
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spaced functionalization of poly(ionic liquid)s for boosting the catalytic conversion of CO2 into cyclic carbonates†
Qianmeng
Zhao
ab,
Shaifei
Liu
d,
Wen
Liu
bc,
Mengqian
Fu
 bc,
Zhenyang
Xu
b,
Qian
Su
*b and
Weiguo
Cheng
*b
bc,
Zhenyang
Xu
b,
Qian
Su
*b and
Weiguo
Cheng
*b
aSinopec Research Institute of Petroleum Processing Co., Ltd, Beijing 100083, China
bBeijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wgcheng@ipe.ac.cn; qsu@ipe.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dAdvanced Energy Science and Technology Guangdong Laboratory, Huizhou, Guangdong 516003, China
First published on 12th February 2025
Abstract
Preparation of cyclic carbonates from CO2 and epoxides via cycloaddition is a well-established synthetic route, which is not only economical but also in line with the atomic economy. To overcome the problem of the cluster formation of hydrogen-bond donors in functionalized poly(ionic liquid)s, which reduces their catalytic activity, a series of spacer-functionalized poly(ionic liquid) catalysts were developed. In poly(ionic) liquids, the hydrogen-donating effect enhances the intrinsic catalytic performance of the active sites and the long-chain alkyl groups prevent interactions between hydrogen-bond-donor groups, thus increasing the utilization of the active sites. Among the developed poly(ionic liquid) catalysts, the poly(ionic liquid) P[AC12VIM][Br] containing amino groups demonstrated the highest catalytic activity (propylene oxide conversion up to 99%), which was comparable with that of bulky ILs. The best catalytic performance of P[AC12VIM][Br] was attributed owing to its multiple functions in not only activating CO2 and epoxides but also stabilizing Br−. Furthermore, the amphiphilicity of amino-functionalized poly(ionic liquid)s boosted their catalytic suitability for epoxide substrates with lipophilic edge groups, which was better than that of conventional poly(ionic liquid)s. Through XPS and NMR analyses, a mechanism of operation is proposed in which imidazole and hydrogen donor groups act co-operatively in epoxy during the reaction to assist in ring-opening. Thus, this study provides a new approach for improving the catalytic performance of poly(ionic liquid) catalysts from the viewpoint of an intrinsic reaction and utilization of the active sites.
Sustainability spotlightThe conversion and utilization of carbon dioxide are very important. The preparation of cyclic carbonates from carbon dioxide is a very effective route for the utilization of carbon dioxide. However, poly(ionic liquid)s, which are the current environmentally friendly catalysts used to catalyze this reaction, need to improve their catalytic performance and reduce the energy consumption. In this regard, we improved the catalytic activity by introducing long alkyl chains in functionalized poly(ionic liquid) catalysts. Furthermore, this work adheres to UN sustainable development goals SDG 9 (industry, innovation, and infrastructure) and SDG 13 (climate action). |
1 Introduction
The increasing CO2 emissions is seriously affecting our planet's ecosystem.1–4 Therefore, the capture and utilization of CO2 have become a worldwide research topic, with the preparation of cyclic carbonates through the cycloaddition reaction of CO2 and epoxides considered a feasible and mature carbon utilization method.5,6 Cyclic carbonates are used in battery electrolytes, polar solvents, chemical intermediates, etc.7 Many types of catalysts, including metal complexes,8 metal salts,9 and ionic liquids (ILs), have been developed for cycloaddition reactions.10 Among these, ILs are considered promising catalysts owing to not only their high catalytic performance but also their advantages of eco-friendliness, high solubility, and customizable structure, which provide a broad space for the application of ILs.11Previous studies have shown that ILs could achieve higher catalytic activity by introducing functionalized hydrogen-donating groups, enabling a high conversion of epoxides under mild reaction conditions.12 However, homogeneous ILs are difficult and costly to separate, which limits their application in industrial production.13 Thus, the dehomogenization of ILs is essential for further promoting their industrial application.14 ILs immobilized on a support can effectively simplify their separation, but there is a possibility in the support causing mass transfer hindrance, leading to a lowered activity compared with that of bulky ILs.15
Poly(ionic liquid)s (PILs) offer the advantages of ILs in terms of eco-friendliness and customizable structure and have higher structural flexibility than immobilized ILs and higher stability than IL monomers.16 Therefore, they could serve as a good alternative to heterogeneous catalysts for cycloaddition reactions. The early application of PILs as catalysts for cycloaddition reactions was reported by Han et al.,17 who developed PILs via free radical polymerization, achieving a 97.4% yield of cyclic carbonate at 110 °C, 6 MPa CO2, 0.6 mol% and 7 h. Thereafter, many researchers have focused on PIL catalytic cycloaddition reactions. However, conventional PILs typically face issues with a low catalytic efficiency due to their simple structure and functions.1 To further improve the catalytic performance of PILs, some researchers have tried to introduce functional groups into the polymeric skeleton, obtaining functionalized PIL catalysts.18 For instance, Yang et al.19 prepared PILs with carboxyl functional groups for cycloaddition reactions, obtaining increased yields from 76% to 97%. Shi et al.20 synthesized a series of hydroxyl- and carboxyl-functionalized PILs for cycloaddition reactions, achieving a high yield of nearly 99%. Studies have shown that functionalized groups can form hydrogen bonds with epoxides, which then induces ring-opening of the epoxide. In addition, functionalized groups can form synergistic effects with Br− in ILs, increasing the catalytic activity of Br−. Therefore, functionalized PILs generally have better catalytic properties. However, functionalized groups tend to form clusters among themselves, leading to the aggregation of active sites. This aggregation of active sites is not conducive to the full utilization of the active sites, so incorporating spacer groups was proposed as a means to improve the utilization of the active sites.21 Thus, to improve the intrinsic activity and promote the utilization of the active sites, an important strategy is to introduce both functional groups and spacer groups into polymeric skeleton.
