
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompetition between dissolution and ion exchange during low temperature synthesis of LiCoO2 on porous carbon scaffolds†
Ishita Kamboj,
Seongbak Moon,
Hannah Denhartog and
Veronica Augustyn *
*
Dept. of Materials Science and Engineering, North Carolina State University, Raleigh, NC 27695, USA. E-mail: vaugust@ncsu.edu
First published on 7th March 2025
Abstract
The intentional design of ionic and electronic pathways in battery electrode architectures is one strategy to optimize battery performance and maximize the utilization of expensive and/or scarce electrode active materials. Porous carbon scaffolds are particularly attractive for advanced electrode architectures due to their light weight and low cost. One major challenge for insertion-type Li-ion battery electrodes utilizing porous carbon scaffolds is direct electrical wiring of commercially relevant electrode materials. In particular, lithium metal oxide cathode materials require high synthesis temperatures (>700 °C in air) that exceed the stability of carbon (∼450 °C). In this work, we studied the mechanism of LiCoO2 deposition onto porous carbon scaffolds from a low temperature (<300 °C) process involving electrodeposition, hydrothermal synthesis, and heat treatment (<300 °C). We determined how variables during hydrothermal synthesis, such as pressure, temperature, duration, and LiOH concentration, influence the synthesis mechanism and resulting LCO crystal structure and microstructure. We found that low hydrothermal pressure and high LiOH concentration favor an ion-exchange mechanism and the formation of nanoflake LiCoO2, while high hydrothermal pressure and low LiOH concentration led to a dissolution–precipitation mechanism and nanoscale LiCoO2. We further demonstrated the versatility of the ion exchange mechanism to deposit LiCoO2 on a variety of monolithic porous carbon scaffolds. Overall, this research provides insight into the versatility, and limitations, of soft chemistry strategies to deposit commercially relevant Li-ion oxide cathode materials directly onto unique porous carbon scaffolds.
Introduction
Electrification of the transportation sector is projected to account for most (4.3 TW h) of the 4.7 TW h total demand for Li-ion batteries (LIBs) by 2030.1 This large projected increase in energy storage demand for transportation could stress supply chains for LIB materials. While new battery chemistries, such as those based on Na, may help alleviate these issues,2 it will also be necessary to further develop LIBs for optimum utilization of electrochemically active materials. At the battery electrode level, intentional design of ion and electron transport pathways in thick electrodes could enable new electrode geometries for energy and power dense LIBs.3–7 Porous current collectors are interesting for deterministically designed electrodes due to their high surface areas for active material deposition and electrolyte infiltration compared to planar metal foils. Porous carbon scaffolds are particularly attractive because carbon is lightweight, abundant, and can be engineered to take on favorable mechanical properties such as structural reinforcement or flexible batteries.8,9 A critical challenge in utilizing porous carbon scaffolds for LIBs is the deposition of commercially relevant lithium transition metal oxide cathode materials. As of 2024, over half of the market for Li-ion cathode materials comes from layered lithiated transition metal oxides such as LiCoO2 or LiNi1/3Mn1/3Co1/3O2.10 These oxides are synthesized using a combined precipitation/solid-state method under high temperatures (>700 °C) in air or oxygen-rich atmospheres.11 Since porous carbon scaffolds are only oxidatively stable to ∼450 °C in air, this makes them incompatible with the standard lithiated metal oxide synthesis methods.12 To circumvent this issue, Zhang et al. utilized a molten salt route to electroplate LIB cathode materials onto porous scaffolds, including carbon foam, at 260 °C.13 However, the requirement for an oxygen-free atmosphere can be an impediment for scaling up the method for manufacturing.Therefore, there is a need to develop processing methods for integrating LIB cathode materials onto porous carbon scaffolds. Electrodeposition is a scalable method for depositing metal oxides with good adhesion onto a conductive substrate.14 The ideal electrodeposition method for LIB electrode materials from the standpoint of scalability and cost would utilize an aqueous electrolyte. This is challenging for state-of-the-art intercalation-type LIB cathode materials that require high synthetic temperatures. In the case of LiCoO2 (LCO), the layered polymorph (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) typically requires high synthesis temperatures (>700 °C) under ambient atmospheres and is sometimes termed “HT-LCO.” On the other hand, the spinel polymorph (Fdm), sometimes termed “LT-LCO”, can be formed at temperatures as low as 20 °C.15–20 The challenge therefore lies in depositing phase-pure HT-LCO at low temperatures and ambient conditions that favor porous carbon scaffolds. In this work, the term “LCO” will refer to HT-LCO, and LT-LCO will be specified as such.
m) typically requires high synthesis temperatures (>700 °C) under ambient atmospheres and is sometimes termed “HT-LCO.” On the other hand, the spinel polymorph (Fdm), sometimes termed “LT-LCO”, can be formed at temperatures as low as 20 °C.15–20 The challenge therefore lies in depositing phase-pure HT-LCO at low temperatures and ambient conditions that favor porous carbon scaffolds. In this work, the term “LCO” will refer to HT-LCO, and LT-LCO will be specified as such.
This challenge has been considered by prior work that combined electrodeposition and hydrothermal treatment methods to produce LCO coatings on porous carbon scaffolds.21 LCO can be obtained from hydrothermal syntheses using cobalt hydroxide precursors at <200 °C. The possibility for a low temperature synthesis of LCO suitable for carbon scaffolds is driven by the structural similarity of layered Rm cobalt oxyhydroxide (CoOOH) and LCO phases (Fig. 1). Indeed, Amatucci and Larcher showed the synthesis of LCO powders via cationic exchange of CoOOH and LiOH·H2O sealed in a quartz ampule and autoclave with water, respectively.22,23 The hypothesis was that the elevated pressure of the hydrothermal method lowers the synthesis temperature for LCO. Over the following decades, a variety of reaction conditions, solvents, oxidizing agents, and cobalt precursors were employed in hydrothermal reactions to produce LCO powders or films with varying morphologies and phase purities.18,24–29 In one instance, Xia et al. proposed that ion-exchange and dissolution–precipitation mechanisms compete during hydrothermal synthesis of LCO.21 The authors utilized this understanding to develop a combined electrodeposition–hydrothermal method route to deposit LCO onto a carbon cloth scaffold at 380 °C. Open questions remain regarding the influence of other hydrothermal parameters (beyond temperature) on the LCO structure and microstructure, and the applicability of the combined electrodeposition–hydrothermal synthesis method to other porous conductive substrates.
 | ||
| Fig. 1 The ion exchange (H+/Li+) of cobalt hydroxide in a LiOH aqueous solution is made possible by the structural similarity of the hydroxides and LCO: (a) α-Co(OH)2, (b) β-Co(OH)2, (c) CoOOH, and (d) HT-LiCoO2. Metastable α-Co(OH)2 can convert to β-Co(OH)2 under alkaline or oxidizing environments and CoOOH and LiCoO2 are isostructural but contain different interlayer ions (H+ vs. Li+). | ||
Here, we investigated the dueling mechanisms of LCO formation onto a range of commercially available porous carbon scaffolds from a combined aqueous electrodeposition–hydrothermal method at temperatures less than 300 °C (Fig. 2). We investigated the influence of the Co(OH)2 precursor phase, the hydrothermal reaction conditions, and the type of porous carbon scaffold on the LCO structure, microstructure, and electrochemical properties. We first considered the aqueous chemistry of β-Co(OH)2 powders and α- and β-Co(OH)2 electrodeposits in concentrated LiOH solutions. We assessed the LiOH concentration regimes necessary for dissolution of Co(OH)2 and oxidation to CoOOH, and propensity for ion-exchange between H+ in the solid (Co(OH)2/CoOOH) and Li+ in the aqueous solution. We then added other driving forces (electric potential, hydrothermal treatment) to determine the mildest synthesis conditions necessary to obtain nanoflake LCO on porous carbon scaffolds. We observed a competition between a dissolution–precipitation and ion-exchange mechanism for LCO formation and delineated the influence of each hydrothermal parameter on the preferred mechanism. We show that while temperature and synthesis duration can modulate particle size/thickness, the internal hydrothermal vessel pressure (controlled by the proportion of vessel volume occupied by LiOH, also called “vessel fill”) and the concentration of LiOH are the most important variables in determining the dominant reaction mechanism. Finally, we applied the ion-exchange synthesis method to deposit nanoflake LCO onto a variety of porous carbon scaffolds. The resulting processing method leads to the deposition of nanoflake LCO onto a range of porous carbon scaffolds using only four feedstock materials (cobalt nitrate, lithium hydroxide, carbon scaffold, water) at <300 °C and without additional oxidizing, chelating, or dispersing agents.
