
Peptide-mediated liquid–liquid phase separation and biomolecular condensates†
Guangle
Li
a,
Chengqian
Yuan
*a and
Xuehai
Yan
 *abc
*abc
aState Key Laboratory of Biopharmaceutical Preparation and Delivery, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: cqyuan@ipe.ac.cn; yanxh@ipe.ac.cn
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
cCenter for Mesoscience, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China
First published on 15th February 2025
Abstract
Liquid–liquid phase separation (LLPS) is a cornerstone of cellular organization, driving the formation of biomolecular condensates that regulate diverse biological processes and inspire innovative applications. This review explores the molecular mechanisms underlying peptide-mediated LLPS, emphasizing the roles of intermolecular interactions such as hydrophobic effects, electrostatic interactions, and π–π stacking in phase separation. The influence of environmental factors, such as pH, temperature, ionic strength, and molecular crowding on the stability and dynamics of peptide coacervates is examined, highlighting their tunable properties. Additionally, the unique physicochemical properties of peptide coacervates, including their viscoelastic behavior, interfacial dynamics, and stimuli-responsiveness, are discussed in the context of their biological relevance and engineering potential. Peptide coacervates are emerging as versatile platforms in biotechnology and medicine, particularly in drug delivery, tissue engineering, and synthetic biology. By integrating fundamental insights with practical applications, this review underscores the potential of peptide-mediated LLPS as a transformative tool for advancing science and healthcare.
 Guangle Li | Guangle Li received his PhD degree from the Institute of Chemistry, Chinese Academy of Sciences (CAS). He then joined Prof. Yi Zuo's group as a postdoc at the University of Hawaii at Manoa. Currently, he is an associate professor at the Institute of Process Engineering (IPE), CAS. His research interests are focused on liquid–liquid phase separation, biointerfaces, and bioinspired materials. |
 Chengqian Yuan | Chengqian Yuan received his PhD degree from the Institute of Chemistry, Chinese Academy of Sciences. He then joined Prof. Xuehai Yan's group as a postdoc at the State Key Laboratory of Biopharmaceutical Preparation and Delivery, IPE, CAS. Currently, he is an associate professor at the IPE, CAS. His research interests include peptide self-assembly and engineering, biomolecular condensates and glass, and phase separation. |
 Xuehai Yan | Xuehai Yan is a full professor at the Institute of Process Engineering, Chinese Academy of Sciences. Currently, he is the deputy director of the State Key Laboratory of Biopharmaceutical Preparation and Delivery and the Center of Mesoscience, IPE, CAS. His research interests are focused on peptide self-assembly and engineering, supramolecular colloids and crystals, phase evolution and dynamic transition, as well as phototherapy and immunotherapy. |
1. Introduction
1.1. Definition and significance of liquid–liquid phase separation (LLPS)
Liquid–liquid phase separation (LLPS) is a fundamental physicochemical process in which a homogeneous solution of macromolecules, such as polymers or biomacromolecules, spontaneously separates into two distinct liquid phases: one enriched in these macromolecules and the other depleted of them.1,2 This process results in the formation of dense, liquid-like droplets suspended within a more dilute surrounding phase. The phase behavior of LLPS can be represented in phase diagrams, which map the conditions under which phase separation occurs.2,3 The study of LLPS in polymers has yielded significant insights into material science, particularly by enabling the design of advanced materials with tunable properties derived from phase-separated structures.4 More recently, the significance of LLPS in cell biology has been recognized as it offers a novel framework for understanding intracellular organization and compartmentalization.5–7 Unlike traditional membrane-bound organelles, LLPS-driven self-assembly generates distinct cytoplasmic and nucleoplasmic compartments, forming biomolecular condensates, some of which evolve into functionally specialized membraneless organelles.8,9 These condensates exhibit dynamic, liquid-like properties, such as rapid component exchange, fusion, and reformation, which are essential for numerous cellular functions.10–12 Membraneless organelles formed through LLPS are involved in a broad range of physiological functions, such as the formation of heterochromatin, storage of nucleic acids, regulating gene expression, stress response, and signal transduction.11,13–17 These structures are critical for maintaining cellular homeostasis and orchestrating molecular transport and cell division.18–21 The realization that LLPS is a fundamental mechanism underlying the formation of these compartments has revolutionized our understanding of cell biology, prompting a reevaluation of numerous biological processes.The recognition of LLPS has profound implications for human health, as it has become increasingly clear that this fundamental biological process is intimately linked to the pathogenesis of various diseases.7,22 Aberrant phase separation has been implicated in a range of conditions, including neurodegenerative disorders, cancer, and infectious diseases.23–26 In neurodegenerative diseases like Alzheimer's disease and amyotrophic lateral sclerosis (ALS), the dysregulation of LLPS can result in the formation of pathological protein aggregates, often due to the transition of biomolecular condensates from liquid-like to solid-like states, as depicted in Fig. 1.6,7,27 These aggregates are believed to arise from the inappropriate or irreversible maturation of dynamic biomolecular condensates, ultimately causing cellular dysfunction and neurodegeneration.28 Similarly, in cancer, misregulated LLPS can alter the behavior of condensates involved in gene expression and cell cycle control, contributing to uncontrolled cell proliferation and tumor development.24 In infectious disease, pathogens may hijack host cells' LLPS mechanisms to enhance their replication or to evade immune detection, further illustrating the broad relevance of LLPS in disease processes.22,29 The growing body of evidence underscores the importance of LLPS not only as a fundamental mechanism for normal cellular function but also as a critical factor in disease pathogenesis. Understanding the nuances of LLPS dysregulation opens new avenues for therapeutic intervention, as targeting the specific molecular interactions that govern phase separation could provide novel strategies for treating disease associated with aberrant condensate formation.30 Thus, LLPS represents a promising area of research with significant potential to inform the development of new therapies to restore normal cellular function and to prevent disease progression.
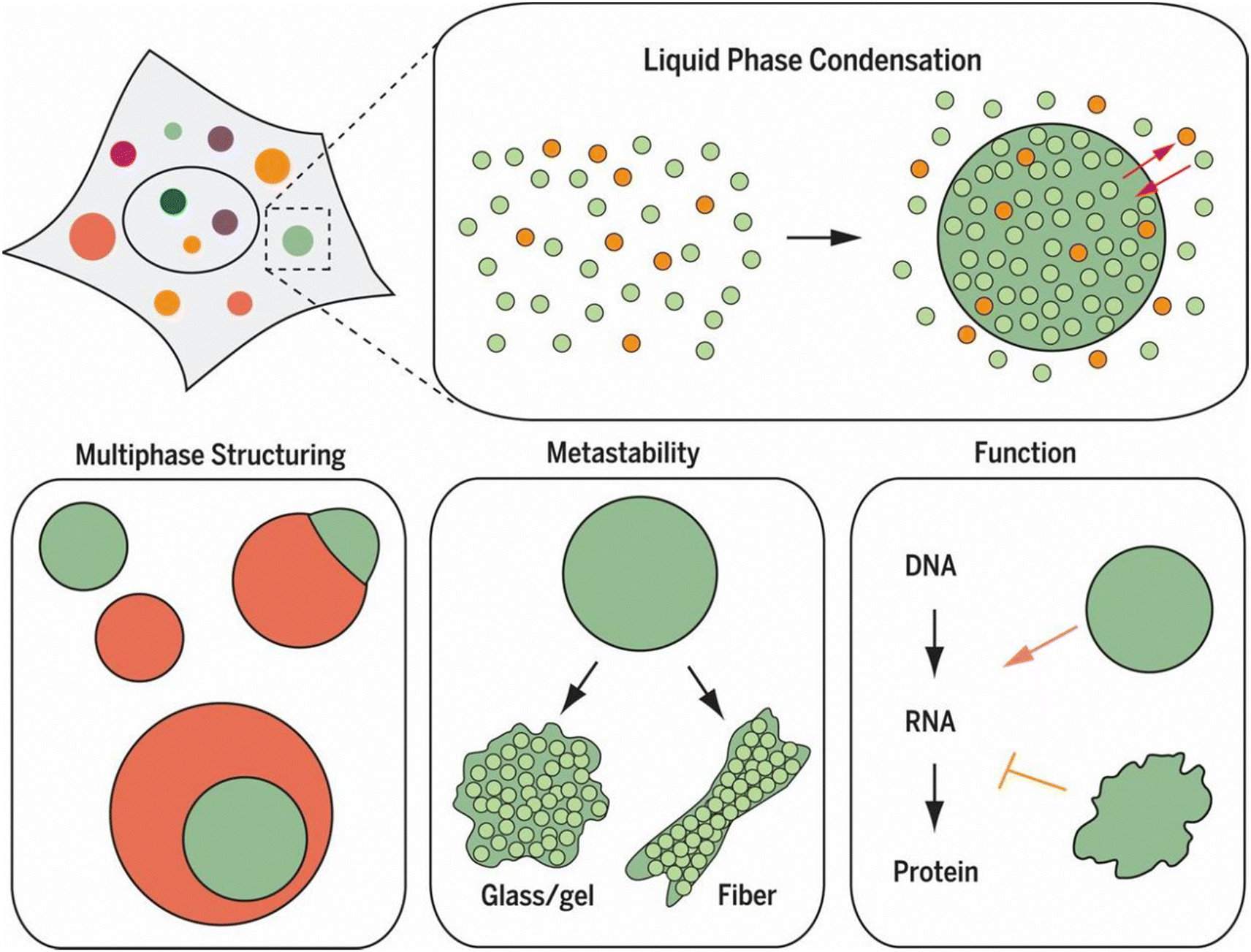 | ||
| Fig. 1 Liquid–liquid phase separation (LLPS) as a fundamental mechanism of cellular organization. LLPS enables the formation of dynamic biomolecular condensates, or membraneless organelles, that selectively concentrate specific molecules through controlled phase separation. These condensates exhibit diverse behaviors, including multiphase structuring and transitions from liquid-like to solid-like states. Such condensates play significant roles in regulating cellular functions, including RNA transcription and protein translation, by creating specialized microenvironments that facilitate specific biochemical reactions and information flow within the cell. Reprinted with permission.6 Copyright 2017, The American Association for the Advancement of Science. | ||
1.2. Protein-mediated LLPS and biomolecular condensates
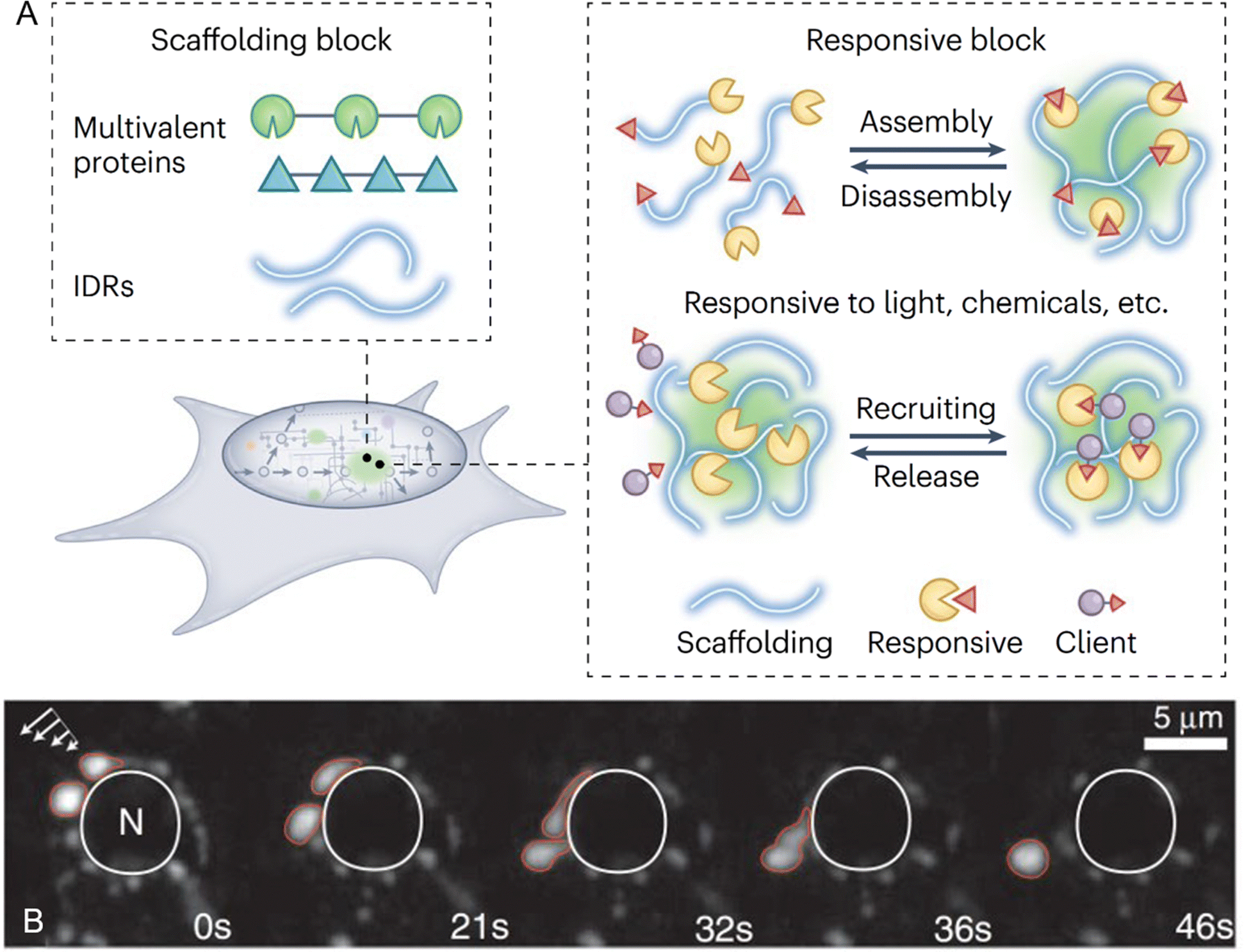 | ||
| Fig. 2 Protein-mediated liquid–liquid phase separation and biomolecular condensates. (A) Building blocks and functional responsiveness of biomolecular condensates. Multivalent proteins or intrinsically disordered regions (IDRs) act as scaffolds to drive phase separation and condensate formation. These condensates exhibit responsiveness to environmental stimuli, enabling controlled assembly and disassembly as well as the selective recruitment and release of client proteins, highlighting their adaptability to dynamic biological processes. Reprinted with permission.32 Copyright 2022 Springer Nature. (B) Dynamic liquid-like properties of P granules in Caenorhabditis elegans germ cells. Time-lapse imaging demonstrates the deformation, dripping, and fusion of P granules (outlined in red) under shear force (indicated by arrows, top left). These perinuclear granules, surrounding the nucleus (N, outlined in white), exhibit characteristic liquid-like behaviors over time, as shown at 0, 21, 32, 36, and 46 seconds. Reprinted with permission.36 Copyright 2009, The American Association for the Advancement of Science. | ||
The concept of scaffold and client proteins is fundamental to understanding the molecular mechanisms that govern the formation, function, and regulation of biomolecular condensates.37 Scaffold proteins are critical for initiating and maintaining phase separation, acting as the structural backbone of condensates by mediating multivalent interactions through multiple interaction motifs or domains. These interactions drive phase separation and provide a robust network that sustains LLPS. Additionally, scaffold proteins play a key role in recruiting client proteins, which, although unable to phase-separate independently, can be incorporated into pre-existing condensates. Often, these interactions involve RNA, particularly in the formation of RNA/protein-rich membraneless organelles.38 RNA, especially those with repetitive sequences, enhances the multivalent interactions necessary for LLPS, acting as both structural components and functional regulators within condensates.11 The architecture of scaffold proteins typically includes either multiple folded domains that interact with short linear motifs in other proteins or IDRs with multiple interacting motifs or “stickers”.35,39 These IDRs mediate weak multivalent interactions essential for driving phase separation. Studies have shown that scaffold valency lowers the saturation concentration required for phase separation, reinforcing their pivotal role in condensate formation.2
In gene regulation, LLPS drives the formation of transcriptional condensates at specific genomic loci. These condensates concentrate transcription factors, RNA polymerase, and other regulatory proteins, thereby enhancing the efficiency and specificity of gene expression.19 The ability of these condensates to assemble and disassemble in response to signaling cues ensures that gene expression can be dynamically modulated to meet changing cellular demands.48 LLPS also contribute to the formation of heterochromatin, where it organizes and silences large genomic regions. This LLPS-mediated chromatin organization restricts access to specific genes, enabling cells to regulate gene expression at a broader, genome-wide scale.14 Biomolecular condensates are equally critical for cellular stress responses.49 Stress granules, for instance, form under stress conditions such as heat shock or oxidative stress.42 These condensates temporarily sequester mRNA and translation-related proteins, preventing mRNA degradation and conserving cellular resources. Once the stress is alleviated, stress granules disassemble, releasing the sequestered mRNA for translation and enabling a rapid recovery of protein synthesis. This transient and reversible sequestration mechanism highlights the adaptive flexibility of condensates in protecting cellular functions under stress. Beyond gene regulation and stress responses, biomolecular condensates are also involved in the regulation of signaling pathways.50 Signaling condensates form at the plasma membrane in response to receptor activation, where they concentrate signaling molecules to amplify and spatially constrain signaling events and ensure signaling precision.
The dynamic and responsive nature of biomolecular condensates makes them indispensable for maintaining cellular homeostasis. However, when the mechanisms governing LLPS become dysregulated, condensates can transition from functional liquid-like states to pathological solid-like aggregates.28 This aberrant phase behavior has been implicated in neurodegenerative diseases such as ALS, Alzheimer's disease, frontotemporal dementia (FTD), and Huntington's disease.6,22 Understanding the molecular mechanisms that regulate LLPS is therefore critical not only for uncovering fundamental cellular principles but also for developing therapeutic approaches to mitigate diseases arising from condensate dysfunction.
1.3. Peptide-mediated LLPS and coacervates
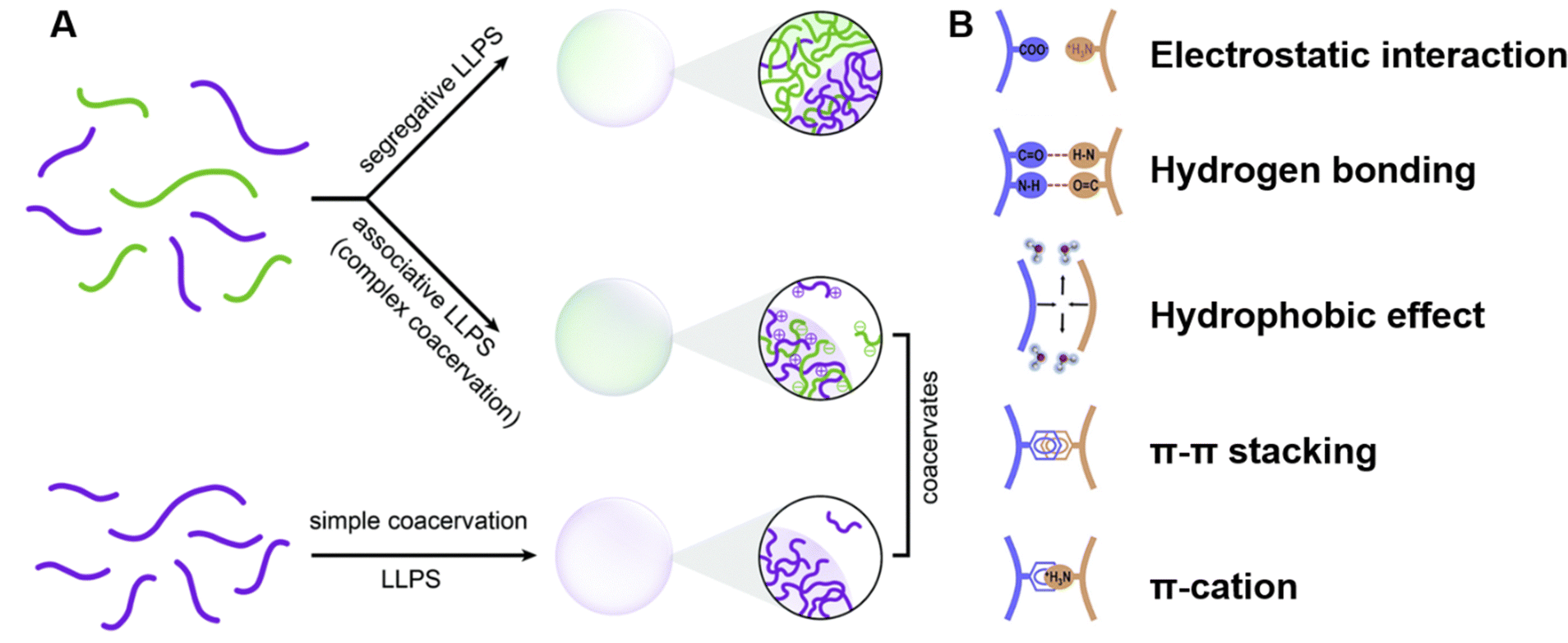 | ||
| Fig. 3 Peptide-mediated liquid–liquid phase separation (LLPS) and coacervates. (A) Schematic representation of the types of LLPS that lead to the formation of coacervates. Segregative LLPS arises from repulsive interactions between distinct solute molecules, resulting in separate solute-enriched compartments. Associative LLPS, also known as complex coacervation, is driven by attractive interactions between solutes, forming coacervates enriched with multiple components. Simple coacervation involves the self-association of a single molecular species into dense, dynamic droplets. Reprinted with permission.53 Copyright 2021 Royal Society of Chemistry. (B) Key molecular interactions driving peptide LLPS. The schematic highlights the primary interactions, including electrostatic interactions between charged residues, hydrogen bonding involving polar groups, hydrophobic interactions between nonpolar residues, π–π stacking between aromatic residues, and π-cation interactions between aromatic and positively charged residues. These interactions collectively determine the phase behavior and functional properties of peptide coacervates. Reprinted with permission.51 Copyright 2023 Elsevier. | ||
Peptide-based coacervates can effectively encapsulate a variety of molecules, including nucleic acids, proteins, and small drugs, protecting them from degradation and facilitating their release in response to specific environmental stimuli.62–65 The tunability of peptide sequences further enhances the utility of these systems, allowing precise control over coacervate formation and dissolution to meet diverse functional requirements. Furthermore, the inherent biocompatibility and straightforward synthesis of peptides make them ideal candidates for developing new biomaterials and therapeutic platforms.66,67 Their ability to form dynamic, responsive scaffolds that mimic the extracellular matrix supports cell growth and tissue regeneration, making them suitable for use in wound healing, cartilage repair, and other regenerative therapies. Additionally, the biodegradability of peptide-based materials ensures that they can be safely integrated into biological systems and eventually resorbed by the body.
