
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradable water-soluble polymer prodrugs for subcutaneous delivery of irritant anticancer drugs†
Léa Guerassimoff a,
Jingming Caoa,
Michaella Augustea,
Amaury Bossiona,
Chen Zhua,
Dao Lea,
Catherine Cailleaua,
Safa Mohamed Ismaila,
Françoise Mercier-Noméb and
Julien Nicolas*a
a,
Jingming Caoa,
Michaella Augustea,
Amaury Bossiona,
Chen Zhua,
Dao Lea,
Catherine Cailleaua,
Safa Mohamed Ismaila,
Françoise Mercier-Noméb and
Julien Nicolas*a
aUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, 91400 Orsay, France. E-mail: julien.nicolas@universite-paris-saclay.fr; Tel: +33 1 80 00 60 81
bUniversité Paris-Saclay, IPSIT, INSERM UMR 996, 91400 Orsay, France
First published on 10th July 2025
Abstract
Chemotherapy is primarily administered intravenously (IV), but this route poses significant challenges (e.g., high costs, patient discomfort, logistical difficulties, side effects such as infections from catheter use). Although oral and subcutaneous (SC) routes are preferred for their convenience and have the potential for better patient comfort and cost reduction, oral chemotherapy faces issues like poor bioavailability and adherence, while SC delivery is unsuitable for irritant or vesicant drugs due to local toxicity. To overcome these limitations, the polymer prodrug strategy has been explored, where drugs are linked to a polymer, reducing toxicity and enhancing drug delivery. Recent work has focused on creating water-soluble polymer prodrugs for SC delivery of paclitaxel (Ptx), a hydrophobic and vesicant drug, which was successfully conjugated to polyacrylamide (PAAm), a very hydrophilic biocompatible polymer, resulting in safer SC injection and enhanced therapeutic efficacy in tumor-bearing mice. However, this strategy's potential depends on adapting it to other vesicant anticancer drugs. Making the polymer degradable for facilitated excretion would also be a key improvement. In this work, this approach has been successfully extended to gemcitabine (Gem), a widely used but irritant anticancer drug, and to a degradable PAAm-based promoiety, having cleavable ester groups in the main chain. The resulting Gem-based prodrugs featured upper critical solution temperature to ensure complete solubility at the temperature of the SC tissue, sustained Gem release, significant degradation under physiological conditions, improved systemic toxicity and absence of local toxicity compared to free Gem. Remarkably, Gem-PAAm polymer prodrugs exhibited significant anticancer efficacy in mice bearing Mia Pa-Ca 2 tumors, outperforming Gemzar®, the commercial formulation of Gem. These advances suggest the potential of these hydrophilic polymer prodrugs to transform SC chemotherapy, enabling the use of a broader range of anticancer drugs while reducing side effects and improving patient outcomes.
1. Introduction
Chemotherapies are almost exclusively administered by the intravenous (IV) route.1 Nonetheless, this route poses a number of major problems in terms of logistics, patient comfort and cost. For example, the high cost of IV chemotherapy comes from outpatient hospital visits, surgical interventions and long hospital stays due to repeated administration cycles or long infusions.2 The IV route is also restrictive for the patient who must be hospitalized regularly, and is invasive due to the need for a central catheter and an implantable chamber, which often leads to serious side effects (e.g., infections),3,4 further prolonging hospital stays and increasing costs.5 Although the oral and subcutaneous (SC) routes are preferred, notably for their ease of use,6–8 oral delivery suffers from poor and variable bioavailability, as well as compliance problems. The SC route is also unsuitable for many vesicant/irritant drugs because of their local toxicity9 at the injection site,10 such as skin reactions (e.g., alopecia, hyperpigmentation, irritation, necrosis) as well as SC cell dysfunction and death after repeated injections.11 This is unfortunate as SC administration could offer an ideal compromise due to its high bioavailability and rapid drug absorption,12 while being able to be implemented at home, resulting in lower costs and greater comfort for the patient.13Although a wide range of nanoscale drug delivery systems have been developed for SC administration,14 those dedicated to cancer therapy are limited to non-irritant and non-vesicant anticancer drugs and biomacromolecules (e.g., therapeutic proteins, antibodies). To alleviate these limitations, it is possible to take advantage of the polymer prodrug approach, which involves coupling drugs to a polymer scaffold via a cleavable linker, resulting in transient drug inactivation.15–19 In this context, we have recently reported the design and the preclinical evaluation of water-soluble polymer prodrugs suitable for the SC delivery of vesicant and irritant anticancer drugs.20 The principle is based on the “drug-initiated” synthesis of well-controlled polyacrylamide (PAAm) chains carrying one drug molecule at the chain end through the use of a drug-bearing controlling agent to perform controlled radical polymerization of AAm. PAAm was selected for its high water-solubility, biocompatibility and stealth properties,21 and we applied this approach to paclitaxel (Ptx), a highly hydrophobic and vesicant anticancer drug used in the formulation of Taxol, as a proof to validate our strategy. The obtained Ptx-PAAm prodrugs enabled safe SC injection without inducing local toxicity and outperformed Taxol during efficacy study in tumor-bearing mice, due to a higher maximum tolerated dose (MTD).20
However, the robustness and the versatility of this strategy, and therefore its future potential for drug delivery, depend primarily on its applicability to other irritant/vesicant drugs. Indeed, transforming this approach into a SC drug delivery platform by adapting it to other anticancer drugs could considerably broaden its field of application and pave the way for the treatment of different types of cancer. In addition, making the polymer promoiety degradable under physiological conditions to facilitate excretion and prevent accumulation in the body would undoubtedly be a key improvement with a view to possible clinical transposition.
Herein, we addressed both challenges by developing PAAm prodrugs for SC administration based on the irritant anticancer drug gemcitabine (Gem) and by making these prodrugs degradable under physiological conditions (Fig. 1).9,22,23 Gem is a nucleoside analogue with proven activity against a broad range of solid tumors (e.g., colon, lung, pancreatic, breast, bladder, ovarian cancers).24 However, like many nucleoside analogues, Gem suffers from serious limitations that often restrict its use, such as its irritant nature, short plasma half-life, rapid metabolism (due to deamination), induction of resistance, and the advent of severe side effects.25 A polymer prodrug strategy based on Gem would therefore not only avoid local toxicity after SC injection, but also prolong its half-life time by avoiding early degradation and improve therapeutic efficacy. As for the degradable nature of Gem-based polymer prodrugs, it was achieved by inserting ester groups in the PAAm backbone by controlled radical ring-opening polymerization (rROP).26–30 More specifically, we copolymerized AAm and 5,6-benzo-2-methylene-1,3-dioxepane (BMDO) as a cyclic ketene acetal (CKA) monomer, during the “drug-initiated” synthesis of the polymer prodrug. Interestingly, the resulting Gem-P(AAm-co-BMDO) copolymers exhibited an upper critical solution temperature (UCST),31 governed by an equilibrium between polymer–polymer interactions and polymer–aqueous medium interactions,32 which was finely tuned to produce fully water-soluble polymer prodrugs at SC tissue temperature (33–35 °C),33 thus preventing early degradation of the copolymer and drug release. Gem-P(AAm-co-BMDO) copolymer prodrugs demonstrated sustained release of Gem in human serum, significant in vitro cytotoxicity on a pancreatic cancer cell line and did not induce any local or systemic adverse effect at high doses after SC injection to mice, conversely to free Gem. These innovative prodrugs also resulted in higher survival rates and greater anticancer efficacy in tumor-bearing mice, compared with IV injection of Gemzar, the commercial formulation of Gem.
 | ||
| Fig. 1 Design and preclinical development of (degradable) polyacrylamide (PAAm)-based prodrugs for the SC administration of the anticancer drug gemcitabine (Gem). | ||
2. Experimental part
2.1 Materials
Azobisisobutyronitrile (98%, AIBN) and acrylamide (>99%, AAm) were purchased from Sigma-Aldrich and recrystallized from ethanol and chloroform, respectively. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (>97%, EDC·HCl), N-hydroxysuccinimide (98%, NHS), cyano-4-[(dodecylsulfanylthiocarbonyl) sulfanyl]pentanoic acid (97%, CDP), anhydrous dimethylsulfoxide (≥99.9%, DMSO), anhydrous dimethylformamide (≥99.9%, DMF), triethylamine (≥99%, TEA), anhydrous theophylline (≥99%), tetrahydrouridine (THU), potassium hydroxide (90%, KOH), Dulbecco's Phosphate Buffer Saline (PBS), Dulbecco's Modified Eagle Medium (DMEM) and human serum were purchased from Sigma-Aldrich and used as received. Gemcitabine·HCl (>98%, Gem) was purchased from TCI (Europe). Deuterated DMSO (DMSO-d6) was obtained from Eurisotop. Methanol (HPLC analytical grade, MeOH) and diethyl ether (HPLC analytical grade) were purchased from Carlo Erba. All other solvents were purchased from Sigma-Aldrich at the highest grade. ROTI®Histofix 4% (formaldehyde, pH 7) was purchased from Roth.2.2 Analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) preceded by a guard column from Agilent Technologies (PL PolarGel-M, 7.5 × 50 mm; bead diameter 8 μm) and a triple detection system (Viscotek TDA/GPCmax from Malvern) with a differential refractive index detector, low and right-angle light scattering detectors and a differential viscometer detector. The eluent was DMSO with 100 mM lithium bromide (LiBr) and 0.4 wt% of 2,6-di-tert-butyl-4-methylphenol (BHT) as a flowrate marker at a flow rate of 0.7 mL min−1. The system was calibrated using poly(methyl methacrylate) (PMMA) standards (peak molar masses, Mp = 540–342 900 g mol−1) from Agilent Technologies. This allowed the determination of the number-average molar mass (Mn), the weight-average molar mass (Mw) and the dispersity (Đ = Mw/Mn). All samples were filtered over 0.22 μm Nylon filters prior to injection. Data were collected and processed with OmniSEC 4.0 software.
