
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModerate-temperature fabrication of BaZrS3 thin films via dithiocarbamate-based solution processing and oxygen-sink boron sulfurization†
Jaewook Leea,
Neul Haa,
Jisu Junga,
GwangHee Leeb,
Sunil V. Barmaa,
Jae-Hwan Kimb,
Jung Kyu Kim ab,
Sae Byeok Joacd,
Jin-Wook Leebcd and
Wooseok Yang
*acd
ab,
Sae Byeok Joacd,
Jin-Wook Leebcd and
Wooseok Yang
*acd
aSchool of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: wooseok.yang@skku.edu
bDepartment of Nano Engineering, Department of Nano Science and Technology, SKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon 16419, Republic of Korea
cSKKU Institute of Energy Science and Technology (SIEST), Sungkyunkwan University, Suwon 16419, Republic of Korea
dDepartment of Future Energy Engineering (DFEE), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
First published on 27th February 2025
Abstract
Chalcogenide perovskites are increasingly recognized as promising light-absorbing materials because of their low toxicity, cost-effectiveness, and abundance. In this class, BaZrS3 is particularly attractive because of its outstanding optoelectronic properties. However, practical device applications are limited by the high-temperature synthesis (>900 °C), creating an ongoing demand for low-temperature, solution-processable methods. Existing low-temperature approaches are often constrained to nanocrystal- or slurry-based synthesis or involve complex sulfurization steps with toxic gases, limiting their scalability and reproducibility. This paper introduces a dithiocarbamate-based molecular ink strategy that enables the use of soluble metal precursors and lower processing temperatures. This method allows the formation of a homogeneous solution that facilitates the fabrication of BaZrS3 thin films at moderate temperatures via a simplified sulfurization process using boron sulfide within a graphite box. The approach was used to fabricate pure BaZrS3 thin films at 650 °C that exhibit a distinct photocurrent response. This straightforward method not only highlights the influence of chemical coordination in solution but also represents a significant advancement in BaZrS3 fabrication, offering an accessible pathway toward scalable production for solar cell applications.
 Wooseok Yang | Wooseok Yang is an Assistant Professor of the school of chemical engineering at Sungkyunkwan University (SKKU) in South Korea. He earned his PhD in Materials Science and Engineering from Yonsei University in 2018. From 2019 to 2021, he worked as a postdoctoral researcher with Prof. David Tilley at the University of Zurich before joining SKKU in 2022. His current research focuses on novel chalcogenide light absorbers (AgBiS2, BaZrS3, etc), (photo-)electrochemical energy conversion to produce H2 and NH3, electrochemical impedance spectroscopy for battery analysis, and electrochemical CO2 capture. |
Introduction
The most important building block in solar energy conversion devices is the light-absorbing semiconductor.1–3 Over the past decade, halide perovskites with an ABX3 structure, – in which the A site hosts an organic or metal component, the B site is occupied by Pb or Sn, and the X site consists of halides, – have emerged as highly attractive candidates.4 This appeal stems not only from their intriguing properties but also from their relatively simple, low-temperature processing methods, which have enabled extensive research and development in this field.5 Consequently, the use of halide perovskites has led to rapid and continuous improvements in efficiency. Recently, single-junction efficiencies of 26.7% and tandem device efficiencies of 34.6% have been recorded, demonstrating the remarkable progress that has been achieved in a short time.6,7 However, despite the promising properties of halide perovskites, the commercialization of perovskite-based solar cells and optoelectronic devices has been hindered by significant challenges. The primary issues are the toxicity of Pb-based perovskites and the vulnerability of their ionic crystal structures to heat and moisture.8 Although various strategies such as encapsulating or substituting Sn with Pb are being investigated to mitigate these issues,9–11 ultimately, the development of new materials that can overcome the intrinsic limitations of halide perovskites is essential.Recently, chalcogenide perovskites, which incorporate chalcogen elements at the anionic positions of the ABX3 structure, have emerged as promising alternatives to halide perovskites.12,13 In chalcogenide perovskites, the B-site cations are coordinated with six X-site anions in an octahedral arrangement, and eight of these octahedrally shared corners form a three-dimensional network.14 Representative chalcogenide perovskites such as CaZrS3, CaHfS3, BaZrS3, and BaHfS3 exhibit this corner-sharing BX6 octahedral structure.15 Among these, BaZrS3 has garnered the most attention and has recently been the primary focus of research.16–18 As a promising alternative to halide perovskites, BaZrS3 offers a Pb-free composition, significantly reducing the environmental toxicity concerns associated with Pb-based materials. Additionally, BaZrS3 is composed of earth-abundant elements that enhance its sustainability and scalability for potential large-scale applications in optoelectronic devices. In addition to these compositional advantages, BaZrS3 possesses a high absorption coefficient and a bandgap of approximately 1.8 eV, which makes it suitable for use as the top cell in tandem solar cells.17 The bandgap of BaZrS3 can be tuned through doping; for instance, the bandgaps for BaZr0.75Ti0.25S3 and BaZr(S0.6Se0.4)3 are 1.43 and 1.63 eV, respectively.19 Additionally, the material exhibits distinct photoluminescence signals at room temperature, indicating its potential for applications in high-performance solar cells and optoelectronic devices.20
Despite these advantages, although the synthesis of BaZrS3 powder was established several decades ago, significant challenges persist in fabricating BaZrS3-based devices.21 One of the most significant obstacles is the excessively high processing temperature. The first BaZrS3 film was generated by using pulsed laser deposition (PLD) to produce a BaZrO3 thin film with subsequent sulfurization at 1000 °C using carbon disulfide (CS2).22 However, this high temperature is unsuitable for solar cell applications for which a conductive substrate is essential. Mo, as a commonly used substrate for solar cells, is known for its exceptional thermal stability, tolerating temperatures up to 600 °C.23 Therefore, to enable the development of BaZrS3 solar cells, it is crucial to reduce the processing temperature to a maximum of 600 °C. Recent studies have shown some progress by demonstrating the synthesis of BaZrS3 nanocrystals at relatively low temperature. For instance, Yang et al. synthesized BaZrS3 nanocrystals using barium dibutyldithiocarbamate (BaDBuDTC) and zirconium diethyldithiocarbamate (ZrDEtDTC) as precursors in the form of Ba–S and Zr–S compounds at 330 °C.24 However, the conversion of nanocrystal-based solutions into films poses significant challenges. This process requires the removal of ligands, which prevents nanocrystal precipitation, and subsequent high-temperature treatment to induce interconnections among the nanocrystals. This post-heat treatment can result in non-uniform crystal aggregation, composition loss, and the formation of secondary phases. Therefore, a direct method to fabricate chalcogenide thin films from homogeneous molecular inks is required. Agrawal's research group has advanced this concept from slurry-based solutions to molecular-ink-based approaches.25–27 Nevertheless, these studies primarily focus on the synthetic process, and the approach remains limited in terms of production versatility. Furthermore, details of the chemistry occurring within the solution and the mechanism of the underlying sulfurization have yet to be systematically studied, both of which are essential for a comprehensive understanding of the mechanism of BaZrS3 formation. Given that the rapid advances in halide perovskites have been largely driven by the simplicity of the solution-based film fabrication process,28–30 developing a similar but simpler solution-processing method for chalcogenide perovskite thin films is essential to accelerate progress in this field.