In the present study, new kinds of PIL catalysts were constructed with long-chain alkyl groups introduced to the separation site. To explore the roles of the functional groups in PILs for catalytic processes, three kinds of hydrophilic hydrogen-donating groups (carboxyl, hydroxyl, and amino) were introduced into the PIL skeleton. Consequently, a series of spaced functionalized PIL catalysts, P[XC12VIM][Br] (C,H,A), were developed for catalyzing cycloaddition reactions. Analytical characterization and activity evaluation of the catalysts were performed, allowing comparison of the effects of different functional groups on the overall catalytic performance of the PILs, and the best catalyst was thus obtained and optimized. The preferred catalysts were then evaluated for assessing their recyclability and substrate suitability. Finally, the roles of different functional groups in the reaction process and their relationship with the catalytic activity were revealed. This study provides new ideas for the design and development of heterogeneous PIL catalysts from the viewpoint of their intrinsic activity and utilization of the active sites.
2 Materials and methods
2.1 Materials
1-Vinylimidazole, 99%, Shanghai Aladdin Biochemical Technology Co.; divinylbenzene (DVB), 99%, Shanghai Aladdin Biochemical Technology Co.; azobisisobutyronitrile (AIBN), 98%, Sinopharm Chemical Reagent Co.; 1-bromododecane, 99%, Shanghai Aladdin Biochemical Technology Co.; anhydrous ethanol, analytically pure, Beijing Chemical Factory; propylene oxide (PO), 99%, Shanghai Aladdin Biochemical Technology Co.; propylene carbonate (PC), 99%, Shanghai Aladdin Biochemical Technology Co.; carbon dioxide, 99%, Beijing Huatong Jingke Gas Chemical Co.; epichlorohydrin, AR, Sinopharm Chemical Reagent Co.; chloropropenyl carbonate, 99%, Sinopharm Group Chemical Reagent Co.; styrene oxide, 98%, Shanghai McLean Biochemical Technology Co.; styrene carbonate, 98%, Shanghai Macklin Biochemical Technology Co.; cyclohexyl epoxide, 99%, Shanghai Macklin Biochemical Technology Co.; cyclohexene carbonate, 98%, Shanghai Macklin Biochemical Technology Co.; sodium hydroxide, AR, Shanghai Aladdin Biochemical Technology Co.; cyclooctane carbonate 99%, Shanghai Aladdin Biochemical Technology Co.; octenyl carbonate, 99%, Sinopharm Chemical Reagent Co.; 2-bromoethanol, 95%, Shanghai McLean Biochemical Technology Co.; 2-bromoacetic acid, 98%, Shanghai McLean Biochemical Technology Co.; 2-bromoacetic acid, 98%, Shanghai McLean Biochemical Technology Co.2.2 Catalyst synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[C12VIM][Br]:DVB = 1:1:3), and 50 mL acetonitrile were added to a 100 mL three-neck flask with condensation reflux, and stirred until completely dissolved. Then, azobisisobutyronitrile (AIBN) (0.98 g, 0.3 mmol, 0.6%) was added and stirred until it was completely dissolved. The air was displaced by the Schlenk technique to obtain an N2 environment. The reaction solution was heated up to 75 °C to initiate the reaction. After 24 h, the crude product was obtained and was washed with water and ethanol repeatedly three times using a mixture of ethanol and water in a 4:1 ratio. Afterwards, the product was placed in a 50 °C vacuum drying oven for 48 h to obtain the white-powdered P[HC12VIM][Br]. Carboxy-functionalized P[CC12VIM][Br] and amino-functionalized P[AC12VIM][Br] were also synthesized by the same method.
:[C12VIM][Br]:DVB = 1:1:3), and 50 mL acetonitrile were added to a 100 mL three-neck flask with condensation reflux, and stirred until completely dissolved. Then, azobisisobutyronitrile (AIBN) (0.98 g, 0.3 mmol, 0.6%) was added and stirred until it was completely dissolved. The air was displaced by the Schlenk technique to obtain an N2 environment. The reaction solution was heated up to 75 °C to initiate the reaction. After 24 h, the crude product was obtained and was washed with water and ethanol repeatedly three times using a mixture of ethanol and water in a 4:1 ratio. Afterwards, the product was placed in a 50 °C vacuum drying oven for 48 h to obtain the white-powdered P[HC12VIM][Br]. Carboxy-functionalized P[CC12VIM][Br] and amino-functionalized P[AC12VIM][Br] were also synthesized by the same method.
2.3 Catalytic activity test
The synthesis of propylene carbonate was carried out in a 25 mL microreactor equipped with pressure and temperature sensors. First, 1.72 g propylene oxide (PO) and a certain amount of PIL catalyst were added. After quickly sealing the reactor, the air inside the reactor was replaced with CO2 and then the reactor was heated up. When the temperature of the reactor reached the target temperature, the pressure was increased to the target value and maintained until the end of the reaction. After the reaction was completed, the reactor was cooled to room temperature. The pressure valve was carefully screwed open to bleed off excess gas from the reactor, and then the reactor was opened and the product collected with a pipette and analyzed by gas chromatography.2.4 Catalyst characterization
The thermal stability of the samples was analyzed using a DTG-60H thermal analyzer (Shimadzu, Japan) in an N2 atmosphere with a temperature programming rate of 10 °C min−1. The surface morphology of the samples was analyzed by scanning electron microscopy (SEM, JSM6700 F, JEOL). The Fourier transform infrared (FT-IR) spectra of the PILs were recorded using a Thermo Nicolet 380 instrument and KBr as a reference for characterizing the specific groups in the samples. The elemental composition (i.e., C/H/N) was analyzed on a Vario EL Cube elemental analyzer to estimate the content of ILs in the PILs. An Agilent 7890 gas chromatography (GS) system with an FID detector and HP-5 capillary column (30 m × 0.25 mm × 0.25 µm) was used to determine the conversion and selectivity. The chemical environment of hydrogen on the samples was analyzed using a Bruker AVANCE III 600 MHz spectrometer with DMSO as the deuterium reagent and tetramethylsilane (TMS) as the internal standard. The ion binding energy of the sample surface was determined by X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi). A physical adsorption meter (ASAP 2020 HD88) was used to test the CO2 adsorption properties of the samples. The swelling properties were measured by weighing the weight of the sample before and after swelling, with propylene carbonate used as the solvent.3 Results and discussion
