
Adsorption enhanced catalytic degradation of airborne formaldehyde in bifunctional composite materials
Xiaomin
Liu
 ,
Wenjing
Miao
,
Linhua
Xie
,
Bin
Li
and
Yufeng
Wu
*
,
Wenjing
Miao
,
Linhua
Xie
,
Bin
Li
and
Yufeng
Wu
*
Institute of Circular Economy, College of Materials Science and Engineering, Beijing University of Technology, Beijing 100124, PR China
First published on 21st January 2025
Abstract
Formaldehyde (HCHO) is one of the well-known carcinogenic VOCs, which poses a serious threat to the environment and human health. Bifunctional composite materials (BFCMs) have great application prospects in the field of HCHO removal due to their impressive adsorption-promoted catalytic synergies and good catalytic stability. However, the urgency of efficient degradation of HCHO at near room temperature poses many challenges to the design of BFCMs, especially the design of components in BFCMs, such as support components (SCs) and catalytic components (CCs). In addition, the influence of multiple components in BFCMs on the degradation of HCHO is highly complex, especially the component synergies and the adsorption-promoted catalysis mechanism. To understand these effects and improve the degradation efficiency of HCHO, we first discuss the encouraging research outcomes in recent years, encompassing the design, preparation, and multilevel synergies in BFCMs (e.g. adsorption-catalytic synergies, heterogeneous synergies within SCs or CCs, structural synergies, and mass and heat transfer synergies). Subsequently, we concentrate on analysing the impacts of support component factors (e.g. types, crystal phases, particle sizes, morphologies, specific surface areas, and acidity) and catalytic component factors (e.g. shapes, morphologies, particle sizes, metal chemistry states, and loading capacities) on the HCHO removal performances. Finally, the degradation mechanism of HCHO and the catalytic stability of BFCMs are concluded.
1 Introduction
As a type of air pollutant with high carcinogenicity and mutagenicity, HCHO is found from many sources (e.g., interior decoration, organic solid waste incineration, and electronic solid waste pyrolysis), and its removal has received extensive attention.1–3 A considerable number of HCHO removal technologies has been developed, mainly including physical adsorption, chemical absorption, catalytic oxidation, plasma decomposition, and biotechnological decomposition.4–6 Among these technologies, catalytic oxidation at room temperature is regarded as the most promising HCHO removal technology.7–9 Besides, various HCHO catalysts (e.g., noble metal catalysts and transition metal oxides/TMO) for application in different humidity and temperature scenarios have been reported. Among them, most noble metal (e.g., Pd, Pt, and Au) based catalysts have been demonstrated to possess excellent HCHO degradation performance at room temperature.2 Unfortunately, they often exhibit the characteristics of low abundance, high cost, poor thermal stability, and susceptibility to toxic inactivation.10 In contrast, TMOs (e.g., MnO2, CeO2, and Co3O4) have been studied to serve as alternatives of noble metal-based catalysts due to their accessibility, competitive price, superior catalytic activity, and environmental friendliness.11 However, most TMO-based catalysts also have their own limitations for HCHO oxidation, such as low catalytic activity at room temperature.12 The possible issues mentioned above constitute a significant obstacle to the further application of catalysts in the degradation of HCHO.To alleviate these problems, noble metals and/or TMO-based catalysts have been studied in depth, such as (i) expanding the range of noble metal species (e.g., Pt, Au, Pd, Ag, and Rh),13 (ii) modifying the particle sizes of catalysts (e.g., mm-Al2O3, μm-MnO2, and nm-Al2O3),14 (iii) investigating the bulk geometry of catalysts (e.g., pellets, monoliths and foams),2 (iv) exploring the crystalline phases of catalysts (e.g., α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2),15 (v) exploring the morphologies of catalysts (e.g., α-MnO2-nanowire, α-MnO2-nanotube, α-MnO2-nanorods, and α-MnO2-nanoflower),16 (vi) exploring the mechanical mixing preparation of different components (e.g., MnOx + UiO-66, MnOx + GO, and Fe2O3 + Mn3O4),17–19 and (vii) designing and synthesizing bifunctional composite materials (BFCMs, e.g., porous carrier-based composite MnO2/UiO-66-NH220 and oxide carrier-based composite Pt/Fe2O3@SnO2).21 Among them, one of the most prominent approaches is the discovery of BFCMs (e.g., 1.5 wt% Pt/TiO2-H200, 0.887 Pt-VO-ZrO2, δ-MnOx/activated carbon, three-dimensionally ordered macroporous (3DOM)∼3 wt% Au/CeO2-Co3O4, 1 wt% MnO2/UIO-66-NH2, 3DOM∼3 wt% Au/CeO2, and GO/MnOx) (Table 1), whose alluring advantages, such as diverse components, structural design, and adjustable functions, have impelled numerous researchers to undertake horizontal and immersive longitudinal studies. Notably, BFCMs specifically refer to the materials that exhibit two or more distinct functionalities, encompassing various combinations of physical, chemical, and biological properties.64 From a transverse perspective, the influence of support components (SCs), catalytic components (CCs), and different preparation processes (e.g., solvothermal method, impregnation method, in situ deposition method, and high-temperature calcination method) on the structure and properties of BFCMs was investigated.17,31,65 Among them, the SCs encompassed various carbon materials (e.g., granular activated carbon, activated carbon fiber, and graphene oxide),12 glass fibers,26 polymers (e.g., silicone-based polymer),7 and MOF materials (such as UIO-66-NH2),20 while the CCs included noble metals, metal oxides, element-doped catalysts, and atomic substitution catalysts. Additionally, the impacts of diverse factors on the structure–activity relationship of BFCMs were investigated from a longitudinal perspective. This mainly included the type of SCs, the specific surface areas of SCs, the crystal phase of CCs/SCs (e.g., Pt/α-MnO2@L-MnO2),66 the loading capacity of CCs in BFCMs, and the doping of different elements in CCs (e.g., N-MnNxO2−x and 0.05 wt% Pt–Na/nano-Al2O3).14,67 Moreover, the enhanced degradation of HCHO was analysed to be associated with several potential factors, such as facilitating the enrichment of gas substrates (e.g., HCHO and O2), augmenting the specific surface areas of CCs, enhancing the dispersion of CCs, increasing interfacial bonding,68 and promoting electron transfer.57 Particularly, the heterogeneous interface based on the synergistic interaction of adsorption-catalytic sites significantly expedited the degradation of HCHO by establishing electron transport channels for catalytic oxidation.24
| BFCMs | BFCMs quality | T (°C) | GHSV (h−1 or mL (g h)−1) | [HCHO]I (ppm or V%) | HCHO removal activity (η′/%) | Ref. |
|---|---|---|---|---|---|---|
| a Coconut shell AC (40–60 mesh). b [HCHO]I (inlet HCHO concentration), η′ (HCHO conversion), and RH (relative humidity). | ||||||
| At room temperature (T ≤ 40°C) | ||||||
| 0.08 wt%Pt–NC/TiO2 (Pt size of 1 nm) | 100 mg | 20 °C | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL (g h)−1 000 mL (g h)−1 |
100 ppm | η′ = 100% (with 50% RH) | 22 |
| F-0.08 wt%Pt/TiO2 | 300 mg | 20 °C | 50000 mL (g h)−1 |
200 ppm | η′ = 89% (with 19% RH) | 23 |
| F-0.1 wt%Pt/TiO2 | 300 mg | 20 °C | 50000 mL (g h)−1 |
200 ppm | η′ = 97% (with 19% RH) | 23 |
| 0.51 wt%Pt-VO-ZrO2 | 100 mg | 20 °C | 600000 mL (g h)−1 |
100 ppm | η′ = 27% (with 30% RH) | 24 |
| 0.87 wt%Pt-VO-ZrO2 | 100 mg | 20 °C | 600000 mL (g h)−1 |
100 ppm | η′ = 95% (with 30% RH) | 24 |
| 1.32 wt%Pt-VO-ZrO2 | 100 mg | 20 °C | 600000 mL (g h)−1 |
100 ppm | η′ = 91% (with 30% RH) | 24 |
| 1 wt%Pt–ZrO2 | 100 mg | 20 °C | 600000 mL (g h)−1 |
100 ppm | η′ = 33% (with 30% RH) | 24 |
| 1 wt%Pd/CeO2 | N/A | 20 °C | 120000 mL (g h)−1 |
600 ppm | η′ = 98% | 25 |
| 0.5 wt%Rh/TiO2 | N/A | 20 °C | 30000 mL (g h)−1 |
140 ppm | η′ = 37% | 13 |
| 1 wt%Rh/TiO2 | N/A | 20 °C | 30000 mL (g h)−1 |
140 ppm | η′ = 88% | 13 |
| 5 wt%Ag@MnO2 | 100 mg | 24 °C | 36000 mL (g h)−1 |
420 ppm | η′ = 100% | 26 |
| 0.015 wt%PtOx@BIT-72-DE | 50 mg | 25 °C | 60000 mL (g h)−1 |
25 ppm | η′ = 100% (with 25% RH) | 27 |
| 0.05 wt%Pt–Na/nano-Al2O3 | 30 mg (60 mesh) | 25 °C | 200000 mL (g h)−1 |
120 ppm | η′ = 100% (with 35% RH) | 28 |
| 0.09 wt%PtSA–MnOOH/MnO2 | 1000 mg | 25 °C | 30000 mL (g h)−1 |
15 ppm | 98% | 29 |
| 0.2 wt%Pt@BEA zeolite | 50 mg | 25 °C | 360000 mL (g h)−1 |
80 ppm | η′ = 95% (with 10% RH) | 30 |
| 0.2 wt%Pt/CeO2-7 | 300 mg | 25 °C | 300000 mL (g h)−1 |
100 ppm | η′ = 92% (with 50% RH) | 31 |
| 0.5 wt%Pt/TiO2-NS | 300 mg | 25 °C | N/A | 253 ppm | η′ = 100% | 32 |
| 1.5 wt%Pt/TiO2-H200 | 10 mg | 25 °C | 600000 mL (g h)−1 |
120 ppm | η′ = 100% (with 50% RH) | 33 |
| 1.5 wt%Pt/SBT-400 | 300 mg | 25 °C | 12600 h−1 |
100 ppm | η′ = 100% | 34 |
| 1 wt%Au/γ-Al2O3 | 10 mg | 25 °C | 600000 mL (g h)−1 |
80 ppm | η′ = 89% (with 50% RH) | 35 |
| 1 wt%Au/SiO2 | 10 mg | 25 °C | 600000 mL (g h)−1 |
80 ppm | η′ = 80% (with 50% RH) | 35 |
| 1 wt%Au/HZSM-5 | 10 mg | 25 °C | 600000 mL (g h)−1 |
80 ppm | η′ = 43% (with 50% RH) | 35 |
| 1 wt%Au/CeO2 | 10 mg | 25 °C | 600000 mL (g h)−1 |
80 ppm | η′ = 31% (with 50% RH) | 35 |
| 1 wt%Au/FeOx | 10 mg | 25 °C | 600000 mL (g h)−1 |
80 ppm | η′ = 18% (with 50% RH) | 35 |
| 1 wt%Pd/KTO–NB | 50 mg | 25 °C | 20000 h−1 |
140 ppm | η′ = 95% (with 25% RH) | 1 |
| 2 wt%Na-1 wt%Pd/TiO2 | 60 mg | 25 °C | 95000 h−1 |
140 ppm | N/A | 36 |
| 3.42 V%MnOx/ACa | 100 mg | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 80.9 (with 30% RH) | 37 |
| 3.58 V%MnOx/AC-N1a | 100 mg | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 90.3 (with 30% RH) | 37 |
| 3.91 V%MnOx/AC-N2a | 100 mg | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 97.1 (with 30% RH) | 37 |
| 4.68 V%MnOx/AC-N3a | 100 mg | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 83.2 (with 30% RH) | 37 |
| α-MnO2/GO–M2∼20 wt% | 100 mg | 25 °C | 120000 mL (g h)−1 |
112 ppm | η′ = 100% (with 70% RH) | 38 |
| 21.55 wt%MnO2@PMIA-2 | N/A | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 71% (with 55% RH) | 39 |
| 23.08 wt%MnO2@PMIA-6 | N/A | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 95.43% (with 55% RH) | 39 |
| 23.54 wt%MnO2@PMIA-10 | N/A | 25 °C | 60000 mL (g h)−1 |
100 ppm | η′ = 78% (with 55% RH) | 39 |
| δ-MnO2@GO-1 wt% | 200 mg | 25 °C | 72000 mL (g h)−1 |
10 ppm | η′ = 98% (with 50% RH) | 40 |
| 25 wt%Ce–MnOx | 300 mg | 25 °C | N/A | 0.97–1.03 ppm | η′ = 73% | 41 |
| (CH3COO)2Co/PAN∼58.79 wt% | 100 mg | 25 °C | 60000 mL (g h)−1 |
10–12 ppm | η′ = 99% | 42 |
| MnO2@SiO2–TiO2 | N/A | 25 °C | N/A | 200 ppm | η′ = 100% (with 40% RH) | 43 |
| Pt/MnOx–CeO2 (Pt–N catalyst) | 200 mg (40–60 mesh) | 25 °C | 30000 mL (g h)−1 |
<100 ppm | η′ = 100% | 44 |
| Pt/MnOx–CeO2 (no hydrogen prereduction) | 200 mg (40–60 mesh) | 25 °C | 30000 mL (g h)−1 |
580 ppm | η′ = 54% | 44 |
| 0.1 wt%Pd/TiO2 | 60 mg | 25 °C | 120000 mL (g h)−1 |
10 ppm | η′ = 97% | 45 |
| 0.1 wt%Pt/TiO2–NS | 100 mg | 30 °C | 210000 mL (g h)−1 |
10 ppm | η′ > 95% (with 50% RH) | 46 |
| Au@Co3O4 | 10 mg | 30 °C | 300000 mL (g h)−1 |
100 ppm | η′ = 92% (with 45% RH) | 47 |
| 0.5 wt%Pd/R-TiO2-30 | 500 mg | 30 °C | 130000 h−1 |
20 ppm | η′ = 94% (with 50% RH) | 48 |
| 1 wt%MnO2/UiO-66-NH2 | 100 mg | 30 °C | 597134 h−1 |
100 ppm | η′ = 88% | 20 |
| 8 wt%Ag/APETS/MCM-41 | N/A | 30 °C | 9000 mL (g h)−1 | N/A | η′ = 6% | 49 |
| 2 wt%Au/CeO2-550 | 50 mg | 36 °C | 36000 mL (g h)−1 |
220 ppm | η′ = 100% | 50 |
| 3DOM∼3%Au/(2.5:1) CeO2–Co3O4 |
200 mg | 39 °C | 15000 mL (g h)−1 |
8 ppm | η′ = 100% | 51 |
| 3DOM∼3%Au/CeO2 | 200 mg | 39 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 100% | 52 |
| 2.30 wt%Au/CeO2 | 50 mg | 40 °C | 35400 h−1 |
0.05 V% | η′ = 80% | 53 |
| 2.22 wt%Au/TiO2 | 50 mg | 40 °C | 35400 h−1 |
0.05 V% | η′ = 5% | 53 |
| 3.52 wt%Au/Al2O3 | 50 mg | 40 °C | 35400 h−1 |
0.05 V% | η′ = 39% | 53 |
| 1.70 wt%Au/SiO2 | 50 mg | 40 °C | 35400 h−1 |
0.05 V% | η′ = 1% | 53 |
| 0.1 wt%Pd/beta zeolites | N/A | 40 °C | 50000 h−1 |
40 ppm | η′ = 25% | 54 |
| 0.25 wt%Pd/Al2O3 | N/A | 40 °C | 50000 h−1 |
40 ppm | η′ = 80% | 55 |
| 3 wt%Ag/MnOx–CeO2 | N/A | 40 °C | 30000 mL (g h)−1 |
580 ppm | η′ = 12% | 56 |
|
||||||
| 40°C < T ≤ 100°C (at relatively low temperatures) | ||||||
| Cu–Ce catalyst | 100 mg | 60 °C | 600000 mL (g h)−1 |