Developing an effective solution-processing method for chalcogenide thin films requires several key criteria to be met. First, sulfur should be included in the homogeneous solution together with Ba and Zr. In previous studies based on the PLD-CS2 process, incorporating the sulfur component by substituting the BaZrO3 powder used in PLD to BaZrS3 powders resulted in a decrease in the synthesis temperature from 900 to 550 °C.22,31 Similarly, in solution-based processes, the pre-formation of Ba–Zr–S bonds at the molecular level can reduce the energy required to crystallize the perovskite structure. Second, it is important to minimize sulfur loss and prevent oxidation during the sulfurization step. Sulfur loss is common during the formation of multinary sulfides at high temperatures because of its high volatility. Considering the high processing temperature in BaZrS3 synthesis, careful management of sulfur loss is essential. Furthermore, oxidation is one of the biggest obstacles in BaZrS3 synthesis. The oxidation product BaZrO3 exhibits high chemical stability and requires substantial energy and high temperatures to be converted back into BaZrS3. Although many studies have used CS2 and H2S gases to mitigate oxidation and continuously supply sulfur, these gases are toxic and require special handling measures.17–19,22
Here, we propose a new method for the low-temperature fabrication of BaZrS3 thin films using a molecular-ink-based system and devise a sulfurization technique that is straightforward and less toxic than the conventional CS2 and H2S sulfurization methods. First, homogeneous solutions containing Ba, Zr, and S were prepared using a dithiocarbamate (DTC)-based molecular ink. This molecular ink can form metal–DTC complexes within the solution, enabling metal–sulfur coordination at the molecular level. This ink was converted into a BaZrS3 thin film through a process involving spin coating followed by graphite box sulfurization. To prevent oxidation and minimize sulfur loss during the sulfurization step, boron sulfurization was used as an “oxygen sink” to remove oxygen from the film while supplying sulfur.32,33 Our research not only demonstrates the effectiveness of boron sulfurization in comparison to elemental sulfur sulfurization but also suggests a new homogeneous ink system containing Ba, Zr, and S. These advances were enabled by gaining comprehensive insights into the fundamental chemistry occurring within the solution. Additionally, we conducted a systematic study on the sulfur loss during the sulfurization process and perovskite phase formation. Finally, we highlight the optical properties and performance of the fabricated films for photodetector applications.
Results and discussion
As illustrated in Fig. 1, the fabrication process begins with the formation of a precipitate-free homogeneous solution containing Ba, Zr, and S components. This is followed by spin-coating and drying in an N2-filled environment and subsequent sulfurization to produce the BaZrS3 thin films. DTC was selected as the sulfur source because its sulfur-containing structure can be synthesized by reacting CS2 with an amine compound. The DTC system exhibits superior solubility and enhanced metal–sulfur bond stability compared to alternative molecular ink systems, such as thiol–amine or thiourea-based approaches, making it a more effective precursor for controlled thin-film synthesis.34,35 In this study, DTC was prepared using N-butylamine and CS2 in pyridine as the solvent. The bidentate sulfur atoms in DTC carry a partially negative charge, which readily attracts positively charged ions in the solution (Fig. 2a).34 To understand the molecular interactions within the solution, liquid Raman spectroscopy was performed. Pure CS2 exhibits a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretch mode (ν1, 650 cm−1) and a bend mode (2ν2, 795 cm−1).36 While the stretch mode remains unchanged when CS2 interacts with other molecules or solvents, the bending mode typically shows a positive shift.37,38 In our DTC solution (Fig. 2b), the CS bending mode shifts positively from 795 to 807 cm−1 upon formation of DTC from CS2 and N-butylamine. This shift is attributed to the partially negative charge on the sulfur component.
S stretch mode (ν1, 650 cm−1) and a bend mode (2ν2, 795 cm−1).36 While the stretch mode remains unchanged when CS2 interacts with other molecules or solvents, the bending mode typically shows a positive shift.37,38 In our DTC solution (Fig. 2b), the CS bending mode shifts positively from 795 to 807 cm−1 upon formation of DTC from CS2 and N-butylamine. This shift is attributed to the partially negative charge on the sulfur component.
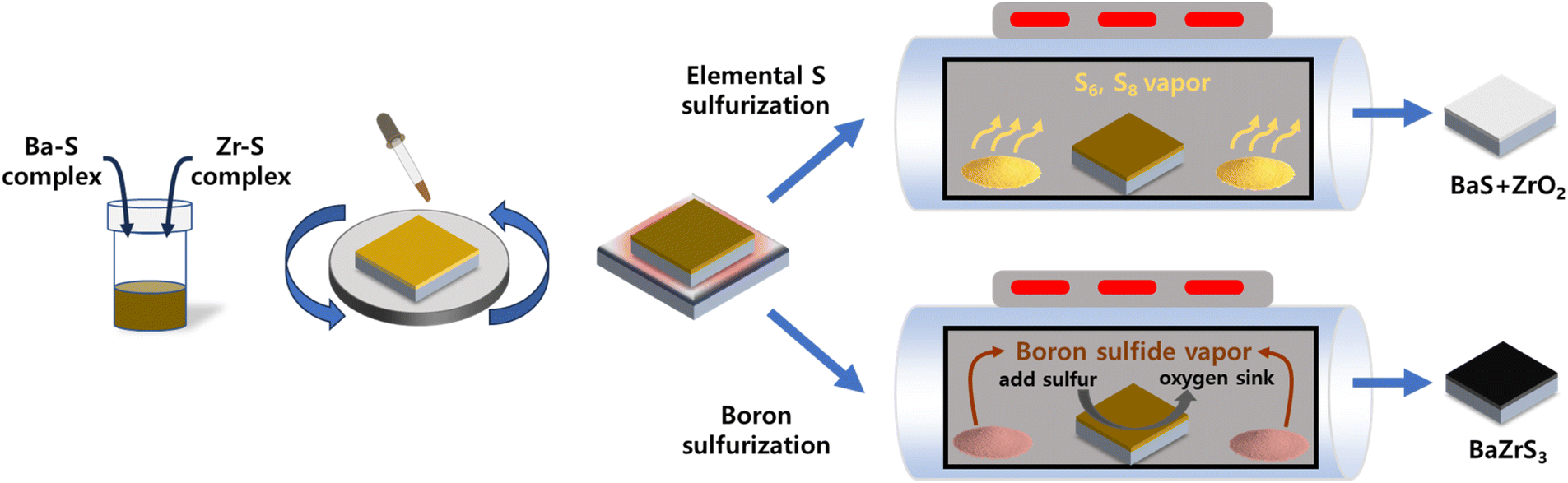 | ||
| Fig. 1 Schematic illustration of the overall process from homogeneous molecular ink to final BaZrS3 thin film comparing elemental sulfur sulfurization and boron sulfurization. | ||
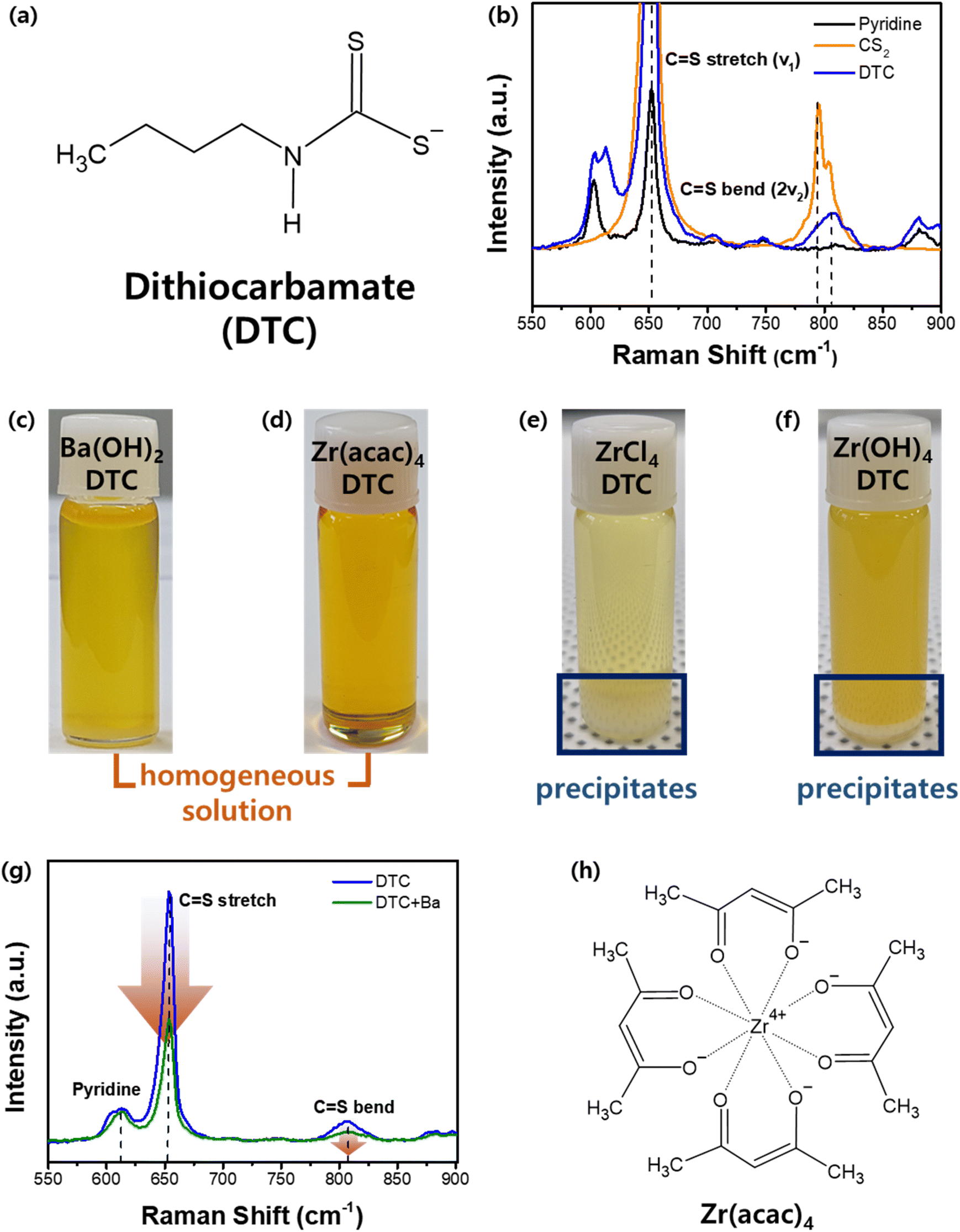 | ||
| Fig. 2 (a) Structure of the DTC compound. (b) Liquid Raman spectroscopy of DTC compared to CS2 and pyridine. (c and d) Homogeneous solutions from optimal metal precursors. (e and f) Precipitated solutions from highly reactive Zr precursors. (g) Liquid Raman spectroscopy of DTC + Ba solution. (h) Structure of Zr acetylacetonate. | ||