3.1 Structural characterization of P[XC12VIM][Br]
In order to determine the monomer structure, the synthesized ILs were characterized by electrospray ionization (ESI) and nuclear magnetic resonance (NMR). The ESI characterization results are shown in Fig. 1, where three peaks could be observed corresponding to the molecular weights of the three monomers: [VHEIM][Br]-139; [VCEIM][Br]-153; [VAEIM][Br]-138. The 1H NMR results of the monomer [VXEIM][Br] are shown in Fig. 2, and the peak position and integration area in the spectrum are consistent with the results in the literature. The above findings confirmed that the synthesized monomer [VXEIM][Br] was consistent with the desired target structure. | ||
| Fig. 1 ESI spectra of [VXEIM][Br]. | ||
 | ||
| Fig. 2 1H NMR spectra of [VXEIM][Br]. | ||
To further determine the structural information of the synthesized monomers and polymers, the materials were analyzed by FT-IR, as shown in Fig. 3. The characteristic peak at 1550 cm−1 presented in all three spectra represented the vibrational peak of the imidazole ring, while the peak at 1170 cm−1 was related to the stretching vibrations of the C–N bond on the imidazole attached to the alkyl chain, proving that the imidazole group in the monomer remained after polymerization.22 The characteristic peaks near 3000 cm−1 were the C–H and stretching vibration peaks of C–H and H–C–H on the main chain, which became larger in the FT-IR spectra of the PILs obtained after polymerization, verifying the successful introduction of long alkyl chains in the PILs. Another peak at 1650 cm−1 was attributed to the characteristic peak of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– while the peak at 970 cm−1 was the vibrational peak of unsaturated C–H. These two peaks were significantly reduced after polymerization with respect to the vibrational peak of the imidazole ring (1550 cm−1), indicating that the successful polymerization of the monomer. For the amino-functionalized P[AC12VIM][Br] and the corresponding monomer [VAEIM][Br], the FT-IR characterization results are shown in Fig. 3(a). In the spectrum of [VAEIM][Br], the peaks at 3140 and 835 cm−1 belonged to the stretching vibration peak and twisting vibration peak of amino groups, respectively, which proved that the amino functional group was successfully introduced, and the above two amino characteristic peaks were retained in the FT-IR spectrum of P[AC12VIM][Br] after the polymerization.23 For the hydroxyl-functionalized P[HC12VIM][Br] and the corresponding monomer [VHEIM][Br], the FT-IR characterization results are shown in Fig. 3(b). It could be seen that new characteristic peaks at 1080 and 881 cm−1 appeared in the spectra, which were attributed to the stretching and bending vibrational peaks of C–O, respectively, which proved the successful incorporation of hydroxyl functional groups.24,25 The FT-IR spectra of the carboxyl-functionalized P[CC12VIM][Br] and the corresponding monomer [VCEIM][Br] are shown in Fig. 3(c). The characteristic peak at 1760 cm−1 was due to the stretching vibration of CO, which proved the successful introduction of the carboxyl functional group.26 The above results indicate that all the functionalized ILs monomers were successfully synthesized and polymerized with long alkyl chains to obtain the targeted PILs. In addition, solid-state nuclear magnetic and elemental analyses were also conducted on P[XC12VIM][Br], with the results shown in Fig. S1 and Table S1,† proving that the synthesized structure was consistent with the desired target structure.
C– while the peak at 970 cm−1 was the vibrational peak of unsaturated C–H. These two peaks were significantly reduced after polymerization with respect to the vibrational peak of the imidazole ring (1550 cm−1), indicating that the successful polymerization of the monomer. For the amino-functionalized P[AC12VIM][Br] and the corresponding monomer [VAEIM][Br], the FT-IR characterization results are shown in Fig. 3(a). In the spectrum of [VAEIM][Br], the peaks at 3140 and 835 cm−1 belonged to the stretching vibration peak and twisting vibration peak of amino groups, respectively, which proved that the amino functional group was successfully introduced, and the above two amino characteristic peaks were retained in the FT-IR spectrum of P[AC12VIM][Br] after the polymerization.23 For the hydroxyl-functionalized P[HC12VIM][Br] and the corresponding monomer [VHEIM][Br], the FT-IR characterization results are shown in Fig. 3(b). It could be seen that new characteristic peaks at 1080 and 881 cm−1 appeared in the spectra, which were attributed to the stretching and bending vibrational peaks of C–O, respectively, which proved the successful incorporation of hydroxyl functional groups.24,25 The FT-IR spectra of the carboxyl-functionalized P[CC12VIM][Br] and the corresponding monomer [VCEIM][Br] are shown in Fig. 3(c). The characteristic peak at 1760 cm−1 was due to the stretching vibration of CO, which proved the successful introduction of the carboxyl functional group.26 The above results indicate that all the functionalized ILs monomers were successfully synthesized and polymerized with long alkyl chains to obtain the targeted PILs. In addition, solid-state nuclear magnetic and elemental analyses were also conducted on P[XC12VIM][Br], with the results shown in Fig. S1 and Table S1,† proving that the synthesized structure was consistent with the desired target structure.