 | ||
| Fig. 2 Schematic representation of the electrodeposition–hydrothermal process studied in this work. (a) Electrodeposition from an aqueous cobalt nitrate solution leads to α-Co(OH)2 nanoflakes on carbon scaffolds. (b) Hydrothermal treatment of these coated scaffolds in concentrated aqueous LiOH solution converts Co(OH)2 to LCO. The vessel pressure (P) and aqueous LiOH solution concentration ([LiOH]) influence the competition between the dissolution and ion exchange formation mechanism of LCO. Other variables include Co(OH)2 phase, hydrothermal temperature, and time. (c) All carbon paper electrodes were finally calcined at 300 °C in air for 8 hours. | ||
Experimental methods
Chemicals
LiOH (anhydrous, 98%), KOH (≥85.0%), Co(OH)2 (99.9%), and dimethyl carbonate (DMC; anhydrous, 99%) were purchased from Thermo Scientific and used as received. Na2SO4 (>99%), Co(NO3)2 (>98%), LiClO4 (99.99%), and propylene carbonate (PC; anhydrous, 99.7%) were purchased from Millipore Sigma and used as received. Li metal chips (battery grade) were purchased from TMAX and used as received. Ultrapure deionized (DI) water (18.2 MΩ cm−1) was used for all aqueous solutions. Carbon foam (CFOAM25) and graphite foam (CFOAM35 HTC) were purchased from CFOAM and used after plasma cleaning. Carbon foams (RVC 10 PPI, RVC 30 PPI, RVC 60 PPI, and RVC 100 PPI) were purchased from Duocel and used after plasma cleaning. Carbon paper (AvCarb MGL190) was purchased from Fuel Cell Earth and used after plasma cleaning. Carbon felt was made by Fiber Materials, Inc. and used after plasma cleaning. CNT foam was made by the research group of Philip Bradford at NC State University and used after plasma cleaning.Influence of LiOH
We made a series of solutions with varied molar ratios of Li+/Co2+ (Table S1†). LiOH was added to each vial in quantities described in Table S1,† along with 10 mL of DI water. LiOH was dissolved via magnetic stirring, and the pH of each solution was measured using a pH probe (Mettler-Toledo FiveEasy). After the initial pH measurement, 40 mg of Co(OH)2 was added to each vial, and the vials were left to stir at 1000 rpm for 7 days at room temperature, followed by 15 days at 60 °C. We took pH measurements after the time intervals listed in Table 1.| Time of pH measurement (days) | Description |
|---|---|
| 0 | Before Co(OH)2 addition |
| 14 | After stirring for 7 days at room temperature and 7 days at 60 °C |
| 22 | After stirring for 7 days at room temperature and 15 days at 60 °C |
We used UV-vis spectroscopy (Ocean Insight OCEANHDX with a Quantum Northwest QPOD3e) to characterize the suspended powders and supernatant in each vial. To collect the samples for analysis, the vials were left on the benchtop for 3 days such that all solids settled to the bottom of the vial. The supernatant was extracted from the top of each vial and transferred to a quartz cuvette (PerkinElmer, 10 mm). To repeat the measurement for the powders, the solutions were shaken and samples diluted by adding one drop of the solution to DI water. The spectra were collected from samples containing ∼200 μL of powder solution from each vial added to 3 mL of DI water in a quartz cuvette.
We used X-ray diffraction (XRD) to characterize the powders after treatment in LiOH. Selected solutions were centrifuged and washed with DI water until the supernatant was pH neutral. The supernatant was discarded, and remaining powders dried at room temperature under vacuum overnight. The XRD patterns were collected using the method described in the Physical characterization section.
Carbon scaffold preparation & electrochemical surface area determination
The carbon scaffolds listed in Table 2 were cut into 2 × 1 cm2 rectangles and plasma cleaned before electrodeposition (Harrick Plasma PDC-32G). The plasma intensity and duration varied depending on the scaffold. Carbon nanotube foam electrodes were plasma cleaned for 3 minutes on low intensity, and all other scaffolds were cleaned for 5 minutes on high intensity.| Scaffold type | Mass of the scaffold (M) [mg] | Double layer capacitance (Cdl) [mF] | Specific electrochemical surface area (AM) [m2 g−1] | Electrodeposition current [mA] |
|---|---|---|---|---|
| CFOAM25 carbon foam | 208.81 | 1.95 | 0.02 | −2 |
| Duocel RVC 10 PPI | 27.43 | 0.30 | 0.03 | −3 |
| CFOAM35 HTC graphite foam | 483.53 | 6.11 | 0.03 | −3 |
| Duocel RVC 30 PPI | 21.94 | 0.35 | 0.04 | −4 |
| Duocel RVC 60 PPI | 34.97 | 1.18 | 0.09 | −8 |
| Fuel Cell Earth AvCarb MGL190 carbon paper | 16.87 | 1.47 | 0.22 | −20 |
| Duocel RVC 100 PPI | 36.35 | 3.48 | 0.24 | −22 |
| Fiber Materials, Inc. carbon felt | 58.83 | 13.27 | 0.56 | −52 |
| CNT foam30,31 | 5.71 | 9.04 | 6.64 | −145 |
The electrochemical surface area (A) of each carbon scaffold was determined from cyclic voltammetry. The electrochemical cell was contained in a 25 mL three-neck glass round bottom flask (Millipore Sigma). The working electrode was a ∼1 cm2 (geometric area) piece of the plasma-cleaned carbon scaffold, the counter electrode was a Pt coil (BioLogic), and the reference electrode was Ag/AgCl in saturated KCl (Pine Research Instrumentation). The electrolyte was 1 M Na2SO4 in DI water. Cyclic voltammetry (CV) was performed between 0 and 0.6 V vs. Ag/AgCl for three cycles from 10–100 mV s−1 using a potentiostat (BioLogic MPG) for all scaffolds except CFOAM25 and the carbon nanotube (CNT) foam, which were cycled between 0 and 0.4 V vs. Ag/AgCl. The double layer capacitance (Cdl) of all scaffolds except CFOAM25 was calculated from the anodic sweep of the second cycle at 50 mV s−1 in a 200 mV stretch of the voltammogram where the anodic current signal was largely capacitive (Fig. S1†). Cdl for CFOAM25 was obtained by a similar cycling method but using the capacity from the cathodic sweep to avoid the current contribution from side reactions. The electrochemical surface area, A, was then calculated by assuming a surface-area normalized capacitance (Cs) of 40 μF cm−2 for carbon:32,33
 | (1) |
Electrodeposition of α-Co(OH)2 on carbon scaffolds
After preparation of the carbon scaffold, metal foil (either stainless steel or nickel) was wrapped around one end of the scaffold. Stainless steel foil was sufficient for carbon paper, but nickel tape was required for more complex carbon scaffolds. The electrodeposition procedure was adapted from Yan et al.34 The electrochemical cell consisted of a 25 mL three-neck glass round-bottom flask containing ∼25 mL of 0.1 M Co(NO3)2 in DI water. A 1 cm2 piece of each carbon scaffold served as the working electrode, Pt coil as the counter electrode, and Ag/AgCl in saturated KCl as the reference electrode. Chronopotentiometry was used to deposit α-Co(OH)2 on the carbon scaffold working electrodes. The electrodeposition procedure was optimized on carbon paper, and consisted of 3 minutes at −20 mA, 30 seconds of rest at open-circuit potential, and another 3 minutes at −20 mA. This protocol produced mass loadings of ∼2–4 mg per cm2 α-Co(OH)2 on carbon paper (α-Co(OH)2@CP). For electrodeposition on other carbon scaffolds, the same protocol was repeated but with the applied current density of −20 mA cm−2 scaled to the experimentally obtained electrochemical surface area values shown in Table 2. The electrodeposition on the CNT foam was done using an applied current of −145 mA.After electrodeposition, the electrode was rinsed thoroughly with DI water and left to dry for at least two hours before hydrothermal treatment to convert the hydroxide to LCO. For the thick scaffolds with smaller pores (carbon felt, CNT foam) the electrodes were soaked in 100 mL of DI water for an hour to completely remove the electrodeposition solution, and dried in a 60 °C oven for at least two hours.