In synthetic biology, peptide-mediated LLPS has been utilized to create artificial compartments and functional biomaterials.65,68 These synthetic systems can be designed to perform specific tasks, such as catalyzing reactions or sequestering molecules, in a manner similar to natural cellular processes. The ability to engineer peptide-based coacervates with specific properties opens up new possibilities for the development of advanced materials that mimic or enhance natural biological functions. As research into peptide-mediated LLPS continues to advance, the potential applications of these systems in biotechnology and medicine are expected to grow, offering innovative solutions for drug delivery, tissue engineering, synthetic biology, and beyond. The ability to design and manipulate these systems at the molecular level holds great promise for enhancing our understanding of biological processes and developing novel therapeutic interventions.
1.4. Scope and objectives of the review
This review aims to provide a comprehensive overview of peptide-mediated LLPS, examining its implications in both biological systems and synthetic applications. The review seeks to elucidate the thermodynamic and molecular mechanisms driving peptide-mediated LLPS, explore how environmental factors such as pH, temperature, and ionic strength, influence phase separation, and analyze the transition of peptide coacervates from liquid-like to solid-like states. Furthermore, it will assess how external stimuli modulate LLPS and discuss the physicochemical properties of peptide coacervates, and their applications in drug delivery, tissue engineering, and synthetic biology. Through this detailed exploration, the review aims to enhance our understanding of peptide-mediated LLPS and underscore its potential for driving innovation in biotechnology and medicine.2. Mechanisms of peptide-mediated LLPS
2.1. Thermodynamics and driving forces
The thermodynamics of peptide-mediated LLPS can be described using free energy considerations, where the system seeks to minimize its total free energy. The relevant free energy is the Gibbs free energy of mixing (ΔGm), determined by both enthalpic (ΔHm) and entropic (ΔSm) contributions:| ΔGm = ΔHm − TΔSm | (1) |
The enthalpy reflects the interaction potentials between peptide and solvent, while the entropy represents the available degrees of freedom for both peptide and solvent molecules. It is also crucial to recognize that the enthalpic contribution can be driven by specific molecular interactions, such as hydrogen bonding, aromatic stacking, or hydrophobic effects, depending on the peptide's sequence composition.69 The mixing entropy specifically accounts for the entropic cost associated with confining peptide molecules in a dense phase. For phase separation to occur, the Gibbs free energy (ΔGm) must be negative, indicating that the system can lower its energy by separating into two distinct phases, a peptide-rich dense phase, and a peptide-poor dilute phase. In LLPS, entropy generally favors a well-mixed state due to the greater number of possible configurations. However, the formation of a condensed phase results in a decrease in entropy, which must be compensated by a favorable enthalpic contribution, often driven by strong peptide–peptide interactions that replace less favorable peptide-solvent interactions. The thermodynamics of LLPS are influenced by the sequence-specific properties of the peptides, which can modulate the strength and nature of these interactions.69 Additionally, the release of water molecules from the peptide surfaces into the bulk phase during condensate formation contributes an entropy gain that partially offsets the overall entropy loss, thereby facilitating phase separation.70
The Flory–Huggins theory extends the classical Gibbs free energy approach to describe the phase behavior of peptides in solution.71,72 This theory considers the size difference between peptide and solvent molecules, as well as the specific interactions between them. In this model, the free energy of mixing is given by:
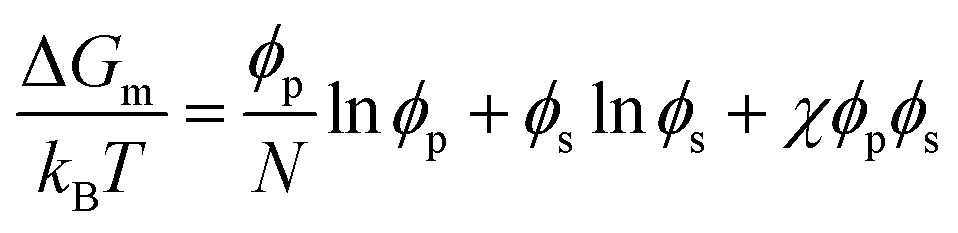 | (2) |
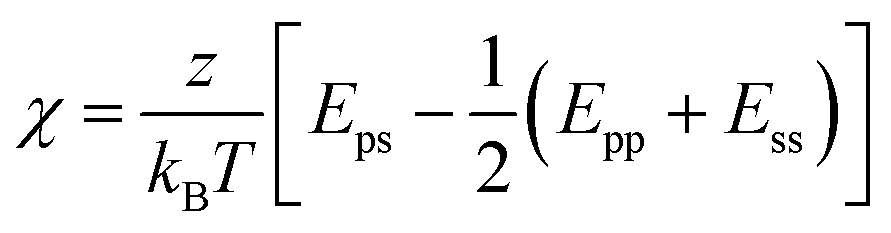 | (3) |
The first two terms in eqn (2) account for the entropy change due to mixing, which is generally positive and favors mixing. However, for large polymers and peptides, this entropic contribution is relatively small due to the limited number of configurations available to these molecules. The third term represents the enthalpy of mixing, which may favor or oppose mixing depending on the sign and magnitude of χ. This parameter is highly sensitive to the specific sequence and chemical nature of the peptides involved, influencing their propensity for demixing under various conditions.
When charged peptides are involved, long-range electrostatic interactions significantly affect phase behavior. The Overbeek and Voorn extension of the Flory–Huggins model introduces a term related to charge density (σ) to account for these effects.73
 | (4) |
Here, σ represents the charge density, calculated according to Debye–Hückel theory, with α as a solvent constant determined by thermal energy kBT and solvent molar volume. Strong interactions, typically lead to a first-order phase transition, resulting in two coexisting liquid phases. The extent of the two-phase region is modulated by interaction strength, which is further influenced by factors such as temperature, pH, peptide chemical groups, and salt concentration. In simple coacervation, the strength is primarily governed by χ, while in complex coacervation, it is determined by ασ3/2. Although the mean-field approach is a fundamental framework, it overlooks various factors like sequence specificity and nuanced interactions. Recent studies show that the sequence-dependent patterns of charged and aromatic residues can significantly alter the phase behavior, introducing complexities beyond the classical model.69 While more advanced theories have been developed to address these complexities, they do not yet fully explain all types of LLPS in peptides, proteins, and polymers.74 Consequently, the classical mean-field approach remains a valuable tool for understanding phase separation.
2.2. Molecular interactions in peptide-mediated LLPS
The multivalent forces driving peptide-mediated LLPS involve various molecular interactions, including electrostatic interactions between charged residues, hydrogen bonds involving polar groups, hydrophobic interactions between nonpolar residues, aromatic stacking (π–π) interactions between aromatic residues, and cation–π interactions between positively charged residues and aromatic groups (Fig. 3B).5,51,75 These forces collectively contribute to the assembly and stabilization of peptide coacervates under different environmental conditions.2.3. Role of amino acid sequence
While individual amino acids contribute to molecular interactions that drive and sustain LLPS, the sequence and arrangement of amino acids also determine the phase behavior and material properties of the resulting condensates.57,89–92 Recent studies have revealed that proteins undergoing LLPS are intrinsically disordered or contain IDRs characterized by low-complexity sequences and enriched in polar, aromatic, proline, and glycine residues.69,93–97 For example, peptides with repetitive arginine-glycine motifs exhibit a strong propensity for LLPS due to the molecular flexibility provided by glycine and the electrostatic interactions facilitated by arginine.57 However, the number and distribution of these motifs are critical, as excessive repeats may increase electrostatic repulsion, hindering phase separation.57,89,98 Histidine-rich beak proteins (HBPs) provide a notable example, with the presence of repetitive regions of low complexity amino acid sequence in their C-termini.78 The motif repeat GHGLY drives the LLPS of HBPs and requires at least two copies and a linker sequence for broader phase separation conditions, such as variations in pH and salt concentration. Alternatively, four GHGLY tandem repeats can independently trigger self-coacervation. LLPS can occur even in sequences lacking traditionally considered essential residues, such as charged or aromatic amino acids. For example, the peptide variant of (GRGDSPYS)25 undergoes LLPS despite the absence of charged, aromatic, arginine, or glycine residues, challenging previous assumptions about strict sequence requirements.89 The sequence not only dictates phase behavior but also impacts the material properties of condensates. (GRGDSPYS)25 shows a viscosity of 11 Pa s, while the variants GRGASPYA and GRGNSPWS exhibit viscosities of 2 and 40 Pa s, respectively.Contrary to earlier assumptions that LLPS requires a longer peptide sequence, LLPS can occur with ultrashort peptides, such as tetrapeptide W2R2 and hexapeptide W3R3, suggesting that phase separation is a general property of peptides, independent of sequence length.84 These peptides undergo self-coacervation under specific conditions, such as high pH and sufficient concentration. Even shorter sequences, like dipeptide WR, can achieve LLPS through interactions with ATP. A comprehensive screening of 400 dipeptides derived from the 20 natural amino acids identified QW as a particularly promising LLPS candidate.99 Both experimental and computational studies confirmed its strong phase separation potential. Other short peptides, such as tert-butyl diphenylalanine and methoxylated diphenylalanine derivatives, also exhibit LLPS capacity.65,100 Additionally, the distribution of amino acid residues further influences LLPS behavior. For example, spacer sequences such as GSG, GLG, or SGS can promote, inhibit, or alter phase-separated structures.101 Peptides with GLG spacers form liquid droplets, while those with GSG spacers remain soluble, and those with SGS spacers form aggregates and hydrogels. These findings emphasize the critical role of sequence and arrangement in determining phase behavior and material properties.
3. Methods for studying LLPS and biomolecular condensates
3.1. Experimental techniques
 | ||
| Fig. 4 Experimental techniques used to study liquid–liquid phase separation (LLPS) and biomolecular condensates. (A) Fluorescence recovery after photobleaching (FRAP) for evaluating the material properties and dynamics of biomolecular condensates. Following laser-induced photobleaching, fluorescence within the condensates gradually recovers, with a shorter recovery time indicating higher fluidity. Reprinted with permission.6 Copyright 2017, The American Association for the Advancement of Science. (B) Fluorescence variation within a biomolecular condensate during a FRAP experiment. Scale bar, 5 μm. Reprinted with permission.111 Copyright 2023 Springer Nature. (C) Schematic of a transmission electron microscopy (TEM) liquid cell used to study protein condensate formation and nucleation. Reprinted with permission.112 Copyright 2019, American Chemical Society. (D) Example of nuclear magnetic resonance (NMR) spectroscopy demonstrating the differences in protein structure between the dilute and condensate phases. NMR spectroscopy can distinguish between atomic bond dynamics and overall molecular dynamics. Reprinted with permission under a Creative Commons Attribution 4.0 International License.113 Copyright 2023 by the authors. (E) PhaseScan microfluidic setup for studying biomolecular condensates, where droplets with varying concentrations of proteins, buffer, and phase separation trigger solutions are generated via a flow-focusing technique, trapped in microwells, and analyzed using epifluorescence microscopy. (F) High-resolution phase diagrams generated by the PhaseScan microfluidic setup. (E) and (F) Reprinted with permission under a Creative Commons Attribution 4.0 International License.114 Copyright 2022 by the authors. | ||
Although FRAP is widely used, several limitations and challenges must be considered when interpreting FRAP data. A common misconception is the assumption that the recovery rate solely reflects the exchange rate between the dilute and dense phases. In reality, the recovery rate is also influenced by factors such as the size of the photobleached droplet, the mobility of molecules within the droplet, and the dimensions of the bleached area, which are often overlooked. Additionally, interpreting FRAP results requires careful consideration, especially for heterotypic condensates composed of multiple molecular species. In these cases, different components may exhibit varying recovery behaviors due to distinct intermolecular interactions and spatial heterogeneities. The scaffold-client model explains this differential behavior, with scaffolds, which have multivalent binding capabilities, showing slower mobility compared to clients that interact through monovalent scaffold-binding modules.37,115 Despite these limitations, FRAP remains popular due to its simplicity, accessibility, and convenience, offering a qualitative assessment of molecular diffusivity within condensates. FRAP is often used in conjunction with other techniques, such as fluorescence correlation spectroscopy and single particle tracking, to gain a more detailed understanding of the diffusion behavior and interaction networks within condensates.103,115–117
Advanced imaging techniques, like super-resolution microscopy, extend the capabilities of conventional microscopy by surpassing the diffraction limit, allowing visualization of nanoscale structures within phase-separated droplets. Techniques such as stochastic optical reconstruction microscopy (STORM) provide detailed images of the internal organization of condensates, revealing substructures critical for understanding the molecular mechanisms driving LLPS.121,122 In addition to offering information on physical dimensions like size, shape, and composition, advanced microscopy techniques also uncover material properties.123 For instance, Brillouin microscopy, a noninvasive imaging modality, utilizes Brillouin light scattering principles to examine interactions between light and acoustic phonons, revealing intrinsic mechanical properties of biological samples at the microscopic scale.124,125 This approach has been used to investigate the mechanical properties of protein aggregates formed through LLPS, which are relevant in neurodegenerative diseases.126–128
However, these techniques often rely on antibody staining or fluorescent labeling, which may alter the native state of biomolecular condensates. Electron microscopy (EM) offers an alternative by visualizing condensates with a high resolution (<10 nm) in a label-free manner. Cryo-electron microscopy (cryo-EM) captures snapshots of LLPS at cryogenic temperatures, preserving the native state of condensates and providing insights into their structural heterogeneity.129,130 More recently, in situ liquid transmission electron microscopy (TEM) has been utilized to observe the dynamic process of LLPS in a time-resolved manner at the nanoscale (Fig. 4C).112 This technique allows visualization of nucleation and growth of biomolecular nanoclusters, shedding light on the early stages of LLPS and the mechanisms behind the formation of biomacromolecular complexes. Collectively, these advanced microscopy techniques provide a comprehensive toolkit for studying the spatiotemporal dynamics of LLPS, from the initial nucleation of droplets to their maturation and interactions within the cellular environment.
X-ray scattering techniques, particularly small angle X-ray scattering (SAXS), are invaluable for studying the overall shape, size distribution, and structural organization of peptide or protein complexes within phase-separated droplets.78,138 SAXS provides nanoscale architectural information on parameters such as the radius of gyration, the degree of compactness, and the presence of higher-order assemblies, all critical for understanding the multivalent interactions and network formation within LLPS.139,140 Beyond traditional SAXS, ultra-small angle X-ray scattering (USAXS) extends the range by capturing information about larger structures, bridging the gap between SAXS and conventional light scattering techniques.141,142 USAXS is particularly useful for analyzing samples with features spanning several orders of magnitude in size, offering structural insights over a wide range of length scales. Furthermore, combining SAXS with NMR and cryo-EM allows for a comprehensive understanding of both the static and dynamic aspects of LLPS, integrating structural and functional studies of biomolecular condensates.143 However, challenges remain, such as the need for high sample concentrations to achieve adequate signal strength and the risk of biomacromolecule damage from energetic beam sources. These techniques also struggle to probe highly disordered regions and may not fully capture the size and complexity of some condensates.
A significant advancement in this field is the integration of high-throughput screening capabilities with microfluidic platforms. For example, droplet-based microfluidics generates thousands of microdroplets, each containing different concentrations of peptides. Automated imaging and analysis tools monitor these droplets, enabling comprehensive exploration of phase diagrams across a wide range of conditions using specialized microfluidic chip designs.114,148–151 Arter et al. developed the PhaseScan platform, a combinatorial droplet microfluidic system for rapid, high-resolution acquisition of multidimensional biomolecular phase diagrams (Fig. 4F).114 This platform automates droplet generation, trapping, and imaging, significantly reducing experimental time and minimizing potential errors by eliminating manual preparation and imaging of individual conditions. This high-throughput technique facilitates the mapping of LLPS conditions, accelerating the discovery of new phase-separating systems and optimizing the conditions for specific phase behaviors.
Emerging technologies within microfluidics continue to push the boundaries of LLPS research. Innovations such as serpentine microchannels allow for continuous observation of droplets over extended periods, while controlled shear stress application provides insights into the dynamic behavior of phase-separated systems.114,147,152 In addition, microfluidic platforms enable experimental methods not feasible with traditional bulk assays, such as measuring the zeta potential of individual droplets, a critical parameter for understanding surface charge properties.153 These advancements improve our understanding of the physical properties of condensates, and allow for simulating complex biological environments where LLPS is pivotal.154,155 By combining microfluidics with other high-resolution imaging techniques, it is possible to explore LLPS dynamics under near-physiological conditions and provide a more accurate representation of in vivo systems.
3.2. Computational approaches
Computational approaches have become indispensable in the study of peptide-mediated LLPS, providing insights at scales that are challenging to achieve experimentally. This part focuses on two main computational methodologies: molecular dynamics simulations, and bioinformatics and machine learning tools.99,156,157 Both approaches offer unique advantages in understanding the complex interactions that drive LLPS.Molecular dynamics simulations offer a detailed atomistic view of peptide interactions and their roles in LLPS.89,99 By simulating the behavior of peptides under varying conditions, molecular dynamics simulations allow for the exploration of the thermodynamics and kinetics of phase separation. Yang et al. have employed molecular dynamics simulations to investigate the phase separation of repetitive polyproline and polyarginine peptides with varying lengths and sequences.158 This study highlighted the critical role of sequence order and peptide length in promoting LLPS. It demonstrated that peptides with fewer than ten polyarginine repeats did not exhibit LLPS, even at high salt concentrations. The simulations revealed that both hydrophobic and electrostatic interactions are crucial drivers of phase separation, aligning well with experimental observations. Molecular dynamics simulations can also explore the effects of external factors, such as temperature and salt concentration, on LLPS.158 For example, the same study showed that increasing salt concentration enhances LLPS by reducing electrostatic repulsion between charged residues. Similarly, temperature variations were shown to modulate the conformational entropy of peptides, thereby affecting their ability to undergo phase separation. As the number of known phase-separating proteins and peptides grows, bioinformatics and machine learning tools have become increasingly important for predicting LLPS behavior based on sequence data.157,159,160 These tools leverage large datasets to identify patterns and features that correlate with LLPS propensity, enabling the prediction of novel phase-separating systems. One notable example is the PSPredictor tool, developed by Chu and coworkers.157 PSPredictor uses a machine learning algorithm trained on sequence data from the LLPSDB database to predict phase-separating proteins (PSPs). The tool combines componential and sequential information during protein embedding, achieving a high accuracy of 94.71% in cross-validation tests. By providing predictions that are not dependent on specific protein types, PSPredictor facilitates the identification of novel scaffold proteins for biomolecular condensates and other phase-separated structures. Machine learning approaches like PSPredictor are particularly powerful because they can process vast amounts of sequence data, uncovering hidden relationships between sequence features and phase behavior. These tools are invaluable for guiding experimental studies, identifying candidates for further investigation, and expanding our understanding of the sequence determinants of LLPS.