58 ppm | η′ = 95% | 57 |
| MnO2–graphene | 100 mg | 65 °C | 30000 mL (g h)−1 |
100 ppm | η′ = 100% | 10 |
| 7 wt%Pt/ZrO2–GA–MOF-5 | 200 mg | 70 °C | N/A | 50 ppm | η′ = 95% | 58 |
| 9 wt%Pt/ZrO2–GA–MOF-5 | 200 mg | 70 °C | N/A | 50 ppm | η′ = 85% | 58 |
| 0.25 wt%Au/α-MnO2 | 66 mg | 75 °C | 60000 mL (g h)−1 |
500 ppm | η′ = 100% (with 25% RH) | 15 |
| 3DOM Au∼1 wt%/CeO2 (80 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 100% | 52 |
| 3DOM Au∼1 wt%/CeO2 (130 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 43% | 52 |
| 3DOM Au∼1 wt%/CeO2 (280 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 40% | 52 |
| 3DOM Au∼1 wt%/CeO2 (80 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 100% | 52 |
| 3DOM Au∼2 wt%/CeO2 (80 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 100% | 52 |
| 3DOM Au∼3 wt%/CeO2 (80 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 100% | 52 |
| 3DOM Au∼4 wt%/CeO2 (80 nm pore size) | 200 mg | 75 °C | 66000 mL (g h)−1 |
0.06 V% | η′ = 91% | 52 |
| 0.01 wt%Pt–SAC/TiO2 (Pt–SAC for single-atom catalyst) | 100 mg | 80 °C | 30000 mL (g h)−1 |
160 ppm | η′ = 5% (with 50% RH) | 22 |
| 7.10 wt%Au/Fe2O3 | 200 mg (40–60 mesh) | 80 °C | 54000 mL (g h)−1 |
6.25 mg m−3 | η′ = 100% | 59 |
| 4.85 wt%Au/Fe2O3 | 200 mg (40–60 mesh) | 80 °C | 54000 mL (g h)−1 |
6.25 mg m−3 | η′ = 81% | 59 |
| 2.52 wt%Au/Fe2O3 | 200 mg (40–60 mesh) | 80 °C | 54000 mL (g h)−1 |
6.25 mg m−3 | η′ = 36% | 59 |
| (ε-MnO2/CeO2)@CeO2 (Ce:Mn = 1:4) |
100 mg | 80 °C | 100000 mL (g h)−1 |
180 ppm | η′ = 100% (with 50% RH) | 60 |
| xMnOx–CeO2, x = Mn/(Mn + Ce) = 0.5 | 200 mg (40–60 mesh) | 90 °C | 30000 mL (g h)−1 |
580 ppm | η′ = 90% | 44 |
| 40 wt%MnO2/NCNT | 200 mg | 100 °C | 30000 mL (g h)−1 |
100 ppm | η′ = 100% | 61 |
| 40 wt%MnO2/CNTs | 200 mg | 100 °C | 30000 mL (g h)−1 |
100 ppm | η′ = 100% | 61 |
|
||||||
| T > 100°C (at relatively high temperatures) | ||||||
| 70 wt%MnO2/CNTs | 200 mg | 150 °C | 30000 mL (g h)−1 |
100 ppm | η′ = 100% | 61 |
| Mn0.9Ce0.1/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 100% | 62 |
| Mn0.8Ce0.2/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 83% | 62 |
| Mn0.7Ce0.3/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 78% | 62 |
| Mn0.6Ce0.4/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 76% | 62 |
| Mn0.5Ce0.5/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 62% | 62 |
| Mn/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 41% | 62 |
| CeO2/PG∼85 wt% | 100 mg (40–60 mesh) | 160 °C | 20000 h−1 |
300 ppm | η′ = 2% | 62 |
| CeO2–rGO–NF | 200 mg | 170 °C | 60000 mL (g h)−1 |
250 ppm | η′ = 93% | 63 |
| 0.73 wt%Au/Fe–O | 200 mg (40–60 mesh) | 180 °C | 54000 mL (g h)−1 |
N/A | η′ = 11% | 59 |
Despite numerous outstanding studies being conducted on BFCMs, the intertwined interactions within or among components pose a significant challenge in determining the factors influencing the catalytic activity of HCHO and the degradation mechanism. Specifically, the following uncertainties within BFCMs might severely impact the optimization of their structure–activity relationships and the determination of HCHO degradation pathways. (i) As for SCs, aside from the adsorption capacity, various factors such as support component types, crystal phases, particle sizes, morphologies, pore sizes, specific surface areas and others may also have an impact on the removal of HCHO.69–71 (ii) As for CCs, particularly the activity of noble metal-based catalysts is prone to variation due to their own characteristics (such as loading capacities, particle sizes, dispersion, morphologies, etc.) and reaction conditions.72–74 (iii) Most crucially, the synergistic factors of SCs and CCs as well as the adsorption-catalytic mechanism in BFCMs are complex, and specifically the catalytic activity might be influenced by the decomposition rate of intermediates and the rate of product desorption.73,75,76 Therefore, it is necessary to summarize the influencing factors and synergistic mechanism of each component in BFCMs to provide a theoretical basis for researchers to design new composites for the efficient degradation of HCHO in the future.
Currently, there are hardly any review articles concerning the removal of total VOCs by adsorption/catalytic oxidation.77,78 Different from these studies, this work focuses on the multistage synergistic degradation of HCHO in composite materials from the perspective of adsorption enhanced catalytic degradation of HCHO. On this basis, the influence of various components (such as SCs and CCs) and reaction conditions on the degradation performance of HCHO is further summarized, and the recyclability and degradation paths of BFCMs are analyzed, thereby better evaluating their superiority and practical feasibility among numerous materials. A specialized review of these studies is anticipated to furnish valuable references for the design of low-cost, stable, and high-performance HCHO degrading BFCMs.
2. Recent progress on adsorption enhanced catalytic degradation of HCHO in bifunctional composite materials
Compared to single-component catalysts, BFCMs commonly exhibit several advantages, such as greater design flexibility, enhanced stability, synergistic effects, superior activity, and selectivity.79 Due to the integration of two or more functional components, BFCMs could achieve synergistic effects, enabling simultaneous or sequential execution of multiple reaction steps, thereby enhancing overall reaction efficiency and product selectivity.80 By adjusting the characteristics of SCs and CCs in BFCMs (such as species, structures, crystal phases, particle sizes, pore sizes, specific surface areas, dispersion and loading of CCs), the synergistic effect between SCs and/or CCs could be significantly intensified, thereby enhancing the adsorption and catalytic degradation ability of HCHO. These studies have not only made a breakthrough in enhancing the removal efficiency of HCHO, but also greatly improved the recycling stability, selectivity and environmental friendliness of BFCMs, providing a new direction for future research.2.1 Design of bifunctional composite materials
To date, various highly compelling studies for HCHO removal based on multi-angle design mainly cover the following aspects. On the one hand, based on material combination innovation, different types of SCs and CCs were exquisitely combined to achieve efficient degradation of HCHO by BFCMs. This mainly encompassed the combinations of noble metals and TMOs (e.g., Pt/TiO2, Rh/TiO2, Pd/TiO2, and Au/TiO2),81 the combinations of alkali metal dopant oxides and SCs (e.g., 0.05 wt% Pt–Na/nano-Al2O3),14 or a combination of noble metals and/or TMOs with SCs (e.g., 7 wt% Pt/ZrO2–GA–MOF-5).58 On the other hand, it could be achieved through process innovation (such as in situ deposition at room temperature, chemical in situ deposition, vapor deposition, and bubble-assisted deposition precipitation),52,82–84 structural innovation (e.g., core–shell structure CeO2@LDHs,85 epitaxial structure α-MnO2@L-MnO2,66 and embedding structure MnO2/UiO-66-NH2),20 functional group modification, crystal surface exposure (e.g., {001} surface in Pt/FTiO2-NS),32 alkali metal/nitrogen atom doping (e.g., 3D-CeO2@CN), and heat/acid/base pretreatment37,86,87 to enhance the catalytic activity of BFCMs towards HCHO. All in all, the aforementioned strategies focusing on material type, structural design, element doping, and material pretreatment significantly enhance the degradation efficiency of HCHO by BFCMs.2.2 Evaluation of degradation performance for HCHO
At low temperatures (e.g., T ≤ 40 °C), minimizing the loading capacities (e.g. <0.1 wt%) of noble metals in BFCMs or utilizing low-cost non-noble metals to effectively degrade HCHO was a desirable goal.2 In Table 1, we summarized the HCHO degradation conditions and some indices for multiple types of BFCMs based on the characteristic reaction temperature zones (e.g., T ≤ 40 °C, 40 °C < T ≤ 100 °C, and T > 100 °C). These primary conditions and indicators included the mass of BFCMs, reaction temperatures (T), gas hourly space velocity (GHSV), HCHO concentration at the inlet ([HCHO]I), HCHO removal activity (η′) under specified humidity, etc. In these reports, some studies had elaborately investigated the source/generation of HCHO involved in the evaluation process of HCHO degradation performance, the evaluation scheme of HCHO degradation, and the evaluation indicators (such as HCHO removal rate, conversion rate, and specific rate). For instance, apart from the HCHO standard gas, polluted air with a specific HCHO concentration could also be produced at a certain temperature via the polyformaldehyde layer in a U-shaped tube. To study the impact of water on catalytic efficiency, the first stream was passed through a water generator and the other through a HCHO generator, and subsequently, the two streams were merged together as feedstock gas. Additionally, while the concentration of HCHO in the stream was manually detected by using an htV-M HCHO meter, the composition of the effluent phase could be analysed online by using a gas chromatograph equipped with a methanation furnace and FID.15 In addition, the evaluation schemes of HCHO degradation ability were often classified into the static test and dynamic single test. The removal rate of HCHO and the thermal regeneration stability of BFCMs could be evaluated in the static test. The removal efficiency of HCHO and the continuous catalytic stability of BFCMs (e.g., a catalytic duration of 100 h) could be tested in a dynamic single test. Generally, the static test device could be composed of a closed glass container, circulation system, electric actuator system and HCHO monitoring system. In the dynamic single test, a HCHO standard gas bottle was connected at one end of a catalyst box, and a CO2 concentration tester and HCHO concentration tester were connected at the other end to test the real-time concentrations of HCHO and CO2 respectively. The concentration of HCHO was measured in real time with the aforementioned HCHO being utilized as the pollutant gas. The thermal regeneration stability of BFCMs could be studied by repeated static tests. Eqn (1) and (2) could be utilized to calculate the removal efficiency of HCHO and the conversion efficiency of HCHO to CO2 under the dynamic test.24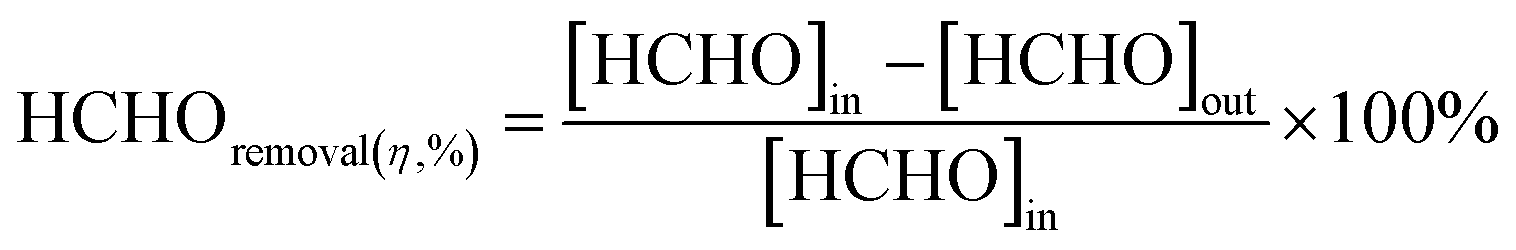 | (1) |
 | (2) |
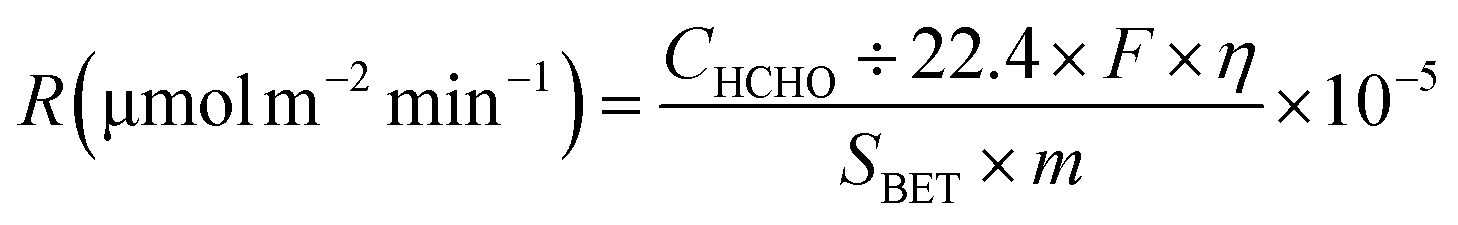 | (3) |
2.3 Adsorption enhanced catalytic degradation of HCHO in bifunctional composite materials
The BFCMs accomplish efficient elimination of HCHO in the same material through the combination of adsorption and catalytic functions. This synergistic effect of adsorption enhanced catalytic degradation significantly enhances the performance and application efficiency of BFCMs. Studies indicated that the synergies in BFCMs mainly include adsorption-catalytic synergies, heterogeneous synergies within SCs or CCs, structural synergies, and heat and mass transfer synergies.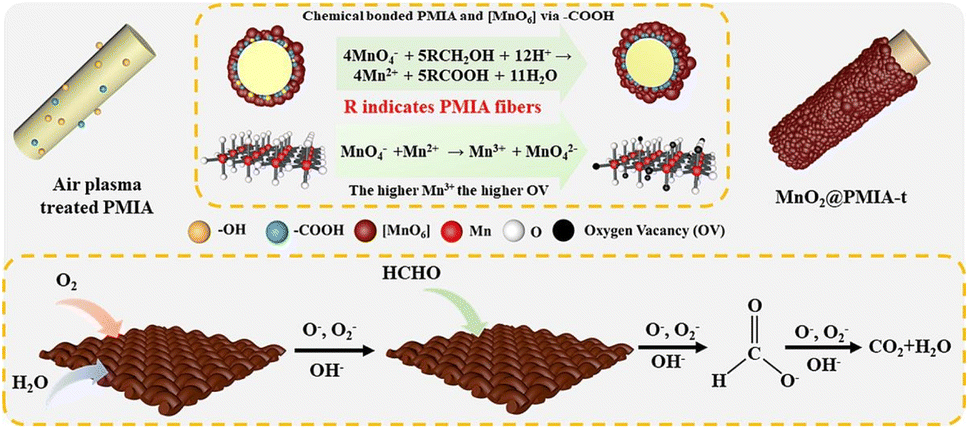 | ||
| Fig. 1 Systematic modulation of the specific surface area, catalytic activity, and laundering durability of fiber-based MnO2 nano-catalysts. Reproduced with permission.39 Copyright 2023, Elsevier. | ||
It is worth mentioning that photo assisted adsorption-catalytic coordination was also a method to enhance the oxidation of HCHO. The pre-deposited graphene oxide on the nickel foam (NF) matrix was utilized as the photothermal conversion layer, which was conducive to the uniform and stable loading of CeO2. The addition of reduced graphene oxide increased near-infrared (NIR) light absorption, and the thermal conductivity of NF improved electron mobility, so that the oxidation of HCHO was stimulated not only by the Olatt from CeO2, but also by the photogenerated electron oxidation to produce the central point O2−. In addition, the central point O2− would reoxidize Ce3+ to Ce4+ in the OVs, accelerating the oxygen consumption and oxygen supply cycle in the lattice, and greatly improving the HCHO removal rate.63 In conclusion, the adsorption-catalytic synergy in BFCMs remarkably enhanced the catalytic performance by integrating different materials and reaction mechanisms. This synergistic effect not only boosted the capture capability of HCHO but also elevated the conversion efficiency of HCHO, offering new ideas and technical approaches for environmental governance.