Our approach involves forming a metal–sulfur complex by leveraging the DTC structure. Achieving a homogeneous solution with DTC requires an appropriate metal precursor containing either Ba or Zr that is capable of forming a stable metal cation–DTC complex that remains dissolved and does not precipitate. The optimal metal–DTC complex was designed based on a detailed understanding of molecular interactions. Various Ba and Zr precursor candidates were evaluated for their solubilities in DTC solutions, as shown in Fig. 2c–f. As described in a previous report in which Ba(OH)2·8H2O was shown to interact with DTC, leading to the formation of a Ba–S complex, a homogeneous solution containing Ba and S was obtained when Ba(OH)2·8H2O was dissolved in a solution of DTC (Fig. 2c).24 Liquid Raman spectroscopy was employed to clarify the interactions and coordination behavior of the Ba and DTC. Fig. 2g illustrates the variations observed before and after dissolving the Ba precursor in the DTC solution. As a solvent in DTC solution, a pyridine peak is observed at 610 cm−1. The CS stretch and bend peaks originating from CS2 are also observed in both solutions. However, whereas the pyridine peak remains largely unchanged upon the dissolution of the Ba precursor in the DTC solution, both the CS stretch and bend peaks exhibited a significant decrease in intensity. As a decrease in the Raman intensity indicates a decrease in the concentration of certain materials, it is evident that the CS bond in DTC was consumed upon reaction with the Ba cation, whereas pyridine had a negligible effect on the dissolution mechanism. As noted previously, DTC contains a partially negative sulfur atom capable of attracting positively charged ions. We also investigated the possibility of an interaction between the N-butylamine in DTC and the Ba cations. N-Butylamine in the DTC structure exhibits a distinct Raman peak at 1296 cm−1 (Fig. S1a†), which does not overlap with peaks from either pyridine or CS2. Analogous to pyridine during the dissolution process, N-butylamine also exhibited no changes in either peak position or intensity before and after the dissolution of the Ba precursor (Fig. S1b†). Thus, we can conclude that the Ba2+ cation is coordinated with the sulfur component in DTC but not with N-butylamine or pyridine in the solution.
To further investigate the dissolution mechanism in the metal–DTC solutions, we conducted additional tests using other metal–DTC systems. Specifically, we examined a DTC-based molecular ink system for fabricating AgBiS2 thin films that utilized the same DTC compound to dissolve the Ag and Bi precursors separately.39 In this study, the binding energy between the metal and DTC was calculated, suggesting a stable metal–DTC complex structure in which the CSS− group coordinates with the metal cations. Using the same Ag and Bi precursors, we prepared Ag–DTC and Bi–DTC solutions with the same concentrations as the Ba–DTC solution and performed liquid Raman spectroscopy. In both the Bi–DTC and Ag–DTC solutions, a decrease in the intensity of the CS stretching and bending modes was observed (Fig. S1c and d†). These consistent results across different metal–DTC complexes reinforce the conclusion that Ba–CSS− coordination takes place.
Unlike Ba, the Zr–DTC solution exhibited a completely different behavior. Our first Zr precursor candidate was ZrCl4. ZrCl4 can react with DTC to form a Zr–DTC complex in colloidal synthesis.24 And it is also utilized in the synthesis of ZrS2 powder, indicating its strong affinity for sulfur components.40,41 However, this affinity has a detrimental effect on the formation of homogeneous solutions. Upon dissolving the ZrCl4 powder in the DTC solution, a white precipitate formed at the bottom of the solution (Fig. 2e). Because the resulting Zr–DTC complex from ZrCl4 was insoluble in pyridine, this precursor was determined to be unsuitable for use as a homogeneous solution. An alternative precursor, Zr(OH)4 was also tested, but white precipitates were also observed (Fig. 2f). To prevent precipitation while achieving a homogeneous solution, the Zr precursor must be dissolved and remain stable in the solution without reacting with DTC. To facilitate this, we introduced chelating ligands to stabilize the Zr in the solution. Zr acetylacetonate (Zr(acac)4) has four bidentate acetylacetonate molecules surrounding the Zr (Fig. 2h). Recent studies have shown that this precursor can be used in the formulation of molecular inks for the synthesis of BaZrS3 thin films. However, the mechanism underlying this dissolution remains elusive.42 In solubility tests to dissolve Zr(acac)4 in the DTC solution, the solution presented a clear yellowish color without any precipitate (Fig. 2d). This precursor was also readily dissolved in pyridine. This indicates that during the dissolution of Zr(acac)4 in the DTC solution, the solvent pyridine dissolved Zr(acac)4, thereby inhibiting the interaction between DTC and Zr. This represents a favorable result that is aligned with our objective of preventing precipitation by inhibiting the strong interactions between Zr and DTC. Liquid Raman spectroscopy was used to probe the molecular chemistry occurring within the solutions. Upon addition of Zr(acac)4 to pyridine, new peaks emerged at 1440 and 1678 cm−1, providing evidence for the dissolution of Zr(acac)4 by pyridine (Fig. S2a†). Furthermore, no reduction in the CS stretching and bending modes was observed, indicating that there was no interaction between Zr and DTC (Fig. S2b†). This suggests that the coordination between Zr and the CSS− group in DTC was successfully inhibited by the chelating ligands surrounding the Zr4+ cation. Through Raman analysis of Ba and Zr, we determined that the Ba–DTC solution involves Ba–DTC coordination, allowing it to exist in a stable state. Conversely, the stability of the Zr–DTC solution is not attributed to Zr–DTC coordination but rather to the chelating ligands, which inhibit strong interactions between Zr and DTC, thereby maintaining a stable state.