4. Physicochemical properties and environmental effects on peptide coacervates
4.1. Materials properties of peptide coacervates
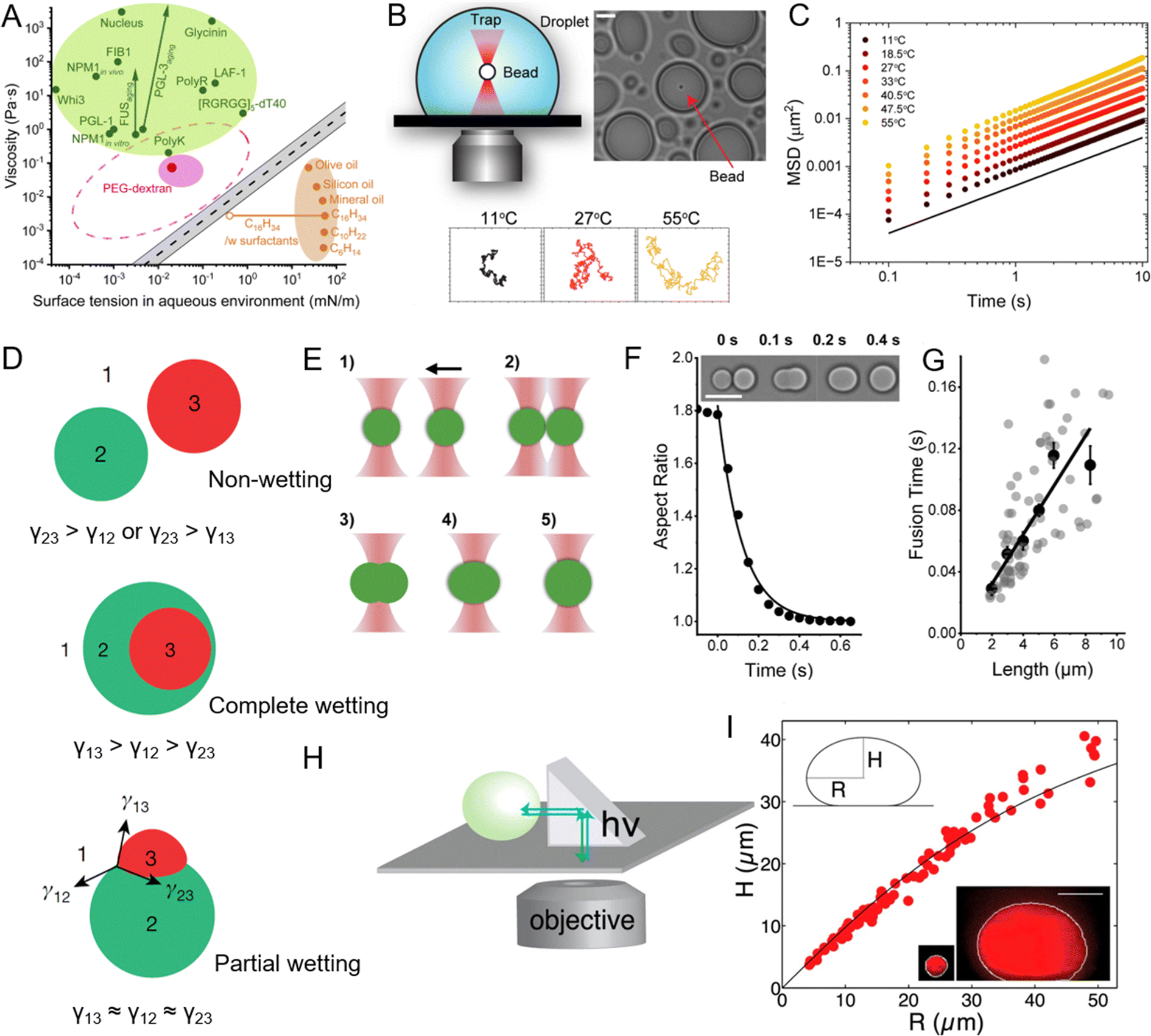 | ||
| Fig. 5 Viscosity and surface tension of peptide coacervates and the experimental methods used to measure these physicochemical properties. (A) Comparison of viscosity and surface tension of biomolecular condensates in aqueous buffer, PEG-dextran coacervate systems, and common “oil droplet” in water. Reprinted with permission under a Creative Commons Attribution 4.0 International License.163 Copyright 2021 by the authors. (B) Schematic of the experimental setup for passive microrheology with optical tweezers to measure the viscosity of peptide coacervates, with representative particle trajectories recorded at three different temperatures. (C) Ensemble-averaged mean squared displacements (MSD) of 200-nm beads within peptide coacervate at varying temperatures. (B) and (C) Reprinted with permission under a Creative Commons Attribution 4.0 International License.169 Copyright 2024 by the authors. (D) Different modes of multiphase droplet structuring based on relative interfacial tensions. In case of high interfacial tension between two droplets (γ23), droplets remain separate (upper). When the 1-3 interface is energetically costly, phase 2 envelopes phase 3 (middle). When the relative energetic costs are balanced, all three phases may share interfaces (lower). Reprinted with permission.167 Copyright 2022, Springer Nature. (E) Illustration of the coacervate fusion experiment using dual-optical traps. (F) Fusion of two peptide coacervates (inset images) shown as a decrease in aspect ratio to 1. Scale bar, 10 μm. (G) Fusion time of peptide coacervates as a function of coacervate size, where the slope of the linear fit corresponds to the inverse capillary velocity, η/γ, with η representing viscosity and γ surface tension. (E)–(G) Reprinted with permission under a Creative Commons Attribution 4.0 International License.163 Copyright 2021 by the authors. (H) and (I) Schematic of XZ imaging of biomolecular condensates using a right-angle prism (H) and the relationship between height and radius of nucleoli at steady state (I), used to determine the surface tension of the condensates. The shape of a biomolecular condensate resting on a surface is determined by a balance between surface tension, which favors the formation of round droplets, and gravitational forces, which tend to flatten droplets. Surface tension can be determined by analyzing the droplet's profile in the XZ dimension. The black line indicates the fit from the average surface tension for all condensates. Reprinted with permission.109 Copyright 2016, Elsevier. | ||
Several methods have been employed to study the viscosity and elasticity of peptide coacervates. Among these, rheology serves as a primary technique for measuring the flow and deformation behavior of coacervates under various stress or strain conditions.170 Microrheology, which usually combines with optical tweezers, tracks the motion of embedded tracer particles and offers a localized understanding of viscoelastic properties on a microscopic scale (Fig. 5B and C).169,171,172 In addition, micropipette aspiration technique, has enabled direct measurement of viscosity in biomolecular condensates.163,173,174 Advances in microfluidic technology further enable precise manipulation and measurement of coacervate properties under controlled conditions. These platforms allow for the generation of droplets with defined sizes and compositions, where viscosity and elasticity can be systematically varied and measured in real time, providing valuable insights into the mechanical properties of coacervates in environments that closely mimic cellular conditions.154
To study surface tension, several experimental techniques are commonly used. Micropipette aspiration is a key method where a droplet of the coacervate is aspirated into a micropipette, and the pressure required to deform the droplets is measured, allowing for the calculation of surface tension.163 Dual optical traps provide a precise method for measuring the surface tension of biomolecular condensates by studying their fusion dynamics (Fig. 5E–G).163,180,181 In this approach, two condensates are individually controlled using optical traps and brought into contact (Fig. 5E). Upon releasing one condensate, the fusion process occurs under the influence of viscosity and surface tension. High-speed imaging captures the process, and changes in the aspect ratio of the condensates are analyzed to extract the fusion time using a single exponential decay model (Fig. 5F). The geometric mean of the condensate diameters defines their length, allowing the ratio of viscosity to surface tension (inverse capillary velocity) to be estimated from the fusion time versus length relationship (Fig. 5G). The right-angle prism imaging technique is another powerful method for determining the surface tension of biomolecular condensates (Fig. 5H).109 By imaging the XZ plane of condensates, this technique measures their radius and height, enabling the application of the Young-Laplace equation to calculate surface tension.182 As shown in Fig. 5I, when a coalesced droplet flattens at the bottom of the nucleus under gravity, the extent of flattening is countered by surface tension, providing key data for its measurement. Using this approach, the surface tension of the outermost granular component of the nucleolus was determined to be approximately 0.4 μN m−1. Similarly, the method was employed to measure the surface tension of purified nucleophosmin droplets in vitro, yielding a value of around 0.8 μN m−1.109 These remarkably low surface tension values indicate the liquid-like, immiscible behavior of nucleolar subcompartments. Atomic force microscopy (AFM) can also be used to measure the forces required to deform a droplet directly, offering insights into both surface tension and mechanical properties.178 Together, these methods provide a comprehensive understanding of surface properties, guiding the design of coacervate systems with tailored properties for specific applications.
4.2. Environmental effects on LLPS
The formation and stability of peptide coacervates are highly sensitive to environmental conditions such as pH, temperature, ionic strength, and molecular crowding.183–186 Understanding how these factors influence LLPS is crucial for deciphering the mechanisms that regulate coacervate behavior both in vivo and in vitro.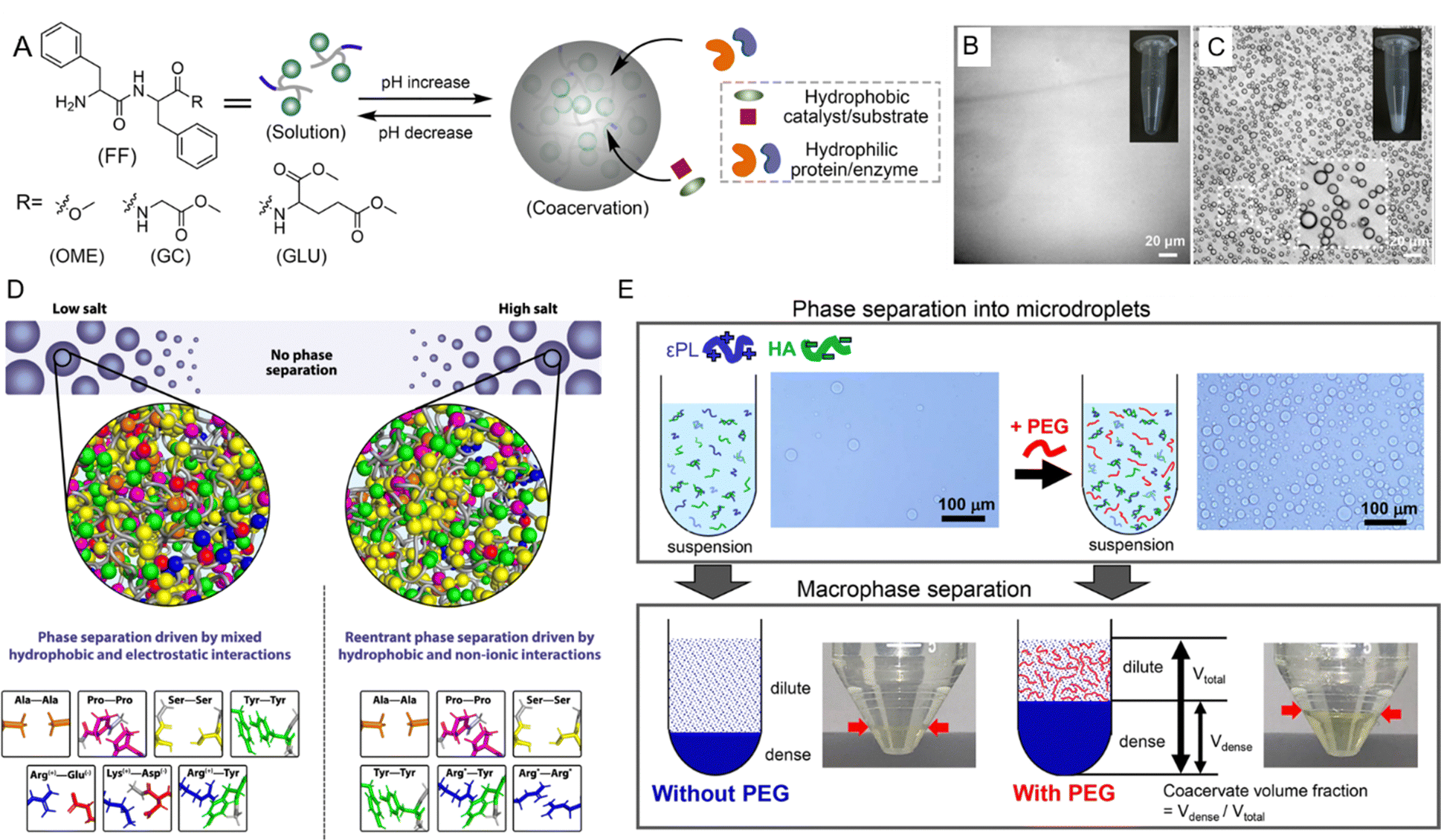 | ||
| Fig. 6 Environmental effects on liquid–liquid phase separation (LLPS) of peptides. (A) Schematic representation of pH-triggered LLPS involving dipeptide components. (B) Micrographs showing diphenylalanine capped with a methoxy group (FF-OMe) in solution at pH 6. (C) Micrographs showing an FF-OMe coacervate dispersion at pH 9. (A)–(C) Reprinted with permission under a Creative Commons Attribution 4.0 International License.65 Copyright 2024 by the authors. (D) Schematic of molecular forces stabilizing condensates in low-salt versus high-salt regimes. In low-salt conditions, phase separation is driven by both electrostatic and hydrophobic interactions, while in high-salt regimes, hydrophobic and nonionic interactions dominate. Asterisks (*) for Arg*-Tyr and Arg*-Arg* indicate that at high salt concentrations, charges are screened, and interactions become predominantly hydrophobic. Reprinted with permission under a Creative Commons Attribution 4.0 International License.188 Copyright 2021 by the authors. (E) Schematic of LLPS in a system of ε-polylysine (ε-PL) and hyaluronic acid (HA) with and without PEG. Microdroplet coacervate suspension was observed via optical microscopy, with macrophase separation occurring after centrifugation. Reprinted with permission under a Creative Commons Attribution 4.0 International License.189 Copyright 2020 by the authors. | ||
4.3. Phase behavior and transition mechanisms
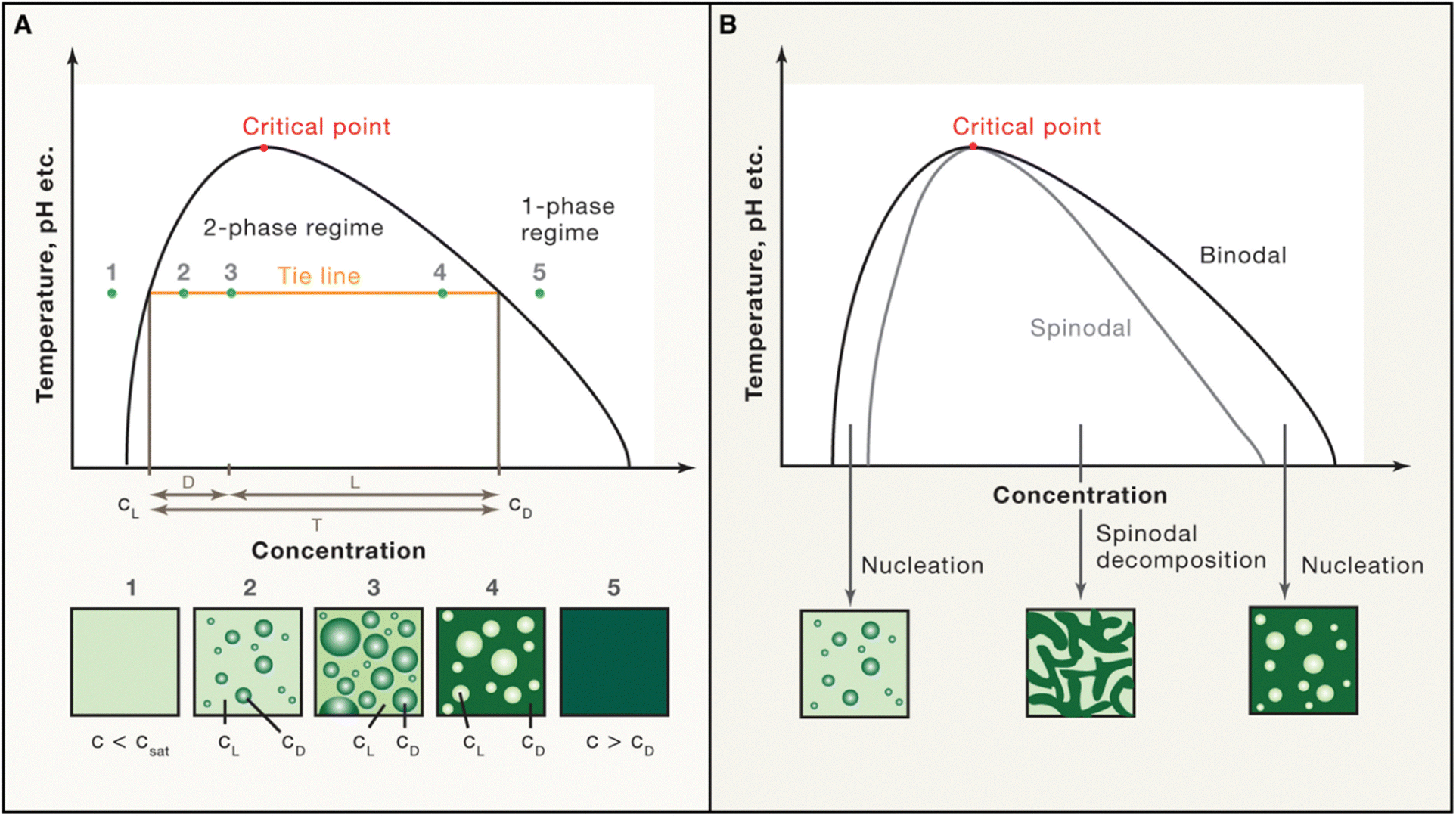 | ||
| Fig. 7 Schematic phase diagram. (A) and (B) The phase diagram shows the coexistence line (black), which delineates the boundary between the one-phase and two-phase regimes, influenced by environmental factors such as temperature and pH. Beyond the critical point, phase separation does not occur. (A) Below the saturation concentration (csat), the system remains in the one-phase regime. In the two-phase regime, the system separates into a light phase (cL) and a dense phase (cD), with fixed concentrations along a tie line (orange). Volume fractions of each phase can be calculated using the lever rule. (B) The spinodal curve (grey) marks the region of instability where spinodal decomposition occurs. Between the coexistence line (binodal) and the spinodal, phase separation requires nucleation. Reprinted with permission.2 Copyright 2019 Elsevier. | ||
The most common phase diagrams used in LLPS studies are temperature-concentration diagrams, where the x-axis represents the concentration of the biomolecules, and the y-axis represents the temperature. These diagrams often feature binodal curves that define the coexistence region, where the system separates into a dense phase and a dilute phase. The binodal curve comprises two arms: the left arm represents the saturation concentration in the dilute phase, while the right arm indicates the concentration in the dense phase (Fig. 7). Constructing accurate phase diagrams for LLPS requires precise experimental techniques to measure biomolecule concentrations in both phases across various temperatures and conditions. Traditional methods include optical microscopy, turbidimetry, and light scattering to determine the cloud-point temperature (the temperature at which phase separation begins at a fixed concentration) or the saturation concentration (the concentration at which phase separation occurs at a fixed temperature).2,204–206 Recent advances have introduced high-throughput techniques like droplet microfluidics, which allow label-free extraction of complete phase diagrams in finite volumes.102,114,145 This method confines solutions in micro-sized compartments, enabling precise measurement of dense phase volume and binodal lines. In addition to experimental approaches, theoretical models, such as the Flory–Huggins theory, are also employed to fit experimental data and predict phase behavior.9 These models account for the free energy of mixing and interaction parameters between different components, providing a framework for understanding the thermodynamics of LLPS. Combining experimental data with theoretical models enables to construct comprehensive phase diagrams that accurately capture the conditions for phase separation.
