
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalyst design strategies towards high performance anion-exchange membrane-based direct ammonia fuel cells
Ho Young
Kim†
a,
Jungki
Kim†
b,
Eunsoo
Lee†
c,
Hyoryeong
Choi
c,
Hyunsoo
Chun
d,
Joyjit
Kundu
 b,
Sang-Il
Choi
*b,
Kwangyeol
Lee
*c and
Jin Young
Kim
*ef
b,
Sang-Il
Choi
*b,
Kwangyeol
Lee
*c and
Jin Young
Kim
*ef
aDepartment of Chemistry and Energy Engineering, Sangmyung University, Seoul 03016, Republic of Korea
bDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: sichoi@knu.ac.kr
cDepartment of Chemistry and Research Institute for Natural Sciences, Korea University, Seoul 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
dDepartment of Mechanical Engineering, Seoul National University, Seoul, 08826, Republic of Korea
eHydrogen·Fuel Cell Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: jinykim@kist.re.kr
fDivision of Energy & Environment Technology, KIST School, Korea National University of Science and Technology (UST), Seoul 02792, Republic of Korea
First published on 23rd January 2025
Abstract
Direct ammonia fuel cells (DAFCs) with anion-exchange membranes (AEMs) are considered a valuable contributor to a carbon-neutral clean energy society, benefitting from the existence of long-established ammonia infrastructure. However, the relatively low cell performance of DAFCs compared to the hydrogen-based fuel cells and the critical issues related to catalyst poisoning limit the widespread use of DAFCs and have spurred multi-directional efforts to develop tailored catalyst compositions and structures specific to DAFCs. In this review, we outline recent progress in the development of electrocatalysts for DAFCs. First, we summarize the operating principles of DAFCs and address critical challenges in electrode reactions, the ammonia oxidation reaction (AOR) and oxygen reduction reaction (ORR). Subsequently, we present an overview of recent endeavours to enhance activity, selectivity, and durability of catalysts for each electrode reaction. We categorize the electrocatalytic exemplars into platinum group metal (PGM) and non-PGM compositions and provide systematic comparisons of each strategy to provide a more comprehensive understanding of catalyst design. Lastly, this review highlights remaining challenges and offers insights into future directions for optimizing DAFC performance.
1 Introduction
The growing interest in carbon neutrality has motivated the search for robust strategies to produce clean energy.1–3 Fuel cells, representative eco-friendly energy conversion devices, have taken a prominent position and currently play a pivotal role in our future energy landscape.4–6 They generate electricity in a clean and sustainable manner through the electrochemical transformation of small molecules (e.g., hydrogen, alcohol, ammonia). Among various fuel cell types, polymer electrolyte membrane fuel cells (PEMFCs) have attracted interest for widespread application because of their high energy density and low-temperature operation. Multitudes of fuel molecules can be used in PEMFCs, but hydrogen is considered the most suitable energy carrier due to its high gravimetric energy density of 142 MJ kg−1, which is significantly higher than that of gasoline at 47 MJ kg−1. Nevertheless, the practical integration of hydrogen into PEMFCs presents substantial challenges, primarily due to the absence of any well-established infrastructure to support the hydrogen economy.7–10 Currently, 95% of hydrogen production relies on hydrocarbon extraction processes such as reforming, coal gasification, and cracking, leading to significant CO/CO2 emissions. Additionally, the common methods of hydrogen storage, such as compression, liquefaction, and solid-state storage, require large-volume systems that limit the efficient onboard application of hydrogen. Furthermore, the flammability of hydrogen poses significant risks, and concerns about its stability during storage and transportation persist.Within this context, ammonia is emerging as a promising alternative to hydrogen, primarily because of its production and distribution advantages.11–15 The long-established Haber–Bosch process is capable of fulfilling demand for mass scale ammonia consumption, which facilitates its entry into the market as a carbon-neutral fuel source. Also, compared with gaseous hydrogen, liquid ammonia has a higher mass density, allowing for a lower internal pressure in the fuel tank, lighter-weight tank hardware, and thus more efficient transportation. Although its gravimetric energy density of 22.5 MJ kg−1 is much lower than that of hydrogen gas, the energy density of a liquid ammonia tank (15.6 MJ L−1) is estimated to be higher than that of a gaseous hydrogen tank at 700 bar (5.6 MJ L−1), allowing efficient energy distribution. The properties of ammonia as a fuel are summarized in Table 1.
| Fuels | Advantages | Disadvantages |
|---|---|---|
| Hydrogen | - Zero emission | - Expensive onboard storage |
| - High energy density | ||
| - Non-toxic | - Low stored energy density | |
| Alcohol | - Easy to convert to hydrogen onboard vehicle | - Corrosiveness |
| - Renewable resources | - Hard to use in cold weather | |
| - Biodegradable | - Miscible in water | |
| Ammonia | - Zero emission | - Toxic |
| - High production capacity | ||
| - Abundant feedstocks | - Low stored energy density | |
| - Well established production and transport infrastructure |
However, the utilization of ammonia fuels in PEMFC systems, commonly referred to as direct ammonia fuel cell (DAFC) technology, remains at a relatively nascent stage of development primarily because they have a lower power density than hydrogen-based PEMFCs.16–23 The inferior performance of DAFC is attributed to a lack of understanding of how to tailor ingredients to configure optimized membrane-electrode assemblies (MEAs) for the DAFC systems. For instance, the perfluorosulfonic acid (PFSA) membrane employed for proton transport in hydrogen-based PEMFCs is vulnerable to contamination and deactivation even at trace ammonia levels.13–23 The detailed comparison between DAFC and PEMFC is summarized in Table 2. In recent studies employing anion exchange membranes (AEMs) with high conductivity, an impressive power output of 420 mW cm−2 was achieved in a single DAFC.17 However, this achievement is still significantly below the performance benchmarks observed in hydrogen-based PEMFCs (approximately 1 W cm−2).24 This performance gap underscores the need to continue efforts to develop highly efficient catalysts for high-performance DAFC. Considering the urgent need to improve DAFC performance with novel design strategies for DAFC catalysts, it is imperative to identify catalysts with optimized compositions and structures, specifically tailored for DAFCs.
| Properties | DAFC | PEMFC |
|---|---|---|
| Fuel | Ammonia | Hydrogen gas |
| Operating temperature | 25–100 °C | 30–80 °C |
| Charge carrier | OH− | H+ |
| Electrolyte membrane | Anion exchange membrane (hydrocarbon) | Proton exchange membrane (PFSA/PTFE) |
| Catalysts | Anode: Pt, Ni, Cu | Anode: Pt |
| Cathode: Pt, Pd, M–Nx/C | Cathode: Pt, Pt–M | |
| Advantages | - Mitigating challenges of hydrogen (storage and transportation) | - High energy efficiency |
| - Fast reaction kinetics | ||
| - Simple system design | ||
| Disadvantages | - Slow reaction kinetics | - Challenges in hydrogen storage and transportation |
| - Crossover issue | - Use of PGM catalyst | |
| - System complexity | - Humidity and thermal management |
In this review, we describe recent advances on electrocatalysis in DAFCs. Among several approaches, we focus on the AEM-based low temperature DAFC systems. First, we summarize the working principles of the DAFC and important challenges with electrode reactions, the ammonia oxidation reaction (AOR) at the anode and oxygen reduction reaction (ORR) at the cathode. We next provide a survey of the electrochemical properties of advanced catalysts suitable for promoting the AOR and ORR. Exemplars of electrocatalysts from two categories are presented: platinum group metal (PGM) and non-PGM compositions. For ORR catalysts previously explored with diverse compositions for PEMFC applications, we focused on non-precious metal catalysts such as metal oxides, perovskites, and single-atom catalysts. These catalysts demonstrate resistance to ammonia poisoning and are better suited for DAFC applications. Finally, we conclude this review by discussing the remaining issues and future perspectives.
2 Electrocatalysis for DAFCs
In this section, we introduce electrocatalytic reactions for the AEM-based DAFC systems operated at low temperatures (50–120 °C) and the critical challenges related to each electrode reaction. A DAFC resembles conventional PEMFC system, composed of an anode, a cathode, and an intervening electrolyte membrane.12–15 In contrast to high-temperature ammonia-based fuel cell platforms, DAFC systems produce electrical power by the electrochemical oxidation of ammonia supplied at the anode, without the need to thermally crack it into hydrogen (Fig. 1).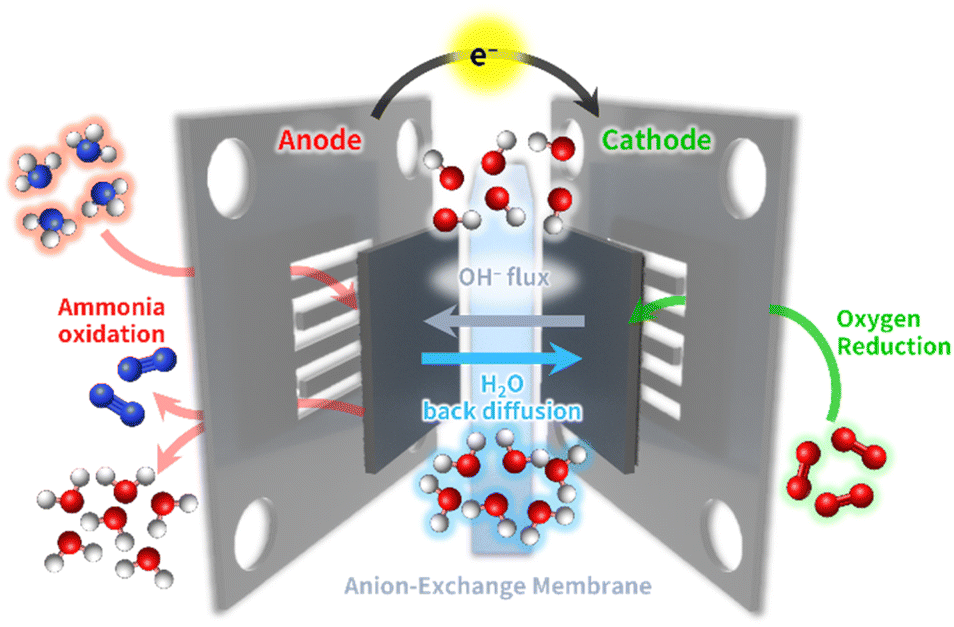 | ||
| Fig. 1 Schematic illustration of direct ammonia fuel cell (DAFC). | ||
Briefly, supplying air and water vapor to the cathode generates hydroxide ions (OH−) by the electroreduction of O2 in the air reacted with water. These OH− ions are conducted to the anode side via ion exchange with quaternary ammonium or pyridinium sites in the hydrated polymer domains of the AEM.18–21 The ammonia fuels in the anode directly react with OH− ions to produce electrons, generating nitrogen and water molecules. The products and unreacted ammonia fuels are emitted through the anode exit stream. The equations for each electrode reaction vs. a standard hydrogen electrode (SHE) in DAFCs and PEMFCs are presented below.
For DAFCs:
| Anode: 4NH3 + 12OH− → 2N2 + 12H2O + 12e−, E0 = −0.77 V |
| Cathode: 3O2 + 6H2O + 12e− → 12OH−, E0 = 0.40 V |
| Overall reaction: 4NH3 + 3O2 → 2N2 + 6H2O, E0 = 1.17 V |
For PEMFCs (in alkaline electrolyte):
| Anode: 2H2 + 4OH− → 4H2O + 4e−, E0 = −0.83 V |
| Cathode: O2 + 2H2O + 4e− → 4OH−, E0 = 0.40 V |
| Overall reaction: 2H2 + O2 → 2H2O, E0 = 1.23 V |
The theoretical voltage achieved by DAFCs (1.17 V) is comparable to that of PEMFCs (1.23 V). Nonetheless, in practice DAFCs exhibit a more pronounced voltage loss than PEMFCs, which is attributed to the more substantial kinetic constraints governing their electrode reactions. This voltage loss primarily arises from the characteristics of the anodic reactions responsible for electron generation, since the cathodic reaction in both systems is ORR.11,13,15 In PEMFCs, electrons are generated through the hydrogen oxidation reaction (HOR), which follows a straightforward reaction pathway. It is widely acknowledged that the HOR proceeds through either the Tafel–Volmer or the Heyrovsky–Volmer mechanisms.25,26 When hydrogen molecules approach catalytic surfaces in alkaline media, they can either undergo a Tafel step, splitting into hydrogen adsorbents (H*) on the catalytic surfaces (H2 → 2H*) or participate in a Heyrovsky step, reacting with hydroxides in the electrolyte to produce water molecules and H* (H2 + OH− → H* + H2O + e−). Subsequently, the produced H* further reacts with hydroxides through the Volmer step, generating water and electrons (H* + OH− → H2O + e−).
In contrast, AOR unfolds through multiple intricate processes, including the sequential dehydrogenation of ammonia (generation of 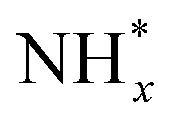 ,
, 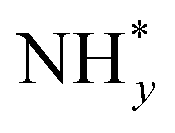 intermediates),
intermediates), 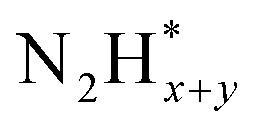 formation
formation 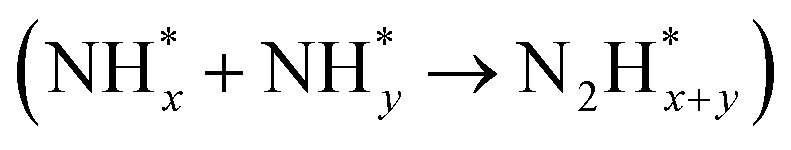 , and the subsequent dehydrogenation of
, and the subsequent dehydrogenation of 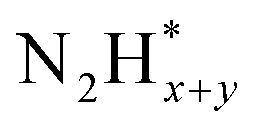 to yield N2, with sluggish reaction kinetics (Fig. 2).27–31 Consequently, the DAFC anode, which requires a high overpotential, suffers significant voltage loss in the overall DAFC system, highlighting the need to develop high-performance catalysts to promote AOR.
to yield N2, with sluggish reaction kinetics (Fig. 2).27–31 Consequently, the DAFC anode, which requires a high overpotential, suffers significant voltage loss in the overall DAFC system, highlighting the need to develop high-performance catalysts to promote AOR.
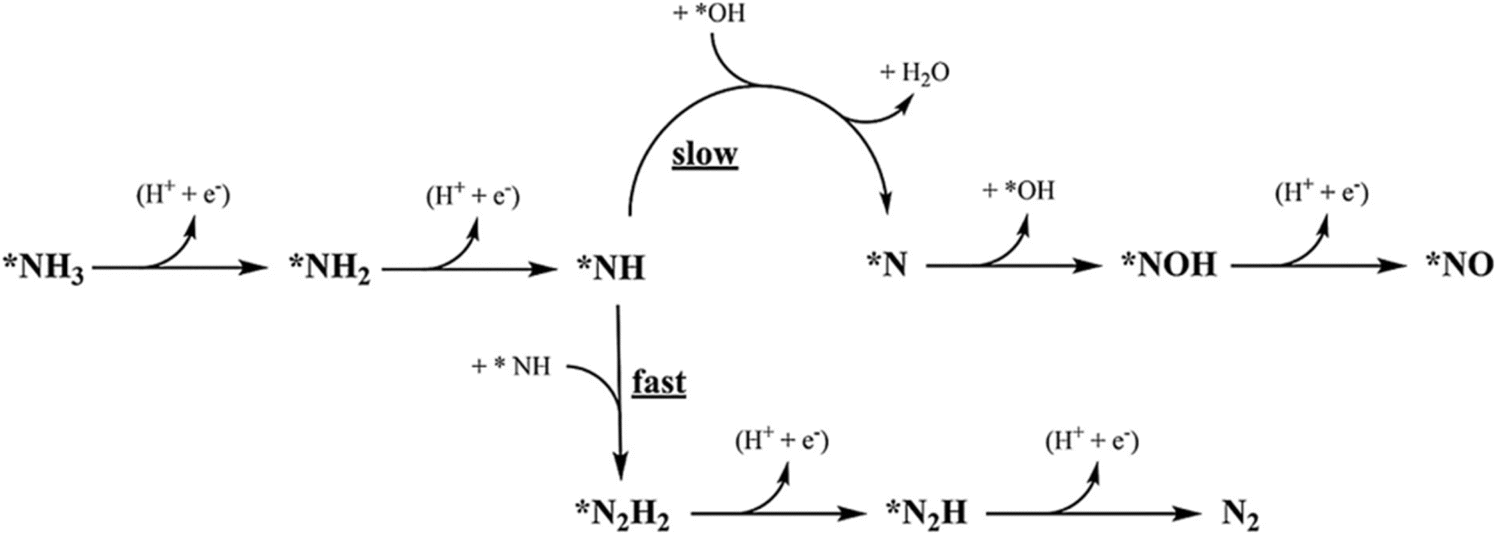 | ||
| Fig. 2 Proposed scheme for the most feasible steps during the electrochemical AOR on Pt. The star denotes adsorbed species. Reproduced from ref. 63 with permission. Copyright 2018 Elsevier. | ||
At the same time, the cathode, which frequently suffers from catalyst deactivation due to ammonia cross-over, is also responsible for voltage loss.13–15 During DAFC operation, a portion of the unreacted ammonia fuel inevitably migrates from the anode to the AEM. Subsequently, this diffused ammonia within the AEM permeates towards the cathodes, triggering an unintended AOR. This inadvertent AOR process disrupts the smooth progression of the ORR at the cathode, leading to the concurrent AOR and ORR. This coexisting AOR and ORR generates a mixed potential at the cathode, which is considerably lower than the anticipated potential for complete ORR, ultimately leading to a substantial decline in DAFC cell voltage. For this reason, the cathode catalysts must have a robust tolerance to ammonia, allow high ORR activity and durability, all while demonstrating remarkable ORR selectivity.
In the following sections, we focus on the progress and electrochemical properties of catalysts for these two electrode reactions. Notable examples from the synthesis strategies for advanced catalysts, and the underlying mechanisms for enhancing activity and durability, are discussed in detail.
3 Electrocatalysts for AOR: PGM vs. non-PGM
According to Sabatier's principle, the rate of catalytic reactions is maximized when the binding strength between the catalytic surfaces and reactants or reaction intermediates is optimal. If the binding strength is too strong, the adsorbed products cannot desorb from the catalytic surfaces. Conversely, the catalytic reaction will not proceed if it is too weak. AOR catalysts have also been developed considering the Sabatier's principle, with a particular focus on the binding strength and adsorption/desorption behaviours of N atoms on catalytic surfaces. In the following sections, we introduce the advances in diverse AOR catalysts developed by multidirectional efforts.3.1. Platinum (Pt) monometallic AOR electrocatalysts
Pt has been extensively studied as an AOR catalyst due to its superior activity among monometallic systems.32–38 In most prior research, Pt was the preferred electrocatalyst for optimizing experimental parameters that enhance AOR while minimizing the formation of byproducts such as nitrate ions and NOx. For example, Müller and Spitzer reported in 1905 that Pt electrodes produce N2 gas during AOR in NaOH solution containing ammonia.32 Later, in 1963, Katan and Galiotto identified a link between anode potential and the oxidation degree of AOR products.33 They showed that a Pt black anode selectively generates pure N2 within a specific potential range (−0.31 to −0.11 vs. SHE). In 1994, Wasmus and colleagues further demonstrated the voltage-dependent behaviour of AOR using differential electrochemical mass spectrometry (DEMS).35 Their DEMS-cyclic voltammetry (CV) experiments in 0.5 M KOH revealed that N2 was the sole product at an anode potential of ∼0.7 V vs. RHE, while NO and nitrate ions formed at potentials above 0.8 V vs. RHE.The high AOR activity of Pt is attributed to its optimal binding strength with atomic nitrogen adsorbates (N*) and 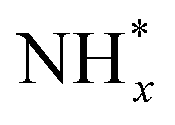 species. Gerischer and Mauerer proposed that N2 evolution on Pt proceeds via the formation of
species. Gerischer and Mauerer proposed that N2 evolution on Pt proceeds via the formation of 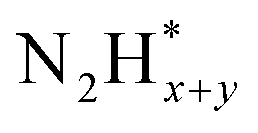 , resulting from the recombination of
, resulting from the recombination of 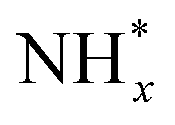 species.27–31 Koper et al. further explored the relationship between N* binding energy and AOR activity, offering insights into Pt catalysts (Fig. 3a).39 They summarized the trend in N* binding energy across various metals as follows: Ru > Rh > Pd > Ir > Pt ≫ Au, Ag, Cu. For Ru, Rh, and Pd, their strong N* binding strength allows ammonia oxidation at significantly lower potentials than Pt. However, these metals deactivate quickly due to rapid deprotonation of
species.27–31 Koper et al. further explored the relationship between N* binding energy and AOR activity, offering insights into Pt catalysts (Fig. 3a).39 They summarized the trend in N* binding energy across various metals as follows: Ru > Rh > Pd > Ir > Pt ≫ Au, Ag, Cu. For Ru, Rh, and Pd, their strong N* binding strength allows ammonia oxidation at significantly lower potentials than Pt. However, these metals deactivate quickly due to rapid deprotonation of 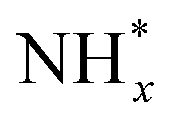 species and irreversible N* adsorption. Conversely, Au, Ag, and Cu lack sufficient N* binding strength to effectively initiate AOR. Pt, positioned between these extremes, exhibits an optimal binding strength that supports a low onset potential. Mavrikakis et al. corroborated this through DFT calculations, predicting N* binding energies for 12 transition metals and confirming Pt's superior AOR activity due to its ideal binding energy (Fig. 3b).40
species and irreversible N* adsorption. Conversely, Au, Ag, and Cu lack sufficient N* binding strength to effectively initiate AOR. Pt, positioned between these extremes, exhibits an optimal binding strength that supports a low onset potential. Mavrikakis et al. corroborated this through DFT calculations, predicting N* binding energies for 12 transition metals and confirming Pt's superior AOR activity due to its ideal binding energy (Fig. 3b).40
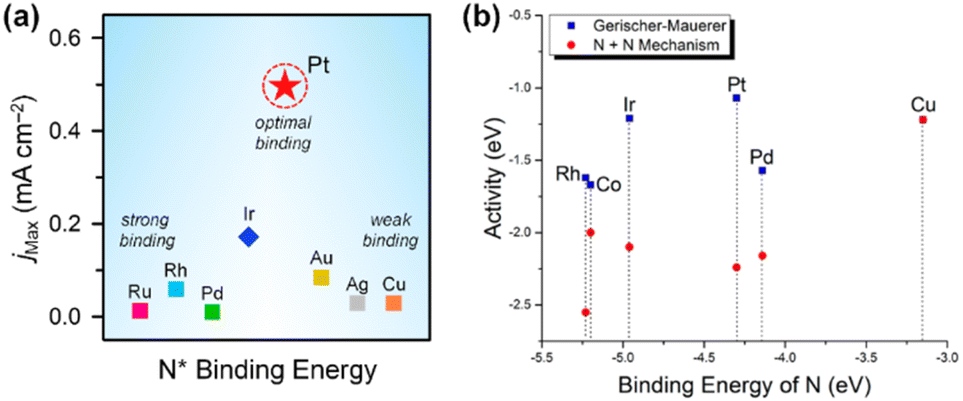 | ||
| Fig. 3 (a) AOR maximum current density and N* binding energy of eight metal elements. Modified from ref. 39 with permission. Copyright 2001 Elsevier. (b) Activity as predicted by Sabatier analysis for both mechanisms at 0 V vs. RHE. Less negative activity is better. The activity is calculated from the thermochemistry of electrochemical steps and the activation energy of N–N bond-forming steps. It is a measure of the maximum resistance of the minimum-energy pathway for each mechanism. Reproduced from ref. 40 with permission. Copyright 2015 American Chemical Society (ACS). | ||
Nevertheless, the high cost of Pt catalysts has driven further investigations aimed at enhancing catalytic performance while reducing the usage of Pt.11,41–43 Among these, nanostructuring strategies can increase the electrochemically active surface areas (ECSAs) of catalytic surfaces, multiplying the number of active sites and thereby boosting the mass activity of the catalysts. For example, nanostructured Pt thin-film (TF) electrode surfaces provided clues for the AOR activity enhancement of Pt catalysts.44–46 Hu and Cheng demonstrated that the electrodeposited Pt TFs have much higher ECSAs than bulk Pt electrodes and exhibited improved AOR activity.44 In addition, they tuned the roughness factor and ECSA of deposited TFs by controlling the current density of the electrodeposition process. The electrodeposition process at 5 mA cm−2 generates a sheet-like dendritic structure composed of small Pt particles with a few nm scales, while large Pt particles of several hundreds of nm with smooth surfaces are formed at 0.12 mA cm−2. Apart from changing the morphology, higher depositing current density leads to a higher Pt loading amount, resulting in a higher activity. Subsequent studies further suggested that the high roughness factor of the deposited TFs directly contributes to the augmentation of ECSA, thereby enhancing AOR activity.45,46 Further efforts focused on support engineering, such as the structural tuning of carbon supports,47,48 doping heteroatoms within support materials,49–51 and exploiting inorganic supports,52,53 have also contributed to the enhancement of ECSAs and AOR activity.