000 g mol−1) preceded by a guard column from Agilent Technologies (PL PolarGel-M, 7.5 × 50 mm; bead diameter 8 μm) and a triple detection system (Viscotek TDA/GPCmax from Malvern) with a differential refractive index detector, low and right-angle light scattering detectors and a differential viscometer detector. The eluent was DMSO with 100 mM lithium bromide (LiBr) and 0.4 wt% of 2,6-di-tert-butyl-4-methylphenol (BHT) as a flowrate marker at a flow rate of 0.7 mL min−1. The system was calibrated using poly(methyl methacrylate) (PMMA) standards (peak molar masses, Mp = 540–342 900 g mol−1) from Agilent Technologies. This allowed the determination of the number-average molar mass (Mn), the weight-average molar mass (Mw) and the dispersity (Đ = Mw/Mn). All samples were filtered over 0.22 μm Nylon filters prior to injection. Data were collected and processed with OmniSEC 4.0 software.2.3 Synthesis procedures
:MeOH 20:1) to give 170 mg of a yellow oil (30% yield). 1H-NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.25 (d, J = 7.6 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 6.29 (d, J = 6.5 Hz, 1H), 6.18 (t, J = 7.5 Hz, 1H), 5.26 (t, J = 5.3 Hz, 1H), 4.18 (m, J = 8.1 Hz, 1H), 3.89 (m, J = 8.0 Hz, 1H), 3.83–3.61 (m, 2H), 3.42–3.35 (t, 2H), 1.86 (s, 2H), 1.68–1.60 (m, 2H), 1.24 (s, 16H), 0.86 (t, J = 6.5 Hz, 3H). 19F-NMR (400 MHz, DMSO-d6) δ ppm: 116.7–117.1 (s, 1F). 13C-NMR (100 MHz, DMSO-d6) δ 218.26, 173.04, 171.96, 162.72, 154.16, 144.53, 122.82, 118.51, 95.73, 84.00, 80.76, 58.66, 36.40, 32.65, 31.84, 31.27, 28.93, 28.86, 28.69, 28.59, 28.36, 28.06, 27.24, 25.22, 23.86, 22.07, 13.86. High-resolution mass spectrometry (ESI+): m/z = [M + H+]. Calculation = 648.299, found = 649.237.:50 (P0), 55:45 (P1), 56:44 (P2), 57:43 (P3) and 60:40 (P4), respectively). For Gem-P(AAm-co-BMDO) with a AAm:BMDO molar ratio of 56:44 (P2), AAm (112 eq., 4.48 mmol, 0.32 g), BMDO (88 eq., 3.52 mmol, 0.57 g) (total mole = 8 mmol), Gem-CDP (1 eq., 0.04 mmol, 26 mg) and AIBN (0.6 eq., 0.024 mmol, 3.9 mg) were dissolved in anhydrous DMSO (10 mL). The solution was bubbled with dry argon to remove dissolved oxygen for 20 min at room temperature and then immersed in a preheated oil bath at 70 °C for 16 h. The solution was then rapidly cooled under air. The copolymer was precipitated into cold methanol and then washed three times with cold methanol followed by centrifugation (5000 rpm, 10 min). The copolymer prodrug was then dried under high vacuum to give 179 mg of a white powder. The purified copolymer prodrugs were characterized by NMR and SEC. 1H-NMR (400 MHz, DMSO-d6) δ ppm: 0.82–0.88 (t, 3H–C12), 1.22–1.25 (s, H–C12), 1.27–1.54 (m, 2H-AAm), 2.00–2.27 (m, 1H-AAm), 2.68–2.71 (d; 2H-BMDO), 3.76–3.93 (m, 2H-Gem), 4.43–4.74 (m, 4H-closed BMDO), 4.93–5.17 (m, 2H-opened BMDO), 5.21–5.35 (m, 1H-Gem), 6.14–6.33 (m, 1H-Gem), 6.66–7.45 (m, 2H-AAm + 4H-BMDO + 1H-Gem), 8.12–8.27 (dd, 2H Gem),11.03–11.12 (t, 1H-Gem).19F-NMR (400 MHz, DMSO-d6) δ ppm: 116.7–117.1 (s, 1F). The same procedure was carried out for other copolymer prodrugs, except that P4 was precipitated into cold THF and P0–P3 were precipitated into cold methanol due to different amounts of BMDO. Note that P0 and P4 were analyzed by 1H-NMR (300 MHz, DMSO-d6).:BMDO molar ratios (56:44 and 57:43) were tested. For P(AAm-co-BMDO) 56:44 (P5), AAm (112 eq., 4.48 mmol, 0.32 g), BMDO (88 eq., 3.52 mmol, 0.57 g) (total mole = 8 mmol), CDP (1 eq., 0.04 mmol, 16.1 mg) and AIBN (0.6 eq., 0.024 mmol, 3.9 mg) were dissolved into anhydrous DMSO (10 mL). The solution was bubbled with dry argon to remove dissolved oxygen for 20 min at room temperature and then immersed in a preheated oil bath at 70 °C for 16 h. The solution was then rapidly cooled under air. The copolymer was precipitated into cold methanol and then washed three times with cold methanol followed by centrifugation (5000 rpm, 10 min). The copolymer was then dried under high vacuum to give 185 mg of a white powder. The purified copolymers were characterized by 1H-NMR and SEC. 1H-NMR (300 MHz, DMSO-d6) δ ppm: 0.82–0.89 (t, 3H–C12), 1.20–1.31 (m, H–C12), 1.31–1.73 (m, 2H-AAm), 2.01–2.25 (m, 1H-AAm), 4.55–4.77 (m, 4H-closed BMDO), 4.97–5.21 (m, 2H-opened BMDO), 6.59–7.52 (m, 6H: 2H-AAm + 4H-BMDO).000 g mol−1 (P10) and 20000 g mol−1 (P11)] were synthesized by varying the initial amount of AAm, the amounts of the other reagents being fixed (AAm = 0.63, 1.26 and 2.53 g, for P9–P11, respectively). For P9, the synthesis was as follows: AAm (78 eq., 8.91 mmol, 0.63 g), Gem-CDP (1 eq., 0.115 mmol, 74.9 mg) and AIBN (0.6 eq., 0.069 mmol, 11.4 mg) were dissolved in anhydrous DMSO (6 mL). The solution was bubbled with dry argon to remove dissolved oxygen for 20 min at room temperature and then immersed in a preheated oil bath at 70 °C for 16 h. The solution was then rapidly cooled under air. The polymer was precipitated into cold methanol and then washed three times with cold methanol followed by centrifugation (5000 rpm, 10 min). It was then dried under high vacuum to give 505 mg of a white powder. The purified polymer was characterized by 1H-NMR and SEC. 1H-NMR (400 MHz, DMSO-d6) δ ppm: 0.82–0.88 (t, 3H–C12), 1.18–1.28 (s, H–C12), 1.30–1.73 (m, 2H-AAm), 1.96–2.27 (m, 1H-AAm), 3.57–3.71(m, 1H-Gem), 3.76–3.93 (m, 2H-Gem), 4.06–4.25 (m, 1H-Gem), 5.21–5.35 (m, 1H-Gem), 6.15–6.20 (t, 1H-Gem), 6.30–6.32 (d, 1H-Gem), 6.41–7.65 (m, 2H-AAm), 8.23–8.25 (d, 1H Gem), 11.01–11.13 (t, 1H-Gem). The same procedure was followed for P10 and P11, yielding 1.1 g of P10 and 1.3 g of P11 after drying under high vacuum.2.4 Degradation procedures
2.5 In vitro evaluations
:methanol (90:10, v/v), followed by ultracentrifugation (13200 rpm, 20 min). The supernatant was then evaporated to dryness under a nitrogen flow at 30 °C and the released drug was quantified by reverse-phase HPLC. The chromatographic system was composed of a Waters 1525 Binary HPLC pump, a Waters 2707 Autosampler, a C18 Uptisphere column (3 μm, 150 × 4.6 mm; Interchim), HPLC column temperature controllers (model 7950 column heater and chiller; Jones Chromatography, Lakewood, CO) and a Waters 2998 programmable photodiode-array detector. The HPLC column was maintained at 30 °C and detection was monitored at 270 nm. The HPLC mobile phase was a mixture of methanol:water with 0.05 M sodium acetate (pH 5.0, eluent A: 5:95, v/v; eluent B: 97:3, v/v). The residues were dissolved in 100 μL of eluent A and centrifuged (13200 rpm, 5 min) before analysis. Elution was performed at a flow rate of 0.8 mL min−1 isocratically for 8 min with eluent A followed by a linear gradient (1 min) to 75% eluent A and kept isocratically for 6 min at 75% eluent A. A linear gradient (1 min) to 100% eluent B was followed by 10 min of isocratic gradient at 100% eluent B. After a linear gradient (1 min) to 100% eluent A, the system was held for 7 min for equilibration back to initial conditions. HPLC graphs and calibration curves can be found in Fig. S1–3,† respectively.2.6 In vivo evaluations