Phase diagrams have far-reaching applications in biological research and material science.3,207,208 In biology, they are used to understand the formation and regulation of membraneless organelles. Mapping the phase behavior of peptides and proteins under various conditions provides insights into the mechanisms driving the assembly and disassembly of these condensates.3,207 In material science, phase diagrams guide the design of smart materials, such as temperature-responsive polymers and drug delivery systems, which leverage LLPS for encapsulating and releasing therapeutic agents in response to environmental triggers.209,210
Overall, phase diagrams are indispensable for understanding the conditions under which LLPS occurs, providing a window into the thermodynamic and kinetic properties of phase-separating systems. The construction and interpretation of these diagrams, through both experimental and theoretical approaches, are crucial for advancing our knowledge of biomolecular condensates in cells and for designing materials with novel functionalities. As research continues to refine these tools, phase diagrams will continue to be central to LLPS studies, offering critical insights into the behavior of complex systems in both natural and synthetic contexts.
The aging of biomolecular condensates is governed by structural and biochemical factors. Proteins with IDRs promote LLPS through transient molecular interactions that maintain fluidity.217 Post-translational modifications, such as phosphorylation and methylation, further stabilize these interactions, delaying the transition to solid-like states.88,218–220 In their absence, condensates age more rapidly, leading to aggregation and functional decline.2,187 Structural changes, including the formation of β-sheet-rich regions or multiphase architectures like gel-core/liquid-shell configurations, contribute to aging by introducing localized molecular order.221 These transitions, driven by enhanced interprotein interactions, progressively stabilize the solid-like state and alter the dynamics of condensates. Proper regulation of aging is critical for preserving the functionality of biomolecular condensates. Besides the posttranslational modification, the loss of the liquid-like character of condensates during aging can be modulated by biochemical processes and amino acid sequence mutations.106,212,213 Understanding the regulatory mechanisms is crucial for developing therapeutic strategies to mitigate pathological aging. By targeting molecular interactions, biochemical pathways, or energy-dependent processes, it may be possible to prevent or reverse aberrant transitions, preserving the dynamic and functional nature of biomolecular condensates.
Peptide self-assembly through LLPS provides a unique opportunity to explore the transition from liquid-like to solid-like states in synthetic systems.51,56,222–225 Unlike the classical theory describing a direct liquid-to-solid transition, Fig. 8A illustrates the nucleation and growth process of supramolecular nanofibrils mediated by LLPS.130 Initially, a homogeneous solution of amphiphilic amino acids or short peptides undergoes phase separation into solute-rich and solute-poor liquid phases. The solute-rich liquid droplets serve as precursors for nanofibril nucleation, with hydrated nanoclusters acting as nucleation loci. This phase separation is primarily entropy-driven, with the favorable entropy contribution likely originating from the expulsion of water from the liquid droplets. These solute-rich liquid droplets create specialized microenvironments that, lower the nucleation barrier compared to classical nucleation pathways and facilitate the assembly of highly ordered nanostructures. The transition from metastable liquid droplets to thermodynamically stable nanofibrils is mediated by enthalpic interactions and follows Ostwald's step rule.226Fig. 8B shows the dynamic evolution of Ag+-coordinated 9-fluorenylmethoxycarbonyl (Fmoc)-Ala nanofibrils from liquid droplets to solidified intermediates and ultimately to mature nanofibrils.130 Cryo-TEM images show the formation of dense liquid droplets that coalesce over time, while solidified droplets exhibit increased contrast, signifying structural reorganization. Fibril-like protrusions emerge from the droplet, marking the transition to more stable, thermodynamically favorable nanofibrils. The inset SEM image further corroborates the elongation of fibrils from the droplet surface, demonstrating the progression from metastable liquid intermediates to ordered fibrillar structures.
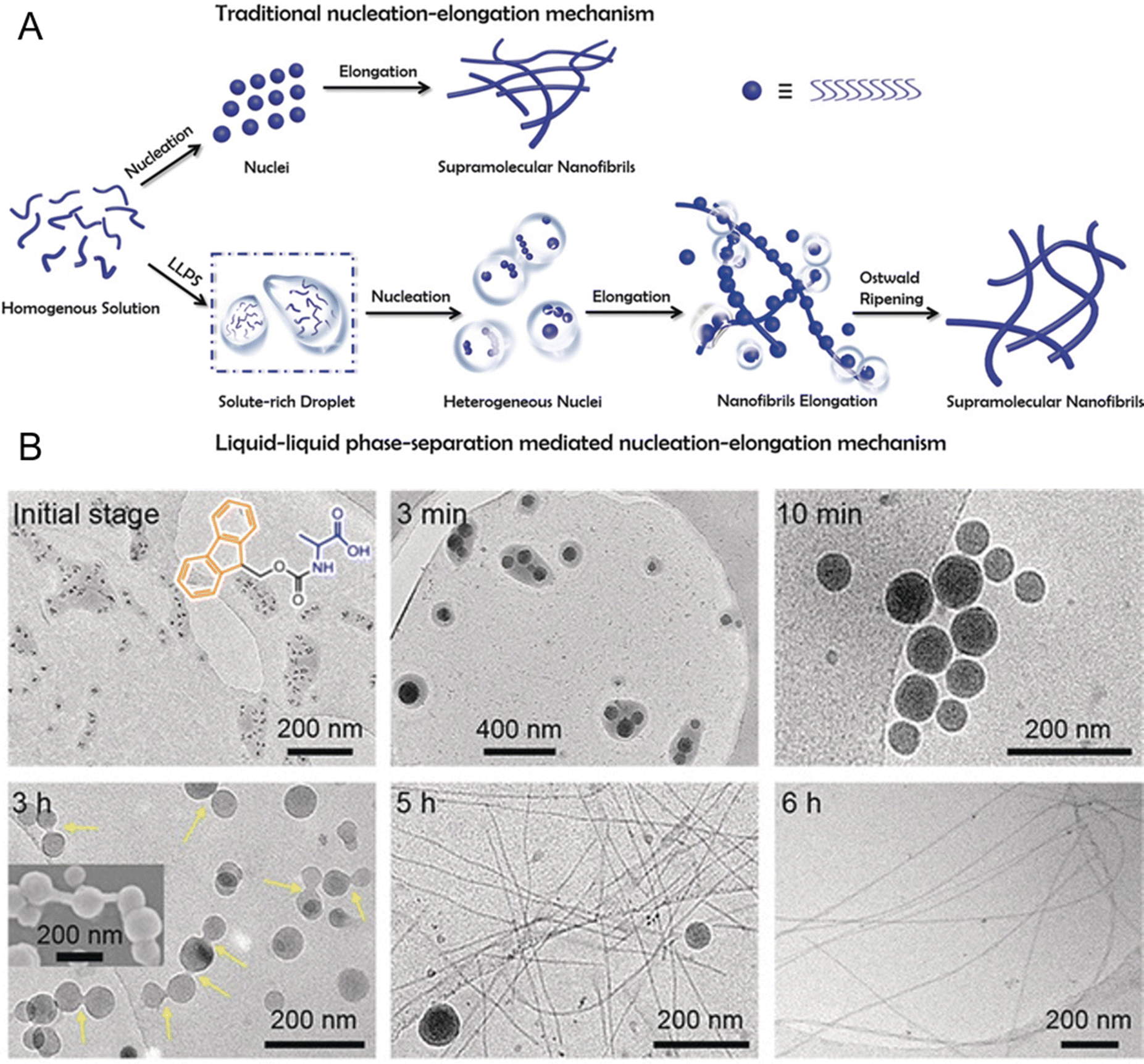 | ||
| Fig. 8 Formation of supramolecular nanofibrils via liquid–liquid phase separation (LLPS)-mediated nucleation and self-assembly of peptides. (A) Schematic of the self-assembly process of supramolecular nanofibrils from amino acids or short peptides, where LLPS precedes nucleation, with solute-rich droplets serving as nucleation precursors for fibril formation following Ostwald's step rule, which is different from the traditional nucleation-elongation mechanism. (B) Cryo-TEM images of the dynamic evolution process of Ag+-coordinated 9-fluorenylmethoxycarbonyl (Fmoc)-Ala self-assembling nanofibrils over time. These images demonstrate the structural evolution from the amino acid-dense liquid droplets to the metastable solidified droplets and, finally, to thermodynamically favorable nanofibrils through LLPS, nucleation, and subsequent nucleation-elongation processes. Reprinted with permission.130 Copyright 2019 Wiley-VCH Verlag KGaA. | ||
The self-assembly of peptides into nanostructures through LLPS is influenced by multiple factors. Recent studies using synthetic coacervates and in vitro reconstitution systems have provided valuable insights into the mechanisms underlying the liquid-to-solid transition in LLPS.53,106 For example, chemically fueled reaction cycles have been utilized to control the liquidity of droplets, allowing precise tuning of the material state of condensates.227 These systems highlight the importance of both thermodynamic and kinetic factors to fully understand and predict the behavior of LLPS under different conditions. Additionally, advanced imaging techniques, such as FRAP and super-resolution microscopy, provide real-time insights into the dynamics of condensates as they transition between liquid-like and solid-like phases.228 These techniques reveal that even within a single condensate, different regions may display varying degrees of liquidity or solidity, reflecting the complex and heterogeneous nature of these structures.
The study of peptide self-assembly through LLPS not only advances our understanding of fundamental processes driving phase separation but also offers insights into the design of biomimetic materials with tunable properties. By controlling environmental conditions such as temperature, pH, and ionic strength, the material state of condensates can be fine-tuned, making them useful for applications in biomaterial engineering, drug delivery, and disease treatment. The transition from liquid-like to solid-like states in LLPS is governed by a complex interplay of molecular interactions, environmental conditions, and cellular regulation. Understanding how these transitions occur is essential for elucidating the dynamics of biomolecular condensate and developing therapeutic interventions for diseases linked to aberrant phase transitions.
4.4. Influence of external stimuli on LLPS
LLPS is a highly dynamic process that cells can finely tune in response to various external stimuli, allowing rapid adaption to changing environmental conditions. Among the most impactful stimuli are light, redox changes, and mechanical forces, each capable of modulating the assembly, disassembly, and material properties of phase-separated condensates.152,229–231 By influencing these properties, external stimuli play a pivotal role in the structural and functional regulation of biomolecular condensates.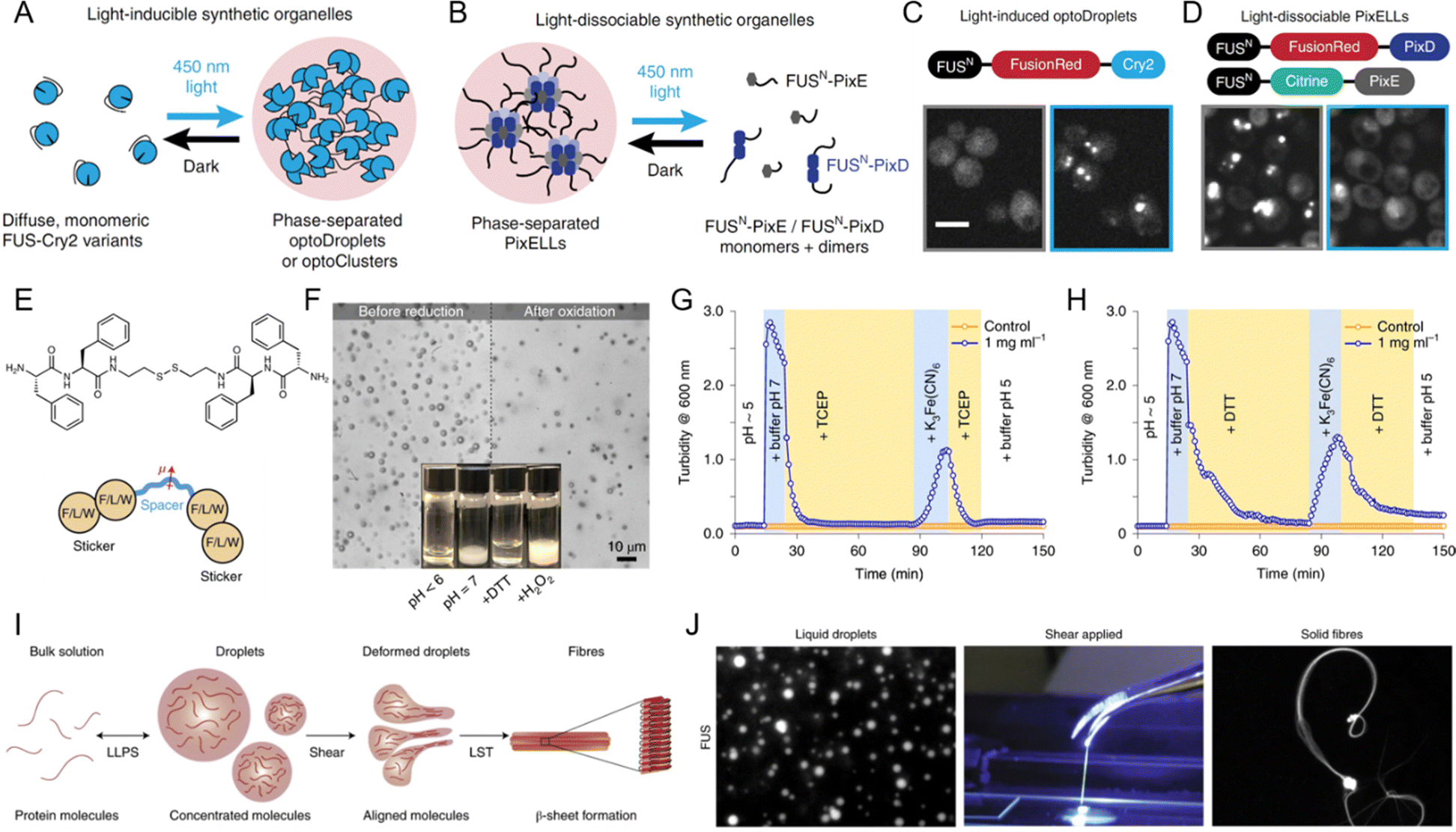 | ||
| Fig. 9 Influence of external stimuli on liquid–liquid phase separation (LLPS). (A)–(D) Light-mediated control of biomolecular condensate formation. (A) and (C) FUS proteins fused to variants of the Cry2 optogenetic system trigger phase separation in the optoDroplet and optoCluster systems upon exposure to 450 nm light. (B) and (D) FUS fusion to the PixD/E proteins enables the formation of the PixELL optogenetic system, which dissociates under 450 nm light. Scale bar, 5 μm. (A)–(D) Reprinted with permission.230 Copyright 2019 Springer Nature. (E)–(H) Redox regulation of LLPS. (E) Structure of bis(phenylalanyl–phenylalanyl) cystamine (FFssFF) and schematic illustration of the synthon motif with two dipeptide stickers and a polar spacer. (F) Formation and dissolution of FFssFF coacervate droplets before reduction and after oxidation. (G) Kinetics of coacervate dissolution upon reduction with tris(2-carboxyethyl) phosphine (TCEP) and reformation through condensation upon oxidation with K3Fe(CN)6, as measured by turbidity. (H) Similar kinetics observed with dithiothreitol (DTT) as a reducing agent. (E)–(H) Reprinted with permission.238 Copyright 2021 Springer Nature. (I) and (J) Influence of shear force on LLPS-driven biomolecular condensates. (I) Schematic and experimental data showing the formation of β-sheet-rich fibers from condensate droplets under shear force. (J) Formation of FUS solid fibers under shear forces. Reprinted with permission.152 Copyright 2020 Springer Nature. | ||
In synthetic systems, redox changes are similarly crucial for modulating LLPS and controlling the formation of peptide coacervates. Redox-sensitive groups, such as disulfide bonds, undergo reduction or oxidation, causing structural changes in proteins or peptides that influence their phase separation behavior. Fig. 9E illustrates the structure of bis(phenylalanyl–phenylalanyl) cystamine (FFssFF) peptide derivative, which consists of two phenylalanine dipeptide “stickers” and a polar cystamine “spacer”.238 This peptide derivative undergoes redox-induced phase separation, with the reduction and oxidation of disulfide bonds playing a critical role in driving coacervation. Fig. 9F shows the reversible behavior of FFssFF coacervates, with droplets dissolving after reduction and reforming after oxidation. Fig. 9G demonstrates the reduction of the disulfide bond using tris (2-carboxyethyl) phosphine (TCEP), resulting in the dissolution of coacervates into a clear solution. Upon oxidation with potassium ferricyanide (K3Fe(CN)6), the coacervates reform, demonstrating the reversibility of this redox-induced coacervation. This process is not limited to TCEP, as shown in Fig. 9H, where dithiothreitol (DTT) is used as the reducing agent and the same reversible coacervation behavior is observed. These findings underscore the versatility of redox-responsive systems, where the ability to control phase separation in synthetic systems mirrors the dynamic behavior seen in biological condensates. By leveraging redox chemistry, researchers can design systems with precisely tunable LLPS behavior, expanding the potential applications of biomolecular condensates in both fundamental research and therapeutic strategies.
5. Applications of peptide-mediated LLPS and biomolecular condensates
5.1. Biomaterials and tissue engineering
Peptide-mediated LLPS and biomolecular condensates have gained significant attention in recent years as promising tools in the field of biomaterials and tissue engineering. These systems offer unique properties such as environmental responsiveness, tunable mechanical strength, and the ability to encapsulate bioactive molecules, which make them highly suitable for various applications, including bioadhesives, tissue repair, biocomposites, and dynamic scaffolds.155,247–252 Inspired by natural processes, peptide coacervates have the potential to create highly adaptable materials that can be customized to meet the specific needs of different biological applications.One of the most promising applications of peptide or protein-based biomolecular condensates is their use as bioadhesives for soft and hard tissue repair.253,254 Traditional adhesives often fall short of meeting the mechanical and biological requirements for effective tissue integration. However, peptide coacervates offer a solution by mimicking the adhesive properties of natural systems. Fig. 10A shows the bioinspired condensates composed of recombinant mussel adhesive protein (rMAP) and hyaluronic acid (HA).247 These rMAP-HA condensates not only outperform their individual components in terms of adhesive performance but also promote cell proliferation when applied to implant surfaces. This cell growth is essential for tissue integration, a key factor in the success of medical devices or implants. Further modifications, such as the fusion of rMAP with low-complexity mammalian sequences, improve the adhesive strength by forming stronger, amyloid-like nanofibrils that firmly adhere to surfaces.255 In addition to enhancing adhesive strength, these coacervates also support tissue repair. Their viscous, water-immiscible nature makes them suitable for sealing tissues and promoting healing. For example, rMAP-HA condensates have been successfully used to seal urinary fistulas, which is a difficult clinical issue, and have shown potential as skin grafts for wound healing.253,256 Moreover, these condensates have been applied as binders for bone xenografts, aiding in bone regeneration.254,257 Their ability to encapsulate and release growth factors, such as bone morphogenetic protein 2, further enhances their regenerative potential by simulating tissue regeneration and promoting healing at the site of injury.254 This versatility makes bioinspired coacervates a powerful tool for advanced tissue repair technologies.