After preparing homogeneous solutions, we tested the effectiveness of each metal–DTC solution in forming sulfide films. The solutions were spin-coated onto quartz substrates, followed by annealing on a heating plate in an N2-filled glove box to prevent oxidation. For the successful formation of metal sulfide films from the metal–DTC solutions, the carbamate structure in DTC must decompose at a specific temperature, allowing the metal to bond with sulfur. X-ray diffraction (XRD) patterns of films annealed at various temperatures were analyzed to determine the optimal crystallization temperature. At 180 °C, films derived from the Ba–DTC solution retained their original Ba(OH)2·8H2O and oxide form (BaO2), indicating that this temperature was insufficient to crystallize Ba–DTC into BaS (Fig. 3a). As the temperature was increased to 400 °C in the N2-filled glove box, a completely white BaS film formed (Fig. 3b, inset) without any secondary phase. Based on these findings, we conclude that even after Ba–DTC coordination, the hydroxide group from the Ba precursor is still reactive and forms an oxide or its original state at low temperatures. Elevated temperatures destabilized the oxide state, promoting the integration of Ba and DTC, thereby facilitating the synthesis of the BaS film. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) were used to determine the microstructure and composition (Fig. 3b–d). These analyses revealed that the BaS film has defined morphological patterns that were covered with Ba and S. Additional experiments were conducted to determine the optimal ratio of Ba to DTC in the solution (Fig. S3a†). From 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:10 and 1:20, the XRD peaks of BaS faded, and there were no apparent peaks for the 1:20 film. The solution was stable for up to 8 days at a 1:5 ratio, confirming its homogeneous state and stable Ba–DTC complex (Fig. S3b†). An overall schematic of the conversion from the Ba–DTC solution to the BaS film is shown in Fig. 3e.
:5 to 1:10 and 1:20, the XRD peaks of BaS faded, and there were no apparent peaks for the 1:20 film. The solution was stable for up to 8 days at a 1:5 ratio, confirming its homogeneous state and stable Ba–DTC complex (Fig. S3b†). An overall schematic of the conversion from the Ba–DTC solution to the BaS film is shown in Fig. 3e.
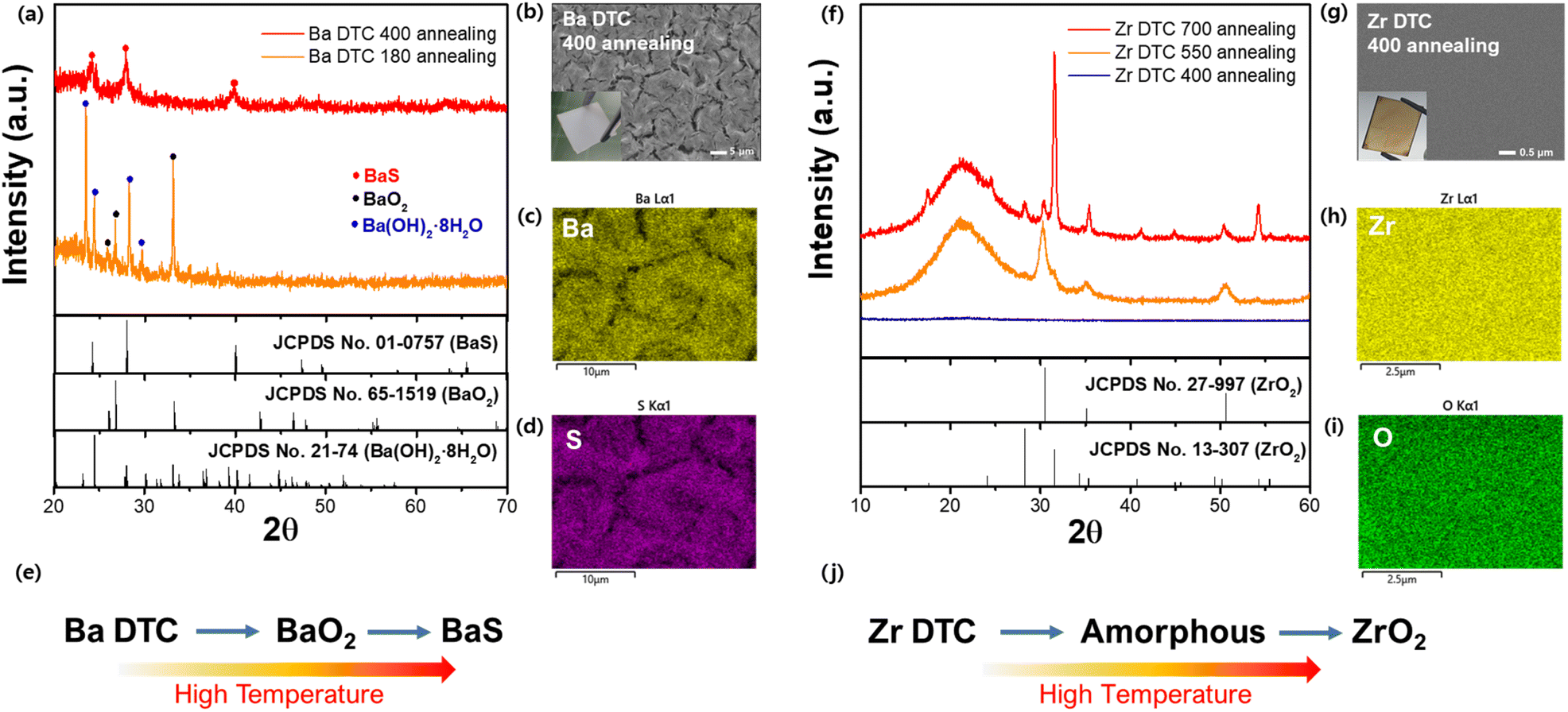 | ||
| Fig. 3 (a) XRD analysis of the Ba–DTC film annealed at 180 and 400 °C. (b) SEM of the Ba–DTC film annealed at 400 °C with an inset image of the actual film. (c and d) EDX mapping of Ba and S in the Ba–DTC film annealed at 400 °C. (e) Overall reaction pathway from Ba–DTC solution to BaS film. (f) XRD analysis of the Zr–DTC film annealed at 400, 550, and 700 °C. (g) SEM of the Zr–DTC film annealed at 400 °C with an inset image of the actual film. (h and i) EDX mapping of Zr and O in the Zr–DTC film annealed at 400 °C. (j) Overall reaction pathway from Zr–DTC solution to ZrO2 film. | ||
The behavior of Zr–DTC was distinct from that of Ba–DTC. As seen in Fig. 3f, the Zr–DTC film remains in an amorphous phase even at 400 °C; a temperature at which the Ba–DTC film had transformed into a BaS film. The resulting film was clear brown without any observable crystalline structures on its surface (Fig. 3g, inset). The SEM images and EDX mapping confirmed that the film did not have a crystallized structure, exhibiting an amorphous phase, and only Zr and O were observed on the film, with no S being identified (Fig. 3g–i). This is likely because the DTC, containing sulfur, is not coordinated with the metal in the DTC solution, allowing it to volatilize readily when heat is applied. To further examine the behavior of Zr under higher energy conditions, the Zr–DTC film was transferred from the heating plate to a tube furnace for intensive thermal treatment. The applied temperature ranged from 550 to 700 °C and was maintained for 9 h. Under these conditions, the film crystallized into ZrO2, leaving no evidence of sulfurization (Fig. 3f). The overall schematic for the conversion of the Zr–DTC solution into the ZrO2 film is illustrated in Fig. 3j.