 | ||
| Fig. 3 FT-IR spectra of (a) [VAEIM][Br] and P[AC12VIM][Br], (b) [VHEIM][Br] and P[HC12VIM][Br], and (c) [VCEIM][Br] and P[CC12VIM][Br]. | ||
In order to investigate the thermal stability of the synthesized PILs, thermogravimetric analysis (TGA) was next performed on the synthesized P[XC12VIM][Br]. The results are shown in Fig. 4. From the TGA curves, it could be seen that the initial decomposition temperature of all the PILs was around 230 °C, which was the starting decomposition temperature of the imidazole group. After that, another loss occurs at 330 °C, which was the complete decomposition temperature of the imidazole group. The decomposition continued when the temperature was raised to around 400 °C, which was the decomposition temperature of the benzene ring in the crosslinker DVB, which was fully decomposed by 450 °C. The TGA results prove that the synthesized PILs had high thermal stability.26
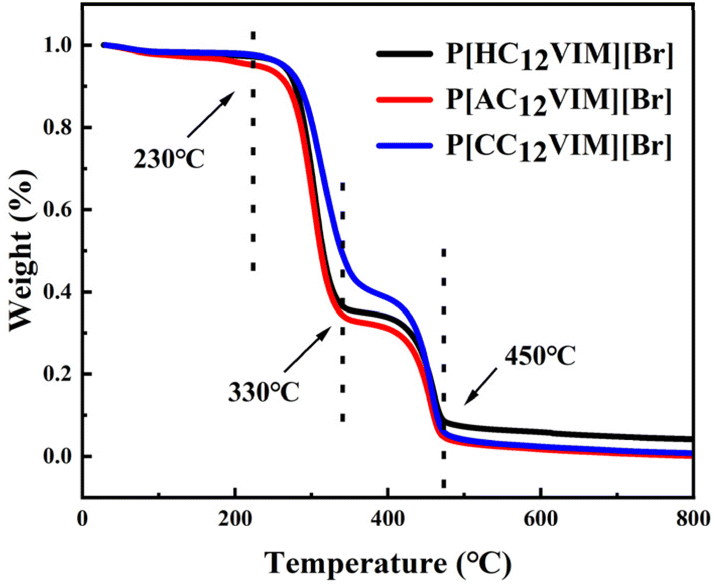 | ||
| Fig. 4 TGA curves of P[XC12VIM][Br]. | ||
Furthermore, the morphology, crystalline, and swelling properties of P[XC12VIM][Br] were also characterized, as shown in Fig. S2–S4 and Table S2.† Both the SEM (Fig. S2†) and XRD (Fig. S3†) characterizations showed that P[XC12VIM][Br] had an amorphous structure. The characterization results for the contact angle (CA) between P[XC12VIM][Br] and water are shown in Fig S4,† indicating that the introduction of long alkyl chains could effectively improve the lipophilicity of P[XC12VIM][Br]. As shown in Table S2,† the BET and swelling test results of P[XC12VIM][Br] indicated that only a small amount of large pores was formed by accumulation of the three PILs. Among all the prepared P[XC12VIM][Br] compounds, P[AC12VIM][Br] containing amino groups demonstrated the highest swelling.
To explore the CO2 adsorption capacity of all the prepared P[XC12VIM][Br] catalysts, CO2 adsorption tests were conducted, as shown in Fig. 5. As can be seen from the curves, all the functionalized PILs had a certain adsorption capacity for CO2, with the amino-functionalized P[AC12VIM][Br] showing a slightly stronger CO2 adsorption capacity than that of the other two polymers (P[HC12VIM][Br] and P[CC12VIM][Br]), which may be due to the stronger interaction between the amino group and CO2.27 The higher CO2 adsorption capacity could facilitate the capture of CO2 as a reactant molecule, thus improving the catalytic efficiency.
 | ||
| Fig. 5 CO2 adsorption isotherm curves of P[XC12VIM][Br]. | ||
In order to further explore the reason for the higher CO2 adsorption capacity of amino-functionalized P[AC12VIM][Br], P[AC12VIM][Br] was mixed with CO2 for a period of time, and FT-IR characterization of the samples before and after mixing was performed, and the results are shown in Fig. 6 (left). The results showed that after mixing, the spectrum of P[AC12VIM][Br] showed a distinct new characteristic peak at around 1780 cm−1, which was attributed to the stretching vibration of the ester group –CO. It can be seen in the partial enlargement in Fig. 6 (right) that the N–H torsional vibration peak was shifted from 831 cm−1 to 849 cm−1, and the N–H stretching vibration peak was shifted from 3440 cm−1 to 3650 cm−1, and also that the peak shape was obviously broadened. The above changes in the two N–H peaks indicate that the hydrogen atoms on the amino group formed hydrogen bonds with CO2. In addition, the N–C stretching vibrational peak of the amino group was observed around 1050 cm−1, with a significantly increased signal intensity, indicating the increase in the connected functional groups. Also, a new peak representing –OH emerged around 3000 cm−1. The above evidence prove that the chemical environment of the amino group was changed significantly, rather than just a simple physical adsorption effect. It is presumed that the amino group interacted with CO2 to form the intermediate carbamate and CO2 was activated.28 This explains the better CO2 adsorption performance of P[AC12VIM][Br] than the other two samples. The higher CO2 adsorption capacity, and the formation of carbamate intermediates are both favorable for the adsorption and activation of CO2 during the reaction process, thus improving the catalytic activity.
 | ||
| Fig. 6 FT-IR spectra of P[AC12VIM][Br] after and before interaction with CO2. | ||
3.2 Evaluation of the catalytic performance of P[XC12VIM][Br]
The catalytic performances of the synthesized PILs (P[XC12VIM][Br]) and the corresponding IL monomers ([VXEIM][Br]) were evaluated using propylene oxide (PO) as a model substrate, and the results are shown in Table 1. First, the IL monomers were evaluated and compared. It could be seen that all three IL monomers demonstrated high catalytic efficiency (>95%) and selectivity (>99%) under the reaction conditions at 120 °C (Table 1, entries 1, 3 and 5). To further compare the catalytic activity of the three monomers, the temperature was lowered to 80 °C, whereby the order of the three monomers' activity was [VAEIM][Br] (62%, Table 1, entry 6) > [VHEIM][Br] (42%, Table 1, entry 2) > [VCEIM][Br] (40%, Table 1, entry 4). This order may be due to the fact that the amino group not only acted as a hydroxyl bond donor to facilitate ring-opening of the epoxide, but also served to capture CO2.28 The effect of the hydrogen donor group on the catalytic performance of the ILs was analyzed. It was found that the PO conversion of the three functionalized ILs [VXEIM][Br] at 1 h and 80 °C was much higher than that of the ILs [VIM][Br] without the hydrogen donor group (35%, Table 1, entry 7). Furthermore, the catalytic performance of [C12VIM][Br] (Table 1, entry 12) was compared with that of [VXEIM][Br] containing a hydrogen-bond donor at 120 °C, and the PO conversion of [VXEIM][Br] containing a hydrogen-bond donor at 1 h (>95%) was found to be higher than the PO conversion of [C12VIM][Br] in 2 h (94%). All the above phenomena proved that the introduction of hydrogen donor groups could effectively improve the catalytic performance of PILs. This is consistent with the results in the previous literature.29| Entrya | Catalyst | Conv. (%) | Sel. (%) | TOFe (h−1) |
|---|---|---|---|---|
| a Reaction conditions: 1 h, 120 °C, 2.5 MPa, 2 mol%. b Reaction conditions: 1 h, 80 °C, 2.5 MPa, 2 mol%. c Reaction conditions: 2 h, 120 °C, 2.5 MPa, 2 mol%. d Reaction conditions: 2 h, 120 °C, 2.0 MPa, 1.0 mol%. e Turnover frequency (TOF): molar amount of PO converted per unit time per unit molar of catalyst. | ||||
| 1 | [VHEIM][Br] | 96 | 99 | 51 |
| 2b | [VHEIM][Br] | 42 | 99 | 22 |
| 3 | [VCEIM][Br] | 96 | 99 | 51 |
| 4b | [VCEIM][Br] | 40 | 99 | 21 |
| 5 | [VAEIM][Br] | 95 | 99 | 50 |
| 6b | [VAEIM][Br] | 62 | 99 | 32 |
| 7b | [VIM][Br] | 35 | 99 | 18 |
| 8 | P[HC12VIM][Br] | 93 | 99 | 49 |
| 9 | P[CC12VIM][Br] | 89 | 99 | 47 |
| 10 | P[AC12VIM][Br] | 96 | 99 | 51 |
| 11c | P[AC12VIM][Br] | 99 | 99 | 26 |
| 12c | [C12VIM][Br]30 | 94 | 99 | 25 |
| 13c | P[C12VIM][Br]30 | 65 | 99 | 13 |
| 14d | 2b 16 | 44 | 100 | 23 |
In addition, the differences in the catalytic activity of the PILs were compared. Among the three functionalized PILs, the catalytic activity of the amino-functionalized P[AC12VIM][Br] (96%, Table 1, entry 10) was found to be slightly higher than that of the other two polymers, P[CC12VIM][Br] (89%, Table 1, entry 9) and P[HC12VIM][Br] (93%, Table 1, entry 8), which was in line with the order of the IL monomers' catalytic activity. Through comparison of the functionalized PILs with conventional PILs, it was found that the PO conversion increased from that of conventional PILs, 65% (P[C12VIM][Br], Table 1, entry 13), to that of functionalized PILs, 99% (P[AC12VIM][Br], Table 1, entry 11), with a significant increase in the catalytic activity, which was in agreement with the trend in the catalytic activity of the IL monomers. This may be due to the fact that propylene oxide is a weakly polar molecule, and the functionalized amino, carboxyl, and hydroxyl groups are hydrophilic, so that the hydrogen donor improves the affinity between the PILs and the reactive substrate, promoting the utilization of the active sites, and thus improving the catalytic activity of the PILs. It could thus be concluded that the introduction of hydrogen donor groups helped to improve the catalytic activity, both for the IL monomers and PILs. Among all the functionalized PILs, the amino-functionalized PILs showed the highest catalytic activity among the three PIL catalysts synthesized, while the carboxyl-functionalized PILs had the weakest catalytic activity. Furthermore, comparing the catalytic performance of conventional hydrogen bond-functionalized PIL (TOF of 23 h−1, Table 1, entry 14) with that of the long chain alkyl-functionalized PIL synthesized in this paper (TOF 49 h−1, Table 1, entry 8), it was found that long chain alkyl groups could substantially improve the catalytic performance of PIL, indicating that the strategy of isolating the hydrogen bond donors with long chain alkyl groups is effective.
Finally, the catalytic activity of the synthesized functionalized PILs (Table 1, entries 8, 9 and 10) were compared with the monomers (Table 1, entries 1, 3 and 5), whereby the polymerized P[AC12VIM][Br] achieved a 96% PO conversion with a TOF of 51 h−1 (Table 1, entry 10), which was comparable with the amino-functionalized [VAEIM][Br] showing 95% PO conversion (Table 1, entry 5). Meanwhile, the other two PILs showed a slight decrease in catalytic activity compared to the corresponding monomers, whereby the PO conversion of the hydroxyl-functionalized catalyst decreased from 96% ([VHEIM][Br], Table 1, entry 1) to 93% (P[HC12VIM][Br], Table 1, entry 8), and that of the carboxyl-functionalized catalyst decreased from 96% ([VCEIM][Br], Table 1, entry 3) to 89% ([CC12VIM][Br], Table 1, entry 9), which may be attributed to the higher swelling properties of the amino-functionalized P[AC12VIM][Br], leading to the full utilization of the active sites (Table S1†). Therefore, the optimal catalyst was P[AC12VIM][Br]. In addition, by extending the reaction time to 2 h, the optimal catalyst P[AC12VIM][Br] exhibited a high catalytic activity with a PO conversion of 99%.
The optimized PIL catalyst P[AC12VIM][Br] was examined to assess its recyclability. As the results show in Fig. 7, after washed with ethyl acetate three times after the reaction and dried for the next use, the catalyst maintained high catalytic performance for five cycles.
 | ||
| Fig. 7 Recyclability of P[AC12VIM][Br]. | ||
The structural changes of the catalysts after recycling were examined by FT-IR characterization and the results are shown in Fig. 8. It could be seen that the main active site imidazole groups (1160, 1550 cm−1) and amino groups (835 cm−1) in the PIL catalysts were retained. In addition, the TG curves obtained before and after use of P[AC12VIM][Br] were compared, and were basically consistent, as shown in Fig. S4.† Indicating that the optimized catalyst had good recyclability.
 | ||
| Fig. 8 FT-IR spectra of P[AC12VIM][Br] before and after use. | ||
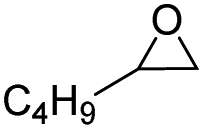
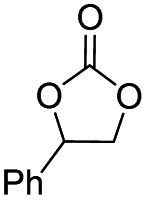
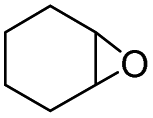
In order to investigate the substrate suitability of the optimized PIL catalyst P[AC12VIM][Br], the activity of P[AC12VIM][Br] was evaluated using different reaction substrates and the results are shown in Table 2. The results showed that P[AC12VIM][Br] had good substrate suitability. Specifically, P[AC12VIM][Br] demonstrated high catalytic activity for epichlorohydrin (Table 2, entry 1), and its catalytic performance was significantly better than that of conventional PILs for reaction substrates with lipophilic groups, such as oxidized styrene and epoxycyclohexane. The conversion of epoxycyclohexane was increased from less than 60% with conventional PILs (Table 2, entries 6–8) to more than 80% with P[AC12VIM][Br] (Table 2, entries 3 and 5). For oxidized styrene, the conversion was increased from 57% with conventional PILs (Table 2, entry 7) to 85% with P[AC12VIM][Br] (Table 2, entry 3). This may be due to the fact that the amphiphilic groups in P[AC12VIM][Br] improve the compatibility of the catalysts in the reaction substrate and thus increase the catalytic activity. In addition, P[AC12VIM][Br] also showed high catalytic activity for epoxides with long alkyl chains. Among them, butyl and octyl groups have large steric hindrance, which is not conducive to contact between the active sites and the reactants, although the catalytic activity of P[AC12VIM][Br] still reached 97% (Table 2, entry 2) and 84% (Table 2, entry 4), respectively, which also indicates that P[AC12VIM][Br] had better compatibility with the reaction substrate.










3.3 In-depth study of the P[XC12VIM][Br] structure–performance relationship
A series of characterizations were conducted on the macroscopic and long-range structure of P[XC12VIM][Br], as shown in the ESI,† which showed there were no significant differences compared to conventional PILs. Therefore, in order to further explore the reason for the improvement in the catalytic performance of P[XC12VIM][Br] from the perspective of its molecular structure, 1H NMR and XPS characterizations were carried out to study the interactions between the intramolecular hydrogen-donating groups and the imidazole groups, and their interactions with the reaction substrate (PO). Since PILs are essentially insoluble in solvents, the IL monomers were used to represent P[XC12VIM][Br]. The four IL monomers, the reaction substrate PO, and their mixtures were characterized by 1H NMR and the results are shown in Fig. 9. First, the interactions between the hydrogen-donating groups and imidazole groups were analyzed. The changes in the chemical environment after introducing hydrogen-donating groups into the imidazole group were compared (Fig. 9(a)). Here, the peaks between 9.25–10.00 ppm represented C2–H on the imidazole. It was found that the C2–H peaks of hydroxyl-functionalized [VHEIM][Br] (9.70 ppm) and amino-functionalized [VAEIM][Br] (9.69 ppm) changed less with the introduction of the hydrogen-donating group compared to that of the non-functionalized [VEIM][Br] (9.70 ppm), whereas the carboxyl-functionalized [VCEIM][Br] (9.55 ppm) C2–H peak was shifted by 0.15 ppm to higher field, which represents a decrease in the electron-withdrawing capacity of C2–H on imidazole due to the introduction of the carboxyl group, which is unfavorable for the reaction to take place. | ||
| Fig. 9 1H NMR spectra of PO with and without the ILs (a) [VEIM][Br]; (b) [VCEIM][Br]; (c) [VAEIM][Br]; and (d) [VHEIM][Br]. | ||
It could be seen that after the ILs were mixed with PO, the peaks representing the C2–H on the imidazole and the four hydrogens on PO were shifted by different degrees (around 0.1 ppm), suggesting that there was an interaction between the imidazole group of the ILs and PO, which was likely due to the formation of hydrogen bonds between the C2–H in the imidazole and the oxygen in PO. This hydrogen bond may play a role in inducing ring-opening in the reaction. Among the three functioned ILs, upon mixing the carboxyl-functionalized [VCEIM][Br] (Fig. 9(b)) with PO, shifts in the peaks representing the four hydrogens on PO (ca. 0.01 ppm) were noted, but were all smaller than those seen with the other two functionalized ILs (ca. 0.03 ppm, Fig. 9(c and d)), indicating that the interaction of [VCEIM][Br] with PO was weaker. Combined with the findings above, it is speculated that there was a weaker interaction between the carboxyl-functionalized [VCEIM][Br] with PO due to the reduced electron-withdrawing capacity of the C2–H on the imidazole group, which is in agreement with the activity evaluation showing that the carboxyl-functionalized [VCEIM][Br] had the weakest catalytic performance.
Finally, the interaction between the three hydrogen-donating groups and PO were investigated (Fig. 9(b–d)), and it could be seen that the peaks representing the hydrogen on the three hydrogen-donating groups were shifted by varying degrees after mixing with PO, wherein the peaks for [VAEIM][Br], [VCEIM][Br], [VHEIM][Br] were shifted by 0.005, 0.2, and 0.05 ppm, respectively. Therefore, it is presumed that the three hydrogen-donating groups can also form hydrogen bonds with the oxygen in PO and assist the C2–H on the imidazole group to activate PO, which explains the enhancement of the catalytic activity of the ILs after the introduction of hydrogen-donating groups.
