2011, 44, 1203–1210 CrossRef CAS.
- K. Nishi, M. Chijiishi, Y. Katsumoto, T. Nakao, K. Fujii, U. Chung, H. Noguchi, T. Sakai and M. Shibayama, J. Chem. Phys., 2012, 137, 224903 CrossRef PubMed.
- K. Nishi, H. Noguchi, T. Sakai and M. Shibayama, J. Chem. Phys., 2015, 143, 184905 CrossRef PubMed.
- K. Nishi, K. Fujii, M. Chijiishi, Y. Katsumoto, U. Chung, T. Sakai and M. Shibayama, Macromolecules, 2012, 45, 1031–1036 CrossRef CAS.
- K. Nishi, K. Fujii, Y. Katsumoto, T. Sakai and M. Shibayama, Macromolecules, 2014, 47, 3274–3281 CrossRef CAS.
- K. Nishi, H. Asai, K. Fujii, Y. S. Han, T.-H. Kim, T. Sakai and M. Shibayama, Macromolecules, 2014, 47, 1801–1809 CrossRef CAS.
- X. Li, K. Hirosawa, T. Sakai, E. Gilbert and M. Shibayama, Macromolecules, 2017, 50, 3655–3661 CrossRef CAS.
- H. Kamata, U. Chung, M. Shibayama and T. Sakai, Soft Matter, 2012, 8, 6876–6879 RSC.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U.-I. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- S. Kondo, T. Hiroi, Y. S. Han, T. H. Kim, M. Shibayama, U. I. Chung and T. Sakai, Adv. Mater., 2015, 27, 7407–7411 CrossRef CAS PubMed.
- K. Oshima, T. Fujimoto, E. Minami and Y. Mitsukami, Macromolecules, 2014, 47, 7573–7580 CrossRef CAS.
- K. Morishima, X. Li, K. Oshima, Y. Mitsukami and M. Shibayama, J. Chem. Phys., 2018, 149, 163301 CrossRef PubMed.
- K. Fujii, H. Asai, T. Ueki, T. Sakai, S. Imaizumi, U. Chung, M. Watanabe and M. Shibayama, SoftMatter, 2012, 8, 1756–1759 RSC.
- H. Asai, K. Fujii, T. Ueki, T. Sakai, U. Chung, M. Watanabe, Y. S. Han, T. H. Kim and M. Shibayama, Macromolecules, 2012, 45, 3902–3909 CrossRef CAS.
- K. Hashimoto, K. Fujii, K. Nishi, T. Sakai and M. Shibayama, Macromolecules, 2016, 49, 344–352 CrossRef CAS.
- D. Stauffer, Introduction to Percolation Theory, Taylor and Francis, London, 1985 Search PubMed.
- D. Stauffer and A. Aharony, Introduction to percolation theory, Taylor and Francis, 1994, vol. 2, p. 181 Search PubMed.
- A. Coniglio, H. E. Stanley and W. Klein, Phys. Rev. Lett., 1979, 42, 518–522 CrossRef CAS.
- M. Takeda, T. Norisuye and M. Shibayama, Macromolecules, 2000, 33, 2909–2915 CrossRef CAS.
- S. Feng, M. F. Thorpe and E. Garboczi, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 276–280 CrossRef CAS PubMed.
- K. Nishi, F. Fujii, U. Chung, M. Shibayama and T. Sakai, Phys. Rev. Lett., 2017, 119, 26801 CrossRef PubMed.
- P. G. de Gennes, J. Phys., Lett., 1976, 37, 1–2 CrossRef.
- M. Daoud and A. Coniglio, J. Phys. A, 1981, 14, L301–L306 CrossRef CAS.
- T. Sakai, Physics of Polymer Gels, Wiley-VCH Verlag GmbH & Co. KGaA, 2020 Search PubMed.
- T. Fujiyabu, F. Toni, X. Li, U. Chung and T. Sakai, Chem. Commun., 2018, 54, 6784–6787 RSC.
- J. Kim, T. Fujiyabu, N. Sakumichi, T. Katashima, Y. Yoshikawa, U. Chung and T. Sakai, Macromolecules, 2020, 53, 7717–7725 CrossRef CAS.
- T. Fujiyabu, Y. Yoshikawa, J. Kim, N. Sakumichi, U. Chung and T. Sakai, Macromolecules, 2019, 52, 9613–9619 CrossRef CAS.
- T. Fujiyabu, Y. Yoshikawa, U.-I. Chung and T. Sakai, Sci. Technol. Adv. Mater., 2019, 20, 608–621 CrossRef PubMed.
- X. Li, S. Nakagawa, Y. Tsuji, N. Watanabe and M. Shibayama, Sci. Adv., 2019, 5, eaax8647 CrossRef CAS PubMed.
- S. Corezzi, D. Fioretto and F. Sciortino, Soft Matter, 2012, 8, 11207–11216 RSC.
- M. Okumoto, Y. Nakamura, T. Norisuye and A. Teramoto, Macromolecules, 1998, 31, 1615–1620 CrossRef CAS.
- B. Hammouda, D. L. Ho and S. Kline, Macromolecules, 2004, 37, 6932–6937 CrossRef CAS.
- J. Wang, Macromolecules, 2015, 48, 1614–1620 CrossRef CAS.
- Y. S. Han, Z. Xu and C. Gao, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef CAS.
- B. M. Venkatesan and R. Bashir, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef CAS PubMed.
- M. Goldberg, R. Langer and X. Jia, J. Biomater. Sci., Polym. Ed., 2007, 241–268 CrossRef CAS PubMed.
- T. Tanaka, Experimental Methods in Polymer Science. Modern Methods in Polymer Research and Technology, Academic Press, London, 2000 Search PubMed.
- M. Shibayama, S. Ozeki and T. Norisuye, AIP Conf. Proc., 2000, 519, 158–163 CrossRef CAS.
- T. Kureha, H. Minato, D. Suzuki, K. Urayama and M. Shibayama, Soft Matter, 2019, 15, 5390–5399 RSC.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakayama and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- X. Li and J. P. Gong, Nat. Rev. Mater., 2024, 9, 380–398 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- H. Yamakawa, Modern Theory of Polymer Solutions, Harper and Row, 1971 Search PubMed.
- L. R. G. Treloar, The Physics of Rubber Elasticity, Clarendon Press, Oxford, 1975 Search PubMed.
- J. D. Ferry, Viscoelastic Properties of Polymers, Wiley, New York, 3rd edn, 1980 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here.