:100), and mouse anti-Ly6G (BioLegend, France, clone A8; diluted 1:50), followed by staining with appropriate secondary antibodies, Alexa FluorTM 488 (Invitrogen, ThermoFisher Scientific A-11008; diluted 1:250) and Alexa FluorTM 594 (Invitrogen, ThermoFisher Scientific A-11012; diluted 1:250). DNA was visualized upon Hoechst counterstaining (Invitrogen, ThermoFisher Scientific H3570). Slides were scanned using NanoZoomer 2.0-RS digital slide scanner (Hamamatsu, Japan). Images were digitally captured from the scanned slides using NDP.view2 software (Hamamatsu).3. Results and discussion
3.1 Polymer prodrug synthesis and physicochemical evaluation
The copolymer prodrugs are composed of AAm units, to confer hydrophilicity, and BMDO units, as precursors of ester groups in the copolymer backbone. This copolymerization system was chosen because of the proven UCST properties of P(AAm-co-BMDO) copolymers, typically between 23 and 55 °C depending on the BMDO content.31 They also exhibit rapid hydrolytic degradation under physiological conditions, faster than that of traditional aliphatic polyesters such as polycaprolactone (PCL), polylactide (PLA) and even poly(lactic-co-glycolic acid) (PLGA), which are still regarded as benchmarks in the field of biodegradable polymers. These two properties therefore guarantee: (i) complete water-solubility in SC tissue ensured by a correctly adjusted Tcp value, a prerequisite for reaching systemic circulation and (ii) efficient excretion of low molar mass copolymer fragments during degradation in vivo. Importantly, due to the rapid degradation of these copolymers, we reasoned that triggering water-solubility only after injection in the SC tissue would prevent early degradation of the copolymer during formulation and storage, and hence early release of Gem, which would be caused by increased solvation of the Gem-polymer linker. The rationale on developing UCST polymer prodrug is summarized in Fig. S4.† | ||
| Fig. 2 Synthesis and characterization of Gem-P(AAm-co-BMDO) copolymer prodrugs. (a) RAFT-mediated copolymerization of AAm and BMDO from Gem-CDP; (b) SEC chromatograms in DMSO of Gem-P(AAm-co-BMDO) copolymer prodrugs (P0–P4, Table 1); (c) 1H-NMR spectra (300 and 400 MHz, DMSO-d6) in the 0–11.5 ppm region of Gem-P(AAm-co-BMDO) copolymer prodrugs (P0–P4, Table 1). | ||
A small library of Gem-P(AAm-co-BMDO) copolymer prodrugs was obtained by varying the AAm:BMDO molar ratio (from 50:50 to 60:40) and by targeting an overall number-average degree of polymerization (DPn) of 200 (P0–P4, Table 1). The copolymerizations exhibited AAm conversions ranging from 40 to 90%, depending on the BMDO content (the higher fBMDO,0, the lower the AAm conversion). They were well-controlled, with Mn,NMR values ranging from 6020 to 9140 g mol−1 and fairly low dispersities obtained (Đ = 1.3–1.5) (Table 1 and Fig. 2b). 1H-NMR and 19F-NMR spectra confirmed the presence of Gem on the copolymer structures, due to the presence of specific proton peaks in the 5–11 ppm region (Fig. 2c), as well as a peak characteristic of their two fluorine atoms at 120 ppm (Fig. S5†). Two Gem-free P(AAm-co-BMDO) copolymers (P5 and P6, Mn,NMR = 8840 and 8860 g mol−1, Đ = 1.2 and 1.1, AAm:BMDO = 56:44 and 57:43, respectively) were also synthesized as controls by RAFT copolymerization of AAm and BMDO from CDP, under the same experimental conditions as for the synthesis of Gem-P(AAm-co-BMDO) (Fig. S6† and Table 1).
| Entry | Initial monomer feed, f0 (mol%) | Copolymer composition, Fa (mol%) | Open BMDOb (mol%) | AAm conversionc (mol%) | Mn,NMRd (g mol−1) | Mn,SECe (g mol−1) | Đe | Drug loadinga (wt%) | Tcpg from UV (°C) | Tcpi from DLS (°C) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAm | BMDO | AAm | BMDO | Heating | Cooling | ||||||||
| a Determined by 1H-NMR after purification, according to: MW Gem/(MW Gem + Mn,NMR polymer prodrug).b Determined by 1H-NMR after precipitation by integrating the 2H (–NH2) of AAm, the 4H (aromatic protons) of open and closed BMDO (6.5–7.5 ppm), the 2H of open BMDO (4.9–5.2 ppm) and the 4H of closed BMDO (4.5–4.8 ppm). n.a. = not applicable.c Determined by 1H-NMR (300 MHz) by integrating the 2H of AAm (6.02–6.24 ppm) at t = 0 and 16 h.d Determined by 1H NMR after purification by integrating the 3H from the CH3 moiety of the RAFT agent C12 alkyl chain (0.86 ppm), the 1H of AAm (2.1 ppm), the 2H of open BMDO (4.9–5.2 ppm) and the 4H of closed BMDO (4.5–4.8 ppm). Note that this method is only accurate for high living chain fractions.e Determined by SEC in DMSO.f Copolymers obtained from the Gem-free CDP RAFT agent.g Determined from the maximum of the first derivative of the heating and cooling curves obtained by UV-Vis spectroscopy at 1 °C min−1 and at 10 mg mL−1 in MilliQ water.h No thermosensitivity in absence of BMDO units.i Determined by DLS from the inflection point of the Dz vs. temperature curve upon cooling at 10 mg mL−1 in MilliQ water.j Not determined. | |||||||||||||
| P0 | 50 | 50 | 84.6 | 15.4 | 90 | 40 | 7300 | 10000 |
1.20 | 4.1 | 64 | 62 | —j |
| P1 | 55 | 45 | 90.2 | 9.8 | 91 | 70 | 6020 | 8310 | 1.25 | 5.0 | 55 | 56 | 41 |
| P2 | 56 | 44 | 90.6 | 9.4 | 90 | 87 | 9000 | 10300 |
1.26 | 3.3 | 37 | 27 | 31 |
| P3 | 57 | 43 | 92.2 | 7.8 | 88 | 90 | 8300 | 9100 | 1.32 | 3.6 | 22 | 22 | 26 |
| P4 | 60 | 40 | 92.7 | 7.3 | 89 | 61 | 9140 | 10880 |
1.52 | 3.3 | 8 | 8 | 6 |
| P5 | 56 | 44 | 90.7 | 9.3 | 91 | 80 | 8840 | 10010 |
1.20 | —f | 40 | 34 | —j |
| P6 | 57 | 43 | 92.1 | 7.9 | 90 | 88 | 8860 | 10890 |
1.14 | —f | 18 | 18 | —j |
| P7 | 100 | 0 | 100 | 0 | n.a. | 79 | 9700 | 17250 |
1.52 | 3.1 | —h | —h | —h |
| P8 | 100 | 0 | 100 | 0 | n.a. | 97 | 8930 | 16210 |
1.24 | —f | —h | —h | —h |
| P9 | 100 | 0 | 100 | 0 | n.a. | 97 | 7700 | 6180 | 1.34 | 3.9 | —h | —h | —h |
| P10 | 100 | 0 | 100 | 0 | n.a. | 94 | 12410 |
12260 |
1.26 | 2.4 | —h | —h | —h |
| P11 | 100 | 0 | 100 | 0 | n.a. | 91 | 27490 |
21340 |
1.29 | 1.1 | —h | —h | —h |
The composition of the different purified copolymers as well as the average percentage of ring-opened and closed BMDO units were determined by 1H-NMR spectroscopy. All copolymer prodrugs (P0–P4, Fig. 2c and Table 1) and drug-free copolymers (P5–P6, Fig. S6† and Table 1) exhibited very high molar fractions of ring-opened BMDO (88–91 mol%), suggesting a great susceptibility to hydrolysis. When the initial molar fraction of BMDO (fBMDO,0) is varied from 0.4 to 0.5, the BMDO contents in the copolymers (FBMDO) was in the 0.07–0.15 range, as expected from the unfavorable reactivity ratios (rBMDO = 0.23 and rAAm = 13.02)31 (Table 1).