Beyond increasing ECSAs, boosting the mass activity of catalysts is pivotal, contingent upon improving the intrinsic activity of each active site (specific activity). For AOR, the strategies to enhance specific activity revolve around understanding catalyst poisoning and alleviating its impact. The poisoning phenomena refers to the deactivation of catalytic surfaces by N* species that are generated as intermediates during the AOR. These N* species are inert after N2 formation on the catalytic surfaces because they strongly bind to catalytic surfaces, impeding OH− adsorption. These site-blocking species accordingly diminish catalytic activity. Experimental evidence has demonstrated the impacts of N* species on catalytic surfaces, providing useful insights into the deactivation of catalysts. For example, Botte et al. demonstrated the poisoning effect of N* species on Pt/C catalysts with an average particle size of 2.7 nm, prepared using a polyol method (Fig. 4a and b).54 The polyol method is a versatile and cost-effective technique for synthesizing controlled, high-purity nanoparticles. They examined the AOR properties of Pt/C catalysts in 1 M KOH and 1 M KOH + 1 M NH3 solutions at room temperature. As shown in the CV curves of Pt/C, the maximum current density of AOR (∼6 mA cm−2) decreased continuously as the number of CV scans increased (Fig. 4c). This activity loss stems from the residual poisonous N* adsorbates on the catalytic surfaces rather than the limited diffusion of ammonia reactant molecules. In addition, the current density of reductive peaks around −0.7 V vs. SHE (presented in the black dashed ellipse area) simultaneously increased when the AOR activity decreased. These reductive peaks are associated with the adsorption of site-blocking N* species, implying the poisoning of Pt catalytic surfaces. Furthermore, CVs before and after the chronoamperometry (CA) test in 1 M KOH + 1 M NH3 also supported the poisoning effect of N* species (Fig. 4d). Open circuit potential (OCV) holding, which allows diffusion of ammonia into the catalytic surfaces, did not recover its initial activity after 10 min. However, CA operation at −0.8 V vs. SHE for 1 min perfectly recovered the initial AOR activity of the catalyst by reducing the adsorbed N* species on the Pt surfaces. In addition, as revealed by Choi et al., the byproducts of N2 formation during AOR (NOx species, including NO, NO2−, NO3−) also can act as site-blocking species for N–N bond formation on the Pt catalytic surfaces.55
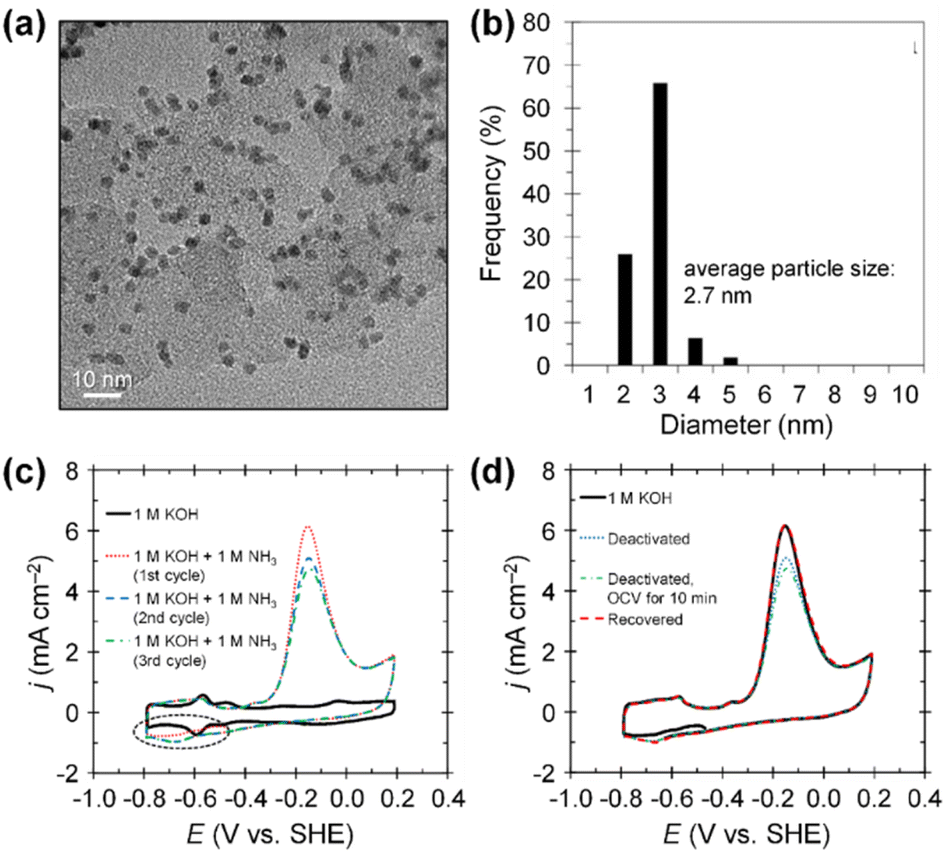 | ||
| Fig. 4 (a) TEM image and (b) particle size histogram of Pt/C catalyst. (c) AOR CV curves of Pt/C catalyst in 1 M KOH with and without 1 M NH3 at a scan rate of 20 mV s−1. (d) AOR curves of fresh, deactivated, and recovered Pt/C catalysts at a scan rate of 20 mV s−1. Reproduced from ref. 54 with permission. Copyright 2017 Elsevier. | ||
The detailed understanding of catalyst deactivation in ammonia environments has driven efforts to optimize Pt catalyst structures to mitigate poisoning, emphasizing control over crystal facets. In catalytic research, single-crystal model studies have revealed surface structure-dependent properties. Aldaz et al. examined the electrocatalytic performance of Pt(100), Pt(110), and Pt(111) toward AOR (Fig. 5a and b).56 Tests in 0.1 M NaOH + 10 mM NH3 demonstrated the sensitivity of AOR activity to surface structure. Pt(100) showed a distinct oxidation peak at 0.57 V with a peak current density of 1.018 mA cm−2, while Pt(110) and Pt(111) exhibited negligible activity. Further studies on stepped surfaces, such as Pt(311), Pt(11,1,1), and Pt(19,1,1), corresponding to [2(100) × (111)], [6(100) × (111)], and [10(100) × (111)], respectively, revealed reduced AOR activity as the width of (100) terraces decreased, evidenced by lower peak current densities and shifts in peak potential to more positive values (Fig. 5c and d). These results highlight that AOR predominantly occurs on (100) sites. Follow-up research with complex crystal planes,57 nanoparticles, and nanostructured Pt thin films with preferential (100) orientation,58–60 confirmed that surfaces with a higher proportion of (100) planes exhibit enhanced AOR activity.
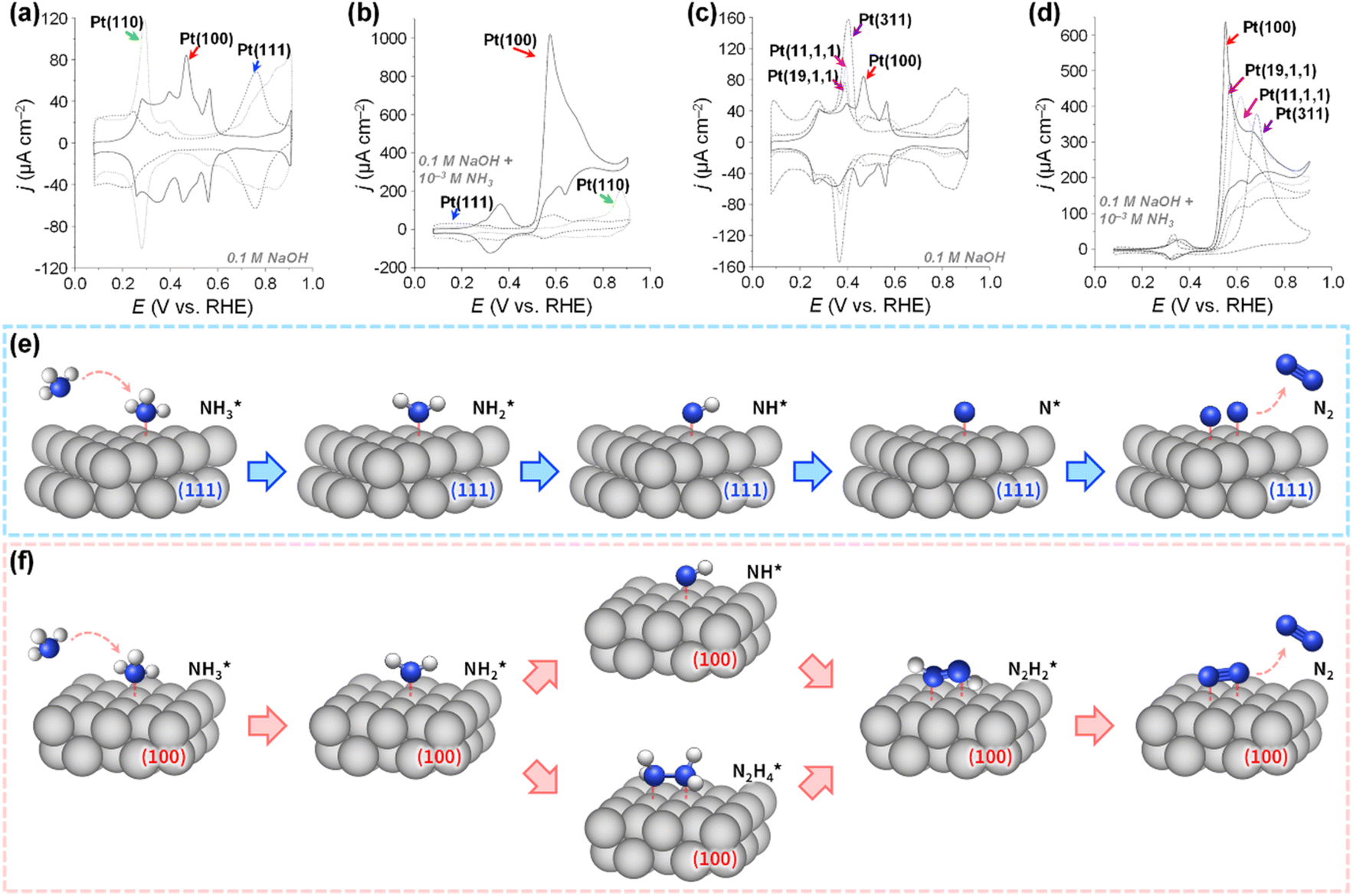 | ||
| Fig. 5 CV curves of Pt(100), Pt(111), and Pt(110) electrodes in (a) 0.1 M NaOH and (b) 0.1 M NaOH + 10−3 M NH3. CV curves of Pt(100), Pt(19,1,1), Pt(11,1,1), and Pt(311) electrodes in (c) 0.1 M NaOH and (d) 0.1 M NaOH + 10−3 M NH3. Reproduced from ref. 56 with permission. Copyright 2003 Elsevier. Schematic illustrations for the AOR on the (e) Pt(111) surfaces and (f) Pt(100) surfaces. | ||
Koper et al. further demonstrated the superior AOR performance of Pt(100) over Pt(111) through comprehensive electrochemical investigations, including voltammetry and kinetic analyses.61 They found that AOR on Pt(100) begins with NH3 dehydrogenation, forming  intermediates within the Hupd region (0.05–0.4 V vs. RHE). These intermediates dimerize to produce hydrazine
intermediates within the Hupd region (0.05–0.4 V vs. RHE). These intermediates dimerize to produce hydrazine 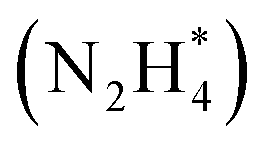 in the rate-determining step, followed by rapid N2 evolution characterized by a Tafel slope of 30 mV dec−1. The high activity of Pt(100) is attributed to its ability to stabilize
in the rate-determining step, followed by rapid N2 evolution characterized by a Tafel slope of 30 mV dec−1. The high activity of Pt(100) is attributed to its ability to stabilize  adsorbates. In contrast, AOR on Pt(111) occurs in the double-layer region (0.4–0.6 V vs. RHE), generating NH* and N* adsorbates through multi-electron transfer reactions with a Tafel slope of 40 mV dec−1. The strong adsorption of NH* and N* on Pt(111) leads to sluggish kinetics and lower AOR activity.
adsorbates. In contrast, AOR on Pt(111) occurs in the double-layer region (0.4–0.6 V vs. RHE), generating NH* and N* adsorbates through multi-electron transfer reactions with a Tafel slope of 40 mV dec−1. The strong adsorption of NH* and N* on Pt(111) leads to sluggish kinetics and lower AOR activity.
Notably, their discovery of  stabilization on Pt(100) and NH* stabilization on Pt(111) surfaces during electrochemical AOR mirrors the ammonia oxidation observed in gas-phase or ultra-high vacuum (UHV) conditions. Subsequently, it was observed that the relationship between the potential and AOR rate on the Pt(100) surface had a strong pH dependency.62 It was found that pH increases lead to a downshifting of the AOR onset potential. Furthermore, N2 evolution occurred on the Pt(100) through a coupling of two NH* adsorbates (Gerischer–Mauerer mechanism). These results were based on standard electrochemical methods combined with online electrochemical mass spectrometry (OLEMS), ion chromatography (IC), Fourier transform-infrared (FT-IR) spectroscopy, and theoretical studies based on DFT calculations.63
stabilization on Pt(100) and NH* stabilization on Pt(111) surfaces during electrochemical AOR mirrors the ammonia oxidation observed in gas-phase or ultra-high vacuum (UHV) conditions. Subsequently, it was observed that the relationship between the potential and AOR rate on the Pt(100) surface had a strong pH dependency.62 It was found that pH increases lead to a downshifting of the AOR onset potential. Furthermore, N2 evolution occurred on the Pt(100) through a coupling of two NH* adsorbates (Gerischer–Mauerer mechanism). These results were based on standard electrochemical methods combined with online electrochemical mass spectrometry (OLEMS), ion chromatography (IC), Fourier transform-infrared (FT-IR) spectroscopy, and theoretical studies based on DFT calculations.63
Model studies on catalytic surfaces have provided valuable guidance for designing nanostructured catalysts with enhanced AOR activity. The development of colloidal synthesis, enabling precise control over nanocrystal size and shape, has significantly advanced electrocatalyst design. Strong surfactants, including polyvinylpyrrolidone (PVP), cetyltrimethylammonium bromide (CTAB), cetyltrimethylammonium chloride (CTAC), and trioctylphosphine oxide (TOPO), allow thermodynamic control of nanocrystal size and morphology, facilitating the production of facet-controlled nanocrystals with highly reactive surfaces.64–66 However, these surfactants often bind tightly to the particles, passivating the surface and reducing catalytic activity. Chemical or electrochemical treatments are commonly used to remove surfactants, but these methods can lead to the formation of inactive oxide layers, metal dissolution, and structural deformation, ultimately degrading the catalytic properties.64–68 Consequently, developing sophisticated nanocrystals without relying on strong stabilizing surfactants has emerged as a critical challenge.
For example, Peng et al. addressed this issue by synthesizing carbon-supported Pt nanocubes (PtNCs/C) possessing a high ratio of (100) by annealing carbon black containing Pt(acac)2 at 200 °C for 1 hour under an H2/CO atmosphere.69 The prepared PtNCs/C exhibited 5.5 times higher AOR peak current density than a commercial Pt/C catalyst in 0.1 M NH3 + 1 M KOH electrolyte solution. In addition, it demonstrated higher stability than Pt/C in the stability test performed by CA at 0.6 V vs. RHE. In addition, Feliu et al. synthesized Pt nanocubes by reducing H2PtCl6 with sodium borohydride (NaBH4) in the water-in-oil microemulsion comprising water (3 vol%), polyethylene glycol dodecyl ether (16.5 vol%), and n-heptane (80.5 vol%).70 Depending on the concentration of HCl in the emulsion, the shape of the nanocrystals could be controlled to produce spherical NPs (0–10 wt%), cubic NPs (10–25 wt%), and concave cubic NPs (25–37 wt%) (Fig. 6a). Among the samples, the nanocrystals prepared with 25 wt% HCl exhibited the highest ratio of (100) (Fig. 6b) and nanocube shapes (Fig. 6c). Importantly, the highest AOR activity of these prepared nanocubes was 2 mA cm−2 in 2 M NaOH + 0.1 M NH3 (Fig. 6d).
 | ||
| Fig. 6 (a) Voltametric profile for Pt nanoparticles prepared by water-in-oil microemulsion in the presence of different amounts of HCl. (b) Amount of (100) sites measured by Ge irreversible adsorption (solid black squares) and by hydrogen adsorption/desorption deconvolution (open red circles) for different HCl% in the aqueous phase of a water-in-oil microemulsion. (c) TEM image of platinum nanoparticles prepared by water-in-oil microemulsion in the presence of 25% HCl in the aqueous phase. (d) Voltametric profiles for ammonia oxidation with Pt nanoparticles prepared by water-in-oil microemulsion in the presence of different amounts of HCl. The inset shows the current density of the main peak versus the amount of HCl. Reproduced from ref. 70 with permission. Copyright 2014 ACS. | ||
In addition, Zhong et al. synthesized Pt NPs (cubic Pt, prickly Pt, and spherical Pt) on the electrode surfaces using the electrodeposition method (Fig. 7a–c). The electrodeposition process was performed with a periodic square-wave potential in 5 mM H2PtCl6 + 0.5 M HCl solution. Briefly, applying −0.3 V (no square wave) formed cubic Pt particles (Fig. 7d).71 Prickly Pt and spherical Pt particles were synthesized by applying a potential from −0.3 to 1 V (square wave) and −0.1 to 1 V (square wave), respectively. The Pt(100) facet distribution measured by the Ge adsorption technique for cubic Pt was 47.8%, which was higher than those of the prickly Pt (26.9%) and spherical Pt (13.8%) NPs. The AOR activity of catalysts 1 M KOH + 0.1 M NH3 showed the following trend, which is commensurate with the ratio of Pt(100): cubic (1.24 mA cm−2) > prickly (0.70 mA cm−2) > spherical (0.34 mA cm−2).
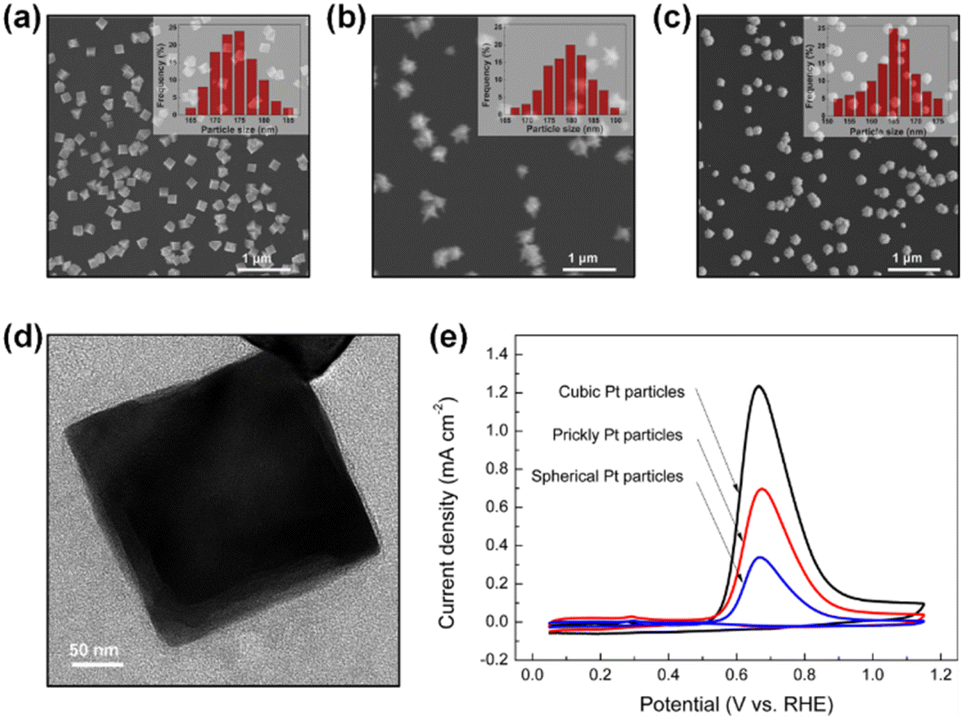 | ||
| Fig. 7 SEM images of the surface morphology of (a) cubic Pt, (b) prickly Pt, and (c) spherical Pt particles. The inset shows the corresponding particle size distribution. (d) TEM image of a cubic Pt particle. (e) AOR CVs measured on cubic, prickly, and spherical Pt particles in 1 M KOH + 0.1 M NH3 solution. Reproduced from ref. 71 with permission. Copyright 2017 ACS. | ||
Furthermore, a recent notable effort by Yoo and Kim et al. successfully addressed the surfactant issue using a wet-chemical process.72 They reported a direct, surfactant-free strategy for synthesizing Pt nanocubes by controlling the reaction kinetics with a hot-separation method (Fig. 8a and b). The synthesized Pt nanocubes were exploited as self-supported catalysts for AOR in a 0.5 M KOH + 0.5 M NH4OH solution and exhibited higher specific and mass activities than those of commercial Pt black catalysts by 13.5 and 9.6 times, respectively (Fig. 8c). Furthermore, a single DAFC device based on a Pt nanocube anode showed 2.3 times higher maximum power density than the Pt black-based one (Fig. 8d). This result indicates that the nanostructuring of catalytic materials has resulted in great improvements in AOR activity as well as practical applicability.