Preparation of β-Co(OH)2 on carbon paper
To prepare β-Co(OH)2 on carbon paper (β-Co(OH)2@CP), the electrodeposited α-Co(OH)2@CP was soaked in 5 mL of 6 M KOH for 12 hours. Then, the electrode was soaked in 1000 mL of DI water for several hours, while the pH was monitored using a pH probe (Mettler-Toledo FiveEasy) every few hours, and replacing the DI water until the pH was neutral. After removing the electrode from the water bath, it was vigorously rinsed with DI water and dried on a Kimwipe for 1 hour before transferring to a vacuum oven to dry for 12 hours at 60 °C.Synthesis of LCO
A hydrothermal reaction was used to convert the electrodeposited cobalt hydroxide on carbon scaffolds to LCO on carbon scaffolds. A 45 mL Teflon-lined acid digestion vessel (Parr Instrument Company) was used for all hydrothermal treatments. The hydroxide-coated carbon scaffolds were added to the Teflon vessel along with a solution of LiOH in DI water. The electrodeposited portion of the scaffold was completely submerged in the solution. The vessel was sealed and placed in a temperature-controlled oven (Baxter Constant Temperature Oven DN-63). We tested the influence of precursor phase (α or β), temperatures of 140 °C and 200 °C, durations of 15 hours and 120 hours, concentrations of 2 M and 4.4 M LiOH, and solution volumes of 5 and 36 mL (denoted as 11% and 80% vessel fill, respectively) on the morphology and electrochemistry of LCO formed directly on carbon paper. The exact parameters employed in each iteration of the experiment are specified where the data is presented in the discussion below.After the hydrothermal treatment, the vessel was removed from the oven and left to cool to room temperature for 2 hours in a closed fume hood. Once cooled, the electrodes were extracted from the vessel and soaked in 1000 mL of DI water for 12 hours. The DI water was replaced as many times as necessary until the solution was pH neutral. The electrodes were removed from the water bath and dried for 1 hour on a Kimwipe and subsequently at 60 °C for 8 hours in air. Finally, all hydrothermally treated carbon paper electrodes were heated at 300 °C for 8 hours in air in a box furnace (Thermo Scientific Lindberg Blue M).
Physical characterization
X-ray diffraction (XRD) was used to characterize the structure of the LiOH treated cobalt hydroxide powders, electrodeposited cobalt hydroxides and LCO@CP electrodes, using either a PANalytical Empyrean or X'Pert Pro X-ray diffractometer in the standard Bragg–Brentano geometry with Cu-Kα (λ = 0.54 Å) radiation. All XRD data shown in the main text was taken using a PANalytical Empyrean diffractometer (45 kV, 40 mA, sealed Cu X-ray tube, Kα1, Kα2 λ = 1.5406 Å, 1.5444 Å), and the instrument used to collect data shown in the ESI† is specified where presented in text. The references for structures used are: α-Co(OH)2 (ref. 35), β-Co(OH) (CIF 1548810), Co3O4 (CIF 1526734), CoOOH (CIF 9009884), HT-LiCoO2 (PDF 000500653), and LT-LiCoO2 (PDF 010803830). The electrode microstructure was determined using a high-resolution scanning electron microscope (SEM; Field Emission FEI Verios 460L or Hitachi SU8700.) Nanoparticle diameter and nanoflake thickness were estimated by taking an average of 10 measurements from SEM images.Electrochemical characterization
Electrochemical characterization was performed in both flooded three-electrode cells and two-electrode coin cells. The data in Fig. 6 was collected from coin cells, and all other electrochemistry data was collected from flooded cells. Any electrodes that were not heat treated at 300 °C were dried in the vacuum oven for 12 hours at 60 °C before transfer to an argon-filled glovebox (<1 ppm H2O and O2). The three-electrode electrochemical cells consisted of a 25 mL three-neck round bottom flask with the deposited carbon scaffold as the working electrode, and 3 × 1 cm2 Li metal reference and counter electrodes. The electrolyte was 25 mL of 1 M LiClO4 in PC. Cyclic voltammetry (CV) was performed at 0.1 mV s−1 for ten cycles using a potentiostat (BioLogic VMP3). After cycling, the electrodes were rinsed thoroughly with DMC and left to dry on a Kimwipe overnight inside the glovebox. After the electrodes were completely dry, they were removed from the glovebox for further characterization.The coin cells consisted of LCO deposited on carbon paper as the cathodes and Li metal as the anodes. After vacuum drying, 1 cm diameter electrodes were punched from the carbon paper with deposited LCO. These electrodes were assembled into 2032 stainless steel coin cells in a glovebox with <1 ppm H2O and O2. The coin cells also consisted of a Li metal chip (TMAX, battery grade) as the anode, a glass fiber separator (Whatman), a stainless steel 316 spring (MTI), two stainless steel 316 spacers (MTI, 0.5 mm) and 200 μL of 1 M LiClO4 in PC electrolyte. The cells were crimped with 0.8 torr of pressure using a digital pressure controlled electric crimper (MSK-160E, MTI). Excess electrolyte was wiped away with a DMC-soaked Kimwipe and the cell was removed from the glovebox. Outside of the glovebox, the electrodes were wiped once again with an ethanol-soaked Kimwipe and cycled on a BioLogic VMP potentiostat using cyclic voltammetry with a scan rate of 0.1 mV s−1.
Discussion
Aqueous chemistry of Co(OH)2 in alkaline solutions
The low temperature synthesis method involves hydrothermal treatment of Co(OH)2 in concentrated LiOH solution at elevated temperatures. We conducted an experiment to understand the interactions of β-Co(OH)2 with aqueous LiOH solutions as a function of the LiOH concentration (Fig. 3). The LiOH concentrations and corresponding pHs are shown in Table S1.† We measured the pH of the solutions before addition of β-Co(OH)2 powder (black curve), after 7 days stirring the solutions at room temperature and 7 days at 60 °C (day 14, blue curve), and after an additional 8 days of stirring at 60 °C (day 22, red curve). We identified three concentration regimes corresponding to the different colors of the final powder solutions (Fig. S2†). These regions of interest are color coded in Fig. 3: region 1 is green (low LiOH concentration, <0.002 mM), region 2 is yellow (intermediate LiOH concentration, 0.002–43 mM), and region 3 is pink (high LiOH concentration, >43 mM LiOH). In region 1, we observed a decrease in the pH of the solutions on day 14 (Fig. 3a, green region, blue curve). After adding the powders and stirring for a total of 22 days, the pH of the solutions settled between 8.5 and 9.5 and the powders turned black. The powders remained suspended in the supernatant immediately after stirring, settling completely after three days of rest to enable separate characterization of the powder and supernatant. XRD (Fig. 3b) showed that β-Co(OH)2 converted to spinel Co3O4. The presence of Co3O4 and decrease in pH indicates deprotonation and partial oxidation of β-Co(OH)2 in dilute LiOH solutions in region 1. UV-vis results (Fig. S2†) showed no significant concentration of absorbing species in the powders or supernatant, indicating no dissolution at low LiOH concentrations. | ||
| Fig. 3 Interaction of powder β-Co(OH)2 with various LiOH concentrations, demonstrating concentration regimes for the formation of Co3O4 (green), partial dissolution of Co(OH)2 (yellow), and complete oxidation to CoOOH (pink) at room temperature and 60 °C. (a) pH as a function of LiOH concentration showing the initial pH of the LiOH solutions (black squares), pH after 7 days of stirring the LiOH solutions with β-Co(OH)2 at room temperature and 7 days at 60 °C (day 14, blue circles), and pH after 8 more days of stirring at 60 °C (day 22, red triangles). LiOH concentration is plotted on a logarithmic axis on the main plot, and on a linear axis in the inset to include the data at 0 mM LiOH. (b) XRD of the powder from all vials on day 22 compared to pristine β-Co(OH)2 in air (black graph). | ||
In region 2, after β-Co(OH)2 powders were added and stirred for 22 days the pH decreased to ∼9.5–11.5 (Fig. 3a, yellow region, red curve compared to black). The magnitude of the pH decrease became smaller for higher LiOH concentrations, suggesting less change in OH− concentration. Over the course of stirring, the β-Co(OH)2 powders changed color from pink to brown. The XRD patterns of the powders showed β-Co(OH)2 as the majority phase. This is consistent with UV-vis of the suspended powders indicating presence of Co2+ through absorption around ∼420 nm (Fig. S2b†). Between 1.08 and 2.15 mM LiOH, UV-vis showed no dissolved Co2+ in the supernatant, indicating that β-Co(OH)2 remained mostly in the solid state (Fig. S2c†). At higher concentrations of 21.5–43 mM LiOH, the UV-vis spectra of the supernatant showed absorption around ∼420 nm indicative of Co2+ species present in the solution. Pralong et al. reported that in alkaline solutions, Co(OH)2 forms the dicobaltite anion, Co(OH)42−, which appears blue.36 The authors reported a solubility limit of 0.048 mg mL−1 for β-Co(OH)2 in 5 M KOH. Therefore, relatively small amounts of Co(OH)42− are present from the spontaneous dissolution of β-Co(OH)2. Within region 2, we hypothesize β-Co(OH)2 partially dissolved in the aqueous LiOH solution to form CoOOH− and raise solution pH. The extent of dissolution increases with LiOH concentration, and CoOOH− becomes detectable via UV-vis between 21.52 and 43.03 mM LiOH (Fig. S2†).