 | ||
| Fig. 2 Schematic illustration for the synergy mechanism of the (ε-MnO2/CeO2)@CeO2 catalyst. Reproduced with permission.60 Copyright 2020, American Chemical Society. | ||
3. Analysis of the effects of SCs/CCs in BFCMs and reaction conditions on HCHO removal
Although single-component catalysts (such as MnOx) possess the advantages of low cost and a simple preparation process, their removal performances on HCHO are easily limited by their characteristics. More and more SCs with excellent adsorption properties or supporting ability (such as AC, MOF, etc.) are introduced to combine with CCs to form BFCMs.30,92 Nevertheless, the influence of BFCMs on the degradation performance of HCHO is rather complex, involving various factors such as the preparation process, SCs, CCs, and reaction conditions. Regarding the preparation processes of BFCMs, interested readers may refer to the synthesis methods described in these studies.93,943.1 Support components in bifunctional composite materials
Besides maintaining catalyst dispersion, SCs could also serve as adsorption or active sites or indirectly engage in catalytic reactions through interfaces in BFCMs.68 Specifically, SCs could affect the catalytic activity of BFCMs towards HCHO through their crystal phases,15 types,83 morphology,95 pore sizes,51 surfactant groups,39 specific surface areas,96 and stability. In practical scenarios, some of these factors might interact in a synergistic or intertwined manner, thereby complicating the determination of the factors that affect catalytic activity.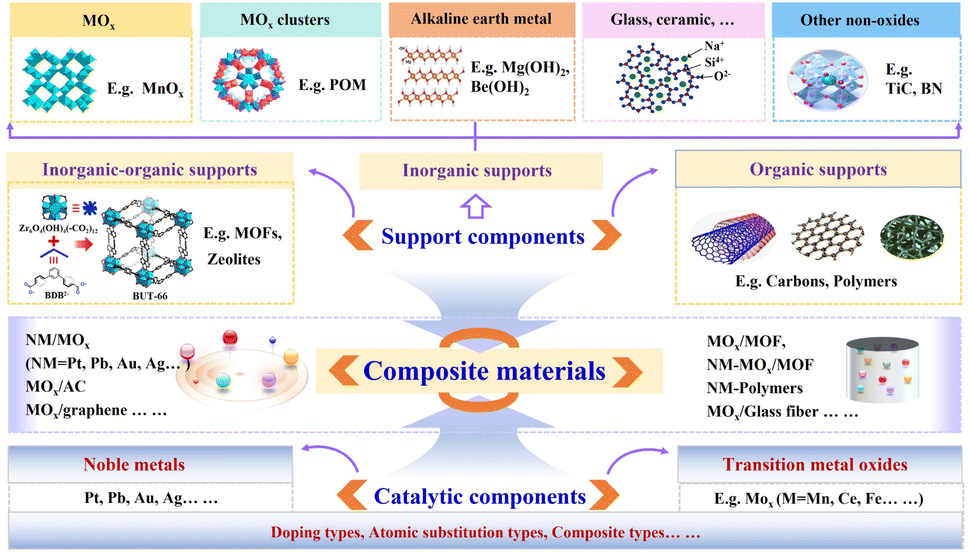 | ||
| Fig. 3 Types of support components and catalytic components in BFCMs. | ||
Additionally, the impact of the support component structure on the catalytic performance of BFCMs encompassed some factors such as their internal structure, surface functional groups, and support component thickness. For instance, compared with powder SCs, the SCs with a 3DOM structure were utilized to load Au nanoparticles, which were more conducive to the dispersion of catalysts and the improvement of catalytic activities.51 Regarding surface functional groups, their introduction could exert an influence on the degradation route of HCHO in BFCMs. After integrating the MnO2 catalyst into graphene nanosheets, the augmented active surface and substantial amount of surface OH-matter significantly streamlined the decomposition path of HCHO without generating CO by-products.10 From the perspective of support component thickness, some developed two-dimensional materials (with thickness <5 nm) enhanced their catalytic performance through short diffusion paths, high specific surface areas, and a large number of active sites.105 For example, ultra-thin (∼4 nm thickness) Bi2WO6 nanosheets loaded with Pt showed good catalytic removal efficiency for HCHO at room temperature, with a removal rate of 93% for 200 ppm HCHO within 1 hour.90 In short, the type and structure of SCs in BFCMs may affect the degradation of HCHO to some extent through the interaction between SCs and CCs, electronic properties and the microstructure. And these factors should be taken into account to optimize the catalytic performance in the design and preparation of BFCMs.
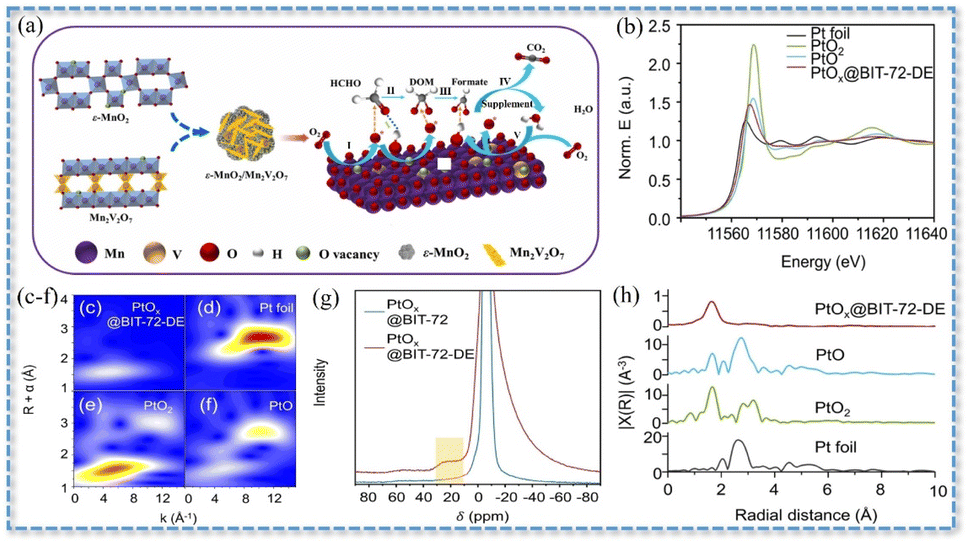 | ||
| Fig. 4 (a) ε-MnO2/Mn2V2O7 synergistic catalysis mechanism. Reproduced with permission.106 Copyright 2023, Elsevier. Pt L3-edge (b) XANES spectra and (c–f) EXAFS spectra. (g) WT analyses of PtOx@BIT-72-DE, PtO, PtO2, and Pt foil. (h) Al MAS NMR spectra of PtOx@BIT-72 and PtOx@BIT-72-DE. Reproduced with permission.27 Copyright 2022, Wiley. | ||
In addition to defect engineering, SCs with large pore sizes could also be obtained through process and technological innovations. Techniques such as the colloidal crystal template method52 and bubbling-assisted deposition precipitation method51 had been developed to prepare 3DOM structures to obtain interconnected networks with adjustable pore sizes and spherical voids. In the 3DOM structure, noble metal nanoparticles were easily distributed and aggregate less, and had good catalytic activities for HCHO. However, a larger aperture was not necessarily more conducive to the improvement of catalytic activities, because the aperture may affect the specific surface areas of the samples and the distribution of noble metals. For example, studies on the catalytic activities of 3DOM Au/CeO2–Co3O4 with pore sizes of 80 nm, 130 nm and 240 nm indicated that the samples with pore sizes of 80 nm had the highest catalytic activity for HCHO oxidation. The reason was the fact that it had the largest specific surface area, smaller pore size and a more uniform distribution of Au nanoparticles.51 Similarly, 3DOM Au/CeO2 with a pore size of 80 nm showed a higher catalytic activity than the catalyst with a pore size of 130 nm. The reason was the fact that Au/CeO2 catalysts with a pore size of 80 nm had a larger surface area than catalysts with other pore sizes. However, the specific surface areas would gradually decrease with the increase in loading of Au nanoparticles.52 These BFCMs with 3DOM structures are expected to be valuable catalyst systems for HCHO removal.
It was notable that the specific surface areas of BFCMs might also be influenced by the loading of CCs. Studies revealed that the specific surface areas decreased as the loading of CCs increased. For instance, the specific surface areas of MnO2/UiO-66-NH2 declined with the increase in MnO2 loading (Fig. 5a–c). When the loading capacity of MnO2 increased from 1 wt% to 20 wt%, the specific surface areas of BFCMs dropped from 883 m2 g−1 to 505 m2 g−1, and the conversion of HCHO (100 ppm) at room temperature decreased from 88% to 47% (Fig. 5d).20 The reason was that the excessive loading of MnO2 would not only clog the pores of BFCMs and restrict the access of HCHO molecules to the catalytically active sites, but also cause the catalytic sites to be buried in BFCMs instead of on the active surface. It had also been demonstrated that when the loading of TMO was substantial enough, the specific surface areas of SCs might initially decrease and then increase with the loading.61 The reason was that as the material was further oxidized, more and more MnO2 was interconnected to form porous structures. When the MnO2 loading reached 70%, the specific surface areas of MnO2/NCNT presented an increasing trend, while the average pore diameter and total pore volume also exhibited a trend of initially increasing and then decreasing. Additionally, there were certain cases where the specific surface areas would increase along with the increase in CC loading capacity. For example, the specific surface areas of MnO2@SiO2–TiO2-4, obtained through a combination of electrospinning and liquid phase synthesis, gradually increased from 4.95 to 48.59 m2 g−1 as the loading capacity of MnO2 increased with the increase in the synthesis period from 0 to 5 cycles.43 This was attributed to the fact that the layered structure of the fiber surface augmented the specific surface area of the material, thereby providing more active sites for HCHO oxidation reactions.