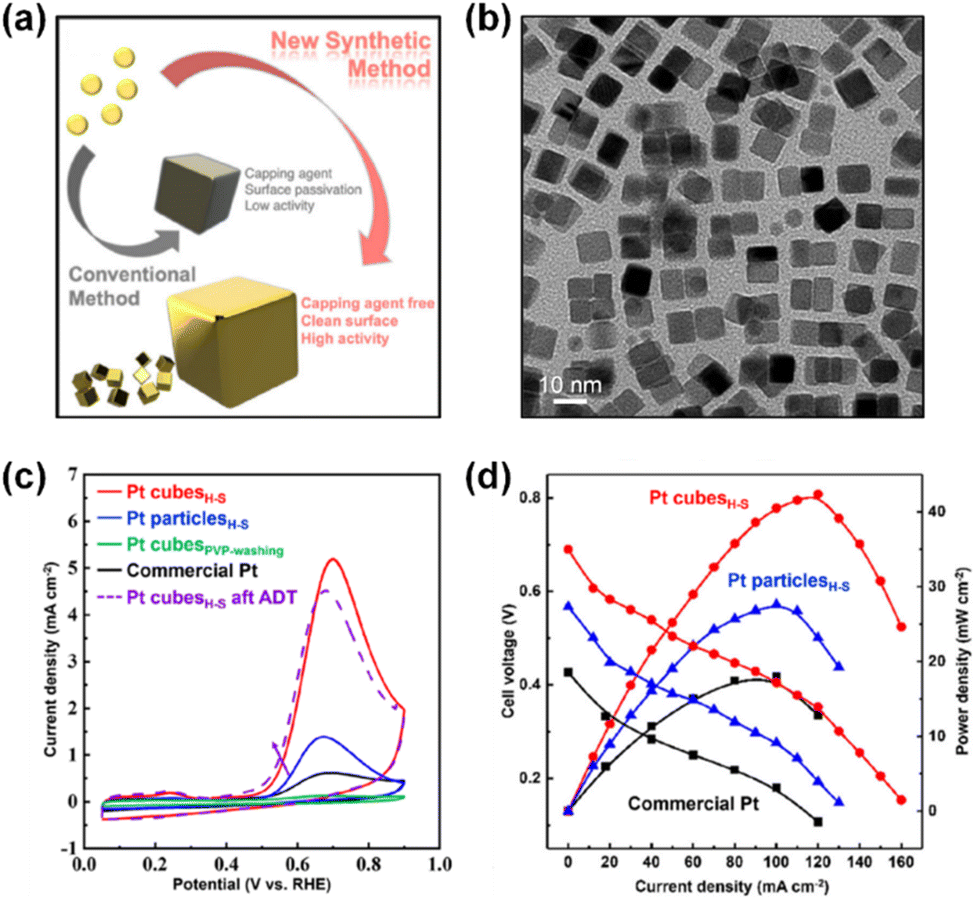 | ||
| Fig. 8 (a) Comparison of shape-controlled nanoparticles prepared from conventional and newly developed synthetic methods. (b) TEM image of as-prepared Pt cubesH–S. (c) CVs of Pt cubesH–S, Pt particlesH–S, Pt cubesPVP, and commercial Pt for the potential region of 0.05 to 0.9 V (vs. RHE) at the scan rate of 100 mV s−1 in a N2-saturated 0.5 M KOH + 0.5 M NH4OH. (d) DAFC polarization curves and power densities of Pt cubesH–S, Pt particlesH–S, and commercial Pt in a cell with 0.5 M NH4OH at the anode (flow rate of 1 cm3 min−1) and O2 at the cathode (flowrate of 400 cm3 min−1). The cell temperature was 50 °C. Reproduced from ref. 72 with permission. Copyright 2021 Wiley-VCH. | ||
3.2. Pt-based multimetallic AOR electrocatalysts
Introducing additional metal species into Pt catalysts can further improve catalytic activity by altering the electronic structure and fundamental physicochemical properties of Pt catalytic surfaces (Table 3). Here we introduce exemplars which optimized Pt catalytic surfaces through Pt surface decoration, and alloying strategies to reduce the use of Pt and modulate the electronic structures.| Catalysts | Morphology | Size (nm) | Synthesis method | Electrolyte | Onset potential | Mass activity (A mgPt−1) | Specific activity (mA cmECSA−2) | Stability | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| Pt-monometallic catalyst | Commercial Pt/C | — | — | — | 1 M KOH + 0.1 M NH3 | 0.496 V (vs. RHE) | 0.063 | — | ∼90% decrease after 2000 cycles of CV | 86 |
| Pt/C | — | — | Polyol reduction method | 0.1 M KOH + 0.1 M NH3 | — | 0.04 | 0.1 | ∼21.3% decrease after 500 cycles of CV | 83 | |
| Pt/C | Cubic | 7.98 | Colloidal method | 0.1 M KOH + 0.1 M NH3 | — | 0.253 | 0.937 | — | 84 | |
| Platinum particle | Cubic | 12–14 | Microemulsion | 0.2 M NaOH + 0.1 M NH3 | ∼0.5 V (vs. RHE) | — | — | — | 70 | |
| Pt particles | Cubic | 175 | Electrochemical method | 1 M KOH + 0.1 M NH3 | — | — | 1.24 | — | 71 | |
| Pt particles | Urchin-like | — | Electrochemical method | 1 M KOH + 0.1 M NH3 | — | — | 0.7 | — | 71 | |
| Pt particles | Spherical | — | Electrochemical method | 1 M KOH + 0.1 M NH3 | — | — | 0.34 | — | 71 | |
| Pt cubesH–S | Cubic | 7.8 | Wet-chemical | 0.5 M KOH + 0.5 M NH4OH | −0.38 V (vs. Hg/HgO) | 0.351 | ∼2.7 | 13.1% decrease after 500 cycles of CV | 72 | |
| Pt NCs/C | Cubic | 10.8 | Colloidal method | 0.1 M KOH + 0.1 M NH3 | — | 0.104 | 0.58 | ∼95% decreased @ 0.6 V after 100 s (vs. RHE) | 76 | |
| Pt-substrate modified | Pt/N–C 5 | Spherical | 3.5 | Alcohol reduction method | 1 M KOH + 0.5 M NH4OH | — | ∼0.28 | — | ∼78% decrease @ −0.35 V after 15 min (vs. Hg/HgO) | 49 |
| Pt/B-CNOuw | Spherical | 4.4 | Alcohol reduction method | 0.5 M NaOH + 1 M NH4OH | −0.384 V (vs. Ag/AgCl) | — | — | ∼83% decrease @ −0.3 V after 100 s (vs. Ag/AgCl) | 50 | |
| erGOx/Pt | Spherical | 2.5 | Electrodeposition | 1 M KOH + 1 M NH4OH | −0.3 V (vs. Ag/AgCl) | — | — | — | 47 | |
| Pt/MPC137 | Spherical | 30–50 | Colloidal method | 1 M NH4OH | — | — | — | ∼48% decrease @ 0.6 V after 15 s (vs. NHE) | 48 | |
| Pt/TiO2 | Flower-like | — | Electrodeposition | 1 M KOH + 0.1 M NH3 | — | — | ∼0.5 | ∼57% decrease @ −0.67 V after 100 s (vs. MSE) | 52 | |
| Pt/C-ITO | Spherical | 4.7 | Electrochemical reduction | 1 M KOH + 1 M NH4OH | 0.5 V (vs. Ag/AgCl) | — | — | ∼63% decrease @ −0.35 V after 1800 s (vs. Ag/AgCl) | 53 | |
| Pt/PBI/MWCNT-CeO2 | Spherical | 3.8 | Polyol method | 1 M KOH + 0.1 M NH3 | — | — | ∼0.52 | ∼76% decrease @ 0.7 V after 30 s (vs. RHE) | 73 | |
| CeO2-modified Pt/Si | — | — | Electroless deposition | 1 M KOH + 0.1 M NH3 | — | — | ∼0.175 | — | 75 | |
| Ir-decorated Pt–NCs/C | Cubic | 10.4 | Colloidal synthesis | 0.1 M KOH + 0.1 M NH3 | 0.43 V (vs. RHE) | 0.263 | 1.25 | ∼90% decreased @ 0.6 V after 100 s. (vs. RHE) | 76 | |
| Ni(OH)2–Ir decorated Pt NCs/C | Cubic | 10.4 | Colloidal synthesis | 0.1 M KOH + 0.1 M NH3 | — | 0.137 | 0.72 | ∼66% decreased @ 0.6 V after 100 s. (vs. RHE) | 76 | |
| Pt-alloy modified | PtEu/C | Spherical | 7.2 | Polyol method | 1 M KOH + 0.1 M NH3 | — | 0.1428 | ∼1.4 | ∼7.2% decrease after 500 cycles of CV | 83 |
| PtZn | Cubic | 9.05 | Colloidal method | 0.1 M KOH + 0.1 M NH3 | ∼0.45 V (vs. RHE) | 0.405 | 1.685 | — | 84 | |
| PtIrNi/SiO2-CNT-COOH | Irregular | 9.3 | Borohydride reduction | 1 M KOH + 0.1 M NH3 | 0.399 V (vs. RHE) | 0.013 | — | ∼48% decrease after 2000 cycles of CV | 86 | |
| Pt6Ru–NCs | Cubic | 9.9 | Hydrothermal reaction | 1 M KOH + 0.5 M NH4OH | 0.49 V (vs. RHE) | 0.192 | 1.02 | ∼49% decrease @ 0.67 V after 500 s (vs. RHE) | 87 | |
| Pt90Ir | — | — | Electrodeposition | 1 M KOH + 0.1 M NH3 | −0.54 V (vs. Hg/HgO) | — | ∼0.35 | ∼90% decrease @ −0.25 V after 100 s (vs. Hg/HgO) | 77 | |
| Pt/Ir/MWCNT | Irregular | 4.3 | Improved Cu short circuit deposition | 0.1 M KOH + 0.1 M NH3 | 0.38 V (vs. RHE) | — | ∼0.23 | ∼57% decrease @ 0.45 V–0.6 V potential step (vs. RHE) | 78 | |
| PtIr | — | — | Electrodeposition | 0.5 M KOH + 0.1 M NH4OH | 0.41 V (vs. RHE) | — | — | — | 79 | |
| Pt9Rh | Spherical | 4.1 | Borohydride reduction | 1 M KOH + 1 M NH4OH | ∼−0.54 V (vs. Hg/HgO) | ∼0.09 | — | ∼62% decrease @ −0.3 V after 20 min (vs. Hg/HgO) | 80 | |
| (100)Pt85Pd15/rGO | Cubic | 5.5 | Wet chemical method | 1 M KOH + 0.1 M NH3 | 0.467 V (vs. RHE) | 0.165 | — | ∼92% decrease @ 0.65 V after 100 s (vs. RHE) | 81 | |
| Pt7Au3 | Spherical | 5.8 | Borohydride reduction | 1 M KOH + 1 M NH4OH | — | 0.09 | — | ∼39% decrease @ −0.3 V after 100 s (vs. Hg/HgO) | 82 | |
 isomerization into
isomerization into 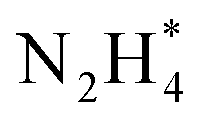 on the Pt surfaces using Y2O3, which suppressed the formation of N* monomers and the poisoning of catalysts.74 Briefly, an ionomer solution containing Y2O3 was dropped onto the surface of a Pt-deposited Si prism (Pt/Si). As shown in Fig. 9a, the CVs of bare Pt (ionomer-coated Pt/Si) and the Y2O3-modified Pt (Y2O3-modified Pt/Si) were nearly identical, indicating that Y2O3 decoration did not change the inherent adsorption properties of H* or OH* on the Pt surfaces. In terms of AOR activity, remarkably, the Y2O3-modified Pt/Si exhibited approximately 1.7 times higher specific activity than the ionomer-coated Pt/Si (Fig. 9b). To reveal the origin of the enhanced AOR activity, they measured changes in the band area of NH3, NO, and N2H4 species according to the applied potential using electrochemical in situ attenuated total reflection-IR (ATR-IR) spectroscopy (Fig. 9c). Compared with ionomer-coated Pt/Si, the Y2O3-modified Pt/Si exhibited a selective increase in the quantity of N2H4 intermediates (Fig. 9d and e). These findings suggest that the Y2O3-modified Pt surface promptly formed N2H4 and mitigated the poisoning of the catalytic surfaces, increasing AOR activity.
on the Pt surfaces using Y2O3, which suppressed the formation of N* monomers and the poisoning of catalysts.74 Briefly, an ionomer solution containing Y2O3 was dropped onto the surface of a Pt-deposited Si prism (Pt/Si). As shown in Fig. 9a, the CVs of bare Pt (ionomer-coated Pt/Si) and the Y2O3-modified Pt (Y2O3-modified Pt/Si) were nearly identical, indicating that Y2O3 decoration did not change the inherent adsorption properties of H* or OH* on the Pt surfaces. In terms of AOR activity, remarkably, the Y2O3-modified Pt/Si exhibited approximately 1.7 times higher specific activity than the ionomer-coated Pt/Si (Fig. 9b). To reveal the origin of the enhanced AOR activity, they measured changes in the band area of NH3, NO, and N2H4 species according to the applied potential using electrochemical in situ attenuated total reflection-IR (ATR-IR) spectroscopy (Fig. 9c). Compared with ionomer-coated Pt/Si, the Y2O3-modified Pt/Si exhibited a selective increase in the quantity of N2H4 intermediates (Fig. 9d and e). These findings suggest that the Y2O3-modified Pt surface promptly formed N2H4 and mitigated the poisoning of the catalytic surfaces, increasing AOR activity.
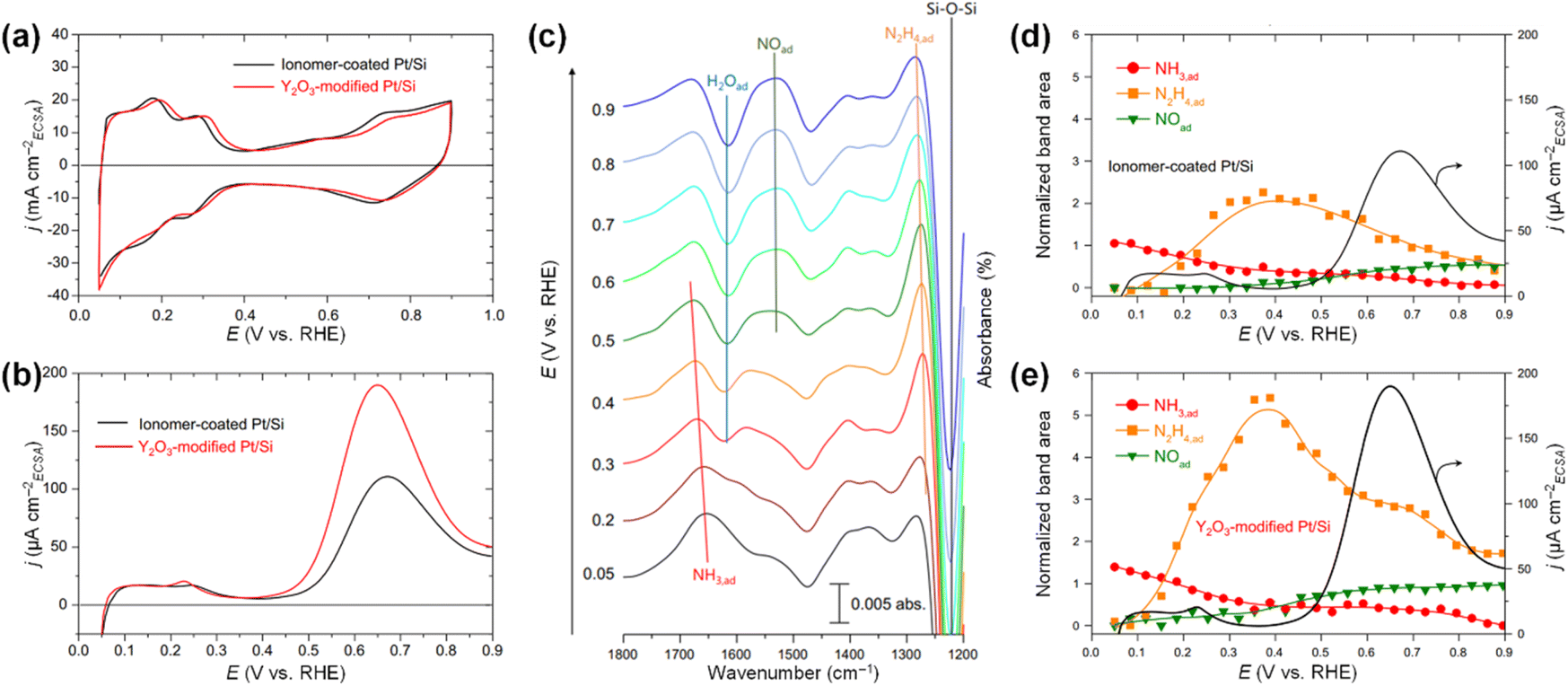 | ||
Fig. 9 CVs of ionomer-coated and Y2O3-modified Pt/Si electrodes in (a) 1 M KOH and (b) 1 M KOH + 0.1 M NH3. (c) Time-resolved IR spectra of the Y2O3-modified Pt surface acquired simultaneously with the linear sweep voltammogram in 1 M KOH + 0.1 M NH3. Potential dependence of the current and the normalized band area of  , , 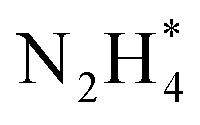 , and NO* of (d) ionomer-coated and (e) Y2O3-modified Pt/Si electrodes. Reproduced from ref. 73 with permission. Copyright 2016 Elsevier. , and NO* of (d) ionomer-coated and (e) Y2O3-modified Pt/Si electrodes. Reproduced from ref. 73 with permission. Copyright 2016 Elsevier. | ||
At the same time, Pt decoration can modulate the ability of the catalytic surfaces to adsorb OH*, enhancing AOR activity.75 As suggested by the Gerischer–Mauerer mechanism, OH* can promote the dehydrogenation of  into
into 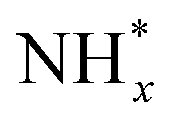 . Using a similar approach, the same research group also demonstrated that decorating Pt with strongly oxophilic CeO2 can improve AOR activity by promoting OH* supply onto the catalytic surfaces. While the CVs of Pt catalysts decorated with CeO2 (CeO2-modified Pt/Si) and without CeO2 (ionomer-coated Pt/Si) were virtually identical, the CeO2-modified Pt/Si exhibited enhanced activity compared to the ionomer-coated Pt/Si by 30%. Analysing the changes in Pt–OH, NH3, NO, and N2H4 in relation to the potential increase using ATR-IR, CeO2-modified Pt showed greater increases in the quantities of NO, N2H4, and Pt–OH species than those of ionomer-coated Pt/Si.
. Using a similar approach, the same research group also demonstrated that decorating Pt with strongly oxophilic CeO2 can improve AOR activity by promoting OH* supply onto the catalytic surfaces. While the CVs of Pt catalysts decorated with CeO2 (CeO2-modified Pt/Si) and without CeO2 (ionomer-coated Pt/Si) were virtually identical, the CeO2-modified Pt/Si exhibited enhanced activity compared to the ionomer-coated Pt/Si by 30%. Analysing the changes in Pt–OH, NH3, NO, and N2H4 in relation to the potential increase using ATR-IR, CeO2-modified Pt showed greater increases in the quantities of NO, N2H4, and Pt–OH species than those of ionomer-coated Pt/Si.
Furthermore, the surface decoration strategy can facilitate the adsorption of 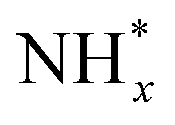 of catalytic surfaces. Choi et al. demonstrated that a trace amount of Ir can improve the AOR activity of Pt nanocubes (Pt NCs) by enhancing the adsorption of
of catalytic surfaces. Choi et al. demonstrated that a trace amount of Ir can improve the AOR activity of Pt nanocubes (Pt NCs) by enhancing the adsorption of  and lowering the energy barrier of the rate-determining step.76 They first prepared Ir-decorated Pt NCs (Fig. 10a and b) and Ir and Ni(OH)2-decorated Pt NCs (Fig. 10c and d). The three prepared Pt NCs samples exhibited nearly identical peak positions in the XRD patterns (Fig. 10e), indicating that the wet-chemical processes did not form alloy phases. The AOR activity test (Fig. 10f) demonstrated activity was enhanced by the Ir or Ir and Ni(OH)2-decoration, and showed the Ir-decorated Pt NCs/C (1.25 mA cmPt−2), Ir and Ni(OH)2-decorated Pt NCs/C (0.72 mA cmPt−2) had higher specific activities than that of Pt NCs/C (0.58 mA cmPt−2). Decorating Ir, or Ir and Ni(OH)2 can also improve AOR stability, evidenced by much slower rates in current decay of Ir-decorated Pt NCs/C and Ir and Ni(OH)2-decorated Pt NCs/C than that of the Pt NCs in the CA measurement (Fig. 10g).
and lowering the energy barrier of the rate-determining step.76 They first prepared Ir-decorated Pt NCs (Fig. 10a and b) and Ir and Ni(OH)2-decorated Pt NCs (Fig. 10c and d). The three prepared Pt NCs samples exhibited nearly identical peak positions in the XRD patterns (Fig. 10e), indicating that the wet-chemical processes did not form alloy phases. The AOR activity test (Fig. 10f) demonstrated activity was enhanced by the Ir or Ir and Ni(OH)2-decoration, and showed the Ir-decorated Pt NCs/C (1.25 mA cmPt−2), Ir and Ni(OH)2-decorated Pt NCs/C (0.72 mA cmPt−2) had higher specific activities than that of Pt NCs/C (0.58 mA cmPt−2). Decorating Ir, or Ir and Ni(OH)2 can also improve AOR stability, evidenced by much slower rates in current decay of Ir-decorated Pt NCs/C and Ir and Ni(OH)2-decorated Pt NCs/C than that of the Pt NCs in the CA measurement (Fig. 10g).
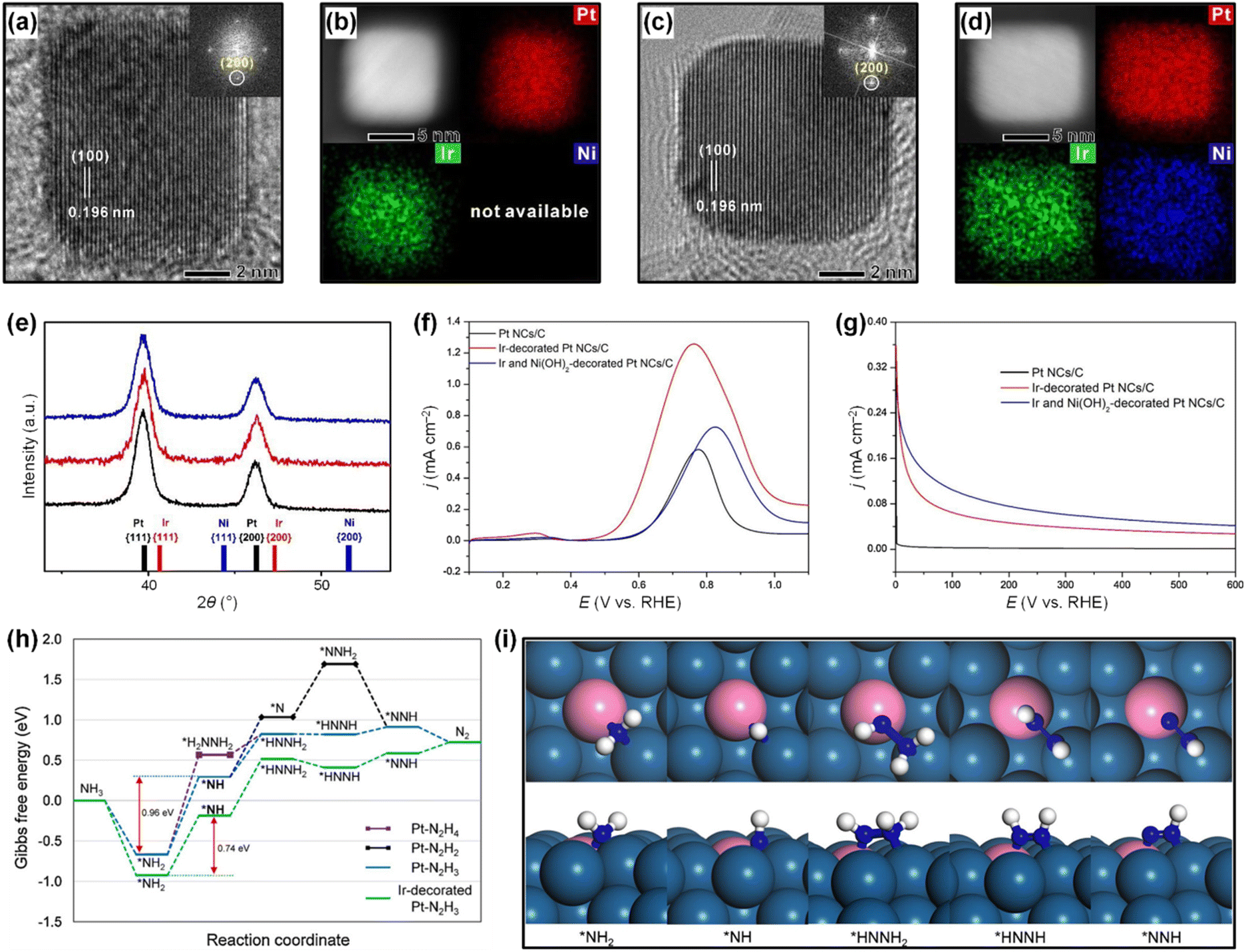 | ||
| Fig. 10 High-resolution TEM images: the insets are SAED patterns of (a) an Ir-decorated Pt NC, and (c) an Ir and Ni(OH)2-decorated Pt NC. HAADF-STEM images and corresponding EDS mapping images of (b) an Ir-decorated Pt NC and (d) an Ir and Ni(OH)2-decorated Pt NC. (e) XRD patterns of Pt NCs/C, Ir-decorated Pt NCs/C, and Ir and Ni(OH)2-decorated Pt NCs/C. Reference peaks for each element are denoted as bar graphs. (f) LSVs recorded on Pt NCs/C, Ir-decorated Pt NCs/C and Ir and Ni(OH)2-decorated Pt NCs/C, in 0.1 M KOH + 0.1 M NH3 solution. (g) CA measurements at 0.60 V (vs. RHE) for Pt NCs/C, Ir decorated Pt NCs/C, and Ir and Ni(OH)2-decorated Pt NCs/C in 0.1 M KOH + 0.1 M NH3 solution. (h) Gibbs free energy curves for AOR pathways on Pt(100) and Ir-decorated Pt(100). (i) Optimized structures (top and side views) of reaction intermediates during AOR on Ir-decorated Pt(100) following the N2H3 pathway. Color code: Pt, dark blue; Ir, pink; N, blue; H, white. Reproduced from ref. 76 with permission. Copyright 2020 Elsevier. | ||
The enhanced AOR activity of Ir-decorated Pt NCs/C were rationalized with DFT calculations. The calculated binding energy of 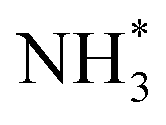 on the Ir-decorated Pt(100) surface (−1.68 eV) was stronger than that of the bare Pt(100) surface (−1.44 eV), indicating the promoted AOR kinetics on the catalytic surfaces with stronger adsorption of NHx. The Gibbs free energy diagrams of reaction pathways on both surfaces (Fig. 10h) also revealed that the energy barrier for the
on the Ir-decorated Pt(100) surface (−1.68 eV) was stronger than that of the bare Pt(100) surface (−1.44 eV), indicating the promoted AOR kinetics on the catalytic surfaces with stronger adsorption of NHx. The Gibbs free energy diagrams of reaction pathways on both surfaces (Fig. 10h) also revealed that the energy barrier for the  -to-NH* step was reduced on the Ir-decorated Pt(100). The optimized structures of AOR intermediates on Ir-decorated Pt(100) are summarized in Fig. 10i.