In region 3, the sample in 215 mM LiOH, became a dark powder after 22 days of stirring with negligible change in pH. The XRD pattern of this powder showed the Rm structure of CoOOH (Fig. 3b), and UV-vis shows no dissolved species in the supernatant (Fig. S2c†). The negligible change in pH during stirring indicates that both protons in Co(OH)2 could not have been expelled into the solution. We hypothesize that a proton-coupled electron transfer reaction took place with the oxidation of Co2+ to Co3+ and corresponding loss of one H+ to the solution. Since there is no significant pH change observed after 22 days of stirring for any concentrations above 86.07 mM LiOH, we hypothesize that oxidation of β-Co(OH)2 to CoOOH began here and continued with higher extents of completion up to 215 mM LiOH (Fig. 3a). We conclude that high concentrations of Li+ (>86.07 mM LiOH) are necessary to prevent dissolution of β-Co(OH)2 and promote the formation of the Rm phase of CoOOH isostructural with LCO.
Ion exchange of electrodeposited Co(OH)2 on carbon paper
We first electrodeposited Co(OH)2 onto carbon paper (CP), which served as a model porous carbon scaffold (Fig. S3†). Yan et al. proposed that the mechanism for Co(OH)2 electrodeposition from an aqueous solution of cobalt(II) nitrate involves the reduction of nitrate and water, which increases the concentration of OH− at the interface. Dissolved Co2+ is proposed to react with OH− and heterogeneously nucleate onto the carbon scaffold. Here, the electrodeposition yielded a blue-green solid, characterized as α-Co(OH)2 from XRD (Fig. 4a) with a nanoflake microstructure (Fig. 2 and S4†) on the carbon paper, which we designate as α-CoOH2@CP. As described in Fig. 1, α-Co(OH)2 is metastable and can readily convert to β-Co(OH)2 under oxidizing conditions (including air).37 Therefore, some β-Co(OH)2 impurities are present and appear in the XRD scan with lower intensities. The most intense XRD reflections come from the carbon paper substrate (Fig. S5†), however, reflections from the electrodeposit do not overlap with those of the scaffold and are still well resolved at lower intensities. SEM shows that the α-Co(OH)2 nanoflakes grew radially outward from the surface to conformally coat the carbon fibers and appeared well-adhered to the scaffold (Fig. S4†). To transform α-CoOH2@CP into LCO@CP at ambient temperature, we attempted three methods: (1) ion exchange in 4.4 M LiOH, (2) conversion to β-Co(OH)2@CP followed by ion exchange in 4.4 M LiOH, and (3) electrochemical ion exchange from a non-aqueous Li+ electrolyte. In the first ion exchange method, the resulting XRD pattern (Fig. 4b) shows a mixed phase of β-Co(OH)2, CoOOH and LCO, indicating incomplete exchange. We also used cyclic voltammetry to further identify the products because different crystallographic phases of LCO show distinct electrochemical signatures in a non-aqueous Li+ electrolyte. Layered Rm HT-LCO or spinel Fdm LT-LCO have similar XRD reflections but different cyclic voltammetry features: layered LCO has one reversible redox couple ∼ 3.9 V, and spinel has one reversible couple at ∼3.7 V and one irreversible cathodic peak at ∼3.2–3.4 V.38,39 The CV of the α-Co(OH)2@CP electrode soaked in 4.4 M LiOH showed an oxidation peak at 3.8 V (1), attributed to Co3+ oxidation and Li+ removal from an octahedral site (Fig. S6, gray curve†). Upon applying reducing potentials, Li+ did not reinsert into the material.
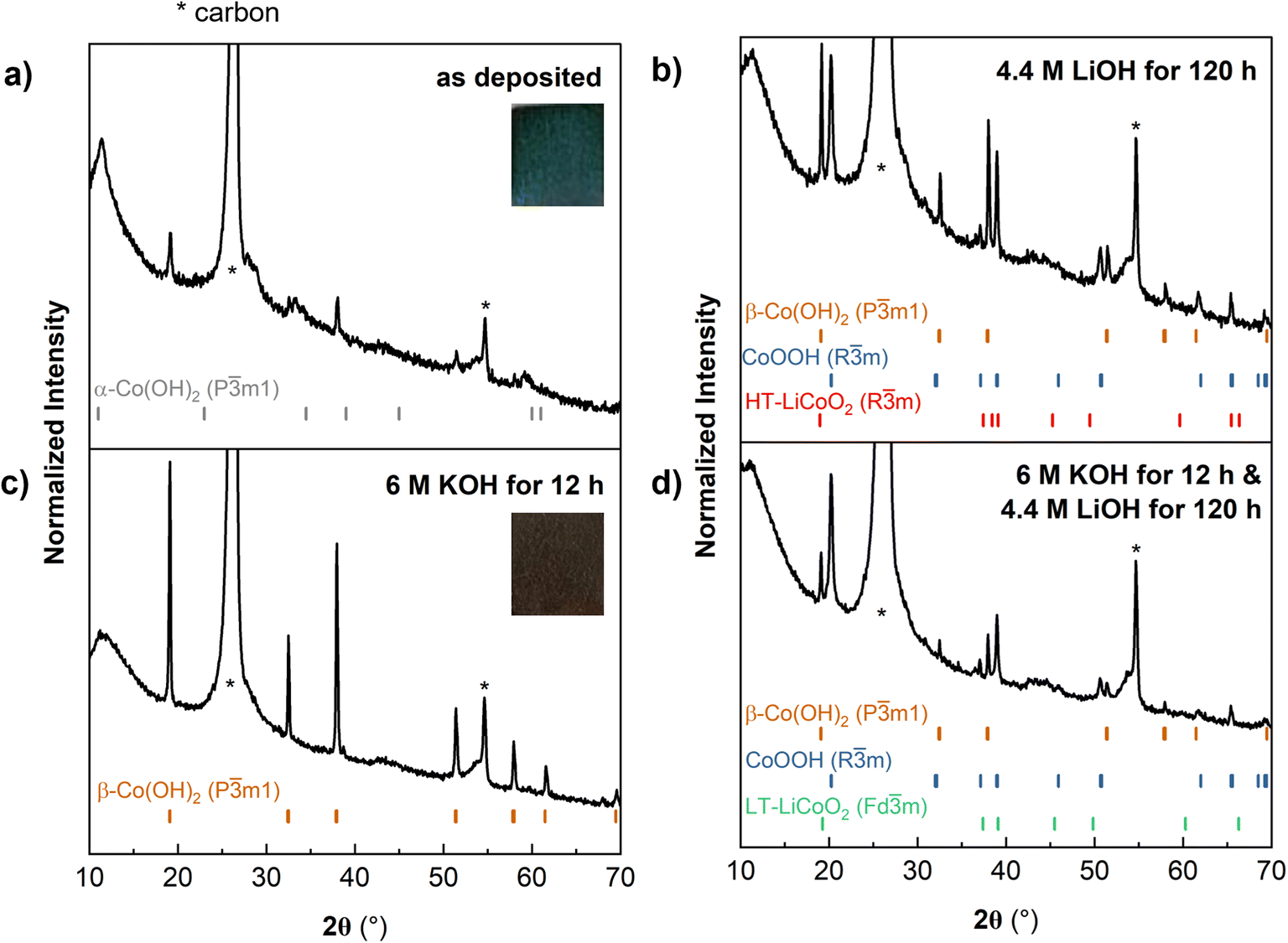 | ||
| Fig. 4 (a) XRD of as-electrodeposited Co(OH)2 shows predominantly α-Co(OH)2 with some β-Co(OH)2. (b) Soaking as-electrodeposited-Co(OH)2 in 4.4 M LiOH for 120 h leads to partial conversion to a mixed phase of CoOOH and LCO. (c) Soaking as-electrodeposited-Co(OH)2 in 6 M KOH for 12 h leads to the conversion of α-Co(OH)2 to β-Co(OH)2. (d) Soaking as-electrodeposited-Co(OH)2 in 6 M KOH for 12 h followed by 4.4 M LiOH for 120 h leads to a mixed phase of CoOOH and LCO. * indicates peaks from the carbon paper substrate, referenced in Fig. S5.† | ||
In the second ambient temperature ion exchange method, we converted α-CoOH2@CP to β-Co(OH)2@CP prior to the Li+/H+ exchange. Strongly alkaline conditions drive the conversion of α-Co(OH)2 to β-Co(OH)2 as water and other molecules are expelled from the interlayer.37 Soaking α-CoOH2@CP in 6 M KOH leads to a color change from blue-green to brown, characteristic of β-Co(OH)2. XRD confirmed this conversion (Fig. 4c). β-Co(OH)2@CP was then soaked in 4.4 M LiOH to drive the exchange of H+ with Li+. XRD (Fig. 4d) showed that the product was a mixed phase of CoOOH and LCO, similar to α-Co(OH)2@CP. However, the CV of this electrode in 1 M LiClO4 in PC was different, with an oxidation peak (1) corresponding to Li+ removal from an octahedral site, and reduction peaks (1′ and 1′′) corresponding to Li+ insertion and restructuring in tetrahedral sites to form the Fdm structure (Fig. S6, orange curve†). These results show that by exposing different polymorphs of Co(OH)2 to high concentrations of LiOH under aqueous, ambient conditions, ion exchange between H+ and Li+ to form a mixed phase of LCO and CoOOH is possible, albeit kinetically sluggish.