 | ||
| Fig. 3 Degradation studies of Gem-P(AAm-co-BMDO) copolymer prodrug. (a) Evolution of the SEC chromatograms at t = 0 h (solid line) and t = 1 h (dotted line) during hydrolytic degradation under accelerated conditions (aqueous KOH 5 wt% solution, room temperature) of Gem-P(AAm-co-BMDO) copolymer prodrugs P0–P4 (Table 1); (b) evolution of the SEC chromatograms at different times (0, 24, 72 and 168 h) during hydrolytic degradation under physiological conditions (PBS, pH 7.4, 37 °C) of Gem-P(AAm-co-BMDO) prodrug P1–P3 (Table 1). | ||
| Entry | Mn,SECa (g mol−1) | Đa | Mn,deg. accel.b (g mol−1) (% Mn loss) | Đdeg accel.b | Mn,deg. physio.c (g mol−1) (% Mn loss) | Đdeg. physio.c | Mn,deg.theo.d (g mol−1) (% Mn loss) |
|---|---|---|---|---|---|---|---|
| a Determined by SEC in DMSO.b Determined by SEC in DMSO after hydrolytic degradation in aqueous KOH 5 wt% solution for 1 h.c Determined by SEC in DMSO after degradation under physiological conditions (PBS, 37 °C, 7 days).d Determined according to: Mn,deg. theo. = ([1/(open BMDO × FBMDO)] − 1) × MW(AAm) + MW(BMDO), with MW being the molecular weight of the considered monomers.e The degradation was not carried out. | |||||||
| P0 | 10000 |
1.20 | 600 (−94%) | 1.78 | —e | —e | 1400 (−86%) |
| P1 | 8310 | 1.25 | 1050 (−87%) | 2.04 | 1460 (−82%) | 1.82 | 870 (−90%) |
| P2 | 10300 |
1.26 | 1140 (−89%) |
2.17 | 1700 (−83%) | 1.94 | 970 (−91%) |
| P3 | 9100 | 1.32 | 860 (−91%) | 2.27 | 2080 (−77%) | 1.83 | 1100 (−88%) |
| P4 | 10880 |
1.52 | 1000 (−91%) | 2.02 | —e | —e | 2200 (−80%) |
Overall, Gem-P(AAm-co-BMDO) prodrugs P0–P4 exhibited sharp UCST transitions upon cooling and heating in water in the 8–64 °C range depending on FBMDO (Fig. 4a), confirming that the BMDO contents investigated (FBMDO = 0.073–0.154) were sufficient to confer thermosensitivity. The transmittance curves of P0–P4 shifted towards higher Tcp values as function of FBMDO (Fig. 4a), following an exponential plateau curve and suggesting some predictability of the Tcp value when varying the copolymer composition (Fig. 4b). These values were around 10 to 20 °C higher than those obtained in the absence of Gem for similar BMDO contents.31 It is also worth noting the sensitivity of the system, since a wide range of Tcp values was obtained by reducing the BMDO content from 15.4% to just 7.3% (Fig. 4b and Table 1). Interestingly, the copolymer prodrug P2 (FBMDO = 0.094, Table 1) has a Tcp value that spans the temperature of the SC tissue (Tcp = 27 and 37 °C upon cooling and heating, respectively), with hysteresis between heating and cooling cycles similar to that observed by DLS (Fig. 4a and c). This hysteresis could be attributed to the onset of polymer degradation during measurement, altering the balance between polymer–polymer and polymer–water interactions. To better understand the evolution of Tcp over time, the Tcp of P3 in water was followed by UV transmittance measurement for 3 days (Fig. 4d). Tcp shifted from 27 to 5 °C after 24 h and the UCST properties disappeared completely after 72 h, confirming the influence of polymer degradation on thermosensitivity.
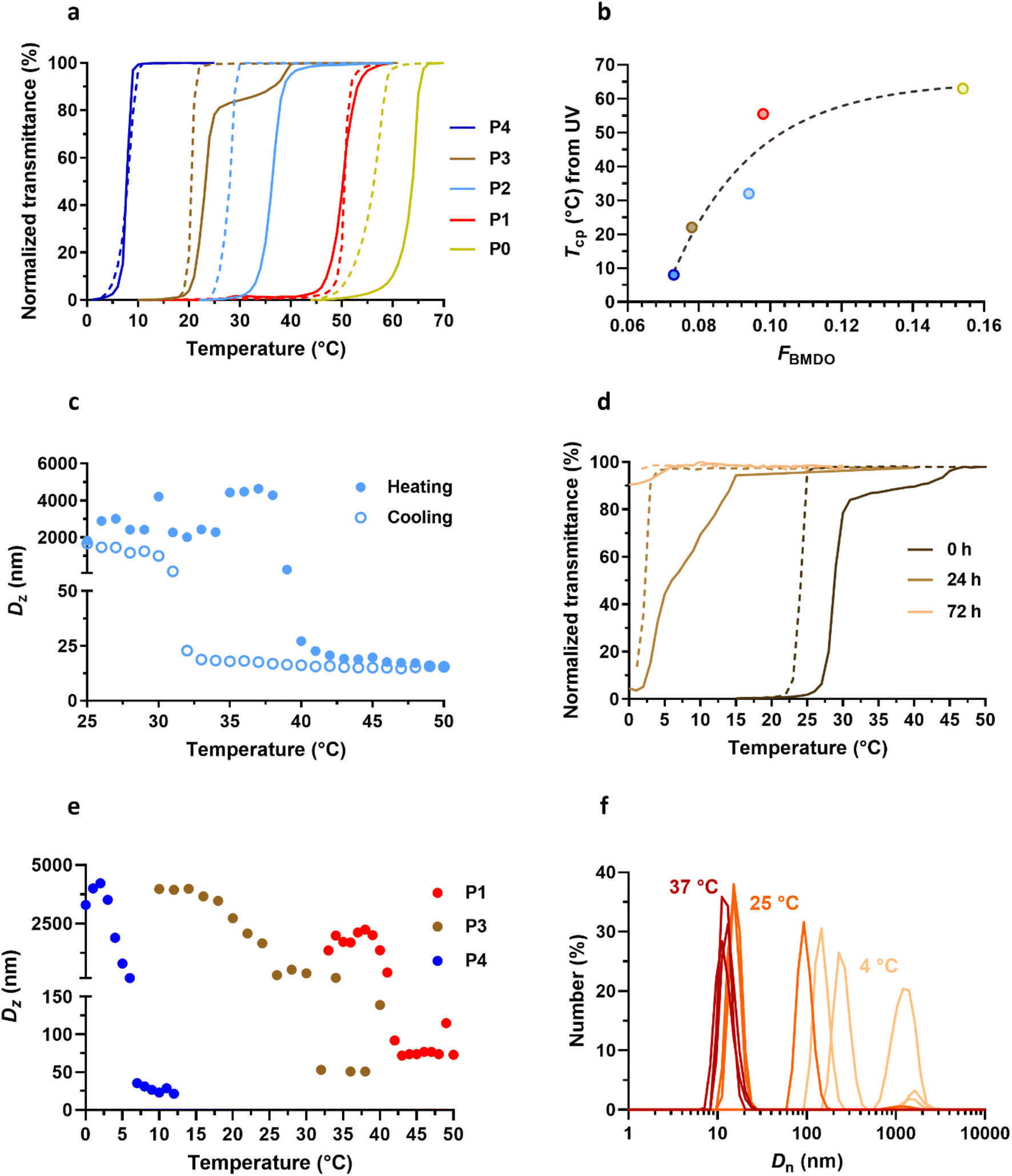 | ||
| Fig. 4 Thermosensitive properties of Gem-P(AAm-co-BMDO) copolymer prodrugs. (a) Variation of the UV transmittance in MilliQ water as function of the temperature of Gem-P(AAm-co-BMDO) copolymer prodrugs P0–P4 solutions at 10 mg mL−1 upon heating (solid lines) and cooling (dotted lines); (b) evolution of Tcp measured by UV (average between heating and cooling values, see Table 1) as a function of FBMDO for copolymers P0–P4; (c) DLS evolution of the intensity-weighted mean diameter (Dz) with temperature of Gem-P(AAm-co-BMDO) copolymer prodrug P2 at 10 mg mL−1 in MilliQ water upon cooling (empty dots) and heating (plain dots); (d) variation of the UV transmittance with temperature of Gem-P(AAm-co-BMDO) copolymer prodrug P3 solution upon heating (solid lines) and cooling (dotted lines) at 10 mg mL−1 in MilliQ water at t = 0, 24 and 72 h; (e) DLS evolution of Dz with temperature of Gem-P(AAm-co-BMDO) copolymer prodrugs P1, P3 and P4 at 10 mg mL−1 in MilliQ water upon cooling; (f) evolution of the number-weighted mean diameter (Dn) (n = 3) of Gem-P(AAm-co-BMDO) copolymer prodrug P3 at 10 mg mL−1 in MilliQ water at T = 4 °C (beige), 25 °C ∼ Tcp (orange) and 37 °C (red). | ||
Measurement of Dz by DLS of P1–P4 at different temperatures gave Tcp values in agreement with those determined by UV transmittance measurements (Table 1 and Fig. 4c and e). In addition, the evolution of Dn with temperature confirmed the solubility of copolymer prodrugs above Tcp, as shown by the presence of 10 nm-unimers, while much larger objects were measured at lower temperatures (T ≪ Tcp) (Fig. 4f). Note also the coexistence of 15 nm-unimers and 100 nm-nano-objects at T = Tcp for P3, suggesting partial (or ongoing) solubilization of the copolymer prodrug.