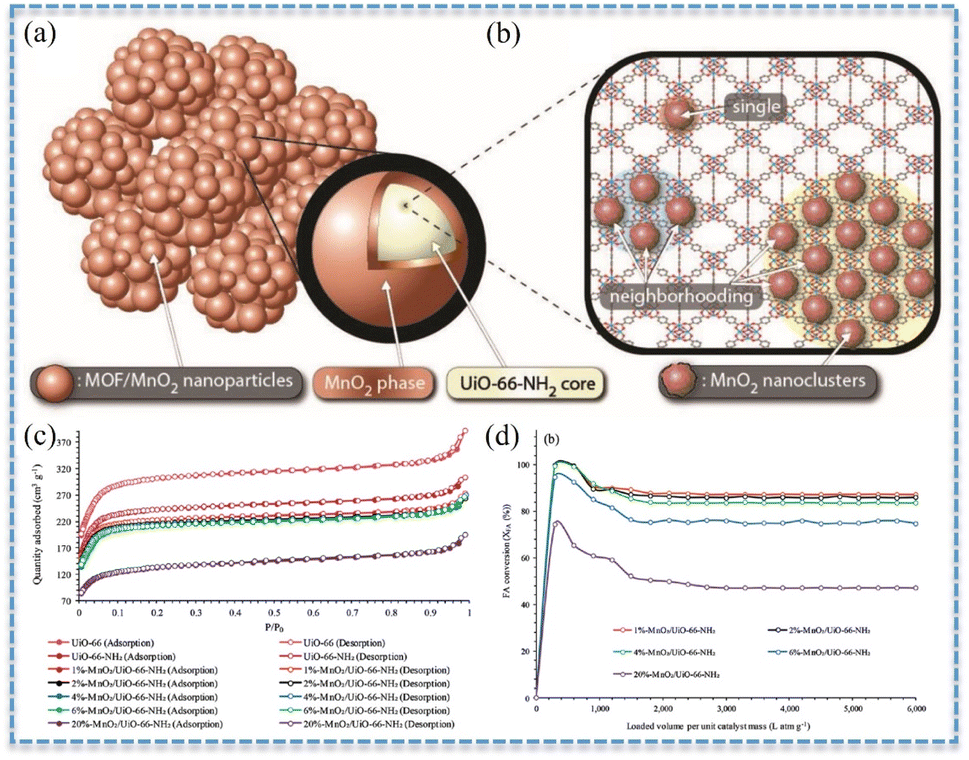 | ||
| Fig. 5 (a) Combined representation of the MnO2/UiO-66-NH2 phases. (b) MnO2 clusters formed in the UiO-66-NH2 pore structure. (c) N2 adsorption–desorption isotherms. (d) HCHO conversion (HCHO: 100 ppm in air, flow rate: 500 mL min−1, catalyst mass: 5 mg, GHSV: 597134 h−1, RH: 0%, and TOS: 1 h). Reproduced with permission.20 Copyright 2022, Wiley. | ||
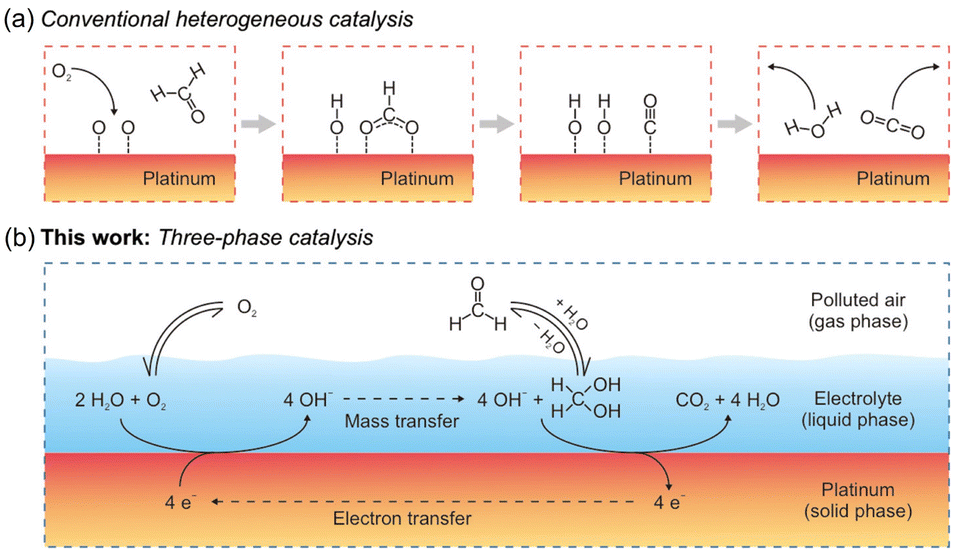 | ||
| Fig. 6 Reaction schemes for the oxidation of HCHO on Pt. (a) Conventional heterogeneous catalysis involves adsorption, diffusion, reaction, and desorption, constrained on a two-dimensional surface. (b) Three-phase catalysis speeds up the process by introducing a new liquid phase, with mass transfer and electron transfer facilitated by the electrolyte and Pt, respectively. Reproduced with permission.122 Copyright 2020, American Chemical Society. | ||
3.2 Catalytic components in bifunctional composite materials
The efficient catalytic oxidation of HCHO at room temperature is one of the most promising technologies. The influence of catalyst components on the degradation performance of HCHO is commonly related to the types, crystal phases, morphologies, shapes, particle sizes, dispersion, metal chemical states and loading capacities of CCs.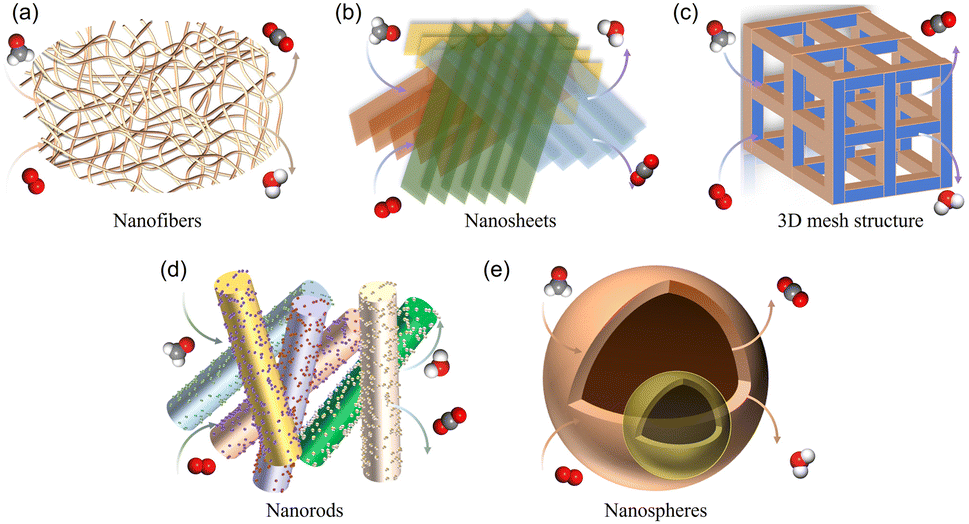 | ||
| Fig. 7 Schematic diagram of external diffusion of HCHO in five typical MnO2 morphologies: (a) nanofibers, (b) nanosheets, (c) three-dimensional mesh structure, (d) nanorods, and (e) nanospheres. | ||
In terms of catalyst shapes, materials with ultra-thin two-dimensional nanosheet shapes presented great potential in generating surface defects. Ultra-thin two-dimensional nanosheets not only had the advantages of large specific surface areas, ultra-thin thickness, abundant active sites and a short diffusion path, but also had a large number of surface defects, including dangling bonds and coordination unsaturated atoms at the edge.46 These factors played a key role in the excellent catalytic performance and stability of catalysts. Moreover, the shape of catalysts may also vary the catalytic performance by affecting gas mass transfer, and so on. Compared with nanosheets, monolayer MnO2 made the external diffusion of HCHO on the catalyst easier, and improved the mass transfer ability and catalytic activities of HCHO. In addition, compared with bulk catalysts, powder shaped MnO2 greatly limited its scope of usage due to its easy agglomeration and tendency to result in the blockage of porous channels for SCs.
000 h−1 GHSV.62 In addition, there were some special cases in which the bulk shape would also provide some benefits to catalytic activities. For example, for silver, which was prone to surface oxidation, the bulk shape prevented excessive oxidation of silver inside, causing the bulk silver to exhibit higher activity than smaller silver nanoparticles. It was believed that the shell prevented the further oxidation of silver in a biphasic silver block (Ag@AgOx) composed of a metal core and oxide shell.26
In addition to particle sizes, particle dispersion would also affect catalytic activities. Stability studies of transition metal atoms (TM/MXene, TM = Au, Ag, Ru, Rh, Pt, Pd, Ti, V, Cr, Co, Ni, Cu, Zn, Cd and Hg) modified on MXene monolayers indicated that Ti/Ti3C2O2 was found to be the most stable system compared to other transition metals. The reason was that Ti atoms were more easily dispersed on the surface of Ti3C2O2 in the form of single atoms without aggregating, which significantly improved their oxidation activity to HCHO.135 In addition, studies on HCHO catalyzed by different BFCMs loaded with Au (such as Au/SiO2, Au/HZSM-5 and Au/γ-Al2O3) indicated that the reaction rate was linearly related to the Au particle sizes, indicating that the smaller the particle sizes and the higher the dispersion of Au, the higher the oxidation activity of HCHO.
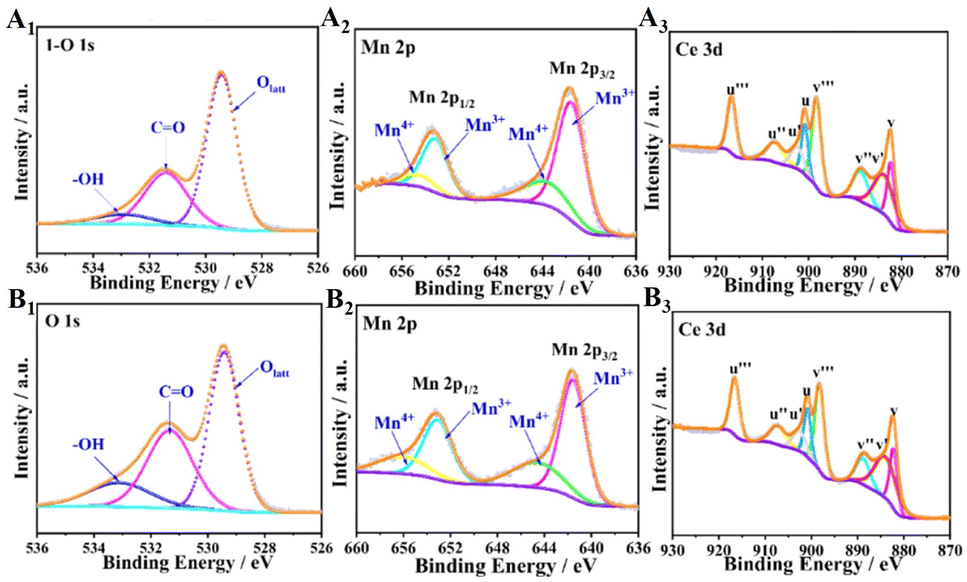 | ||
| Fig. 8 (1) O 1s, (2) Mn 2p, and (3) Ce 3d spectra of the catalysts: (A) C-T100 and (B) C-T120. Reproduced with permission.60 Copyright 2020, American Chemical Society. | ||
The chemical state of metal could be regulated by support component-assisted electron transfer, ammonia modification, element doping, and so on. For example, manganese oxides were thought to be the catalysts with the highest HCHO oxidation activity among TMOs.137 This activity was analyzed to derive from the REDOX cycle between manganese species (Mn4+/Mn3+), resulting in a continuous oxygen activation and oxygen transfer process that promoted electron transfer on catalyst surfaces.56 Considering enhancing the electron transfer and conductivity of manganese oxide, graphene was introduced as a support component to form graphene–MnO2 nanocomposite materials. The additional interface of the hybrid region facilitated charge transfer between the different oxidation states of Mn4+/Mn3+, resulting in high reaction rates and good catalytic performance.10 Similarly, the content of Mn3+ and OV concentration in BFCMs could be adjusted by ammonia modification. A study of HCHO degradation by MnOx/AC–N2 at room temperature indicated that the excellent catalytic performance of BFCMs was attributed to the regulation of OVs by ammonia modification, while the increased Mn3+ content in catalysts led to the formation of more surface OVs to maintain electrostatic equilibrium. The existence of abundant OVs promoted the adsorption, activation and migration of O2 molecules to form more ROS.10 Different from modification, doping was another method to achieve the regulation of metal oxidation states in BFCMs. For example, the doping of Ce or Co elements could replace some Mn4+ sites to increase the content of Mn3+ and surface vacancy defects in catalysts, thereby improving the catalytic performance of materials.138 It should be noted that the oxidation state of metals in BFCMs could be varied by regulating the type of precipitants (such as NH3·H2O, KOH, NH4HCO3, K2CO3 and KHCO3).139
In terms of reaction conditions and the conversion rate, the conversion rate of HCHO did not increase linearly with the increase in noble metal loading capacities. Instead, there existed an optimum loading capacity. For instance, when the loading capacity of Pt increased from 0.3 wt% to 1 wt%, the conversion of Pt/TiO2 to HCHO at room temperature escalated to 100%. Nevertheless, when the loading capacity of Pt was augmented from 1 wt% to 2 wt%, the conversion rate of HCHO remained scarcely variable. Hence, in this study, the optimal loading capacity of Pt on TiO2 was ascertained to be 1 wt%.128 The optimal loading capacity of Pt in diverse reaction systems exhibited more or fewer dissimilarities. In contrast to Pt/TiO2, the removal rates of HCHO by Pt-VO-ZrO2 with varying Pt loading capacities were in the sequence of 0.87 Pt-VO-ZrO2 > 1.32 Pt-VO-ZrO2 > 0.51 Pt-VO-ZrO2 > ZrO2.24 Before attaining the optimal loading capacity, the augmentation of noble metal content was beneficial for enhancing the activity of BFCMs. However, once this optimal loading capacity was surpassed, the continuous increase in noble metal loading capacity might result in a decline in the activity of BFCMs. The reason was that the excessive loading would probably cause adverse effects such as accumulation of active substances, specific surface reduction, and pore blockage. Compared with 1 wt% Pt/sepiolite, the overloading of Pt on 2 wt% Pt/sepiolite led to the aggregation of Pt particles and an increase in catalyst sizes, resulting in a decrease in active sites and specific surface areas of Pt nanoparticles. In addition, the pores and channels of sepiolite nanofibers may also be blocked by the aggregated Pt nanoparticles, resulting in the reduction of their specific surface areas.76 Similarly, 7 wt% Pt/ZrO2–GA–MOF-5 presented the highest activity due to its optimal loading of Pt, while the over-loading 9 wt% Pt led to a decrease in catalytic efficiency. The reason was the accumulation of Pt nanoparticles caused by excessive loading, resulting in a decrease in specific surface areas, thus reducing the catalytic effect.140
3.3 Effect of reaction conditions on HCHO degradation
In addition to the above factors, the degradation of HCHO would also be affected by a variety of reaction conditions, mainly including heat, light, electricity, the initial concentration of HCHO, relative humidity, environmental media, pH and gas flow rate. In practical applications, these parameters need to be optimized according to the conditions to improve the degradation efficiency of HCHO.3.3.1.1 Thermal catalysis. In the catalytic process of HCHO, temperature was one of key factors affecting the catalytic activities and stability of BFCMs. At a lower temperature, some reaction intermediates/products adsorbed on the catalyst surface tended to block the active sites. In contrast, an increased temperature was prone to facilitate desorption of intermediates and/or products to promote the release of active sites or improve product selectivity.19 For example, while the removal efficiency of HCHO on MnOx/AC–N2 decreased slightly with increasing temperature, the CO2 selectivity increased significantly. This was attributed to the fact that the increased temperature made it easier for CO2 to release from intermediates and/or products.92 However, excessive temperature conditions would also have some adverse influence on the reaction, including increasing energy consumption or causing some other problems in the nanomaterial composite process. For example, for small-size nanoclusters with high interfacial energy and poor thermal stability, relatively high temperatures tended to cause irreversible growth and agglomeration.23 In addition, high temperature conditions also tended to lead to pore collapse and lower specific surface areas, resulting in a poor HCHO removal performance (Fig. 9e and f).141
 | ||
| Fig. 9 (a) Graphene-enhanced photothermal decomposition of carcinogenic HCHO by α-MnO2, (b) XRD patterns of different samples, (c) α-MnO2 and (d) α-MnO2/GO-M1. Reproduced with permission.38 Copyright 2022, Elsevier. HCHO removal (e) and CO2 production (f) over the MIL-88B(Fe) nanorods (sample T100) and the control samples. Reproduced with permission.141 Copyright 2021, American Chemical Society. | ||
3.3.1.2 Photocatalysis and photothermal catalysis. Owing to low energy consumption, non-toxicity, cost-effectiveness and mild reaction conditions, photocatalysis is regarded as a promising technology for HCHO degradation. However, photocatalysts were frequently affected by electron–hole recombination, significantly reducing their photocatalytic performance. To alleviate this problem, researchers had proposed various strategies. Firstly, the construction of heterogeneous structures or composite materials had been demonstrated to be an effective strategy to inhibit electron–hole pair recombination and enhance electron transport, thereby improving photocatalytic activity under visible light. For instance, the Bir/P25-2 composite exhibited a photoelectric value that was 4.59 times higher and a band gap that was 1.7 times wider than those of birnessite, leading to significantly enhanced charge separation efficiency.101 The Bir/P25-2 composite demonstrated superior photocatalytic activity, achieving the removal of 80 ppm of gaseous HCHO within 50 minutes under visible light irradiation. This enhanced performance was attributed to the increased number of OVs in Bir/P25-2 compared to birnessite, which facilitated the generation of more superoxide free radicals and hydroxyl free radicals, thereby accelerating pollutant decomposition. Secondly, the band gap of photocatalysts could be narrowed through metal ion or non-metallic element doping, extending its response range into the visible light spectrum.142 Thirdly, the usage of auxiliary catalysts or sacrificial agents could also aid in the separation of electron–hole pairs, further enhancing photocatalytic efficiency.143 Beyond their application in HCHO degradation, BFCMs (e.g. Fe–PEI–CN) based on photocatalysis exhibited significant potential for wastewater purification.144,145
In order to reduce the energy consumption caused by heating, a light-regulated photothermal catalysis technique was developed to improve the catalytic activities of HCHO.11,38,146 It is well known that manganese oxides had a strong thermal catalytic activity, but also had a generally low photocatalytic activity.147–149 Studies indicated that the incorporation of graphene oxide (GO) with strong light absorption ability and high photothermal conversion efficiency could enhance electron transport and effective separation of electrons from holes.18 The degradation rate of HCHO by the resultant nanocomposite α-MnO2/GO at room temperature and in light was as high as 100%, and its PXRD and SEM are shown in Fig. 9a–d.38 This work not only provided a new strategy for the combination of catalysts and photothermal materials to utilize solar energy, but also expanded the research on the photocatalysis of manganese oxides in HCHO.