In conclusion, while the Ba–DTC complex in Ba–DTC film decomposes at 400 °C to form BaS, the behavior of Zr–DTC differs significantly. At 400 °C, the sulfur content within the Zr–DTC film undergoes complete volatilization. Concurrently, at elevated thermal conditions, the remaining acetylacetonate reacts, resulting in the formation of ZrO2.
To prevent oxidation and ensure sufficient sulfur incorporation to replace the volatilized DTC in the Zr–DTC film, we introduced a sulfurization process using a graphite box, avoiding gas-phase sulfurization methods. The graphite box was chosen for its scalability and practicality, as previously used ampoule process requires vacuum sealing, limiting film size. It enables large-area sulfurization without CS2 gas while offering a simpler, reusable design that eliminates complex sealing steps. Elemental S powder was placed with 400 °C annealed Zr–DTC film inside a graphite box, which was then closed with a cover and annealed at 700 °C in a tube furnace. However, the resulting film was identical to the film annealed without S, indicating that this sulfurization technique is ineffective for fabricating Zr sulfide films. This can also be interpreted in terms of hard–soft acid–base (HSAB) theory.43 HSAB theory, grounded in Pearson's absolute hardness, classifies elements as “hard” or “soft” based on their polarizability.44 When an element has high polarizability, it tends to be soft, whereas elements with low polarizability are generally hard. Group 3 and 4 elements, including Ti, Zr, and Hf, are hard acids. An important aspect of this theory is that hard acids interact more favorably with hard bases in order to stabilize the metal complex. A comparative analysis of O and S revealed that O acts as a relatively hard base, whereas S functions as a relatively soft base. Consequently, as a hard acid, Zr preferentially interacts with O rather than S. The oxophilicity of Zr is evident not only from the perspective of the HSAB theory but also in terms of the dissociation enthalpy. The tendency of a metal to be oxidized (oxophilicity) or sulfurized (thiophilicity) is determined by the difference between the dissociation enthalpies of the metal oxides and metal sulfides; this tendency is quantified as a value between 0 and 1. On this scale, Zr has 0.8 oxophilicity and 0.2 thiophilicity, indicating that it is highly oxophilic.45 The strong thermodynamic tendency of Zr toward oxidation can be demonstrated through the theoretical frameworks of the HSAB theory and the balance between oxophilicity and thiophilicity, and is further corroborated by our experimental results. To mitigate this challenge of unwanted oxidation, we formulated a strategic approach that involves not only the incorporation of sulfur but also the elimination of oxygen within the film. Sulfurization using boron sulfide was achieved by simply adding boron powder together with the sulfur powder. Boron sulfide has been utilized for the sulfurization of various materials, including V2O5, RuO2, and Ga2O3.46,47 Moreover, the approach has been used to convert BaZrO3 powder into BaZrS3 powder.33 These methods all utilize a mixture of boron and sulfur powders, which react together with increasing temperature. As the temperature reaches 120 °C, sulfur starts to sublime and yields B2S3 or BS2.48 Above 300 °C, B2S3 and BS2 start to sublime and these gaseous boron sulfides react with the film.47 The sulfurization of the oxide material using boron sulfide follows reaction (1).33
| AO2 + nBxSy → AS2 + mB2O3 | (1) |
The effectiveness of boron sulfide derives from a difference between the thermodynamic stability of the byproduct B2O3  and reactants B2S3
and reactants B2S3  , BS2
, BS2 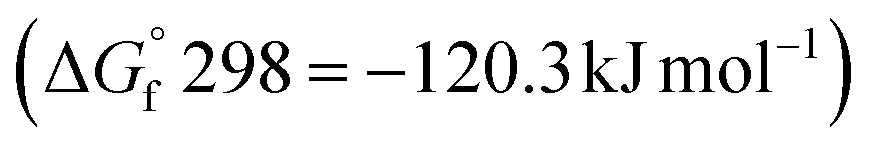 .33 This difference between the byproducts and reactants is greater than the difference between CS2 and H2S sulfurization, making it thermodynamically favorable.48
.33 This difference between the byproducts and reactants is greater than the difference between CS2 and H2S sulfurization, making it thermodynamically favorable.48
We adopted the boron sulfurization method (b-S) and applied it to Ba–DTC and Zr–DTC films. The elemental sulfurization method (e-S) was also employed to validate the effect of b-S, and XRD analysis was conducted to compare the differences between the two approaches. First, Ba–DTC films, annealed at 400 °C, were sulfurized for 3 h using both methods at temperatures of 350, 380, and 400 °C. In both methods, the BaS film resulting from annealing at 400 °C was sulfurized to BaS3 at 350 °C, while at 380 °C, BaS2 emerged alongside BaS3. At 400 °C, BaS2 was the sole remaining phase with few traces of BaS3 (Fig. 4a). The sulfur incorporation into the film was completed at 350 °C; above this temperature, only sulfur detachment was observed. These findings indicate that the e-S method is sufficient to sulfurize the BaS film even at relatively low temperatures and that b-S exhibits a similar tendency to e-S.
 | ||
| Fig. 4 (a) XRD analysis of Ba–DTC film sulfurized at 350, 380, and 400 °C with elemental sulfurization (e-S) and boron sulfurization (b-S). (b) XRD analysis of Zr–DTC film sulfurized at 350, 380, and 400 °C with e-S and b-S. (c and d) SEM images of Ba–DTC films sulfurized at 350 °C with e-S and b-S. (e and f) SEM images of Zr–DTC films sulfurized at 700 °C with e-S and b-S. (g and h) Overall reaction pathways of Ba–DTC and Zr–DTC followed by e-S and b-S. | ||
In contrast, the Zr–DTC films exhibited significantly different results. The amorphous Zr–DTC film, annealed at 400 °C, was sulfurized to ZrS2 after 9 h at 700 °C exclusively with the b-S method, whereas the e-S method produced ZrO2, indicating no sulfurization effect (Fig. 4b). This finding suggests that the b-S method is more effective for sulfurizing Zr–DTC films. In the e-S method, the removal of oxygen from the Zr–DTC film is challenging, which is a disadvantage of the sulfurization process. However, as described in reaction (1), oxygen serves as a reactant in the b-S method, facilitating the completion of the reaction. During the b-S process, boron sulfide utilizes oxygen on the film surface and it is converted into B2O3, thereby acting as an “oxygen sink” by absorbing oxygen. While HfH2 is also recognized for its role as an oxygen sink, boron was selected due to its lower cost and safer handling.49 The reaction of HfH2 with sulfur powder generates toxic H2S gas, requiring strict safety precautions, whereas boron enables a safer and more controllable sulfurization process.
Unlike the e-S method, oxygen plays a critical role in the b-S method. Within the Zr–DTC film, oxygen originating from the acetylacetonate-chelating ligands remains and provides sufficient oxygen to react with boron sulfide during sulfurization. The distinction between the resulting films was also observed using SEM analysis. Given the negligible difference between the e-S and b-S methods in the sulfurization of Ba–DTC films, the morphological features of the films produced using both techniques were nearly indistinguishable (Fig. 4c and d). In contrast, the structural differences between ZrS2 synthesized using the b-S method and ZrO2 synthesized using the e-S method were significant (Fig. 4e and f). In conclusion, BaS films synthesized from a Ba–DTC solution can be converted into Ba polysulfides (BaS3 and BaS2) via both the e-S and b-S sulfurization methods (Fig. 4g). However, the amorphous film derived from the Zr–DTC solution can be successfully transformed into ZrS2 only through the b-S method, with the e-S method proving ineffective in promoting this conversion (Fig. 4h).