-to-NH* step was reduced on the Ir-decorated Pt(100). The optimized structures of AOR intermediates on Ir-decorated Pt(100) are summarized in Fig. 10i.
 | ||
| Fig. 11 (a) HRTEM image of PtEu/C catalyst. (b) LSVs of as-prepared catalysts in 1 M KOH + 0.1 M NH3. CVs in 1 M KOH + 0.1 M NH3 for 500 cycles of (c) Pt/C and (d) PtEu/C catalysts. Reproduced from ref. 83 with permission. Copyright 2017 Elsevier. | ||
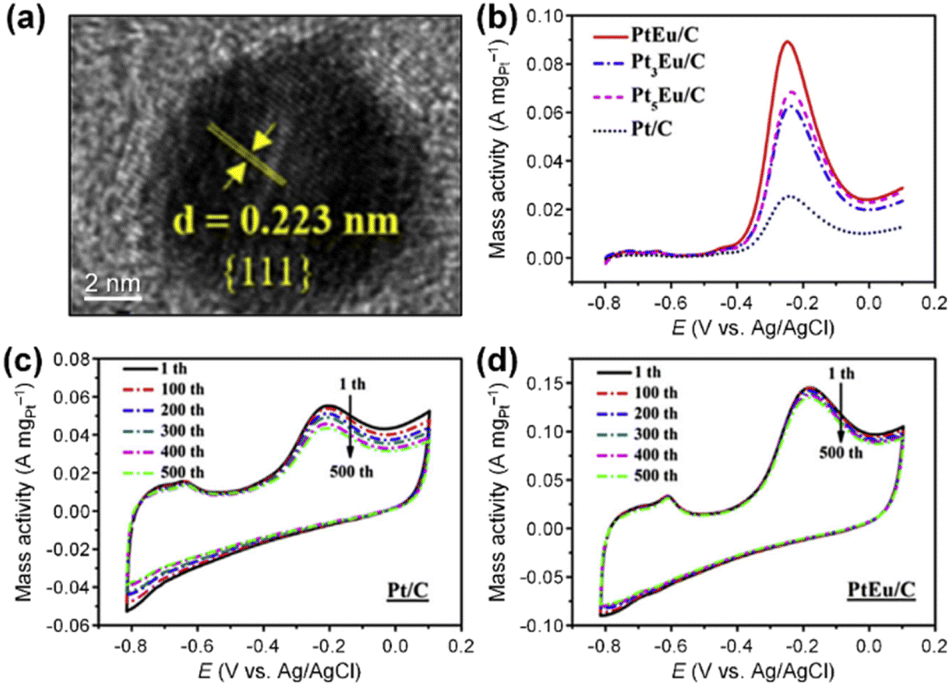
More importantly, Pt–M alloys with earth-abundant transition metal elements have demonstrated notable achievements as highly active AOR catalysts. For example, Shao et al. investigated the electrocatalytic properties of Pt–M alloy NCs with various compositions (M = Fe, Co, Ni, Zn).84 As shown in Fig. 12a, among the Pt–M alloys, Pt–Zn NCs exhibited the highest AOR mass activity (0.405 A mgPt−1), followed by Pt–Fe (0.352 A mgPt−1), Pt–Co (0.349 A mgPt−1), Pt–Ni (0.270 A mgPt−1) and Pt NCs (0.253 A mgPt−1). These enhanced activities of Pt–M NCs is related to the reduced oxophilicity of catalytic surfaces that occurs when introducing M elements. Alloying Pt with less electronegative M can induce a partial charge transfer from M to Pt, decreasing vacancies in the Pt electronic structure. Subsequently, the oxygen in OH* loses its ability to transfer 2π electrons to Pt, decreasing the adsorption of OH*. Because the OH* and 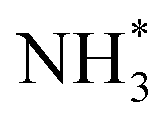 are competitively adsorbed on the Pt surfaces during the AOR process, diminishing the adsorption of OH* can promote
are competitively adsorbed on the Pt surfaces during the AOR process, diminishing the adsorption of OH* can promote 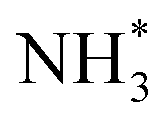 adsorption and increase AOR activity. We note that the AOR activity of the Pt electrode in a non-aqueous environment (without OH−) can be higher than that measured in an aqueous solution containing OH−, as reported by Peng and coworkers.85 Accordingly, the CVs of catalysts obtained in a 0.1 M KOH solution indicated the ability to adsorb OH* was diminished on the Pt–M nanocubes as indicated by OH− adsorption peaks whose peak potentials were positively shifted compared to the Pt NCs (Fig. 12b). Pt 4f XPS analysis also confirmed a negative shift of 0.1 eV in the binding energy of the Pt–Zn NCs compared to Pt NCs, implying a partial charge transfer from Zn to Pt (Fig. 12c).
adsorption and increase AOR activity. We note that the AOR activity of the Pt electrode in a non-aqueous environment (without OH−) can be higher than that measured in an aqueous solution containing OH−, as reported by Peng and coworkers.85 Accordingly, the CVs of catalysts obtained in a 0.1 M KOH solution indicated the ability to adsorb OH* was diminished on the Pt–M nanocubes as indicated by OH− adsorption peaks whose peak potentials were positively shifted compared to the Pt NCs (Fig. 12b). Pt 4f XPS analysis also confirmed a negative shift of 0.1 eV in the binding energy of the Pt–Zn NCs compared to Pt NCs, implying a partial charge transfer from Zn to Pt (Fig. 12c).
 | ||
| Fig. 12 (a) AOR CVs of Pt-based nanocubes in 0.1 M KOH + 0.1 M NH3. (b) Voltage profiles of Pt-based nanocubes in 0.1 M KOH. (c) Pt 4f XPS spectra of Pt and Pt–Zn nanocubes. Reproduced from ref. 84 with permission. Copyright 2020 Tsinghua University Press and Springer-Verlag GmbH Germany, part of Springer Nature. | ||
Wu et al. introduced ternary PtIrNi alloy catalysts supported on CNT + SiO2 composites and compared them with Pt and PtIr analogues (Fig. 13a and b).86 The ternary catalyst was synthesized using an ultrasonication-assisted sodium borohydride reduction method, where acoustic cavitation accelerates the reduction process. This facilitated the formation of small, uniform nanoparticles on the supports and the successful synthesis of a ternary PtIrNi alloy. In AOR tests (Fig. 13c), PtIrNi/SiO2-CNT-COOH exhibited an onset potential at 0.399 V, which is much lower than those of Pt/SiO2-CNT-COOH (0.496 V) and PtIr/SiO2-CNT-COOH (0.446 V). As the same support materials were employed, the enhanced AOR activities of the alloy catalysts can be attributed to the alloy compositions. The change in the calculated Gibbs free energy at 0.3 V on the Pt(100), Pt3Ir(100), and PtIrNi(100) facets for respective AOR pathways suggest the origin of the activity (Fig. 13d). On Pt(100) facets, the transition from 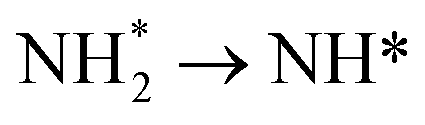 in the AOR pathway is non-spontaneous. In contrast, on the (100) facets of the alloy catalysts, all of the transitions were predicted to be spontaneous, in agreement with the experimental results, which showed lower onset potentials. Furthermore, in the calculation of density of states (DOS), compared with the Pt(100) facet (−2.25 eV), PtIr(100) and PtNiIr(100) exhibited upshifts in the Pt d-band centers by 0.28 eV (−1.97 eV) and 0.38 eV (−1.87 eV), respectively (Fig. 13e).
in the AOR pathway is non-spontaneous. In contrast, on the (100) facets of the alloy catalysts, all of the transitions were predicted to be spontaneous, in agreement with the experimental results, which showed lower onset potentials. Furthermore, in the calculation of density of states (DOS), compared with the Pt(100) facet (−2.25 eV), PtIr(100) and PtNiIr(100) exhibited upshifts in the Pt d-band centers by 0.28 eV (−1.97 eV) and 0.38 eV (−1.87 eV), respectively (Fig. 13e).
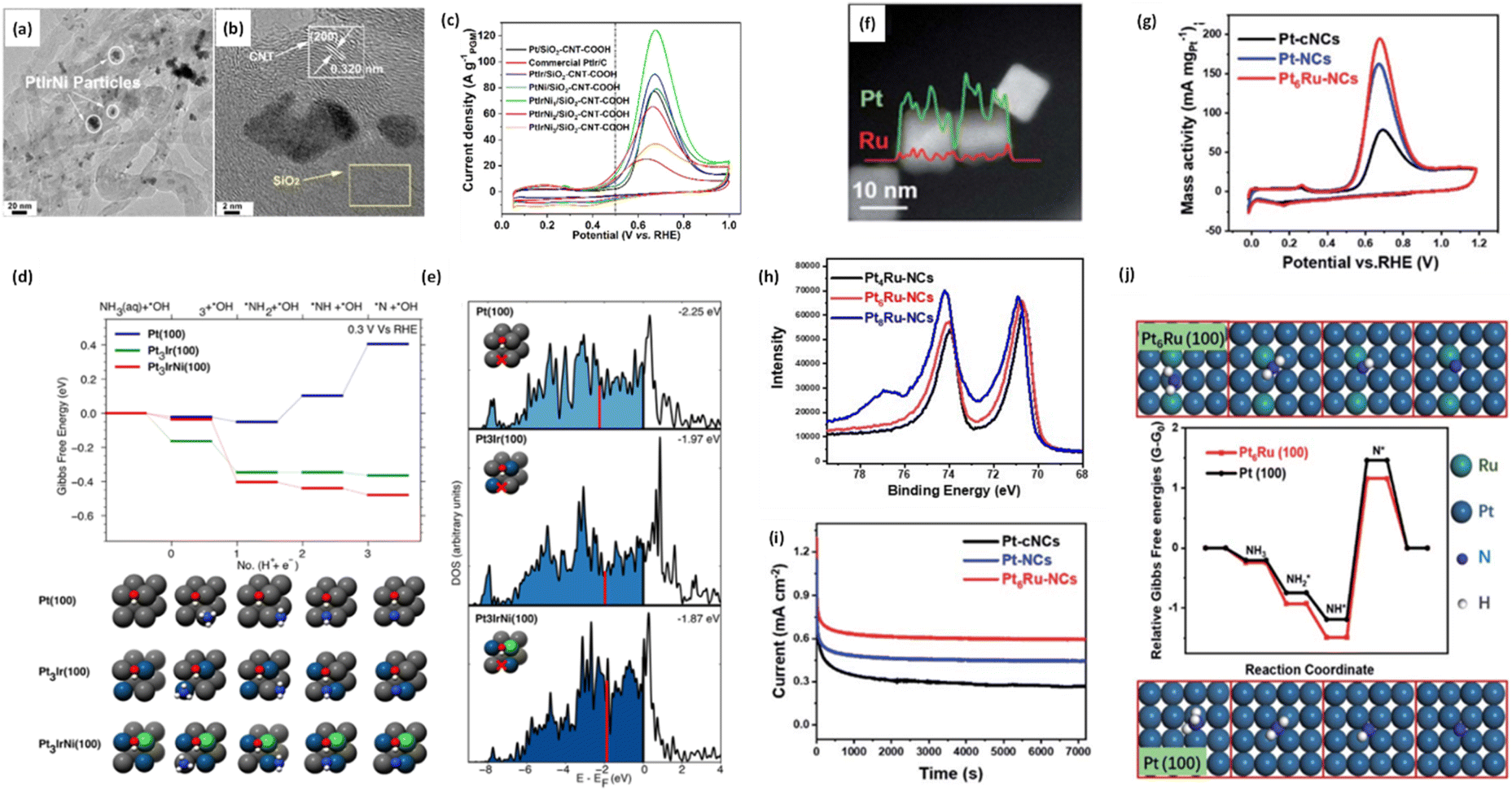 | ||
| Fig. 13 (a and b) TEM images of the PtIrNi/SiO2-CNT-COOH, (c) CV curves of the as-prepared catalysts in 1 M KOH + 0.1 M NH3 solution, (d) DFT calculations of the initial ammonia dehydrogenation steps at 0.3 V on Pt(100), Pt3Ir(100), Pt3IrNi(100). All surfaces have co-adsorbed *OH (1/4 ML). The geometries for each surface are shown. (e) Group d-orbital density of states (DOS) projected onto the bridge site of model systems (marked via red cross). The vertical red solid lines indicate a d-band center. Reproduced from ref. 86 with permission. Copyright 2020 ACS. (f) STEM image, EDS line scan spectrum and the corresponding elemental mapping of Pt6Ru–NCs, (g) mass activity of the AOR of Pt-cNCs, Pt–NCs, Pt6Ru–NCs in 1 M KOH + 0.1 M NH3 electrolyte, (h) Pt 4f XPS spectra of Pt4Ru–NCs, Pt6Ru–NCs and Pt8Ru–NCs, (i) stability tests of Pt-cNCs, Pt–NCs, Pt6Ru–NCs at 0.67 V, (j) Gibbs free energy charges for the adsorption of AOR intermediates on Pt(100) and Pt6Ru(100) planes. Reproduced from ref. 87 with permission. Copyright 2021 RSC. | ||
The upshifts in the d-band centers result in fewer antibonding states for Pt, leading to stronger NH* adsorption and increased AOR activity. Chen et al. synthesized PtRu alloy-based NCs (Fig. 13f) and showed the onset potentials of 0.5 V, 0.51 V, and 0.55 V for PtRu–NCs, Pt–NCs, and Pt-cNCs (c: crystalline), respectively (Fig. 13g).87 The mass activities presented the following trend: PtRu–NCs (192 mA mgPt−1) > Pt–NCs (165 mA mgPt−1) > Pt-cNCs (75 mA mgPt−1). The incorporation of Ru supplied OH to Pt and regulated the adsorption of NH3 and intermediates, facilitating NH3 dehydrogenation and enhancing AOR activity. Additionally, as the content of Ru increased, the Pt 4f binding energy shifted negatively (Fig. 13h), allowing stronger π-Back donation bonds to reactive species due to electron-rich Pt. Stability tests showed that Pt6Ru–NCs were the most stable, followed by Pt–NCs and Pt–cNCs (Fig. 13i). DFT calculations also indicated that Pt6Ru possesses stronger adsorption strength for the AOR intermediates compared to the pure Pt, resulting in increased activity and stability, as it exhibits weaker adsorption strength for poisoning N* (Fig. 13j).
3.3. Non-PGM-based AOR electrocatalysts
Although the Pt-based catalysts boasted low overpotential and high N2 selectivity in AOR catalysis, their widespread application is impeded by the cost and rapid deactivation during the process.88 In response to these critical constraints, significant endeavours have been made to design effective non-PGM-based AOR catalysts that incorporate earth-abundant transition metal elements (Table 4). To optimize the efficiency of non-PGM catalysts, it is imperative to account for the binding strength between the catalytic surfaces and intermediate species, akin to the approach employed with precious metal catalyst development.40| Catalysts | Electrolyte | Onset potential (VRHE) | Stability | Peak current density (mA cm−2) | Ref. | |
|---|---|---|---|---|---|---|
| Ni modified catalysts | NiCu DHTs | 0.1 M NaOH + 0.05 M NH4OH | 1.31 | 0.5 → 0.4 mA cm−2 (−20%) @ 1.5 V (vs. RHE) after 1 h | 11 | 100 |
| NiCu/CP | 0.5 M NaOH + 0.055 M NH4Cl | 1.47 | 8.5 → 8.2 mA cm−2 (−3.5%) @ 0.55V(vs. Ag/AgCl) after 2 h | 50 | 103 | |
| Ni1Cu3–S-T/CP | 1 M NaOH + 0.2 M NH4Cl | 1.69 | 90 → 84 mA cm−2 (−6.6%) @ 1.64 V (vs. RHE) after 24 h | 130 | 101 | |
| NiCuFe alloy | 0.5 M KOH + 0.055 M NH4Cl | 0.62 | 10 mA cm−2 @ 0.5 V (vs. Ag/AgCl) for 9 h | 58 | 107 | |
| NiCuFe oxyhydroxide | 0.5 M KOH + 0.055 M NH4Cl | 1.43 | 10 mA cm−2 at 0.55 V(vs. SCE) for 12 h | 52 | 108 | |
| NiCo | 0.5 M KOH +0.33 mM NH3 | 0.83 | — | 9 | 109 | |
| Ni4Cu1Co1.5-BP | 1 M KOH + 0.1 M NH3 | 0.3 | 6 → 5 mA cm−2 (−16.6%) @ 0.65 V (vs. Hg/HgO) after 5 h | 104.08 | 105 | |
| Ni(OH)2–Cu2O@CuO | 1 M KOH + 1 M NH3 | 1.37 | 78 → 75 mA cm−2 (−3.8%) @ 0.6 V (vs. Hg/HgO) after 32 h | 60 | 99 | |
| NiCuCo–S-T/CP | 1 M NaOH + 0.2 M NH4Cl | 1.24 | 105 mA cm−2 @ 1.64 V (vs. RHE) for 12 h | 139 | 105 | |
| Ag/Ni | 1.5 M NaOH + 0.5 M NH3 | 1.37 | 10 mA cm−2 @ 0.55 V (vs. Hg/HgO) for 350 h | 45 | 112 | |
| Ni–Sn | 0.5 M K2SO4 + 10 mM NH3 | 1.51 | — | 4 | 110 | |
| Cu-modified catalysts | CuCo/NF | 10 mM Na2SO4 + 0.05 M NH3 | — | — | 9 | 115 |
| CuSn(OH)6 | 0.5 M K2SO4 + 10 mM NH3 | 1.56 | — | 3.4 | 116 | |
| CuO | 1 M KOH + 1 M NH3 | 1.19 | 200 mA cm−2 @ 0.6 V (vs. Hg/HgO) for 400 h | 120 | 114 | |
| Ag3CuS2 | 0.5 M NaOH + 0.1 M NH4Cl | 1.37 | 10 mA cm−2 @ 1.5 V (vs. RHE) for 10 h | 100 | 118 | |
| CuCo/CC | 0.5 M K2SO4 + 10 mM NH3 | 1.12 | — | 8.7 | 113 | |
| Cu9Cr | 0.1 M KOH + 0.1 M NH4OH | 1.1 | — | 22 | 117 | |
| Other metal modified catalysts | TiO | 0.5 M NaOH + 0.1 M NH3 | 0.4 | 0.02 → 0.01 mA cm−2 (−50%) @ 1.1 V (vs. RHE) after 2 h | 0.02 | 119 |
| (Mn, Fe, Co, Ni, Cu)3O4 | 0.1 M KPF6 + NH3 saturated | 0.8 | 5 → 4.8 mA cm−2 (−0.04%) @ 1.0 V (vs. NHE) after 1 h | — | 96 | |
| SrCo0.8Cu0.1Nb0.1O3−δ | 1 M NaOH + 1 M NH3 | 0.35 | — | 0.9 | 170 | |
In this context, employing Ni-based multimetallic compositions have been considered an effective strategy to improve N2 selectivity as well as AOR activity.96 In particular, the synergistic effect between Ni and Cu has been widely employed to promote AOR.97–104 For example, Tao et al. prepared NiCu catalysts on a carbon paper (CP) electrode using the electrodeposition method.103 As shown in a scanning electron microscopy (SEM) image, the prepared catalysts of Ni and Cu particles with a size of around 100 nm exhibited uniform dispersion on CP (Fig. 14a). The XPS analyses of the NiCu catalysts confirmed the formation of NiOOH and Cu(OH)2 during electrochemical activation (Fig. 14b and c). In AOR tests in 0.5 M NaOH, Ni/CP showed no significant changes in the CV curves regardless of the presence of NH4Cl (Fig. 14d). In contrast, under the same condition, NiCu/CP exhibited a dramatic rise in current density at an onset potential of approximately 0.47 V vs. Ag/AgCl after NH4Cl addition, indicating the synergy between Ni and Cu (Fig. 14e). A CA stability test at 0.55 V vs. Ag/AgCl also suggested the beneficial effect of Cu incorporation, where the NiCu/CP exhibited a much higher stabilized AOR current density of (8.5 mA cm−2) than that of Ni/CP (0.8 mA cm−2). Importantly, in the ammonia electrolysis cell (AEC) test based on the two-electrode cell configuration, the NiCu/CP anode exhibited excellent AOR current density, which was two-fold higher than that of the Pt/C anode (Fig. 14f).
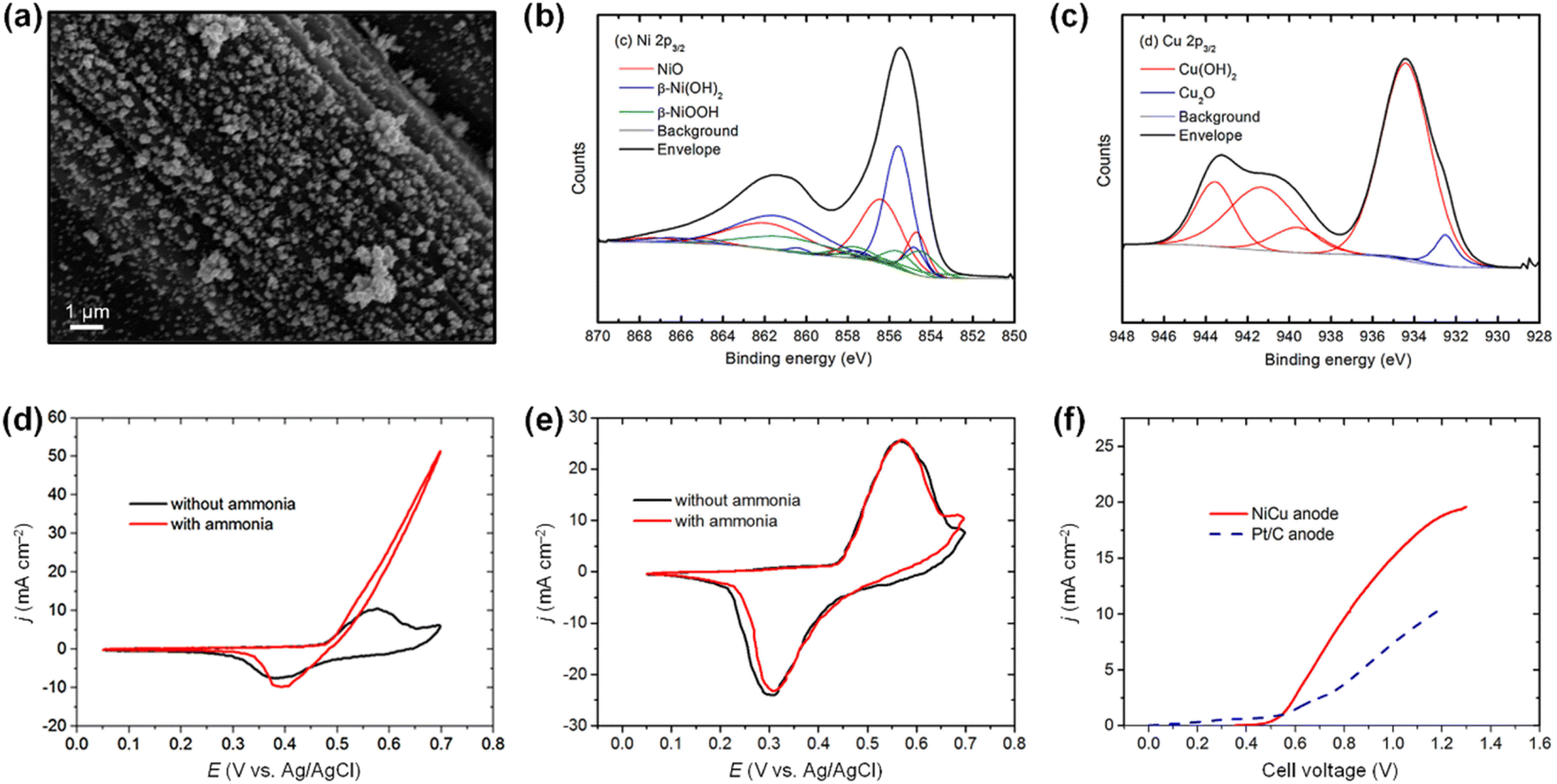 | ||
| Fig. 14 (a) SEM images of the NiCu/CP electrode. (b) and (c) XPS spectra of NiCu/CP after electrochemical reaction. (d) CV plots of NiCu/CP electrode and (e) Ni/CP in 0.5 M NaOH with and without 55 mM NH4Cl. (f) The LSV data of NiCu/CP anode and Pt/C anode for AEC reproduced from ref. 103 with permission. Copyright 2018 Elsevier. | ||
Beyond the binary NiCu systems, incorporating third metal elements could improve the AOR activity of NiCu catalysts.105–108 For example, Ming et al. developed Co-doped Ni4Cu1 (Ni4Cu1Cox-BP; x = 0, 0.5, 1.0, 1.5, and 2.0) catalysts using a hydrothermal method.106 Interestingly, the increase in Co doping led to a continuous decrease in the AOR onset potential of the catalysts, implying AOR kinetics were improved by Co incorporation. When Ni4Cu1-BP catalysts were compared, the optimized Ni4Cu1Co1.5-BP exhibited notable improvements in AOR activity (Fig. 15a), activation energy reduction (Fig. 15b), and enhanced stability (Fig. 15c), showcasing the favourable outcomes of Co doping. The DFT calculations revealed that Co doping can elevate the electron density around O, facilitating the adsorption of intermediates (Fig. 15d). This contributed to the reduction in the Gibbs free energy associated with AOR, resulting in enhanced AOR performance. As shown in Fig. 15e, the DAFC fabricated using a Ni4Cu1Co1.5-BP anode and Mn-Co-BP2000 cathode achieved an OCV of 0.75 V and a notable power density of 115.7 mW cm−2. This performance stands among the foremost achievements of DAFCs utilizing Pt-free electrodes, underscoring the competitiveness of Ni4Cu1Co1.5-BP as an anode catalyst, comparable to Pt-based catalysts.