3.3.1.3 Electrocatalysis and photoelectron catalysis. Conventional electrocatalytic processes necessitate an external power source, which greatly restricts their commercial viability. In recent years, the electrocatalytic technology combined with the piezoelectric effect has attracted more and more attention. When subjected to mechanical stress, piezoelectric materials would generate an internal electric field that could modulate the charge migration behaviour of catalysts, thereby enhancing electrocatalytic efficiency. For instance, the piezoelectric effect could degrade HCHO by inducing polarization in two-dimensional piezoelectric material 2H–MoS2, inhibiting photogenerated electron–hole pair recombination and promoting the generation of hydroxyl radicals.150 Furthermore, self-powered triboelectric technology represents an alternative approach for achieving electrocatalytic HCHO degradation by leveraging the contact electrification effect and the principle of electrostatic induction. This method harnesses mechanical energy from the ambient environment and converts it into electrical energy for utilization. For instance, MnOx–PMMA self-powered triboelectric catalysts with a three-dimensional nanocomposite structure, driven by wind-induced friction between PMMA fibers and SSM, generated triboelectric charges that promoted reactive oxygen species (ROS) formation, thus accelerating the catalytic oxidation of HCHO and intermediates. At ambient temperature and under a 15 ppm HCHO atmosphere, this composite achieved a CO2 yield increase of 78.93% compared to MnOx catalysts.151 This study provided a new strategy for efficient, long-term, low-energy HCHO degradation at ambient temperature. On this basis, photo-electron catalysis combines the advantages of both photocatalysis and electrocatalysis, improving photogenerated carrier separation efficiency and regulating electron directional transfer via the applied voltage, thereby enhancing reaction rates and selectivity.152 A nano-catalyst Pt/TiO2–ZnO prepared using the sol–gel method and supported on a stainless steel mesh removed 74% of HCHO through photo-electron catalytic oxidation within 120 minutes.81
In short, thermal catalytic oxidation commonly necessitates elevated reaction temperatures, resulting in substantial energy consumption and rendering it unsuitable for natural environments. Nonetheless, this method retains potential in specific industrial applications, particularly where rapid treatment of large volumes of pollutants is required. In contrast, photo-catalysis may attract much attention due to its high efficiency and environmental protection, while photo-thermal catalysis and photo-electron catalysis technologies would also present important application prospects in the field of HCHO degradation due to their higher potential and flexibility.
| CWP = −r′Rp2ρc/CsDe | (4) |
| r′ = (cin − cout)G/m | (5) |
| Kn = λ/γ | (6) |
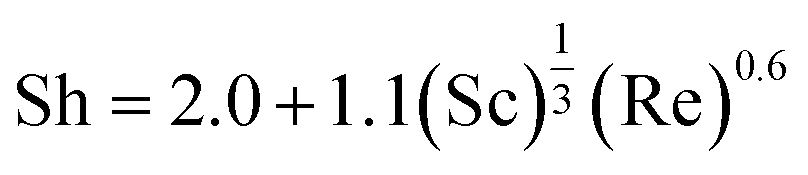 | (7) |
| Sh = d·κm/D | (8) |
When the residence time was different, the catalyst efficiency decreased with the increase in flow rate, and it was closely related to the contact time between HCHO and the catalyst surface. It is worth mentioning that, unlike the contact time, the residence time was calculated as the depth of filling catalyst divided by speed. The longer the contact time, the higher the pollutant removal efficiency. The reduction of airflow speed allowed the reactant to fully come into contact with the active sites.167 In other words, the mass transfer coefficient varied with contact time, which depended on airflow speed.
3.4 The degradation pathways of HCHO
Density functional theory (DFT) simulations were often combined with other analytical measurements to analyze the mechanisms associated with the HCHO adsorption/oxidation pathway, including in situ diffuse infrared Fourier transform spectroscopy (DRIFTS) and X-ray photoelectron spectroscopy (XPS).168–171 The adsorption behavior of reactants (O2 and HCHO) and subsequent oxidation path of HCHO could be illustrated by density functional theory (DFT) calculation and in situ diffuse infrared Fourier transform (DRIFT) spectroscopy analysis.24By analogy with the VOC oxidation mechanism, three kinetic mechanisms of HCHO oxidation were analyzed to elucidate the reaction pathways, namely the Mars-van Krevelen (MVK) mechanism, Langmuir–Hinshelwood (L–H) mechanism, and Eley–Ridea (E–R) mechanism (Fig. 10). (i) As for the MVK mechanism, the adsorbed HCHO molecules first reacted with the Olatt on the catalyst surface to form OVs. Then, compensation of airborne oxygen or oxygen atoms in the bulk phase regenerated the reduction center.54,172,173 (ii) As for the L–H mechanism, the first step was the adsorption of oxygen and HCHO on catalysts, followed by the REDOX process. According to whether HCHO and oxygen were adsorbed at the same active site, the L–H model could be divided into the single-point L–H model and two-site L–H model.174 (iii) In the E–R mechanism, the catalytic process occurred between adsorbed oxygen and gas-phase HCHO molecules, or between adsorbed HCHO molecules and gas-phase oxygen.77,175,176
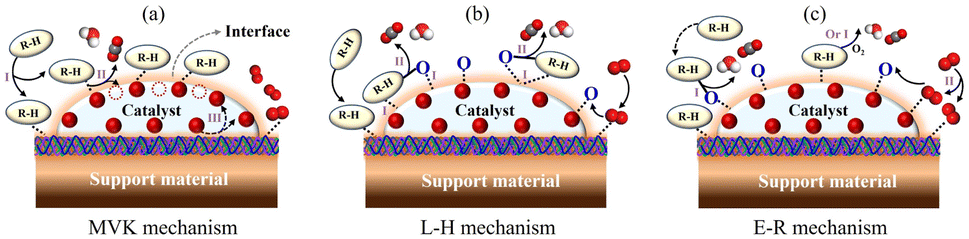 | ||
| Fig. 10 Kinetic model of VOC oxidation on BFCMs: the (a) MVK mechanism, (b) L–H mechanism, and (c) E–R mechanism. | ||
Among the three mechanisms, most studies had indicated that the oxidation pathway of HCHO followed the MVK mechanism, in which Olatt played a key role. The main reactions involved in these studies are listed in Table 2. For example, the lattice O in Au/Fe2O3 was activated by the loaded Au and further participated in the MVK process to oxidize HCHO.177 Similarly, MnO2/UiO-66-NH2 also followed the HCHO oxidation mechanism similar to MVK. During oxidation, HCHO was first adsorbed onto the lattice by interacting with Zr in the lattice, while O2 molecules were split into O atoms at the same location, and then HCHO and O atoms react to form CO2 and H2O. Among them, there were three types of possible products which formed after the combination of HCHOads with *O2, and each type of product further reacted to produce CO2 and H2O as final products.20 In addition to surface Olatt, chemisorbed ROS (such as O2−, O− or OH) were demonstrated to play an active role in the activation of HCHO and O2, and also the oxidation of HCHO and intermediates.180 Surface oxygen preferentially located at OVs (such as the defect of MnO2) could not only activate molecular oxygen as an electron donor to form adsorbed substances lacking electrons (such as O2− and O−), but also react with H2O to form surface hydroxyl groups (O2−, O− + H2O → –OH) to assist the adsorption and degradation of HCHO.181 Among the other two possible mechanisms (i.e. L–H mechanism and E–R mechanism) of HCHO oxidation, the selection of a specific mechanism was speculated to be related to the structure of BFCMs and their surface catalytic reaction. For example, in the catalytic oxidation reaction of HCHO on the surface of FeN3-graphene, the L–H mechanism was believed to dominate the oxidation of HCHO.182 Similarly, the oxidation of HCHO catalyzed by δ-MnO2@GO mainly followed the L–H mechanism.40 By comparing the L–H and E–R oxidation mechanisms of HCHO on Ti/Ti3C2O2, it was suggested that the E–R mechanism had a lower energy barrier in the rate-limiting step. It made the huge energy release of HCHO dissociative adsorption on Ti/Ti3C2O2 with activated O2 promote the subsequent reaction steps as well as good catalytic cycle efficiency.135
| Equation no. | Reaction class | Elementary reactions | Ref. |
|---|---|---|---|
| 1 | ROS generation | O2 + e− = O2− | 177 |
| 2H2O + 4h+ → O2 + 4H+ | |||
| O2− + 2H+ + e− = H2O2 | |||
| H2O2 + e− = OH− + ·OH | |||
| OH− + h+ → ·OH | |||
| 2 | ROS propagation | O* + H2O → 2*OH | 24 |
| HCHO + O* → *HCOOH | |||
| *HCOO + *OH → CO2 + H2O | |||
| 3 | ROS propagation | O2(ads) → O2(ads)− → O(ads)− → O2(ads/lattice)− | 50 |
| 4 | ROS generation, ROS propagation, and HCHO oxidation | □ + hv → e− + h+ | 178 |
| h+ + OH(ads)− or H2O(ads) → ·OH + H+ | |||
| e− + O2(ads) → ·O2− | |||
| HCHO + ·OH → DOM | |||
| DOM + ·OH → HCOOH | |||
| HCOOH + ·O2− → HCO3− + ·OH | |||
| HCOOH + ·OH → HCOO− + H2O | |||
| HCOO− + ·OH → CO2 + H2O | |||
| 5 | ROS generation, ROS propagation, and HCHO oxidation | HCHO⋯OH + H2O → H2C(OH)2 | 19 |
| H2C(OH)2 + O* → H2COO⋯OH + H2O | |||
| H2COO⋯OH + O* → HCOOH⋯OH | |||
| HCOOH⋯OH + O* → ·CO3⋯OH + H2O | |||
| 2H+ + ·CO3⋯OH → CO2 + H2O | |||
| 6 | HCHO oxidation | FAads + *O2 → *O + *H2CO2 | 20 |
| FAads + *O2 → *O + *HCOOH | |||
| FAads + *O2 → *OH + *HCOO− | |||
| 7 | HCHO oxidation | HCHO → DOM (dioxymethylene) | 179 |
| →formates → carbonates → CO2 | |||
| 8 | HCHO oxidation | HCHO → DOM → HCOOH/HCOO− | 46 |
| →H2CO3/HCO3− → CO2 + H2O |