After confirming the effectiveness of b-S for sulfurizing the Zr–DTC film, the next step was to validate this approach using a mixed solution of Ba–DTC and Zr–DTC to produce the BaZrS3 crystal structure. A homogeneous mixture of each solution was prepared and subjected to spin coating followed by sulfurization. Both e-S and b-S methods were conducted within a temperature range of 600 to 700 °C for a fixed duration of 6 h. Initially, the processing time was varied between 1 and 9 h, and 6 h was determined to be the minimum time required to achieve distinct crystallinity (Fig. S4a†). As anticipated from the previous results, the e-S method did not result in the formation of the BaZrS3 phase, with the BaS and ZrO2 phases persisting at all temperatures (Fig. 5a). Moreover, the crystallinities of BaS and ZrO2 increased with increasing temperature without any evidence of the formation of the target material, BaZrS3. However, as observed for the Zr–DTC film, b-S was also effective for the Ba + Zr DTC film. Although identical phases were observed with e-S in b-S method at 600 °C, complete sulfurization of ZrO2 occurred at 650 and 700 °C, successfully yielding a pure BaZrS3 phase without any secondary phases (Fig. 5b). Furthermore, the b-S method effectively reduced the sulfurization time to 3 h at both temperatures (Fig. S4b†). Because of the high volatility of the Ba component, the use of equimolar amounts of Ba and Zr resulted in the formation of a secondary phase associated with ZrS2 (Fig. S5a†). To suppress this secondary phase and achieve the correct stoichiometry for BaZrS3, an excess of the Ba precursor relative to Zr was necessary. A Ba to Zr ratio of 1.3:1 resulted in the highest crystallinity, establishing it as the optimal stoichiometric ratio for the preparation of mixed Ba–DTC and Zr–DTC solutions (Fig. S5b†). The SEM and EDX mapping images of resulting films from 650 and 700 °C, presented in Fig. S6 and S7,† show that Ba, Zr, and S collectively form a well-defined BaZrS3 crystalline structure, with O residing independently of this structure. This distinct separation confirms the purity of the BaZrS3 phase, with no incorporation of oxygen impurities within the structure.
 | ||
| Fig. 5 (a) XRD analysis of Ba + Zr DTC film annealed at 600, 650, and 700 °C with the e-S method. (b) XRD analysis of Ba + Zr DTC film annealed at 600, 650, and 700 °C with the b-S method with an inset image of BaZrS3 film. (c) Raman analysis of the BaZrS3 film obtained by the b-S method at 650 °C. (d) Light absorption coefficient of BaZrS3 film obtained by the b-S method at 650 °C. (e) Tauc plot and room-temperature PL spectra of the BaZrS3 film obtained by the b-S method at 650 °C. | ||
Fig. 5c shows the room-temperature Raman spectra of the film obtained by the b-S method at 650 °C for 3 h, which reveals several peaks that are assigned to A2g + B13g, A4g, A6g, B62g, B51g, and B72g vibrational modes of BaZrS3.50 The optical properties of these films were systematically evaluated. A light absorption coefficient of over 1.0 × 105 cm−1 at 420 nm was confirmed by UV-vis spectroscopy measurements (Fig. 5d). Fig. 5e shows the photoluminescence (PL) spectrum excited at 400 nm and the Tauc plot of BaZrS3. The PL spectrum displays a distinct emission peak at approximately 2 eV corresponding to the band-to-band radiative recombination in BaZrS3. This emission closely aligns with the excitonic absorption edge observed in the Tauc plot, also at approximately 2 eV, suggesting the presence of a strong excitonic effect in this material. Although XRD and SEM analyses confirmed the presence of a BaZrS3 phase that was free of secondary phases at both 650 and 700 °C, depth-profile X-ray photoelectron spectroscopy (XPS) was subsequently performed with an etching range from 0 to 700 s to achieve a more comprehensive assessment (Fig. S8a and b†). The depth-profile XPS analysis revealed peaks corresponding to Ba 3d, Zr 3d, and S 2p at both temperatures.51 Additionally, B2O3, an indicator of boron sulfurization and an oxygen sink, was detected at all film depths, implying that the gaseous form of boron sulfide was fully capable of penetrating the entire depth of the film.52 At 650 °C, however, minimal concentrations of Ba, Zr, and S were found at the surface, transitioning to a uniform distribution at increased depths (Fig. S8a†). In contrast, at 700 °C, these elements maintained a homogeneous distribution across all depths, suggesting a more consistent compositional profile within the film structure (Fig. S8b†). This variation is attributed to the differences in the carbon concentration across the film depth (Fig. S8c†). The 650 °C sample exhibits substantial carbon accumulation on the surface, which gradually decreases with increasing depth, in contrast to the 700 °C sample. Cross-sectional SEM images further support these findings, revealing that amorphous carbon residues are observed in the upper layers of the 650 °C sample, obscuring the BaZrS3 grain structure (Fig. S8d†). Conversely, in the 700 °C sample, BaZrS3 grains are discernible in the deeper regions, with carbon residues progressively diminishing toward the surface (Fig. S8e†). This trend is also evident after prolonged sulfurization (Fig. S9a and c†). At 700 °C, 1 h sulfurization resulted in carbon residues covering the upper layer; however, as the sulfurization time was increased to 3 and 6 h, these residues dissipated, allowing a clear visualization of the crystalline structure. These results suggest that the crystallization of BaZrS3 initiates in the lower layers following boron sulfide infiltration, promoting growth from the bottom to the top. Furthermore, elevated annealing temperatures and extended times effectively reduced the surface carbon residues, enabling the formation of a more distinct crystalline morphology.
To investigate the feasibility of using this material for application in optoelectronic devices, photodetectors employing BaZrS3 thin films were produced. The lateral device structure of Au/PMMA/BaZrS3/glass was used to fabricate the device (Fig. 6a). The additional PMMA (polymethyl methacrylate) layer acts as a surface passivation layer, minimizing defects and trap states to enhance charge transport and photocurrent response. In our experiment, it effectively stabilized the photocurrent by reducing current fluctuations. Samples formed at 650 °C (3 h) and 700 °C (3 h) were prepared and evaluated under 10 V bias voltage with different illumination power densities (Fig. 6b and c). Interestingly, the 700 °C sample demonstrated negligible photocurrent compared to the 650 °C sample, which is likely attributable to intrinsic defects within the film. Theoretical and computational analyses suggested that sulfur vacancies are the most thermodynamically facile trap states to form during BaZrS3 crystallization.53,54 Consistent with this, a relative sulfur ratio (S/Ba + Zr + S) of 0.576 was measured for the 650 °C sample, whereas for the 700 °C sample, sulfur vaporization during the high temperature process resulted in a lower ratio of 0.563. The 650 °C sample was further evaluated under increasing illumination power densities, proving a positive correlation with photocurrent by reaching 0.369 nA at an illumination intensity of 99.95 mW cm−2 (Fig. 6d and e). The device showed a responsivity of 5.27 μA under an illumination power density of 7.895 mW cm−2 (Fig. 6f). In prolonged stability measurements at 99.95 mW cm−2, the device maintained a consistent photocurrent for at least 160 s (Fig. 6g). Despite the limited number of studies on BaZrS3 photodetectors, some have made notable progress.31,42,55–57 One study which used a device structure similar to ours, reported a photocurrent exceeding 2 nA, although this was achieved using BaZrS3 powder as the source in a pulsed layer deposition process.31 In contrast, within molecular-ink-based systems, to our knowledge, only a single study has been documented, reaching a photocurrent of approximately 0.5 nA, which is comparable to our findings.42
 | ||
| Fig. 6 (a) A schematic of the device architecture for an Au/PMMA/BaZrS3/glass photodetector. I–V curve under 10 V bias voltage and different illumination power densities of photodetectors sulfurized at (b) 700 °C (3 h) and (c) 650 °C (3 h). (d) Time-dependent photocurrent curve of the 650 °C (3 h) photodetector under 10 V bias voltage and different illumination powder densities. (e) Photocurrent and (f) responsivity of the 650 °C (3 h) photodetector as a function of irradiance intensities under 10 V bias. (g) Time-dependent pulsed photocurrent curves of the 650 °C (3 h) photodetector under 10 V bias voltage with 99.95 mW cm−2 illumination powder density. | ||
Although we have successfully reduced the processing temperature to moderate, this is still high to be used with conductive substrates. Therefore, further research is required to lower the processing temperature through synthesis optimization and structural refinement. Moreover, investigating the defect chemistry by strategically controlling various trap states is critical for achieving enhanced photocurrent performance. The DTC molecular ink method combined with boron sulfurization offers a versatile approach with significant potential for synthesizing other chalcogenide perovskites. As the molecular ink methodology is still in its early stages for BaZrS3 research, numerous future studies are expected to focus on these foundational strategies.