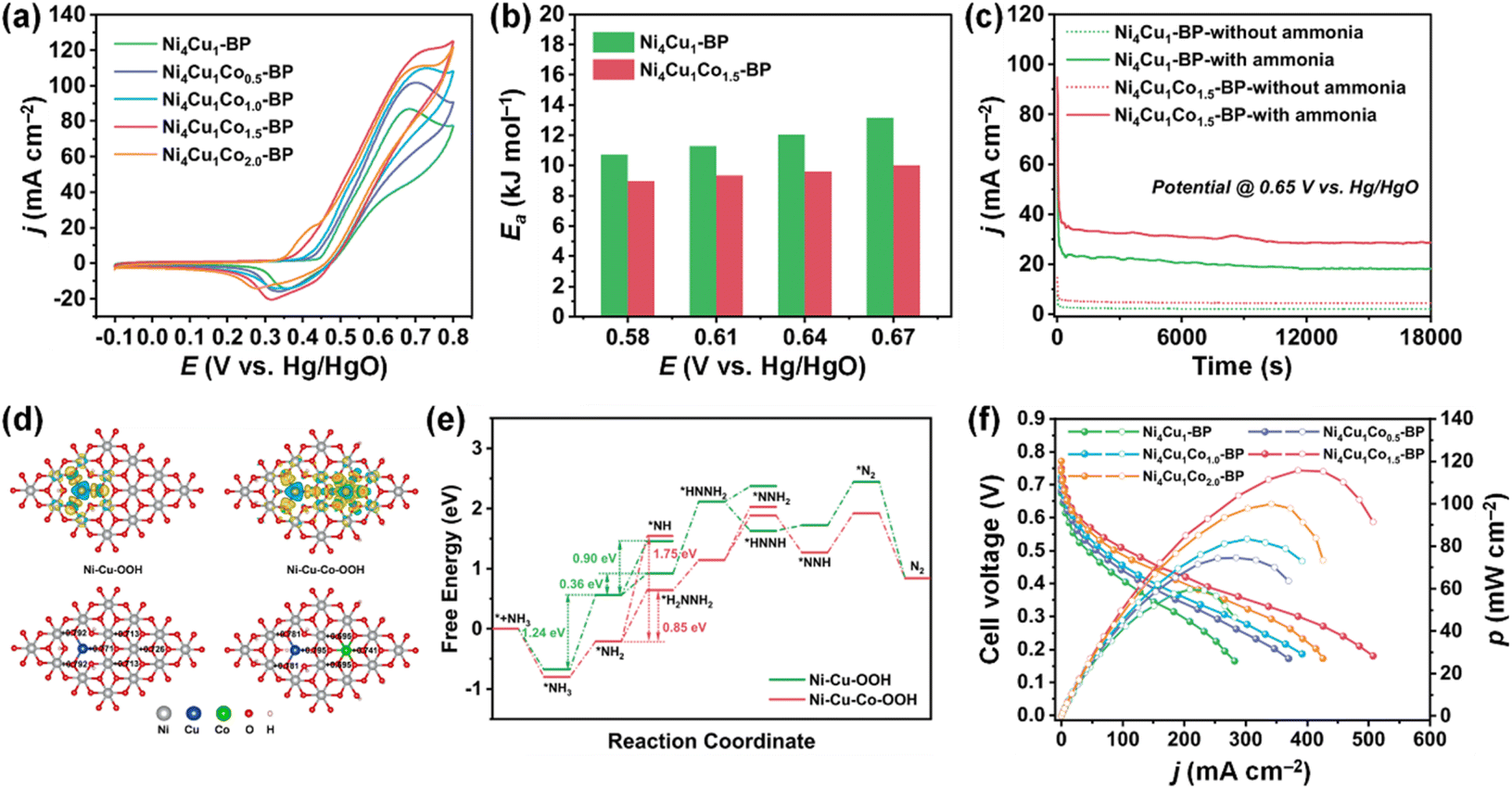 | ||
| Fig. 15 (a) AOR CVs of catalysts in 1 M KOH + 0.1 M NH3. (b) A comparison of the apparent activation energy (Ea) of Ni4Cu1-BP and Ni4Cu1Co1.5-BP in 1 M KOH + 0.1 M NH3. (c) CA measurements of Ni4Cu1-BP and Ni4Cu1Co1.5-BP at 0.65 V vs. Hg/HgO in 1 M KOH + 0.1 M NH3. (d) Charge density difference and charge distribution of the Ni–Cu–OOH and Ni–Cu–Co–OOH catalysts, where the yellow and cyan areas indicate electron accumulation and depletion, respectively. (e) The Gibbs free energy change diagrams of AOR on Ni–Cu–OOH and Ni–Cu–Co–OOH catalysts toward N2. (f) Polarization and power density curves of DAFCs based on different anode electrodes from this work with anode fuel of 3 M KOH + 7 M NH3. Reproduced from ref. 106 with permission. Copyright 2023 Elsevier. | ||
Doping Fe into the NiCu electrode can also enhance AOR activity. Tao et al. developed a bifunctional Ni4Cu5Fex/C (x = 0, 0.5, 1, 3, and 5) alloy catalyst using the solvothermal method.107 Among the Ni4Cu5Fex/C catalysts, the Ni4Cu5Fe1 catalyst showed the highest AOR activity and stability in an alkaline solution (Fig. 16a). In addition, the AOR activity of the Ni4Cu5Fe1/C catalyst increased at higher pH values and ammonia concentration (Fig. 16b and c). In addition, the Ni4Cu5Fe1/C catalyst also promotes ORR, even with NH4Cl in the electrolyte solution, indicating that this ternary alloy catalyst has the potential to serve as both the cathode and anode catalysts in a DAFC (Fig. 16d). When this bifunctional catalyst electrode was assembled with a commercial AEM in a single cell, the symmetric DAFC exhibited an OCV of 0.62 V and power density of 8.9 mW cm−2 at 80 °C (Fig. 16e and f). Moreover, using DFT calculations, Xue et al. revealed that Fe dopants significantly reduce the energy barrier of 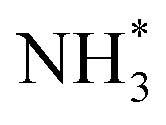 adsorption onto the Ni–Cu OOH surface.108
adsorption onto the Ni–Cu OOH surface.108
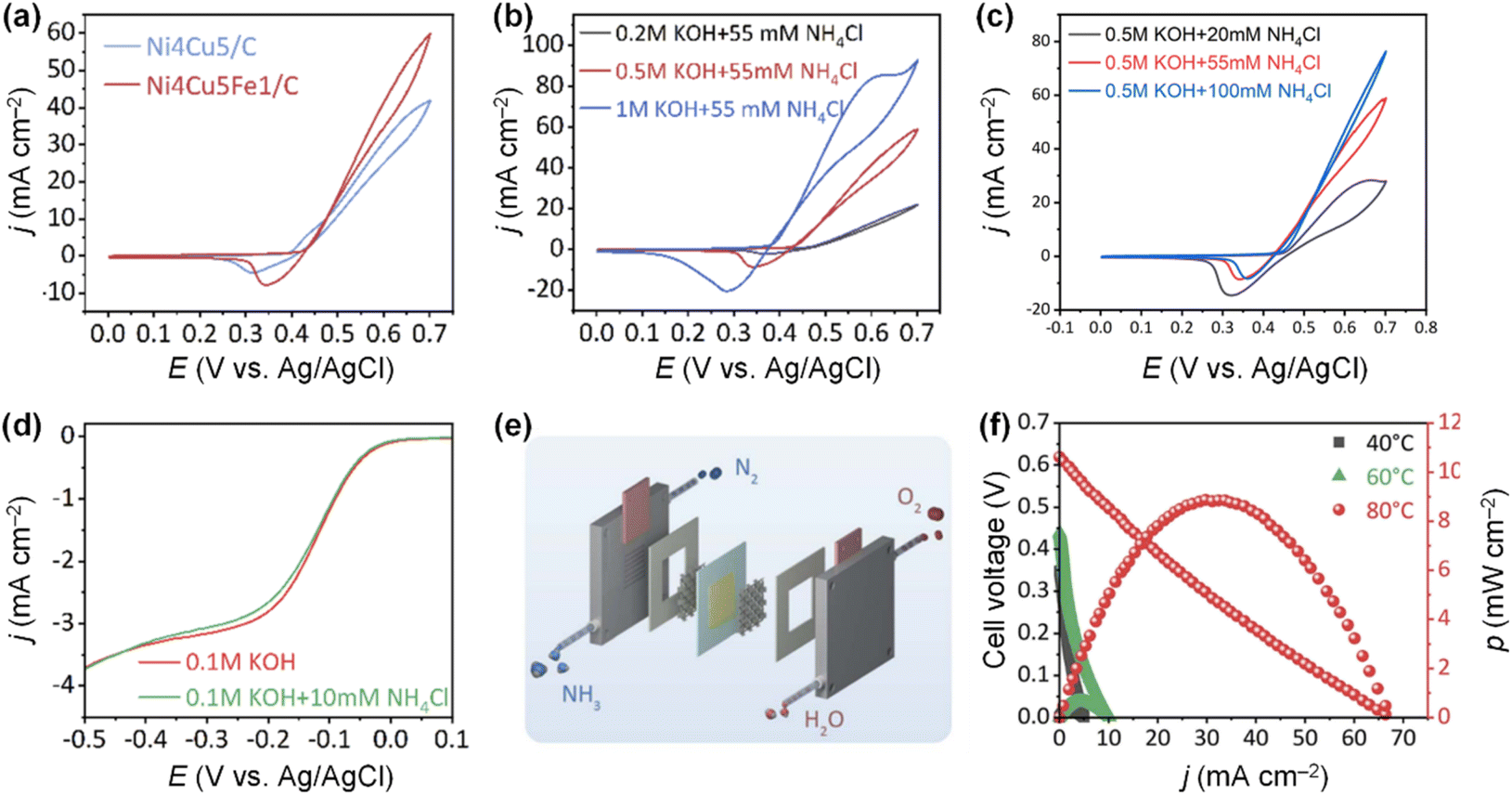 | ||
| Fig. 16 (a) AOR CVs of Ni4Cu5/C and Ni4Cu5Fe1/C in 0.5 M KOH + 55 mM NH4Cl. AOR CVs of Ni4Cu5Fe1/C in various (b) pH values and (c) ammonia concentrations. (d) LSV curves of Ni4Cu5Fe1/C in O2-saturated 0.1 M KOH and 0.1 M KOH + 10 mM NH4Cl. (e) Schematic illustration of DAFC. (f) Polarization curves and power density curves of symmetric DAFC based on the Ni4Cu5Fe1/C at various temperatures. Reproduced from ref. 107 with permission. Copyright 2022 Royal Society of Chemistry (RSC). | ||
In addition to Ni–Cu compositions, interactions between Ni and other transition metal elements can have a positive impact on AOR.109–111 Almomani et al. synthesized Co/Ni–C bimetallic catalysts, consisting of Ni(OH)2 and Co(OH)2, with varying Co contents (0, 10, 20, and 40 wt%) using the solvothermal method.109 In the electrochemical measurement, the AOR maximum power densities of the Ni–C catalysts (0.55 mW cm−2) were inferior to those of Co10/Ni–C (0.85 mW cm−2), Co20/Ni–C (1.1 mW cm−2), and Co40/Ni–C (0.93 mW cm−2). When the Co concentration exceeded 40%, Co species blocked the Ni active sites, leading to a decrease in AOR activity.
Wang et al. also prepared Ag/Ni electrocatalysts via a facile hand-mixing method.112 The AOR activities of Ag/Ni were tested in 1.5 M NaOH + 0.5 M NH3 and compared with pristine Ag and Ni electrocatalysts. As shown in Fig. 17a, Ag/Ni outperformed monometallic Ag and Ni electrodes. The Ag electrode displayed no catalytic activity, while the Ni electrode showed higher onset potential (0.6 V vs. Hg/HgO) than the Ag/Ni electrode (0.46 V vs. Hg/HgO), indicating a slower initiation of the AOR process. Notably, Ag/Ni predominantly produced N2 with a faradaic efficiency of 59.8% during NH3 electrolysis at 0.7 V vs. Hg/HgO for 2 h. In contrast, for the Ni electrode, a dominant product of NH3 electrolysis was O2, implying that the oxygen evolution reaction (OER) is favoured over AOR on the Ni electrode. CV analysis of the Ag electrode in electrolytes with and without NH3 elucidated the interaction between Ag and Ni. As shown in Fig. 17b, the peaks related to the oxidation of Ag+ to Ag2+ vanished upon anodic scanning in the presence of NH3. This disappearance suggests that a strong Ag+–NH3 complex formed from the oxidized Ag+, hindering the over-oxidation of NH3. The pristine Ni showed consistent CV results irrespective of the presence of NH3, due to the competitive OER. In other words, the role of Ag in Ag+–NH3 complex formation and Ni as OH− adsorption were decoupled to promote N2 selectivity above 0.4 V, vs. Hg/HgO (Fig. 17c).
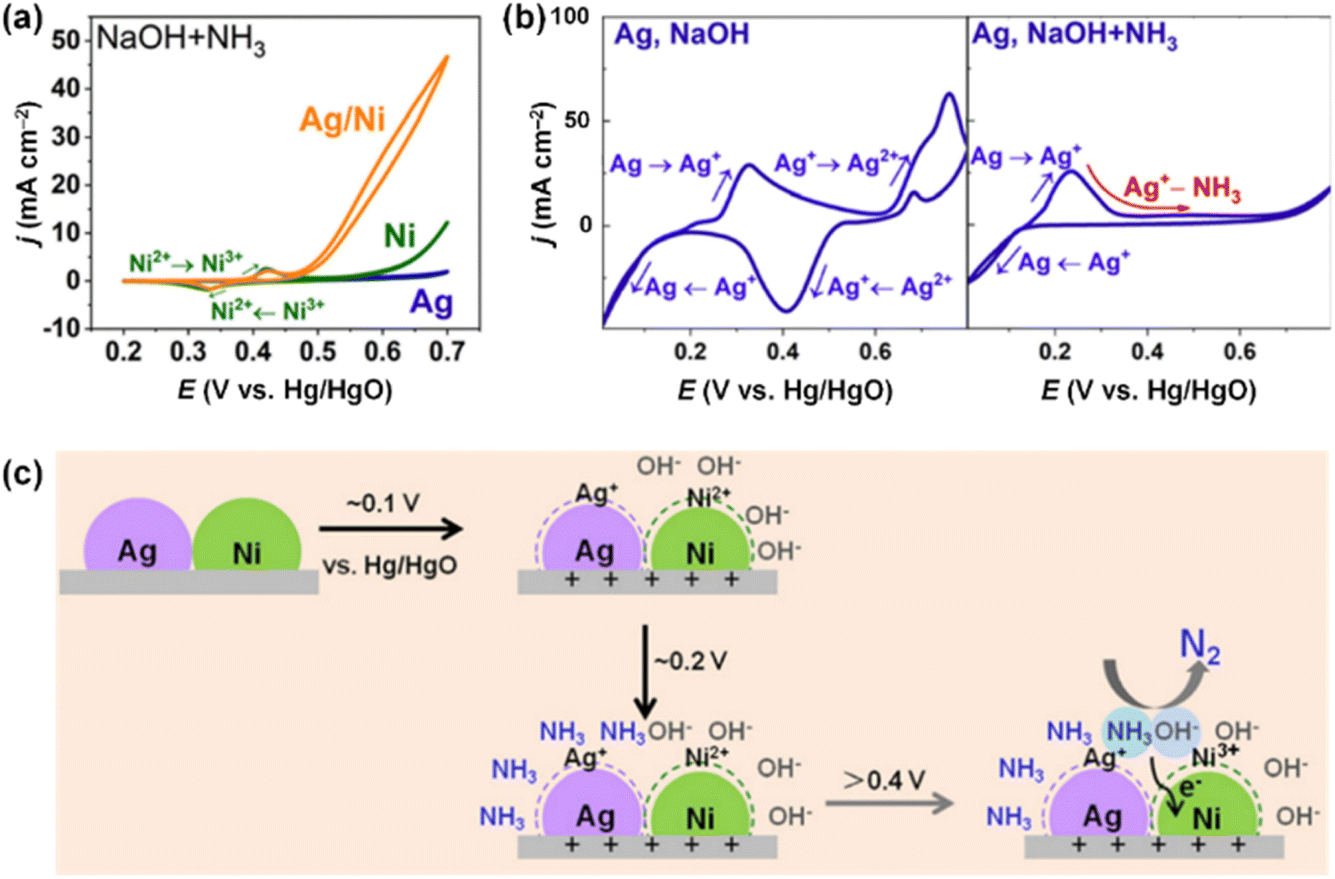 | ||
| Fig. 17 (a) AOR CVs of Ag/Ni, Ag and Ni in 1.5 M NaOH + 0.5 M NH3. (b) CVs of Ag in 1.5 M NaOH with 0.5 M NH3 (left)/without NH3 (right). (c) Illustration of mediation of Ag and Ni in AOR. Reproduced from ref. 112 with permission. Copyright 2022 RSC. | ||
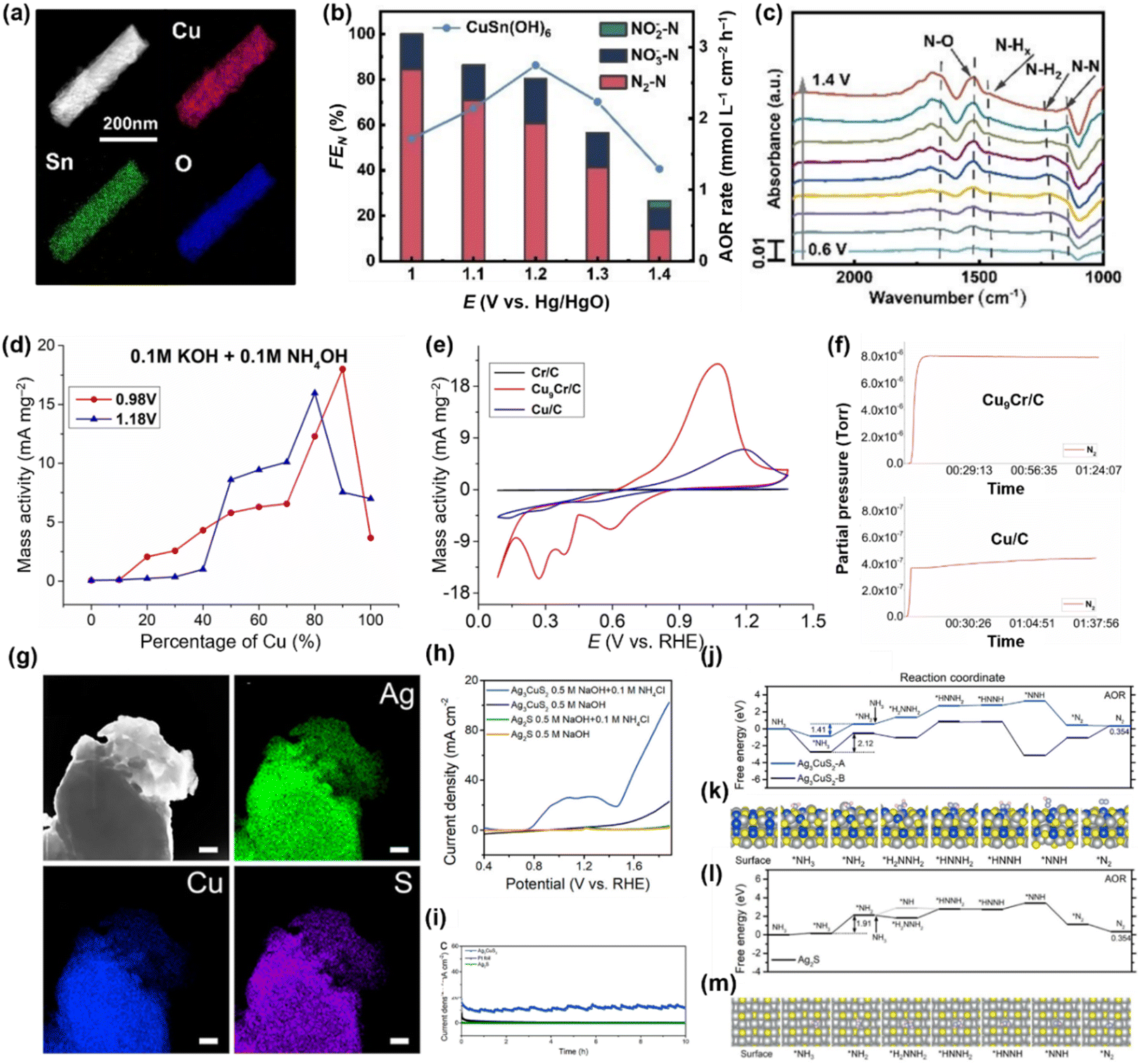 | ||
| Fig. 18 (a) HADDF-STEM and EDS elemental mapping images of CuSn(OH)6. (b) faradaic efficiencies and AOR rate for CuSn(OH)6. (c) In situ IR spectra of AOR on CuSn(OH)6 surface in pH 11 with 10 mM of NH3–N. Reproduced from ref. 110 with permission. Copyright 2022 Wiley-VCH. (d) AOR mass activity with different Cu ratio at 0.98 and 1.18 V vs. RHE. (e) AOR CVs of Cr/C, Cu/C, Cu9Cr/C catalysts in 0.1 M KOH + 0.1 M NH4OH. (f) Mass spectroscopy data corresponding with chronoamperometry at 1.1 (V vs. RHE) for Cu9Cr (top) and Cu (bottom). Reproduced from ref. 117 with permission. Copyright 2023 Wiley-VCH. (g) EDS mapping for the individual elemental for Ag3CuS2 (h) AOR polarization curves for Ag3CuS2 and Ag2S in 0.5 M NaOH and 0.5 M NaOH + 0.1 M NH4Cl (i) CA test of Ag3CuS2, Ag2S, Pt/C at 1.5 V for 10 h. Calculated free energy in reaction pathway for (j) Ag3CuS2, (l) Ag2S and optimized 3d structure in reaction pathway for (k) Ag3CuS2 and (m) Ag2S. Reproduced from ref. 118 with permission. Copyright 2022 ACS. | ||
The critical effect of Cu and Sn integration in AOR catalysts was validated, with Cu acting as the catalytic centre and Sn adsorbing NH3. In addition, a high valence cation like Cr6+ can improve the catalyst selectivity by converting intermediates including hydrazine to N2.117 As shown in Fig. 18d, bimetallic CuCr alloy catalysts with less than 50% Cr ratio displayed remarkably higher mass activity than metallic Cu, and the best performance was achieved at the optimal ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr = 9:1 (Fig. 18e). Mass spectroscopy analysis identified the improved selectivity of the CuCr catalyst towards N2 compared with metallic Cu (Fig. 18f). Besides, Zhang et al. synthesized a foamed Ag3CuS2 film that demonstrated superior catalytic activities and durability for AOR (Fig. 18g).118 The Ag3CuS2 film catalysts exhibited a low onset potential of 0.7 V and achieved a current density of 100 mA cm−2 at 1.85 V (vs. RHE) (Fig. 18h). Furthermore, the catalysts maintained long-term durability at 1.5 V (vs. RHE) for 10 hours (Fig. 18i), underscoring their remarkable potential for practical ammonia electrolysis applications. Theoretical studies revealed that the Ag3CuS2 surface results in a reduced energy barrier for the conversion of *NH3 to *NH2 (rate-determining step), compared to Ag2S (Ag3CuS2: 1.41 eV, Ag2S: 1.91 eV). Beyond that, N2 desorption occurred spontaneously on Ag3CuS2, suggesting AOR process was more favourable on Ag3CuS2 than on Ag2S (Fig. 18j–m).