In practical applications, the oxidation pathway and products of HCHO may be affected by calcination temperature and RH. Taking Au/CeO2 as an example, according to the difference of pretreatment calcination temperatures (such as 350 °C, 550 °C and 750 °C), the intermediate products could be divided into three categories: (i) DOM and formate; (ii) formate, DOM and carbonate species; (iii) formate and carbonate species. On CeO2-based BFCMs, the oxidation of HCHO mainly followed the MVK mechanism, that is, it mainly oxidized HCHO to CO2 and H2O through surface Olatt.63 In addition to calcination temperature, the degradation pathway of HCHO may also vary due to an increase in ambient RH. For example, under dry conditions, [O]s reacted with HCHO to form adsorbed formate, and the separated hydrogen atoms reacted with [O]s to form [OH]s, which would further react with formate to form [H2CO3], and produced the final products CO2 and H2O. However, as the reaction occurred, the adsorbed water in the PtOx@BIT-72-DE channel gradually increased. Under wet conditions, [O]s could directly react with H2O to form [OH]s, resulting in a higher catalytic activity of PtOx@BIT-72-DE under wet conditions than that under dry conditions.27
4. Long-term stability, regeneration performance and recyclability
In addition to evaluating the influence of SCs, catalysts and reaction conditions on the catalytic performance of HCHO, it was also crucial to consider the catalytic stability and regeneration performance capability of BFCMs in practical applications.183–1854.1 Long-term stability of bifunctional composite materials
Due to the uncertainty of RH in the surrounding environment, the study on the stability and water resistance of BFCMs was of great significance in practical applications. In order to study the reproducibility and durability of Pt-related BFCMs, the retention ratios of Pt/Fe2O3@SnO2, Pt/Fe2O3 and Pt/SnO2 to the removal capacity of HCHO were still as high as 94.40, 98.77 and 95.58%, respectively, after repeated oxidation at room temperature 5 times without any regeneration treatment. This implied that Pt-based catalysts exhibited outstanding stability and high efficiency for HCHO removal (Fig. 11).21 Similarly, when RH was 30%, the initial conversion rate of HCHO on 0.87Pt-VO-ZrO2 was 95%. The conversion rate remained at 93% after 720 min of reaction, which meant that the BFCMs had good stability.24 In addition, after the 250 min cycle test, the removal ability of MnOx/AC–N2 to HCHO did not decrease significantly, indicating that the BFCMs had good durability. The good durability of BFCMs was attributed to the stability of surface OVs as the catalytically active sites for the cyclic reaction of catalysts.92 With the increase in RH (20–50%), MnOx/AC–N2 had no significant influence on the removal rate of HCHO (97.5–87.1%), indicating that the BFCMs had a good water resistance.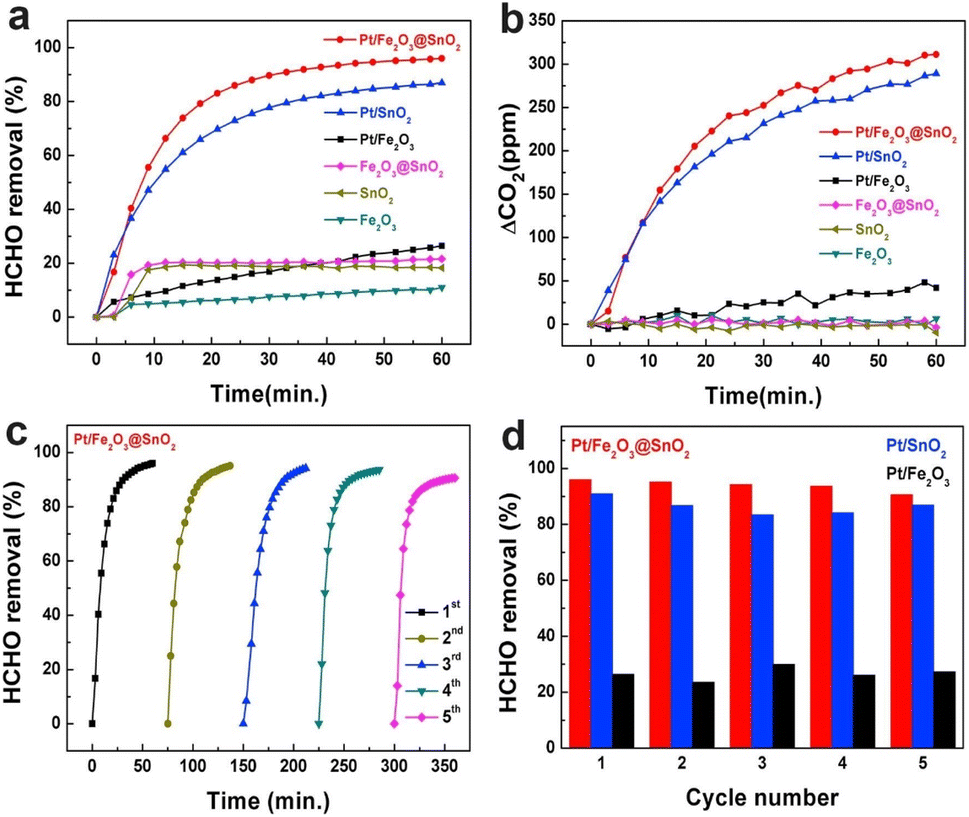 | ||
| Fig. 11 (a) HCHO removal capability of the samples; (b) variations of CO2 concentrations during HCHO removal; (c) consecutive 5-cycle HCHO removal treatment of Pt/Fe2O3@SnO2; (d) comparison of the reusability of the samples. Reproduced with permission.21 Copyright 2018, Elsevier. | ||
4.2 Regeneration of bifunctional composite materials
In general, BFCMs suitable for large-scale applications of HCHO removal should be easy to regenerate in addition to high activity and long-term stability. The existing research indicated that there were many approaches for regeneration, such as temperature control regeneration, alkali washing regeneration, soaking regeneration and so on.In addition to low temperature heating, the regeneration of BFCMs could also be achieved through high temperature calcination. A study indicated that Fe–Mn binary metal oxides could achieve regeneration and recovery of catalytic activities after being calcined at 250 °C in air for 1 h.60 It is worth mentioning that in some cases, the degree of regeneration of BFCMs could be judged based on their adsorption isotherm. For example, if there was no significant hysteresis in the water absorption isotherm of a sample, it may mean that its regeneration process was relatively easy to achieve.186 In conclusion, the thermal regeneration of BFCMs was a sophisticated process, encompassing numerous technologies and approaches. Through rational control of temperature and other circumstances, the activity of catalysts could be effectively restored to minimize resource waste and prevent environmental pollution.
4.3 Recyclability of bifunctional composite materials
The recyclability of BFCMs constituted an important research direction, aiming at improving catalytic efficiency, prolonging the service life of catalysts, and minimizing the waste of resources. From a cost perspective, recyclability was an essential issue worthy of consideration in practical applications. If there were no significant alterations in phases, structure or crystallinity after several repeated uses, it indicated that the sample had high recyclable value. For instance, after 7 cycles of experiments, the XRD peak of ZT/GF0.2 hardly varied, and its removal rate of HCHO (94.38%) and CO2 production rate scarcely declined, indicating that this sample possessed good recyclability and significant practical application value.189 Similarly, 1.8 wt% Au/CeO2 demonstrated no significant loss of activity after 6 cycles of catalytic reaction, suggesting that the sample exhibited multiple cycles of oxidation of HCHO and recyclability.167 The spent MnO2/PET could be simply regenerated by heating at 105 °C for 30 min, indicating that MnO2/PET could be utilized as a recyclable material for the removal of toxic HCHO from indoor air. In conclusion, the recyclability of BFCMs is a multi-faceted matter, involving the structure, phase, catalytic activity of the catalyst, and specific recovery technology. Through continuous research and technological improvement, it is anticipated that more efficient and environmentally friendly BFCM recycling strategies would be achieved in the future.5 Conclusions and future perspectives
Bifunctional composite materials have made great breakthroughs and progress with their superior synergies, becoming one of the most promising materials in the field of VOC removal. In this study, the research progress of adsorption-enhanced catalytic degradation of HCHO based on BFCMs was summarized in detail. In this paper, we first discuss the design and preparation of BFCMs, as well as the multistage synergies (such as adsorption-catalytic synergies, heterogeneous synergies within SCs or CCs, structural synergies, and heat and mass transfer synergies) in BFCMs. These synergetic effects significantly enhanced the adsorption of HCHO, increased the exposure of active sites, and accelerated mass transfer. The activation of O2 and surface adsorbed water at these sites generated an abundance of surface ROS and hydroxyl groups, which was beneficial for further enhancing the activity and stability of BFCMs to degrade HCHO. Meanwhile, to further clarify the influencing factors of HCHO degradation, the impacts of SCs, CCs, and reaction conditions (such as initial concentration of HCHO, light/heat/electricity, relative humidity, environmental media and pH) on HCHO removal in BFCMs are analyzed. Based on the conversion rate of HCHO and specific rate, the degradation performances of BFCMs are evaluated under dynamic and static circumstances, and the degradation pathways of HCHO are also analyzed in combination with DFT calculation. Finally, considering the feasibility of BFCMs in catalytic applications, the relevant issues such as long-term stability, renewability, and recyclability of BFCMs are summarized. In short, the study of adsorption enhanced catalytic degradation of airborne HCHO by BFCMs holds practical feasibility and universality. This study could offer a valuable reference for the design, development, and optimization of regulatory factors for HCHO removal materials in air.In the future, in addition to thermal catalysis, photocatalysis, photothermal catalysis and photoelectric catalysis, ultrasonic catalysis and/or ultrasonic assisted catalysis may be utilized as an emerging environmental treatment technology for the HCHO removal. It can not only enhance the exposure of active sites on the catalyst surface, but also facilitate improved mass and heat transfer between reactants and catalysts, thereby enhancing catalytic efficiency. The integration of these technologies would provide comprehensive and efficient solutions for HCHO treatment in the future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships.Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (No. 2022YFC3902604), the National Natural Science Foundation of China (No. 52425004 and 22305011), the R&D Program of Beijing Municipal Education Commission (No. KM202210005009), and Beijing Natural Science Foundation (No. 2242002).References
- L. Zhou, S. He, Y. Sang, X. Zhang, H. Liu, C. Jia and X. Xu, Catal. Commun., 2020, 142, 106034 CrossRef CAS
.