Given the limited research on BaZrS3-based optoelectronic devices, this study makes a pivotal contribution by elucidating the molecular interactions within a homogeneous solution and providing a comprehensive examination of the boron sulfurization mechanism. As such, the study provides a cornerstone for the exploration and application of this material.
Conclusions
In this study, we introduce a DTC-based molecular ink approach for the fabrication of BaZrS3 thin films at moderate temperatures. The molecular ink system was based on Ba–DTC coordination complexes formed inside a solution, with the Zr–DTC complexes being stabilized via chelating ligands surrounding the Zr cations. During film formation, while Zr–DTC solutions typically result in ZrO2 rather than ZrS2, Ba–DTC complexes can be effectively sulfurized into Ba polysulfides. To alleviate this oxidation issue in the Zr–DTC film, boron sulfide was employed as an oxygen sink, facilitating sulfurization. This boron sulfurization proved to be effective for converting Zr–DTC films into ZrS2 and enabled the synthesis of BaZrS3 films from combined Ba + Zr DTC solutions. BaZrS3 films were successfully synthesized at 650 °C with a sulfurization duration of 3 h using boron sulfide. Characterization by XRD, XPS, SEM, and Raman spectroscopy confirmed the crystallinity and structural integrity of the BaZrS3 films and revealed that crystallization initiates from the substrate–film interface. In photodetector applications, the BaZrS3 films exhibited a photocurrent level comparable to previously reported results, demonstrating stability for several minutes. This study represents the first application of boron sulfurization for BaZrS3 film fabrication via a molecular ink system, offering a streamlined and simplified alternative to existing methodologies. This approach is expected to lay the groundwork for future advancements in BaZrS3-based solar cells, providing a highly accessible and relatively low-temperature fabrication pathway.Experimental
Materials
Barium hydroxide octahydrate powder (Ba(OH)2·8H2O, 98%; Sigma-Aldrich), zirconium(IV) acetylacetonate powder (Zr(acac)4, 98%; Sigma-Aldrich), zirconium(IV) chloride powder (ZrCl4, 99.5%; Thermo Fisher Scientific), zirconium(IV) hydroxide powder (Zr(OH)4, 97%; Sigma-Aldrich), boron powder (B, amorphous, 95%; Sigma-Aldrich), sulfur powder (S, 99.98%; Sigma-Aldrich), pyridine (C2H5N, anhydrous, 99.8%; Sigma-Aldrich), N-butylamine (CH3(CH2)3NH2, 99.5%; Sigma-Aldrich), and carbon disulfide (CS2, 99.9%; Sigma-Aldrich) were used.Ba–DTC and Zr–DTC molecular inks were prepared by dissolving the Ba or Zr precursor in the DTC solution (6 mL). All dissolution processes were performed inside an N2-filled glove box. For the Ba–DTC solution, Ba(OH)2·8H2O powder (0.4732 g) was dissolved in DTC solution to give a Ba/DTC ratio of 1:5. Similarly, for the Zr–DTC solution, Zr(acac)4 powder (0.7314 g) was dissolved in the DTC solution to give a Zr/DTC ratio of 1:5. More than 6 h were required to completely dissolve the metal precursors in the DTC solution. Subsequently, the final molecular ink was prepared by mixing the two metal–DTC solutions. Under some conditions where excess Ba was required, less Zr powder was used than Ba (e.g., for a Ba/Zr ratio of 1.3:1, 0.5617g of Zr(acac)4 powder was used). The final solution was stirred for 10 h to completely blend the metal and DTC compounds. The solution remained unchanged, with no precipitate, for more than 10 h, confirming the homogeneous nature of the molecular ink.
Data availability
The data will be provided upon reasonable request.Author contributions
J. Lee – conceptualization, data curation, formal analysis, investigation, writing – original draft; N. Ha – investigation; J. Jung – investigation; G. Lee – formal analysis; S. V. Barma – formal analysis; J.-H. Kim – formal analysis; S. Jo – supervision; J.-W Lee. – supervision, resources; W. Yang – supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant No. RS-2023-0025177). J.–W. L. acknowledges financial support from ‟Leaders in Industry-university Cooperation 3.0” Project (LINC 3.0, No. 1345370621), supported by the Ministry of Education and NRF.References
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- W. Yang, X. Zhang and S. D. Tilley, Chem. Mater., 2021, 33, 3467–3489 CrossRef CAS.
- Q. A. Akkerman and L. Manna, ACS Energy Lett., 2020, 5, 604–610 CrossRef CAS PubMed.
- R. Sharif, A. Khalid, S. W. Ahmad, A. Rehman, H. G. Qutab, H. H. Akhtar, K. Mahmood, S. Afzal and F. Saleem, Nanoscale Adv., 2023, 5, 3803–3833 RSC.
- M. A. Green, E. D. Dunlop, M. Yoshita, N. Kopidakis, K. Bothe, G. Siefer, D. Hinken, M. Rauer, J. Hohl-Ebinger and X. Hao, Prog. Photovolt.: Res. Appl., 2024, 32, 425–441 CrossRef.
- PV magazine online, https://www.pv-magazine.com/2024/09/12/longi-achieves-34-6-efficiency-for-two-terminal-tandem-perovskite-solar-cell-prototype/, accessed november 2024.
- M.-G. Ju, M. Chen, Y. Zhou, J. Dai, L. Ma, N. P. Padture and X. C. Zeng, Joule, 2018, 2, 1231–1241 CrossRef CAS.
- J. Zhou, Z. Liu, P. Yu, G. Tong, R. Chen, L. K. Ono, R. Chen, H. Wang, F. Ren and S. Liu, Nat. Commun., 2023, 14, 6120 CrossRef CAS PubMed.
- R. Hosseinian Ahangharnejhad, Z. Song, T. Mariam, J. J. Gardner, G. K. Liyanage, Z. S. Almutawah, B. M. Anwar, M. Junda, N. J. Podraza and A. B. Phillips, ACS Appl. Energy Mater., 2021, 4, 7571–7578 CrossRef CAS.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu and M. Xiao, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- N. Thakur, K. Aly, M. Mohery, M. Ebrahium, P. Kumar and P. Sharma, J. Alloys Compd., 2023, 957, 170457 CrossRef CAS.
- M. Suhail, H. Abbas, M. B. Khan and Z. H. Khan, J. Nanopart. Res., 2022, 24, 142 CrossRef CAS.
- D. Tiwari, O. S. Hutter and G. Longo, J. Phys. Energy, 2021, 3, 034010 CrossRef CAS.
- S. J. Adjogri and E. L. Meyer, Materials, 2021, 14, 7857 CrossRef CAS PubMed.
- S. P. Ramanandan, A. Giunto, E. Z. Stutz, B. Reynier, I. T. F. M. Lefevre, M. Rusu, S. Schorr, T. Unold, A. F. I. Morral and J. A. Márquez, J. Phys. Energy, 2023, 5, 014013 CrossRef.
- K. V. Sopiha, C. Comparotto, J. A. Márquez and J. J. Scragg, Adv. Opt. Mater., 2022, 10, 2101704 CrossRef CAS.