:Cr = 9:1 (Fig. 18e). Mass spectroscopy analysis identified the improved selectivity of the CuCr catalyst towards N2 compared with metallic Cu (Fig. 18f). Besides, Zhang et al. synthesized a foamed Ag3CuS2 film that demonstrated superior catalytic activities and durability for AOR (Fig. 18g).118 The Ag3CuS2 film catalysts exhibited a low onset potential of 0.7 V and achieved a current density of 100 mA cm−2 at 1.85 V (vs. RHE) (Fig. 18h). Furthermore, the catalysts maintained long-term durability at 1.5 V (vs. RHE) for 10 hours (Fig. 18i), underscoring their remarkable potential for practical ammonia electrolysis applications. Theoretical studies revealed that the Ag3CuS2 surface results in a reduced energy barrier for the conversion of *NH3 to *NH2 (rate-determining step), compared to Ag2S (Ag3CuS2: 1.41 eV, Ag2S: 1.91 eV). Beyond that, N2 desorption occurred spontaneously on Ag3CuS2, suggesting AOR process was more favourable on Ag3CuS2 than on Ag2S (Fig. 18j–m).
Ti-based materials have been used as AOR electrocatalysts.119 The defect-rich TiO nanofibers on Ti foil were designed using corrosion and a subsequent annealing process. As-synthesized TiO/Ti foil displayed superior AOR activity and stability in alkaline ammonia solution with reduced onset potentials of 0.4 V. DFT calculations revealed that the oxygen vacancies in TiO2 provide the lower energy barrier of the rate-determining *HNNH2 formation step and facilitate N2 desorption.
4 Electrocatalysts for ORR: PGM vs. non-PGM
This section discusses the catalytic materials for promoting ORR, a cathodic reaction for the DAFC. ORR catalysts have been most intensively studied in electrocatalytic research fields, and their performance in DAFC systems is anticipated to improve by employing optimized ORR catalysts with high activity and durability. The following sections describe examples of ORR catalysts utilized in the cathode of DAFC.4.1. PGM ORR electrocatalysts
As in conventional low-temperature fuel cell systems, Pt-based materials have been widely tested as cathode catalysts for fuel cell devices employing ammonia fuels because they have optimal binding energy with ORR intermediates (Table 5).| Catalyst | Electrolyte | Activity | Stability | Ref. |
|---|---|---|---|---|
| Pt/C | 1 M KOH | E 1/2: ∼0.91 V | — | 19 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.85 V | |||
| Pt/C | 1 M KOH | E 1/2: 0.864 V | 0.833 → 0.772 V (−7.3%) after 10000 CV (0.6–1.1 V) cycles |
165 |
| 1 M KOH +0.1 M NH3 | E 1/2: 0.833 V | |||
| Pd/C | 0.1 M KOH | j @ 0.85 V: ∼−1.6 mA cm−2 | - No initial activity in the ammonia solution | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−0.2 mA cm−2 | |||
| Au/C | 0.1 M KOH | j @ 0.85 V: ∼−0.7 mA cm−2 | - No initial activity in the ammonia solution | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−0.1 mA cm−2 | |||
| Ag/C | 0.1 M KOH | j @ 0.85 V: ∼−0.093 mA cm−2 | - No initial activity in the ammonia solution | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−0.0125 mA cm−2 |
From the early ammonia–oxygen fuel cell systems of the 1960s to the recently reported AEM-based DAFC stack, Pt catalysts are recognized as the most fundamental cathode catalysts.18–20,34,120–124 For example, Eguchi et al. tested the performance of MEA-based DAFC configured by AEM (A201, Tokuyama Co.; thickness = 28 μm) and PGM electrocatalysts.121 In this work, the cathode layers of each MEA consisted of Pt/C catalysts, while anode catalyst layers were configured using three types of anode catalysts (Pt/C, Ru/C, PtRu/C). The Pt/C-based cathode layers were employed as a counter electrode to evaluate the influence of anode materials on DAFC performance.
As another example, Siddiqui et al. developed DAFC MEAs using quaternary ammonium-functionalized AEMs (thickness = 500 μm) consisting of polystyrene and divinylbenzene.18 Both electrodes were fabricated by coating Pt/C catalysts on the gas diffusion layers based on carbon fibre paper. At 25 °C, the DAFC exhibited 0.28 V of OCV and 0.64 mW cm−2 of maximum power density at 1 bar of pressure of gaseous ammonia fuel. The performance of the developed DAFC was also tested at various temperatures and relative humidity (RH). Subsequently, the DAFC stack with five single-cells was fabricated.20 The MEAs for the DAFC stack were configured using the same compartment as in the above work, and the developed DAFC stack exhibited 1.25 V of OCV and 1.34 mW cm−2 of maximum power density at 25 °C.
The effective optimization strategies utilized in the PEMFC studies, including compositional tuning,125–128 shape control,129–131 and strain engineering,132–134 suggest crucial insights into high-performance catalysts for DAFC. However, Pt-based catalysts are critically unsatisfactory as DAFC cathode materials because they are vulnerable to ammonia crossover during the DAFCs operation.135–137 Nitrogen-containing species strongly bind with Pt surfaces, so the cathode catalysts are prone to poisoning and deactivation.
Model catalytic studies on polycrystalline Pt electrodes, performed with a rotating ring-disk electrode (RRDE) system, revealed the impact of ammonium ions on ORR activity and selectivity on Pt catalytic surfaces.135,136 For instance, Halseid et al. tested the ORR properties of polycrystalline Pt electrodes in the 0.5 M HClO4 electrolyte solutions containing ammonium ions at different concentrations (0, 0.1, 0.25 M).135 The series of ORR tests demonstrated that ammonium ions lead to a significant deterioration in ORR activity (Fig. 19a). More importantly, in the 0.5 M HClO4 solutions containing ammonium ions, the selectivity for the four-electron ORR process decreased, resulting in higher yields of H2O2 and radical species (Fig. 19b). As these chemical entities decompose the polymer structure of the membrane and ionomers and promote metal dissolution and catalyst disintegration, they are widely recognized as primary factors contributing to MEA oxidation and performance deterioration.137–139 This ORR activity degradation of Pt-based catalysts by NH3 poisoning was also confirmed in the single-cell test based on MEAs. Eguchi and coworkers fabricated MEAs comprising Pt/C cathode and measured the open circuit voltage (OCV) under H2 and NH3 feeding. After NH3 feeding, the Pt/C-based MEA showed rapid OCV degradation due to the NH3 crossover followed by catalyst poisoning (Fig. 19c). Verdaguel-Casadevall et al.136 predicted that the crossover of only 500 ppm of ammonium ions from the anode to the cathode in an MEA can degrade the performance of a DAFC by 50% at a cell voltage of 0.7 V (Fig. 19d).
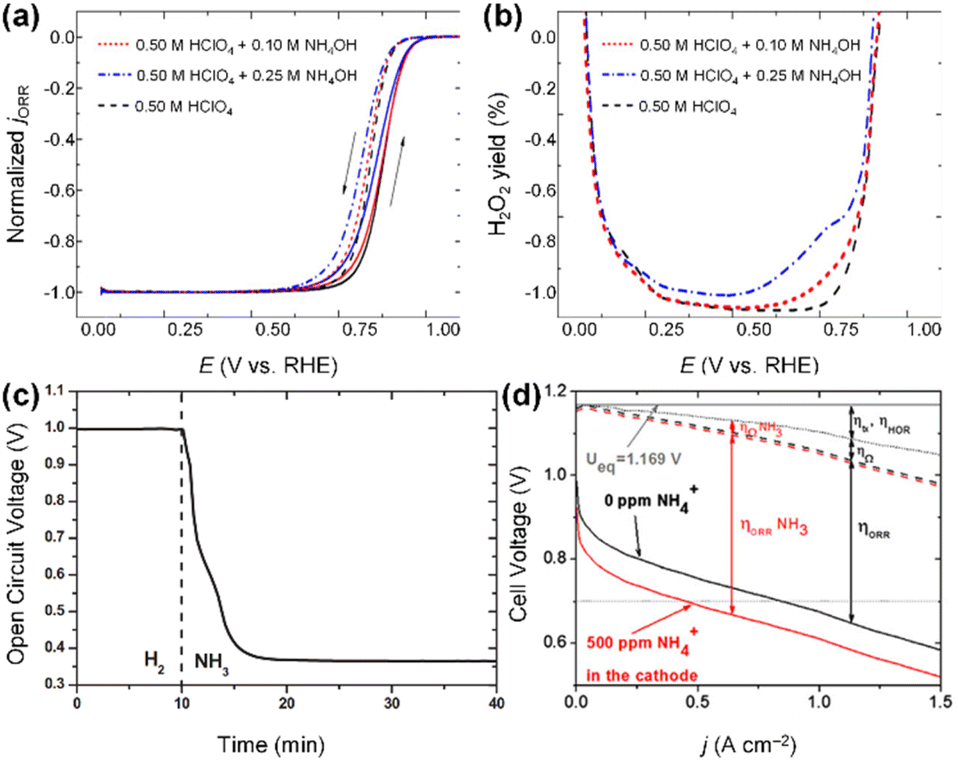 | ||
| Fig. 19 (a) Normalized ORR current on a polycrystalline Pt electrode. (b) Relative yield of H2O2 in 0.50 M HClO4 solution containing NH4OH with various concentrations. Reproduced from ref. 135 with permission. (c) Temporal open circuit voltage (OCV) for the DAFC with Pt/C cathode. After ammonia feeding, OCV rapidly decreases. Copyright 2012 Elsevier. Reproduced from ref. 121 with permission. (d) Predicted effects of ammonia poisoning in DAFC. Ueq represents the theoretical equilibrium potential for the overall fuel cell reaction. Copyright 2012 Elsevier. Copyright 2008 Elsevier. Reproduced from ref. 136 with permission. | ||
In addition, an in situ FT-IR spectroscopy study revealed ammonia and nitric oxide adsorbed on the Pt surfaces. Further, the catalyst deactivation and H2O2 formation by ammonium ions was more severe in the sulfuric acid electrolyte, which is a similar environment to the PFSA-based polymer electrolyte membrane. This highlights the inappropriateness of PFSA-based PEMs in DAFC applications. Garzon et al. also reported similar degradation phenomena of Pt surfaces by ammonium ions in the half-cell tests, using the commercial Pt/C catalyst.140 More critically, in DAFC, a significant rate of ammonia crossover from the anodes to the cathodes can generate a reverse current at the cathode, leading to a significant reduction in OCV and performance.141,142
In alkaline media, the rate of Pt dissolution is not negligible, and this can influence catalyst deformation and subsequently degrade activity.143,144 Zadick and coworkers investigated the degradation of Pt/C in an alkaline medium through identical location transmission electron microscopy (ILTEM) analyses, combined with electrochemical measurements.143 Along with the morphological changes, the degradation of the Pt/C catalysts was evaluated using the ECSA loss of Pt/C catalysts as a criterion. The ECSA loss of the Pt/C catalyst in alkaline media was three times larger than that in acidic media, indicating severe catalyst degradation in alkaline media. Koper et al. also revealed that the accelerated Pt/C degradation at high pH originates from promoted Ostwald ripening, Smoluchowski agglomeration, and nanoparticle detachment.144 The comprehensive study using TEM and in situ X-ray absorption (XAS) analyses suggested that these processes are driven by Pt-catalysed carbon corrosion as well as the dissolution/redeposition of Pt NPs. Although these processes occur on the Pt/C catalysts regardless of the pH of electrolytes, an alkaline environment accelerates the degradation and catalyst deactivation. Besides these stability issues, high cost and scarcity of Pt have also motivated researchers to explore efficient alternative catalytic materials for Pt-based catalysts, similar to the AOR catalyst research field.
4.2. Other PGM-based ORR electrocatalysts
As alternatives to Pt-based catalysts, Pd-based catalysts exhibiting high stability in alkaline environments are considered promising cathode catalysts for DAFC single-cells. These catalysts can deliver high alkaline ORR activity close to those of Pt/C catalysts.145–148 Notably, Yan et al. revealed the catalytic behaviour of commercial Pd/C catalysts when exposed to ammonia through a comparative study of various ORR catalysts.149 Although Pd/C underwent a decrease in ORR specific activity after its catalytic surface being poisoned, similar to Pt/C, the activity loss observed in Pd/C was lower than that of Pt/C. Additionally, because the Pd is known to be inactive for AOR, it is anticipated that Pd-based catalysts can be used to configure robust cathodes with high ORR selectivity that prevent the generation of reverse current and resulting OCV loss.Yan et al. also demonstrated the appropriateness of Pd/C cathodes by comparing the DAFC performance of a Pd/C cathode-based MEA with a Pt/C cathode-based version.150 In the MEA fabrication, hydroxide exchange membranes (HEMs; PAP-TP-85, PiperION; thickness = 18 μm) were exploited to attain high performance. The Pd/C cathode-based DAFC delivered 304 mW cm−2 of maximum power density at 95 °C (Fig. 20a), which was much higher than that of the Pt/C cathode-based one (197 mW cm−2). In addition, after continuous operation at 300 mA cm−2, the Pd/C cathode-based DAFC demonstrated high durability, with a slow decay rate of ∼1 mV h−1 in 36 h (Fig. 20b). In addition, An et al. recently developed a DAFC composed of a commercial AEM (FAS-30, Fumasep; thickness = 30 μm), a PtRu/C anode, and a Pd/C cathode.151 The developed DAFC exhibited 0.67 V of OCV and 20.7 mW cm−2 of maximum power density at 95 °C, demonstrating the practical applicability of Pd/C as a DAFC cathode catalyst.
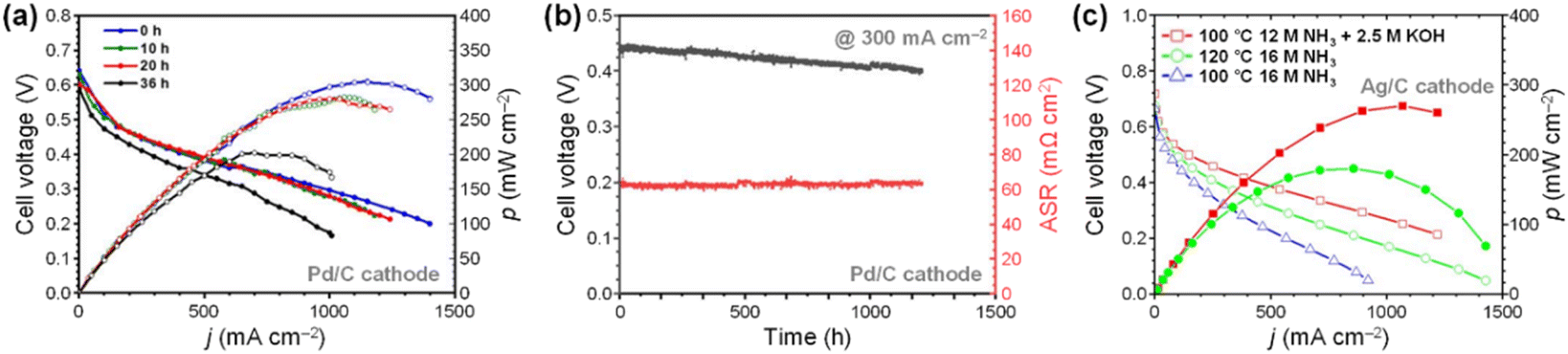 | ||
| Fig. 20 (a) Polarization curves and (b) cell voltage and ASR as a function of time at 300 mA cm−2 of a DAFC based on a Pd/C cathode. Reproduced from ref. 150 with permission. Copyright 2021 Elsevier. (c) Polarization curves of DAFC based on Ag/C cathode. Reproduced from ref. 154 with permission. Copyright 2020 Elsevier. | ||
Moreover, among the PGM materials, Ag has special significance as an alkaline ORR catalyst in terms of its low cost as well as high performance, which is close to that of Pt.149,152–154 In particular, Ag-based catalysts were recently applied in DAFC systems.154 The developed DAFC, configured by HEM (Tokuyama; thickness = 30 μm), with an anode with Pt1Ir10/C, and a cathode with Ag-based dispersed electrocatalyst, was tested in a KOH-free environment (Fig. 20c). Because DAFC operation without an alkaline aqueous solution can improve fuel management as well as prevent intercell shorting current (shunt currents) and the corrosion of compartments in the DAFC stack, the KOH-free environment is of interest in terms of practical use. The developed DAFC showed 180 mW cm−2 of maximum power density at 120 °C, the best performance among the previously reported KOH-free DAFC systems. Better understanding of optimization strategies toward high-performance Ag-based ORR catalysts is expected to further accelerate the widespread use of Ag-based catalytic nanomaterials in DAFCs. However, since Ag also shows an intrinsic loss in specific activity, developing catalytic materials that experience minimal poisoning of catalytic surfaces by ammonia remains a formidable challenge.149 In the next section, we discuss examples that successfully addressed this issue by using DAFC cathode catalysts composed of an earth-abundant, non-PGM composition.
4.3. Non-PGM-based ORR electrocatalysts
In this section, we present recent progress in highly efficient non-PGM catalysts to promote ORR under DAFC operating conditions. The primary advantage of a fuel cell operated with an alkaline environment is that it opens the possibility of using non-PGM catalysts (Table 6), which can effectively reduce the cost of the fuel cell configuration.155–157 At a high pH, the ORR, which generally entails the highest irreversibility in fuel cells, is also facilitated because the polarization losses are lower compared to an acidic environment.158–161 Accordingly, efficient catalysts have been developed to achieve high performance in single-cell configurations based on the AEMs.155,161,162| Catalyst | Electrolyte | Activity | Stability | Ref. |
|---|---|---|---|---|
| Acta 4020 | 1 M KOH | E 1/2: ∼0.91 V | — | 19 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.9 V | |||
| Pajarito | 0.1 M KOH | j @ 0.85 V: ∼−2 mA cm−2 | ∼−2 → ∼−1.75 mA cm−2 (−12.5%) after 1 h CA @ 0.85 V | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−2 mA cm−2 | |||
| Fe-ZIF | 0.1 M KOH | j @ 0.85 V: ∼−1.25 mA cm−2 | ∼−1.25 → ∼−1.2 mA cm−2 (−4%) after 1 h CA @ 0.85 V | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−1.25 mA cm−2 | |||
| Fe,Co-ZIF | 0.1 M KOH | j @ 0.85 V: ∼−1.25 mA cm−2 | ∼−1.4 → ∼−1.3 mA cm−2 (−7%) after 1 h CA @ 0.85 V | 149 |
| 0.1 M KOH + 0.1 M NH3 | j @ 0.85 V: ∼−1.25 mA cm−2 | |||
| Fe–N–C | 1 M KOH | E 1/2: ∼0.92 V | — | 154 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.9 V | |||
| MCS-BP2000 | 1 M KOH | E 1/2: ∼0.84 V | 0.837 → 0.829 V (−1%) after 10000 CV (0.6–1.1 V) cycles |
165 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.837 V | |||
| MCS-XC72R | 1 M KOH | E 1/2: ∼0.834 V | 0.828 → 0.816 V (−1.5%) after 10000 CV (0.6–1.1 V) cycles |
165 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.828 V | |||
| MCS-MWCNTs | 1 M KOH | E 1/2: ∼0.826 V | 0.823 → 0.809 V (−1.5%) after 10000 CV (0.6–1.1 V) cycles |
165 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.823 V | |||
| ZIF-NC-0.5Fe–3N | 1 M KOH | E 1/2: ∼0.887 V | ∼−0.55 → ∼−0.45 mA cm−2 (−18.2%) after 1 h CA @ 0.85 V | 178 |
| 1 M KOH + 0.1 M NH3 | E 1/2: ∼0.883 V | |||
| FeNC-50 | 1 M KOH | E 1/2: 0.890 V | 0.880 → 0.880 V (−0%) after 10000 CV (0.6–1.1 V) cycles |
179 |
| 1 M KOH + 0.1 M NH3 | E 1/2: 0.880 V |
As another example, the research groups of Ming and Xiao developed Mn–Co spinel (MCS) nanocrystals supported on three different carbon supports (Fig. 21), such as BP2000, Vulcan XC-72R, and multi-walled carbon nanotubes.165 In the half-cell tests, the developed MCS catalysts showed nearly identical activity regardless of the presence of ammonia in the electrolyte solution, in contrast to the Pt/C catalysts that underwent a decrease in activity in the ammonia-containing electrolyte solution (Fig. 21a and d). Notably, the best-performing MCS/BP2000 catalyst even showed a higher ORR half-wave potential (E1/2) than that of Pt/C in the 1 M KOH + 0.1 M NH3. In addition, after 10000 cycles in an accelerated durability test (ADT), it exhibited a much smaller loss in E1/2 of MCS/BP2000 (8 mV) compared to that of Pt/C (61 mV), indicating the high durability of the MCS/BP2000 catalysts (Fig. 21b and e). Further, a DAFC fabricated by pairing the MCS/BP2000 cathode with a commercial PtIr/C anode showed a higher maximum power density (128.2 mW cm−2) than the Pt/C cathode-based DAFC (100.1 mW cm−2) at 80 °C (Fig. 21c and f). Recently, using the developed MCS/BP2000 catalyst, the same research groups performed a systematic optimization of the DAFC cathode for the first time.166 They investigated the effects of three factors, including catalyst loading, ionomer binder type, and binder content, on the DAFC cell performance. The influence of each factor was in the following order: binder type > binder content > catalyst loading. The best-performing DAFC single-cell exhibited 158.6 mW cm−2 of maximum power density under 2 mg cm−2 of cathode catalyst loading, PTFE binder, and 15 wt% of binder content. These efforts suggest useful guidelines for designing and optimizing the catalyst layer for high-performance DAFCs.