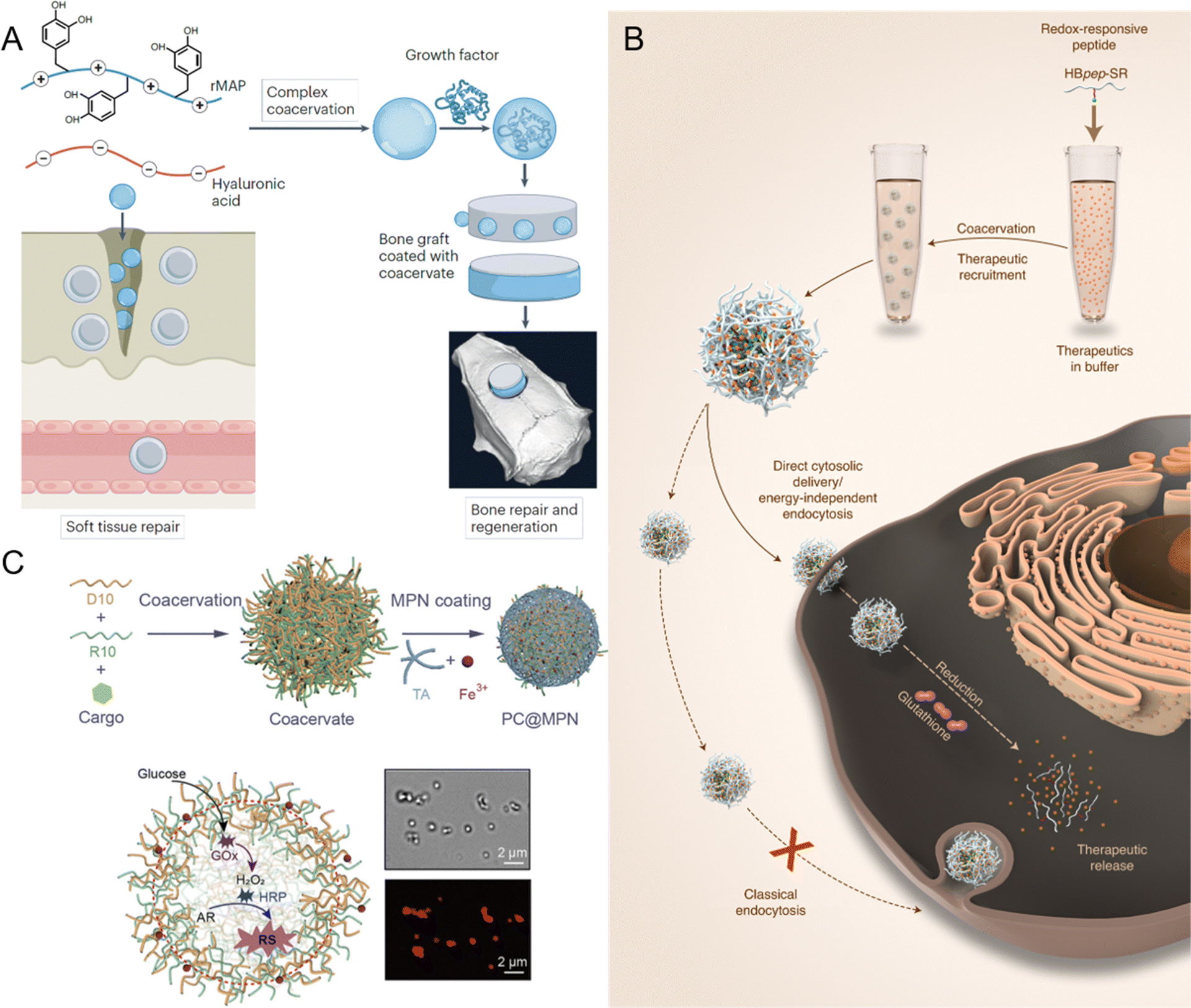 | ||
| Fig. 10 Applications of peptide-mediated liquid–liquid phase separation (LLPS) and biomolecular condensates in various fields. (A) Application of recombinant mussel adhesive protein (rMAP) and hyaluronic acid (HA) coacervates for tissue engineering, including soft tissue repair and bone regeneration via growth factor recruitment. Reprinted with permission.247 Copyright 2024 Springer Nature. (B) Design of redox-responsive peptide coacervates for direct cytosolic drug delivery. Histidine-rich beak peptides conjugated with a lysine residue and a self-immolative moiety (HBpep-SR) undergo coacervation, encapsulating macromolecular therapeutics. Upon incubation with cells, the therapeutics-loaded coacervates enter the cytosol, where intracellular glutathione triggers coacervate disassembly and drug release. Reprinted with permission.258 Copyright 2022 Springer Nature. (C) Fabrication of membrane-bound protocells (PC@MPN) with emergent properties, achieved by coating peptide coacervates (PCs) with metal-phenolic networks (MPNs). The protocell facilitates enzymatic cascade reactions, where glucose oxidase (GOx) converts glucose to H2O2 and horseradish peroxidase (HRP) catalyzes the oxidation of Amplex Red (AR) to resorufin (RS), as indicated by red fluorescence. Reprinted with permission.259 Copyright 2023 American Chemical Society. | ||
Peptide coacervates also offer significant potential as dynamic scaffolds in tissue engineering.260 The mechanical properties, such as stiffness and flexibility, can be tuned by modifying the peptide sequence, allowing for the creation of scaffolds tailored to specific tissue types. HBPs derived from squid have shown exceptional mechanical strength due to their repetitive peptide motifs, which facilitate strong intermolecular interactions.261 These properties make them well-suited for applications such as bone tissue engineering, where scaffolds must withstand mechanical forces. Elastin-like polypeptides, on the other hand, are widely used for soft tissue scaffolds such as skin and vascular scaffolds, owing to their excellent biocompatibility, low immunogenicity, and bioactive properties.250,262 In tissue engineering, scaffolds must not only provide mechanical support but also mimic the extracellular matrix to promote cell adhesion, proliferation, and differentiation.263 Peptide coacervates offer a unique advantage in this regard due to their structural resemblance to the extracellular matrix. This enables them to provide a supportive environment conducive to cell growth and tissue regeneration. Furthermore, the properties of coacervates can be tailored to meet the specific requirements of different tissues. For example, the mechanical strength and degradation rate of coacervate-based scaffolds can be adjusted to optimize them for bone, cartilage, or soft tissue applications. This customization is achieved by modifying the peptide sequences or coacervation conditions, enabling scaffolds to degrade at rates matching the regeneration needs of different tissues.247,264 Integrating peptide coacervates into composite materials provides an opportunity to create hybrid scaffolds that combine the mechanical stability of synthetic polymers with the biological activity and tunability of peptides.247 These hybrid systems retain the strength of traditional biomaterials while incorporating bioactive components that support tissue regeneration. Such composites enable the development of scaffolds that can endure mechanical forces yet offer the biochemical cues essential for tissue repair, making them suitable for applications in bone and cartilage engineering where both mechanical strength and biological responsiveness are required.
Despite the promising applications of peptide coacervates in tissue engineering and biomaterials, challenges remain in optimizing their properties for clinical use. One of the main challenges is achieving precise control over the coacervation process to ensure reproducibility and consistency in material properties. Variations in coacervate formation can lead to inconsistencies in scaffold performance, which could impact their effectiveness in clinical applications. Additionally, the long-term stability and biocompatibility of peptide-based materials must be carefully evaluated to ensure their suitability for in vivo use. These materials must degrade at a rate that matches tissue regeneration, and their degradation products must be non-toxic and biocompatible. Future research should focus on improving the design and functionality of peptide coacervates for specific tissue engineering applications. This includes exploring novel peptide sequences that enhance mechanical strength, environmental responsiveness, and bioactivity. Integrating peptide coacervates with other biomaterials, such as hydrogels or bioactive ceramics, may yield hybrid systems with enhanced performance, offering multifunctionality for applications across different tissue types.
5.2. Drug delivery systems
Peptide coacervates have emerged as a transformative platform for drug delivery, offering unique advantages over traditional systems like liposomes, polymers, and lipid nanoparticles.265–270 While lipid-based carriers offer high biocompatibility and versatile drug encapsulation, they often suffer from low drug loading and require complex surface modification to address stability issues, endosomal entrapment, and high hepatic or splenic uptake.268,271 Similarly, polymers face challenges such as particle aggregation and potential toxicity.268 Peptide coacervates may overcome these hurdles through LLPS, producing micrometer-sized, membraneless droplets capable of sequestering a wide range of therapeutic molecules, including proteins, nucleic acids, and small molecules.64,272,273 Encapsulation relies on non-covalent interactions, including electrostatic forces, hydrogen bonding, and hydrophobic interactions.266 These interactions are crucial for maintaining the integrity and efficacy of therapeutic agents, particularly sensitive biomolecules like proteins and nucleic acids, which are susceptible to degradation in the biological environment.62,239 By creating a stable microenvironment, peptide coacervates effectively shield therapeutic agents from premature degradation, ensuring that they reach their target site intact. Additionally, the amino acid-based composition allows for simple, precise tuning of physicochemical properties through single amino acid mutations, providing unparalleled adaptability and scalability compared to the labor-intensive fabrication of liposomes and polymers.162 Furthermore, peptide coacervates exhibit negligible cytotoxicity and excellent biocompatibility, avoiding the safety concerns associated with inorganic nanoparticles and synthetic polymers.258A defining feature of peptide coacervates-based drug delivery systems is their capacity for controlled and stimuli-responsive release of encapsulated therapeutics.61,239,274 Redox-responsive peptide coacervates, for example, are designed to release their cargo in response to the redox conditions prevalent in specific physiological environments, such as the tumor microenvironment, which is often characterized by high levels of reducing agents.62,239 This targeted release is particularly beneficial in cancer therapy, where minimizing systemic exposure and off-target effects is crucial. Wang et al. demonstrated that redox-responsive peptide coacervates could be engineered to selectively release tissue plasminogen activator in response to the reducing conditions found at thrombus sites, significantly enhancing the therapeutic efficacy and reducing off-target effects.62 Similarly, another study by Sun et al. explored the potential of redox-responsive coacervates in the delivery of CRISPR-Cas9 complexes, where the coacervates disassembled in response to the intracellular reducing environment, allowing for efficient cytosolic delivery of the genome-editing tool.239 These findings underscore the versatility of redox-responsive coacervates for site-specific drug delivery, making them a powerful platform for developing next-generation therapeutics. Fig. 10B illustrates the design of redox-responsive peptide coacervates, histidine-rich beak peptides conjugated with a lysine residue and a self-immolative moiety (HBpep-SR), that can directly deliver a wide range of macromolecular therapeutics into the cytosols.258 In a reducing environment, such as glutathione-rich cytosol, HBpep-SR is reduced, leading to self-catalytic cleavage of the SR moiety and disassembling of the coacervates. In addition to their redox-responsiveness, peptide coacervates also exhibit a high degree of modularity and adaptability, which can be leveraged to design sophisticated drug delivery systems. A notable aspect of this adaptability is the ability to encapsulate multiple substances within a single coacervate. Lim et al. developed glucose-responsive peptide coacervates that co-encapsulate insulin and glucose oxidase, providing a coordinated release of insulin in response to rising blood glucose levels.64 This concept could extend other conditions, such as cancer or metabolic disorders, where multiple therapeutic targets require a coordinated response.275,276 These multifunctional platforms enable the treatment of complex diseases through the simultaneous and responsive release of complementary drugs, potentially increasing treatment efficacy while minimizing side effects.
The interaction of peptide coacervates with biological membranes is another critical factor in their effectiveness as drug delivery systems.277,278 Unlike traditional delivery vehicles that rely on lipid membranes, peptide coacervates can interact directly with the cytosol, facilitating efficient drug release inside cells. This is achieved through electrostatic attractions that promote cellular uptake via endocytosis or, in some cases, enable direct cytosolic delivery.277 The work of Sun et al. exemplifies this potential by demonstrating the use of micrometer-sized peptide coacervates as delivery vesicles capable of crossing cell membranes without relying on endocytosis (Fig. 10B).258 These coacervates, designed to be both pH- and redox-responsive, disassemble within the intracellular environment, releasing their cargo directly into the cytosol. This capability challenges the conventional belief that only nanosized vesicles can penetrate cell membranes, suggesting that larger coacervates may also serve as effective delivery vehicles. This work highlighted the potential of peptide coacervates in delivering macromolecular therapeutics, including proteins, peptides, and RNAs, with both precision and efficiency, showcasing their utility in diverse therapeutic applications.
While peptide coacervates offer numerous advantages in drug delivery, challenges remain in translating them into clinical applications, primarily related to stability.279 Without a traditional membrane, they are susceptible to a rapid coalescence or collapse in biological fluids.280,281 Their stability is significantly influenced by environmental factors such as pH, temperature, and ionic strength.56,190,219 For example, high ionic conditions can disrupt the interactions that maintain coacervate structure, causing premature release of encapsulated drugs.219 Several strategies have been developed to address these challenges. One prominent approach is encasing coacervates within stabilizing membranes made of terpolymers, phospholipids, or polysaccharides.282–284 These membranes can mitigate environmental sensitivity while retaining the ability to respond to external stimuli, although they may reduce permeability to large biomolecules. Cross-linking at the droplet interface is another effective approach.63,285,286 The incorporation of stabilizing agents like polyphenols (e.g., tannic acid) forms supramolecular networks, improving stability without compromising functionality.63 Exposing coacervates to deionized water induces counterion extraction and physical crosslinking, resulting in the formation of viscoelastic interfaces that prevent fusion and enhance long-term stability.286,287 Increasing the surface charge density, as indicated by high zeta potential, has also been reported to reduce droplet fusion through enhanced electrostatic repulsion.153 Additionally, peptide coacervates have been stabilized against Ostwald ripening through a combination of attractive electrostatic interactions and translational entropy within the coacervates, where the charged nature of the components creates an energetic barrier that effectively suppresses droplet coarsening.280 Beyond stability, another consideration is the potential immunogenicity of peptide coacervates, which must be carefully evaluated to avoid unwanted immune responses that could compromise their efficacy or safety.288 Scalability of production is also a challenge, as consistent and reproducible manufacturing processes are essential for regulatory approval and clinical use. Advances in manufacturing techniques, including microfluidics and high-throughput screening offer promising solutions to streamline large-scale production.
Peptide coacervates represent a highly promising approach to drug delivery, offering controlled and targeted delivery of various therapeutic agents. Their ability to interact effectively with biological membranes, combined with customizability and multifunctionality, makes them a valuable tool in the development of next-generation drug delivery systems. As research in this field continues to advance, overcoming current challenges related to stability, immunogenicity, and scalability will be crucial to fully realizing the potential. Ongoing studies that delve into the mechanisms underlying coacervate formation, stability, and interaction with biological systems will be essential in advancing this field. The pioneering work on direct cytosolic delivery by micrometer-sized coacervates without endocytosis underscores the transformative potential of these systems, particularly for challenging applications such as cancer therapy and the delivery of macromolecular drugs.258 Continued exploration in these areas will drive the transition of peptide coacervate-based systems from laboratory research to clinical applications, offering innovative solutions for treating complex diseases.
5.3. Synthetic biology
The concept of artificial cells, or protocells, represents a key frontier in synthetic biology, where researchers aim to build cell-like systems capable of replicating essential functions of living organisms.53,289–294 Peptide coacervates have emerged as a promising platform for constructing these artificial cells due to their ability to create membraneless compartments that closely resemble the internal organization of natural cells.53,295–297 This section examines the various roles of peptide coacervates in artificial cell development, emphasizing their role in mimicking cellular functions, supporting biochemical processes, and enabling complex synthetic biology applications.One of the fundamental features of living cells is their compartmentalization, which allows for the segregation of biochemical processes and the establishment of specialized microenvironments. Peptide coacervates inherently possess the ability to form discrete, membraneless compartments, making them ideal candidates for constructing artificial cells.53 These coacervates can encapsulate diverse biomolecules, such as nucleic acids, enzymes, and metabolites, thereby creating microenvironments that facilitate specific biochemical reactions.298–300 The selective permeability of coacervate droplets is another critical attribute that mirrors the behavior of natural cellular membranes.285 Coacervates can selectively sequester or exclude certain molecules based on their size, charge, or hydrophobicity. This property enables the coacervates to function as primitive organelles, where they can concentrate substrates, protect sensitive molecules, or control the flow of ions and other small molecules. Such selective permeability is crucial for maintaining the internal environment of artificial cells and ensuring that the encapsulated biochemical processes proceed efficiently.65
While coacervates naturally form membraneless compartments, they can also be integrated with structured membranes to create hybrid systems.259,294,301 In these hybrid constructs, coacervates function as primitive organelles within structured membranes, providing localized environments for specific reactions, while the membranes not only prevent aggregation and coalescence but also regulate the selective biomolecule sequestration and chemical exchange across the membrane.284Fig. 10C illustrates hybrid PC@MPN protocells, where peptide coacervates (PCs), mimicking the cytosol, are coated with a metal-phenolic network (MPN) membrane.259 The PCs are formed by the self-assembly of oppositely charged oligopeptides (R10 and D10), while the MPN membrane is generated through coordination between metal ions (e.g., Fe3+) and polyphenols (e.g., tannic acid). This hybrid design provides structural stability while preserving the dynamic properties of coacervates, enabling the construction of artificial cells that can selectively exchange molecules and maintain internal biochemical processes. Furthermore, the integration of coacervates with structured membranes also enables the construction of more sophisticated artificial cells that can interact with their environment in a controlled manner.302 Coacervate-membrane hybrids can be engineered to respond to changes in pH, temperature, or the presence of specific molecules, triggering the release of encapsulated contents or a change in the physical properties of the cell.53,303,304
The high degree of macromolecular crowding within coacervates mimics the crowded interior of living cells, which is known to significantly influence the kinetics and thermodynamics of biochemical reactions.305 This crowded environment enhances the binding affinity between enzymes and substrates, accelerates reaction rates, and can stabilize proteins that would otherwise be unstable in dilute solutions.300,306,307 One of the most striking demonstrations of the ability of coacervates to support biochemical reactions is their use in gene expression systems.298 Coacervates have been shown to significantly enhance the transcription rates of genes, with rates that are comparable to those observed in vivo.298 This enhancement is largely due to the increased binding constant of DNA to transcription factors and RNA polymerase in the crowded environment of the coacervate. The coacervate droplets can thus function as efficient microreactors for gene expression, supporting both transcription and translation within a confined space. Furthermore, coacervates have been used to construct artificial cells that can carry out more complex functions, such as the synthesis of proteins from DNA templates.298,308,309 These artificial cells can encapsulate the entire transcription and translation machinery, enabling the production of functional proteins within the coacervate droplets. The ability to carry out such complex biochemical processes within coacervates highlights their potential as building blocks for more advanced synthetic cells.
Beyond simply supporting biochemical reactions, peptide coacervates can be engineered to simulate specific cellular processes, making them valuable for studying cellular dynamics and creating functional synthetic systems. Coacervates have been used to replicate aspects of cellular metabolism, such as the compartmentalization of metabolic pathways.230,305,310 By confining different enzymes in separate coacervate droplets, it is possible to establish synthetic metabolic networks that carry out sequential reactions in a controlled manner. Fig. 10C demonstrates the capability of PC@MPN protocells to compartmentalize and sustain enzymatic cascade reactions. These protocells are loaded with enzymes such as glucose oxidase (GOx) and horseradish peroxidase (HRP), as well as the substrate Amplex Red (AR).259 GOx catalyzes the oxidation of glucose to produce H2O2, which acts as a substrate for HRP to convert AR into resorufin (RS). The bright-field and fluorescence microscopy images confirm the success of this reaction, with red fluorescence indicating the production of RS. This example highlights how peptide coacervates can serve as compartments for synthetic metabolic networks, enabling sequential reactions within confined spaces. In addition to metabolic processes, coacervates have been used to model the dynamics of intracellular organelles.295 Their liquid-like nature of coacervates allows them to undergo fusion and fission, similar to the behavior of natural membraneless organelles such as P granules.36 This dynamic behavior can be harnessed to create artificial cells with controllable internal organization, where the distribution and interaction of coacervate-based organelles can be modulated in response to external stimuli.100,296,311,312 Another key cellular process that can be mimicked using peptide coacervates is the assembly of nucleic acids and proteins into functional complexes.300 By engineering coacervates to selectively encapsulate specific proteins and nucleic acids, it is possible to recreate these types of structures within artificial cells. This capability opens new avenues for studying the principles of cellular organization and for designing synthetic cells with custom-built compartments.
The unique properties of peptide coacervates make them highly suitable for various applications in biotechnology, particularly in the development of cell-free systems and biosensors.313,314 In cell-free protein synthesis, for example, coacervates can be used to create microreactors that enable the efficient production of proteins without requiring living cells. By creating optimized conditions within coacervates, these systems can be tailored to produce specific proteins or to screen gene expression constructs in high-throughput applications.298,315 In biosensing, coacervate can encapsulate enzymes or other reactive molecules, forming droplets that respond to target analytes with high sensitivity. These biosensors can be used in a variety of applications, including medical diagnostics, environmental monitoring, and industrial process control.
Peptide coacervates offer a powerful and versatile platform for the construction of artificial cells. Their ability to mimic cellular compartmentalization, support complex biochemical reactions, and simulate dynamic cellular processes makes them invaluable for designing responsive and functional synthetic systems. By integrating coacervates with lipid membranes and other materials, sophisticated artificial cells could be created to interact dynamically with their environment and perform a wide range of functions.