- A. Swarnkar, W. J. Mir, R. Chakraborty, M. Jagadeeswararao, T. Sheikh and A. Nag, Chem. Mater., 2019, 31, 565–575 CrossRef CAS.
- S. Sharma, Z. Ward, K. Bhimani, K. Li, A. Lakhnot, R. Jain, S.-F. Shi, H. Terrones and N. Koratkar, ACS Appl. Electron. Mater., 2021, 3, 3306–3312 CrossRef CAS.
- S. Niu, H. Huyan, Y. Liu, M. Yeung, K. Ye, L. Blankemeier, T. Orvis, D. Sarkar, D. J. Singh and R. Kapadia, Adv. Mater., 2017, 29, 1604733 Search PubMed.
- Y. Wang, N. Sato and T. Fujino, J. Alloys Compd., 2001, 327, 104–112 Search PubMed.
- X. Wei, H. Hui, C. Zhao, C. Deng, M. Han, Z. Yu, A. Sheng, P. Roy, A. Chen and J. Lin, Nano Energy, 2020, 68, 104317 CrossRef CAS.
- D. Payno, S. Kazim, M. Salado and S. Ahmad, Sol. Energy, 2021, 224, 1136–1143 CrossRef CAS.
- R. Yang, A. D. Jess, C. Fai and C. J. Hages, J. Am. Chem. Soc., 2022, 144, 15928–15931 Search PubMed.
- J. W. Turnley, K. C. Vincent, A. A. Pradhan, I. Panicker, R. Swope, M. C. Uible, S. C. Bart and R. Agrawal, J. Am. Chem. Soc., 2022, 144, 18234–18239 CrossRef CAS PubMed.
- K. C. Vincent, S. Agarwal, J. W. Turnley and R. Agrawal, Adv. Energy Sustainability Res., 2023, 4, 2300010 CrossRef CAS.
- A. A. Pradhan, M. C. Uible, S. Agarwal, J. W. Turnley, S. Khandelwal, J. M. Peterson, D. D. Blach, R. N. Swope, L. Huang and S. C. Bart, Angew. Chem., 2023, 135, e202301049 CrossRef.
- C. Xu, L. Cheng, Z. Li, X. Zheng, S. Shan, T. Chen, W. Fu, Y. Yang, L. Zuo and H. Chen, Adv. Energy Mater., 2023, 13, 2300168 CrossRef CAS.
- M. Feng, S. You, N. Cheng and J. Du, Electrochim. Acta, 2019, 293, 356–363 CrossRef CAS.
- W. Xu, G. Lei, C. Tao, J. Zhang, X. Liu, X. Xu, W. Y. Lai, F. Gao and W. Huang, Adv. Funct. Mater., 2018, 28, 1802320 CrossRef.
- Z. Yu, X. Wei, Y. Zheng, H. Hui, M. Bian, S. Dhole, J.-H. Seo, Y.-Y. Sun, Q. Jia and S. Zhang, Nano Energy, 2021, 85, 105959 CrossRef CAS.
- L. S. Breton, V. V. Klepov and H.-C. Zur Loye, J. Am. Chem. Soc., 2020, 142, 14365–14373 CrossRef CAS PubMed.
- R. Bystrický, S. K. Tiwari, P. Hutár, L. u. r. Vančo and M. Sykora, Inorg. Chem., 2022, 61, 18823–18827 CrossRef PubMed.
- J. C. Sarker and G. Hogarth, Chem. Rev., 2021, 121, 6057–6123 CrossRef CAS PubMed.
- J. Tan, X. Zhang, J. Suh, N. Ha, J. Lee, S. D. Tilley and W. Yang, Mater. Today Energy, 2023, 34, 101288 CrossRef CAS.
- V. Vento, S. Tarrago Velez, A. Pogrebna and C. Galland, Nat. Commun., 2023, 14, 2818 CrossRef CAS PubMed.
- N. Gong, X. He, M. Zhou, L. He, L. Fan, W. Song, C. Sun and Z. Li, Mater. Res. Bull., 2017, 85, 104–108 CrossRef CAS.
- D. Li, S. Sun, C. Sun, X. Jiang, S. Gao and Z. Li, Spectrochim. Acta, Part A, 2012, 96, 193–199 CrossRef CAS PubMed.
- N. Ha, G. Lee, J. Park, J. H. Lee, J. Jung, S. V. Barma, J. Kim, J. H. Kim, J. K. Kim and S. J. Kwon, Adv. Energy Mater., 2025, 15, 2402099 CrossRef CAS.
- M. Zhang, Y. Zhu, X. Wang, Q. Feng, S. Qiao, W. Wen, Y. Chen, M. Cui, J. Zhang and C. Cai, J. Am. Chem. Soc., 2015, 137, 7051–7054 CrossRef CAS PubMed.
- D. Yoo, M. Kim, S. Jeong, J. Han and J. Cheon, J. Am. Chem. Soc., 2014, 136, 14670–14673 CrossRef CAS PubMed.
- S. Agarwal, K. C. Vincent, J. W. Turnley, D. C. Hayes, M. C. Uible, I. Durán, A. S. M. Canizales, S. Khandelwal, I. Panicker and Z. Andoh, Adv. Funct. Mater., 2024, 34, 2405416 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- L.-M. Wu and D.-K. Seo, J. Am. Chem. Soc., 2004, 126, 4676–4681 CrossRef CAS PubMed.
- M.-J. Zhang, X.-M. Jiang, L.-J. Zhou and G.-C. Guo, J. Mater. Chem. C, 2013, 1, 4754–4760 RSC.
- H. U. Huerter, B. Krebs, H. Eckert and W. Mueller-Warmuth, Inorg. Chem., 1985, 24, 1288–1292 CrossRef CAS.
- S. Agarwal, J. W. Turnley, A. A. Pradhan and R. Agrawal, J. Mater. Chem. C, 2023, 11, 15817–15823 RSC.
- N. Gross, Y.-Y. Sun, S. Perera, H. Hui, X. Wei, S. Zhang, H. Zeng and B. Weinstein, Phys. Rev. Appl., 2017, 8, 044014 CrossRef.
- Y. Han, J. Xu, Y. Liang, X. Chen, M. Jia, J. Zhang, L. Lian, Y. Liu, X. Li and Z. Shi, Chem. Eng. J., 2023, 473, 145351 CrossRef CAS.
- J. O. Tijani, U. O. Momoh, R. B. Salau, M. T. Bankole, A. S. Abdulkareem and W. D. Roos, Environ. Sci. Pollut. Res., 2019, 26, 19942–19967 CrossRef CAS PubMed.
- X. Wu, W. Gao, J. Chai, C. Ming, M. Chen, H. Zeng, P. Zhang, S. Zhang and Y.-Y. Sun, Sci. China Mater., 2021, 64, 2976–2986 CrossRef CAS.
- Z. Yuan, D. Dahliah, R. Claes, A. Pike, D. P. Fenning, G.-M. Rignanese and G. Hautier, PRX Energy, 2024, 3, 033008 CrossRef.
- S. Dhole, X. Wei, H. Hui, P. Roy, Z. Corey, Y. Wang, W. Nie, A. Chen, H. Zeng and Q. Jia, Photonics, 2023, 10, 366 CrossRef CAS.
- T. Gupta, D. Ghoshal, A. Yoshimura, S. Basu, P. K. Chow, A. S. Lakhnot, J. Pandey, J. M. Warrender, H. Efstathiadis and A. Soni, Adv. Funct. Mater., 2020, 30, 2001387 CrossRef CAS.
- B. Zhao, H. Chen, R. Ahsan, F. Hou, E. R. Hoglund, S. Singh, M. Shanmugasundaram, H. Zhao, A. V. Krayev and H. Htoon, ACS Photonics, 2024, 11, 1109–1116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08848d |
| This journal is © The Royal Society of Chemistry 2025 |