3.2 In vitro characterization
The in vitro biological evaluation (i.e., release, cytotoxicity and injectability) was performed using P2 and P3 polymer prodrugs as they exhibit both lower Tcp values than that of the SC tissue (Tcp,DLS = 31 and 26 °C, respectively) and the highest ester contents, making them promising candidates for future in vivo applications. This study has been completed by the evaluation of non-degradable (i.e., BMDO-free) polymer prodrug Gem-PAAm P7 and its drug-free counterpart PAAm P8. | ||
| Fig. 5 In vitro biological evaluation of Gem-P(AAm-co-BMDO), Gem-PAAm, P(AAm-co-BMDO) and PAAm. (a) HPLC release profiles of Gem from Gem-P(AAm-co-BMDO) P2 determined at 37 °C in MilliQ water (blue) and in human serum (orange); (b) cell viability (MTT test) of Mia PaCa-2 cells after incubation for 72 h with increasing concentrations of Gem, Gem-P(AAm-co-BMDO) P2 and P3 and Gem-PAAm P7; (c) determination of the IC50 for the four tested conditions (unpaired two-tailed t test; *p < 0.05, P2, P7 vs. P3); (d) number-weighted mean diameter (Dn) of aqueous solutions of P2, P3 and P7 at 1.23 mg mL−1 stored at 37 °C; (e) cell viability (MTT test) of Mia PaCa-2 cells after incubation for 72 h with increasing concentrations of P(AAm-co-BMDO) P5 and P6, and PAAm P8; (f) force needed using a syringe fitted with a 26 G needle to inject an aqueous solution of P2 and P7 at different concentrations (20–100 mg mL−1 for P2 and 100–1500 mg mL−1 for P7). The horizontal dashed line of 30 N is considered as the maximum acceptable injection force for SC administration.46 The values are expressed as the means ± SD. | ||
Both P2 and P3 led to significant cytotoxicity, with IC50 values of 180 nM and 770 nM, respectively (Fig. 5b and c). As is often the case with polymer prodrugs, P2 and P3 exhibited higher IC50 values than that of free Gem (IC50 = 18 nM), due to the cleavage step of the Gem-polymer covalent linkage to release the parent drug. Importantly, P3 was significantly less cytotoxic than P2, with an IC50 value 4-times higher than that of P2 (Fig. 5c). As expected from their different Tcp values, DLS evaluation of P2 and P3 in solution at 37 °C showed different colloidal properties, as P2 was characterized by coexistence of nano-objects of different sizes (Dn = 190 nm and 340 nm), whereas P3 gave unimers of ∼10 nm in size (Fig. 5d). This can have an impact on cellular uptake mechanisms and eventually on cytotoxicity, as nano-objects have been shown to follow different endocytosis pathways depending on their size44 and Mia PaCa-2 are associated with enhanced macropinocytosis (i.e., which favors internalization of nano-objects >250 nm in size).45 Moreover, fully soluble Gem-P(AAm-co-BMDO) unimers P3 are likely to be much prone to degradation throughout the MTT assay, producing small oligomer prodrugs, compared to Gem-P(AAm-co-BMDO) aggregates P2. We also showed that Gem-free copolymers P(AAm-co-BMDO) P5 and P6 were not cytotoxic up to at least 5 μM, ruling out potential cytotoxicity from the copolymer itself (Fig. 5e).
To study independently the influence of degradation on cytotoxicity, we also synthesized and evaluated two non-degradable, BMDO-free copolymers of similar chain length: a Gem-PAAm polymer prodrug P7 (Mn,NMR = 9700 g mol−1, Đ = 1.52) and its Gem-free counterpart PAAm P8 (Mn,NMR = 8930 g mol−1, Đ = 1.24) (Fig. S8† and Table 1). As expected, due to the absence of BMDO units in the polymer chains, P7 and P8 did not exhibit UCST behavior. Whereas PAAm P8 was also not cytotoxic up to at least 5 μM (Fig. 5e), Gem-PAAm P7 produced ∼10 nm-unimers (Fig. 5d), but exhibited the same IC50 value as Gem-P(AAm-co-BMDO) P2 aggregates and thus remained more cytotoxic than Gem-P(AAm-co-BMDO) P3 unimers (Fig. 5b and c). This result could be attributed to the persistence of the PAAm chain in Gem-PAAm P7 compared to the rapid degradation of Gem-P(AAm-co-BMDO) P3 into small oligomers.
The injectability of aqueous solutions of Gem-P(AAm-co-BMDO) P2 and Gem-PAAm P7 was measured as function of the concentration with a 26 G × 1/2′′ needle, which is within the size range suitable for humans (25–27 G). Previous work demonstrated that the force needed to inject PAAm (Mn = 37000 g mol−1) remained well below the maximum tolerated force of 30 N (<5 N at 200 mg mL−1), whereas injection of Ptx-PAAm considerably increased the required force (∼20 N at 100 mg mL−1), which even exceeded the 30 N limit at 200 mg mL−1.20 This was assigned to intermolecular hydrophobic interactions between the Ptx moieties and/or between the Ptx moieties and the C12 alkyl chain from the RAFT end groups. Herein, P2 and P7 were designed with a hydrophilic drug and a lower Mn (∼10000 g mol−1), to maintain low viscosity and allow high doses to be administered subcutaneously. At a concentration up to 100 mg mL−1, which corresponds to a dose in Gem of ∼50 mg kg−1, the SC injection of both polymer prodrugs required a very low force of ∼1 N (Fig. 5f), which is comparable to the force required for PAAm alone.20 Not only this result confirms the absence of hydrophobic interactions between the Gem groups and/or between the Gem groups and the terminal C12 alkyl chains, but it also ruled out detrimental effect of hydrophobic BMDO units on the viscosity of P2. The Gem-based polymer prodrugs synthesized in this work can therefore be injected more easily via the subcutaneous route than the Ptx-PAAm prodrugs. To assess injectability at very high concentrations, P7 was injected at 1500 mg mL−1 (corresponding to a Gem equivalent dose of ∼650 mg kg−1), which led to a 3-fold increase of the force value (3.6 N), but remained well below the limit of 30 N. Therefore, high doses of water-soluble, Gem-based polymer prodrugs can be easily administered subcutaneously.
3.3 In vivo evaluations
As the MTD of Gem injected intravenously (GemIV) is about 80 mg kg−1 after four IV injections,47 increasing concentrations of Gem from 80 to 160 mg kg−1 were injected subcutaneously (GemSC) to healthy mice following two different injection protocols: (i) a single injection at day 0, or (ii) four injections of a quarter dose each at days 0, 4, 7 and 11. Notably, such doses of GemSC did not lead to body weight loss, either using one unique or four injections (Fig. S9a–c†). Thus, higher doses of GemSC from 200 to 1000 mg kg−1 were injected using the most convenient single injection protocol, as no difference was observed between the two protocols. While doses from 200 to 650 mg kg−1 were well tolerated, a significant body weight loss was observed at 1000 mg kg−1 (Fig. 6a), indicating a MTD for GemSC ranging between 650 and 1000 mg kg−1. These results showed that the SC route achieved higher MTD values than the IV route, probably due to a reduction in the Cmax value (defined as the highest concentration of a drug), caused by the time required for the Gem to diffuse from the SC tissue into the systemic circulation.