6. Conclusions
Peptide-mediated liquid–liquid phase separation (LLPS) represents a rapidly evolving area of research with profound implications for both fundamental biology and applied sciences. Through this review, we have explored the intricate molecular mechanisms driving LLPS, the critical role of environmental factors in modulating phase behavior, and the diverse material properties of peptide coacervates. These insights highlight the versatility of LLPS as a mechanism for cellular organization and its potential for innovative applications in biotechnology and medicine. The dynamic nature of LLPS allows cells to compartmentalize and regulate biochemical processes without the need for traditional membrane-bound organelles, offering a flexible means of adapting to changing environmental conditions. This adaptability, however, also underscores the potential for dysregulation, which can lead to pathological states, including neurodegenerative diseases and cancer. Understanding the factors that govern the transition between liquid-like and solid-like states in biomolecular condensates is crucial for developing therapeutic strategies to mitigate these diseases. From a practical perspective, the ability to engineer peptide coacervates with tailored properties opens new avenues for applications in drug delivery, tissue engineering, and synthetic biology. The modularity and responsiveness of these systems to environmental stimuli make them ideal candidates for developing next-generation biomaterials and therapeutic platforms. However, challenges remain in ensuring the stability, biocompatibility, and scalability of peptide coacervates, which are essential for translating these promising technologies into clinical applications. In conclusion, LLPS is a powerful and versatile mechanism with significant potential to advance our understanding of cellular biology and to drive innovation in various scientific and medical fields. Continued research in this area will likely yield further breakthroughs, enabling the development of novel materials and therapeutic strategies that leverage the unique properties of glassy structures.316–318Author contributions
Guangle Li: conceptualization, funding acquisition, investigation, writing – original draft, writing – review & editing; Chengqian Yuan: conceptualization, funding acquisition, writing – review & editing; Xuehai Yan: conceptualization, funding acquisition, project administration, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by National Key R&D Program of China (2023YFA0915300), National Natural Science Foundation of China (No. 22232006, 22025207 and 22402204), International Partnership Program of the Chinese Academy of Sciences (039GJHZ2023064GC and 039GJHZ2023058FN), Youth Innovation Promotion Association of CAS (No. 2022049), and IPE Project for Frontier Basic Research (No. QYJC-2022-011).References
- A. Ianiro, H. Wu, M. M. J. van Rijt, M. P. Vena, A. D. A. Keizer, A. C. C. Esteves, R. Tuinier, H. Friedrich, N. A. J. M. Sommerdijk and J. P. Patterson, Nat. Chem., 2019, 11, 320–328 CrossRef CAS PubMed
.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed
- D. Bracha, M. T. Walls, M.-T. Wei, L. Zhu, M. Kurian, J. L. Avalos, J. E. Toettcher and C. P. Brangwynne, Cell, 2018, 175, 1467–1480 CrossRef CAS PubMed
- F. Wang, P. Altschuh, L. Ratke, H. Zhang, M. Selzer and B. Nestler, Adv. Mater., 2019, 31, 1806733 CrossRef PubMed
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef PubMed
- S. Alberti and D. Dormann, Annu. Rev. Genet., 2019, 53, 171–194 CrossRef CAS PubMed
- E. Gomes and J. Shorter, J. Biol. Chem., 2019, 294, 7115–7127 CrossRef CAS PubMed
- Timothy J. Nott, E. Petsalaki, P. Farber, D. Jervis, E. Fussner, A. Plochowietz, T. D. Craggs, David P. Bazett-Jones, T. Pawson, Julie D. Forman-Kay and Andrew J. Baldwin, Mol. Cell, 2015, 57, 936–947 CrossRef CAS PubMed
- Y. Dai, L. You and A. Chilkoti, Nat. Rev. Bioeng., 2023, 1, 466–480 CrossRef CAS PubMed
- T. Hirose, K. Ninomiya, S. Nakagawa and T. Yamazaki, Nat. Rev. Mol. Cell Biol., 2023, 24, 288–304 CrossRef CAS PubMed
- Y. Dai, Z. Zhou, W. Yu, Y. Ma, K. Kim, N. Rivera, J. Mohammed, E. Lantelme, H. Hsu-Kim, A. Chilkoti and L. You, Cell, 2024, 187, 5951–5966 CrossRef CAS PubMed
- X. Su, J. A. Ditlev, E. Hui, W. Xing, S. Banjade, J. Okrut, D. S. King, J. Taunton, M. K. Rosen and R. D. Vale, Science, 2016, 352, 595–599 CrossRef CAS PubMed
- A. R. Strom, A. V. Emelyanov, M. Mir, D. V. Fyodorov, X. Darzacq and G. H. Karpen, Nature, 2017, 547, 241–245 CrossRef CAS PubMed
- C. Roden and A. S. Gladfelter, Nat. Rev. Mol. Cell Biol., 2021, 22, 183–195 CrossRef CAS PubMed
- M.-T. Wei, Y.-C. Chang, S. F. Shimobayashi, Y. Shin, A. R. Strom and C. P. Brangwynne, Nat. Cell Biol., 2020, 22, 1187–1196 CrossRef CAS PubMed
- J. A. Riback, C. D. Katanski, J. L. Kear-Scott, E. V. Pilipenko, A. E. Rojek, T. R. Sosnick and D. A. Drummond, Cell, 2017, 168, 1028–1040 CrossRef CAS PubMed
- N. N. Noda, Z. Wang and H. Zhang, J. Cell Biol., 2020, 219, e202004062 CrossRef CAS PubMed
- A. S. Lyon, W. B. Peeples and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2021, 22, 215–235 CrossRef CAS PubMed
- X. Wu, Q. Cai, Z. Feng and M. Zhang, Dev. Cell, 2020, 55, 18–29 CrossRef CAS PubMed
- J. Y. Ong and J. Z. Torres, Mol. Cell, 2020, 80, 9–20 CrossRef CAS PubMed
- B. Wang, L. Zhang, T. Dai, Z. Qin, H. Lu, L. Zhang and F. Zhou, Signal Transduction Targeted Ther., 2021, 6, 290 CrossRef PubMed
- E. Astoricchio, C. Alfano, L. Rajendran, P. A. Temussi and A. Pastore, Trends Biochem. Sci., 2020, 45, 706–717 CrossRef CAS PubMed
- S. Mehta and J. Zhang, Nat. Rev. Cancer, 2022, 22, 239–252 CrossRef CAS PubMed
- S. Yang, W. Shen, J. Hu, S. Cai, C. Zhang, S. Jin, X. Guan, J. Wu, Y. Wu and J. Cui, Front. Immunol., 2023, 14, 1162211 CrossRef CAS PubMed
- G. Pei, H. Lyons, P. Li and B. R. Sabari, Nat. Rev. Mol. Cell Biol., 2024 DOI:10.1038/s41580-024-00789-x
- S. Elbaum-Garfinkle, J. Biol. Chem., 2019, 294, 7160–7168 CrossRef CAS PubMed
- B. S. Visser, W. P. Lipiński and E. Spruijt, Nat. Rev. Chem., 2024, 8, 686–700 CrossRef CAS PubMed
- S. Wang, T. Dai, Z. Qin, T. Pan, F. Chu, L. Lou, L. Zhang, B. Yang, H. Huang, H. Lu and F. Zhou, Nat. Cell Biol., 2021, 23, 718–732 CrossRef CAS PubMed
- T. Liang, Y. Dong, I. Cheng, P. Wen, F. Li, F. Liu, Q. Wu, E. Ren, P. Liu, H. Li and Z. Gu, Nat. Biomed. Eng., 2024, 8, 1469–1482 CrossRef CAS PubMed
- Z. Wang and H. Zhang, Trends Cell Biol., 2019, 29, 417–427 CrossRef CAS PubMed
- Z.-G. Qian, S.-C. Huang and X.-X. Xia, Nat. Chem. Biol., 2022, 18, 1330–1340 CrossRef CAS PubMed
- R. K. Das, K. M. Ruff and R. V. Pappu, Curr. Opin. Struct. Biol., 2015, 32, 102–112 CrossRef CAS PubMed
- T. S. Harmon, A. S. Holehouse, M. K. Rosen and R. V. Pappu, eLife, 2017, 6, e30294 CrossRef PubMed
- J. Wang, J.-M. Choi, A. S. Holehouse, H. O. Lee, X. Zhang, M. Jahnel, S. Maharana, R. Lemaitre, A. Pozniakovsky, D. Drechsel, I. Poser, R. V. Pappu, S. Alberti and A. A. Hyman, Cell, 2018, 174, 688–699 Search PubMed
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Jülicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS PubMed
- S. F. Banani, A. M. Rice, W. B. Peeples, Y. Lin, S. Jain, R. Parker and M. K. Rosen, Cell, 2016, 166, 651–663 CrossRef CAS PubMed
- Y. Lin, David S. W. Protter, Michael K. Rosen and R. Parker, Mol. Cell, 2015, 60, 208–219 CrossRef CAS PubMed
- P. Li, S. Banjade, H.-C. Cheng, S. Kim, B. Chen, L. Guo, M. Llaguno, J. V. Hollingsworth, D. S. King, S. F. Banani, P. S. Russo, Q.-X. Jiang, B. T. Nixon and M. K. Rosen, Nature, 2012, 483, 336–340 CrossRef CAS PubMed
- D. L. J. Lafontaine, J. A. Riback, R. Bascetin and C. P. Brangwynne, Nat. Rev. Mol. Cell Biol., 2021, 22, 165–182 CrossRef CAS PubMed
- C. P. Brangwynne, T. J. Mitchison and A. A. Hyman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4334–4339 CrossRef CAS PubMed
- D. S. W. Protter and R. Parker, Trends Cell Biol., 2016, 26, 668–679 CrossRef CAS PubMed
- B. Van Treeck, D. S. W. Protter, T. Matheny, A. Khong, C. D. Link and R. Parker, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2734–2739 CrossRef CAS PubMed
- Q. Wang, I. A. Sawyer, M.-H. Sung, D. Sturgill, S. P. Shevtsov, G. Pegoraro, O. Hakim, S. Baek, G. L. Hager and M. Dundr, Nat. Commun., 2016, 7, 10966 CrossRef CAS PubMed
- J. L. Watson, E. Seinkmane, C. T. Styles, A. Mihut, L. K. Krüger, K. E. McNally, V. J. Planelles-Herrero, M. Dudek, P. M. McCall, S. Barbiero, M. Vanden Oever, S. Y. Peak-Chew, B. T. Porebski, A. Zeng, N. M. Rzechorzek, D. C. S. Wong, A. D. Beale, A. Stangherlin, M. Riggi, J. Iwasa, J. Morf, C. Miliotis, A. Guna, A. J. Inglis, J. Brugués, R. M. Voorhees, J. E. Chambers, Q.-J. Meng, J. S. O’Neill, R. S. Edgar and E. Derivery, Nature, 2023, 623, 842–852 CrossRef CAS PubMed
- Z. Gao, W. Zhang, R. Chang, S. Zhang, G. Yang and G. Zhao, Front. Microbiol., 2021, 12, 751880 CrossRef PubMed
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin, L. Van Den Bosch, P. Tompa and M. Fuxreiter, Trends Cell Biol., 2018, 28, 420–435 CrossRef CAS PubMed
- A. Boija, I. A. Klein, B. R. Sabari, A. Dall’Agnese, E. L. Coffey, A. V. Zamudio, C. H. Li, K. Shrinivas, J. C. Manteiga, N. M. Hannett, B. J. Abraham, L. K. Afeyan, Y. E. Guo, J. K. Rimel, C. B. Fant, J. Schuijers, T. I. Lee, D. J. Taatjes and R. A. Young, Cell, 2018, 175, 1842–1855 CrossRef CAS PubMed
- S. Alberti and A. A. Hyman, Nat. Rev. Mol. Cell Biol., 2021, 22, 196–213 CrossRef CAS PubMed
- K. Jaqaman and J. A. Ditlev, Curr. Opin. Cell Biol., 2021, 69, 48–54 CrossRef CAS PubMed
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Chem, 2023, 9, 2425–2445 CAS
- A. Lampel, Chem, 2020, 6, 1222–1236 CAS
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC
- S. Song, T. Ivanov, D. Yuan, J. Wang, L. C. da Silva, J. Xie and S. Cao, Biomacromolecules, 2024, 25, 5468–5488 CrossRef CAS PubMed
- M. Naz, L. Zhang, C. Chen, S. Yang, H. Dou, S. Mann and J. Li, Commun. Chem., 2024, 7, 79 CrossRef PubMed
- A. Sathyavageeswaran, J. Bonesso Sabadini and S. L. Perry, Acc. Chem. Res., 2024, 57, 386–398 CrossRef CAS PubMed
- A. Baruch Leshem, S. Sloan-Dennison, T. Massarano, S. Ben-David, D. Graham, K. Faulds, H. E. Gottlieb, J. H. Chill and A. Lampel, Nat. Commun., 2023, 14, 421 CrossRef CAS PubMed
- D. Priftis, L. Leon, Z. Song, S. L. Perry, K. O. Margossian, A. Tropnikova, J. Cheng and M. Tirrell, Angew. Chem., Int. Ed., 2015, 54, 11128–11132 CrossRef CAS PubMed
- L. Ma, X. Fang and C. Wang, Front. Bioeng. Biotechnol., 2023, 10, 1100365 CrossRef PubMed
- R. Augustine, N. Kalva, H. A. Kim, Y. Zhang and I. Kim, Molecules, 2019, 24, 2961 CrossRef CAS PubMed
- J. D. Tang, S. R. Caliari and K. J. Lampe, Biomacromolecules, 2018, 19, 3925–3935 CrossRef CAS PubMed
- J. Wang, M. Abbas, Y. Huang, J. Wang and Y. Li, Commun. Chem., 2023, 6, 243 CrossRef CAS PubMed
- W. Yim, Z. Jin, Y.-C. Chang, C. Brambila, M. N. Creyer, C. Ling, T. He, Y. Li, M. Retout, W. F. Penny, J. Zhou and J. V. Jokerst, Nat. Commun., 2024, 15, 7295 CrossRef CAS PubMed
- Z. W. Lim, Y. Ping and A. Miserez, Bioconjugate Chem., 2018, 29, 2176–2180 CrossRef CAS PubMed
- S. Cao, T. Ivanov, J. Heuer, C. T. J. Ferguson, K. Landfester and L. Caire da Silva, Nat. Commun., 2024, 15, 39 CrossRef CAS PubMed
- H. Betre, L. A. Setton, D. E. Meyer and A. Chilkoti, Biomacromolecules, 2002, 3, 910–916 CrossRef CAS PubMed
- J. Sun, L. Xiao, B. Li, K. Zhao, Z. Wang, Y. Zhou, C. Ma, J. Li, H. Zhang, A. Herrmann and K. Liu, Angew. Chem., Int. Ed., 2021, 60, 23687–23694 CrossRef CAS PubMed
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720–724 CrossRef CAS PubMed
- E. W. Martin and T. Mittag, Biochemistry, 2018, 57, 2478–2487 CrossRef CAS PubMed
- S. Mukherjee and L. V. Schäfer, Nat. Commun., 2023, 14, 5892 CrossRef CAS PubMed
- M. L. Huggins, J. Phys. Chem., 1942, 46, 151–158 CrossRef CAS
- P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS
- J. T. G. Overbeek and M. J. Voorn, J. Cell. Comp. Physiol., 1957, 49, 7–26 CrossRef CAS
- X. Wei, J. Zhou, Y. Wang and F. Meng, Phys. Rev. Lett., 2020, 125, 268001 CrossRef CAS PubMed
- A. C. Murthy, G. L. Dignon, Y. Kan, G. H. Zerze, S. H. Parekh, J. Mittal and N. L. Fawzi, Nat. Struct. Mol. Biol., 2019, 26, 637–648 CrossRef CAS PubMed
- M.-T. Wei, S. Elbaum-Garfinkle, A. S. Holehouse, C. C.-H. Chen, M. Feric, C. B. Arnold, R. D. Priestley, R. V. Pappu and C. P. Brangwynne, Nat. Chem., 2017, 9, 1118–1125 CrossRef CAS PubMed
- A. Gobbi and G. Frenking, J. Am. Chem. Soc., 1993, 115, 2362–2372 CrossRef CAS
- B. Gabryelczyk, H. Cai, X. Shi, Y. Sun, P. J. M. Swinkels, S. Salentinig, K. Pervushin and A. Miserez, Nat. Commun., 2019, 10, 5465 CrossRef PubMed
- Q. Guo, G. Zou, X. Qian, S. Chen, H. Gao and J. Yu, Nat. Commun., 2022, 13, 5771 CrossRef CAS PubMed
- W. Wei, Y. Tan, N. R. Martinez Rodriguez, J. Yu, J. N. Israelachvili and J. H. Waite, Acta Biomater., 2014, 10, 1663–1670 CrossRef CAS PubMed
- Y. Hong, S. Najafi, T. Casey, J.-E. Shea, S.-I. Han and D. S. Hwang, Nat. Commun., 2022, 13, 7326 CrossRef PubMed
- X. Wu, Y. Sun, J. Yu and A. Miserez, Commun. Chem., 2024, 7, 5 CrossRef CAS PubMed
- K. Deepankumar, Q. Guo, H. Mohanram, J. Lim, Y. Mu, K. Pervushin, J. Yu and A. Miserez, Adv. Mater., 2022, 34, 2103828 CrossRef CAS PubMed
- V. Castelletto, J. Seitsonen, A. Pollitt and I. W. Hamley, Biomacromolecules, 2024, 25, 5321–5331 CrossRef CAS PubMed
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed
- X. Ren, Q. Zou, C. Yuan, R. Chang, R. Xing and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 5872–5876 CrossRef CAS PubMed
- D. Su, Y. Yu, L. Zhao, T. Wang and X. Yan, Acta Chim. Sin., 2023, 81, 1486–1492 CrossRef CAS
- S. Qamar, G. Wang, S. J. Randle, F. S. Ruggeri, J. A. Varela, J. Q. Lin, E. C. Phillips, A. Miyashita, D. Williams, F. Ströhl, W. Meadows, R. Ferry, V. J. Dardov, G. G. Tartaglia, L. A. Farrer, G. S. Kaminski Schierle, C. F. Kaminski, C. E. Holt, P. E. Fraser, G. Schmitt-Ulms, D. Klenerman, T. Knowles, M. Vendruscolo and P. St George-Hyslop, Cell, 2018, 173, 720–734 CrossRef CAS PubMed
- S. Rekhi, C. G. Garcia, M. Barai, A. Rizuan, B. S. Schuster, K. L. Kiick and J. Mittal, Nat. Chem., 2024, 16, 1113–1124 CrossRef CAS PubMed
- I. Katzir, E. Haimov and A. Lampel, Adv. Mater., 2022, 34, 2206371 CrossRef CAS PubMed
- Chi W. Pak, M. Kosno, Alex S. Holehouse, Shae B. Padrick, A. Mittal, R. Ali, Ali A. Yunus, David R. Liu, Rohit V. Pappu and Michael K. Rosen, Mol. Cell, 2016, 63, 72–85 CrossRef CAS PubMed
- Y. Lin, S. L. Currie and M. K. Rosen, J. Biol. Chem., 2017, 292, 19110–19120 CrossRef CAS PubMed