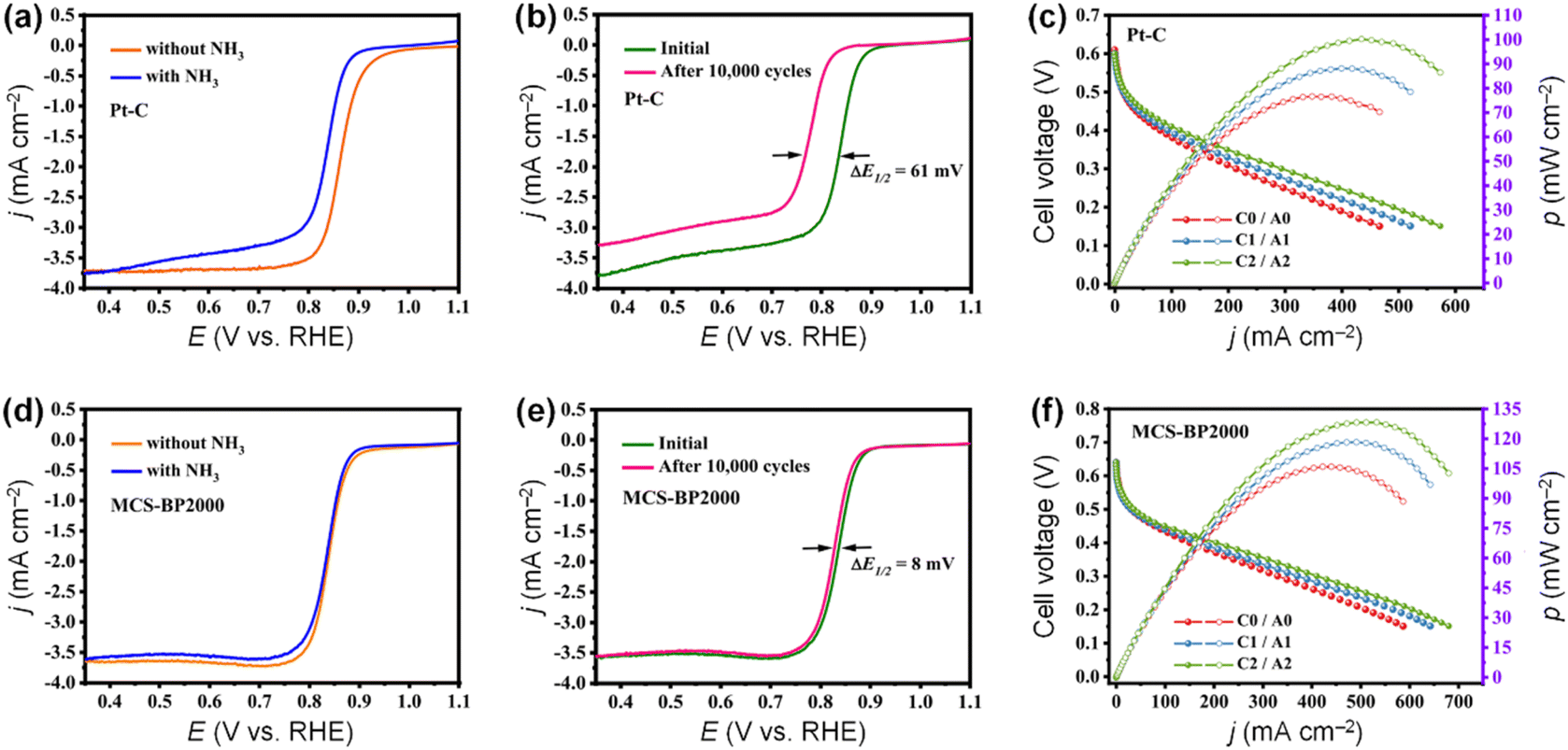 | ||
| Fig. 21 (a and d) ORR polarization curves in O2-saturated 1 M KOH solution with and without 0.1 M NH3 of (a) Pt–C and (d) MCS-BP2000. (b and e) ORR polarization curves before and after 10000 ADT cycles in O2-saturated 1 M KOH + 0.1 M NH3 of (b) Pt–C and (e) MCS-BP2000. (c and f) Polarization and power density curves of DAFCs based on (c) Pt–C cathode and (f) MCS-BP2000 cathode. Reproduced from ref. 165 with permission. Copyright 2021 ACS. | ||
Accordingly, Tao et al. reported several results for perovskite-based DAFC cathodes, including SrCo0.8Cu0.1Nb0.1O3−δ, SrFe0.8Cu0.1Nb0.1O3−δ, and LaCr0.25Fe0.25Co0.5O3−δ.170–173 For example, they developed perovskites based on LaCoO3−δ (LCO), such as LaCr0.5Co0.5O3−δ (LCCO), LaFe0.5Co0.5O3−δ (LFCO), and LaCr0.25Fe0.25Co0.5O3−δ (LCFCO) and compared their ORR performance.172 Electrocatalytic examination of the ORR revealed the pivotal contributions of both Cr and Fe in augmenting activity and influencing structural and electronic properties (Fig. 22a). The introduction of Cr, which is related to the creation of surface oxygen vacancies, resulted in an elevated four-electron ORR pathway. At the same time, the incorporation of Fe, influencing ORR reaction kinetics, enhanced the ORR E1/2. Notably, the LFCFO catalyst prepared at optimized temperature (LCFCO-700) exhibited high ORR activity, surpassing Pt/C in 0.1 M KOH (Fig. 22b). In addition, the LCFCO-700 catalyst showed virtually identical activity in both pure KOH solution and KOH with ammonia solution, indicating excellent ammonia resistance (Fig. 22c). Remarkably, the DAFC based on the LCFCO-700 cathode showed comparable performance to the Pt/C cathode-based one, demonstrating the practical applicability of perovskite as a promising non-PGM cathode catalyst for DAFCs (Fig. 22d). Subsequently, the developed LCFCO catalysts were utilized to optimize perovskite-based DAFC cathode layers to improve DAFC performance.173 By optimizing the carbon support, ionomer, and hydrophilicity of the electrode, the maximum power density of the DAFC increased from 5.92 mW cm−2 to 30.1 mW cm−2. These optimization studies clearly illustrate the importance of three-phase interfaces in the catalyst layer for improving MEA performance.
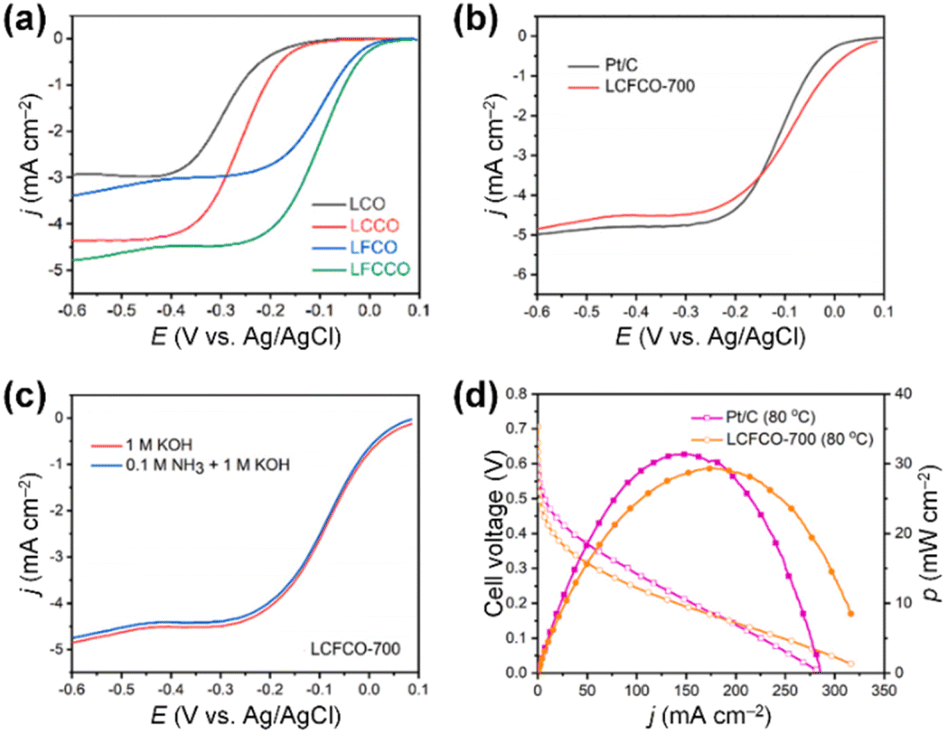 | ||
| Fig. 22 (a) Polarization curves obtained for the LCO, LCCO, LFCO and LCFCO perovskites in O2-saturated 0.1 M KOH solution. (b) Polarization curves of Pt/C and LCFCO-700 electrocatalysts in O2-saturated 0.1 M KOH solution. (c) Polarization curves obtained for the LCFCO-700 in O2-saturated 0.1 M KOH solution with and without 0.1 M NH3. (d) Polarization and power density curves of DAFCs based on Pt/C and LCFCO-700 cathodes. Reproduced from ref. 172 with permission. Copyright 2022 Elsevier. | ||
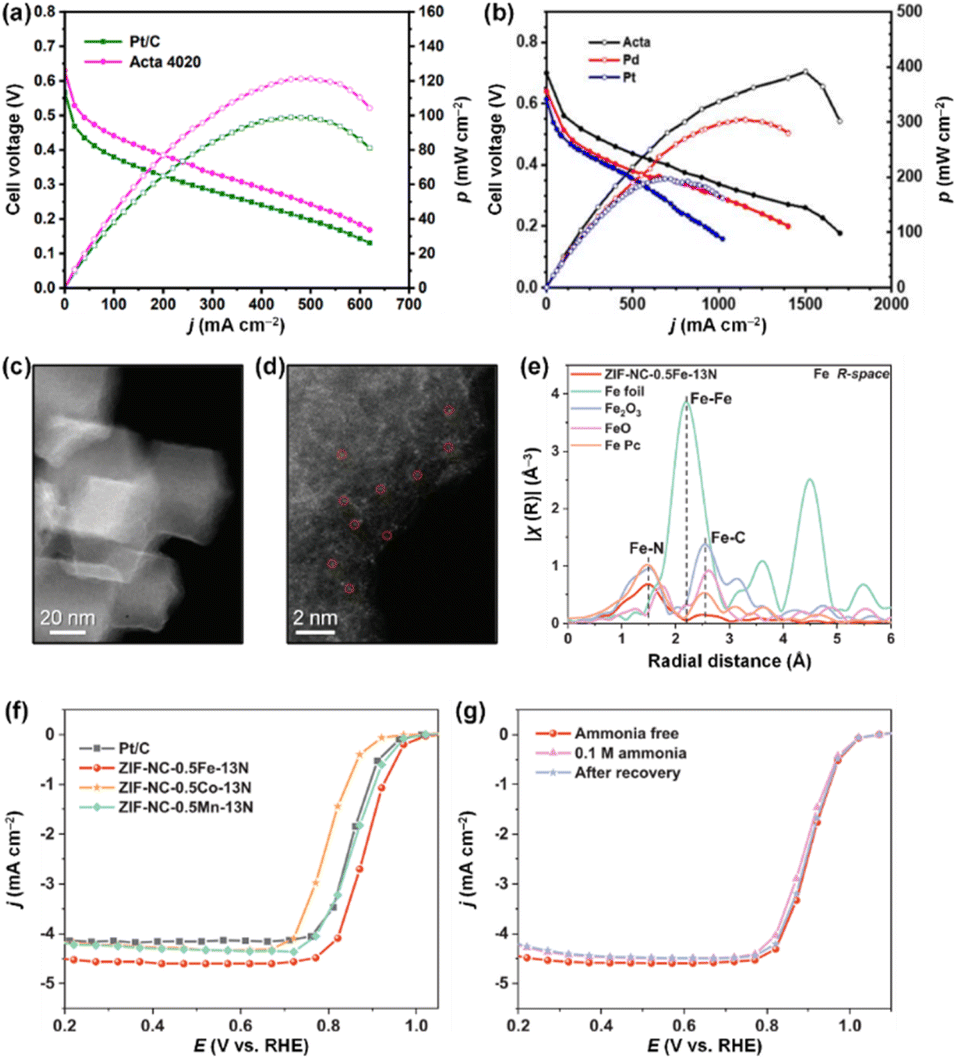 | ||
| Fig. 23 (a) Polarization and power density curves of DAFC prototypes with Pt/C and Acta 4020 cathodes. Reproduced from ref. 19 with permission. Copyright 2019 Cell Press. (b) Polarization and power density curves of optimized DAFC with Pt/C, Pd/C, and Acta 4020 cathodes. Reproduced from ref. 150 with permission. Copyright 2021 Elsevier. (c) TEM and (d) HAADF-STEM images of ZIF-NC-0.5Fe–13N catalyst. (e) Fe K-edge EXAFS spectra of the ZIF-NC-0.5Fe–13N, Fe Pc, and Fe foil. (f) Steady-state ORR polarization curves of ZIF-NC-0.5Fe–13N, ZIF-NC-0.5Co–13N, ZIF-NC-0.5 Mn–13N, and Pt/C in O2-saturated 0.1 M KOH. (g) Steady-state ORR polarization curves of ZIF-NC-0.5Fe–13N in 0.1 M KOH with and without 0.1 M NH3. Reproduced from ref. 178 with permission. Copyright 2023 Wiley-VCH. | ||
These findings underscore the potential of Fe–N/C as a highly promising catalyst for DAFC cathodes, increasing confidence in the prospect of achieving enhanced performance with continued material development. For example, Li et al. synthesized Fe–N/C catalysts with mononuclear centers without the presence of Fe-derived crystalline structures using carbonized ZIF-8 nanocrystals impregnated with Fe ions (Fe3+/ZIF-NC) (Fig. 23c).178 Pyrolysis and a subsequent thermal N-doping step were used to synthesize Fe–N–C–13N catalysts. The mononuclear structure of the Fe–N sites in the resulting Fe–N–C–13N catalysts was identified using high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) analyses (Fig. 23d). Extended X-ray absorption fine structure (EXAFS) spectra showed that the coordination number of Fe–N bonds was 3.7 (Fig. 23e). These physicochemical analyses illustrate that the Fe–N–C–13N catalyst possesses MN4 moieties within the carbon frameworks.
Notably, the well-defined Fe–N–C–13N structure exhibited higher ORR activity and selectivity than the Pt/C catalyst in 0.1 M KOH (Fig. 23f). It also presented high ammonia resistance, showing nearly identical ORR activity regardless of the presence of ammonia in the electrolyte (Fig. 23g). The superior ammonia tolerance of MN4 sites to Pt has been substantiated through density functional theory calculations, elucidating markedly greater adsorption capacity and bonding strength between MN4 and O2 than MN4 and NH3. In contrast, with Pt, the observed adsorption capacity and bonding strength trends diverged from those on MN4 sites, culminating in a notable decline in activity upon exposure to ammonia.
In addition to maintaining precise control of the environment around each single metal atom, modulating the content of metal atoms in the catalyst is also crucial to achieve high performance.179 Joh et al. reported that single atoms could form small metallic nanoclusters or carbide nanocrystals when the content of the metal atom was increased.179 They also found that the Fe–N/C catalyst underwent critical losses in ORR activity, durability, and ammonia resistance by the undesired formation of Fe or Fe3C nanostructures. Hence, to successfully apply Fe–N/C catalysts in DAFC, an optimized loading amount should be selected. These results indicate that the meticulous optimization of fundamental material synthesis can assist in realizing highly active and durable catalysts and ultimately provide insights into efficient and robust electrochemical energy conversion devices.
5 Conclusions
This review summarizes recent progress in the development of DAFC catalysts. We first introduced the operating principles and critical issues in electrode reactions for DAFCs. With PGM catalysts, poisoning issues due to the strong adsorption of N-intermediates and ammonia crossover represent a formidable challenge. For non-PGM catalysts, the high overpotential for each electrode reaction leads to undesirable side reactions and low catalytic performance. We point out that alleviating the catalytic poisoning of PGM catalysts and modifying the electronic structures of non-PGM catalysts are central to catalyst optimization. By categorizing the various structural/compositional optimization strategies for enhancing catalytic activity, we illustrate the current status of catalyst development for AOR and ORR. We also provided several activity comparison tables exhibiting each approach and its enhancement of activity and stability, providing a comprehensive understanding of catalyst design strategies.Although many electrocatalysts have been developed using rational design principles, including logical choices in the composition and catalyst structures, the catalytic performances for AOR and ORR still need to be improved before they can be practically used in DAFC systems. A summary of the reported performance of the DAFC MEA systems is given in Table 7. Here, we suggest several approaches for accelerating the advancement of DAFC catalysts: compositional tuning, structural tuning, functional additive, and protective media.
| Membrane | Fuel | Anode catalyst | Cathode catalyst | Temperature (°C) | Peak power density (mW cm−2) | Ref. |
|---|---|---|---|---|---|---|
| CPPO-PVA blend membrane | Wet hydrogen, aqueous ammonia solution and ammonia gas | Cr-decorated Ni/C | MnO2/C | Room temperature | 16 | 16 |
| Quaternary ammonium functional group-based membrane | Gaseous ammonia | Pt/C | Pt/C | 25 | 6.4 | 18 |
| AMI-7001 anion exchange membrane | Gaseous ammonia | Pt/C | Pt/C | 25 | 6.2 | 20 |
| 80 | 7.1 | |||||
| HEM:PiperION membrane |
NH3 and N2 | PtIr/C | Acta 4020 | 60 | 3.3 | 21 |
| 95 | 17.7 | |||||
| Fumasep FAS-30 | Ammonia and 3 (M) KOH | PtRu/C | Pd/C | 95 | 20.7 | 23 |
| Nafion® 117 | 1 M (KOH + NH4OH) | Pt/C-ITO | Pt/C | 50 | 3.3 | 53 |
| FAA-3-50 (fumapem) | 0.5 M NH4OH solution | Pt cubesH-S | Pt/C | 50 | 42.4 | 72 |
| Nafion® 117 | 3 M (KOH + NH4OH) | Pt9Rh | Pt/C | 50 | 5.37 | 80 |
| Nafion® 117 | 5M NH4OH and 1 M KOH | Pt7Au3 | Pt/C | 40 | 2.64 | 82 |
| PVC | 1 M NH3 and 3 M KOH | Ni50Cu50/CNTs | Pt/C | 25 | 10.94 | 102 |
| Anion exchange membrane | 7 M NH3 + 3 M KOH | Ni4Cu1Co1.5 | Mn-Co-BP2000 | 80 | 115.7 | 106 |
| PiperION PAP-TP-85 | 7 M NH3 + 1.25 M KOH | PtIr/C | Acta 4020 | 95 | 390 | 150 |
| PAP-TP-85 | 16 M NH3 | PtIr/C | Fe–N–C | 120 | 180 | 154 |
| Alkaline polymer electrolyte membrane | 3 M NH3 + 3 M KOH | PtIr/C | Mn-Co-BP2000 | 80 | 128.2 | 165 |
| Fumapem FAA | Concentrated ammonia solution + 1M KOH | NiCu/C | SrCo0.8Cu0.1Nb0.1O3−δ | Room temperature | 0.25 | 170 |
| Co-impregnated ARE-PVA membrane | 5 M NH3 + 1M KOH | Pt/C | SrFe0.8Cu0.1Nb0.1O3−δ | Room temperature | 0.35 | 171 |
| PiperION-A20-HCO3 TP-85 | 7 M NH4OH + 1 M KOH | PtIr/C | LaCr0.25Fe0.25Co0.5O3–δ | 100 | 34 | 172 |
| PiperION-A20-HCO3 TP-85 | 7 M NH4OH + 1 M KOH | PtIr/C | LaCr0.25Fe0.25Co0.5O3–δ | 95 | 30.1 | 173 |
Early investigations of the reaction mechanisms and deep insights on the control of intermediates binding energy by modulating the d-band structure of catalytic surfaces provide a wide range of diverse sources for the design of advanced catalysts, paving the way for catalyst development and electrode preparation. The multimetallic solid-state compounds, including intermetallic phases,180–184 high-entropy alloys,185,186 and interstitial compounds187,188 prepared by compositional modification and phase engineering, offer a myriad of opportunities to tune the electronic structures of catalytic surfaces and modulate the intermediate binding energy, thereby increasing the number of catalytic events. Besides, non-precious metal catalysts, with the exception of Ni-based materials, have yet to undergo extensive compositional exploration, necessitating further efforts in this area. High-throughput computational screening and machine learning can expedite the discovery of new candidates by predicting activity and stability, reducing dependence on trial-and-error methods.
Structural tuning is also an important direction for optimizing catalytic activity. As it is well-known that Pt(100) is highly active and selective for AOR, three-dimensional (3D) nanoarchitectures with a high proportion of Pt(100) facets (such as cubic nanocages) can be viable for promoting AOR.189,190 Furthermore, for non-PGM catalysts, 3D nanoarchitectures can provide rapid improvement in performance through high specific surface area and nanoscale confinement effects, and are expected to reduce overpotential toward both AOR and ORR effectively. In the case of SAC, the way of attaining high loadings of atomically dispersed sites is crucial.191 Furthermore, it is essential to develop scalable synthetic methods to maintain these advanced catalysts' structural/compositional integrity at mass production. These methods can bridge the gap between laboratory-scale catalysis studies and realistic device performance. A synthetic flow-reactor or advanced sol–gel process could be useful.
To ensure that the catalysts perform optimally under practical operating conditions, catalyst development should align closely with advancements in DAFC system designs, such as innovative membranes and additives. Particularly, functional additives can be pivotal, to suppress the ammonia-induced initial performance drop. For example, related to AOR, highly active catalysts such as Pt nanocubes can be improved by incorporating or combining other functional inorganic compounds. The tandem catalyst particles or electrodes, which employ functional additives that mitigate the poisoning Pt surfaces or cocatalysts by scavenging the reaction intermediates strongly adsorbed on the Pt surfaces, are anticipated to boost AOR selectivity and help stabilize the AOR process.192,193 For the ORR, Pt-based catalysts in alkaline environments often suffer severe disintegration. Using non-PGM additives with high selectivity toward AOR in the ORR catalysts or electrodes is expected to mitigate the catalyst poisoning caused by ammonia crossing over from the anode.194 Additives with antioxidation efficacy can also directly reduce the oxophilicity of Pt catalytic surfaces. The reduced oxophilicity can lower the frequency of redox reaction of Pt surfaces during the electrochemical process, leading to retarded dissolution of the Pt species and enhanced stability.139 Establishing a universal stability metric is also crucial to ensuring the stable application of these materials at the system level.
A protective medium is considerably important to enhancing durability. For AOR, coating thin carbon atomic layers195 or porous protective layers196 onto the surfaces of highly active nanoarchitectures like Pt nanocubes can suppress the structural deformation of the catalyst particles, helping achieve long-term stability. For the ORR, highly porous carbon support materials can limit the migration of Pt nanoparticles, Ostwald ripening, and particle detachment via spatial shielding, resulting in the long-term operation of fuel cell cathodes.197
Given the current state of efforts, the prospect of developing an efficient and robust catalyst for operating DAFC appears promising in the near future. Furthermore, we anticipate that our findings will offer valuable insights for the design of advanced catalysts. With continued interdisciplinary efforts and novel approaches, we believe that the development of efficient and durable DAFC catalysts is within reach, paving the way for a sustainable energy society.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KIST Institutional Program (2E33281). This work was also supported by the National Research Foundation of Korea (NRF) grants funded by the Ministry of Science and ICT (MSIT), which are (NRF-2022M3J1A1085384), (No. RS-2023-00256106), (No. RS-2023-00207831), (No. RS-2024-00346153), Future Hydrogen Original Technology Development Program (NRF-2021M3I3A1082879). Furthermore, this work was supported by the Korea Institute Energy Technology Evaluation and Planning (KETEP) grant funded by the Ministry of Trade, Industry, and Energy (MOTIE) (no. 20213030030260). This research was supported by the Nano & Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (RS-2024-00449629).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Y. Lei, Z. Wang, D. Wang, X. Zhang, H. Che, X. Yue, C. Tian, J. Zhong, L. Guo, L. Li, H. Zhou, L. Liu and Y. Xu, Nat. Clim. Change, 2023, 13, 693–700 CrossRef.
- A. Slameršak, G. Kallis and D. W. O'Neill, Nat. Commun., 2022, 13, 6932 CrossRef PubMed.
- R. F. Service, Science, 2004, 303, 29 CrossRef CAS PubMed.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462–474 CrossRef CAS.
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- S. van Renssen, Nat. Clim. Change, 2020, 10, 799–801 CrossRef.
- D. Castelvecchi, Nature, 2022, 611, 440–443 CrossRef CAS PubMed.
- M. B. Bertagni, S. W. Pacala, F. Paulot and A. Porporato, Nat. Commun., 2022, 13, 7706 CrossRef CAS PubMed.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- O. Siddiqui and I. Dincer, Therm. Sci. Eng. Prog., 2018, 5, 568–578 CrossRef.
- R. Abbasi, B. P. Setzler, J. Wang, Y. Zhao, T. Wang, S. Gottesfeld and Y. Yan, Curr. Opin. Electrochem., 2020, 21, 335–344 CrossRef CAS.
- Y. Guo, Z. Pan and L. An, J. Power Sources, 2020, 476, 228454 CrossRef CAS.
- G. Jeerh, M. Zhang and S. Tao, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- R. Lan and S. Tao, Electrochem. Solid-State Lett., 2010, 13, B83–B86 CrossRef CAS.
- S. Gottesfeld, J. Electrochem. Soc., 2018, 165, J3405–J3412 CrossRef CAS.
- O. Siddiqui and I. Dincer, Fuel Cells, 2018, 18, 379–388 CrossRef CAS.
- Y. Zhao, B. P. Setzler, J. Wang, J. Nash, T. Wang, B. Xu and Y. Yan, Joule, 2019, 3, 2472–2484 CrossRef CAS.
- O. Siddiqui and I. Dincer, Chem. Eng. Sci., 2019, 200, 285–293 CrossRef CAS.
- Y. Zhao, T. Wang, B. P. Setzler, R. Abbasi, J. Wang and Y. Yan, ACS Energy Lett., 2021, 6, 1996–2002 CrossRef CAS.
- S. Chatterjee, R. K. Parsapur and K.-W. Huang, ACS Energy Lett., 2021, 6, 4390–4394 CrossRef CAS.
- Y. Liu, Z. Pan, O. C. Esan, X. Xu and L. An, Energy Fuels, 2022, 36, 13203–13211 CrossRef CAS.
- Y. Sun, S. Polani, F. Luo, S. Ott, P. Strasser and F. Dionigi, Nat. Commun., 2021, 12, 5984 CrossRef CAS PubMed.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- H. Kim, J. M. Yoo, D. Y. Chung, Y. Kim, M. Jung, M. S. Bootharaju, J. Kim, S. Koo, H. Shin, G. Na, B. S. Mun, J. H. Kwak, S.-E. Sung and T. Hyeon, ACS Nano, 2022, 16, 16529–16538 CrossRef CAS PubMed.
- K. Siddharth, Y. Chan, L. Wang and M. Shao, Curr. Opin. Electrochem., 2018, 9, 151–157 CrossRef CAS.
- H. Gerischer and A. Mauerer, J. Electroanal. Chem. Interfacial Electrochem., 1970, 25, 421–433 CrossRef CAS.
- K. Endo, Y. Katayama and T. Miura, Electrochim. Acta, 2005, 50, 2181–2185 CrossRef CAS.