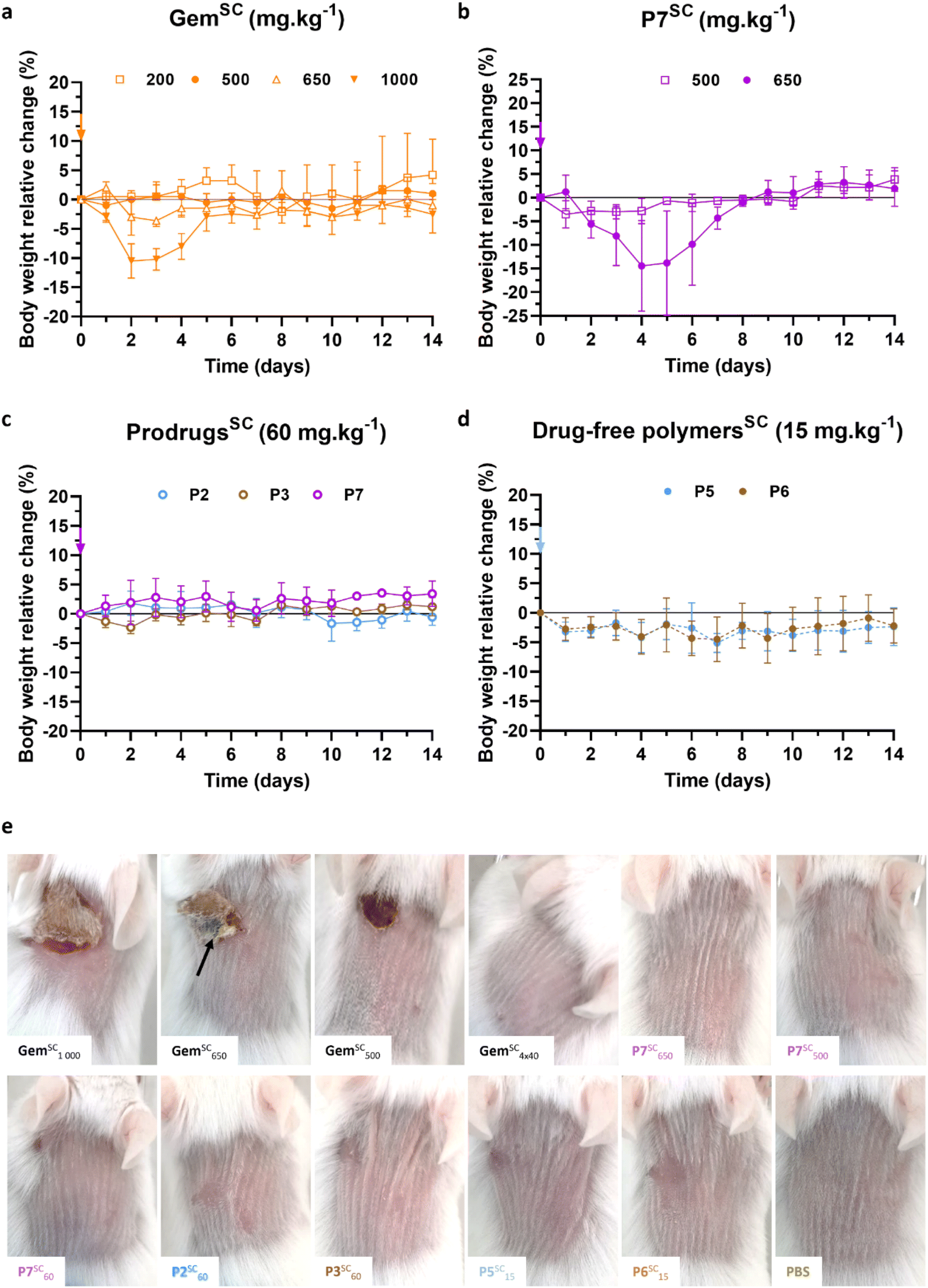 | ||
| Fig. 6 SC injection of Gem, Gem-based polymer prodrugs and control polymers to mice. Relative body weight changes of mice as a function of time after single SC injection of: (a) free Gem at different doses (200, 500, 650 and 1000 mg kg−1); (b) Gem-PAAm P7 at 500 and 650 mg kg−1; (c) Gem-P(AAm-co-BMDO) P2, P3 and Gem-PAAm P7 at 60 mg kg−1, and (d) drug-free P(AAm-co-BMDO) P5 and P6 at 15 mg kg−1 (equiv. Gem; that is 85 mg mL−1 in terms of polymer concentration). The values are expressed as the means ± SD (n = 3); (e) representative pictures of mice 8 days after SC injection of Gem at 1000, 650, 500 mg kg−1 (single injection) and 160 mg kg−1 (4 injections), Gem-PAAm P7 at 650, 500 and 60 mg kg−1 (single injection), Gem-P(AAm-co-BMDO) P2 and P3 at 60 et 15 mg kg−1 (single injection), drug-free P(AAm-co-BMDO) P5 and P6 at 15 mg per kg equiv. Gem (single injection), and PBS (single injection). The black arrow indicates necrosis zone. | ||
Gem-PAAm P7 was then injected subcutaneously (P7SC) at 500 and 650 mg kg−1 to healthy mice via a single injection (Fig. 6b) to determine its MTD. P7SC did not induce significant body weight loss (<5%) at 500 mg kg−1, which can already be considered as a very high drug concentration for SC administration. However, a transient decrease in body weight of 14% (which returned to normal after 3 days for all mice) was observed at 650 mg kg−1 after two days, suggesting similar MTD than GemSC.
Gem-P(AAm-co-BMDO) P2 and P3, and Gem-PAAm P7 were then SC-injected at 60 mg kg−1 (Fig. 6c), while drug-free P(AAm-co-BMDO) copolymers P5 and P6 were SC-injected at 15 mg per kg equiv. Gem (that is 85 mg mL−1 in terms of polymer concentration) (Fig. 6d). This injection protocol was carried out to evaluate their toxicity at doses comparable to clinical use of GemIV, known under the brand name Gemzar® (Gemzar) from Lilly France SA,48 that is 1000 mg.m−2, which is equivalent to 20 mg kg−1 for standard human weight and body surface. Importantly, none of these prodrugs and drug-free copolymers led to significant body weight loss compared to untreated mice which received PBS (Fig. S9d†).
 | ||
| Fig. 7 Histological evaluation of the injection site after SC administration of Gem, Gem-PAAm, Gem-P(AAm-co-BMDO) and PBS to mice. Representative HES-stained sections of skin sections after 14 days of mice at the injection site after SC administration of: (a) Gem and (b) Gem-PAAm P7 at 500 mg kg−1. Representative immunofluorescence images of skin sections after 14 days of mice at the injection site after SC administration of: (c) Gem and (d) Gem-PAAm P7 at 500 mg kg−1, using primary antibodies anti-CD3 and anti-Ly6G, and toluidine blue staining for the detection of mast cells respectively. (e) Histopathological scoring (H-Score) of tissular inflammation in mice after SC injection of Gem at 1 000, 650 and 500 mg kg−1, Gem-PAAm P7 at 650, 500 and 60 mg kg−1, Gem-P(AAm-co-BMDO) P2 and P3 at 60 mg kg−1, P(AAm-co-BMDO) P5 and P6 at 15 mg kg−1 and PBS. The values are expressed as the means ± SD (n = 3). Unpaired two-tailed t-test; *p < 0.01 (Table S2†). (f) Histopathological scoring (H-Score) of necrotic changes in mice after SC injection of Gem at 1000, 650 and 500 mg kg−1, Gem-PAAm P7 at 650, 500 and 60 mg kg−1 and PBS. The values are expressed as the means ± SD (n = 3). Unpaired two-tailed t-test; ****p < 0.0001 (Table S2†). (g) Measurement of the epidermis' thickness from HES-stained sections of skin sections after 14 days after SC administration of Gem at 1 000, 650 and 500 mg kg−1, Gem-PAAm P7 at 650, 500 and 60 mg kg−1, Gem-P(AAm-co-BMDO) P2 and P3 at 60 mg kg−1, P(AAm-co-BMDO) P5 and P6 at 15 mg kg−1 and PBS. The values are expressed as the means ± SD after 10 measurements per sample (n = 3). (h) Representative HES-stained sections of skin sections after 14 days of mice at the injection site after SC administration of PBS. | ||
Remarkably, Gem-PAAm P7SC did not induce local toxicity at or near the injection site at the highest doses tested of 500 and 650 mg kg−1, as shown on representative pictures at days 1, 8 and 14 (Fig. 6e and S10†). Moreover, HES-stained sections of skin samples after 14 days did not show significant histopathological lesion or hyperplasia at 500 mg kg−1 (Fig. 7b and e–g), while one mouse out of 3 treated by Gem-PAAm P7SC at the MTD (650 mg kg−1) exhibited moderate inflammation. Immunofluorescence of skin sections after SC injection of Gem-PAAm P7SC at 500 mg kg−1 confirmed the absence of inflammation (Fig. 7d). Taken together, these promising results pave the way for safe SC administration of Gem at high doses under the form of water-soluble polymer prodrugs, since the cutaneous toxicity of free Gem has been suppressed.
Lower, more clinically relevant doses of Gem-P(AAm-co-BMDO) P2, P3 and Gem-PAAm P7 (60 mg kg−1), and drug-free counterparts P(AAm-co-BMDO) P5 and P6 (15 mg kg−1) did not induce any local toxicity at and near the injection site, as illustrated by pictures at days 1, 8 and 14 (Fig. 6e and S10†). Slight transient skin irritations appeared in isolated cases but did not persist after a few days. HES-stained sections of skin samples after 14 days (Fig. S11†) showed that the integrity of the SC tissue was preserved and that there was no inflammation (Fig. 7e) or necrosis (Fig. 7f), with the exception of a very small epidermal hyperplasia (Fig. 7g). This study also highlighted the harmlessness of adding BMDO to the polymer backbone, as no difference in toxicity was observed with or without BMDO in the copolymer (see P2 and P3 vs. P7).
410 g mol−1, Đ = 1.26 and P11: 27490 g mol−1, Đ = 1.29, see Fig. S12† and Table 1) was synthesized on a gram scale and high yield (58–84%), demonstrating the robustness of the synthesis protocol for obtaining large quantities. Chain length variation was intended to investigate the influence of polymer chain length on anticancer efficacy, as this parameter it is expected to have an impact on biodistribution, body excretion and interaction with the immune system.49 Importantly, all molar masses were chosen to be below the kidney filtration threshold (∼6 nm in size, which correspond to ∼40 kg mol−1 for PEG50–52), to facilitate renal excretion. Varying the chain length also gave the possibility to target different drug loadings (3.9, 2.4 and 1.1 wt% for P9–P11, respectively).SC injections of water-soluble Gem-PAAm prodrugs P9–P11 were carried out once a week during 3 consecutive weeks (i.e., days 17, 24 and 31) at 60 mg per kg Gem equiv. dose (which corresponds to the MTD of Gemzar) and benchmarked against the IV-injection of the commercial formulation of Gem (GemzarIV) at the same dose, in mice bearing Mia PaCa-2 pancreatic xenografts, which is a relevant model for Gem. The efficacy of the treatments was evaluated by following the evolution of tumor volume (Fig. 8a) and the survival rate (Fig. 8b).