- E. W. Martin and A. S. Holehouse, Emerging Top. Life Sci., 2020, 4, 307–329 CrossRef CAS PubMed
- F. G. Quiroz and A. Chilkoti, Nat. Mater., 2015, 14, 1164–1171 CrossRef CAS PubMed
- E. W. Martin, A. S. Holehouse, I. Peran, M. Farag, J. J. Incicco, A. Bremer, C. R. Grace, A. Soranno, R. V. Pappu and T. Mittag, Science, 2020, 367, 694–699 CrossRef CAS PubMed
- B. S. Schuster, G. L. Dignon, W. S. Tang, F. M. Kelley, A. K. Ranganath, C. N. Jahnke, A. G. Simpkins, R. M. Regy, D. A. Hammer, M. C. Good and J. Mittal, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11421–11431 CrossRef CAS PubMed
- A. Bremer, M. Farag, W. M. Borcherds, I. Peran, E. W. Martin, R. V. Pappu and T. Mittag, Nat. Chem., 2022, 14, 196–207 CrossRef CAS PubMed
- A. Netzer, A. Baruch Leshem, S. Veretnik, I. Edelstein and A. Lampel, Small, 2024, 20, 2401665 CrossRef CAS PubMed
- Y. Tang, S. Bera, Y. Yao, J. Zeng, Z. Lao, X. Dong, E. Gazit and G. Wei, Cell Rep. Phys. Sci., 2021, 2, 100579 CrossRef CAS
- R. Kubota, S. Torigoe and I. Hamachi, J. Am. Chem. Soc., 2022, 144, 15155–15164 CrossRef CAS PubMed
- D. Sementa, D. Dave, R. S. Fisher, T. Wang, S. Elbaum-Garfinkle and R. V. Ulijn, Angew. Chem., Int. Ed., 2023, 62, e202311479 CrossRef CAS PubMed
- K. W. Y. Chan, M. Navi, J. Kieda, T. Moran, D. Hammers, S. Lee and S. S. H. Tsai, Lab Chip, 2022, 22, 2647–2656 RSC
- N. O. Taylor, M.-T. Wei, H. A. Stone and C. P. Brangwynne, Biophys. J., 2019, 117, 1285–1300 CrossRef CAS PubMed
- A. Molliex, J. Temirov, J. Lee, M. Coughlin, A. P. Kanagaraj, H. J. Kim, T. Mittag and J. P. Taylor, Cell, 2015, 163, 123–133 CrossRef CAS PubMed
- S. Elbaum-Garfinkle, Y. Kim, K. Szczepaniak, C. C.-H. Chen, C. R. Eckmann, S. Myong and C. P. Brangwynne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7189–7194 CrossRef CAS PubMed
- A. Patel, Hyun O. Lee, L. Jawerth, S. Maharana, M. Jahnel, Marco Y. Hein, S. Stoynov, J. Mahamid, S. Saha, Titus M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, Loic A. Royer, M. Weigert, Eugene W. Myers, S. Grill, D. Drechsel, Anthony A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed
- I. Alshareedah, T. Kaur, J. Ngo, H. Seppala, L.-A. D. Kounatse, W. Wang, M. M. Moosa and P. R. Banerjee, J. Am. Chem. Soc., 2019, 141, 14593–14602 CrossRef CAS PubMed
- P. R. Banerjee, A. N. Milin, M. M. Moosa, P. L. Onuchic and A. A. Deniz, Angew. Chem., Int. Ed., 2017, 56, 11354–11359 CrossRef CAS PubMed
- M. Feric, N. Vaidya, T. S. Harmon, D. M. Mitrea, L. Zhu, T. M. Richardson, R. W. Kriwacki, R. V. Pappu and C. P. Brangwynne, Cell, 2016, 165, 1686–1697 CrossRef CAS PubMed
- S. Jain, Joshua R. Wheeler, Robert W. Walters, A. Agrawal, A. Barsic and R. Parker, Cell, 2016, 164, 487–498 CrossRef CAS PubMed
- N. Galvanetto, M. T. Ivanović, A. Chowdhury, A. Sottini, M. F. Nüesch, D. Nettels, R. B. Best and B. Schuler, Nature, 2023, 619, 876–883 CrossRef CAS PubMed
- H. Le Ferrand, M. Duchamp, B. Gabryelczyk, H. Cai and A. Miserez, J. Am. Chem. Soc., 2019, 141, 7202–7210 CrossRef CAS PubMed
- S. Guseva, V. Schnapka, W. Adamski, D. Maurin, R. W. H. Ruigrok, N. Salvi and M. Blackledge, J. Am. Chem. Soc., 2023, 145, 10548–10563 CrossRef CAS PubMed
- W. E. Arter, R. Qi, N. A. Erkamp, G. Krainer, K. Didi, T. J. Welsh, J. Acker, J. Nixon-Abell, S. Qamar, J. Guillén-Boixet, T. M. Franzmann, D. Kuster, A. A. Hyman, A. Borodavka, P. S. George-Hyslop, S. Alberti and T. P. J. Knowles, Nat. Commun., 2022, 13, 7845 CrossRef CAS PubMed
-
I. Alshareedah, T. Kaur and P. R. Banerjee, in Methods in Enzymology, ed. C. D. Keating, Academic Press, 2021, vol. 646, pp. 143–183 Search PubMed
- I. Alshareedah, G. M. Thurston and P. R. Banerjee, Biophys. J., 2021, 120, 1161–1169 CrossRef CAS PubMed
- R. S. Fisher and S. Elbaum-Garfinkle, Nat. Commun., 2020, 11, 4628 CrossRef CAS PubMed
- X. Zhang, H. Li, Y. Ma, D. Zhong and S. Hou, APL Bioeng., 2023, 7, 021502 CrossRef CAS PubMed
- S. Ambadipudi, J. Biernat, D. Riedel, E. Mandelkow and M. Zweckstetter, Nat. Commun., 2017, 8, 275 CrossRef PubMed
- N. M. Kanaan, C. Hamel, T. Grabinski and B. Combs, Nat. Commun., 2020, 11, 2809 CrossRef CAS PubMed
- H. Zhang, S. Shao and Y. Sun, Biophys. Rep., 2022, 8, 2–13 CAS
- S. V. Ulianov, Artem K. Velichko, M. D. Magnitov, Artem V. Luzhin, A. K. Golov, N. Ovsyannikova, I. I. Kireev, Alexey S. Gavrikov, Alexander S. Mishin, A. K. Garaev, A. V. Tyakht, Alexey A. Gavrilov, O. L. Kantidze and S. V. Razin, Nucleic Acids Res., 2021, 49, 10524–10541 CrossRef CAS PubMed
- K. A. Ibrahim, A. S. Naidu, H. Miljkovic, A. Radenovic and W. Yang, ACS Nano, 2024, 18, 10738–10757 CrossRef CAS PubMed
- G. Scarcelli and S. H. Yun, Nat. Photonics, 2008, 2, 39–43 CrossRef CAS PubMed
- R. Prevedel, A. Diz-Muñoz, G. Ruocco and G. Antonacci, Nat. Methods, 2019, 16, 969–977 CrossRef CAS PubMed
- R. Schlüßler, K. Kim, M. Nötzel, A. Taubenberger, S. Abuhattum, T. Beck, P. Müller, S. Maharana, G. Cojoc, S. Girardo, A. Hermann, S. Alberti and J. Guck, eLife, 2022, 11, e68490 CrossRef PubMed
- G. Antonacci, V. de Turris, A. Rosa and G. Ruocco, Commun. Biol., 2018, 1, 139 CrossRef PubMed
- R. De Santis, V. Alfano, V. de Turris, A. Colantoni, L. Santini, M. G. Garone, G. Antonacci, G. Peruzzi, E. Sudria-Lopez, E. Wyler, J. J. Anink, E. Aronica, M. Landthaler, R. J. Pasterkamp, I. Bozzoni and A. Rosa, Cell Rep., 2019, 27, 3818–3831 CrossRef CAS PubMed
- S. Kim, J. Huang, Y. Lee, S. Dutta, H. Y. Yoo, Y. M. Jung, Y. Jho, H. Zeng and D. S. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E847–E853 CrossRef CAS PubMed
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed
- J. E. Bramham and A. P. Golovanov, Nat. Commun., 2022, 13, 1767 CrossRef CAS PubMed
- S. E. Reichheld, L. D. Muiznieks, F. W. Keeley and S. Sharpe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4408–E4415 CrossRef CAS PubMed
- H. Cai, B. Gabryelczyk, M. S. S. Manimekalai, G. Grüber, S. Salentinig and A. Miserez, Soft Matter, 2017, 13, 7740–7752 RSC
- S. Da Vela, M. K. Braun, A. Dörr, A. Greco, J. Möller, Z. Fu, F. Zhang and F. Schreiber, Soft Matter, 2016, 12, 9334–9341 RSC
- N. Salvi, A. Abyzov and M. Blackledge, Angew. Chem., Int. Ed., 2017, 56, 14020–14024 CrossRef CAS PubMed
- C. F. Pantoja, A. Ibáñez de Opakua, M.-S. Cima-Omori and M. Zweckstetter, Angew. Chem., Int. Ed., 2023, 62, e202218078 CrossRef CAS PubMed
- T. H. Kim, B. J. Payliss, M. L. Nosella, I. T. W. Lee, Y. Toyama, J. D. Forman-Kay and L. E. Kay, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104897118 CrossRef CAS PubMed
- E. W. Martin, T. S. Harmon, J. B. Hopkins, S. Chakravarthy, J. J. Incicco, P. Schuck, A. Soranno and T. Mittag, Nat. Commun., 2021, 12, 4513 CrossRef CAS PubMed
- G. Bianchi, S. Longhi, R. Grandori and S. Brocca, Int. J. Mol. Sci., 2020, 21, 6208 CrossRef CAS PubMed
- S. Maiti, A. Singh, T. Maji, N. V. Saibo and S. De, Curr. Res. Struct. Biol., 2024, 7, 100138 CrossRef CAS PubMed
- O. Matsarskaia, S. Da Vela, A. Mariani, Z. Fu, F. Zhang and F. Schreiber, J. Phys. Chem. B, 2019, 123, 1913–1919 CrossRef CAS PubMed
- S. L. Burg, A. Washington, D. M. Coles, A. Bianco, D. McLoughlin, O. O. Mykhaylyk, J. Villanova, A. J. C. Dennison, C. J. Hill, P. Vukusic, S. Doak, S. J. Martin, M. Hutchings, S. R. Parnell, C. Vasilev, N. Clarke, A. J. Ryan, W. Furnass, M. Croucher, R. M. Dalgliesh, S. Prevost, R. Dattani, A. Parker, R. A. L. Jones, J. P. A. Fairclough and A. J. Parnell, Commun. Chem., 2019, 2, 100 CrossRef
- C. A. Brosey and J. A. Tainer, Curr. Opin. Struct. Biol., 2019, 58, 197–213 CrossRef CAS PubMed
- N. A. Erkamp, R. Qi, T. J. Welsh and T. P. J. Knowles, Lab Chip, 2023, 23, 9–24 RSC
- M. Linsenmeier, M. R. G. Kopp, S. Stavrakis, A. de Mello and P. Arosio, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118823 CrossRef CAS PubMed
- T. Beneyton, C. Love, M. Girault, T.-Y. D. Tang and J.-C. Baret, ChemSystemsChem, 2020, 2, e2000022 CrossRef CAS
- M. Linsenmeier, M. R. G. Kopp, F. Grigolato, L. Emmanoulidis, D. Liu, D. Zürcher, M. Hondele, K. Weis, U. Capasso Palmiero and P. Arosio, Angew. Chem., Int. Ed., 2019, 58, 14489–14494 CrossRef CAS PubMed
- M. R. G. Kopp, M. Linsenmeier, B. Hettich, S. Prantl, S. Stavrakis, J.-C. Leroux and P. Arosio, Anal. Chem., 2020, 92, 5803–5812 CrossRef CAS PubMed
- A. Bremer, T. Mittag and M. Heymann, Lab Chip, 2020, 20, 4225–4234 RSC
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19243–19248 CrossRef CAS PubMed
- P. Li, X. Zeng, S. Li, X. Xiang, P. Chen, Y. Li and B.-F. Liu, Anal. Chem., 2022, 94, 687–694 CrossRef CAS PubMed
- Y. Shen, F. S. Ruggeri, D. Vigolo, A. Kamada, S. Qamar, A. Levin, C. Iserman, S. Alberti, P. S. George-Hyslop and T. P. J. Knowles, Nat. Nanotechnol., 2020, 15, 841–847 CrossRef CAS PubMed
- T. J. Welsh, G. Krainer, J. R. Espinosa, J. A. Joseph, A. Sridhar, M. Jahnel, W. E. Arter, K. L. Saar, S. Alberti, R. Collepardo-Guevara and T. P. J. Knowles, Nano Lett., 2022, 22, 612–621 CrossRef CAS PubMed
- N. Taylor, S. Elbaum-Garfinkle, N. Vaidya, H. Zhang, H. A. Stone and C. P. Brangwynne, Soft Matter, 2016, 12, 9142–9150 RSC
- Q. Ma, Y. Song, W. Sun, J. Cao, H. Yuan, X. Wang, Y. Sun and H. C. Shum, Adv. Sci., 2020, 7, 1903359 CrossRef CAS PubMed
- M. Tsanai, Pim W. J. M. Frederix, C. F. E. Schroer, P. C. T. Souza and S. J. Marrink, Chem. Sci., 2021, 12, 8521–8530 RSC
- X. Chu, T. Sun, Q. Li, Y. Xu, Z. Zhang, L. Lai and J. Pei, BMC Bioinf., 2022, 23, 72 CrossRef CAS PubMed
- X. Yang, Y. Wang and G. Yang, Sci. Rep., 2024, 14, 13382 CrossRef CAS PubMed
- H. Cai, R. M. Vernon and J. D. Forman-Kay, Biomolecules, 2022, 12, 1131 CrossRef CAS PubMed
- K. Yu, Z. Liu, H. Cheng, S. Li, Q. Zhang, J. Liu, H.-Q. Ju, Z. Zuo, Q. Zhao, S. Kang and Z.-X. Liu, Briefings Bioinf., 2023, 24, bbac550 CrossRef PubMed
- I. Kaminker, W. Wei, A. M. Schrader, Y. Talmon, M. T. Valentine, J. N. Israelachvili, J. H. Waite and S. Han, Soft Matter, 2017, 13, 9122–9131 RSC
- S. H. Hiew, Y. Lu, H. Han, R. A. Gonçalves, S. R. Alfarano, R. Mezzenga, A. N. Parikh, Y. Mu and A. Miserez, J. Am. Chem. Soc., 2023, 145, 3382–3393 CrossRef CAS PubMed
- H. Wang, F. M. Kelley, D. Milovanovic, B. S. Schuster and Z. Shi, Biophys. Rep., 2021, 1, 100011 CAS
- G. Li, X. Xu and Y. Y. Zuo, J. Colloid Interface Sci., 2023, 630, 21–27 CrossRef CAS PubMed
- G. Li, X. Xu and Y. Y. Zuo, Biophys. J., 2023, 122, 1772–1780 CrossRef CAS PubMed
- G. Li, X. Xu and Y. Y. Zuo, Biophys. J., 2023, 122, 3099–3107 CrossRef CAS PubMed
- B. Gouveia, Y. Kim, J. W. Shaevitz, S. Petry, H. A. Stone and C. P. Brangwynne, Nature, 2022, 609, 255–264 CrossRef CAS PubMed
- Y. Sun, X. Wu, J. Li, M. Radiom, R. Mezzenga, C. S. Verma, J. Yu and A. Miserez, Nat. Commun., 2024, 15, 10094 CrossRef CAS PubMed
- I. Alshareedah, A. Singh, S. Yang, V. Ramachandran, A. Quinn, D. A. Potoyan and P. R. Banerjee, Sci. Adv., 2024, 10, eadi6539 CrossRef CAS PubMed
- R. S. Fisher and A. C. Obermeyer, Chem. Sci., 2024, 15, 19795–19804 RSC
- A. R. Strom, Y. Kim, H. Zhao, Y.-C. Chang, N. D. Orlovsky, A. Košmrlj, C. Storm and C. P. Brangwynne, Cell, 2024, 187, 5282–5297 CrossRef CAS PubMed
- L. Zhu, Y. Pan, Z. Hua, Y. Liu and X. Zhang, J. Am. Chem. Soc., 2024, 146, 14307–14317 CrossRef CAS PubMed
- J. V. Roggeveen, H. Wang, Z. Shi and H. A. Stone, Biophys. J., 2024, 123, 1393–1403 CrossRef CAS PubMed
- D. Sundaravadivelu Devarajan, J. Wang, B. Szała-Mendyk, S. Rekhi, A. Nikoubashman, Y. C. Kim and J. Mittal, Nat. Commun., 2024, 15, 1912 CrossRef CAS PubMed
- Y. Wang, S. Li, M. Mokbel, A. I. May, Z. Liang, Y. Zeng, W. Wang, H. Zhang, F. Yu, K. Sporbeck, L. Jiang, S. Aland, J. Agudo-Canalejo, R. L. Knorr and X. Fang, Nature, 2024, 634, 1204–1210 CrossRef CAS PubMed
- L. D. Zarzar, V. Sresht, E. M. Sletten, J. A. Kalow, D. Blankschtein and T. M. Swager, Nature, 2015, 518, 520–524 CrossRef CAS PubMed
- J. Marthelot, E. F. Strong, P. M. Reis and P. T. Brun, Nat. Commun., 2018, 9, 4477 CrossRef CAS PubMed
- E. Spruijt, J. Sprakel, M. A. Cohen Stuart and J. van der Gucht, Soft Matter, 2010, 6, 172–178 RSC
- A. Mangiarotti, N. Chen, Z. Zhao, R. Lipowsky and R. Dimova, Nat. Commun., 2023, 14, 2809 CrossRef CAS PubMed
- Y. Zhang, R. Prasad, S. Su, D. Lee and H.-X. Zhou, Cell Rep. Phys. Sci., 2024, 5, 102218 CrossRef CAS PubMed
- A. Ghosh, D. Kota and H.-X. Zhou, Nat. Commun., 2021, 12, 5995 CrossRef CAS PubMed
- F. K. Hansen, J. Colloid Interface Sci., 1993, 160, 209–217 CrossRef CAS
- H. Cinar, Z. Fetahaj, S. Cinar, R. M. Vernon, H. S. Chan and R. H. A. Winter, Chem. - Eur. J., 2019, 25, 13049–13069 CrossRef CAS PubMed
- S. Gudlur, F. V. Ferreira, J. S. M. Ting, C. Domene, S. Maricar, A. P. Le Brun, N. Yepuri, M. Moir, R. Russell, T. Darwish, A. Miserez and M. Cárdenas, Front. Soft Matter, 2024, 3 Search PubMed
- A. A. M. André and E. Spruijt, Int. J. Mol. Sci., 2020, 21, 5908 CrossRef PubMed
- S. S. Ribeiro, N. Samanta, S. Ebbinghaus and J. C. Marcos, Nat. Rev. Chem., 2019, 3, 552–561 CrossRef CAS
- J. A. Villegas, M. Heidenreich and E. D. Levy, Nat. Chem. Biol., 2022, 18, 1319–1329 CrossRef CAS PubMed
- G. Krainer, T. J. Welsh, J. A. Joseph, J. R. Espinosa, S. Wittmann, E. de Csilléry, A. Sridhar, Z. Toprakcioglu, G. Gudiškytė, M. A. Czekalska, W. E. Arter, J. Guillén-Boixet, T. M. Franzmann, S. Qamar, P. S. George-Hyslop, A. A. Hyman, R. Collepardo-Guevara, S. Alberti and T. P. J. Knowles, Nat. Commun., 2021, 12, 1085 CrossRef CAS PubMed
- S. Park, R. Barnes, Y. Lin, B.-J. Jeon, S. Najafi, K. T. Delaney, G. H. Fredrickson, J.-E. Shea, D. S. Hwang and S. Han, Commun. Chem., 2020, 3, 83 CrossRef CAS PubMed
- T. Lu, K. K. Nakashima and E. Spruijt, J. Phys. Chem. B, 2021, 125, 3080–3091 CrossRef CAS PubMed
- G. L. Dignon, W. Zheng, Y. C. Kim and J. Mittal, ACS Cent. Sci., 2019, 5, 821–830 CrossRef CAS PubMed
- J. B. Otis and S. Sharpe, Biomacromolecules, 2022, 23, 5225–5238 CrossRef CAS PubMed
- K. M. Ruff, S. Roberts, A. Chilkoti and R. V. Pappu, J. Mol. Biol., 2018, 430, 4619–4635 CrossRef CAS PubMed
- X. Zeng, C. Liu, M. J. Fossat, P. Ren, A. Chilkoti and R. V. Pappu, APL Mater., 2021, 9, 021119 CrossRef CAS PubMed
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, Nat. Chem., 2020, 12, 814–825 CrossRef CAS PubMed
- J. R. Simon, N. J. Carroll, M. Rubinstein, A. Chilkoti and G. P. López, Nat. Chem., 2017, 9, 509–515 CrossRef CAS PubMed
- L. D. Muiznieks, S. Sharpe, R. Pomès and F. W. Keeley, J. Mol. Biol., 2018, 430, 4741–4753 CrossRef CAS PubMed
- S. Roberts, M. Dzuricky and A. Chilkoti, FEBS Lett., 2015, 589, 2477–2486 CrossRef CAS PubMed
- N. K. Dutta, M. Y. Truong, S. Mayavan, N. Roy Choudhury, C. M. Elvin, M. Kim, R. Knott, K. M. Nairn and A. J. Hill, Angew. Chem., Int. Ed., 2011, 50, 4428–4431 CrossRef CAS PubMed