Since ion exchange in concentrated LiOH yielded partial exchange of H+ with Li+, we investigated whether electrochemical de-insertion of H+ followed by electrochemical insertion of Li+ would yield LCO by cycling α-Co(OH)2@CP and β-Co(OH)2@CP in a non-aqueous Li+ electrolyte (1 M LiClO4 in PC). The electrode was first oxidized to de-intercalate H+, then reduced to drive Li+ intercalation. The results in Fig. 5a demonstrate negligible current response (<0.001 mA) for α-Co(OH)2@CP over the course of four cycles, indicating no H+ de-intercalation/Li+ intercalation. However, cycling β-Co(OH)2@CP similarly showed oxidation and reduction peaks corresponding to insertion/deinsertion of Li+ from the spinel Fdm structure of LT-LCO (Fig. 5a, orange). The magnitude of the oxidation and reduction peaks increased with cycling, suggesting increased utilization of the electrode. Fig. 5b shows the ex situ XRD pattern of β-Co(OH)2@CP after cycling 1 M LiClO4 in PC, which depicts an almost complete transformation of the electrode to spinel LT-LCO. From this and Fig. 4, we observed that β-Co(OH)2@CP was able to electrochemically insert Li+ and α-Co(OH)2@CP could not, even when α-Co(OH)2@CP soaked in LiOH had partial H+/Li+ exchange. These results suggest that the presence of interlayer molecules in α-Co(OH)2 prevent electrochemical H+ de-insertion and inhibit Li+ insertion.
 | ||
| Fig. 5 (a) Cyclic voltammetry of α-Co(OH)2@CP and β-Co(OH)2@CP in 1 M LiClO4 in PC at 0.1 mV s−1. (b) XRD pattern of β-Co(OH)2@CP after cycling in 1 M LiClO4 in PC showing partial transformation to spinel LT-LCO. | ||
Combining electrodeposition, hydrothermal synthesis, and heat treatment to synthesize LCO@CP
In the previous sections, we established that Co(OH)2 can undergo partial oxidation and exchange of H+ with Li+ at room temperature in concentrated LiOH, or using an electrochemical method. Prior work showed that LT- and HT-LCO can be synthesized by hydrothermally treating Co(OH)2 with concentrated aqueous LiOH.21,25 However, there has not been a detailed investigation into the factors influencing the transformation of Co(OH)2 into layered LCO under hydrothermal conditions. Here, we discuss the influence of four hydrothermal synthesis parameters employed in this study (pressure or vessel fill, LiOH concentration, temperature, and duration of hydrothermal treatment) on the resulting synthesis mechanism, morphology, and electrochemistry of LCO formed directly from α-Co(OH)2 on carbon paper (LCO@CP). We utilized cyclic voltammetry to discern differences in crystal structure (LT- vs. HT-LCO) and morphology by analyzing features such as the peak current shape, potential, peak potential separation (ΔV), capacity, and coulombic efficiency (CE). For example, cyclic voltammetry revealed that calcination did marginally improve the capacity retention and coulombic efficiency of LCO@CP electrodes (Fig. S7a†). However, we did not observe differences in crystallographic structure or morphology before and after calcining (Fig. S7b–d†). For this reason, the calcination was used on all carbon paper electrodes described here, but not on other carbon scaffolds, to prevent unnecessary scaffold embrittlement.Xia et al. reported a three-step electrodeposition, hydrothermal synthesis, and heat treatment process to obtain LCO on carbon cloth (380 °C).21 Here, we began by treating electrodeposited α-Co(OH)2@CP electrode with similar hydrothermal conditions (80% reactor fill, 2 M LiOH, 15 hours at 200 °C) followed by heat treatment at 300 °C in air to yield LCO@CP. There is a distinct change in morphology after hydrothermal synthesis as the α-Co(OH)2 nanoflakes transform into a dense agglomeration of nanoparticles on carbon paper (Fig. 6a and b). We observed the nanoparticles were approximately 91 ± 22 nm in diameter from SEM in Fig. 6b. The stark change in morphology suggests a dissolution–precipitation reaction took place during hydrothermal treatment. The CV response of LCO@CP in 1 M LiClO4 in PC showed a redox couple at ∼3.9 V with a peak potential separation of 40 mV (Fig. 6c), characteristic of layered LCO. These peaks correspond to coupled Li+/e− transfer from/to the oxide during the anodic/cathodic cycles. The sharpness of the peaks and small peak potential hysteresis indicate little dispersion in the insertion site energies and good reversibility, indicative of a well-crystallized LCO material. The nanoscale microstructure should allow for shorter electron transport and Li+ solid-state diffusion distances, which facilitate fast kinetics. However, the current diminished rapidly upon cycling, as demonstrated by the low first cycle CE of 50%. We hypothesize that this decline in signal comes from detachment of the LCO nanoparticles to the carbon paper matrix, leading to progressively decreased active material utilization with cycle number.
 | ||
| Fig. 6 Influence of the hydrothermal synthesis pressure (% vessel fill) and LiOH concentration on the microstructure and performance of LCO@CP. SEM images and cyclic voltammograms of LCO@CP electrodes made using (a–c) 80% fill and 2 M LiOH, (d–f) 11% fill and 2 M LiOH, and (g–i) 11% fill and 4.4 M LiOH. The temperature and duration of hydrothermal synthesis were kept constant at 200 °C and 15 hours, respectively. | ||
The synthesis conditions for LCO@CP electrodes in Fig. 6a–c led to dissolution of α-Co(OH)2. Next, we decreased the hydrothermal synthesis pressure by decreasing the vessel filling from 80% to 11% and repeated the hydrothermal synthesis with all else held equal. Under these conditions, the synthesis yielded two distinct morphologies (Fig. 6d and e): irregularly shaped micron size particles surrounded by nanoscale (<50 nm), roughly spherical particles. The cyclic voltammetry of LCO@CP made under these conditions particles is shown in Fig. 6f. The first cycle CE was higher (66%) than for the electrode made from the high 80% fill volume synthesis (CE = 50%). This suggests that while the low fill volume synthesis yielded LCO particles that were better adhered to the carbon paper, cycling stability was still a problem. The first cycle CV displays two sets of redox couples: 1/1′ at ∼3.9 V, and 2/2′ at ∼4.1 V and ∼4.2 V that correspond to Li+ ordering to form a superstructure in LCO.40 Narrow peak separation implies fast electrochemical kinetics of the active material as described for Fig. 6c, and we attribute this CV contribution to the nanoparticles in Fig. 6d and e. Upon cycling, the 1/1′ peak separation and width increased, and the peak current decreased, while the 2/2′ peaks became less defined and eventually disappeared. This suggests a change from an electrode with fast-ion insertion kinetics and structural homogeneity of the Li+ active sites in the solid to an electrode with sluggish diffusion and poor utilization of the active material. Assuming DLi+ of 6.5 × 10−11 cm2 s−1 41 and linear diffusion with a potential-independent scan rate, the estimated Li+ diffusion distance in LCO is ∼0.13 μm. This diffusion distance agrees with the scale of the micron-sized LCO particles obtained in these synthesis conditions.
Next, we maintained the lower fill volume while increasing the concentration of LiOH from 2 M to 4.4 M. This synthesis yielded LCO@CP with the LCO particles forming an interconnected nanoflake matrix (Fig. 6g and h), with nanoflakes averaging 20 ± 5 nm thick. In the corresponding CV (Fig. 6i), this electrode had a higher first cycle CE (71%) than the electrodes from the other two syntheses, despite having larger peak breadth and separation for 1/1′ (130 mV). The improved CE and cycling stability suggest that the adhesion of the LCO to the carbon paper was better. As a result, hydrothermal conditions yielding LCO nanoflakes resulted in the most favorable morphology for carbon paper-based electrodes.
The pressure and LiOH concentration in the hydrothermal vessel significantly influence the morphology and electrochemical behavior of LCO@CP electrodes. Given the two types of microstructures (nanoflakes and nanoparticles), we hypothesize that there are two different reaction mechanisms possible for the formation of LCO@CP from electrodeposited α-Co(OH)2@CP. First, we consider the nanoflake morphology of LCO@CP formed under low pressure and high LiOH concentration. Since this is similar to the microstructure of α-Co(OH)2@CP, we hypothesize that these conditions favor a topotactic ion exchange mechanism: oxidation of Co(OH)2 to CoOOH and exchange of H+ and Li+ result in the formation of LCO. During the experiments described in Fig. 3, in the absence of hydrothermal conditions we observed via XRD the oxidation of commercial powders of β-Co(OH)2 to CoOOH when stirred in an aqueous solution of concentration 215 mM LiOH. When α-Co(OH)2@CP was left soaking in 4.4 M LiOH at room temperature and pressure, we observed a partial exchange of H+ with Li+ (Fig. 4b).
Hydrothermal synthesis at high pressures and/or low LiOH concentrations leads to LCO nanoparticle formation that is different from the nanoflake α-Co(OH)2@CP precursor. Under these conditions, we hypothesize that the mechanism involves dissolution of α-Co(OH)2 and precipitation of LCO. Control experiments (Fig. 3) confirmed the dissolution of β-Co(OH)2 powders in dilute LiOH (21.5–43 mM LiOH). Under hydrothermal conditions, we propose that soluble CoOOH− reacts with Li+ to form LCO. In terms of cycling performance, the nanoflake LCO morphology formed via the proposed ion exchange mechanism is more favorable than the micron-scale LCO crystals from the proposed dissolution–precipitation mechanism.
Consequently, the next experiments investigated the influence of the hydrothermal treatment time (15 to 120 h) and temperature (140 °C) while holding the pressure (11% reactor fill) and solution concentration (4.4 M LiOH) constant. Fig. 6g–i shows the CV and microstructure of the LCO@CP electrode produced from a 15 h hydrothermal reaction whereas Fig. 7a and b shows the corresponding results for an electrode produced from a 120 h hydrothermal reaction. The shorter timescale yielded exclusively the nanoflake LCO morphology, while longer hydrothermal treatment led to a mixed microstructure containing both nanoflakes and nanoparticles. When the α-Co(OH)2@CP electrodes are submerged in LiOH solution in the hydrothermal treatment vessel, any Co(OH)2 flakes that come loose are suspended in solution. We hypothesize that during the 120 h synthesis under these mild hydrothermal treatment conditions, suspended Co(OH)2 flakes can react with LiOH in solution to precipitate smaller LiCoO2 particles on the electrode surface also via an ion exchange mechanism. Decreasing the hydrothermal temperature to 140 °C for the same duration of 120 h led to the formation of nanoflakes that were thinner than those formed at 200 °C (Fig. 7a vs. Fig. 7c). This finding confirms that the hydrothermal temperature could be used to modulate LCO nanoflake thickness, as suggested by Xia et al.21
 | ||
| Fig. 7 Influence of hydrothermal temperature and Co(OH)2 precursor phase on the synthesis of LCO@CP. SEM images and cyclic voltammograms of hydrothermal syntheses performed using (a and b) α-Co(OH)2@CP at 200 °C, (c and d) α-Co(OH)2@CP at 140 °C, and (e and f) β-Co(OH)2@CP at 140 °C. The vessel fill, LiOH concentration, and synthesis time were held constant at 11%, 4.4 M LiOH, and 120 h, respectively. | ||
Finally, we considered the influence of the precursor Co(OH)2 phase on the synthesis of LCO@CP by performing syntheses with either α-Co(OH)2@CP or β-Co(OH)2@CP. The SEM images of products from both syntheses (Fig. 7c and e) show no significant difference in morphology between the resulting LCO. Since α-Co(OH)2 converts to β-Co(OH)2 in alkaline environments (Fig. 4a and c), it is likely that the conversion occurs “in situ” in the 4.4 M LiOH solution during hydrothermal synthesis. Therefore, utilizing α-Co(OH)2@CP as the precursor material bypasses the need for an additional soaking step.
Toward deterministic electrode architectures for LIBs
Thus far we established that free-standing, binder-free porous carbon paper electrodes featuring LCO with a nanoflake morphology exhibit superior electrochemical performance (CE and capacity retention) compared to the electrodes with a nanoparticle morphology. Fig. 8a shows the cathodic capacity retention over ten CV cycles for LCO@CP electrodes as a function of hydrothermal treatment temperature and duration (while holding the vessel fill and LiOH concentration constant at 11% and 4.4 M, respectively). The electrode produced from the 120 h at 200 °C synthesis (Fig. 8a, black curve) starts with a cathodic capacity close to the practical capacity for LCO (140 mA h g−1 or 0.5 Li+/e− per LCO). This electrode has better capacity retention than the electrode made from 15 h of hydrothermal synthesis at 200 °C. For a hydrothermal synthesis duration of 120 h, the electrode produced at 200 °C exhibits over 20 mA g−1 more capacity than the electrode made at 140 °C during the first cycle, but by the 10th cycle, the performance gap narrows such that the 200 °C electrode is only marginally better. The narrow gap between the electrodes' cathodic capacity retention persisted after 20 CV cycles (Fig. S8†), but the electrode produced at 200 °C for 120 h exhibited slightly superior performance. All electrodes show a slight improvement in CE during cycling, with the higher CEs for those synthesized at 120 h (Fig. 8b).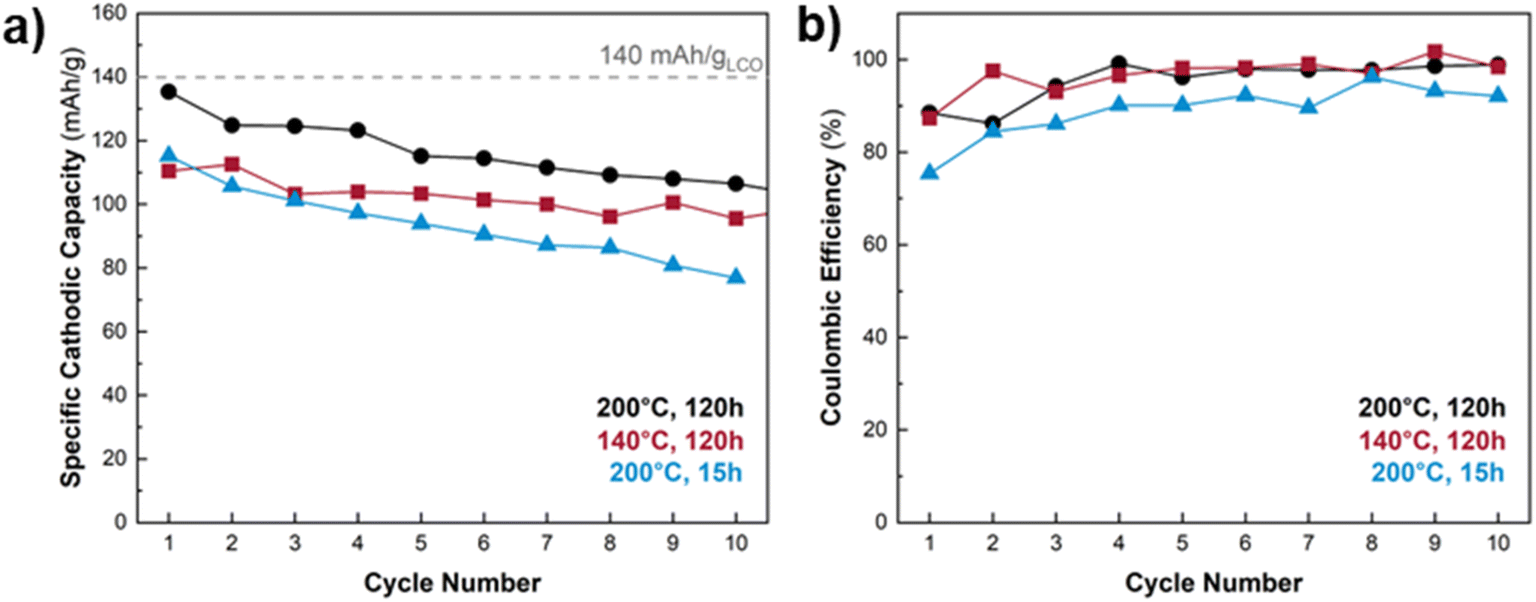 | ||
| Fig. 8 (a) Cycling stability and (b) coulombic efficiency of LCO@CP electrodes made using different hydrothermal synthesis temperature and duration while holding pressure and LiOH concentration constant (11% vessel fill and 4.4 M LiOH). The dashed horizontal line indicates the theoretical specific capacity of LCO. The 120 h treatment at 200 °C produces an electrode with near-theoretical cathodic capacity during the first cycle, and superior capacity retention over electrodes produced at 140 °C for 120 h or 200 °C for 15 h. | ||
All LCO@CP electrodes showed a capacity decline within the first ten cycles. We used SEM to qualitatively examine the microstructure of the LCO@CP electrodes before and after electrochemical cycling (Fig. S9 and S10†). It revealed that for 15 h and 120 h treatments, some LCO nanoflakes detached from the matrix during cycling. These detached nanoflakes formed agglomerates found on the surface of the nanoflake matrix, further from the carbon paper. This likely increased the electronic resistance for electron transfer to/from the LCO, leading to worse cycling performance. Furthermore, we observed a difference in the continuity and adhesion of the LCO nanoflake matrix to the carbon paper when the hydrothermal treatment length increased from 15 h to 120 h. Fig. S9† shows that for the 15 h synthesis, the nanoflake matrix remained fully covering the carbon paper scaffold after cycling. The detached LCO nanoflakes decorated the matrix's surface and did not protrude out from the electrode. Fig. S10† shows that for the 120 h synthesis, the nanoflake matrix did not remain as a continuous coating on the carbon paper scaffold, with patches of carbon paper visible in the low magnification images (Fig. S10a and c†). The higher magnification images in Fig. S10e and g† reveal that there were small gaps between the nanoflakes and carbon fibers where portions of the matrix were interconnected with itself but not contacting the carbon scaffold. This detachment of the nanoflake matrix from the scaffold was even more pronounced after cycling (Fig. S10f and h†). Compared to the electrode hydrothermally treated for 15 h in Fig. S9,† in the 120 h case there were more nanoflake agglomerates decorating the surface of the matrix and stacking on top of one another to protrude far from the matrix's surface in Fig. S10.† The morphological rearrangement and nanoflake matrix detachment from the carbon paper both result in disruptions to the electrode's electronic percolation network and can limit the achievable capacity during electrochemical cycling. However, despite worse adhesion of LCO to the carbon scaffold after the longer hydrothermal treatment, the 120 h electrode experiences a higher initial capacity and less capacity fade (21%) compared to the 15 h case (33%). We hypothesize that the longer synthesis improves the electrochemical properties of the LCO itself by improving electronic/ionic conductivity and/or allowing for more conversion of precursor to LCO. The higher capacity is accompanied by greater volume change of the LCO particles during cycling, which can lead to additional exfoliation observed for the 120 h synthesis in Fig. S10.†
Further work optimizing this method for low surface area and aspect ratio scaffolds such as carbon paper should focus on tailoring the amount of precursor α-Co(OH)2 and hydrothermal treatment time necessary to form a stable coating of LCO on the electrodeposited scaffolds. While we utilized plasma cleaning to functionalize the carbon paper before electrodeposition, further investigation of methods that better adhere the LCO to the carbon scaffold is warranted to improve the cycling stability.
Energy considerations
An important consideration for the commercial viability of this method is the overall energy consumption relative to currently used solid-state synthesis of cathode materials followed by slurry-cast electrode preparation. Attributes of the electrodeposition–hydrothermal-heat treatment method described here are that it directly leads to the direct fabrication of an LCO electrode without the use of fluorinated polymers or toxic solvents. It is an aqueous-based batch process that can make multiple electrodes simultaneously. However, further research is necessary to assess if the method leads to energy savings relative to commercialized battery electrode processes. Prior studies into the energy used for the hydrothermal synthesis of Li ion materials show that although solvothermal processes occur at substantially lower temperatures, the energy required can be comparable or larger than for higher temperature solid state reactions.42–44 In the method developed here, energy-intensive steps include Co(OH)2 electrodeposition and heating of the LiOH solution during hydrothermal treatment. The electrodeposition utilizes an unoptimized three electrode electrochemical cell, where the counter electrode reaction is presumably the energy-intense oxygen evolution reaction. Therefore, strategies to reduce the energy consumption include a more judicious choice of the electrodeposition counter electrode (anodic) reaction.Versatility of the method with other porous carbon scaffolds
Finally, we demonstrate the versatility of the combined electrodeposition–hydrothermal method to produce LCO at 200 °C on eight other commercially available carbon scaffolds. These scaffolds varied in geometry from foam-like to fiber-like microstructures. The processing protocol consisted of electrodepositing α-Co(OH)2 onto each carbon scaffold and hydrothermal synthesis in 4.4 M LiOH for 15 hours at 200 °C with 11% reactor fill. There was no final annealing step at 300 °C. The electrodeposition current was adjusted for each scaffold based on its experimentally determined electrochemical surface area (Table 2). After electrodeposition, all scaffolds were completely coated by α-Co(OH)2 nanoflakes. ESI Fig. S11–S19† show the complete microstructural characterization of each electrode as pristine, electrodeposited, and hydrothermally treated. SEM images for four representative carbon scaffolds coated with LCO are shown in Fig. 9. Cyclic voltammetry confirmed the presence of LCO on seven scaffolds, although with varying degrees of electrochemical reversibility (Fig. S20†). We could not reliably connect electrodes made from the most porous scaffolds (Duocel RVC 10 and 30 PPI) as they became too brittle following the hydrothermal treatment. They both appear to be coated with LCO in SEM images (Fig. S12 and S14†). The low magnification images in the top row of Fig. 9 show the bare carbon scaffolds. The electrochemical surface area and aspect ratio of the scaffold influenced the morphology of the LCO, which became more obviously nanoflake-like with increasing electrochemical surface area and aspect ratio. This is best observed by comparing the morphologies of the lowest electrochemical surface area scaffold (CFOAM25 foam in Fig. 9 and S11†) with the highest (CNT foam, Fig. S19†). | ||
| Fig. 9 SEM images of LCO deposited on four different commercially available porous carbon scaffolds: (a) CFOAM25 foam, (b) Duocel RVC 60 PPI, (c) Duocel RVC 100 PPI, and (d) carbon felt. The top row of low magnification images is of the bare scaffolds. All synthesis involved electrodeposition of α-Co(OH)2 and hydrothermal treatment in 4.4 M LiOH for 15 hours at 200 °C with 11% reactor fill. | ||
The LCO nanoparticles on the lower surface area and aspect ratio scaffolds (CFOAM25 and Duocel RVC 60 PPI, respectively) resembled products of a dissolution–precipitation mechanism. However, after the hydrothermal treatment, the LiOH solution was free of particles, contrary to the dark, cloudy solution in syntheses that followed the dissolution–precipitation mechanism for LCO formation on carbon paper. Therefore, we hypothesize that ion-exchanged LCO was formed on the low surface area scaffolds. However, the lower mass loadings resulted in smaller particles not easily identified as nanoflakes by inspection, as described in the discussion of Fig. 7.
Conclusions
In this work, we developed and characterized a sequential electrodeposition–hydrothermal-heat treatment method to deposit LCO from α-Co(OH)2 on different porous carbon scaffolds at low temperatures (<300 °C). We first investigated the aqueous chemistry of α- and β-Co(OH)2 in concentrated LiOH aqueous solutions under ambient conditions. We demonstrated that oxidation β-Co(OH)2 to CoOOH in bulk powder is possible in LiOH concentrations > 215 mM, and H+/Li+ ion exchange is possible in electrodeposited α- and β-Co(OH)2 in under LiOH concentrations near saturation in water (4.4 M LiOH). The results also showed a pH and Li+ concentration regime for partial dissolution of β-Co(OH)2 to CoOOH−. We found that H+/Li+ exchange could take place electrochemically but yielded spinel LT-LCO.We also presented the influence of hydrothermal synthesis parameters, such as vessel pressure, LiOH concentration, treatment duration, temperature, and Co(OH)2 phase, on the synthesis of LCO@CP electrodes. By independently varying synthesis parameters, we showed the individual effects of each parameter on the resulting LCO morphology and electrochemical behavior. The hydrothermal vessel pressure (controlled by vessel fill) and LiOH concentration are both key determinants of the synthesis mechanism: higher pressures (80% vessel fill) and low LiOH concentrations (2 M LiOH) favor dissolution of the α-Co(OH)2 precursor, followed by precipitation of LCO nanoparticles on the carbon scaffold. While the LCO was well-crystallized as evidenced by cyclic voltammetry and XRD, electrodes with this morphology did not exhibit good electrochemical cycling. We hypothesize that in this case, the LCO nanoparticles did not adhere well to each other or the carbon, leading to severe capacity fade. With lower hydrothermal synthesis pressures (11% vessel fill) and higher LiOH concentrations (4.4 M LiOH), LCO forms via an oxidation and ion exchange mechanism directly on the carbon scaffold. This process preserves the nanoflake morphology of the α-Co(OH)2 precursor, which was interconnected and well-adhered to the scaffold. Changing the duration and temperature of the hydrothermal treatment fine-tunes the morphology: shorter duration minimizes nanoparticle formation and lower temperature leads to thinner nanoflakes. We used this method to deposit nanoflake LCO on nine different commercial carbon scaffolds with varied surface areas, aspect ratios, and porosities without compromising the integrity of the scaffolds, demonstrating that the processing conditions are suitable for a wide range of porous carbon architectures. The ion exchange based LCO deposition process is conducive to designing free-standing electrode architectures under ambient, aqueous conditions.
Data availability
The manuscript data is available at https://zenodo.org/records/14902881.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grant no. 1901906 and a Graduate Research Fellowship to I. K. under grant no. DGE-2137100. The authors thank Dr Noah Holzapfel and Dr Saeed Saeed for useful discussions on the titration and UV-vis experiments, as well as Prof. William Joe Sagues and Dr Yaojing Qiu for insight into the energy consumption of the electrodeposition–hydrothermal method compared to solid-state synthesis of LCO. The authors thank the groups of Prof. Philip Bradford and Prof. Peter Fedkiw at NC State University for CNT foam and carbon felt samples, respectively. This work was performed in part at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-2025064). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI).References
- J. Fleishmann, P. Schaufuss, M. Linder, M. Hanicke, E. Horetsky, D. Ibrahim, S. Jautelat, L. Torscht and A. van de Rijt, Battery 2030: Resilient, Sustainable, and Circular, https://www.mckinsey.com/industries/automotive-and-assembly/our-insights/battery-2030-resilient-sustainable-and-circular, accessed August 1, 2024 Search PubMed.
- C. Vaalma, D. Bucholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- X. Lu, A. Bertei, D. P. Finegan, C. Tan, S. R. Daemi, J. S. Weaving, K. B. O'Regan, T. M. M. Heenan, G. Hinds, E. Kendrick, D. J. L. Brett and P. R. Shearing, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- Y. Kuang, C. Chen, D. Kirsch and L. Hu, Adv. Energy Mater., 2019, 9, 1901457 CrossRef.
- T. Danner, M. Singh, S. Hein, J. Kaiser, H. Hahn and A. Latz, J. Power Sources, 2016, 334, 191–201 CrossRef CAS.
- J. Wu, Z. Ju, X. Zhang, X. Xu, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi and G. Yu, ACS Nano, 2022, 16(3), 4805–4812 CrossRef CAS PubMed.
- D. J. Arnot, K. S. Mayilvahanan, Z. Hui, K. J. Takeuchi, A. C. Marschilok, D. C. Bock, L. Wang, A. C. West and E. S. Takeuchi, Acc. Mater. Res., 2022, 3, 472–483 CrossRef CAS.
- Z. Wu, Y. Wang, X. Liu, C. Lv, Y. Li, D. Wei and Z. Liu, Adv. Mater., 2019, 31, 1800716 CrossRef PubMed.
- L. E. Asp, M. Johansson, G. Lindbergh, J. Xu and D. Zenkert, Funct. Compos. Struct., 2019, 1, 042001 CrossRef CAS.
- Li-Ion Battery Cathode Market Size & Trends, https://www.grandviewresearch.com/industry-analysis/lithium-ion-battery-cathode-market-report, accessed August 1, 2024 Search PubMed.
- B. Huang, L. Cheng, X. Li, Z. Zhao, J. Yang, Y. Li, Y. Pang and G. Cao, Small, 2022, 18, 2107697 CrossRef CAS PubMed.
- G. Sádovská, P. Honcová, J. Morávková, I. Jirka, M. Vorokhta, R. Pilař, J. Rathouský, D. Kaucký, E. Mikysková and P. Sazama, Carbon, 2023, 206, 211–225 CrossRef.
- H. Zhang, H. Ning, J. Busbee, Z. Shen, C. Kiggins, Y. Hua, J. Eaves, J. Davis, T. Shi, Y. T. Shao, J. M. Zuo, X. Hong, Y. Chan, S. Wang, P. Wang, P. Sun, S. Xu, J. Liu and P. V. Braun, Sci. Adv., 2017, 3, e1602427 CrossRef PubMed.
- S. Behboudikhiavi, J. O. Omale, B. Babu, L. Piraux and A. Vlad, J. Electrochem. Soc., 2023, 170, 020509 CrossRef CAS.
- E. Rossen, J. N. Reimers and J. R. Dahn, Solid State Ionics, 1993, 62, 53–60 CrossRef CAS.
- R. J. Gummow, M. M. Thackeray, W. I. F. David and S. Hull, Mater. Res. Bull., 1992, 27, 327–337 CrossRef CAS.
- E. Antolini, Solid State Ionics, 2004, 170, 159–171 CrossRef CAS.
- S. Song, K. Han and M. Yoshimura, J. Am. Ceram. Soc., 2000, 83, 2839–2844 CrossRef CAS.
- H. Porthault, R. Baddour-Hadjean, F. Le Cras, C. Bourbon and S. Franger, Vib. Spectrosc., 2012, 62, 152–158 CrossRef CAS.
- R. J. Gummow, D. Liles and M. Thackeray, Mater. Res. Bull., 1993, 28, 235–246 CrossRef CAS.
- Q. Xia, M. Ni, M. Chen and H. Xia, J. Mater. Chem. A, 2019, 7, 6187–6196 RSC.
- G. G. Amatucci, J. M. Tarascon, D. Larcher and L. C. Klein, Solid State Ionics, 1996, 84, 169–180 CrossRef CAS.
- D. Larcher, M. R. Palacín, G. G. Amatucci and J. M. Tarascon, J. Electrochem. Soc., 1997, 144, 408–417 CrossRef CAS.
- T. Azib, F. Le Cras and H. Porthault, Electrochim. Acta, 2015, 160, 145–151 CrossRef CAS.
- K. Kumar Bokinala, M. Pollet, A. Artemenko, M. Miclau and I. Grozescu, J. Solid State Chem., 2013, 198, 45–49 CrossRef CAS.
- H. Porthault, F. Le Cras, R. Baddour-Hadjean, J. P. Pereira-Ramos and S. Franger, Electrochim. Acta, 2011, 56, 7580–7585 CrossRef CAS.
- Y. Tao, B. Zhu and Z. Chen, J. Alloys Compd., 2007, 430, 222–225 CrossRef CAS.
- S.-W. Song, K.-S. Han, I. Sasagawa, T. Watanabe and M. Yoshimura, Solid State Ionics, 2000, 135, 277–281 CrossRef CAS.
- K. S. Han, S. W. Song, T. Watanabe and M. Yoshimura, Electrochem. Solid-State Lett., 1999, 2, 63–66 CrossRef CAS.
- M. A. Spencer, O. Yildiz, I. Kamboj, P. D. Bradford and V. Augustyn, Energy Fuels, 2021, 35, 16183–16193 CrossRef CAS.
- S. Faraji, O. Yildiz, C. Rost, K. Stano, N. Farahbakhsh, Y. Zhu and P. D. Bradford, Carbon, 2017, 111, 411–418 CrossRef CAS.
- Z. Wang, C. Meng, J. Wang, Z. Song and R. Yu, Eur. J. Inorg. Chem., 2023, 26, e202300014 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors, Springer US, Boston, MA, 1999 Search PubMed.
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS PubMed.
- V. Pralong, A. Delahaye-Vidal, B. Beaudoin and J.-M. Tarascon, J. Mater. Chem., 1999, 9, 955–960 RSC.
- Z. Huang, Y. Zhao, Y. Song, Y. Li, G. Wu, H. Tang and J. Zhao, RSC Adv., 2016, 6, 80059–80064 RSC.
- B. Garcia, J. Farcy, J. P. Pereira-Ramos, J. Perichon and N. Baffier, J. Power Sources, 1995, 54, 373–377 CrossRef CAS.
- C. Y. Yao, T. H. Kao, C. H. Cheng, J. M. Chen and W.-M. Hurng, J. Power Sources, 1995, 54, 491–493 CrossRef CAS.
- S. Y. Vassiliev, E. E. Levin and V. A. Nikitina, Electrochim. Acta, 2016, 190, 1087–1099 CrossRef CAS.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Nat. Energy, 2018, 3, 936–943 CrossRef CAS.
- P. Benedek, N. Wenzler, M. Yarema and V. C. Wood, RSC Adv., 2017, 7, 17763–17767 RSC.
- J. B. Dunn, L. Gaines, J. C. Kelly, C. James and K. G. Gallagher, Energy Environ. Sci., 2015, 8, 158–168 RSC.
- F. Mozaffarpour, N. Hassanzadeh and E. Vahidi, Clean Technol. Environ. Policy, 2022, 24, 3319–3330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta09258a |
| This journal is © The Royal Society of Chemistry 2025 |