 | ||
| Fig. 8 Evaluation of the anticancer efficacy of SC-injected water-soluble Gem-based polymer prodrugs and IV-injected Gemzar®. (a) Tumor growth evolution with time after tumor induction [the values are expressed as the means ± SEM (n = 7–12 per group). Two-way ANOVA, with Tukey's correction for multiple comparisons between GemzarIV and prodrug groups at days 71 and 74; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.002, ****p < 0.0001]; (b) body weight evolution with time after tumor induction [the values are expressed as the means ± SEM (n = 7–12 per group)] and (c) survival rate evolution with time after tumor induction [the values are expressed as the means ± SEM] of mice bearing Mia Pa-Ca 2 xenografts after injection of GemzarIV at 60 mg kg−1, and injection of P9SC–P11SC at 60 mg per kg Gem equiv. dose, on days 17, 24 and 31 (indicated by black arrows in panel a). | ||
Firstly, there were no adverse cutaneous reactions at the injection sites during treatment, demonstrating the absence of local toxicity of polymer prodrugs at such a dose as predicted by previous acute local toxicity study with P7sc (see Fig. 6 and 7). Treatments with P9SC–P11SC showed relatively similar tumor growth rates to GemzarIV up to 24 days after tumor induction (Fig. 8a), with an average tumor volume of ∼200 mm3. Subsequently, the tumor volume curves associated with P9SC–P11SC began to slowly diverge from that of GemzarIV towards significantly lower average tumor volumes, indicating greater anticancer efficacy. In particular, at day 74, the tumor volumes associated with P9SC–P11SC converged to a mean value of 1490 mm3, corresponding to a 22% tumor volume reduction compared with GemzarIV (1904 mm3). In addition, the evolution of the relative body weight loss in mice treated with Gem-PAAm prodrugs showed that the treatment was well tolerated, with mice losing no more than 10% on average of their body weight throughout the study (Fig. 8b). Remarkably, the SC injection of Gem-PAAm prodrugs also significantly increased the overall survival of mice. Indeed, 84 days after tumor induction, no mice survived in the GemzarIV group, whereas survival rates were 17, 36 and 33% for mice treated with P9SC, P10SC and P11SC, respectively (Fig. 8c). The lower survival rate observed with P9SC could be explained by the excessively short PAAm chain length, which led to a too rapid excretion of the prodrug, not allowing a significant release of Gem. In contrast, longer PAAm chain lengths induced a greater stealth effect and thus longer circulation times, allowing sustained release of Gem. Despite similar survival rates observed with P10SC and P11SC, and a lower drug loading for P11SC, we believe that P11SC is the best candidate as it gave a 100% survival rate for much longer than the other two prodrugs.
4. Conclusion
In this work, we have designed SC-injectable, water-soluble polymer prodrugs based on Gem as an irritant anticancer drug and PAAm as a highly water-soluble polymer. These polymer prodrugs were also made degradable by the insertion of ester groups in the main chain through rROP of BMDO with AAm during the “drug-initiated” synthesis. Not only the Gem-P(AAm-co-BMDO) copolymer prodrugs exhibited rapid degradation in a few days under physiological conditions, but they also showed tunable UCST properties depending on the BMDO content, allowing solubilization at body temperature after injection.Sustained drug release was achieved in human serum over a week and in vitro assays on a pancreatic cancer cell line showed significant cytotoxicity of the polymer prodrugs. Degradable and non-degradable polymer prodrugs were easily injected under clinically relevant SC injection conditions, up to high doses. Importantly, SC injection of Gem-PAAm prodrugs and their degradable counterparts at high doses to mice did not induce local toxicity or even inflammation compared to free Gem, which engendered severe inflammation and even necrotic areas. This suggested that these prodrugs can be safely injected subcutaneously without the BMDO units and degradation products being toxic. Remarkably, the SC administration of Gem-PAAm prodrugs to mice bearing Mia Pa-Ca 2 tumor xenografts resulted in anticancer efficacy and survival rates superior to those of the commercial formulation of Gem (Gemzar®), injected intravenously.
Altogether, these results therefore successfully demonstrated the possibility of switching from IV administration of Gemzar® to SC administration of water-soluble, Gem-PAAm prodrugs. They also propose alternative polymer backbones that are degradable, non-cytotoxic, and do not induce any cutaneous toxicities in vivo, opening the path to designing polymeric systems with better body clearance outcomes. Finally, this research work argues in favor of the use of the “drug-initiated” method to design water-soluble polymer prodrugs for SC administration of vesicant/irritant anticancer drugs and pave the way for safer and less costly administration of chemotherapeutics.
Data availability
Data will be made available on request.Author contributions
J. N. conceived and designed the research, and obtained funding; L.G designed the experiments; L. G., J. C., M. A. performed the experiments; A. B., C. Z., D. L., S. M. I., C. C. helped to perform the experiments; F. M. N. performed histology studies; L. G., J. N. wrote the manuscript. All authors contributed to the discussion of the results and to the revision of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 771829). We thank the ERC for the financial support of the PhD thesis of LG. The CNRS and University Paris-Saclay are also acknowledged for financial support. The authors thank Dr Julie Mougin (IGPS, Univ Paris-Saclay), Stéphanie Denis (IGPS, Univ Paris-Saclay) and Valérie Domergue (UMS IPSIT, AnimEx, Univ Paris-Saclay) for technical assistance in HPLC, cell culture and in vivo experiments, respectively.References
- N. Connelly and J. Pritchett, Global Oncology Trends 2023, The IQVIA Institute, 2023 Search PubMed.
- M. Duh, J. Weiner, P. Lefebvre, M. Neary and A. Skarin, Costs Associated with Intravenous Chemotherapy Administration in Patients with Small Cell Lung Cancer: A Retrospective Claims Database Analysis, Curr. Med. Res. Opin., 2008, 24, 967–974 CrossRef PubMed.
- J.-C. Lucet, L. Bouadma, J.-R. Zahar, C. Schwebel, A. Geffroy, S. Pease, M.-C. Herault, H. Haouache, C. Adrie, M. Thuong, A. Français, M. Garrouste-Orgeas and J.-F. Timsit, Infectious Risk Associated with Arterial Catheters Compared to Central Venous Catheters, Crit. Care Med., 2010, 38, 1030–1035 CrossRef PubMed.
- M. Pujol, A. Hornero, M. Saballs, M. J. Argerich, R. Verdaguer, M. Cisnal, C. Peña, J. Ariza and F. Gudiol, Clinical Epidemiology and Outcomes of Peripheral Venous Catheter-Related Bloodstream Infections at a University-Affiliated Hospital, J. Hosp. Infect., 2007, 67, 22–29 CrossRef CAS PubMed.
- S. Vento and F. Cainelli, Infections in Patients with Cancer Undergoing Chemotherapy: Aetiology, Prevention, and Treatment, Lancet Oncol., 2003, 4, 595–604 CrossRef PubMed.
- K. Beusterien, J. Grinspan, I. Kuchuk, S. Mazzarello, S. Dent, S. Gertler, N. Bouganim, L. Vandermeer and M. Clemons, Use of Conjoint Analysis to Assess Breast Cancer Patient Preferences for Chemotherapy Side Effects, Oncologist, 2014, 19, 127 CrossRef CAS PubMed.
- J.-F. Jin, L.-L. Zhu, M. Chen, H.-M. Xu, H.-F. Wang, X.-Q. Feng, X.-P. Zhu and Q. Zhou, The Optimal Choice of Medication Administration Route Regarding Intravenous, Intramuscular, and Subcutaneous Injection, Patient Prefer. Adherence, 2015, 9, 923–942 Search PubMed.
- M. Krohe, D. Eek, I. Mazar, A. Horsfield, F. Pompilus, R. Friebe and A. Shields, Patient-Reported Preferences for Oral versus Intravenous Administration for the Treatment of Cancer: A Review of the Literature, Patient Prefer. Adherence, 2016, 10, 1609–1621 CrossRef PubMed.
- Y. Wengström and A. Margulies, European Oncology Nursing Society extravasation guidelines, Eur. J. Oncol. Nurs., 2008, 12, 357–361 CrossRef.
- S. Huynh Dagher, A. Blom, H. Chabanol and E. Funck-Brentano, Cutaneous Toxicities from Targeted Therapies Used in Oncology: Literature Review of Clinical Presentation and Management, Int. J. Womens Dermatol., 2021, 7, 615–624 CrossRef PubMed.
- S. M. Shenaq, E.-H. A. Abbase and J. D. Friedman, Soft-Tissue Reconstruction Following Extravasation of Chemotherapeutic Agents, Surg. Oncol. Clin., 1996, 5, 825–846 CrossRef CAS.
- D. Leveque, Subcutaneous Administration of Anticancer Agents, Anticancer Res., 2014, 34, 1579–1586 CAS.
- B. Bittner, W. Richter and J. Schmidt, Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities, Biodrugs, 2018, 32, 1–16 CrossRef PubMed.
- L. Tomasini, M. Ferrere and J. Nicolas, Subcutaneous drug delivery from nanoscale systems, Nat. Rev. Bioeng., 2024, 2, 501–520 CrossRef CAS.
- V. Delplace, P. Couvreur and J. Nicolas, Recent Trends in the Design of Anticancer Polymer Prodrug Nanocarriers, Polym. Chem., 2014, 5, 1529–1544 RSC.
- L. Guerassimoff, M. Ferrere, A. Bossion and J. Nicolas, Stimuli-Sensitive Polymer Prodrug Nanocarriers by Reversible-Deactivation Radical Polymerization, Chem. Soc. Rev., 2024, 53, 6511–6567 RSC.
- J. Khandare and T. Minko, Polymer–Drug Conjugates: Progress in Polymeric Prodrugs, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Peptide–Drug Conjugates as Effective Prodrug Strategies for Targeted Delivery, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Polymer–drug conjugate therapeutics: advances, insights and prospects, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- A. Bordat, T. Boissenot, N. Ibrahim, M. Ferrere, M. Levêque, L. Potiron, S. Denis, S. Garcia-Argote, O. Carvalho, J. Abadie, C. Cailleau, G. Pieters, N. Tsapis and J. Nicolas, A Polymer Prodrug Strategy to Switch from Intravenous to Subcutaneous Cancer Therapy for Irritant/Vesicant Drugs, J. Am. Chem. Soc., 2022, 144, 18844–18860 CrossRef CAS PubMed.
- Y. Kaneda, Y. Tsutsumi, Y. Yoshioka, H. Kamada, Y. Yamamoto, H. Kodaira, S. Tsunoda, T. Okamoto, Y. Mukai, H. Shibata, S. Nakagawa and T. Mayumi, The Use of PVP as a Polymeric Carrier to Improve the Plasma Half-Life of Drugs, Biomaterials, 2004, 25, 3259–3266 CrossRef CAS PubMed.
- S. D'Epiro, M. Salvi, C. Mattozzi, S. Giancristoforo, M. Campoli, R. Zanniello, C. Luci, L. Macaluso, S. Giovannoni, R. Iacovelli, S. Calvieri and A. Richetta, Gemcitabine-Induced Extensive Skin Necrosis, Case Rep., 2012, 2012, 831616 Search PubMed.
- I. Kuku, E. Kaya, A. Sevinc and I. Aydogdu, Gemcitabine-Induced Erysipeloid Skin Lesions in a Patient with Malignant Mesothelioma, J. Eur. Acad. Dermatol. Venereol., 2002, 16, 271–272 CrossRef CAS PubMed.
- L. W. Hertel, G. B. Boder, J. S. Kroin, S. M. Rinzel, G. A. Poore, G. C. Todd and G. B. Grindey, Evaluation of the Antitumor Activity of Gemcitabine (2’,2’-Difluoro-2’-Deoxycytidine, Cancer Res., 1990, 50(14), 4417–4422 CAS.
- C. Galmarini, J. Mackey and C. Dumontet, Nucleoside Analogues: Mechanisms of Drug Resistance and Reversal Strategies, Leukemia, 2001, 15, 875–890 CrossRef CAS PubMed.
- S. Agarwal, Chances and Limitations of the Radical Ring-Opening Polymerization of Cyclic Ketene Acetals for the Synthesis of Degradable Polyesters, Polym. Chem., 2010, 1, 953–964 RSC.
- A. W. Jackson, Reversible-Deactivation Radical Polymerization of Cyclic Ketene Acetals, Polym. Chem., 2020, 11, 3525–3545 RSC.
- T. Pesenti and J. Nicolas, 100th Anniversary of Macromolecular Science Viewpoint: Degradable Polymers from Radical Ring-Opening Polymerization: Latest Advances, New Directions, and Ongoing Challenges, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- F. Sbordone and H. Frisch, Plenty of Space in the Backbone: Radical Ring-Opening Polymerization, Chem.–Eur. J., 2024, 30, e202401547 CrossRef CAS PubMed.
- A. Bossion, C. Zhu, L. Guerassimoff, J. Mougin and J. Nicolas, Vinyl Copolymers with Faster Hydrolytic Degradation than Aliphatic Polyesters and Tunable Upper Critical Solution Temperatures, Nat. Commun., 2022, 13, 2873 CrossRef CAS PubMed.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Thermoresponsive polymer nanocarriers for biomedical applications, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- P. Webb, Temperatures of Skin, Subcutaneous Tissue, Muscle and Core in Resting Men in Cold, Comfortable and Hot Conditions, Eur. J. Appl. Physiol., 1992, 64, 471–476 CrossRef CAS PubMed.
- V. Burckbuchler, G. Mekhloufi, A. P. Giteau, J. L. Grossiord, S. Huille and F. Agnely, Rheological and Syringeability Properties of Highly Concentrated Human Polyclonal Immunoglobulin Solutions, Eur. J. Pharm. Biopharm., 2010, 76, 351–356 CrossRef CAS PubMed.
- D. Vinciguerra, M. Jacobs, S. Denis, J. Mougin, G. Yohann, G. Lazzari, C. Zhu, S. Mura, P. Couvreur and J. Nicolas, Heterotelechelic Polymer Prodrug Nanoparticles: Adaptability to Different Drug Combinations and Influence of the Dual Functionalization on the Cytotoxicity, J. Controlled Release, 2019, 295, 223–236 CrossRef CAS PubMed.
- Y. Bao, T. Boissenot, E. Guégain, D. Desmaële, S. Mura, P. Couvreur and J. Nicolas, Simple Synthesis of Cladribine-Based Anticancer Polymer Prodrug Nanoparticles with Tunable Drug Delivery Properties, Chem. Mater., 2016, 28, 6266–6275 CrossRef CAS.
- A. Maksimenko, J. Mougin, S. Mura, E. Sliwinski, E. Lepeltier, C. Bourgaux, S. Lepêtre-Mouelhi, F. Zouhiri, D. Desmaële and P. Couvreur, Polyisoprenoyl Gemcitabine Conjugates Self Assemble as Nanoparticles, Useful for Cancer Therapy, Cancer Lett., 2012, 334, 346–353 CrossRef PubMed.
- P. Couvreur, B. Stella, L. H. Reddy, H. Hillaireau, C. Dubernet, D. Desmaële, S. Lepêtre-Mouelhi, F. Rocco, N. Dereuddre-Bosquet, P. Clayette, V. Rosilio, V. Marsaud, J.-M. Renoir and L. Cattel, Squalenoyl Nanomedicines as Potential Therapeutics, Nano Lett., 2006, 6, 2544–2548 CrossRef CAS.
- N. Aggarwal and B. F. Sloane, Cathepsin B: Multiple Roles in Cancer, PROTEOM.–Clin. Appl., 2014, 8, 427–437 CrossRef CAS.
- D. Dheer, J. Nicolas and R. Shankar, Cathepsin-Sensitive Nanoscale Drug Delivery Systems for Cancer Therapy and Other Diseases, Adv. Drug Delivery Rev., 2019, 151–152, 130–151 CrossRef CAS PubMed.
- A. J. M. D'Souza and E. M. Topp, Release from Polymeric Prodrugs: Linkages and Their Degradation, J. Pharm. Sci., 2004, 93, 1962–1979 CrossRef PubMed.
- A. Tardy, N. Gil, C. M. Plummer, D. Siri, D. Gigmes, C. Lefay and Y. Guillaneuf, Polyesters by a Radical Pathway: Rationalization of the Cyclic Ketene Acetal Efficiency, Angew. Chem., Int. Ed., 2020, 59, 14517–14526 CrossRef CAS PubMed.
- V. Heinemann, Gemcitabine: Progress in the Treatment of Pancreatic Cancer, Oncology, 2001, 60, 8–18 CrossRef CAS PubMed.
- S. Mazumdar, D. Chitkara and A. Mittal, Exploration and insights into the cellular internalization and intracellular fate of amphiphilic polymeric nanocarriers, Acta Pharm. Sin. B, 2021, 11, 903–924 CrossRef CAS.
- C. Commisso, S. M. Davidson, R. G. Soydaner-Azeloglu, S. J. Parker, J. J. Kamphorst, S. Hackett, E. Grabocka, M. Nofal, J. A. Drebin, C. B. Thompson, J. D. Rabinowitz, C. M. Metallo, M. G. Vander Heiden and D. Bar-Sagi, Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells, Nature, 2013, 497, 633–637 CrossRef CAS.
- R. Watt, H. Khatri and A. Dibble, Injectability as a Function of Viscosity and Dosing Materials for Subcutaneous Administration, Int. J. Pharm., 2018, 554, 376–386 CrossRef PubMed.
- A. Maksimenko, J. Caron, J. Mougin, D. Desmaële and P. Couvreur, Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies, Int. J. Pharm., 2015, 482, 38–46 CrossRef CAS PubMed.
- C. Manegold, Gemcitabine (Gemzar®) in non-small cell lung cancer, Expert Rev. Anticancer Ther., 2004, 4, 345–360 CrossRef CAS PubMed.
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. A. Satchi-Fainaro, Distribution, Metabolism and Elimination of Polymer Therapeutics, J. Controlled Release, 2012, 161, 446–460 CrossRef CAS PubMed.
- B. Du, M. Yu and J. Zheng, Transport and Interactions of Nanoparticles in the Kidneys, Nat. Rev. Mater., 2018, 3, 358–374 CrossRef.
- R. Duncan, The Dawning Era of Polymer Therapeutics, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- T. Yamaoka, Y. Tabata and Y. Ikada, Distribution and Tissue Uptake of Poly(Ethylene Glycol) with Different Molecular Weights after Intravenous Administration to Mice, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02967h |
| This journal is © The Royal Society of Chemistry 2025 |