- J. Guo, C. Lin, C. Jiang and P. Zhang, Appl. Surf. Sci., 2019, 475, 237–255 CrossRef CAS
- J. Gong, S. Rong, X. Wang and Y. Zhou, J. Cleaner Prod., 2022, 377, 134242 CrossRef CAS
- Z. Zheng, C. Zhang, J. Li, D. Fang, P. Tan, Q. Fang and G. Chen, J. Hazard. Mater., 2024, 474, 134710 CrossRef CAS PubMed
- X. J. Zheng, Z. Jaued, C. Liu, A. Tanuir, O. Sandhu, H. R. Liu, X. G. Ji, C. Z. Xing, H. Lin and D. L. Du, J. Environ. Sci., 2024, 135, 656–668 CrossRef CAS PubMed
- X. Wang, J. Li, J. Xing, M. Zhang, R. Liao, C. Wang, Y. Hua and H. Ji, J. Colloid Interface Sci., 2024, 656, 104–115 CrossRef CAS PubMed
- S. Niu and H. Yan, J. Hazard. Mater., 2015, 287, 259–267 CrossRef CAS PubMed
- X. Wang, Z. Xu, J. Li, M. Zhang, K. Li, Y. Zheng and H. ji, Appl. Surf. Sci., 2023, 637, 157917 CrossRef CAS
- X. Wang, F. Yang, J. Yi, J. Kong, J. Gong, A. Yuan, Z. Rui and H. Ji, AIChE J., 2022, 69, e17895 CrossRef
- L. Lu, H. Tian, J. He and Q. Yang, J. Phys. Chem. C, 2016, 120, 23660–23668 CrossRef CAS
- T. He, S. Rong, D. Ding, Y. Zhou, N. Zhang and W. He, ACS Catal., 2023, 13, 8049–8062 CrossRef CAS
- Y. Hua, Y. Ahmadi and K.-H. Kim, Adv. Sci., 2023, 10, 2300079 CrossRef CAS PubMed
- X. Sun, J. Lin, H. Guan, L. Li, L. Sun, Y. Wang, S. Miao, Y. Su and X. Wang, Appl. Catal., B, 2018, 226, 575–584 CrossRef CAS
- Z. Zhang, G. He, Y. Li, C. Zhang, J. Ma and H. He, Environ. Sci. Technol., 2022, 56, 10916–10924 CrossRef CAS PubMed
- J. Chen, D. Yan, Z. Xu, X. Chen, X. Chen, W. Xu, H. Jia and J. Chen, Environ. Sci. Technol., 2018, 52, 4728–4737 CrossRef CAS PubMed
- T. He, D. Shao, X. Zeng and S. Rong, Chemosphere, 2020, 261, 127778 CrossRef CAS PubMed
- M. Zhang, B. Huang, H. Jiang and Y. Chen, Front. Chem. Sci. Eng., 2017, 11, 594–602 CrossRef CAS
- Z. Wang, H. Yu, L. Zhang, L. Guo and X. Dong, J. Taiwan Inst. Chem. Eng., 2020, 107, 119–128 CrossRef CAS
- H. Xie, X. Chen, C. Zhang, Z. Lao, X. Liu, X. Xie, R. Semiat and Z. Zhong, Environ. Sci.: Nano, 2022, 9, 767–780 RSC
- K. Vikrant, K. H. Kim, C. He and D. A. Giannakoudakis, Adv. Funct. Mater., 2022, 32, 2107922 CrossRef CAS
- T. Lv, C. Peng, H. Zhu and W. Xiao, Appl. Surf. Sci., 2018, 457, 83–92 CrossRef CAS
- X. Sun, J. Lin, Y. Chen, Y. Wang, L. Li, S. Miao, X. Pan and X. Wang, Commun. Chem., 2019, 2, 27 CrossRef
- H. Wu, Y. Liu, G. Chen, T. Li, W. Jiang, R. Jia, M. Zhang, S. Yuan, L. Shi and L. Huang, ACS Appl. Nano Mater., 2022, 5, 13100–13111 CrossRef CAS
- S. Peng, R. Li, Y. Huang, Y. Zhang, J.-j. Cao and S. Lee, Appl. Surf. Sci., 2022, 600, 154056 CrossRef CAS
- G. Li and L. Li, RSC Adv., 2015, 5, 36428–36433 RSC
- Z. Xu, J. Chen, S. Cai, D. Yan, X. Chen, W. Xu, J. Chen and H. Jia, Mater. Today Energy, 2019, 14, 100343 CrossRef
- L. Ren, Q. Ma, A. Yin, X. Feng, T. Zhang and B. Wang, ChemSusChem, 2022, 15, e202201324 CrossRef CAS PubMed
- G. Zhang, Z. Sun, Y. Duan, R. Ma and S. Zheng, Appl. Surf. Sci., 2017, 412, 105–112 CrossRef CAS
- J. Yu Zheng, K. Ling Zhou, W. Kang Zhao, Y. Wang, J. He, X. Wang, H. Wang, H. Yan and C. Bao Han, J. Colloid Interface Sci., 2022, 628, 359–370 CrossRef PubMed
- X. Liu, Y. Liu, Y. Wu, S. Dong, G. Qi, C. Chen, S. Xi, P. Luo, Y. Dai, Y. Han, Y. Zhou, Y. Guo and J. Wang, J. Hazard. Mater., 2023, 458, 131848 CrossRef CAS PubMed
- J. Xie, S. Wang and F. Wang, Appl. Surf. Sci., 2023, 644, 158709 CrossRef
- L. Nie, P. Zhou, J. Yu and M. Jaroniec, J. Mol. Catal. A: Chem., 2014, 390, 7–13 CrossRef CAS
- M. Chen, W. Wang, Y. Qiu, H. Wen, G. Li, Z. Yang and P. Wang, ACS Catal., 2022, 12, 5565–5573 CrossRef CAS
- M. Huang, Y. Li, M. Li, J. Zhao, Y. Zhu, C. Wang and V. K. Sharma, Environ. Sci. Technol., 2019, 53, 3610–3619 CrossRef CAS PubMed
- B. Chen, X. Zhu, Y. Wang, L. Yu and C. Shi, Chin. J. Catal., 2016, 37, 1729–1737 CrossRef CAS
- C. Zhang, Y. Li, Y. Wang and H. He, Environ. Sci. Technol., 2014, 48, 5816–5822 CrossRef CAS PubMed
- Y. Huang, Y. Liu, W. Wang, M. Chen, H. Li, S.-c. Lee, W. Ho, T. Huang and J. Cao, Appl. Catal., B, 2020, 278, 119294 CrossRef CAS
- X. Zeng, C. Shan, M. Sun, D. Ding and S. Rong, Chin. Chem. Lett., 2022, 33, 4771–4775 CrossRef CAS
- Z. Duan, Y. Zhou, H. Yang, D. Yan, D. Song, H. Liu, B. Deng, S. Peng and W. Xu, Chem. Eng. J., 2023, 472, 145104 CrossRef CAS
- Y. Li, T. Dong, P. Huang, J. Ji and H. Huang, Appl. Catal., B, 2024, 341, 123322 CrossRef CAS
- S. Gong, W. Wang, K. Chen, K. Xiao and Y. Yin, J. Environ. Chem. Eng., 2022, 10, 107571 CrossRef CAS
- Y. Jiao, C. Jing, Y. Wang, F. Yao, G. Ye, X. Wang, G. Zhao, W. Peng, H. Huang and D. Ye, Appl. Surf. Sci., 2023, 639, 158215 CrossRef CAS
- F. Cui, W. Han, Y. Si, W. Chen, M. Zhang, H. Y. Kim and B. Ding, Compos. Commun., 2019, 16, 61–66 CrossRef
- X. Tang, J. Chen, X. Huang, Y. Xu and W. Shen, Appl. Catal., B, 2008, 81, 115–121 CrossRef CAS
- H. Huang, X. Ye, H. Huang, L. Zhang and D. Y. C. Leung, Chem. Eng. J., 2013, 230, 73–79 CrossRef CAS
- S. Sun, X. Wu, Z. Huang, H. Shen, H. Zhao and G. Jing, Chem. Eng. J., 2022, 435, 135035 CrossRef CAS
- Q. Liu, Y. Wang, M. Wen, Y. Guo, Y. Wei, G. Li and T. An, Environ. Sci.: Nano, 2022, 9, 4162–4176 RSC
- M. He, J. Ji, B. Liu and H. Huang, Appl. Surf. Sci., 2019, 473, 934–942 CrossRef CAS
- Z. Qu, D. Chen, Y. Sun and Y. Wang, Appl. Catal., A, 2014, 487, 100–109 CrossRef CAS
- H. Li, S. Fang, G. Jiang and Z. Zhang, Environ. Sci.: Nano, 2023, 10, 80–91 RSC
- B. Liu, Y. Liu, C. Li, W. Hu, P. Jing, Q. Wang and J. Zhang, Appl. Catal., B, 2012, 127, 47–58 CrossRef CAS
- J. Zhang, Y. Jin, C. Li, Y. Shen, L. Han, Z. Hu, X. Di and Z. Liu, Appl. Catal., B, 2009, 91, 11–20 CrossRef CAS
- H. Li, X. Liu, C. Guo, T. Liu and J. Lu, Chin. J. Catal., 2009, 30, 1001–1006 CAS
- S. J. Park, I. Bae, I.-S. Nam, B. K. Cho, S. M. Jung and J.-H. Lee, Chem. Eng. J., 2012, 195–196, 392–402 CrossRef CAS
- B. Bai and J. Li, ACS Catal., 2014, 4, 2753–2762 CrossRef CAS
- X. Tang, J. Chen, Y. Li, Y. Li, Y. Xu and W. Shen, Chem. Eng. J., 2006, 118, 119–125 CrossRef CAS
- X. Zhu, X. Gao, R. Qin, Y. Zeng, R. Qu, C. Zheng and X. Tu, Appl. Catal., B, 2015, 170, 293–300 CrossRef
- H. Tan, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Chem.–Eur. J., 2019, 25, 16718–16724 CrossRef CAS PubMed
- C. Li, Y. Shen, M. Jia, S. Sheng, M. O. Adebajo and H. Zhu, Catal. Commun., 2008, 9, 355–361 CrossRef CAS
- S. Zhang, H. Wang, H. Si, X. Jia, Z. Wang, Q. Li, J. Kong and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 40285–40295 CrossRef CAS PubMed
- S. Peng, X. Yang, J. Strong, B. Sarkar, Q. Jiang, F. Peng, D. Liu and H. Wang, J. Hazard. Mater., 2020, 396, 122750 CrossRef CAS PubMed
- C. Wang, H. Liu, T. Chen, C. Qing, X. Zou, J. Xie and X. Zhang, Appl. Clay Sci., 2018, 159, 50–59 CrossRef CAS
- Y. Li, P. Sun, T. Liu, L. Cheng, R. Chen, X. Bi and X. Dong, Sep. Purif. Technol., 2023, 311, 123236 CrossRef CAS
- J. Wang, Q. Huang, W. Zhou, Y.-Y. Luo, J.-H. Shi, F.-X. Huang and J. Shi, Chem. Eng. J., 2025, 504, 158892 CrossRef CAS
- H. Zhiwei, G. Xiao, C. Qingqing, H. Pingping, H. Jiming, L. Junhua and T. Xingfu, Angew. Chem., Int. Ed., 2012, 57, 4198–4203 Search PubMed
- J. Zhou, L. Qin, W. Xiao, C. Zeng, N. Li, T. Lv and H. Zhu, Appl. Catal., B, 2017, 207, 233–243 CrossRef CAS
- T. He, X. Zeng and S. Rong, J. Mater. Chem. A, 2020, 8, 8383–8396 RSC
- C. Ma, G. Pang, G. He, Y. Li, C. He and Z. Hao, J. Environ. Sci., 2016, 39, 77–85 CrossRef CAS PubMed
- J. Ye, M. Zhou, Y. Le, B. Cheng and J. Yu, Appl. Catal., B, 2020, 267, 118689 CrossRef CAS
- L. Nie, S. Xin, C. Fang, H. Chen and Y. Yang, Chem. Eng. J., 2024, 479, 147830 CrossRef CAS
- M. Wang, L. Zhang, W. Huang, T. Xiu, C. Zhuang and J. Shi, Chem. Eng. J., 2017, 320, 667–676 CrossRef CAS
- G. Wang, B. Huang, Z. Lou, Z. Wang, X. Qin, X. Zhang and Y. Dai, Appl. Catal., B, 2016, 180, 6–12 CrossRef CAS
- J. Wang, G. He, C. Wang, X. Chen, X. Liu, Y. Li, W. Shan and H. He, Appl. Catal., B, 2024, 347, 123787 CrossRef CAS
- W. Tan, S. Xie, X. Zhang, K. Ye, M. Almousawi, D. Kim, H. Yu, Y. Cai, H. Xi, L. Ma, S. N. Ehrlich, F. Gao, L. Dong and F. Liu, ACS Appl. Mater. Interfaces, 2023, 16, 454–466 CrossRef PubMed
- D. C. Fang, J. Y. Zheng, C. B. Han, W. K. Zhao, Y. G. Lu, B. C. Sun, L. Sun, X. Wang and H. Yan, Appl. Catal., B, 2023, 334, 122837 CrossRef CAS
- Y. Ma and G. Zhang, Chem. Eng. J., 2016, 288, 70–78 CrossRef CAS
- C. Yang, G. Miao, Y. Pi, Q. Xia, J. Wu, Z. Li and J. Xiao, Chem. Eng. J., 2019, 370, 1128–1153 CrossRef CAS
- S. K. Mondal, P. Aina, A. A. Rownaghi and F. Rezaei, ChemPlusChem, 2024, 89, e202300419 CrossRef CAS PubMed
- Q. Yu, Y. Feng, J. Wei, X. Tang and H. Yi, Chin. Chem. Lett., 2022, 33, 3087–3090 CrossRef CAS
- A. Keunecke, M. Dossow, V. Dieterich, H. Spliethoff and S. Fendt, Front. Energy Res., 2024, 12, 1344179 CrossRef
- C. Nie, L. Liu and R. He, Sep. Purif. Technol., 2018, 206, 316–323 CrossRef CAS
- Y. Chen, Z. Mai, S. Fan, Y. Wang, B. Qiu, Y. Wang, J. Chen and Z. Xiao, J. Membr. Sci., 2021, 628, 119233 CrossRef CAS
- A. Farhad, M. Hossein, R. Mashallah, T. Shima, B. Addie, K. Frank, M. A. Tejraj, L. Jian-Rong and A. Mohammad, Prog. Mater. Sci., 2021, 125, 100904 Search PubMed
- Y. Shi, X. Guo, Y. Wang, F. Kong and R. Zhou, Green Energy Environ., 2023, 8, 1654–1663 CrossRef CAS
- S. Xia, G. Zhang, Y. Meng, C. Yang, Z. Ni and J. Hu, Appl. Catal., B, 2020, 278, 119266 CrossRef CAS
- T. Lv, C. Peng, H. Zhu and W. Xiao, Appl. Surf. Sci., 2018, 457, 83–92 CrossRef CAS
- L. Nie, J. Yu, X. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777–2783 CrossRef CAS PubMed
- W. Jinlong, L. Jinge, J. Chuanjia, Z. Peng, Z. Pengyi and Y. Jiaguo, Appl. Catal., B, 2016, 204, 147–155 Search PubMed
- H. Chen, Z. Rui, X. Wang and H. Ji, Catal. Today, 2015, 258, 56–63 CrossRef CAS
- K. Vellingiri, K. Vikrant, V. Kumar and K.-H. Kim, Chem. Eng. J., 2020, 399, 125759 CrossRef CAS
- W.-J. Qiang, Q. Huang, J.-H. Shen, Q.-F. Ke, J.-Y. Lü and Y.-P. Guo, J. Cleaner Prod., 2022, 368, 133089 CrossRef CAS
- Y. Huang, Y. Liu, W. Wang, M. Chen, H. Li, S.-c. Lee, W. Ho, T. Huang and J. Cao, Appl. Catal., B, 2020, 278, 119294 CrossRef CAS
- G. S. Zhu, J. Y. Zheng, C. Bao Han, H. Zhang, P. Chang, W. Kang Zhao, J. He, Y. Xia, X. Song and H. Yan, ChemistrySelect, 2023, 8, 4060 Search PubMed
- S. Zhu, J. Zheng, S. Xin and L. Nie, Chem. Eng. J., 2022, 427, 130951 CrossRef CAS