To further compare the effect of different hydroxyl-donating groups on the imidazole group, the chemical states of two elements, namely N and Br, in the synthesized functionalized PIL catalysts P[XC12VIM][Br] were characterized by XPS, and the results are shown in Fig. 10. By fitting the plot of N 1s (Fig. 10(a)), two peaks were generated with binding energies of 401.21–401.36 eV and 399.20–399.35 eV, which were attributed to N on the imidazole ring and electron transfer due to strong interactions with the surrounding charged ions, respectively.32 By fitting the Br 3d plot (Fig. 10(b)), peaks with binding energies of 68.09–68.96 eV and 67.08–67.74 eV were obtained, which were attributed to Br− in the ILs. The binding energies of Br− in P[AC12VIM][Br] was greater than that of the other two polymers, increasing from 68.09 and 67.08 eV for P[HC12VIM][Br] to 68.09 and 67.10 eV for P[CC12VIM][Br] and 68.96 and 67.74 eV for P[AC12VIM][Br]. This increasing trend may be due to the fact that the amino group plays a role in stabilizing Br−, leading to an increase in the binding energy of Br−. The binding energy of N in P[AC12VIM][Br] is smaller than that of the other two polymers (Fig. 10(a)), which suggests that the amino group contributes to the reduction of the binding energy of Br− to the imidazole cation. (P[HC12VIM][Br]: 401.29 eV and 399.20 eV; P[CC12VIM][Br]: 401.36 eV and 399.35 eV; P[AC12VIM][Br]: 401.21 eV and 399.20 eV, with an overall decrease of about 0.15).18
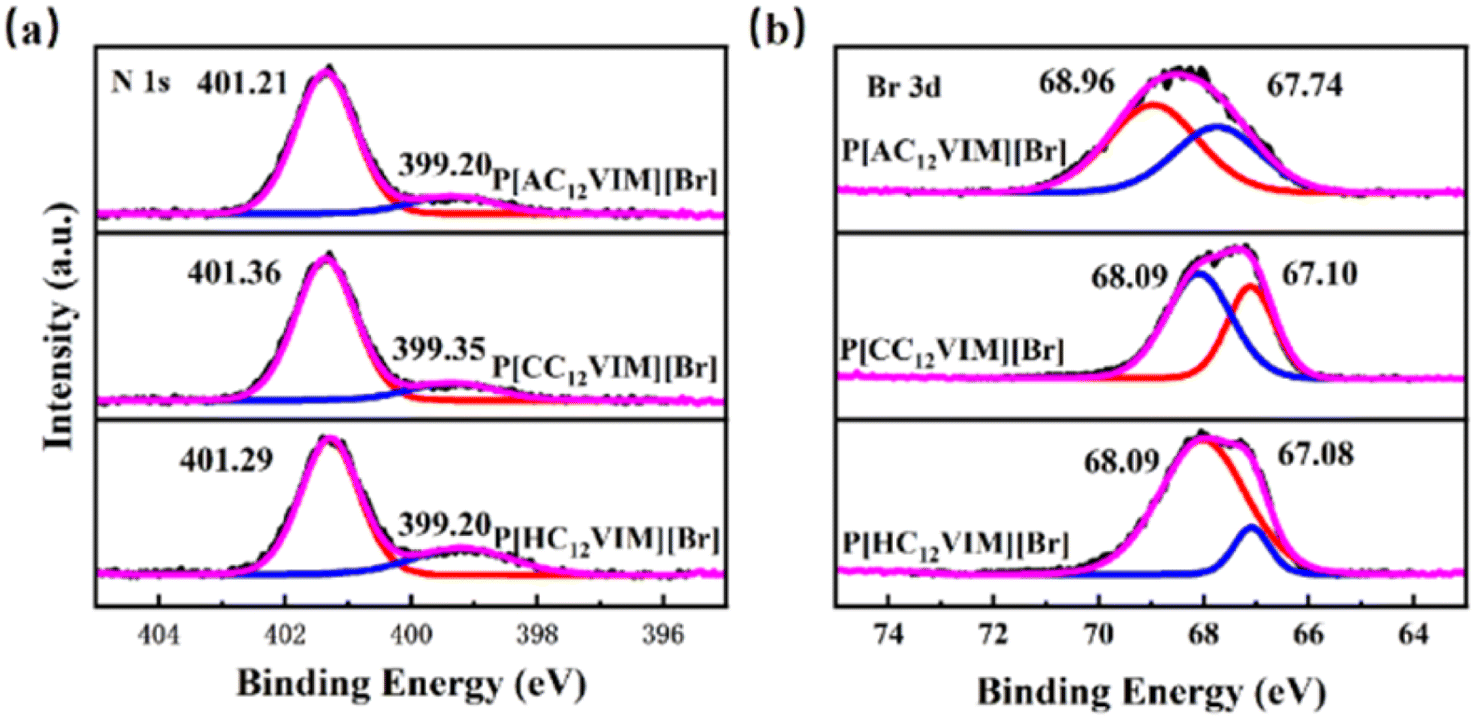 | ||
| Fig. 10 XPS spectra of P[XC12VIM][Br] (a) N 1s; (b) Br 3d. | ||
In order to further verify the interaction between the active sites and PO, DFT simulations were carried out on the structure of the ILs before and after combining with PO, as shown in Fig. 11. First, the bond lengths of PO after the interaction between PO and the ILs were compared, and it was found that the addition of ILs led to the elongation of the PO bond lengths by about 0.02 ([CC12VIM][Br] + PO (1.45 and 1.46 Å) (Fig. 11(b)); [HC12VIM][Br] + PO (1.45 and 1.45 Å) (Fig. 11(c)); [AC12VIM][Br] + PO (1.45 and 1.45 Å) (Fig. 11(a)); PO (1.43 and 1.44 Å)), which proved that there was an interaction between PO and the ILs. This is consistent with the results in the 1H NMR characterization. The charge densities of Br− in the three ILs were simulated and the results showed the following trend: ([CC12VIM][Br] (−0.618) (Fig. 11(e)) < [HC12VIM][Br] (−0.644) (Fig. 11(f)) < [AC12VIM][Br] (−0.677) (Fig. 11(d))). Larger charge densities favored the occurrence of interactions between Br− and PO during the reaction, which was consistent with the results from the activity evaluation. The bond length between Br− and the imidazole ring was also calculated and the result showed the following trend: ([CC12VIM][Br] (3.55 Å) (Fig. 11(e)) < [HC12VIM][Br] (3.88 Å) (Fig. 11(f)) ≈ [AC12VIM][Br] (3.88 Å) (Fig. 11(d))). The bond in [CC12VIM][Br] was found to be shorter than in the other two ILs, while the bond length difference between [HC12VIM][Br] and [AC12VIM][Br] was not significant. The longer bond facilitated freeing Br−, thus promoting the reaction. This is consistent with the results showing that [CC12VIM][Br] had the worst activity in the evaluation.
 | ||
| Fig. 11 Structures determined by DFT of (a) [AC12VIM][Br] + PO; (b) [CC12VIM][Br] + PO; (c) [HC12VIM][Br] + PO; (d) [AC12VIM][Br]; (e) [CC12VIM][Br]; (f) [HC12VIM][Br]. | ||
Based on the 1H NMR characterization results and previous studies,26 and taking the best amino-functionalized P[AC12VIM][Br] as an example, a possible catalytic mechanism is proposed, as shown in Fig. 12. First, the C2–H of the imidazole on the cation of the PILs interacts with the O in the epoxide to form a hydrogen bond, while the amino group also forms a hydrogen bond with the O, thus synergistically activating the epoxide. At the same time Br− is freed from the PILs and nucleophilically attacks the less hindered C in the epoxide, leading to ring-opening of the epoxide, during which the amino group also plays a role in stabilizing Br−. The carbamate intermediate formed by amino-activated CO2 is inserted into the alkyl carbonate anion formed by the ring-opening, while the amino group is regenerated. Finally, the alkyl carbonate anion undergoes intramolecular cyclization to form a cyclic carbonate while the catalyst is regenerated.
 | ||
| Fig. 12 Proposed catalytic mechanism for the cycloaddition reaction. | ||
4 Conclusion
A series of space-functionalized PIL catalysts with both hydrophilic hydrogen-donating groups (carboxyl, hydroxyl, and amino) and long-chain alkyl groups was successfully synthesized in this study and used to catalyze cycloaddition reactions. It was found that the introduction of hydrogen donor groups could enhance the intrinsic catalytic performance of the active sites, and the introduction of separating groups could effectively improve the utilization of hydrogen-donating groups. Among the synthesized PIL catalysts, the amino-functionalized PIL catalyst P[AC12VIM][Br] exhibited the highest catalytic performance, which was comparable with that of bulky ILs. This could be mainly attributed to the multiple roles of the amino group in stabilizing the bromide ion and activating CO2 while acting as a hydrogen donor for the ring-opening process. In addition, the catalyst demonstrated good recyclability and better substrate suitability for epoxide substrates with lipophilic edge groups than conventional PIL catalysts. Finally, a possible mechanism for the cycloaddition reaction was proposed. This study provides new ideas for the design of PIL catalysts from the viewpoint of the intrinsic reaction and utilization of the active sites.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Qianmeng Zhao: investigation, conceptualization, methodology, writing – review & editing, formal analysis, writing – original draft, experiment; Shaifei Liu: writing – review & editing, investigation; Wen Liu: simulation, review; Mengqian Fu: visualization; Zhenyang Xu: review; Qian Su: formal analysis, methodology, resources, writing – review & editing, funding acquisition; Weiguo Cheng: formal analysis, methodology, resources, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We sincerely acknowledge the National Natural Science Foundation of China (No. 22378395, 22078329), Young Elite Scientists Sponsorship Program by CAST (2022QNRC001).References
- J. Wang, S. Zhang and G. Cui, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- S. Lian, C. Song, Q. Liu, E. Duan, H. Ren and Y. Kitamura, Acta Sci. Circumstantiae, 2021, 1, 281–295 Search PubMed.
- R. Luo, X. Liu, M. Chen, B. Liu and Y. Fang, ChemSusChem, 2020, 13, 3945–3966 CrossRef CAS PubMed.
- D. Damiani, J. Litynski, H. McIlvried, D. Vikara and R. D. Srivastava, Greenhouse Gases:Sci. Technol., 2012, 2, 9–16 CrossRef CAS.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 45, 631–675 RSC.
- M. Mikkelsen, M. Jrgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- R. Paddock, Y. Hiyama, J. McKay and S. Nguyen, Tetrahedron Lett., 2004, 45, 2023–2026 CrossRef CAS.
- K. Sumar and L. Suman, Ind. Eng. Chem. Res., 2014, 53, 541–546 CrossRef.
- F. Jutz, J. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322–353 CrossRef CAS PubMed.
- T. Mu and Y. Chen, Green Chem., 2019, 21, 2544–2574 RSC.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- S. Lai, J. Gao, H. Zhang, L. Cheng and X. Xiong, J. CO2 Util., 2020, 38, 148–157 CrossRef CAS.
- S. Jagtap, V. Raje, S. Samant and B. Bhanage, J. Mol. Catal. A: Chem., 2007, 266, 69–74 CrossRef CAS.
- T. Shi, J. Wang, J. Sun, M. Wang, W. Cheng and S. Zhang, RSC Adv., 2013, 3, 3726–3732 RSC.
- J. He, T. Wu, Z. Zhang, K. Ding, B. Han, Y. Xie, T. Jiang and Z. Liu, Chem.–Eur. J., 2007, 13, 6992–6997 CrossRef CAS PubMed.
- L. Ji, Z. Luo, Y. Zhang, R. Wang, Y. Ji, F. Xia and G. Gao, Mol. Catal., 2018, 446, 124–130 CrossRef CAS.
- Y. Yang, Y. Guo, C. Gao, M. North, J. Yuan, H. Xie and Q. Zheng, ACS Sustain. Chem. Eng., 2023, 11, 2634–2646 CrossRef CAS.
- T. Shi, J. Wang, J. Sun, M. Wang and S. Zhang, RSC Adv., 2013, 3, 3726–3732 RSC.
- T. Chang, L. He, L. Bian, H. Han and X. Gao, RSC Adv., 2013, 4, 727–731 RSC.
- J. Chang, Y. Liu, Q. Su, L. Liu, L. Deng, T. Ying, L. Dong, Z. Luo, Q. Li and W. Cheng, ChemistrySelect, 2021, 6, 6380–6387 CrossRef CAS.
- Z. Guo, X. Cai, J. Xie, X. Wang and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CrossRef CAS PubMed.
- J. Lu, M. Zhang, B. Li, L. Dong and M. Fan, J. South China Norm. Univ., Nat. Sci. Ed., 2021, 53, 35–42 Search PubMed.
- J. Sun, S. Zhang and W. Cheng, Tetrahedron Lett., 2008, 39, 3588–35912 CrossRef.
- H. Gou, Synthesis of cyclic carbonates from CO2 catalyzed by functional polymerized ionic liquids, University of Chinese Academy of Sciences, 2021 Search PubMed.
- Y. Zou, Y. Ge, Q. Zhang, W. Liu, X. Li, G. Cheng and H. Ke, Catal. Sci. Technol., 2022, 12, 273–281 RSC.
- L. Ji, Z. Luo, F. Xia and G. Guohua, Mol. Catal., 2016, 446, 124–130 CrossRef.
- S. Liu, Q. Su, M. Fu, L. Deng, Y. Wang, L. Dong, Y. Liu, X. Ma and W. Cheng, Catal. Lett., 2023, 153, 2429–2442 CrossRef CAS.
- J. C. Q. Zhao, L. Deng, Y. Li, Z. Yang, Q. Su and W. Cheng, Appl. Mater. Today, 2023, 35, 101970 CrossRef.
- Q. Zhao, M. Fu, Z. Xu, L. Deng, Y. Li, X. Meng, Q. Su and W. Cheng, Mol. Catal., 2023, 551, 113651 CrossRef CAS.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00661e |
| This journal is © The Royal Society of Chemistry 2025 |