- K. Julius, J. Weine, M. Gao, J. Latarius, M. Elbers, M. Paulus, M. Tolan and R. Winter, Macromolecules, 2019, 52, 1772–1784 CrossRef CAS
- Y. Akahoshi, H. Sugai, M. Mimura, Y. Shinkai, R. Kurita, K. Shiraki and S. Tomita, Biomacromolecules, 2023, 24, 704–713 CrossRef CAS PubMed
- T. Kaur, I. Alshareedah, W. Wang, J. Ngo, M. M. Moosa and P. R. Banerjee, Biomolecules, 2019, 9, 71 CrossRef CAS PubMed
- O. Annunziata, N. Asherie, A. Lomakin, J. Pande, O. Ogun and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14165–14170 CrossRef CAS PubMed
- P. Dogra, A. Joshi, A. Majumdar and S. Mukhopadhyay, J. Am. Chem. Soc., 2019, 141, 20380–20389 CrossRef CAS PubMed
- E. G. P. Stender, S. Ray, R. K. Norrild, J. A. Larsen, D. Petersen, A. Farzadfard, C. Galvagnion, H. Jensen and A. K. Buell, Nat. Commun., 2021, 12, 7289 CrossRef CAS PubMed
- D. Q. P. Reis, S. Pereira, A. P. Ramos, P. M. Pereira, L. Morgado, J. Calvário, A. O. Henriques, M. Serrano and A. S. Pina, Nat. Commun., 2024, 15, 9368 CrossRef CAS PubMed
- M. Heidenreich, J. M. Georgeson, E. Locatelli, L. Rovigatti, S. K. Nandi, A. Steinberg, Y. Nadav, E. Shimoni, S. A. Safran, J. P. K. Doye and E. D. Levy, Nat. Chem. Biol., 2020, 16, 939–945 CrossRef CAS PubMed
- S. C. Weber, Curr. Opin. Cell Biol., 2017, 46, 62–71 CrossRef CAS PubMed
- H. Tao, C. Rigoni, H. Li, A. Koistinen, J. V. I. Timonen, J. Zhou, E. Kontturi, O. J. Rojas and G. Chu, Nat. Commun., 2023, 14, 5277 CrossRef CAS PubMed
- L. S. Taylor and G. G. Z. Zhang, Adv. Drug Delivery Rev., 2016, 101, 122–142 CrossRef CAS PubMed
- J. Chen, A. Tsuchida, A. D. Malay, K. Tsuchiya, H. Masunaga, Y. Tsuji, M. Kuzumoto, K. Urayama, H. Shintaku and K. Numata, Nat. Commun., 2024, 15, 527 CrossRef CAS PubMed
- X. Gui, F. Luo, Y. Li, H. Zhou, Z. Qin, Z. Liu, J. Gu, M. Xie, K. Zhao, B. Dai, W. S. Shin, J. He, L. He, L. Jiang, M. Zhao, B. Sun, X. Li, C. Liu and D. Li, Nat. Commun., 2019, 10, 2006 CrossRef PubMed
- M. Linsenmeier, M. Hondele, F. Grigolato, E. Secchi, K. Weis and P. Arosio, Nat. Commun., 2022, 13, 3030 CrossRef CAS PubMed
- A. Boija, I. A. Klein and R. A. Young, Cancer Cell, 2021, 39, 174–192 CrossRef CAS PubMed
- I. Alshareedah, W. M. Borcherds, S. R. Cohen, A. Singh, A. E. Posey, M. Farag, A. Bremer, G. W. Strout, D. T. Tomares, R. V. Pappu, T. Mittag and P. R. Banerjee, Nat. Phys., 2024, 20, 1482–1491 Search PubMed
- L. Jawerth, E. Fischer-Friedrich, S. Saha, J. Wang, T. Franzmann, X. Zhang, J. Sachweh, M. Ruer, M. Ijavi, S. Saha, J. Mahamid, A. A. Hyman and F. Jülicher, Science, 2020, 370, 1317–1323 CrossRef CAS PubMed
- A. Abyzov, M. Blackledge and M. Zweckstetter, Chem. Rev., 2022, 122, 6719–6748 CrossRef CAS PubMed
- Z. Monahan, V. H. Ryan, A. M. Janke, K. A. Burke, S. N. Rhoads, G. H. Zerze, R. O'Meally, G. L. Dignon, A. E. Conicella, W. Zheng, R. B. Best, R. N. Cole, J. Mittal, F. Shewmaker and N. L. Fawzi, EMBO J., 2017, 36, 2951–2967 CrossRef CAS PubMed
- W. M. Aumiller and C. D. Keating, Nat. Chem., 2016, 8, 129–137 CrossRef CAS PubMed
- F. Luo, X. Gui, H. Zhou, J. Gu, Y. Li, X. Liu, M. Zhao, D. Li, X. Li and C. Liu, Nat. Struct. Mol. Biol., 2018, 25, 341–346 CrossRef CAS PubMed
- A. Garaizar, J. R. Espinosa, J. A. Joseph, G. Krainer, Y. Shen, T. P. J. Knowles and R. Collepardo-Guevara, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119800119 CrossRef CAS PubMed
- R. Chang, C. Yuan, P. Zhou, R. Xing and X. Yan, Acc. Chem. Res., 2024, 57, 289–301 CrossRef CAS PubMed
- C. Yuan, R. Xing, J. Cui, W. Fan, J. Li and X. Yan, CCS Chem., 2024, 6, 255–265 CrossRef CAS
- P. Zhou, R. Xing, Q. Li, J. Li, C. Yuan and X. Yan, Matter, 2023, 6, 1945–1963 CrossRef CAS
- R. Dec, W. Dzwolak and R. Winter, J. Am. Chem. Soc., 2024, 146, 6045–6052 CrossRef CAS PubMed
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS PubMed
- C. Donau, F. Späth, M. Sosson, B. A. K. Kriebisch, F. Schnitter, M. Tena-Solsona, H.-S. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS PubMed
- J. Ji, W. Wang and C. Chen, Acta Biochim. Biophys. Sin., 2023, 55, 1023–1033 CrossRef CAS PubMed
- Y. Bao, H. Chen, Z. Xu, J. Gao, L. Jiang and J. Xia, Angew. Chem., Int. Ed., 2023, 62, e202307045 CrossRef CAS PubMed
- E. M. Zhao, N. Suek, M. Z. Wilson, E. Dine, N. L. Pannucci, Z. Gitai, J. L. Avalos and J. E. Toettcher, Nat. Chem. Biol., 2019, 15, 589–597 CrossRef CAS PubMed
- M. Mondal, P. E. Jankoski, L. D. Lee, D. M. Dinakarapandian, T.-Y. Chiu, W. S. Swetman, H. Wu, A. K. Paravastu, T. D. Clemons and V. Rangachari, J. Am. Chem. Soc., 2024, 146, 25299–25311 CrossRef CAS PubMed
- L. Jia, S. Gao and Y. Qiao, Small Methods, 2024, 8, 2301724 CrossRef CAS PubMed
- N. Schneider, F.-G. Wieland, D. Kong, A. A. M. Fischer, M. Hörner, J. Timmer, H. Ye and W. Weber, Sci. Adv., 2021, 7, eabd3568 CrossRef CAS PubMed
- Y. Shin, J. Berry, N. Pannucci, M. P. Haataja, J. E. Toettcher and C. P. Brangwynne, Cell, 2017, 168, 159–171 CrossRef CAS PubMed
- E. H. Brumbaugh-Reed, Y. Gao, K. Aoki and J. E. Toettcher, Nat. Commun., 2024, 15, 6717 CrossRef CAS PubMed
- N. Martin, L. Tian, D. Spencer, A. Coutable-Pennarun, J. L. R. Anderson and S. Mann, Angew. Chem., Int. Ed., 2019, 58, 14594–14598 CrossRef CAS PubMed
- N. Ikeuchi, T. Komachi, K. Murayama, H. Asanuma, A. Maruyama and N. Shimada, ACS Appl. Mater. Interfaces, 2021, 13, 5652–5659 CrossRef CAS PubMed
- M. Abbas, W. P. Lipiński, K. K. Nakashima, W. T. S. Huck and E. Spruijt, Nat. Chem., 2021, 13, 1046–1054 CrossRef CAS PubMed
- Y. Sun, X. Xu, L. Chen, W. L. Chew, Y. Ping and A. Miserez, ACS Nano, 2023, 17, 16597–16606 CrossRef CAS PubMed
- Y. Dai, C. F. Chamberlayne, M. S. Messina, C. J. Chang, R. N. Zare, L. You and A. Chilkoti, Chem, 2023, 9, 1594–1609 CAS
- X. Wang, J. Liu, C. Mao and Y. Mao, Cell Commun. Signaling, 2024, 22, 143 CrossRef PubMed
- K. Guo, J. Zhang, P. Huang, Y. Xu, W. Pan, K. Li, L. Chen, L. Luo, W. Yu, S. Chen, S. He, Z. Wei and C. Yu, Cell Rep., 2023, 42, 113321 CrossRef CAS PubMed
- S. Yang, C. Liu, Y. Guo, G. Li, D. Li, X. Yan and X. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122420119 CrossRef CAS PubMed
- S. Yang, Y. Yu, S. Jo, Y. Lee, S. Son and K. H. Lee, Nat. Commun., 2024, 15, 10394 CrossRef CAS PubMed
- R. Niwayama, H. Nagao, T. S. Kitajima, L. Hufnagel, K. Shinohara, T. Higuchi, T. Ishikawa and A. Kimura, PLoS One, 2016, 11, e0159917 CrossRef PubMed
- A. Brown, Nat. Rev. Mol. Cell Biol., 2000, 1, 153–156 CrossRef CAS PubMed
- M. J. Harrington, R. Mezzenga and A. Miserez, Nat. Rev. Bioeng., 2024, 2, 260–278 CrossRef CAS
- Y. Song, Biomater. Sci., 2024, 12, 1943–1949 RSC
- J. Liu, F. Zhorabek and Y. Chau, Matter, 2022, 5, 2787–2812 CrossRef CAS
- G. C. Yeo, B. Aghaei-Ghareh-Bolagh, E. P. Brackenreg, M. A. Hiob, P. Lee and A. S. Weiss, Adv. Healthcare Mater., 2015, 4, 2530–2556 CrossRef CAS PubMed
- A. K. Varanko, J. C. Su and A. Chilkoti, Annu. Rev. Biomed. Eng., 2020, 22, 343–369 CrossRef CAS PubMed
- A. Huang and L. Su, Acc. Mater. Res., 2023, 4, 729–732 CrossRef CAS PubMed
- H. J. Kim, B. H. Hwang, S. Lim, B.-H. Choi, S. H. Kang and H. J. Cha, Biomaterials, 2015, 72, 104–111 CrossRef CAS PubMed
- E. Y. Jeon, S.-H. Um, J. Park, Y. Jung, C.-H. Cheon, H. Jeon and J. J. Chung, Small, 2022, 18, 2200416 CrossRef CAS PubMed
- M. Cui, X. Wang, B. An, C. Zhang, X. Gui, K. Li, Y. Li, P. Ge, J. Zhang, C. Liu and C. Zhong, Sci. Adv., 2019, 5, eaax3155 CrossRef CAS PubMed
- W. H. Park, J. Lee, H. J. Kim, K. I. Joo and H. J. Cha, Chem. Eng. J., 2022, 446, 137272 CrossRef CAS
- H. J. Kim, B.-H. Choi, S. H. Jun and H. J. Cha, Adv. Healthcare Mater., 2016, 5, 3191–3202 CrossRef CAS PubMed
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed
- L. Jiang, Y. Zeng, H. Li, Z. Lin, H. Liu, J. J. Richardson, Z. Gao, D. Wu, L. Liu, F. Caruso and J. Zhou, J. Am. Chem. Soc., 2023, 145, 24108–24115 CrossRef CAS PubMed
- C. Dong and Y. Lv, Polymers, 2016, 8, 42 CrossRef PubMed
- Y. Sun, S. H. Hiew and A. Miserez, Acc. Chem. Res., 2024, 57, 164–174 CrossRef CAS PubMed
- S. Acosta, L. Quintanilla-Sierra, L. Mbundi, V. Reboto and J. C. Rodríguez-Cabello, Adv. Funct. Mater., 2020, 30, 1909050 CrossRef CAS
- S. Chen, Q. Guo and J. Yu, Aggregate, 2022, 3, e293 CrossRef CAS
- F. Jehle, E. Macías-Sánchez, S. Sviben, P. Fratzl, L. Bertinetti and M. J. Harrington, Nat. Commun., 2020, 11, 862 CrossRef CAS PubMed
- B. Wu, P. Ding, M. Wang, M. A. Cohen Stuart and J. Wang, Matter, 2023, 6, 2517–2519 CrossRef CAS
- J. Liu, E. Spruijt, A. Miserez and R. Langer, Nat. Rev. Mater., 2023, 8, 139–141 CrossRef CAS
- D. M. Mitrea, M. Mittasch, B. F. Gomes, I. A. Klein and M. A. Murcko, Nat. Rev. Drug Discovery, 2022, 21, 841–862 CrossRef CAS PubMed
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed
- X. Zhu, J. Yuan, R. Chang, W. Fan, Y. Wang, H. Li, Y. Zhang, P. Zhou and X. Yan, Colloids Surf., A, 2024, 697, 134331 CrossRef CAS
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed
- N. R. Johnson and Y. Wang, Expert Opin. Drug Delivery, 2014, 11, 1829–1832 CrossRef CAS PubMed
- C. Forenzo and J. Larsen, Mol. Pharmaceutics, 2023, 20, 4387–4403 CrossRef CAS PubMed
- Y. Tian, Q. Hu, Z. Sun, Y. Yu, X. Li, T. Tian, X. Bi, Y. Li, B. Niu and Z. Zhang, Small, 2024, 20, 2311890 CrossRef CAS PubMed
- Z. W. Lim, V. B. Varma, R. V. Ramanujan and A. Miserez, Acta Biomater., 2020, 110, 221–230 CrossRef CAS PubMed
- S. Yu, W. Chen, G. Liu, B. Flores, E. L. DeWolf, B. Fan, Y. Xiang and M. J. Webber, J. Am. Chem. Soc., 2024, 146, 7498–7505 CrossRef CAS PubMed
- J. Liu, R. Feng and Y. Chau, Matter, 2022, 5, 1637–1639 CrossRef CAS
- A. Shebanova, Q. M. Perrin, K. Zhu, S. Gudlur, Z. Chen, Y. Sun, C. Huang, Z. W. Lim, E. A. Mondarte, R. Sun, S. Lim, J. Yu, Y. Miao, A. N. Parikh, A. Ludwig and A. Miserez, Adv. Sci., 2024, 11, 2402652 CrossRef CAS PubMed
- H.-X. Zhou, D. Kota, S. Qin and R. Prasad, Chem. Rev., 2024, 124, 8550–8595 CrossRef CAS PubMed
- K. K. Nakashima, M. H. I. van Haren, A. A. M. André, I. Robu and E. Spruijt, Nat. Commun., 2021, 12, 3819 CrossRef CAS PubMed
- A. Ianeselli, D. Tetiker, J. Stein, A. Kühnlein, C. B. Mast, D. Braun and T. Y. Dora Tang, Nat. Chem., 2022, 14, 32–39 CrossRef CAS PubMed
- A. F. Mason, B. C. Buddingh, D. S. Williams and J. C. M. van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef CAS PubMed
- F. Pir Cakmak, A. M. Marianelli and C. D. Keating, Langmuir, 2021, 37, 10366–10375 CrossRef CAS PubMed
- Y. Ji, Y. Lin and Y. Qiao, J. Am. Chem. Soc., 2023, 145, 12576–12585 CrossRef CAS PubMed
- M. Abbas, J. O. Law, S. N. Grellscheid, W. T. S. Huck and E. Spruijt, Adv. Mater., 2022, 34, 2202913 CrossRef CAS PubMed
- A. Agrawal, J. F. Douglas, M. Tirrell and A. Karim, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203483119 CrossRef CAS PubMed
- A. Agrawal, A. Radakovic, A. Vonteddu, S. Rizvi, V. N. Huynh, J. F. Douglas, M. V. Tirrell, A. Karim and J. W. Szostak, Sci. Adv., 2024, 10, eadn9657 CrossRef CAS PubMed
- U. Pramanik, A. Das, E. M. Brown, H. L. Struckman, H. Wang, S. Stealey, M. L. Sprunger, A. Wasim, J. Fascetti, J. Mondal, J. R. Silva, S. P. Zustiak, M. E. Jackrel and J. S. Rudra, bioRxiv, 2024 DOI:10.1101/2024.09.10.612317
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef CAS PubMed
- A. J. Dzieciol and S. Mann, Chem. Soc. Rev., 2012, 41, 79–85 RSC
- Z. Lin, T. Beneyton, J.-C. Baret and N. Martin, Small Methods, 2023, 7, 2300496 CrossRef CAS PubMed
- H. Wu and Y. Qiao, Supramol. Mater., 2022, 1, 100019 Search PubMed
- W. Mu, L. Jia, M. Zhou, J. Wu, Y. Lin, S. Mann and Y. Qiao, Nat. Chem., 2024, 16, 158–167 CrossRef CAS PubMed
- W. Mu, Z. Ji, M. Zhou, J. Wu, Y. Lin and Y. Qiao, Sci. Adv., 2021, 7, eabf9000 CrossRef CAS PubMed
- N. A. Yewdall, A. A. M. André, T. Lu and E. Spruijt, Curr. Opin. Colloid Interface Sci., 2021, 52, 101416 CrossRef CAS
- N. Martin, ChemBioChem, 2019, 20, 2553–2568 CrossRef CAS PubMed
- C. D. Crowe and C. D. Keating, Interface Focus, 2018, 8, 20180032 CrossRef PubMed
- E. Sokolova, E. Spruijt, M. M. K. Hansen, E. Dubuc, J. Groen, V. Chokkalingam, A. Piruska, H. A. Heus and W. T. S. Huck, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11692–11697 CrossRef CAS PubMed
- J. Liu, F. Zhorabek, X. Dai, J. Huang and Y. Chau, ACS Cent. Sci., 2022, 8, 493–500 CrossRef CAS PubMed
- M. Seal, O. Weil-Ktorza, D. Despotović, D. S. Tawfik, Y. Levy, N. Metanis, L. M. Longo and D. Goldfarb, J. Am. Chem. Soc., 2022, 144, 14150–14160 CrossRef CAS PubMed
- T. Y. Dora Tang, C. Rohaida Che Hak, A. J. Thompson, M. K. Kuimova, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2014, 6, 527–533 CrossRef CAS PubMed
- N. Gao, C. Xu, Z. Yin, M. Li and S. Mann, J. Am. Chem. Soc., 2022, 144, 3855–3862 CrossRef CAS PubMed
- T. Lu, S. Javed, C. Bonfio and E. Spruijt, Small Methods, 2023, 7, 2300294 CrossRef CAS PubMed
- C. D. Reinkemeier and E. A. Lemke, Curr. Opin. Chem. Biol., 2021, 64, 174–181 CrossRef CAS PubMed
- S. Lim and D. S. Clark, Trends Biotechnol., 2024, 42, 496–509 CrossRef CAS PubMed
- I. B. A. Smokers, B. S. Visser, A. D. Slootbeek, W. T. S. Huck and E. Spruijt, Acc. Chem. Res., 2024, 57, 1885–1895 CrossRef CAS PubMed
- T. J. Nott, T. D. Craggs and A. J. Baldwin, Nat. Chem., 2016, 8, 569–575 CrossRef CAS PubMed
- A. B. Cook, S. Novosedlik and J. C. M. van Hest, Acc. Mater. Res., 2023, 4, 287–298 CrossRef CAS PubMed
- L. L. J. Schoenmakers, N. A. Yewdall, T. Lu, A. A. M. André, F. H. T. Nelissen, E. Spruijt and W. T. S. Huck, ACS Synth. Biol., 2023, 12, 2004–2014 CrossRef CAS PubMed
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC
- E. Dine, A. A. Gil, G. Uribe, C. P. Brangwynne and J. E. Toettcher, Cell Systems, 2018, 6, 655–663 CrossRef CAS PubMed
- M. Matsuo and K. Kurihara, Nat. Commun., 2021, 12, 5487 CrossRef CAS PubMed
- A. O. Robinson, J. Lee, A. Cameron, C. D. Keating and K. P. Adamala, ACS Biomater. Sci. Eng., 2024, 10, 773–781 CrossRef CAS PubMed
- C. M. Green, D. Sementa, D. Mathur, J. S. Melinger, P. Deshpande, S. Elbaum-Garfinkle, I. L. Medintz, R. V. Ulijn and S. A. Díaz, Commun. Chem., 2024, 7, 49 CrossRef CAS PubMed
- T. Y. Dora Tang, D. van Swaay, A. deMello, J. L. Ross Anderson and S. Mann, Chem. Commun., 2015, 51, 11429–11432 RSC
- R. Xing, C. Yuan, W. Fan, X. Ren and X. Yan, Sci. Adv., 2023, 9, eadd8105 CrossRef CAS PubMed
- S. Cao, W. Fan, R. Chang, C. Yuan and X. Yan, CCS Chem., 2024, 6, 2814–2824 CrossRef CAS
- C. Yuan, W. Fan, P. Zhou, R. Xing, S. Cao and X. Yan, Nat. Nanotechnol., 2024, 19, 1840–1848 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01477d |
| This journal is © The Royal Society of Chemistry 2025 |