- Q. Xu, W. Lei, X. Li, X. Qi, J. Yu, G. Liu, J. Wang and P. Zhang, Environ. Sci. Technol., 2014, 48, 9702–9708 CrossRef CAS PubMed
- H.-F. Li, N. Zhang, P. Chen, M.-F. Luo and J.-Q. Lu, Appl. Catal., B, 2011, 110, 279–285 CrossRef CAS
- J. He, X. Yu, X. Luan, H. Li, S. J. Shah, W. Su, Z. Jia, J. Chen, L. Zhou, J. Deng, Z. Zhao, Z. Huang and Z. Zhao, Chem. Eng. J., 2024, 479, 147828 CrossRef CAS
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2019, 120, 464–525 CrossRef PubMed
- J. Liu, C. Xiong, S. Jiang, X. Wu and S. Song, Appl. Catal., B, 2019, 249, 282–291 CrossRef CAS
- W. Yuan, C. Wang, T. Qi, Y. Wan, S. Zhang, Y. Wu, K. Hu, G. Peng, B. Zhang and S. Shi, Appl. Catal., B, 2022, 318, 121780 CrossRef CAS
- C. Mang, G. Li, J. Luo, M. Rao, Z. Peng and T. Jiang, J. Environ. Chem. Eng., 2022, 10, 108740 CrossRef CAS
- C. Ma, D. Wang, W. Xue, B. Dou, H. Wang and Z. Hao, Environ. Sci. Technol., 2011, 45, 3628–3634 CrossRef CAS PubMed
- J. Gong, M. Zhang, J. Li, X. Wang, Y. Zhou, C. Yang, Y. Hua, C. Wang and A. Yuan, Appl. Surf. Sci., 2024, 669, 160513 CrossRef CAS
- C. Wang, X. Liu, J. Wang, Y. Li, S. Xie, F. Liu, C. Zhang, Y. Zheng, W. Shan and H. He, Chin. Chem. Lett., 2024, 35, 108739 CrossRef CAS
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC
- X. Liu, J. Wu, S. Zhang, Q. Li, Z. Wu and J. Zhang, Appl. Catal., B, 2023, 320, 121994 CrossRef CAS
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS PubMed
- X. Li, M. Chen, G. Li and P. Wang, Appl. Surf. Sci., 2022, 589, 152855 CrossRef CAS
- D. Cericola and M. E. Spahr, Electrochim. Acta, 2016, 191, 558–566 CrossRef CAS
- H. Toda, H. Li, R. Batres, K. Hirayama and H. Fujihara, Acta Mater., 2023, 257, 119188 CrossRef CAS
- V. Viswanathan and F. Y.-F. Wang, Nanoscale, 2012, 4, 5110 RSC
- F. Zhang, S.-W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127–129 CrossRef CAS
- Y. Lee, G. He, A. J. Akey, R. Si, M. Flytzani-Stephanopoulos and I. P. Herman, J. Am. Chem. Soc., 2011, 133, 12952–12955 CrossRef CAS PubMed
- S. Rong, P. Zhang, Y. Yang, L. Zhu, J. Wang and F. Liu, ACS Catal., 2017, 2, 1057–1067 CrossRef
- S. Lai, Y. Qiu and S. Wang, J. Catal., 2006, 237, 303–313 CrossRef CAS
- A. Marcilla, A. Gómez, S. Menargues, J. García-Martínez and D. Cazorla-Amorós, J. Anal. Appl. Pyrolysis, 2003, 68–69, 495–506 CrossRef CAS
- Z. Xu, J. Yu, J. Low and M. Jaroniec, ACS Appl. Mater. Interfaces, 2014, 6, 2111–2117 CrossRef CAS PubMed
- R. Oketani, H. Takahashi, M. Hoquante, C. Brandel, P. Cardinael and G. Coquerel, J. Mol. Struct., 2019, 1184, 36–40 CrossRef CAS
- L. Wang, Y. Hou, X. Zhong, J. Hu, F. Shi and H. Mi, Carbohydr. Polym., 2018, 208, 42–49 CrossRef PubMed
- J. Zhang, L. Xu and W.-Y. Wong, Coord. Chem. Rev., 2017, 355, 180–198 CrossRef
- P. Zhou, X. Zhu, J. Yu and W. Xiao, ACS Appl. Mater. Interfaces, 2013, 5, 8165–8172 CrossRef CAS PubMed
- J. Xu, X. Xiao, Z. Zhang, Y. Wu, D. T. Boyle, H. K. Lee, W. Huang, Y. Li, H. Wang, J. Li, Y. Zhu, B. Chen, W. Mitch and Y. Cui, Nano Lett., 2020, 20, 8719–8724 CrossRef CAS PubMed
- Y. Yazawa, N. Takagi, H. Yoshida, S.-i. Komai, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, Appl. Catal., A, 2002, 233, 103–112 CrossRef CAS
- C. Dong, J.-J. Yang, L.-H. Xie, G. Cui, W.-H. Fang and J.-R. Li, Nat. Commun., 2022, 13, 4991 CrossRef CAS PubMed
- C. Dong, T. He, W. Wu, G.-R. Si, L.-H. Xie and J.-R. Li, J. Mater. Chem. A, 2024, 12, 15055–15062 RSC
- L. Yang, D. Lu, L. Zhu and D. Xia, J. Cleaner Prod., 2023, 413, 137462 CrossRef CAS
- S. K. Mondal, P. Aina, A. A. Rownaghi and F. Rezaei, ChemPlusChem, 2023, 89, e202300419 CrossRef PubMed
- C. Zhang, H. He and K.-i. Tanaka, Appl. Catal., B, 2006, 65, 37–43 CrossRef CAS
- T. He, D. Ding, Y. Zhou and S. Rong, ACS ES&T Eng., 2022, 2, 1403–1413 Search PubMed
- B.-T. Teng, S.-Y. Jiang, Z.-X. Yang, M.-F. Luo and Y.-Z. Lan, Surf. Sci., 2010, 604, 68–78 CrossRef CAS
- Y. He, Y. Zhu, J. Qian, P. Wang, Y. Zhang, K. Xu, B. Lu, Y. Liu and J. Shen, Chem. Eng. J., 2023, 474, 145737 CrossRef CAS
- R. F. André, G. Rousse, C. Sassoye, M. Avdeev, B. Lassalle-Kaiser, B. Baptiste and S. Carenco, Chem. Mater., 2023, 35, 5040–5048 CrossRef
- T. Gan, Z. Shi, Y. Deng, J. Sun and H. Wang, Electrochim. Acta, 2014, 147, 157–166 CrossRef CAS
- S. H. Brodersen, U. Grønbjerg, B. Hvolbæk and J. Schiøtz, J. Catal., 2011, 284, 34–41 CrossRef CAS
- J. Zhou, G. Liu, Q. Jiang, W. Zhao, Z. Ao and T. An, Chin. J. Catal., 2020, 41, 1633–1644 CrossRef CAS
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC
- L. Zhang, S. Wang, C. Ni, M. Wang and S. Wang, Chem. Eng. Sci., 2021, 229, 116011 CrossRef CAS
- Z. Fan, Z. Zhang, W. Fang, X. Yao, G. Zou and W. Shangguan, Chin. J. Catal., 2016, 37, 947–954 CrossRef CAS
- H. Tan, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Chem.–Eur. J., 2019, 25, 16718–16724 CrossRef CAS PubMed
- S. Zhang, Y. Zhuo, C. I. Ezugwu, C.-c. Wang, C. Li and S. Liu, Environ. Sci. Technol., 2021, 55, 8341–8350 CrossRef CAS PubMed
- W. Sun and K. Yang, Mater. Chem. Front., 2020, 8, 76–80 CAS
- D. Kong, Y. Zheng, M. Kobielusz, Y. Wang, Z. Bai, W. Macyk, X. Wang and J. Tang, Mater. Today, 2018, 21, 897–924 CrossRef CAS
- Y. Bai, S. Li, B. Yin, J. Zhao and H. Li, Trans. Tianjin Univ., 2024, 30, 130–139 CrossRef CAS
- C. You, C. Wang, M. Cai, Y. Liu, B. Zhu and S. Li, Acta Phys.-Chim. Sin., 2024, 40, 2407014 CrossRef
- N. Zhang, W. He, Z. Cheng, J. Lu, Y. Zhou, D. Ding and S. Rong, Chem. Eng. J., 2023, 466, 143160 CrossRef CAS
- F. Yuan, R. Yang, C. Li, Y. Tan, X. Zhang, S. Zheng and Z. Sun, Build. Environ., 2022, 219, 109216 CrossRef
- C. Mang, J. Luo, P. Cao, X. Zhang, M. Rao, G. Li and T. Jiang, Chemosphere, 2022, 287, 132293 CrossRef CAS PubMed
- Y. Huang, X. Zhu, D. Wang and S. Hui, Environ. Res., 2023, 238, 117265 CrossRef CAS PubMed
- Y. Zhu, W. Zhao, B. Jing, J. Zhou, B. Cai, D. Li and Z. Ao, Chin. Chem. Lett., 2023, 34, 107816 CrossRef CAS
- W. Kang Zhao, J. Yu Zheng, C. Bao Han, J. Ruan, Y. Lu, K. Ling Zhou, T. Rui Zhai, H. Wang and H. Yan, Chem. Eng. J., 2022, 440, 135877 CrossRef
- S. Xu, Q. Shen, J. Zheng, Z. Wang, X. Pan, N. Yang and G. Zhao, Adv. Sci., 2022, 9, 2203941 CrossRef CAS PubMed
- S. Zhou, Q. Liu, Y. Zhou and Y. Li, Adv. Environ. Prot., 2022, 12, 1041–1048 CrossRef
- S.-Y. Zhang, Z. Li, X. Shen, J. Shan, J. Zhan, H. Zhou, X. Yi, H.-Y. Lian and Y. Liu, Environ. Res., 2023, 235, 116683 CrossRef CAS PubMed
- T. Cai, P. Zhang, X. Shen, E. Huang, X. Shen, J. Shi, Z. Wang and Q. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 37147–37154 CrossRef CAS PubMed
- W. Yuan, S. Zhang, Y. Wu, X. Huang, F. Tian, S. Liu and C. Li, Appl. Catal., B, 2020, 272, 118992 CrossRef CAS
- M. A. Sidheswaran, H. Destaillats, D. P. Sullivan, J. Larsen and W. J. Fisk, Appl. Catal., B, 2011, 107, 34–41 CrossRef CAS
- B. Zhu, X.-S. Li, P. Sun, J.-L. Liu, X.-Y. Ma, X. Zhu and A.-M. Zhu, Chin. J. Catal., 2017, 38, 1759–1769 CrossRef CAS
- B. Zhu, L.-Y. Zhang, M. Li, Y. Yan, X.-M. Zhang and Y.-M. Zhu, Chem. Eng. J., 2020, 381, 122599 CrossRef CAS
- Y.-H. Liu, J.-A. Lv, M.-M. Xu, C. Dong, X.-M. Liu, J.-R. Li and L.-H. Xie, ACS Appl. Nano Mater., 2023, 6, 7794–7801 CrossRef CAS
- Z. Lin, X. Tong, W. Shen, J.-C. Roux and H. Xi, J. Cleaner Prod., 2020, 244, 118863 CrossRef CAS
- H. Huang and D. Y. C. Leung, J. Catal., 2011, 280, 60–67 CrossRef CAS
- X. Zhu, B. Cheng, J. Yu and W. Ho, Appl. Surf. Sci., 2016, 364, 808–814 CrossRef CAS
- D. Li, X. Chen, Y. Huang, G. Zhang, D. Zhou and B. Xiao, J. Hazard. Mater., 2022, 439, 129608 CrossRef CAS PubMed
- Z. Wang, J. Pei and J. Zhang, Build. Environ., 2013, 65, 49–57 CrossRef
- N. Wakao, S. Kaguei and T. Funazkri, Chem. Eng. Sci., 1979, 34, 325–336 CrossRef CAS
- S. Rong, P. Zhang, J. Wang, F. Liu, Y. Yang, G. Yang and S. Liu, Chem. Eng. J., 2016, 306, 1172–1179 CrossRef CAS
- X. Li, H. Li, Y. Huang, J. Cao, T. Huang, R. Li, Q. Zhang, S.-c. Lee and W. Ho, J. Hazard. Mater., 2022, 424, 127217 CrossRef CAS PubMed
- F. Liu, S. Zhang, X. Zhang, J. Shen, L. Wan, A. Bahi and F. Ko, J. Hazard. Mater., 2022, 421, 126769 CrossRef CAS PubMed
- L. Liu, G. Jing, C. Xu, X. Zhang, X. Zhang, L. Guo, Z. Huang, X. Wu, H. Zhao, C.-S. Yuan, H. Shen and W. Xia, Appl. Catal., B, 2024, 344, 123634 CrossRef CAS
- C. Mang, C. Huang, J. Luo, M. Rao, Z. Peng and G. Li, Mol. Catal., 2022, 530, 112643 CrossRef CAS
- J. Chen, M. Jiang, W. Xu, J. Chen, Z. Hong and H. Jia, Appl. Catal., B, 2019, 259, 118013 CrossRef CAS
- L. Zhao, Y. Yang, J. Liu and J. Ding, Chemosphere, 2023, 330, 138754 CrossRef CAS PubMed
- Y. Wang, K. Liu, J. Wu, Z. Hu, L. Huang, J. Zhou, T. Ishihara and L. Guo, ACS Catal., 2020, 10, 10021–10031 CrossRef CAS
- Y. Guo, M. Wen, G. Li and T. An, Appl. Catal., B, 2021, 281, 119447 CrossRef CAS
- Z. Zheng, C. Zhang, J. Li, D. Fang, P. Tan, Q. Fang and G. Chen, Chemosphere, 2024, 356, 142024 CrossRef CAS PubMed
- Z. Lan, Y. Yu, H. Li, L. Zhang and Y. Cao, Mater. Sci. Semicond. Process., 2021, 123, 105547 CrossRef CAS
- G. Li, C. Mang, J. Luo, M. Rao, Z. Peng and T. Jiang, J. Colloid Interface Sci., 2022, 618, 229–240 CrossRef CAS PubMed
- S. Rong, P. Zhang, F. Liu and Y. Yang, ACS Catal., 2018, 8, 3435–3446 CrossRef CAS
- R. Fang, X. Huang, X. a. Luo, Y. Sun, Z. Liu, L. Ao, F. Dong and H. Huang, J. Environ. Chem. Eng., 2023, 11, 109064 CrossRef CAS
- B.-B. Chen, C. Shi, M. Crocker, Y. Wang and A.-M. Zhu, Appl. Catal., B, 2013, 132–133, 245–255 CrossRef CAS
- Z. Liu, D. Zhang, G. Jin and W. Yang, Appl. Surf. Sci., 2020, 534, 147594 CrossRef CAS
- Z. Yan, Z. Xu, J. Yu and M. Jaroniec, Appl. Catal., B, 2016, 199, 458–465 CrossRef CAS
- H. Wang, W. Guo, Z. Jiang, R. Yang, Z. Jiang, Y. Pan and W. Shangguan, J. Catal., 2018, 361, 370–383 CrossRef CAS
- K. Qian, L. Du, X. Zhu, S. Liang, S. Chen, H. Kobayashi, X. Yan, M. Xu, Y. Dai and R. Li, J. Mater. Chem. A, 2019, 7, 14592–14601 RSC
- H. Li, X. Feng, D. Ma, M. Zhang, Y. Zhang, Y. Liu, J. Zhang and B. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 3160–3163 CrossRef CAS PubMed
- P. Zhou, J. Yu, L. Nie and M. Jaroniec, J. Mater. Chem. A, 2015, 3, 10432–10438 RSC
- J. Wang, P. Zhang, J. Li, C. Jiang, R. Yunus and J. Kim, Environ. Sci. Technol., 2015, 49, 12372–12379 CrossRef CAS PubMed
- C. Huang, Y. Ding, Y. Chen, P. Li, S. Zhu and S. Shen, J. Environ. Sci., 2017, 60, 61–69 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |