
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous polymeric nanoparticles for effective treatment of inflammatory diseases: an in vivo study†
Divya
Pareek
a,
Md.
Zeyaullah
b,
Sukanya
Patra
a,
Oviya
Alagu
a,
Gurmeet
Singh
a,
Kirti
Wasnik
a,
Prem Shankar
Gupta
a and
Pradip
Paik
 *a
*a
aSchool of Biomedical Engineering, Indian Institute of Technology, Banaras Hindu University (BHU), Varanasi, Uttar Pradesh 221 005, India. E-mail: paik.bme@iitbhu.ac.in; pradip.paik@gmail.com
bDepartment of Zoology, Banaras Hindu University (BHU), Varanasi, Uttar Pradesh 221005, India
First published on 23rd January 2025
Abstract
Acute inflammatory diseases require suitable medicine over the existing therapeutics. In this line, the present work is focused on developing polymeric nanomedicine for the treatment of inflammatory disorders. Herein, cell viable nanoparticles (GlyNPs) of size 180–250 nm in diameter and pore size of 4–5 nm in diameter, based on glycine and acryloyl chloride, have been developed and proved to be a potential anti-inflammatory agent without using any conventional drugs. These particles exhibit colloidal stability (with a zeta potential of −35.6 mV). A network pharmacology-based computational study has been executed on 9076 genes and proteins responsible for inflammatory diseases, out of which 10 are selected that have a major role in rheumatoid arthritis (RA). In silico docking study has been conducted to find out the targeted efficiency of the GlyNPs considering 10 inflammation-specific markers, namely IL-6, IL-1β, TNF-α, TLR-4, STAT-1, MAPK-8, MAPK-14, iNOS, NF-κβ and COX-2. The results revealed that the GlyNPs could be an excellent anti-inflammatory component similar to aspirin. The in vitro inflammation activity of these GlyNPs has also been checked on an inflammation model generated by LPS in RAW 264.7 macrophages. Then, the in vitro anti-inflammation efficiency has been checked with 10–150 μg mL−1 of GlyNP doses. The treatment efficiency has been checked on inflammation-responsible immune markers (NO level, NF-κβ, INF-γ, IL-6, IL-10, and TNF-α) and it was found that the GlyNPs are an excellent component in reducing inflammation. The in vivo therapeutic response of GlyNPs on the induced rheumatoid arthritis (RA) model has been evaluated by measuring the morphological, biochemical and immune-cytokine and interferon levels responsible for the inflammation, using a 2 g kg−1 dose (sample to weight of rat). The anti-inflammatory efficiency of GlyNPs without using additional drugs was found to be excellent. Thus, GlyNPs could be paramount for the potential treatment of various inflammatory diseases.
1. Introduction
Acute inflammatory diseases are relatively common and can affect people of all ages.1 Inflammation causes rheumatoid arthritis (RA), inflammatory bowel disease, psoriasis, and systemic lupus erythematosus.2 Furthermore, inflammation can cause tissue damage, organ dysfunction, and numerous organ damage.3,4 Researchers are continuing to explore new medicines to suppress inflammation. For example, the development of targeted immunotherapies is gaining traction, emphasizing the need for strategies that can both mitigate inflammatory processes and enhance patient recovery outcomes.5 However, a comprehensive understanding of the inflammatory pathways involved in these diseases is required to identify immunotherapeutic targets.6 Over the past three decades, advancements in treating inflammation have gained limited success, using molecular medicine. However, their cellular mechanism and therapeutic strategies are complicated.7 The strategies involved specific inflammatory cytokines, gene therapies that modulate immune responses, and drug delivery systems based on anti-inflammatory agents.7 These therapies specifically target the biomarker molecules intricate in the inflammatory process. Although these treatment procedures improved the quality of life for many patients to some extent by reducing symptoms, they showed severe side effects.8 Hence, it is urgent to develop suitable medicines to reduce inflammation and resolve the associated issues. Targeted therapies, for example, cell-depleting monoclonal antibodies, rituximab (anti-CD20) and alemtuzumab (anti-CD52), were used against other inflammatory diseases, including RA.9 Anti-cytokine therapies targeting specific inflammatory pathways, e.g., TNF-α, IL-12, IL-17, IL-23, and IL-35-based therapies, have also been reported.10 Similarly, bispecific antibodies and nanobodies,11i.e., recombinant variable domains of heavy chain-only antibodies (which make them effective for therapeutic applications), were also used to target multiple immune mediators and gene therapy approaches to reduce inflammation.2 However, these treatment approaches required surgical interventions, several challenges persist, and the high cost limits access for many patients.Many nanomaterial-based approaches are also used for the treatment of inflammatory diseases.12 Nanoparticles can be designed to target specific cells and tissues involved in inflammation, such as activated macrophages or endothelial cells, present at the site of inflammation.13 Steroid/non-steroid/peptide/biomimetic-based nanoparticles were also reported for anti-inflammation purposes.14 Inorganics such as Au, Ag, ZnO, SiO2, TiO2, CdO, ZnO, Fe2O3etc. NPs are reported for treating inflammatory diseases due to their catalytic properties and ability to directly react to neutralize reactive oxygen/nitrogen species (ROS/RNS) and inhibit the pro-inflammatory cytokines, such as IL-6, INF-γ and TNF-α that drive inflammation by promoting the levels of anti-inflammatory cytokines like IL-4 and IL-10.14,15 Anti-inflammatory drug-loaded nanoparticles are also reported to reduce chronic inflammation,14 such as CeO2 loaded with enzyme,16 PEGylated Pt NPs16 tannic-acid mineral nanoparticles (TMPs) assembled with bioactive calcium and phosphate ions reported for their use in anti-inflammation.17 Although inorganic NPs were used for treating inflammation, their potential to induce inflammation, variability in immune response, and toxicity concerns must be carefully addressed before successful clinical implementation.15 On the other hand, a few polymeric nanoparticles, such as PEGylated liposomes,18 hyaluronic acid-coated solid lipid NPs,19 PCL,20 PEG-b-PLA micelles,21 poly(NIPAm-co-AMPS),22 and chitosan/poly(γ-glutamic acid)23etc. are also reported for the treatment of inflammatory disorders.14 There is also an increasing trend to load anti-inflammatory drugs in polymeric nanoparticles and deliver the same to resolve inflammatory diseases.24 Despite that, there is huge concern about the biodistribution, pharmacokinetics and toxicity of drugs in resolving inflammation-related issues.24 All these medicines limit their use with secondary infections due to the lack of specificity.1 Earlier, we have used glycine and acryloyl chloride-based functional polymeric nanoparticles/nanocapsules without using/loading any conventional drugs for wound healing. However, without loading, no anti-inflammatory drugs/steroids have ever been reported as anti-inflammatory agents for the treatment of RA. These polymeric nanocapsules could be promising therapeutics for the effective treatment of inflammatory diseases such as RA, which is the main focus of this work. There is a lot of scope to improve in this direction.
In the above line, the present work focused on developing mesoporous polymeric nanoparticles (GlyNPs) based on glycine and acryloyl chloride. These NPs were qualified for therapeutic applications through the cell viability study on RAW 264.7 macrophage and PC-12 cell lines with varying concentrations, studying the cellular uptake on RAW 264.7 macrophages. Then, a network pharmacology-based computational study was accomplished to find out the genes and proteins responsible for the major inflammatory diseases, for example RA. Subsequently, in silico docking study has been conducted to find out the targeted efficiency of the synthesized polymeric particles considering ten inflammation-specific markers (e.g., IL-6, IL-1β, TNF-α, TLR-4, STAR-1, MAPK-8, MAPK-14, iNOS, NF-κβ and COX-2) and the results have been compared with the commercially available standard anti-inflammatory drugs (aspirin and diclofenac). Based on the computational results, the efficiency of the synthesis of polymeric partials has been authenticated in an in vitro study by varying the doses of novel polymeric NPs and the inflammation levels of a few immune markers (NO, NF-κβ, INF-γ, IL-6, IL-10, and TNF-α) were verified to be responsible for the inflammation. After receiving the progressive in vitro therapeutic response of the novel polymeric NPs, the work has been extended to an in vivo pristane-induced RA rat model to check the therapeutic proficiency of the synthesized GlyNPs without loading any additional drugs. Then, the in vivo therapeutic efficiency was evaluated by studying the morphology of the rat's paws and ankles, biochemical analysis by blood serum, and immune parameters using ELISA assay. Finally, the effective therapeutic potential of the synthesized glycine-acryloyl chloride-based GlyNPs to control inflammation without using any conventional drug has been established.
2. Experimental methods
2.1 Materials
The following materials were used: Glycine (98%, Sigma Aldrich, Germany), potassium hydroxide (KOH) (Merck), 1,4-dioxane (99%) (Merck), acryloyl chloride stabilized with 400 ppm phenothiazines (96%, Thermofisher-scientific), magnesium sulphate anhydrous (SRL), trimethylamines (≥99.5%, Merck), SDS (sodium dodecyl sulphate) (90%, Merck), hexadecane anhydrous (99%, Sigma Aldrich, Germany), divinylbenzene (DVB) (Alfa-Aesar), ethyl acetate, chloroform, and 1,4-dioxane (99% pure, Merck). 2,2-Azobisisobutyronitrile (AIBN) (98%, SRL), dichloromethane, n-hexane, hydrochloric acid, diethyl ether, dichloromethane, sodium bisulphate, sodium chloride, toluene, DMSO-d6, CdCl3, phosphate buffered saline (PBS) (pH 7.4), isopropanol, phosphotungstic acid, DNS (sodium chloride and dextrose injection IP (0.9% and 5% w/v)) (Jedux), DMEM Cell Clone, fetal bovine serum (Gibco), penicillin–streptomycin cocktail and gentamicin (Himedia), methyl thiazole tetrazolium (MTT), DMSO (Merck), DABCO (Sigma Aldrich), rhodamine B (Sigma), Hoechst 33258 (Cyamann), Nile red (SRL), Quanti blue (Invivogen), Griess reagent (Biotium) and DAPI (Invitrogen). These chemicals were used as received commercially. All samples were prepared using ultrapure water (18.2 MΩ cm) (Sisco Research Laboratories Pvt. Ltd, Mumbai, India).2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were used to recrystallise the solid N-acryloyl glycine monomer (NAG). The yield of the product was obtained to be 63.75%.
:50) repeatedly (5–7 times) through centrifugation (15000 rpm at 4 °C) with a 45-minute cycle followed by lyophilization. Then, the solid white sample was stored for further studies.
:1) were used to recrystallise the solid N-acryloyl glycine monomer (NAG). The yield of the product was obtained to be 63.75%.
:50) repeatedly (5–7 times) through centrifugation (15000 rpm at 4 °C) with a 45-minute cycle followed by lyophilization. Then, the solid white sample was stored for further studies.
2.3 Characterization of the GlyNPs
The chemical functionality of the monomer and the polymers was characterized through FTIR (Nicolet iS5, 4 Thermo Fisher Scientific Inc., USA) (with KBr pellet), 1H-NMR and 13C-NMR (Model: AVH D 500 AVANCE III HD 500 MHz One Bay NMR Spectrometer, BRUKER BioSpin International AG spectrometer, solvent DMSO-d6).The morphology and size of the polymer particles was checked through HRTEM (Tecnai G2 20 TWIN, FEI Company of USA (S.E.A.) PTE, LTD), AFM (NTEGRA Prima, NT-MDT Service & Logistics Ltd) and dynamic light scattering (DLS; Nano-ZS ZEN3600, Malvern, UK) at 25 °C. The zeta potential (ζ) was calculated with the comprehensive option attached to the DLS.
2.4 Biological study of GlyNPs
All cell-based experiments were performed using RAW264.7 (monocyte macrophages) cell lines, and neural crest-originated catecholamine PC12 cells (pheochromocytoma) were obtained from the National Centre for Cell Science, Pune, India. All cells were cultured using DMEM-high glucose media (4.5 g L−1) containing sodium pyruvate and sodium bicarbonate (Sigma-Aldrich), supplemented with FBS (10 vol%) and penicillin and streptomycin cocktail (1 vol%), in a CO2 incubator at 37 °C with 5% CO2. Cell lines were cultured in tissue culture T-75 flasks (gentix), and the culture medium was changed every alternate day.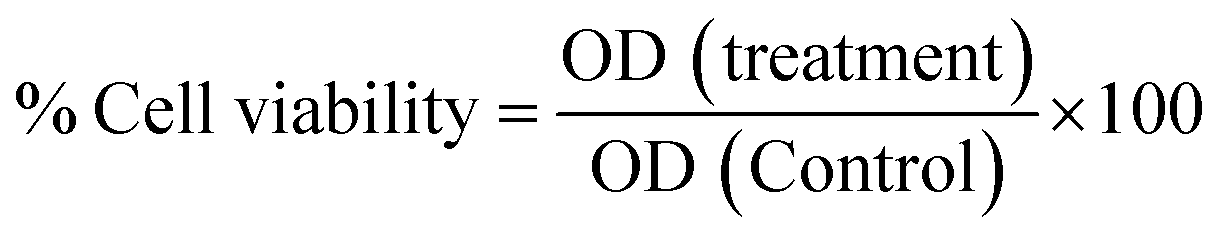 | (1) |
All the experiments were conducted in triplicate and represented with statistical errors.
2.5 In silico studies
To investigate the potential uses of GlyNPs, several in silico studies were carried out. First, network pharmacology predictions applying various important molecular targets associated with anti-inflammatory capacity were identified. Next, molecular docking was performed to examine the interactions between the GlyNPs and the identified target proteins. Then, using the computational results, we illuminated the potential mechanisms of action, therapeutic efficacy, and specificity of the GlyNPs for biological applications. All the software tools used for computational purposes are enlisted (in Table 1 with web references).| Marker | Forward (5′ to 3′) | Reverse primer (3′ to 5′) | Amplicon size |
|---|---|---|---|
| IFN-γ | GAAAGGATGCATTCATG | AAAATTCAAAATAGTGCT | 205 |
| IL-6 | ACAAGAAAGACAAGCCAG | GTCTATTCGACCTCAGTGT | 145 |
| TNF-α | CCACAAGCAGGAATGAG | AAAGACCTCCCTCTACAC | 140 |
| IL-10 | AAAATAAGAGCAAGGC | GTATGCAAATTCATTG | 82 |
| GAPDH | TGGGAGTTGCTGTTGAAGCG | GGTGGAAGAACTACAGT | 103 |
(a) Protein preparation. For docking, the following receptors were selected: COX2 (pdb id 6COX), NFκβ (pdb id ILE5), IL-6 (pdb id 1ALU), IL-1β (pdb id 5I1B), TNF-α (pdb id 2AZ5), iNOS (pdb id 4NOS), TLR-4 (pdb id 2Z62), MAPK-14 (pdb id 4F9Y), MAPK-8 (pdb id 4G1W), and STAT-1 (pdb id 3WWT). The 3D structures of the chosen receptors were retrieved from the RCSB protein Data Bank (https://www.rcsb.org/). Subsequently, water molecules were removed using Autodock 4.2.6, adding polar hydrogen and Kollman's charges. The resulting proteins were then saved in PDBQT format.
(b) Ligand preparation. Molecular docking studies were conducted using AutoDock 4.2 software. For this study, the ligands selected were N-acryloyl glycine. Their 3D structures were in ‘.pdf’ format in PubChem (Table S1, ESI†). Furthermore, ‘pdbqt’ files for the ligands were generated by Open Babel, followed by Grid Generation and Docking Analysis. Following the preparation of ligands and proteins, the molecular docking was conducted using Autodock (4.2.6 software). A grid with dimensions less than 1 Å specific to X, Y, and Z coordinates was established to encompass the protein, as done in the case of blind docking. This grid was centred around the protein to facilitate the most feasible docking conformations. Subsequently, the grid file was saved as a (.gpf) file. Then autogrid was initiated. Then, docking calculations were performed using the Lamarckian genetic algorithm (GA), fixing for 30 runs. The files were saved as a ‘.pdf’; final docking results were obtained after running the auto dock. dlg’ file format showed H-bonding, binding energy and inhibition constants. The best docking conformations of the ligands with the receptors were selected based on the estimated binding energy (Eg) and inhibition constant (Ki). The protein–ligand complex with the lowest binding energy was extracted in the PDBQT format. Furthermore, the interactions between ligands and proteins of these complexes were extracted using LigPlot+, Autodock and ChimeraX in 2D and 3D.
2.6 Immunological studies
2.7 Animal model of pristane-induced RA
In vivo studies were performed according to the Institute Animal Ethical Committee guidelines. A pristane-induced RA model was developed for the study.29 A total of 9 Wistar rats each of weight 185 ± 5 gm were selected by a randomization method for the experiment. All animals were acclimated to the standard animal house environment for 7 days before initiation of the experiments. After the acclimatization, the animals were randomized into three groups based on body weight in individual cages. Herein, three groups of rats were employed. The first group of rats were treated with saline and referred to as the sham group. The second and third groups of rats were administered with 100 μL pristane (MP Biomedicals) through intradermal injection in each paw to induce arthritis. The rats of the second group were kept without treatment, and the rats of the third group were referred to as the treatment group. GlyNPs treatment was performed after full arthritis induction, which took 15 days to develop. The results obtained from the treatment group of rats were compared with the sham and positive control groups. The GlyNPs dose was decided as 2 mg kg−1 of rat (effectively 360 ± 10 μg GlyNPs) against inflammation (RA).Biosafety/ethical permission. All the in vivo studies were conducted according to the Institutional Animal Ethical Committee (IAEC) of Indian Institute of Technology (IIT), Banaras Hindu University, Varanasi guidelines (Registration no. 2123/GO/Re/S/21/CPCSEA) and approval (IAEC Approval No. IIT(BHU)/IAEC/2024/II/028, dated 13/09/2024).
Statistical analysis. The statistical analyses used Origin 2021, Student's t-test and one-way ANOVA. The data have been presented as mean ± standard deviation (S.D.). Statistical significance was calculated at a p-value of 0.05. Data was marked with ‘*’ for p < 0.05, ‘**’ for p < 0.01 and ‘***’ for p < 0.001.
3. Results
Synthesis and characterization of glycine monomer and polymeric nanocapsules
As mentioned in the Method section, the GlyNPs have been synthesized. In the first step, glycine and acryloyl chloride were synthesized by mini emulsion polymerization, followed by a modified method to the desired structure of the GlyNPs.25,30 In the emulsion method, SDS, hexadecane, AIBN, and DVB are used as stabilizers, co-stabilizers of the emulsion, free radical initiators, and cross-linking agents, respectively. SDS and hexadecane were used to prevent the coalescence of the droplets and GlyNPs.The chemical functionalities of the ‘mers’ and polymer particles were characterized through 1H-NMR, 13C-NMR (400 MHz, and solvent DMSO-d6) and FTIR. The characteristic bands for glycine monomers are assigned as follows. 1H-NMR (δ) (Fig S1, ESI†): 8.37 ppm (sec. amide), 6.29 ppm (gem, –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) N), 6.07 ppm (cis, –C(O)N), 6.13 ppm (trans, –C(O)N), 3.45 ppm (2H, d); 13C NMR (δ) (Fig. S2, ESI†): 171.66 ppm (–COOH), 165.79 ppm (amide –CO), 131.54 ppm, 131.31 ppm and 126.66 ppm (alkane adjacent to amide), and 41.37–39.05 ppm (aliphatic –CH). As shown in Fig. S1 and S2 (ESI†), the distinct bands assigned for glycine polymer are as follows. 1H-NMR (δ) (Fig. S3, ESI†): 8.21 ppm (amide, 1H), 4.12–4.00 ppm (–CH2 adjacent to amide) and 3.92–2.47 ppm (alkane –CH); 13C NMR (δ) (Fig. S4, ESI†): (40.04–39.04 ppm) splitting of 2° alkane. For the glycine monomer, the characteristic FTIR bands obtained are at 3342 cm−1 for the secondary amine (–NH–) and overlapping broadband for hydroxyl (–OH), 2930 cm−1 for aliphatic stretching (–CH–), 2535 cm−1 for the carbon–carbon double bond in conjugation with carbonyl (C–CO–), 1719 cm−1 for the carbonyl (–CO) group attached with a hydroxyl group, 1541 cm−1 for the (–CN–) amide band, 1450 cm−1 for the carbon–hydrogen aliphatic band (–C–H), and 1280 cm−1 for the amide band (–CN–). GlyNPs show characteristic bands at 3342 cm−1 for secondary amine (–NH–) and (–OH), 2930 cm−1 for aliphatic alkane stretching (–CH–, –CH2), 1701 cm−1 for the carbonyl of the acid (–COOH) and 1602 cm−1 for the amide group (–CN) (Fig. 1(a)).
O) N), 6.07 ppm (cis, –C(O)N), 6.13 ppm (trans, –C(O)N), 3.45 ppm (2H, d); 13C NMR (δ) (Fig. S2, ESI†): 171.66 ppm (–COOH), 165.79 ppm (amide –CO), 131.54 ppm, 131.31 ppm and 126.66 ppm (alkane adjacent to amide), and 41.37–39.05 ppm (aliphatic –CH). As shown in Fig. S1 and S2 (ESI†), the distinct bands assigned for glycine polymer are as follows. 1H-NMR (δ) (Fig. S3, ESI†): 8.21 ppm (amide, 1H), 4.12–4.00 ppm (–CH2 adjacent to amide) and 3.92–2.47 ppm (alkane –CH); 13C NMR (δ) (Fig. S4, ESI†): (40.04–39.04 ppm) splitting of 2° alkane. For the glycine monomer, the characteristic FTIR bands obtained are at 3342 cm−1 for the secondary amine (–NH–) and overlapping broadband for hydroxyl (–OH), 2930 cm−1 for aliphatic stretching (–CH–), 2535 cm−1 for the carbon–carbon double bond in conjugation with carbonyl (C–CO–), 1719 cm−1 for the carbonyl (–CO) group attached with a hydroxyl group, 1541 cm−1 for the (–CN–) amide band, 1450 cm−1 for the carbon–hydrogen aliphatic band (–C–H), and 1280 cm−1 for the amide band (–CN–). GlyNPs show characteristic bands at 3342 cm−1 for secondary amine (–NH–) and (–OH), 2930 cm−1 for aliphatic alkane stretching (–CH–, –CH2), 1701 cm−1 for the carbonyl of the acid (–COOH) and 1602 cm−1 for the amide group (–CN) (Fig. 1(a)).
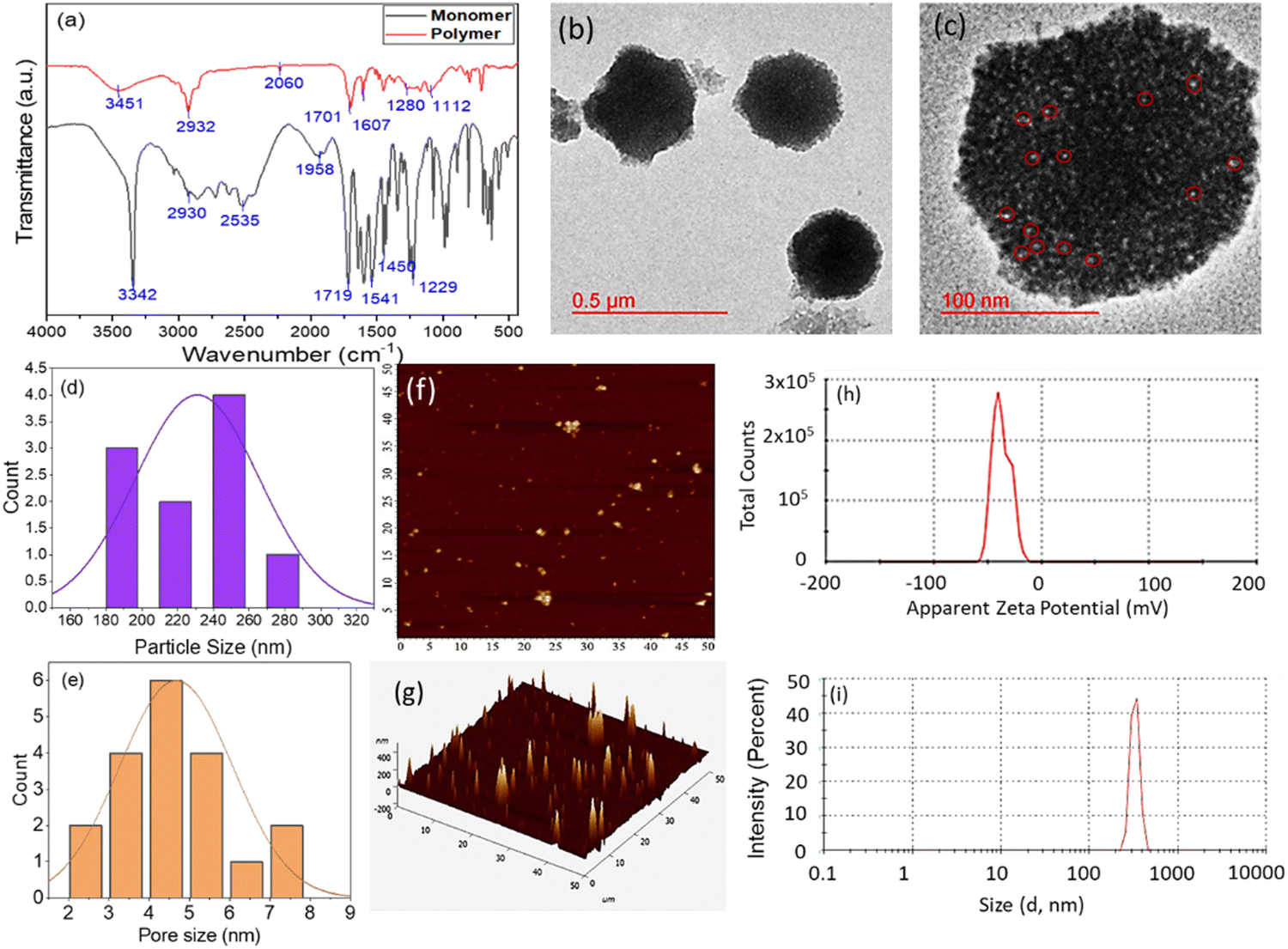 | ||
| Fig. 1 (a) FTIR spectra showing the absorption band for the Gly monomers and polymers, (b) and (c) HRTEM images of GlyNPs at 0.5 μm and at 100 nm scale; (d) particle size distribution calculated from HRTEM images taking 100 particles, (e) pore size distribution of the particles calculated from (c), (f) AFM topography 2D image and (g) 3D AFM image; (h) plot for zeta potential results obtained from DLS and (i) hydrodynamic diameter of the polymeric particles calculated from a Zetasizer. Particles are of size 180–20 nm. The pore size obtained to be avg. 4–5 nm in diameter (highlighted with a red circle). (c) Zeta potential obtained to be −35.6 mV. Scale for the AFM image is in μm. | ||
The morphology and particle size of the polymer particles were studied through HRTEM (Fig. 1(b) and (c)) and AFM (Fig. 1(f) and (g)). HRTEM results confirm that the sizes of the GlyNPs are in the range of 180–280 nm in diameter (Fig. 1(d)). Most of the particles are spherical in nature and many of them are core–shell in nature. The light core and darker shell structure are also observed for the hollow core–shell structure of the polymer particles with a core size of 150 ± 5 nm in diameter and a shell thickness of 65 ± 5 nm (Fig. S5, ESI†). It is also observed that the particles are porous in nature. The white spots highlighted with red circles of the particles are pores (HRTEM image, Fig. 1(c)). The average pore diameter is calculated to be 4–5 nm (Fig. 1(e)). Furthermore, to confirm the morphology of the particles, non-contact mode AFM was conducted and the particle sizes were measured from topographic 2D images and 3D images, and the particle size is calculated to be 137–150 nm in diameter (Fig. 1(f) and (g)), which is matching well with the results obtained from HRTEM. Thus, both the microscopy results revealed that the particles are monodispersed in nature. The monodispersed nature of the particles was also confirmed through DLS (particle size ∼324 nm) (with PDI ∼1.0) (Fig. 1(i)). This larger particle size value from DLS is obtained due to the hydrodynamic diameter. To confirm the colloidal stability of the polymeric particles, the zeta potential (ζ) was measured, and the value obtained was −35.6 mV, comprising the high colloidal stability of the polymeric particles (Fig. 1(h)).
Cell viability
The cell viability of the nanoparticles is crucial for their safe therapeutic applications. Polymeric nanoparticles have unique physicochemical properties that can lead to varying degrees of cytotoxicity depending on their size, shape, and surface chemistry. Therefore, the cell viability of the nanoparticles on RAW 264.7 macrophages (Fig. 2(a)) and PC-12 (Fig. 2(b)) is performed using an MTT assay, as mentioned in the Method section. RAW 264.7 cells, a murine macrophage cell line, are a useful model for evaluating the interaction of nanoparticles with immune cells. For inflammation and immunity, RAW 264.7 is the primary cell line for understanding the cytotoxic effects, and it can provide valuable insights into the biocompatibility and safety of NPs.31 To support the cell friendliness, viability was further checked on PC-12 cells, and it is a widely used model cell line for studying the effects of nanoparticles on neuronal cells.31 It is observed that with increasing the concentration from 10 μg mL−1 to higher, the cell viability for both RAW 264.7 and PC-12 cells varied. Cells are viable even at 100 μg mL−1 concentration of GlyNPs. We can assume that after 24 h of treatment, the % of viability varied due to experiencing contact inhibition by cells in the respective confined space. However, for both the cell lines, GlyNPs are taken in different concentrations such as 10, 20, 40, 80, 100, 125, 150, 200 and 250 μg mL−1, and the % viability values relative to the control groups are found to be 91.4 ± 2.9, 90.9 ± 2.3, 86.2 ± 1.8, 87.4 ± 0.06, 79.6 ± 5.9, 80.2 ± 1.3, 71.4 ± 2.5, 66.01 ± 2.4 and 65.3 ± 4.8, respectively for RAW 264.7 macrophages. For PC-12 cells, the % viability varied from 136 ± 5.8, 134.72 ± 5.98, 119.38 ± 5.44, 111.57 ± 5.33, 90.09 ± 4, 88.71 ± 4.38, 85.97 ± 4.22, 82.52 ± 4.11, respectively. At lower concentrations, the observed higher viability of PC-12 cells compared to the control is attributed to a potential stimulatory or protective effect of the GlyNPs. The GlyNPs at lower concentrations promote cellular activity, such as enhanced metabolic processes leading to increased viability as well as enhance the cell proliferation. | ||
| Fig. 2 The cell viability of GlyNPs with various concentrations. (a) Cell viability results on RAW 264.7 macrophages and (b) viability on the PC-12 cell line. Data was marked with ‘*’ for p < 0.05, ‘**’ for p < 0.01 and ‘***’ for p < 0.001. | ||
However, at higher concentrations of GlyNPs, the compound exceeds the threshold for such stimulatory effects and exhibits excellent cytotoxic effects. These results confirm that the Gly NPs are viable and suitable for therapeutic applications.
Cellular uptake of GlyNPs
The cellular uptake of NPs is a fundamental aspect of their functionality in therapeutic applications with target specificity. It influences the target efficiency of the therapeutic and safety profiles. To check the cellular uptake of GlyNPs, it was stained with Nile red and incubated with RAW 264.7 macrophages for 24 h with a non-toxic dose of 50 μg mL−1 followed by cell fixing and staining with DAPI to detect the nucleus. Then the confocal microscopy images of Nile red-GlyNPs were acquired (Fig. 3). The DAPI-stained cell nucleus was identified using the blue channel (λexc = 405 nm) (Fig. 3(a1)–(a3)) and fluorescence emission for the GlyNPs was observed using the red channel (λexc = 546 nm). The cellular uptake was qualitatively studied by acquiring fluorescence images after 24 h of incubation with Nile red-GlyNPs. From Fig. 3, it is clearly observed that GlyNPs were taken up inside the cell in the cytoplasm within 24 h of incubation. However, a considerable amount of uptake in the nucleus is less evident (Fig. S6, ESI†). This selective internalization suggested that the GlyNPs will further help to deliver biomolecules or drugs inside the cells. As GlyNPs are showing intracellular uptake, it is allowed to interact directly with the internal cellular machinery, which can be leveraged for therapeutic purposes. The uptake of GlyNPs by the immune cells (RAW 264.7) directs us to study their immune behaviour as they may change signalling pathways leading to the reduction of inflammation.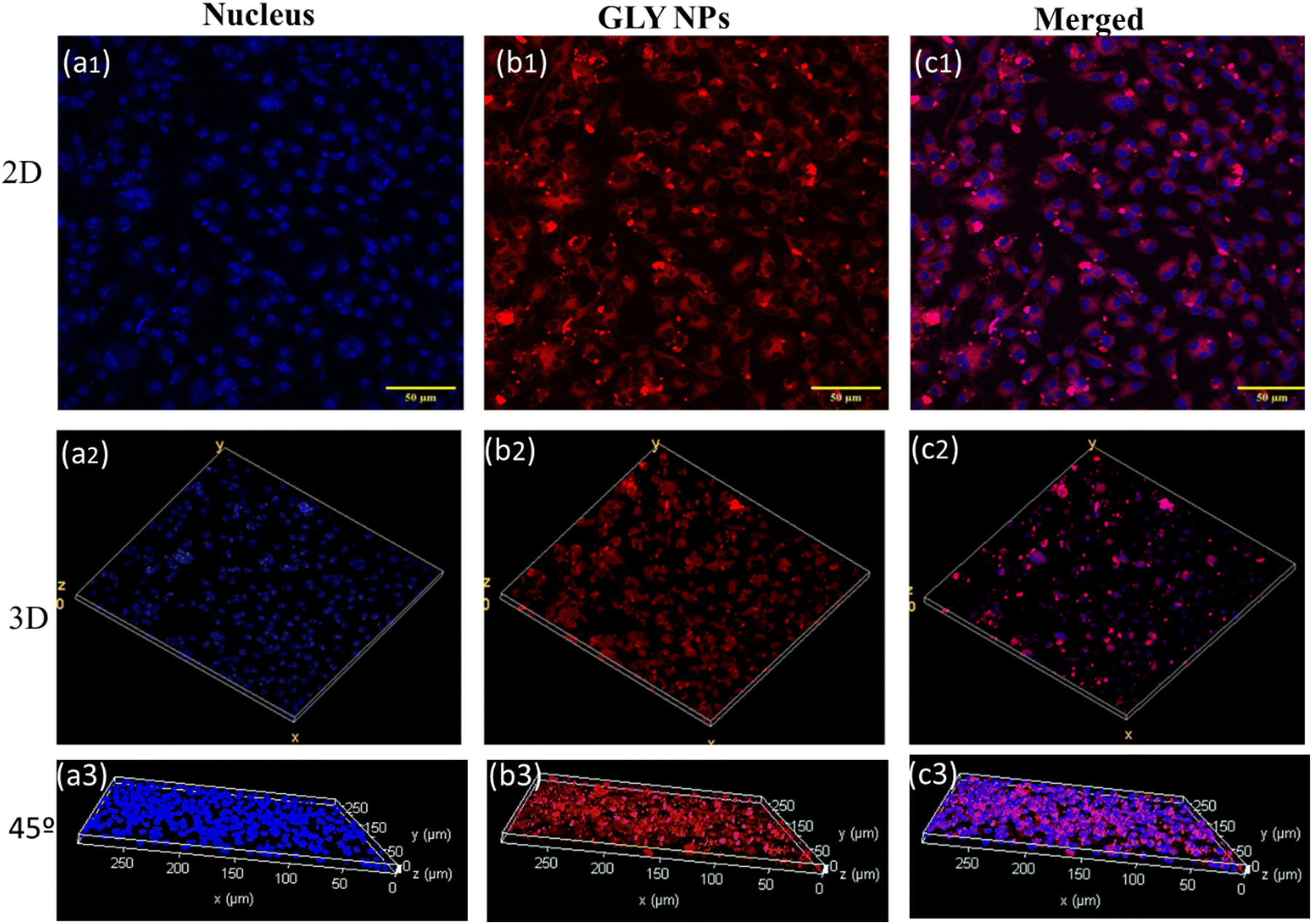 | ||
| Fig. 3 The cellular uptake of GlyNPs, studied through confocal microscopy, (a1) 2D representation of the nucleus that is stained with DAPI, (a2) 3D representation of a1, (a3) 45° representation of cellular uptake where the Z axis shows the nucleus in 3D of the particles. (b1) 2D representation of GlyNPs stained with Nile red, (b2) 3D representation of b1, (b3) 45° representation of cellular uptake, where the Z axis shows internalisation of particles. (c1) 2D representation of GlyNPs stained with Nile red and the nucleus in blue colour, (c2) 3D representation of c1 and (c3) 45° representation of cellular uptake where the Z axis shows the internalisation of the particles. | ||
In silico study of GlyNPs
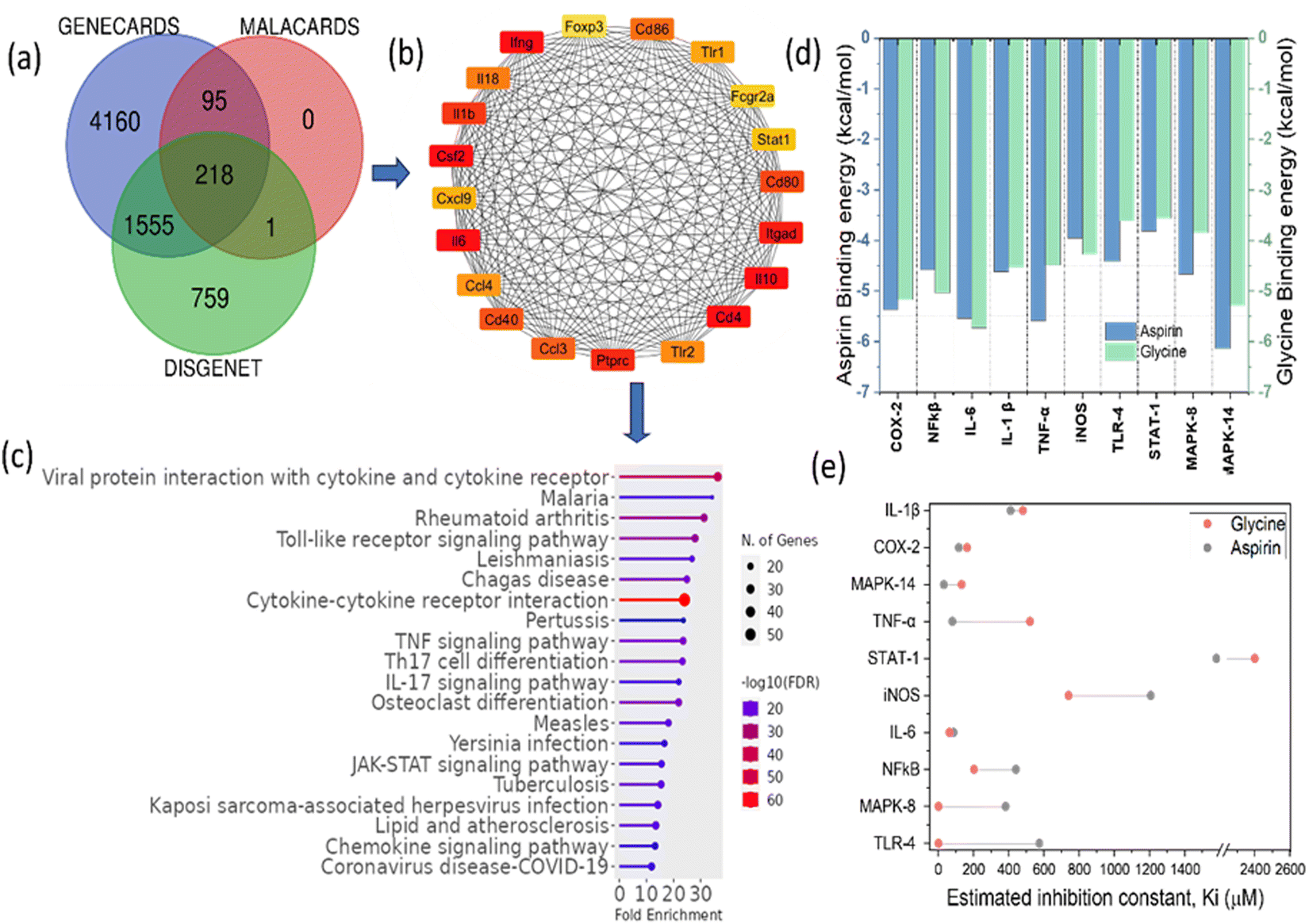 | ||
| Fig. 4 Target network pharmacology and in silico investigation of GlyNPs. (a) Identification and prediction of common targeted genes and proteins utilizing three different databases represented by Venn diagram, (b) protein–protein interaction (PPI) network of the targeted proteins to understand the relationships between the targets (the red to yellow colour box of each target shows the intensity of higher to lower interaction) and (c) Shiny Go (lollipop) plot results obtained for the targeted genes involved in various biological processes including RA and their fold enrichment, (d) binding energy results obtained from the docking for different genes with glycine and aspirin associated to RA and inflammation, and (e) a comparison with the inhibition constant (Ki) of aspirin and GlyNPs in terms of estimation inhibition obtained from molecular docking for different target genes resulting from network pharmacology. | ||
Molecular docking
Molecular docking is a critical computational technique for determining the ligand's optimal orientation and target protein after binding to form a stable complex. The knowledge of the binding modes has been utilized to calculate the binding affinity of the two molecules.33 The docking approach has been utilized to rapidly screen vast databases of potential therapeutic compounds in silico to determine the favourable domains that are most likely to bind to proteins of interest.34,35All the molecular docking studies were performed with specific inflammation-targeted protein receptors based on the network pharmacology analysis and previously reported anti-inflammatory activities. The blind docking results are obtained for all the inflammation-based targeted proteins such as COX2, NFκβ, IL-6, IL-1 β, TNF-α, iNOS, TLR-4, MAPK 14, MAPK 8, and STAT 1. For all the obtained results, the binding energy and interactions are shown in more detail in Tables S3–S12 (ESI†). Here, we have taken G1 (mer), G2 (dimer), G3 (trimer) and G4 (tetramer) of N-acryloyl glycine. We have used well-known anti-inflammatory drugs aspirin and diclofenac as controls.
For primary inflammatory receptor COX2 (pdb id 6COX), the binding energy values with aspirin and diclofenac are calculated to be −5.37 and −5.11 kcal mol−1, respectively. Aspirin forms H-bonds with the HIS90 residue and diclofenac forms H-bonds with HIS90 and ARG513. Meanwhile, for G1, G2, G3, and G4, the binding energy values obtained were −5.17, −4.09, −3.39 and −3.54 kcal mol−1, respectively. The H-bonds are majorly formed with G1 at ASN87, HIS90 and ARG513 residues, for G2 at HIS90 and TYR91, for G3 at HIS90, ASN87 and TYR91, and for G4 at HIS90 and TYR91 (see Table S3, ESI†). For primary inflammatory receptor NF-κβ, the binding energy values with aspirin and diclofenac are calculated to be −4.58 and −4.81 kcal mol−1, respectively. Aspirin forms H-bonds with ARG54 and LYS241 residues and diclofenac forms H-bonds with LYS145 only. Whereas, for G1, G2, G3 and G4, the binding energy values were obtained to be −4.11, −5.04, −5.67 and −4.59 kcal mol−1, respectively. The H-bonds are majorly formed with G1 at LYS114 and ASN136 residues, for G2 at LYS145 and LYS203, for G3 at LYS145 and LYS2023, and for G4 at LYS144 and LYS203 (see Table S4, ESI†).
For primary inflammatory receptor IL-6, the binding energy values with aspirin and diclofenac are calculated to be −5.54 and −6.74 kcal mol−1, respectively. Aspirin forms H-bonds with LYS27 and ARG30 residues and diclofenac forms H-bonds with LYS27 only. Whereas, for G1, G2, G3 and G4, the binding energy values obtained were −4.88, −5.73, −4.55 and −4.80 kcal mol−1, respectively. The H-bonds majorly formed with G1 at LYS27 and ARG30 residues similar to the control drug aspirin. For G2, the H-bonds majorly formed at ASP26 and LYS27 and ARG30, for G3 at LYS27 and ARG30, and for G4 at SER22, ASP26 and ARG182 (see Table S5, ESI†).
Similarly, it is found that IL-1β is also involved in the RA and inflammation. From the docking study, the binding energy values and the H-bond formation with different residues have been investigated and the results are shown in Table S6 (ESI†). The binding energy values with the aspirin and diclofenac are calculated to be −4.62 and −4.95 kcal mol−1, respectively. Aspirin forms H-bonds with VAL3 and LYS93/94 residues and diclofenac forms H-bonds with LYS65 only. For G1, G2, G3 and G4, the binding energy values obtained were −4.53, −4.04, −3.07 and −2.10 kcal mol−1, respectively. The H-bonds are majorly formed with G1 at ARG4, PHE46 and LYS93 residues. For G2, H-bonds prominently formed with ARG4, GLY49 and LYS93; for G3 with VAL3, GLN48 and LYS93; and for G4 at VAL3, GLN48 and LYS93/94 (see Table S6, ESI†). From these results, it is clearly evident that the obtained results are comparable to the aspirin and diclofenac.
The role of TNF-α in inflammation is well-known. The docking study has also been performed for TNF-α, and the results are shown in Table S6 (ESI†) in detail. The binding energy values with aspirin and diclofenac are calculated to be −5.59 and −4.89 kcal mol−1, respectively. Aspirin forms H-bonds with VAL74 and LYS112 residues and diclofenac forms H-bonds with PRO70 and LYS112. For G1, G2, G3 and G4, the binding energy values were obtained to be −4.22, −4.48, −2.69 and −3.35 kcal mol−1, respectively. The H-bonds majorly formed with G1 at PRO70 and LYS112 residues. For G2, the H-bonds majorly formed at LYS112 only, for G3 at CYS69, and for G4 at PRO70 and ARG103 (see Table S7, ESI†).
Inducible nitric oxide synthase (iNOS) is also a major mediator of inflammation. The docking study for iNOS was performed and the results are shown in Table S8 (ESI†) in detail. The binding energy values with aspirin and diclofenac are calculated to be −3.95 and −4.42 kcal mol−1, respectively. Aspirin forms H-bonds with HIS84 and diclofenac with the THR95 residue. For G1, G2, G3 and G4, the binding energy values were obtained to be −4.27, −6.00, −4.57 and −2.50 kcal mol−1, respectively. The H-bonds predominantly formed with G1 at HIS84, for G2 at HIS 84 and ARG86, for G3 at ARG86 and LYS103, and for G4 at ARG86, THR95 and ARG452 (see Table S8, ESI†). Therefore, the HIS84 residue interacts with both the drugs and our GlyNPs.
Toll-like receptor (TLR4) possesses a central role in innate immune signalling and inflammatory responses by modulating the activity of transcription factors. A docking study has been performed and the results are shown in Table S9 (ESI†). The binding energy values with the aspirin and diclofenac are calculated to be −4.42 and −4.7 kcal mol−1, respectively. Aspirin forms H-bonds with the LYS47 and ILE48 residues; for diclofenac, H-bonds formed only with LYS47. For G1, G2, G3 and G4, the binding energy values are calculated to be −3.88, −3.62, −3.26 and −2.74 kcal mol−1, respectively. The H-bonds majorly formed for G1 at GLN281 and LYS254; for G2 at LYS229; for G3 at SER251 and LYS 282, and for G4 at LYS229/282 (see Table S9, ESI†).
Mitogen-activated protein kinase-14 (MAPK-14) (pdb id 4G1W) has an important role in the production of various pro-inflammatory cytokines and is an immune responsive component.36 The docking results are shown in Table S10 (ESI†). The binding energy values with aspirin and diclofenac are calculated to be −6.14 and −6.95 kcal mol−1, respectively. Aspirin forms H-bonds with LYS165 and diclofenac with the HIS126 residue. For G1, G2, G3 and G4, the binding energy values obtained were −4.56, −5.29, −4.44 and −4.64 kcal mol−1, respectively. The H-bonds majorly formed for G1 at GLU160, G2 at HIS126, G3 at HIS126, and G4 at HIS126 sites (see Table S10, ESI†).
The final docking was performed with mitogen-activated protein kinase-8 (MAPK-8) (pdb id 4G1W), which is involved in the production of inflammatory cytokines. The docking results are shown in Table S11 (ESI†). The binding energy values with aspirin and diclofenac are calculated to be −4.66 and −4.12 kcal mol−1, respectively. Aspirin forms H-bonds with LYS220 and diclofenac with LYS252. For G1, G2, G3 and G4, the binding energy values obtained are −3.85, −3.08, −2.85 and −2.49 kcal mol−1, respectively. The H-bonds majorly formed with G1 at LYS203 and GLU204, and for G2, G3, and G4, H-bonds majorly formed with TYR202 and LYS203 residue sites (see Table S11, ESI†).
STAT1 is a key inflammatory gene responsible for inflammation as it is resulted from our network pharmacology analysis. The docking results are shown in Table S12 (ESI†). The binding energy values with the aspirin and diclofenac are calculated to be −3.82 and −3.6 kcal mol−1, respectively. Aspirin forms H-bonds with TYR68 and diclofenac with ARG84. For G1, G2, G3 and G4, the binding energy values obtained are −3.33, −3.57, −2.36 and −1.16 kcal mol−1, respectively. The H-bonds majorly formed with G1 at TYR106 and LYS110 residues. G2 has formed H-bonds majorly at LYS110/114, for G3 at LYS40 and ARG113, and for G4 at ARG113 sites (see Table S12, ESI†).
Furthermore, the binding interactions with GlyNPs (polymer, nanolattices) with 10 different selected targets are shown in the form of 2D and 3D images (Fig. 5). From 2D images, the residues in the hydrogen bonds are represented by green dotted lines and hydrophobic interactions highlighted with red semicircles (Fig. 5). 3D images show the overall groove available and pockets that interact with the ligands. This signalling activation leads to the onset of various inflammatory diseases.
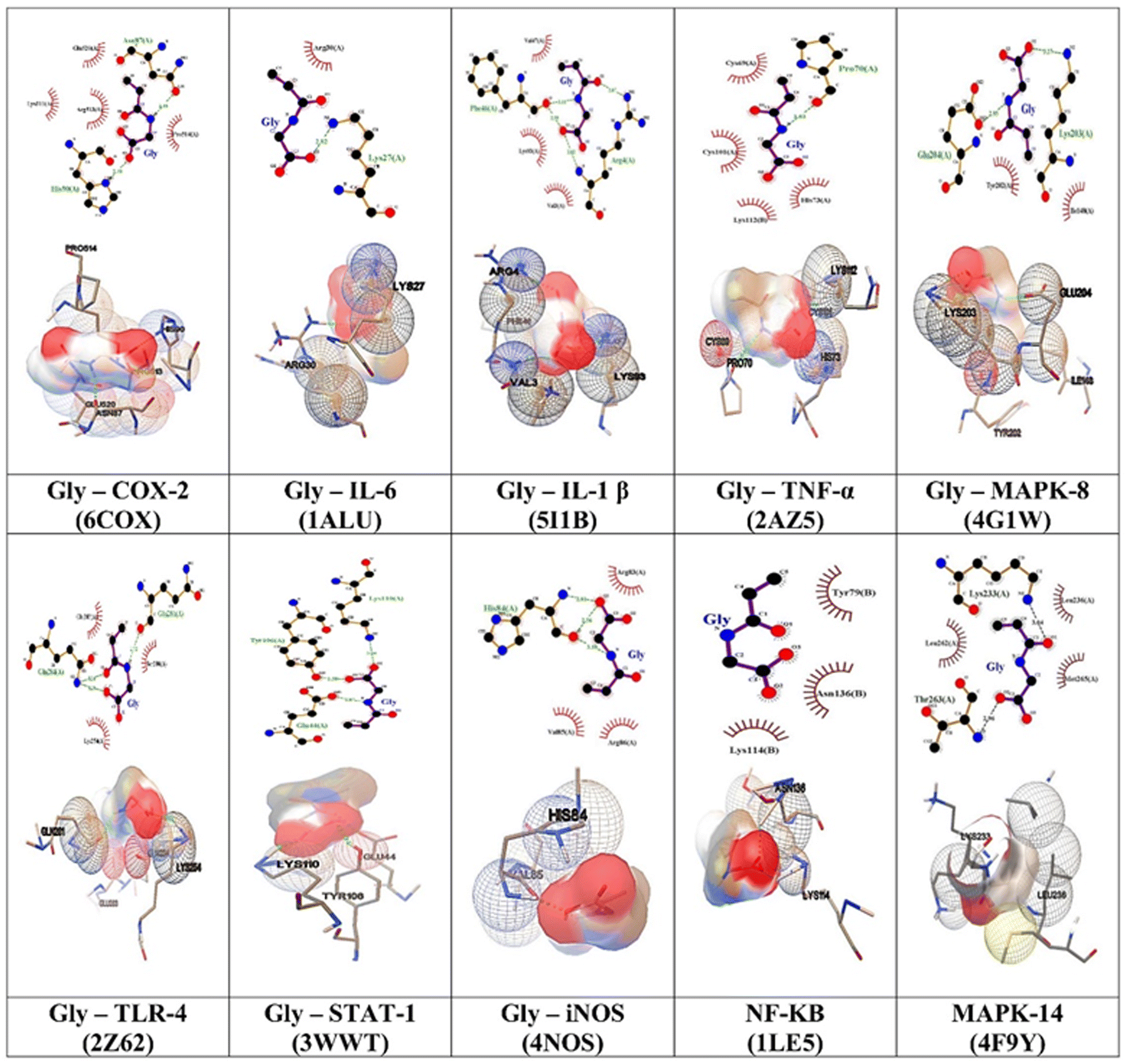 | ||
| Fig. 5 In silico docking study to find out the molecular protein interactions with monomeric units of GlyNPs (G1). The interactions between polymer domain (G1) and molecular protein targets of COX2, NF-κβ, IL-6, IL-1 β, TNF-α, iNOS, TLR-4, MAPK 14, MAPK 8, and STAT1 responsible for the inflammation are presented. The binding interactions are depicted in 2D and 3D formats. Within the 2D images, the presence of residues engaged in hydrogen bonding is illustrated by green dotted lines, while hydrophobic interactions are emphasised with red hemi circles. 3D images comprehensively depict available grooves and pockets interacting with the ligands. The ligplots (2D images) showing the interactions between G1 (polymeric unit, monomer) with interacted residues along with binding energy in kcal mol−1 and inhibition constants (Ki). In the images, the red balls represent oxygen, the black balls represent carbon, and the blue balls represent nitrogen atoms. | ||
Therefore, the network pharmacology results provided important information on the targets involved in the inflammation and the effective interactions of the polymeric particles synthesized in this work to reduce the inflammation. The binding residues and the inhibition constants (Ki) between G1, G2, G3, G4 and reference drug molecules with the targets are shown in (Tables S3–S12, ESI†) in detail.
Immunological responses of GlyNPs
A colorimetric-based assay for detecting NF-κβ activation in macrophages was used to address this issue. The results are highlighted by activating the inflammation pathway with LPS since it is a well-known inflammation inducer. The LPS treatment upregulates NF-κβ concerning the control condition. The inflammatory modulation via the NF-κβ pathway has been compared by the response of macrophages, where other than the control, all the treatment groups are prepared for inflamed conditions with the help of LPS. The treatment condition is tested with different concentrations of GlyNPs, such as 10, 20, 40, 100, and 150 μg mL−1. These concentrations were selected based on cell viability results obtained on macrophages, as shown in Fig. 2(a). The treatment with the GlyNPs shows suppression in inflammation with different concentrations (10, 20, 40, 100 and 150 μg mL−1) and the values were calculated to be 1.72 ± 0.27, 1.70 ± 0.20, 1.32 ± 0.34, 1.15 ± 0.17 and 1.14 ± 0.14, respectively. It can be noted that, for the LPS-treated sample, the value of NF-κβ level obtained was 1.90 ± 0.07 (for 0.2 μg mL−1), which is higher than the value obtained with GlyNPs. Therefore, in addition to NPs to LPS-treated cells, the NF-κβ levels varied, as shown in Fig. 6(a), and it is concentration dependent. These results indicated that the GlyNPs are an anti-inflammatory component (potential postbiotics). From Fig. 6(a), it is evident that the results represented here are statistically significant compared with the control for LPS, 10 μg mL−1 and 20 μg mL−1 of GlyNPs. To identify and confirm the underlying signalling pathways,28 GlyNPs are used to reduce inflammation, so further study is required. However, a decrease in NF-κβ levels or activity would generally lead to reduced inflammatory responses by reducing the production of inflammatory mediators and generating inflammatory T cells. Furthermore, it modulates immune regulation by altering Treg cell development.12
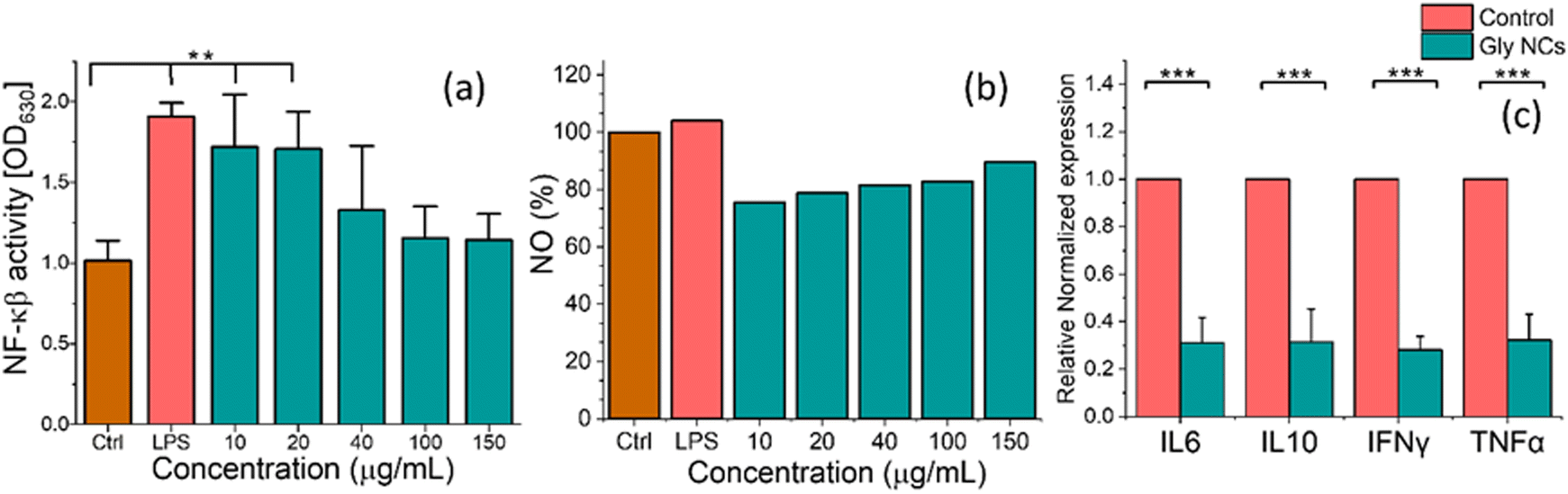 | ||
| Fig. 6 In vitro immune response of GlyNPs and anti-inflammation behaviour studied for control cells, LPS-treated cells and cells treated with varying concentrations of GlyNPs, (a) NF-κβ level study, (b) NO release and response study, and (c) study of the response of inflammatory markers (TNF-α, IFN-γ, IL-6 and IL-10) and the results are compared with the control (only cells, 1 × 105 cells per well) and cells treated with LPS. The standard deviation and the significance of the results were estimated according to the procedure mentioned in the Method section (** for p < 0.01 and *** for p < 0.001). | ||
NO production by GlyNPs
NO is a signalling molecule that plays a key role in the pathogenesis of inflammation and is involved in the generation of inflammatory disorders.38,39 Therefore, NO inhibitors can be an important therapeutic option in managing inflammatory diseases.40,41 To address the role of GlyNPs on NO production, macrophages (RAW 264.7 cells) were taken as model immune cells. Then, its anti-inflammatory effect under normal physiological conditions was determined with varying concentrations such as 10, 20, 40, 100 and 150 μg mL−1. The production of NO by RAW 264.7 cells (taking 1 × 105 number of cells per well) was measured at λmax = 540 nm (absorption band) with the help of Griess reagent, and the results are shown in Fig. 6(b). It is evident that with change in the doses of GlyNPs from 10, 20, 40, 100 and 150 μg mL−1, the extent of NO release changes as 75.5%, 78.78%, 81.59%, 82.83% and 89.49%, respectively. The release of NO for the control sample (only cells) is considered 100%. Whereas, when the cells were treated with 0.2 μg mL−1 LPS using the same number of cells, the NO release enhanced to 104.83%. Thus, it is clearly evident that the rate of release of NO can be modulated with the extent of doses of GlyNPs used. Mechanistically, NO is produced in physiological conditions by nitric oxide synthase,42 which converts into nitric oxide radicals (NO˙) and can modulate inflammatory diseases such as RA. Therefore, controlling the NO production level with varying dosages of GlyNPs could be an alternate strategy for treating inflammatory diseases such as rheumatoid arthritis (RA).Response of the immunological markers
Inflammation is induced in cells, and GlyNPs were used with different concentrations (10, 20, 40, 100, and 150 μg mL−1) to check their effectiveness. The suppression of NF-κβ and NO levels is noticed with the change in the concentration. Therefore, the effective dose is decided below 100 μg mL−1 for checking the status of the prominently involved immunological markers. Therefore, the inflammatory markers such as IL-6, IL-10, TNF-α and IFN-γ have been checked, and the results are shown in Fig. 6(c). RAW 264.7 cells (1 × 105 cells per well) were treated with a selected effective dose of GlyNPs (50 μg mL−1) for 24 h. Then with the help of triazole, the RNA was isolated. RNA concentration was normalized with nuclease-free water for cDNA synthesis, and RT-PCR experiments were performed as mentioned in the Experimental section. The RT-PCR results are shown in Fig. 6(c), with the genetic expression of inflammatory markers for IL-6, IL-10, TNF-α and IFN-γ. IL-6 plays an essential role in cell signalling and acute inflammation, and it is required for the rapid resolution of wound healing. From Fig. 6(c), it is evident that the IL-6 level is decreased from the control to GlyNP-treated sample. According to the heat map gene expression results, there is a 3.237 fold reduction in IL-6, comprising a quite excellent anti-inflammatory effect of the GlyNPs. It is also noted that IL-10 is an anti-inflammatory cytokine. It maintains the equilibrium for the immune response, allowing the clearance of infections. Therefore, the IL-10 expression levels were also calculated using the same approach. From the relative normalized heat map gene expression results (Fig. 6(c)), it is clearly evident that there is a 3.377 fold reduction in IL-10 level, which comprises an excellent anti-inflammatory effect of the GlyNPs. It can also be noted that the IL-10 level possesses an M2-like macrophage phenotype that is strongly suggested for particles’ (GlyNPs) anti-inflammatory properties.43 Their values have been calculated under similar experimental conditions to confirm the normalised gene expression levels of TNF-α and IFN-γ. From Fig. 6(c), it is evident that the TNF-α level decreased from control to GlyNP-treated cells by 2.991 fold; for IFN-γ, it decreased by 3.469 fold. This reduction in the expression of these cytokines can lead to the conclusion that the GlyNPs are not only anti-inflammatory in nature, but can also play an essential role in modulating the host's innate immunity. Therefore, these findings are crucial in reducing inflammation by modulating immune response, such as in RA.44 However, the ratio in the expression of these cytokine levels does not change significantly, showing a balanced reduction in the expression of the overall cytokines, and the results are statistically significant.In vivo anti-inflammatory efficiency of GlyNPs on an RA model
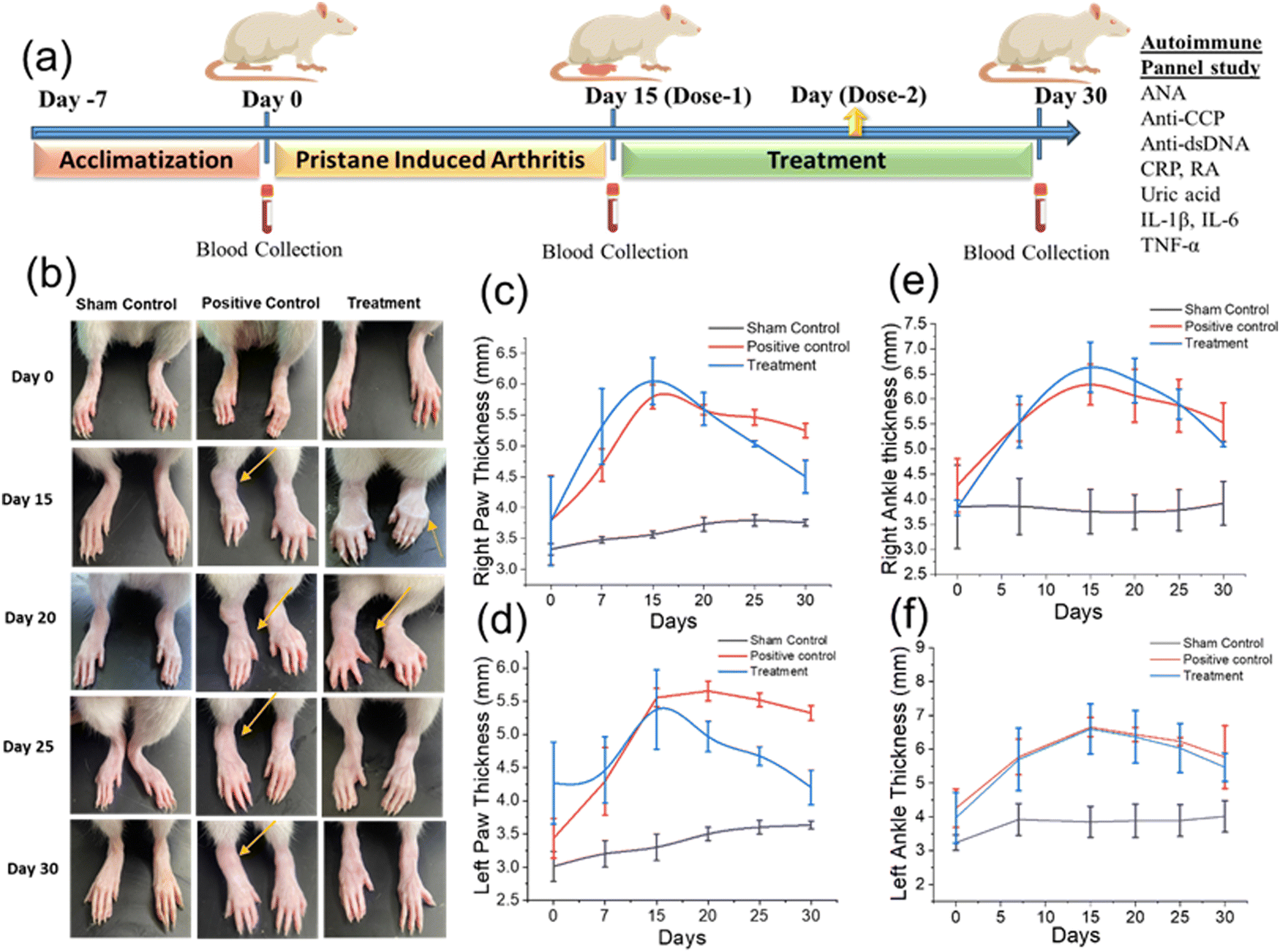 | ||
| Fig. 7 The study of in vivo anti-inflammation (RA) potential of GlyNPs. (a) Schematic plan for the entire in vivo experimental and treatment plan followed by an autoimmune panel study plan. (b) Camera photographic images of the sham control, positive control and GlyNP-treated rats showing the morphological changes in paw oedema at different days to study the anti-inflammation effects. (c) and (d) The change in thickness of the right and left paws, respectively, of the rats on different days. (e) and (f) Change in thickness of the right and left ankles, respectively, of the rats on different days. | ||
Autoimmune and inflammatory marker panel
As mentioned earlier, with the development of inflammation (RA), various biochemical parameters associated with autoimmunity can be changed. Therefore, studying these parameters is crucial for sham, positive control and GlyNP-treated rats. Herein, we have studied the levels of the RA factor, anti-cyclic citrullinated peptide (anti-CCP), antinuclear antibody (ANA), anti-double stranded DNA antibodies (anti-dsDNA), CRP, and uric acid levels on different days of treatment and the results are shown in the subsequent sections (see Fig. 8(a)–(f)). | ||
| Fig. 8 Study of the autoimmune and inflammatory marker levels. (a) RA level, (b) anti-CCP, (c) CRP level, (d) ANA level, (e) anti-dsDNA and (f) serum uric acid levels obtained on day 0, 15 and 30. Statistical significance was calculated at a p-value of 0.05. Data was marked with ‘*’ for p < 0.05, ‘**’ for p < 0.01 and ‘***’ for p < 0.001. | ||
Rheumatoid factor (RF)
RF is an antibody made by our immune system and facilitates the formation of immune complexes, which contribute to the inflammatory characteristic of RA.45 Therefore, it is very important to determine the RF levels of the rats used in the in vivo anti-inflammation study for this work. Hence, the blood serum samples were collected from GlyNP-treated, sham and positive control rats to perform the RF test on day 0 (before inducing pristane to develop RA), 15 (after RA developed and before treatment started) and 30 (after 15 days of post-treatment). Then, the RF values were calculated through the turbidimetric immunoassay on the above-specified days, and the results are shown in Fig. 8(a). The RF levels for all the sham control rats at day 0th, day 15th and day 30th are found to be 1.7 ± 0.26, 1.96 ± 0.20 and 2.03 ± 0.15 IU mL−1, respectively. For the positive control group of rats, the RF levels are found to be 1.94 ± 0.19, 5.56 ± 0.25 and 4.03 ± 0.35 IU mL−1 on day 0, day 15 and day 30, respectively. Whereas, for the treatment group of rats, the RF levels were calculated to be 2.1 ± 0.1 and 5.5 ± 0.4 IU mL−1 on day 0 (i.e. when no inflammation is there) and on day 15 (when inflammation was developed). However, after 15 days of post-treatment with GlyNPs (i.e., on the 30th day), the RF level decreased to 2.66 ± 0.77 IU mL−1. This RF level is almost in the order of the RF level of the sham control group of rats. Therefore, the effectiveness of GlyNPs at the onsite delivery shows wonderful results for the reduction of inflammation and RA. Furthermore, the clinical significance of the RF levels has been studied by the other diagnostic tests that have been reported in the subsequent sections. However, the reduction in the RF level for the positive control rats (rats with RA developed but not treated with GlyNPs) from 5.56 ± 0.25 and 4.03 ± 0.35 IU mL−1 for day 15 and day 30, respectively, signifies the autoimmune response of the body. In conclusion, GlyNPs are effective in reducing RF levels and inflammation.Anti-cyclic citrullinated peptide (anti-CCP)
High anti-CCP levels are usually common in severe RA. Furthermore, the increase in anti-CCP level is directly proportional to the tissue and joint damage. Therefore, the detection of anti-CCP levels is essential to determine the severity of inflammation as well as RA. Hence, the blood serum levels were collected from all the rats under experiments to perform the anti-CCP test on day 0 (before inducing pristane), 15 (after RA developed and before the treatment started) and 30 (15 days post-treatment). Then, the anti-CCP levels were calculated using the ELISA kit on the above-specified days, and the results are shown in Fig. 8(b).The anti-CCP levels for all the sham control rats at day 0, day 15 and day 30 are found to be 2.33 ± 0.20 AU mL−1, 2.53 ± 0.40 AU mL−1 and 2.24 ± 0.89 AU mL−1, respectively. For the positive control group of rats, the anti-CCP levels were found to be 2.06 ± 0.40, 5.19 ± 0.469 and 3.51 ± 0.17 IU mL−1 on day 0, day 15 and day 30, respectively. Whereas, for the treatment group of rats the anti-CCP levels were calculated to be 2.06 ± 0.40 and 5.19 ± 0.469 IU mL−1 on day 0 (i.e., when no inflammation is there) and day 15 (when inflammation was developed), respectively and after 15 days without GlyNP treatment (i.e., on 30th day), the anti-CCP level is decreased to 3.51 ± 0.17 IU mL−1, indicating the decrease in the severity of RA due to autoimmunity. However, after 15 days of post-treatment with GlyNPs (i.e., on the 30th day), the anti-CCP level decreased to 0.920 ± 0.31 IU mL−1. This reduction in the anti-CCP level gives strong evidence for the anti-inflammatory activity of GlyNPs.
Effect of GlyNPs on CRP levels
CRP is an acute-phase reactant pentameric protein synthesized by the liver and is directly related to the inflammation induced by IL-6. It plays a crucial role in the body's immune system by binding to pathogens and damaging cells.46 In RA, CRP serves as a biomarker for analysing its severity. The increase in CRP levels can increase joint inflammation and damage the tissues.47 Therefore, the detection of CRP levels is essential to find out the severity of inflammation as well as RA. To measure the CRP levels, the blood serum samples from all the rats under experiments were collected on day 0 (before inducing pristane), 15 (after RA developed and before the treatment started) and 30 (15 days post-treatment). The CRP levels were calculated using the ELISA kit, as mentioned earlier, and the results are shown in Fig. 8(c). It is clearly evident that for the sham control group of rats, the CRP level changes from 0.28 ± 0.02 to 0.38 ± 0.22 and 0.45 ± 0.1 mg L−1 from day 0 to 15 and 30, respectively. For the positive control group, the CRP level changes from 0.26 ± 0.041 mg L−1 (on day 0th) to 1.93 ± 0.37 mg L−1 (on day 15th), that is, after pristane injection (RA development). Further, it is noticed that for the positive control group of rats, the CRP level reached 2.3 ± 0.3 mg L−1 on the 30th day, demonstrating the increase in severity of inflammation of RA. Interestingly, after 15 days of immunization (post-treatment) with GlyNPs, the CRP level for the treatment group of rats came down from 1.99 ± 0.37 mg L−1 (from severe inflammation state, day 15 of RA) to 0.80 ± 0.45 mg L−1, which comprises strong evidence for the anti-inflammatory and anti-RA potential of GlyNPs.Antinuclear antibody (ANA) levels
Antinuclear antibody (ANA) testing is an important screening tool for autoimmune conditions. ANA binds DNA or associated nucleosome proteins.48 ANA correlates with elevated C-reactive protein (CRP) levels and increased joint inflammation in RA patients. Recent research highlights that ANA-positive patients with RA have different clinical characteristics than their ANA-negative patients. Significantly, a positive ANA level is associated with RA, which can be reduced with anti-rheumatic drugs.49 Hence, ANA levels were checked with the help of an ELISA test on day 0, day 15 (inflammation achieved), and day 30 (treatment), and the results are shown in Fig. 8(d).It is clearly evident that for all three groups of rats, the ANA levels range between 0.22 ± 0.15 AU mL−1 to 0.32 ± 0.09 AU mL−1 on day 0. On day 15, the ANA levels for the sham, positive control and treatment group of rats were found to be 0.38 ± 0.22 AU mL−1, 1.19 ± 0.18 AU mL−1 and 1.09 ± 0.10 AU mL−1, respectively. On day 30, the ANF levels for the sham, positive control and treatment group of rats are found to be 0.28 ± 0.17 AU mL−1, 0.92 ± 0.08 AU mL−1 and 0.37 ± 0.024 AU mL−1, respectively. Therefore, on treatment with GlyNPs, the ANA level decreased approximately to a similar value as on day 0. Whereas for the positive control, in the RA developed rat group, it reduced only by ∼20%, possibly due to the natural immune response. Thus, it can be concluded that the GlyNPs impact the reduction of the ANA levels as well as the inflammation due to RA.
Anti-double-stranded DNA antibodies (anti-dsDNA)
Anti-dsDNA is an antibody that targets double-stranded DNA molecules in the nucleus of cells in the human body and is one of the important antibodies for autoimmune disorders.50 Therefore, checking the anti-dsDNA antibody levels for RA patients is very important. Various studies have reported that 82.92% of RA patients tested positive for anti-dsDNA antibodies.51 Furthermore, anti-dsDNA antibodies can be generated by anti-TNFα, which modulates inflammation status.52 Hence, anti-dsDNA levels were checked with the help of an ELISA test on day 0, day 15 (inflammation achieved), and day 30 (treatment), and the results are shown in Fig. 8(e). It is clearly evident that the anti-dsDNA levels for days 0, 15 and 30 are found to be 8.81 ± 0.65 IU mL−1, 8.53 ± 0.40 IU mL−1 and 8.93 ± 0.33 IU mL−1 for sham control, i.e., there is no change in the values. The positive control rats’ value changed from 11.95 ± 0.38 IU mL−1 (15th day) to 10.71 ± 0.22 IU mL−1 for the 30th day (no treatment performed). However, for the treatment group of rats, the anti-dsDNA level changes from 11.13 ± 0.14 IU mL−1 (15th day) to 8.46 ± 0.52 IU mL−1 for the 30th day (on treatment with GlyNPs). This value is equal to the anti-dsDNA level of the 0th day. Thus, this result is strong evidence that GlyNPs effectively reduce inflammation.Study of the uric acid levels
Uric acid is the final waste product of purine metabolism, and it can lead to gout and kidney stones and potentially affect joint health due to crystal formation in the joints.53 Increasing uric acid levels contribute to inflammation, joint damage, and RA. Hence, the uric acid levels were checked on day 0, day 15 (inflammation achieved), and day 30 (treatment), and the results are shown in Fig. 8(f). The uric acid levels for all the sham control rats at day 0, day 15 and day 30 are found to be 2.8 ± 0.52, 2.73 ± 0.35 and 2.43 ± 0.92 mg dL−1, respectively. For the positive control group of rats, the uric acid levels are calculated to be 2.76 ± 0.30, 5.30 ± 0.3 and 3.7 ± 0.26 mg dL−1 on day 0, day 15 and day 30, respectively. Whereas, for the treatment group of rats, the uric acid levels were calculated to be 2.66 ± 0.41, 5.43 ± 0.30, 3.1 ± 0.20 mg dL−1 on day 0 (i.e., when no inflammation is there) and day 15 (when inflammation was developed), and on day 30 (means 15 days after post-treatment), respectively. Therefore, after 15 days post-treatment with GlyNPs (i.e., on the 30th day), the uric acid level decreased almost to the level of day 0. In conclusion, this radical change in the uric acid level makes strong evidence for the anti-inflammatory and anti-RA activity of GlyNPs.Inflammatory parameter in rats
IL-1β is a potent pro-inflammatory cytokine, and it is crucial for host defence against infection and injuries54 IL-1β also serves an important role in the pathogenesis of inflammation and RA.55,56TNF-α is a potent pro-inflammatory cytokine that modulates facets of macrophage functions, and it is rapidly released after trauma and chronic infections. It has been shown to be one of the most abundant early inflamed tissues.57 Furthermore, it can potentially trigger pathological implications, including autoimmune illnesses.58 Therefore, it is very important to determine the level of immune markers such as IL-1β, TNF-α and IL-6 in healthy sham, positive control and GlyNP-treated rats. To measure these parameters, the blood serum samples from all the groups of rats were collected, and experiments were performed through the ELISA kit, as mentioned in the Method section. The results are shown in Fig. 9(a)–(c). It can be noted that the IL-1β, TNF-α and IL-6 levels were measured on the 30th day, i.e., 15 days post-treatment. It is evident from Fig. 9(a) that the IL-1β levels in the control sham, positive control and GlyNP treated group of rats are found to be 87.56 ± 4.37, 196.32 ± 9.81 and treatment 186.54 ± 9.32 ng L−1, respectively. The IL-6 levels in the control sham, positive control and GlyNP-treated group of rats observed to be 221.10 ± 11.05, 615.42 ± 30.77 and treatment 411.46 ± 20.57 ng L−1, respectively (Fig. 9(b)). The TNF-α levels, as shown in Fig. 9(c) for the sham control group, positive control and GlyNP-treated group of rats, are found to be 121.56 ± 6.07, 547.22 ± 27.36 and 311.20 ± 15.56 ng L−1, respectively. Thus, from all these results, it can be concluded that the GlyNPs potentially reduce inflammation due to RA.
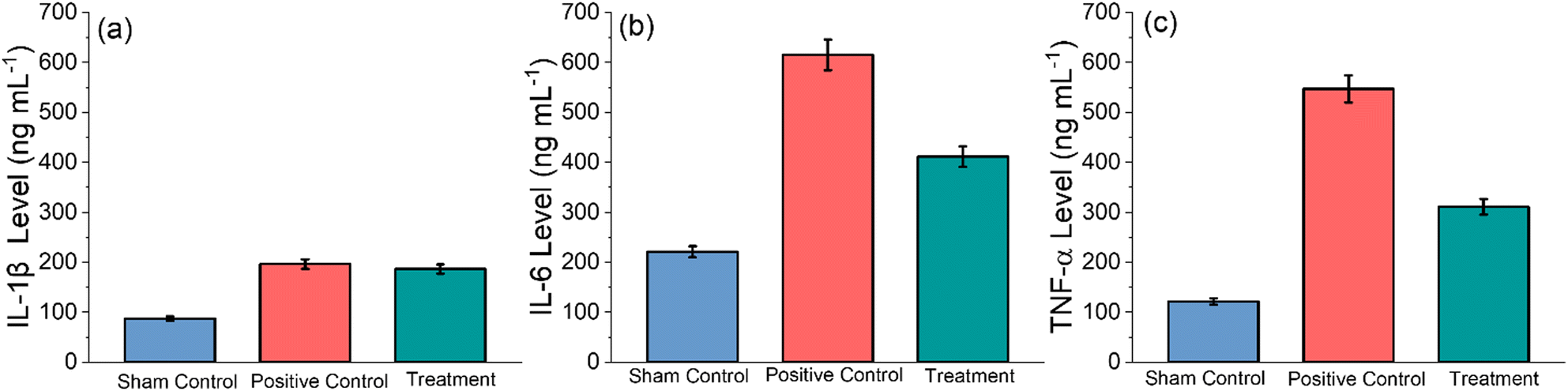 | ||
| Fig. 9 Study of the in vivo immune cytokines to check the inflammatory parameters for the Sham, positive control and treatment group of rats: (a) IL-1β Level, (b) IL-6 and (c) TNF-α level. This study was performed on the 30th day, i.e., after 15 days post-treatment. | ||
4. Discussion
Inflammatory diseases are very common nowadays due to various reasons59,60 and patients suffer a lot, irrespective of age.1 Severe inflammation causes RA, inflammatory bowel disease, psoriasis, and systemic lupus erythematosus.2 The inflammation can cause tissue injury, organ dysfunction, and damage organs severely.3,4 Therefore, there is an urgent need to develop new therapeutics and strategies to mitigate inflammatory diseases and improve patients’ recovery outcomes from the existing treatment options. Extensive research works using inorganic nanoparticles, enzymes, nanobodies, PEGylates, etc.16,17,61 have been conducted. However, over the past three decades, it has not gained remarkable advancements to cure inflammatory disorders. Hence, the present state-of-the-art work is very much promising in this challenging scenario.In this work, we have developed porous GlyNPs of size 180–250 nm with pore size of 4–5 nm in diameter using glycine and acryloyl chloride (Fig. 1(a)–(g)). GlyNPs also exhibit colloidal stability (ζ = −35.6) (Fig. 1(h)). The advantage of these GlyNPs is that they are biocompatible in a broad concentration range (10–250 μg mL−1) (tested on RAW 264.7 macrophages, immune cells, Fig. 2(a), and PC-12 cells, neuronal cells, Fig. 2(b)). GlyNPs are proliferative in nature at a low concentration, such as 80 μg mL−1 (Fig. 2(b)). Based on the proliferative nature, polymeric nanoparticles of glycine have been used earlier for wound healing and to reduce the inflammation of wounds only by our research group where the nanoparticles were of size below 50 nm.25 At lower concentrations, the observed higher viability of PC-12 cells compared to the control is attributed to a potential stimulatory and protective effect of GlyNPs. The GlyNPs at lower concentrations promote cellular activity, such as enhanced metabolic processes leading to increased viability and enhanced cell proliferation (Fig. 2(b))62 resembling its suitability for therapeutic applications. Usually, inorganic/metal NPs below a size of 8 nm in diameter can be easily excreted through the urinary tract.63 Charged NPs can circulate in the blood for a longer period of time due to the coulombic interactions.63 The advantages of using the amino acid polymeric nanoparticles are: (i) they are degradable in nature, and (ii) their degraded products are not toxic (as we have reported earlier for a similar type of polymer),64 and (iii) the debris, which could be excreted easily through the urinary tract.
Different types of nanoparticles can be internalized into the cells (macrophages and endothelial cells) through various processes, such as receptor-mediated or caveolin-mediated endocytosis. The cellular internalization mechanisms strongly depend on the type of nanoparticles, their size and shapes, and surface properties.65 However, the actual mechanisms of internalization of nanoparticles are still elusive. In this study, we have studied the cellular internalization of GlyNPs (Fig. 3) through confocal microscopy, since the internalization of these NPs regulates the secretion of inflammation-specific markers66,67 such as IL-6, IL-1β, TNF-α, TLR-4, STAT-1, MAPK-8, MAPK-14, iNOS, NF-κβ and COX-2, that were identified from Network pharmacology and through the in silico study (Fig. 4 and 5). To validate the secretion of inflammation-specific markers, we have performed in vitro study on the macrophages by varying the concentrations of GlyNPs (Fig. 6) under a cell-based inflammation model. The cell-based inflammation was generated using LPS. The results show that the GlyNPs with different concentrations regulate the secretion levels of NO, NF-κβ, INF-γ, IL-6, IL-10, and TNF-α, comprising the reduction of inflammation (Fig. 6).
To validate the therapeutic efficiency of GlyNPs, an RA model has been developed with rats, and it is observed that controlling the levels of IL-1β, TNF-α and IL-6 within 15 days of post-treatment can reduce the inflammation. The autoimmune and inflammatory markers such as (a) RA, (b) anti-CCP, (c) CRP, (d) ANA, (e) anti-dsDNA and (f) serum uric acid levels observed are reduced compared to the control and sham group of rats within 15 days post-treatment. The results obtained are at a statistically significant level (the statistical significance was calculated at a p-value of 0.05. Data was marked with ‘*’ for p < 0.05, ‘**’ for p < 0.01 and ‘***’ for p < 0.001). Thus, GlyNPs provide excellent anti-inflammatory properties to reduce the impact of RA. It can be noted that complicated autoimmune diseases, such as RA, can cause severe articular damage (cardiovascular problem) and reduce the life quality of patients.45,68 Using GlyNP treatment, the patient's life quality can be improved, for which extensive clinical research is required. A series of nanoparticles were used for the treatment of inflammatory bowel disease.69,70 However, amino acid-based nanoparticles or GlyNPs have never been used, which may have better therapeutic efficiency. Psoriasis is an inflammatory autoimmune disorder of the skin, and to treat psoriasis, different nanoformulations (nanoparticles with drugs) were used. The available treatment options for psoriasis are non-specific and associated with systemic toxicity due to the drug and their difficulty associated with deciding the doses.71 GlyNPs could hold a predictable therapeutic option for treating psoriasis since it is biocompatible, and no additional drugs were used to reduce inflammation. However, the patients are the crucial parameters and extensive research is required with clinical trials. Since the GlyNPs are porous in nature (4–5 nm, see Fig. 1(c) and (e)), many small anti-inflammatory drug molecules can be loaded into them. Furthermore, GlyNPs can be easily internalized by the cells (Fig. 3). These facilities of GlyNPs could be advantageous for targeted delivery to the inflammation-specific cells or to the macrophages to secrete different levels of inflammatory markers and subsequently the inflammation can be reduced through the synergetic effects of GlyNPs and anti-inflammatory drug molecules. Further research has been considered to address all these issues and establish great success with clinical significance.
5. Conclusions
Inflammation causes various diseases, including chronic tissue injury, which demands medicines suitable for the existing therapeutic options. In this work, glycine and acryloyl-based polymeric nanoparticles, GlyNPs, have been synthesized, which provide excellent anti-inflammatory properties. Detailed cell-based studies conclude that these GlyNPs are biocompatible, and studies on macrophages’ immunological response are commendable. Network pharmacology and in silico studies revealed that these GlyNPs possess target-specific immunomodulatory effects and potential for treating inflammatory diseases. Additionally, RT-PCR analysis revealed the downregulation of specified pro-inflammatory cytokines in support of the anti-inflammatory activities of GlyNPs. Thus, network pharmacology, in silico, in vitro and in vivo studies revealed that GlyNPs are an excellent target-specific immunomodulatory agent, which can also be potentially used for targeted drug delivery and could be an alternate therapeutic for treating acute inflammatory diseases like RA. Furthermore, a detailed study with extensive sampling is required, followed by clinical trials. Any potential anti-inflammatory drug can also be loaded in GlyNPs to form a nanoformulation, which may synergistically reduce inflammation and be useful for treating various inflammatory diseases. To address all the issues related to inflammation and to establish great success in treatment with clinical significance, further research has been taken into consideration.Author contributions
P. Paik is the principal project investigator (PI). Ideation, experimental design, experimental results analysis, and manuscript writing performed by D. Pareek and P. Paik. Characterization helped by S. Patra, G. Singh, K. Wasnik, and P. S. Gupta. Molecular docking, helped by O. Alagu. In vivo work helped by Md. Zeyaullah. Manuscript written and finalized by D. Pareek and P. Paik. All the authors checked and approved the manuscript for publication.Data availability
All the original data will be available with this article's communicating author and will be provided as required.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support awarded to Prof. Paik by the I-DAPT foundation (ref. I-DAPT/IT (BHU)/2023-24/Project Sanction/47), Indian Council of Medical Research (ICMR), India (Ref: EMDR/SG/12/2023-4724), STARS-IISc. Bangalore (ref. MoE-STARS/STARS-2/2023-0318) and Anusandhan National Research Foundation (ANRF), India (ref: CRG/2023/005576). D. Pareek acknowledges the DST-INSPIRE fellowship awarded to carry out the PhD work (ref. IF 180928).References
- E. R. Brannon, M. V. Guevara, N. J. Pacifici, J. K. Lee, J. S. Lewis and O. Eniola-Adefeso, Nat. Rev. Mater., 2022, 7, 796–813 CrossRef CAS PubMed.
- I. B. McInnes and E. M. Gravallese, Nat. Rev. Immunol., 2021, 21, 680–686 CrossRef CAS PubMed.
- G. Y. Chen and G. Nuñez, Nat. Rev. Immunol., 2010, 10, 826–837 CrossRef CAS PubMed.
- E. Kolaczkowska and P. Kubes, Nat. Rev. Immunol., 2013, 13, 159–175 CrossRef CAS PubMed.
- J. Khambhati, M. Engels, M. Allard-Ratick, P. B. Sandesara, A. A. Quyyumi and L. Sperling, Atherosclerosis, 2018, 276, 1–9 CrossRef CAS PubMed.
- P. Kong, Z.-Y. Cui, X.-F. Huang, D.-D. Zhang, R.-J. Guo and M. Han, Signal Transduction Targeted Ther., 2022, 7, 131 CrossRef CAS PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2024, 15, 758–760 CrossRef CAS PubMed.
- G. Monteleone, A. Moscardelli, A. Colella, I. Marafini and S. Salvatori, Autoimmun. Rev., 2023, 22, 103410 CrossRef CAS PubMed.
- K. F. Baker and J. D. Isaacs, Ann. Rheum. Dis., 2018, 77, 175 CrossRef CAS PubMed.
- D. Wu, Y. Jin, Y. Xing, M. D. Abate, M. Abbasian, M. Abbasi-Kangevari, Z. Abbasi-Kangevari, F. Abd-Allah, M. Abdelmasseh, M.-A. Abdollahifar, D. M. Abdulah, A. Abedi, V. Abedi, H. Abidi, R. G. Aboagye, H. Abolhassani, K. Abuabara, M. Abyadeh, I. Y. Addo, K. N. Adeniji, A. V. Adepoju, M. A. Adesina, Q. E. Sakilah Adnani, M. Afarideh, S. Aghamiri, A. Agodi, A. Agrawal, C. E. Aguilera Arriagada, A. Ahmad, D. Ahmad, S. Ahmad, S. Ahmad, A. Ahmadi, A. Ahmed, A. Ahmed, J. P. Aithala, A. A. Ajadi, M. Ajami, M. Akbarzadeh-Khiavi, F. Alahdab, M. T. AlBataineh, S. Alemi, A. A. Saeed Al-Gheethi, L. Ali, S. M. Alif, J. U. Almazan, S. Almustanyir, J. S. Alqahtani, I. Alqasmi, I. U. Khan Altaf, N. Alvis-Guzman, N. J. Alvis-Zakzuk, Y. M. Al-Worafi, H. Aly, R. Amani, H. Amu, G. A. Amusa, C. L. Andrei, A. Ansar, H. Ansariniya, A. E. Anyasodor, J. Arabloo, R. Arefnezhad, J. Arulappan, M. Asghari-Jafarabadi, T. Ashraf, J. A. Atata, S. S. Athari, D. Atlaw, M. M. d Wahbi Atout, A. Aujayeb, A. T. Awan, H. Ayatollahi, S. Azadnajafabad, A. Y. Azzam, A. Badawi, A. D. Badiye, S. Bagherieh, A. A. Baig, B. B. Bantie, M. Barchitta, M. Bardhan, S. L. Barker-Collo, F. Barone-Adesi, K. Batra, N. S. Bayileyegn, A. H. Behnoush, U. I. Belgaumi, M. Bemanalizadeh, I. M. Bensenor, K. A. Beyene, A. S. Bhagavathula, P. Bhardwaj, S. Bhaskar, A. N. Bhat, S. Bitaraf, V. R. Bitra, A. Boloor, K. Bora, J. S. Botelho, R. Buchbinder, D. Calina, L. A. Cámera, A. F. Carvalho, J. S. Kai Chan, V. K. Chattu, E. C. Abebe, F. Chichagi, S. Choi, T.-C. Chou, D.-T. Chu, K. Coberly, V. M. Costa, R. A. S. Couto, N. Cruz-Martins, O. Dadras, X. Dai, G. Damiani, A. M. Dascalu, M. Dashti, S. A. Debela, R. P. Dellavalle, A. K. Demetriades, A. A. Demlash, X. Deng, H. D. Desai, R. Desai, S. M. Rahman Dewan, S. Dey, S. D. Dharmaratne, D. Diaz, M. Dibas, R. J. Dinis-Oliveira, M. Diress, T. C. Do, D. K. Doan, M. Dodangeh, M. Dodangeh, D. Dongarwar, J. Dube, A. M. Dziedzic, A. Ed-Dra, H. A. Edinur, N. Eissazade, M. Ekholuenetale, T. C. Ekundayo, N. M. Elemam, M. Elhadi, A. O. Elmehrath, O. A. Abdou Elmeligy, M. Emamverdi, T. I. Emeto, H. L. Esayas, H. B. Eshetu, F. Etaee, A. F. Fagbamigbe, S. Faghani, I. R. Fakhradiyev, A. Fatehizadeh, M. Fathi, A. Feizkhah, G. Fekadu, M. Fereidouni, S.-M. Fereshtehnejad, J. C. Fernandes, P. Ferrara, G. Fetensa, I. Filip, F. Fischer, B. Foroutan, M. Foroutan, T. Fukumoto, B. Ganesan, B. N. Belete Gemeda, S.-H. Ghamari, M. Ghasemi, M. Gholamalizadeh, T. K. Gill, R. F. Gillum, M. Goldust, M. Golechha, P. Goleij, D. Golinelli, H. Goudarzi, S.-Y. Guan, Y. Guo, B. Gupta, V. B. Gupta, V. K. Gupta, R. Haddadi, N. R. Hadi, R. Halwani, S. Haque, I. Hasan, R. Hashempour, A. Hassan, T. S. Hassan, S. Hassanzadeh, M. B. Hassen, J. Haubold, K. Hayat, G. Heidari, M. Heidari, R. Heidari-Soureshjani, C. Herteliu, K. Hessami, K. Hezam, Y. Hiraike, R. Holla, M.-S. Hosseini, H.-H. Huynh, B.-F. Hwang, S. E. Ibitoye, I. M. Ilic, M. D. Ilic, A. Iranmehr, F. Iravanpour, N. E. Ismail, M. Iwagami, C. C. D. Iwu, L. Jacob, M. Jafarinia, A. Jafarzadeh, K. Jahankhani, H. Jahrami, M. Jakovljevic, E. Jamshidi, C. T. Jani, M. D. Janodia, S. K. Jayapal, S. Jayaram, J. Jeganathan, J. B. Jonas, A. Joseph, N. Joseph, C. E. Joshua, K. Vaishali, B. Kaambwa, A. Kabir, Z. Kabir, V. Kadashetti, F. Kaliyadan, F. Kalroozi, V. K. Kamal, A. Kandel, H. Kandel, S. Kanungo, J. Karami, I. M. Karaye, H. Karimi, H. Kasraei, S. Kazemian, S. A. Kebede, L. Keikavoosi-Arani, M. Keykhaei, Y. S. Khader, H. Khajuria, F. Khamesipour, E. A. Khan, I. A. Khan, M. Khan, M. J. Khan, M. A. B. Khan, M. A. Khan, H. Khatatbeh, M. M. Khatatbeh, S. Khateri, H. R. Khayat Kashani, M. S. Kim, A. Kisa, S. Kisa, H. Y. Koh, P. Kolkhir, O. Korzh, A. L. Kotnis, P. A. Koul, A. Koyanagi, K. Krishan, M. Kuddus, V. V. Kulkarni, N. Kumar, S. Kundu, O. P. Kurmi, C. La Vecchia, C. Lahariya, T. Laksono, J. Lám, K. Latief, P. Lauriola, B. K. Lawal, T. T. Thu Le, T. T. Bich Le, M. Lee, S. W. Lee, W.-C. Lee, Y. H. Lee, J. Lenzi, M. Levi, W. Li, V. S. Ligade, S. S. Lim, G. Liu, X. Liu, E. Llanaj, C.-H. Lo, V. S. Machado, A. A. Maghazachi, M. A. Mahmoud, T. A. Mai, A. Majeed, P. M. Sanaye, O. M. Makram, E. M. Rad, K. Malhotra, A. A. Malik, I. Malik, T. H. Mallhi, D. C. Malta, M. A. Mansournia, L. G. Mantovani, M. Martorell, S. Masoudi, S. Z. Masoumi, Y. Mathangasinghe, E. Mathews, A. G. Mathioudakis, A. Maugeri, M. Mayeli, J. R. Carabeo Medina, G. G. Meles, J. J. Mendes, R. G. Menezes, T. Mestrovic, I. M. Michalek, A. C. Micheletti Gomide Nogueira de Sá, E. T. Mihretie, L. H. Nhat Minh, R. Mirfakhraie, E. M. Mirrakhimov, A. Misganaw, A. Mohamadkhani, N. S. Mohamed, F. Mohammadi, S. Mohammadi, S. Mohammed, S. Mohammed, S. Mohan, A. Mohseni, A. H. Mokdad, S. Momtazmanesh, L. Monasta, M. A. Moni, M. Moniruzzaman, Y. Moradi, N. Morovatdar, E. Mostafavi, P. Mousavi, G. D. Mukoro, A. Mulita, G. B. Mulu, E. Murillo-Zamora, F. Musaigwa, G. Mustafa, S. Muthu, F. Nainu, V. Nangia, S. N. Swamy, Z. S. Natto, P. Navaraj, B. P. Nayak, A. Nazri-Panjaki, H. Negash, M. H. Nematollahi, D. H. Nguyen, H. T. Hien Nguyen, H. Q. Nguyen, P. T. Nguyen, V. T. Nguyen, R. K. Niazi, T. K. Nikolouzakis, L. A. Nnyanzi, M. Noreen, C. I. Nzoputam, O. J. Nzoputam, B. Oancea, I.-H. Oh, H. Okati-Aliabad, O. C. Okonji, P. G. Okwute, A. T. Olagunju, M. I. Olatubi, I. I. Olufadewa, M. Ordak, N. Otstavnov, M. O. Owolabi, P. A. Mahesh, J. R. Padubidri, A. Pak, R. Pakzad, R. Palladino, A. Pana, I. Pantazopoulos, P. Papadopoulou, S. Pardhan, A. Parthasarathi, A. Pashaei, J. Patel, A. R. Pathan, S. Patil, U. Paudel, S. Pawar, P. Pedersini, U. Pensato, D. M. Pereira, J. Pereira, M. O. Pereira, R. B. Pereira, M. F. P. Peres, A. Perianayagam, S. Perna, I.-R. Petcu, P. S. Pezeshki, H. T. Pham, A. K. Philip, M. A. Piradov, I. Podder, V. Podder, D. Poddighe, E. J. Sady Prates, I. Qattea, A. Radfar, P. Raee, A. Rafiei, A. Raggi, F. Rahim, M. Rahimi, M. Rahimifard, V. Rahimi-Movaghar, M. O. Rahman, M. H. Ur Rahman, M. Rahman, M. A. Rahman, A. M. Rahmani, M. Rahmani, S. Rahmani, V. Rahmanian, P. Ramasubramani, N. Rancic, I. R. Rao, S. Rashedi, A. M. Rashid, N. Ravikumar, S. Rawaf, E. M. Mohamed Redwan, N. Rezaei, N. Rezaei, N. Rezaei, M. Rezaeian, D. Ribeiro, M. Rodrigues, J. A. Buendia Rodriguez, L. Roever, E. Romero-Rodríguez, A. M. A. Saad, B. Saddik, S. Sadeghian, U. Saeed, A. Safary, M. Safdarian, S. Z. Safi, A. Saghazadeh, D. Sagoe, F. S. Sharif-Askari, N. S. Sharif-Askari, A. Sahebkar, H. Sahoo, M. A. Sahraian, M. R. Sajid, S. Sakhamuri, J. W. Sakshaug, M. A. Saleh, L. Salehi, S. Salehi, A. S. Farrokhi, S. Samadzadeh, S. Samargandy, N. Samieefar, A. M. Samy, N. Sanadgol, R. K. Sanjeev, M. Sawhney, G. K. Saya, A. Schuermans, S. Senthilkumaran, S. G. Sepanlou, Y. Sethi, M. Shafie, H. Shah, I. Shahid, S. Shahid, M. A. Shaikh, S. Sharfaei, M. Sharma, M. Shayan, H. S. Shehata, A. Sheikh, J. K. Shetty, J. I. Shin, R. Shirkoohi, N. A. Shitaye, K. M. Shivakumar, V. Shivarov, P. Shobeiri, S. Siabani, M. M. Sibhat, E. E. Siddig, C. R. Simpson, E. Sinaei, H. Singh, I. Singh, J. A. Singh, P. Singh, S. Singh, M. S. Siraj, A. Al Mamun Sohag, R. Solanki, S. Solikhah, Y. Solomon, M. S. Soltani-Zangbar, J. Sun, M. D. Szeto, R. Tabarés-Seisdedos, S. M. Tabatabaei, M. Tabish, E. Taheri, A. Tahvildari, I. M. Talaat, J. J. L. Lukenze Tamuzi, K.-K. Tan, N. Y. Tat, R. T. Oliaee, A. Tavasol, M.-H. Temsah, P. Thangaraju, S. Tharwat, N. S. Tibebu, J. H. Vera Ticoalu, T. Tillawi, T. Y. Tiruye, A. Tiyuri, M. R. Tovani-Palone, M. Tripathi, G. M. Tsegay, A. R. Tualeka, S. S. Ty, C. S. Ubah, S. Ullah, S. Ullah, M. Umair, S. Umakanthan, E. Upadhyay, S. M. Vahabi, A. G. Vaithinathan, S. V. Tahbaz, R. Valizadeh, S. B. Varthya, T. J. Vasankari, N. Venketasubramanian, G.-I. Verras, J. H. Villafañe, V. Vlassov, D. C. Vo, Y. Waheed, A. Waris, B. G. Welegebrial, R. Westerman, D. P. Wickramasinghe, N. D. Wickramasinghe, B. Willekens, B. Z. Woldegeorgis, M. Woldemariam, H. Xiao, D. Y. Yada, G. Yahya, L. Yang, F. Yazdanpanah, D. K. Yon, N. Yonemoto, Y. You, M. Zahir, S. S. Zaidi, M. Zangiabadian, I. Zare, M. A. Zeineddine, D. T. Zemedikun, N. G. Zeru, C. Zhang, H. Zhao, C. Zhong, M. Zielińska, M. Zoladl, A. Zumla, C. Guo and L.-S. Tam, eClinicalMedicine, 2023, 64, 102193 CrossRef PubMed.
- E. Alexander and K. W. Leong, J. Nanobiotechnol., 2024, 22, 661 CrossRef PubMed.
- Y. Huang, X. Guo, Y. Wu, X. Chen, L. Feng, N. Xie and G. Shen, Signal Transduction Targeted Ther., 2024, 9, 34 CrossRef PubMed.
- A. K. Mishra, L. Rani, R. Singh, H. K. Dewangan, P. K. Sahoo and V. Kumar, J. Drug Delivery Sci. Technol., 2024, 93, 105446 CrossRef CAS.
- R. Brusini, M. Varna and P. Couvreur, Adv. Drug Delivery Rev., 2020, 157, 161–178 CrossRef CAS PubMed.
- R. Mohammapdour and H. Ghandehari, Adv. Drug Delivery Rev., 2022, 180, 114022 CrossRef CAS PubMed.
- F. A. Waliaveettil, J. Jose and E. I. Anila, ACS Appl. Bio Mater., 2025, 9(1), 628–641 CrossRef PubMed.
- H. Jeong, K. Subramanian, J.-B. Lee, H. Byun, H. Shin and J.-H. Yun, Biomater. Sci., 2025, 13, 810–825 RSC.
- C. M. A. van Alem, M. Boonstra, J. Prins, T. Bezhaeva, M. F. van Essen, J. M. Ruben, A. L. Vahrmeijer, E. P. van der Veer, J. W. de Fijter, M. E. Reinders, O. Meijer, J. M. Metselaar, C. van Kooten and J. I. Rotmans, Nephrol., Dial., Transplant., 2018, 33, 44–53 CrossRef CAS PubMed.
- M. Zhou, J. Hou, Z. Zhong, N. Hao, Y. Lin and C. Li, Drug Delivery, 2018, 25, 716–722 CrossRef CAS PubMed.
- Y. Karabey-Akyurek, A. G. Gurcay, O. Gurcan, O. F. Turkoglu, S. Yabanoglu-Ciftci, H. Eroglu, M. F. Sargon, E. Bilensoy and L. Oner, Pharm. Dev. Technol., 2017, 22, 972–981 CrossRef CAS PubMed.
- Q. Wang, J. Jiang, W. Chen, H. Jiang, Z. Zhang and X. Sun, J. Controlled Release, 2016, 230, 64–72 CrossRef CAS PubMed.
- R. L. Bartlett, S. Sharma and A. Panitch, Nanomedicine, 2013, 9, 419–427 CrossRef CAS PubMed.
- R. M. Gonçalves, A. C. L. Pereira, I. O. Pereira, M. J. Oliveira and M. A. Barbosa, J. Mater. Sci.: Mater. Med., 2015, 26, 167 CrossRef PubMed.
- Y.-C. Chen, S. F. Gad, D. Chobisa, Y. Li and Y. Yeo, J. Controlled Release, 2021, 330, 438–460 CrossRef CAS PubMed.
- P. S. Gupta, K. Wasnik, G. Singh, S. Patra, D. Pareek, D. D. Yadav, M. S. Tomar, S. Maiti, M. Singh and P. Paik, Mater. Adv., 2023, 4, 4718–4731 RSC.
- P. S. Gupta, K. Wasnik, S. Patra, D. Pareek, G. Singh, D. D. Yadav, S. Maity and P. Paik, Nanoscale, 2024, 16, 1770–1791 RSC.
- J. Pietsch, S. Riwaldt, J. Bauer, A. Sickmann, G. Weber, J. Grosse, M. Infanger, C. Eilles and D. Grimm, Int. J. Mol. Sci., 2013, 14, 1164–1178 CrossRef CAS PubMed.
- T. Liu, L. Zhang, D. Joo and S.-C. Sun, Signal Transduction Targeted Ther., 2017, 2, 17023 CrossRef PubMed.
- J. Tuncel, S. Haag, M. H. Hoffmann, A. C. Y. Yau, M. Hultqvist, P. Olofsson, J. Bäcklund, K. S. Nandakumar, D. Weidner, A. Fischer, A. Leichsenring, F. Lange, C. Haase, S. Lu, P. S. Gulko, G. Steiner and R. Holmdahl, PLoS One, 2016, 11, e0155936 CrossRef PubMed.
- A. K. Yamala, V. Nadella, Y. Mastai, H. Prakash and P. Paik, Nanoscale, 2017, 9, 14006–14014 RSC.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- S. X. Ge, D. Jung and R. Yao, Bioinformatics, 2020, 36, 2628–2629 CrossRef CAS PubMed.
- G. M. Morris and M. Lim-Wilby, in Molecular Modeling of Proteins, ed. A. Kukol, Humana Press, Totowa, NJ, 2008, pp. 365–382 DOI:10.1007/978-1-59745-177-2_19.
- P. Mark Andrew, A. S. Marisa, L. W. Darby and X. Zhong-Ru, in Molecular Docking, ed. P. V. Dimitrios, IntechOpen, Rijeka, 2018, ch. 8 DOI:10.5772/intechopen.72898.
- P. C. Agu, C. A. Afiukwa, O. U. Orji, E. M. Ezeh, I. H. Ofoke, C. O. Ogbu, E. I. Ugwuja and P. M. Aja, Sci. Rep., 2023, 13, 13398 CrossRef CAS PubMed.
- U. Lo, V. Selvaraj, J. M. Plane, O. V. Chechneva, K. Otsu and W. Deng, Sci. Rep., 2014, 4, 7405 CrossRef CAS PubMed.
- Q. Guo, Y. Jin, X. Chen, X. Ye, X. Shen, M. Lin, C. Zeng, T. Zhou and J. Zhang, Signal Transduction Targeted Ther., 2024, 9, 53 CrossRef CAS PubMed.
- C. H. Evans, Agents Actions Suppl., 1995, 47, 107–116 CAS.
- J. N. Sharma, A. Al-Omran and S. S. Parvathy, Inflammopharmacology, 2007, 15, 252–259 CrossRef CAS PubMed.
- C. Wang, R. Rao, Z. Cai, H. Yang, L. Zhang, S. Ji, C. Zhang, D. Gao, Y. Hu, J. Li, W. Xiong, H. Jiang, J. Chu and D. Wu, ACS Nano, 2023, 17, 9025–9038 CrossRef CAS PubMed.
- J. Yeo, Y. M. Lee, J. Lee, D. Park, K. Kim, J. Kim, J. Park and W. J. Kim, Nano Lett., 2019, 19, 6716–6724 CrossRef CAS PubMed.
- Y. Oh, H. Jeong, S. Lim and J. Hong, Biomacromolecules, 2020, 21, 4972–4979 CrossRef CAS PubMed.
- Z. Strizova, I. Benesova, R. Bartolini, R. Novysedlak, E. Cecrdlova, L. K. Foley and I. Striz, Clin. Sci., 2023, 137, 1067–1093 CrossRef CAS PubMed.
- M. I. Edilova, A. Akram and A. A. Abdul-Sater, Biomed. J., 2021, 44, 172–182 CrossRef CAS PubMed.
- J. S. Smolen, D. Aletaha, A. Barton, G. R. Burmester, P. Emery, G. S. Firestein, A. Kavanaugh, I. B. McInnes, D. H. Solomon, V. Strand and K. Yamamoto, Nat. Rev. Dis. Primers, 2018, 4, 18001 CrossRef PubMed.
- J. E. Pope and E. H. Choy, Semin. Arthritis Rheum., 2021, 51, 219–229 CrossRef CAS PubMed.
- C. Ciurtin, G. A. Helmy, A. C. Ferreira, J. J. Manson, E. C. Jury and T. McDonnell, Clin. Immunol., 2024, 265, 110281 CrossRef CAS PubMed.
- D. S. Pisetsky and P. E. Lipsky, Nat. Rev. Rheumatol., 2020, 16, 565–579 CrossRef CAS PubMed.
- S. S. Paknikar, C. S. Crowson, J. M. Davis and U. Thanarajasingam, ACR Open Rheumatol., 2021, 3, 422–426 CrossRef PubMed.
- A. Kavanaugh, R. Tomar, J. Reveille, D. H. Solomon and H. A. Homburger, Arch. Pathol. Lab. Med., 2000, 124, 71–81 CrossRef CAS PubMed.
- Z. F. Mahdi, S. H. Mohammed and A. R. Hadi, Al-Rafidain J. Med. Sci., 2023, 5, 105–111 CrossRef.
- G. Steiner and J. Smolen, Arthritis Res. Ther., 2002, 4, S1 CrossRef PubMed.
- D. Alkhudir, A. Al-Herz, K. Saleh, A. Alawadhi, W. Al-Kandari, E. Hasan, K. Mokaddem, A. Ghanem, Y. A. Bartella, M. Hussain, N. AlHadhood, Y. Ali, E. Nahar, A. Alenizi, A. Aldei, F. Abutiban, S. Hayat, H. Behbehani, F. Baron, H. Alhajeri, A. Alkadi and A. Alsaber, Open Access Rheumatol.: Res. Rev., 2023, 15, 223–230 CrossRef CAS PubMed.
- N. Caronni, F. La Terza, F. M. Vittoria, G. Barbiera, L. Mezzanzanica, V. Cuzzola, S. Barresi, M. Pellegatta, P. Canevazzi, G. Dunsmore, C. Leonardi, E. Montaldo, E. Lusito, E. Dugnani, A. Citro, M. S. F. Ng, M. Schiavo Lena, D. Drago, A. Andolfo, S. Brugiapaglia, A. Scagliotti, A. Mortellaro, V. Corbo, Z. Liu, A. Mondino, P. Dellabona, L. Piemonti, C. Taveggia, C. Doglioni, P. Cappello, F. Novelli, M. Iannacone, L. G. Ng, F. Ginhoux, S. Crippa, M. Falconi, C. Bonini, L. Naldini, M. Genua and R. Ostuni, Nature, 2023, 623, 415–422 CrossRef CAS PubMed.
- S. Pasi, R. Kant, S. Gupta and A. Surolia, Biomaterials, 2015, 42, 121–133 CrossRef CAS PubMed.
- M. Adachi, S. Okamoto, S. Chujyo, T. Arakawa, M. Yokoyama, K. Yamada, A. Hayashi, K. Akita, M. Takeno, S. Itoh, T. Takii, Y. Waguri-Nagaya, T. Otsuka, K. Hayakawa, K. Miyazawa and K. Onozaki, J. Interferon Cytokine Res., 2013, 33, 297–307 CrossRef CAS PubMed.
- G. van Loo and M. J. M. Bertrand, Nat. Rev. Immunol., 2023, 23, 289–303 CrossRef CAS PubMed.
- D.-i Jang, A. H. Lee, H.-Y. Shin, H.-R. Song, J.-H. Park, T.-B. Kang, S.-R. Lee and S.-H. Yang, Int. J. Mol. Sci., 2021, 22, 2719 CrossRef CAS PubMed.
- D. Furman, J. Campisi, E. Verdin, P. Carrera-Bastos, S. Targ, C. Franceschi, L. Ferrucci, D. W. Gilroy, A. Fasano, G. W. Miller, A. H. Miller, A. Mantovani, C. M. Weyand, N. Barzilai, J. J. Goronzy, T. A. Rando, R. B. Effros, A. Lucia, N. Kleinstreuer and G. M. Slavich, Nat. Med., 2019, 25, 1822–1832 CrossRef CAS PubMed.
- V. P. Chavda, J. Feehan and V. Apostolopoulos, Cells, 2024, 13(22), 1906 CrossRef CAS PubMed.
- Y. Huang, J. Xu, G. Sun, X. Cheng, Y. An, X. Yao, G. Nie and Y. Zhang, Biomaterials, 2025, 314, 122822 CrossRef CAS PubMed.
- J. E. Karbowniczek, K. Berniak, J. Knapczyk-Korczak, G. Williams, J. A. Bryant, N. D. Nikoi, M. Banzhaf, F. de Cogan and U. Stachewicz, J. Colloid Interface Sci., 2023, 650, 1371–1381 CrossRef CAS PubMed.
- K. Ernits, C. K. Saha, T. Brodiazhenko, B. Chouhan, A. Shenoy, J. A. Buttress, J. J. Duque-Pedraza, V. Bojar, J. A. Nakamoto, T. Kurata, A. A. Egorov, L. Shyrokova, M. J. O. Johansson, T. Mets, A. Rustamova, J. Džigurski, T. Tenson, A. Garcia-Pino, H. Strahl, A. Elofsson, V. Hauryliuk and G. C. Atkinson, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305393120 CrossRef CAS PubMed.
- K. Wasnik, P. S. Gupta, S. Mukherjee, A. Oviya, R. Prakash, D. Pareek, S. Patra, S. Maity, V. Rai, M. Singh, G. Singh, D. D. Yadav, S. Das, P. Maiti and P. Paik, ACS Appl. Bio Mater., 2023, 6, 5644–5661 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- C. S. L. Tuttle, L. A. N. Thang and A. B. Maier, Ageing Res. Rev., 2020, 64, 101185 CrossRef CAS PubMed.
- Y. Qu, J. Li, Q. Qin, D. Wang, J. Zhao, K. An, Z. Mao, Z. Min, Y. Xiong, J. Li and Z. Xue, npj Parkinson's Dis., 2023, 9, 18 CrossRef CAS PubMed.
- Q. Wang, X. Qin, J. Fang and X. Sun, Acta Pharm. Sin. B, 2021, 11, 1158–1174 CrossRef CAS PubMed.
- M. Barani, A. Rahdar, S. Sargazi, M. S. Amiri, P. K. Sharma and N. Bhalla, Sens. Bio-Sens. Res., 2021, 32, 100417 CrossRef.
- J. Gao, J. Li, Z. Luo, H. Wang and Z. Ma, Drug Des., Dev. Ther., 2024, 18, 2921–2949 CrossRef PubMed.
- M. Pradhan, A. Alexander, M. R. Singh, D. Singh, S. Saraf, S. Saraf and Ajazuddin, Biomed. Pharmacother., 2018, 107, 447–463 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02012j |
| This journal is © The Royal Society of Chemistry 2025 |