- N. J. Bunce and D. Bejan, Electrochim. Acta, 2011, 56, 8085–8093 CrossRef CAS.
- K. Siddharth, P. Alam, M. D. Hossain, N. Xie, G. S. Nambafu, F. Rehman, J. W. Y. Lam, G. Chen, J. Cheng, Z. Luo, G. Chen, B. Z. Tang and M. Shao, J. Am. Chem. Soc., 2021, 143, 2433–2440 CrossRef CAS PubMed.
- E. Müller and F. Spitzer, Z. Elektrochem., 1905, 11, 917–931 CrossRef.
- T. Katan and R. J. Galiotto, J. Electrochem. Soc., 1963, 110, 1022–1023 CrossRef CAS.
- E. J. Cairns, E. L. Simons and A. D. Tevebaugh, Nature, 1968, 217, 780–781 CrossRef CAS.
- S. Wasmus, E. J. Vasini, H. T. Mishima and W. Vielstich, Electrochim. Acta, 1994, 39, 23–31 CrossRef CAS.
- F. J. Vidal-Igelsias, J. Solla-Gullón, J. M. Feliu, H. Baltruschat and A. Aldaz, J. Electroanal. Chem., 2006, 588, 331–338 CrossRef.
- F. J. Vidal-Igelsias, J. Solla-Gullón, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2006, 8, 102–106 CrossRef.
- W. Peng, L. Xia, B. Huang, L. Zhuang and J. Lu, J. Phys. Chem. C, 2011, 115, 23050–23056 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, J. Electroanal. Chem., 2001, 506, 127–137 CrossRef CAS.
- J. A. Herron, P. Ferrin and M. Mavrikakis, J. Phys. Chem. C, 2015, 119, 14692–14701 CrossRef CAS.
- T. Matsui, S. Suzuki, Y. Katayama, K. Yamauchi, T. Okanishi, H. Muroyama and K. Eguchi, Langmuir, 2015, 31, 11717–11723 CrossRef CAS PubMed.
- H. S. Pillai and H. Xin, Ind. Eng. Chem. Res., 2019, 58, 10819–10828 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, J. M. Pérez and A. Aldaz, Electrochem. Commun., 2006, 8, 102–106 CrossRef CAS.
- C. Zhong, W. B. Hu and Y. F. Cheng, J. Power Sources, 2011, 196, 8064–8072 CrossRef CAS.
- J. Liu, C. Zhong, Y. Yang, Y. T. Wu, A. K. Jiang, Y. D. Deng, Z. Zhang and W. B. Hu, Int. J. Hydrogen Energy, 2012, 37, 8987 Search PubMed.
- J. Liu, W. Hu, C. Zhong and Y. F. Cheng, J. Power Sources, 2011, 196, 8064–8072 CrossRef.
- L. Cunci, C. A. Velez, I. Perez, A. Suleiman, E. Larios, M. Jose-Yacaman, J. J. Watkins and C. R. Cabrera, ACS Appl. Mater. Interfaces, 2014, 6, 2137–2145 CrossRef CAS PubMed.
- C. M. Poventud-Estrada, R. Acevedo, C. Morales, L. Betancourt, D. C. Diaz, M. A. Rodriguez, E. Larios, M. Jose-Yacaman, E. Nicolau, M. Flynn and C. R. Cabrera, Microgravity Sci. Technol., 2017, 29, 381–389 CrossRef CAS.
- V. A. Ribeiro, I. C. de Freitas, A. O. Neto, E. V. Spinacé and J. C. M. Silva, Mater. Chem. Phys., 2017, 200, 354–360 CrossRef CAS.
- L. L. Sikeyi, T. D. Ntuli, T. H. Mongwe, N. W. Maxakato, N. J. Coville and M. S. Maubane-Nkadimeng, Electroanal. Chem., 2022, 917, 116411 CrossRef CAS.
- K. Siddharth, Y. Wang, J. Wang, F. Xiao, G. Nambafu, U. Shahid, F. Yang, E. Delmo and M. Shao, Front. Chem. Sci. Eng., 2022, 16, 930–938 CrossRef CAS.
- J. Liu, B. Liu, Z. Ni, Y. Deng, C. Zhong and W. Hu, Electrochim. Acta, 2014, 150, 146–150 CrossRef CAS.
- J. C. M. Silva, R. M. Piasentin, E. V. Spinacé, A. O. Neto and E. A. Baranova, Mater. Chem. Phys., 2016, 180, 97–103 CrossRef CAS.
- Z.-F. Li, Y. Wang and G. G. Botte, Electrochim. Acta, 2017, 228, 351–360 CrossRef CAS.
- H. Kim, M. W. Chung and C. H. Choi, Electrochem. Commun., 2018, 94, 31–35 CrossRef CAS.
- F. J. Vidal-Iglesias, N. García-Aráez, V. Montiel, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2003, 5, 22–26 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, J. M. Feliu and A. Aldaz, J. Phys. Chem. B, 2005, 109, 12914–12919 CrossRef CAS PubMed.
- J. Solla-Gullón, F. J. Vidal-Iglesias, P. Rodríguez, E. Herrero, J. M. Feliu, J. Clavilier and A. Aldaz, J. Phys. Chem. B, 2004, 108, 13573–13575 CrossRef.
- E. Bertin, C. Roy, S. Garbarino, D. Guay, J. Solla-Gullón, F. J. Vidal-Iglesias and J. M. Feliu, Electrochem. Commun., 2012, 5, 197–199 CrossRef.
- E. Bertin, S. Garbarino, D. Guay, J. Solla-Gullón, F. J. Vidal-Iglesias and J. M. Feliu, J. Power Sources, 2013, 225, 323–329 CrossRef CAS.
- V. Rosca and M. T. M. Koper, Phys. Chem. Chem. Phys., 2006, 8, 2513–2524 RSC.
- I. Katsounaros, T. Chen, A. A. Gewirth, N. M. Markovic and M. T. M. Koper, J. Phys. Chem. Lett., 2016, 7, 387–392 CrossRef CAS PubMed.
- I. Katsounaros, M. C. Figueiredo, F. Calle-Vallejo, H. Li, A. A. Gewirth, N. M. Markovic and M. T. M. Koper, J. Catal., 2018, 359, 82–91 CrossRef CAS.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- M. A. Montiel, F. J. Vidal-Iglesias, V. Montiel and J. Solla-Gullón, Curr. Opin. Electrochem., 2017, 1, 34–39 CrossRef CAS.
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735 CrossRef CAS PubMed.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- H. Yang, Y. Tang and S. Zou, Electrochem. Commun., 2014, 38, 134–137 CrossRef CAS.
- C. Zhang, S. Y. Hwang and Z. Peng, J. Mater. Chem. A, 2013, 1, 14402–14408 RSC.
- R. A. Martínez-Rodríguez, F. J. Vidal-Iglesias, J. Solla-Gullón, C. R. Cabrera and J. M. Feliu, J. Am. Chem. Soc., 2014, 136, 1280–1283 CrossRef PubMed.
- J. Liu, X. Fan, X. Liu, Z. Song, Y. Deng, X. Han, W. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 18856–18864 CrossRef CAS PubMed.
- H. Jin, S. Lee, Y. Sohn, S.-H. Lee, P. Kim and S. J. Yoo, Int. J. Energy Res., 2021, 45, 18281–18291 CrossRef CAS.
- Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Phys. Chem. C, 2015, 119, 9134–9141 CrossRef CAS.
- Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Catal., 2016, 344, 496–506 CrossRef CAS.
- Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Catal., 2016, 6, 2026–2034 CrossRef CAS.
- K. Siddharth, Y. Hong, X. Qin, H. J. Lee, Y. T. Chan, S. Zhu, G. Chen, S.-I. Choi and M. Shao, Appl. Catal., B, 2020, 269, 118821 CrossRef CAS.
- S. Le Vot, L. Roué and D. Bélanger, J. Power Sources, 2013, 223, 221–231 CrossRef CAS.
- S. Morita, E. Kudo, R. Shirasaka, M. Yonekawa, K. Nagai, H. Ota, M. N. Gamo and H. Shiroishi, J. Electroanal. Chem., 2016, 762, 29–36 CrossRef CAS.
- J. Jiang, Electrochem. Commun., 2017, 75, 52–55 CrossRef CAS.
- M. H. M. T. Assumpção, R. M. Piasentin, P. Hammer, R. F. B. De Souza, G. S. Buzzo, M. C. Santos, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Appl. Catal., B, 2015, 174–175, 136–144 CrossRef.
- Z. Liu, Y. Li, X. Zhang, S. Rao, J. Li, W. Wang, Z. Sun and J. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 28816–28825 CrossRef CAS PubMed.
- J. C. M. Silva, S. G. da Silva, R. F. B. De Souza, G. S. Buzzo, E. V. Spinacé, A. O. Neto and M. H. M. T. Assumpção, Appl. Catal., A, 2015, 490, 133–138 CrossRef CAS.
- Y. Kang, W. Wang, J. Li, C. Hua, S. Xue and Z. Lei, Int. J. Hydrogen Energy, 2017, 42, 18959–18967 CrossRef CAS.
- Y. T. Chan, K. Siddharth and M. Shao, Nano Res., 2020, 13, 1920–1927 CrossRef CAS.
- W. Peng, L. Xiao, B. Huang, L. Zhuang and J. Lu, J. Phys. Chem. C, 2011, 115, 23050–23056 CrossRef CAS.
- Y. Li, X. Li, H. Pillai, J. Lattimer, N. Mohd Adli, S. G. Karakalos, M. Chen, L. Guo, H. Xu, J. Yang, D. Su, H. Xin and G. Wu, ACS Catal., 2020, 10, 3945–3957 CrossRef CAS.
- Q. Xue, Y. Zhao, J. Zhu, Y. Ding, T. Wang, H. Sun, F. Li, P. Chen, P. Jin, S. Yin and Y. Chen, J. Mater. Chem. A, 2021, 9, 8444–8451 RSC.
- X. Ren, Y. Wang, A. Liu, Z. Zhang, Q. Lv and B. Liu, J. Mater. Chem. A, 2020, 8, 24284–24306 RSC.
- A. O. Elnabawy, J. A. Herron, S. Karraker and M. Mavrikakis, J. Catal., 2021, 397, 137–147 CrossRef CAS.
- J. Łuczak and M. Lieder, Adv. Colloid Interface Sci., 2023, 319, 102963 CrossRef PubMed.
- A. Kapałka, A. Cally, S. Neodo, C. Comninellis, M. Wächter and K. M. Udert, Electrochem. Commun., 2010, 12, 18–21 CrossRef.
- Y.-J. Shih, Y.-H. Huang and C. P. Huang, Electrochim. Acta, 2018, 281, 410–419 CrossRef CAS.
- S. Zhang, L. Yan, H. Jiang, Y. Zhao, L. Yang, Q. Fu, E. Guo, J. Zhang and X. Zhao, Sci. China Mater., 2023, 66, 2715–2725 CrossRef CAS.
- R. M. Choueiri and L. D. Chen, J. Phys. Chem. C, 2022, 126, 17952–17965 CrossRef CAS.
- R. M. Choueiri, S. W. Tatarchuk, A. Klinkova and L. D. Chen, Electrochem. Sci. Adv., 2022, 2, e2100142 CrossRef CAS.
- S. He, V. Somayaji, M. Wang, S.-H. Lee, Z. Geng, S. Zhu, P. Novello, C. V. Varanasi and J. Liu, Nano Res., 2022, 15, 4785–4791 CrossRef CAS.
- K. Nagita, Y. Yuhara, K. Fujii, Y. Katayama and M. Nakayama, ACS Appl. Mater. Interfaces, 2021, 13, 28098–28107 CrossRef CAS PubMed.
- W. Xu, R. Lan, D. Du, J. Humphreys, M. Walker, Z. Wu, H. Wang and S. Tao, Appl. Catal., B, 2017, 218, 470–479 CrossRef CAS.
- J. Huang, J. Cai and J. Wang, ACS Appl. Energy Mater., 2020, 3, 4108–4113 CrossRef CAS.
- A. Yang, J. Wang, K. Su, W. Lei, X. Qiu and Y. Tang, Chem. - Eur. J., 2021, 27, 4869–4875 CrossRef CAS PubMed.
- H. Zhang, H. Wang, X. Tong, L. Zhou, X. Yang, Y. Wang, M. Zhang and Z. Wu, Chem. Eng. J., 2023, 452, 139582 CrossRef CAS.
- H. M. Zhang, Y. F. Wang, Y. H. Kwok, Z. C. Wu, D. H. Xia and D. Y. C. Leung, ChemSusChem, 2018, 11, 2889–2897 CrossRef CAS PubMed.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. S. Irvine and S. Tao, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- H. K. Vu, T. Mahvelati-Shamsabadi, T. T. Dang, S. H. Hur, S. G. Kang and J. S. Chung, ACS Appl. Nano Mater., 2023, 6, 20688–20699 CrossRef CAS.
- H. Wang, X. Tong, L. Zhou, Y. Wang, L. Liao, S. Ouyang and H. Zhang, Sep. Purif. Technol., 2022, 303, 122293 CrossRef CAS.
- Z. Hu, S. Lu, F. Tang, D. Yang, C. Zhang, Q. Xiao and P. Ming, Appl. Catal., B, 2023, 334, 122856 CrossRef CAS.
- M. Zhang, J. Zhang, G. Jeerh, P. Zou, B. Sun, M. Walker, K. Xie and S. Tao, J. Mater. Chem. A, 2022, 10, 18701–18713 RSC.
- M. Zhu, Y. Yang, S. Xi, C. Diao, Z. Yu, W. S. V. Lee and J. Xue, Small, 2021, 17, 2005616 CrossRef CAS PubMed.
- F. Almomani and M. A. H. S. Saad, Int. J. Hydrogen Energy, 2021, 46, 4678–4690 CrossRef CAS.
- J. Hou, Y. Cheng, H. Pan and P. Kang, Inorg. Chem., 2023, 62, 3986–3992 CrossRef CAS PubMed.
- A. Allagui, S. Sarfraz and E. A. Baranova, Electrochim. Acta, 2013, 110, 253–259 CrossRef CAS.
- Y. Jin, X. Chen and J. Wang, Chem. Commun., 2022, 58, 10631–10634 RSC.
- J. Cui, J. Hou, H. Pan and P. Kang, J. Electroanal. Chem., 2023, 940, 117502 CrossRef CAS.
- J. Huang, Z. Chen, J. Cai, Y. Jin, T. Wang and J. Wang, Nano Res., 2022, 15, 5987–5994 CrossRef CAS.
- M.-H. Tsai, T.-C. Chen, Y. Juang, L.-C. Hua and C. Huang, Electrochem. Commun., 2020, 121, 106875 CrossRef CAS.
- J. Hou, Y. Cheng, H. Pan and P. Kang, ChemElectroChem, 2022, 9, e202101301 CrossRef CAS.
- A. Cleetus, H. Teller and A. Schechter, ChemCatChem, 2023, 15, e202300035 CrossRef CAS.
- S. Zhang, L. Yan, H. Jiang, L. Yang, Y. Zhao, X. Yang, Y. Wang and J. Shen, ACS Appl. Mater. Interfaces, 2022, 14, 9036–9045 CrossRef CAS PubMed.
- S. Zhang, Y. Zhao, L. Yan, H. Jiang, X. Yang, Y. Wang, H. Song and X. Zhao, Int. J. Hydrogen Energy, 2021, 46, 39208–39215 CrossRef CAS.
- E. L. Simons, E. J. Cairns and D. J. Surd, J. Electrochem. Soc., 1969, 116, 556–561 CrossRef CAS.
- S. Suzuki, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2012, 208, 257–262 CrossRef CAS.
- H. Kwon, M. K. Kabiraz, J. Park, A. Oh, H. Baik, S.-I. Choi and K. Lee, Nano Lett., 2018, 18, 2930–2936 CrossRef CAS PubMed.
- M. K. Kabiraz, B. Ruqia, J. Kim, H. Kim, H. J. Kim, Y. Hong, M. J. Kim, Y. K. Kim, C. Kim, W.-J. Lee, W. Lee, G. H. Hwang, H. C. Ri, H. Baik, H.-S. Oh, Y. W. Lee, L. Gao, H. Huang, S. M. Paek, Y.-J. Jo, C. H. Choi, S. W. Han and S.-I. Choi, ACS Catal., 2022, 12, 3516–3523 CrossRef CAS.
- H. C. Kim, R. K. Pramadewandaru, M. K. Kabiraz, G. A. B. Azizar, H. Wahidah, Y. Kim, S.-U. Lee, H.-J. Chae, S.-I. Choi and J. W. Hong, ACS Catal., 2024, 14, 3756–3765 CrossRef CAS.
- J. Zhang, M. B. Vukmirovic, K. Sasaki, A. U. Nilekar, M. Mavrikakis and R. R. Adzic, J. Am. Chem. Soc., 2005, 127, 12480–12481 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, L. Bech and I. Chorkendorff, ChemCatChem, 2012, 4, 341–349 CrossRef CAS.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velazquez-Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. Zhang, H. Z. Yang, J. Y. Fang and S. Z. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu, J. Zhao, Y. Liu, H. Yuan, F. Abild-Pedersen, F. B. Prinz, J. K. Nørskov and Y. Cui, Science, 2016, 354, 1031–1036 CrossRef CAS PubMed.
- R. Halseid, M. Heinen, Z. Jusys and R. J. Behm, J. Power Sources, 2008, 176, 435–443 CrossRef CAS.
- A. Verdaguer-Casadevall, P. Hernandez-Fernandez, I. E. L. Stephens, I. Chorkendorff and S. Dahl, J. Power Sources, 2012, 220, 205–210 CrossRef CAS.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS PubMed.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- H. Y. Kim, Y. Lee, J. Kim, J. Woo, Y. Jun, H. J. Son, K. An and J. Y. Kim, Chem. Eng. J., 2023, 476, 146662 CrossRef CAS.
- T. Lopes, J. Chlistunoff, J.-M. Sansinena and F. H. Garzon, Int. J. Hydrogen Energy, 2012, 37, 5202–5207 CrossRef CAS.
- K. R. Lee, D. Song, S. B. Park and J.-i. Han, RSC Adv., 2014, 4, 5638–5641 RSC.
- S. Ishiyama, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, MRS Bull., 2019, 119, 110561 CrossRef CAS.
- A. Zadick, L. Dubau, N. Sergent, G. Berthomé and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS.
- T. J. P. Hersbach, A. C. Garcia, T. Kroll, D. Sokaras, M. T. M. Koper and A. T. Garcia-Esparza, ACS Catal., 2021, 11, 9904–9915 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2014, 7, 1739–1744 CrossRef CAS PubMed.
- Z. Cui, L. Li, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 7278–7281 CrossRef CAS PubMed.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- R. Abbasi, H. Wang, J. R. C. Lattimer, H. Xu, G. Wu and Y. Yan, J. Electrochem. Soc., 2020, 167, 164510 CrossRef CAS.
- T. Wang, Y. Zhao, B. P. Setzler and Y. Yan, J. Electrochem. Soc., 2021, 168, 014507 CrossRef CAS.
- Y. Liu, Z. Pan, O. C. Esan, X. Xu and L. An, Energy Fuels, 2022, 36, 13203–13211 CrossRef CAS.
- A. Holewinski, J.-C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef CAS PubMed.
- W. Wang, J.-Q. Chen, Y.-R. Tao, S.-N. Zhu, Y.-X. Zhang and X.-C. Wu, ACS Catal., 2019, 9, 3498–3510 CrossRef CAS.
- B. Achrai, Y. Zhao, T. Wang, G. Tamir, R. Abbasi, B. P. Setzler, M. Page, Y. Yan and S. Gottesfeld, J. Electrochem. Soc., 2020, 167, 134518 CrossRef CAS.
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654–2675 RSC.
- M. Piana, S. Catanorchi and H. A. Gasteiger, ECS Trans., 2008, 16, 2045–2055 CrossRef CAS.
- M. H. Robson, A. Serov, K. Artyushkova and P. Atanassov, Electrochim. Acta, 2013, 90, 656–665 CrossRef CAS.
- J. B. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333–5339 RSC.
- Y. J. Sa, D. J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T. Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- R. Lan and S. Tao, Electrochem. Solid-State Lett., 2010, 13, B83–B86 CrossRef CAS.
- M. Zhang, P. Zou, G. Jeerh, S. Chen, J. Shields, H. Wang and S. Tao, ACS Sustainable Chem. Eng., 2020, 8, 12817–12824 CrossRef CAS.
- Z. Hu, Q. Xiao, D. Xiao, Z. Wang, F. Gui, Y. Lei, J. Ni, D. Yang, C. Zhang and P. Ming, ACS Appl. Mater. Interfaces, 2021, 13, 53945–53954 CrossRef CAS PubMed.
- Z. Hu, D. Yang, C. Zhang, P. Ming and Q. Xiao, Energy Fuels, 2023, 37, 8600–8613 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. I. Jung, M. Risch, S. Park, M. G. Kim, G. Nam, H. Y. Jeong, Y. Shao-Horn and J. Cho, Energy Environ. Sci., 2016, 9, 176–183 RSC.
- P. Zou, S. Chen, R. Lan and S. Tao, ChemSusChem, 2019, 12, 2788–2794 CrossRef CAS PubMed.
- P. Zou, S. Chen, R. Lan, J. Humphreys, G. Jeerh and S. Tao, Int. J. Hydrogen Energy, 2019, 44, 26554–26564 CrossRef CAS.
- G. Jeerh, P. Zou, M. Zhang and S. Tao, Appl. Catal., B, 2022, 319, 121919 CrossRef CAS.
- G. Jeerh, P. Zou, M. Zhang and S. Tao, ACS Appl. Mater. Interfaces, 2023, 15, 1029–1041 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- F. Xiao, Y. C. Wang, Z. P. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J. C. Li, C. J. Zhong, Z. Y. Zhou and M. Shao, Adv. Mater., 2021, 33, 2006292 CrossRef CAS PubMed.
- Z. Wu, Y. Wang, Y. Cao, B. Wang, Z. Sun, J. Yang and Y. Li, Adv. Funct. Mater., 2023, 33, 2301084 CrossRef CAS.
- H. W. Jang, S. Y. Lee, J. Y. Lee and H. I. Joh, Carbon Lett., 2023, 33, 215–223 CrossRef CAS.
- H. Y. Kim and S. H. Joo, J. Mater. Chem. A, 2020, 8, 8195–8217 RSC.
- H. Y. Kim, J. Y. Kim and S. H. Joo, Bull. Korean Chem. Soc., 2021, 42, 724–736 CrossRef CAS.
- H. Y. Kim, M. Jun, S. H. Joo and K. Lee, ACS Nanosci. Au, 2023, 3, 28–36 CrossRef CAS PubMed.
- S. Yu and H. Yang, Chem. Commun., 2023, 59, 4852–4871 RSC.
- F. Lin, M. Li, L. Zeng, M. Luo and S. Guo, Chem. Rev., 2023, 123, 12507–12593 CrossRef CAS PubMed.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- F. Xing, J. Ma, K. Shimizu and S. Furukawa, Nat. Commun., 2022, 13, 5065 CrossRef CAS PubMed.
- S. H. Joo and J. S. Lee, J. Catal., 2021, 404, 911–924 CrossRef CAS.
- J. Lee, S. W. Choi, S. Back, H. Jang and Y. J. Sa, Appl. Catal., B, 2022, 309, 121265 CrossRef CAS.
- J. Park, T. Kwon, J. Kim, H. Jin, H. Y. Kim, B. Kim, S. H. Joo and K. Lee, Chem. Soc. Rev., 2018, 47, 8173–8202 RSC.
- H. Y. Kim, M. Jun, K. Lee and S. H. Joo, ACS Catal., 2023, 13, 355–374 CrossRef CAS.
- J. H. Kim, Y. J. Sa, T. Lim, J. Woo and S. H. Joo, Acc. Chem. Res., 2022, 55, 2672–2684 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D. H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C. T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rümmerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS PubMed.
- S. M. Jung, S. W. Yun, J. H. Kim, S. H. You, J. Park, S. Lee, S. H. Chang, S. C. Chae, S. H. Joo, Y. Jung, J. Lee, J. Son, J. Snyder, V. Stamenkovic, N. M. Markovic and Y. T. Kim, Nat. Catal., 2020, 3, 639–648 CrossRef CAS.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y. H. Chung, H. Kim, B. S. Mun, K. S. Lee, N. S. Lee, S. J. Yoo, D. H. Lim, K. Kang, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- D. S. Baek, K. A. Lee, J. Park, J. H. Kim, J. Lee, J. S. Lim, S. Y. Lee, T. J. Shin, H. Y. Jeong, J. S. Son, S. J. Kang, J. Y. Kim and S. H. Joo, Angew. Chem., Int. Ed., 2021, 60, 1441–1449 CrossRef CAS PubMed.
Footnote |
| † H. Y. K., J. K., and E. L. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |