
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed direct regioselective C5–H alkylation reactions of functionalized indoles with α-diazomalonates†
Tomohiro Isono,
Shingo Harada *,
Mai Yanagawa and
Tetsuhiro Nemoto*
*,
Mai Yanagawa and
Tetsuhiro Nemoto*
Graduate School of Pharmaceutical Sciences, Chiba University, Chuo-ku, Chiba 260-8675, Japan. E-mail: Sharada@chiba-u.jp; tnemoto@faculty.chiba-u.jp
First published on 15th July 2025
Abstract
Functionalization of the pyrrole nucleus in indoles has been extensively investigated, whereas benzenoid ring functionalization studies remain scarce. Advances in direct C–H functionalization have been hindered by the inherent low reactivity and need for precise control of regioselectivity. Herein, we developed regioselective C5–H alkylation reactions of indoles bearing carbonyl functionalities at the C3 position using copper-carbene species. This methodology regioselectively affords functionalized heteroarenes bearing synthetically useful structural motifs characteristic of bioactive molecules and natural products. Experimental and computational mechanistic findings underscored the pivotal importance of the copper catalyst system and rationalized the origin of regioselectivity.
Introduction
The chemical structure 2,3-benzopyrrole, known as indole, possessing 10 π-electrons serves as a fundamental core architecture in numerous natural products, pharmaceutical drugs, and functional materials.1 Among the various substitution patterns on the essential scaffold, 5-substituted indole derivatives have attracted considerable attention in recent years due to their unique biological activities and synthetic challenges in organic chemistry.2 Notable examples of C5-functionalized indoles3,4 include the natural products semicochliodinol A,5,6 isolated from rice cultures of the fungus genus Chaetomium, and penitrem D,7 isolated from Penicillium crustosum, as well as the therapeutic agent sumatriptan,8 used to treat migraine headaches.9,10 The C2 and C3 positions of indole exhibit pronounced reactivity, enabling a diverse array of chemical modifications. Conversely, straightforward synthetic methodologies for functionalizing the less reactive C4 to C7 positions are limited.11 Functionalization at the C4–H or C7–H positions of indoles can be achieved through strategic placement of directed metalation groups at the C3 position or the indole nitrogen, respectively. Direct modification of the C5 and C6 positions, however, remains a formidable synthetic challenge.12 In 2024, Ishikawa et al. successfully accomplished a highly selective bromination of indolo[2,3-a]quinolizidine through a unique mechanistic pathway involving the transient in situ generation of aniline derivatives (Fig. 1A).13 More recently, Liang et al. made a groundbreaking contribution by reporting the iridium-catalyzed C5-borylation of indoles, succeeding in the theoretical design of ligands that induce multiple non-covalent interactions, particularly dispersion forces (Fig. 1B).2 In these strategies, the installed halogen14 or boron functionalities served as valuable handles for further diversification through various coupling reactions.15–17 As a landmark work, Shi et al. established catalyst-controlled direct C4–H and C5–H arylation reactions of indoles bearing a removable pivaloyl group at the C3 position.18 The highly efficient strategy enables regioselective construction of C(sp2)–C(sp2) bonds and can serve as a powerful synthetic platform for the assembly of pharmaceutically relevant scaffolds. On the other hand, successful C5-selective alkylation for constructing C(sp3)–C(sp2) bonds remains limited,19–21 often producing only modest to low yields22,23 with poor regioselectivity.24,25 Regiocontrol can be achieved through intramolecular reactions,26–29 but more general and versatile methodologies are needed. Although direct arene alkylation utilizing carbene species30–35 represents a powerful synthetic strategy,36–38 achieving precise regioselectivity control remains a significant challenge (Fig. 1C).39 As an alternative approach, a strategy proceeding through indoline intermediates was developed by exploiting the para-directing nature of aniline derivatives, followed by dehydrogenative reoxidation to indoles.40,41 Within the framework of our research program,42–44 we focused on carbene chemistry45 to develop selective transformations for synthesizing high-value molecules.46 Our group demonstrated the C4-selective functionalization of indoles employing rhodium-carbene species.47 α,β-Unsaturated enones at the C3 positions were utilized as both directing groups and enophiles. Building upon this study, further optimization of the catalytic system enabled remote C5–H alkylation using the same substrate (Fig. 1D). Mechanistic studies indicated that Cu(OAc)(SbF6) is the actual active catalyst for carbene-transfer reactions.48–52 Copper presents an attractive alternative to precious metals in synthetic chemistry due to its environmentally friendly nature, low cost, and abundance.53–56 Herein, we describe the development of a regioselective C5–H functionalization protocol of indoles with α-diazomalonates as carbene sources. Computational studies were also performed to shed light on the origin of the regioselectivity.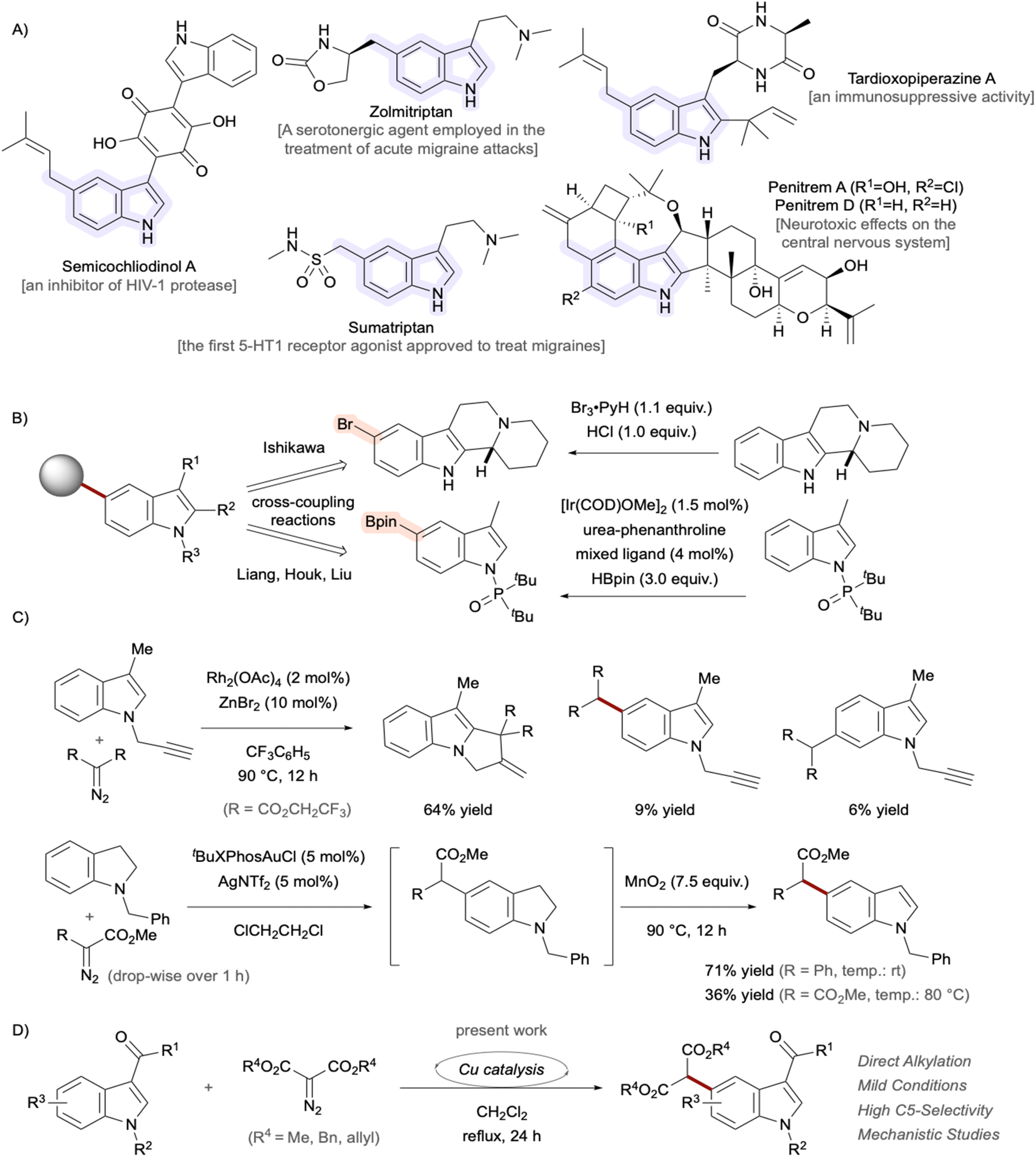 | ||
| Fig. 1 (A) Representative bioactive indole derivatives bearing alkyl substituents at the C5 position. (B) Established stepwise methodologies for accessing C5-functionalized indoles. (C) Reported reactions between indole/indoline derivatives and metal-carbene species. (D) Direct C5–H alkylation under copper catalysis. | ||
Results and discussion
We initiated our investigation using N-benzyl indole 1a bearing an enone functionality as the model substrate, along with dimethyl α-diazomalonates 2a (Table 1). At the outset, a combination of rhodium(II), copper(II), and silver(I) salts was examined based on previous studies, leading to the formation of the C5–H functionalized product 3a in 18% yield (entry 1).47 Trace amounts of the C4–H alkylated product (4a) and its cyclized derivative (5a) were also detected in the crude NMR analysis. To identify the catalytically active species, individual metal salts were examined separately; however, none of the reactions afforded the targeted product 3a (entries 2–4).57 In contrast, evaluation of binary metal salt combinations revealed that the Cu(OAc)2·H2O/AgSbF6 system selectively promoted C5–H alkylation (entries 5–7). Replacing AgSbF6 with AgNTf2 or AgBF4, or substituting Cu(OAc)2·H2O with an alternative Lewis acidic salt hydrate, such as ScCl3·6H2O, did not improve the catalytic activity in this reaction system (entries 8–10).58 Solvent screening demonstrated that CH2Cl2 was the appropriate reaction medium (entries 11 and 12).58 Reducing the solvent volume by approximately half and increasing the concentration to 0.02 M led to a slight enhancement in yield of 3a (77% yield, entry 13). Notably, the yield and regioselectivity were maintained even when the catalyst loading was reduced, affording 3a in 77% yield (entry 14). Rhodium(III)-mediated generation of Rh-carbene species facilitated C4–H alkylation (4a, 37% yield, entry 15). Ultraviolet irradiation-induced generation of a metal-free carbene did not afford 3a (entry 16), suggesting that Cu and Ag salts promote C5 selectivity via an alternative catalytic pathway. Further modifications of supporting ligands and copper valency did not improve the yield of 3a (entries 17–19); thus, the conditions described in entry 14 were determined to be optimal.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Metal salt(s) (mol%) | Temp. (°C) | Solvent (M) | Yield of 3a (%) | Yield of 4a (%) | Yield of 5a (%) |
| a All reactions were conducted with metal salt(s) (0.0025–0.1 mmol), 1a (0.1 mmol) and 2a (0.2 mmol) under the indicated conditions for 24 h without using a syringe pump for slow addition of the diazo compounds. The yield was estimated by 1H NMR analysis using diphenylmethanol as the internal standard. cap, caprolactamate. | ||||||
| 1 | Rh2(cap)4 (2.5), Cu(OAc)2·H2O (100), AgSbF6 (10) | 40 | ClCH2CH2Cl (0.0125) | 18 | Trace | Trace |
| 2 | Rh2(cap)4 (2.5) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 3 | AgSbF6 (10) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 4 | Cu(OAc)2·H2O (100) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 5 | Rh2(cap)4 (2.5), AgSbF6 (10) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 6 | Rh2(cap)4 (2.5), Cu(OAc)2·H2O (100) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 7 | Cu(OAc)2·H2O (100), AgSbF6 (10) | 40 | ClCH2CH2Cl (0.0125) | 62 | Trace | 3 |
| 8 | Cu(OAc)2·H2O (100), AgNTf2 (10) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 9 | Cu(OAc)2·H2O (100), AgBF4 (10) | 40 | ClCH2CH2Cl (0.0125) | 20 | 7 | Trace |
| 10 | ScCl3·6H2O (100), AgSbF6 (10) | 40 | ClCH2CH2Cl (0.0125) | 0 | 0 | 0 |
| 11 | Cu(OAc)2·H2O (100), AgSbF6 (10) | 40 | PhCl (0.0125) | 42 | Trace | 3 |
| 12 | Cu(OAc)2·H2O (100), AgSbF6 (10) | Reflux | CH2Cl2 (0.0125) | 71 | Trace | 4 |
| 13 | Cu(OAc)2·H2O (100), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 77 | 3 | 7 |
| 14 | Cu(OAc)2·H2O (10), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 77 | 5 | 5 |
| 15 | [Cp*RhCl2]2 (5), Cu(OAc)2·H2O (10), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 3 | 37 | Trace |
| 16 | 365 nm UV light instead of metal salt | 40 | CH2Cl2 (0.02) | 0 | 0 | 0 |
| 17 | CuOAc (10), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 28 | Trace | 3 |
| 18 | Cu(acac)2 (10), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 21 | 4 | 2 |
| 19 | Cu(NTf2)2 (10), AgSbF6 (10) | Reflux | CH2Cl2 (0.02) | 42 | Trace | 4 |

With the established conditions exhibiting high regioselectivity in hand, we next explored the generality and limitations of the current C–H functionalization methodology (Table 2). Utilization of dibenzyl malonate slightly enhanced the chemical yield, affording 3b in 79% yield. Replacing the enone functionality with a benzoyl group at the indole 3-position further increased the yield to 91% (3c). Products featuring aryl ketone functionalities with electron-donating and electron-withdrawing groups at the ortho-, meta-, and para-positions were obtained in good to excellent yields (3d–3h, 70–90%). With respect to substituents on the indole nitrogen, methyl, triisopropylsilyl, and para-methoxybenzyl groups, as well as a potentially reactive allyl group in carbene reactions, were well tolerated in the C5-selective functionalization processes (3i–3l, 72–79%). Substrates bearing a phenyl group at the C2 position and a methyl group at the C7 position were amenable to the catalysis (3m and 3p, 70% and 56% yield, respectively). Of note, reactions employing indole variants with substituents at the C6 position proximal to the reaction site proceeded, furnishing the corresponding products 3n and 3o in moderate to good yields. Substitution of the methyl group with a phenyl or secondary alkyl group at the β-position of the unsaturated ketone preserved the reactivity (3q and 3r, 74% yield for both), whereas 3-acetylindole derivatives produced diminished yields (3s, 55% yield). Additionally, non-symmetric malonate diesters, including acid-labile tert-butyl ester, were applicable, furnishing products 3u–3w in 68–75% yields. The Cu/Ag catalysis exhibited suboptimal performance in the C–H functionalization of the hydrocarbazolone variant (3x).

Scheme 1 illustrates the preliminary outcomes of product derivatization. The carbonyl group at the 3-position of indole was removed by heating under acidic conditions (3c → 6). An allylation of the malonate unit proceeded smoothly, affording 7 with the quaternary carbon in 65% yield (Scheme 1b). A Pd-catalyzed decarboxylation reaction facilitated the formation of a primary alkyl group, characteristic of those found in bioactive 5-substituted indoles (Scheme 1c and Fig. 1A).
 | ||
| Scheme 1 Derivatization of the product. Conditions: (a) TsOH (1 eq.), ethylene glycol (1.0 M), C6H6 (0.05 M), reflux, 6 h; (b) allyl bromide (1.5 eq.), NaH (1.2 eq.), DMF (0.2 M), rt, 5 h; (c) H2 (1 atm), Pd/C (10%), AcOEt (0.05 M), rt, 2 h; toluene/AcOEt (0.01 M), reflux, 1 h. | ||
Subsequently, we directed our attention to elucidating the reaction mechanism of the developed catalytic C–H functionalization. Copper and silver salts can react with acceptor-substituted diazo compounds, exhibiting similar reactivity in certain cases.59 However, replacing AgSbF6 with LiSbF6 afforded comparable yields, indicating that Ag-carbene species are not involved in the C5 functionalization process and highlighting the importance of the SbF6 counteranion (Table 3, entry 1). The results obtained using Zn(OAc)2·2H2O or Ni(OAc)2·4H2O demonstrated the essential role of the Cu core in this transformation (entries 2 and 3). Although reactions employing CuCl2/AgSbF6 or Cu(SbF6)2 proceeded, the efficiency was reduced compared to the optimal conditions, suggesting the involvement of the OAc counteranion (entries 4 and 5). Furthermore, mass balance considerations using a stoichiometric amount of Cu(OAc)2·H2O and a catalytic amount of AgSbF6 (Table 1, entry 13) strongly suggest the preferential formation of Cu(OAc)(SbF6) over Cu(SbF6)2.60–62 Next, the effect of Ag salt loading was investigated by varying the amount from 3 to 300 mol%. When 3 or 30 mol% of AgSbF6 was employed, the yield of 3a decreased to 62% or 35%, respectively (entries 6 and 8), compared to the optimized conditions (entry 7). Upon further increasing the AgSbF6 loading beyond 100 mol%, substantial amounts of black insoluble material appeared in the reaction flask, and both 1a and 2a remained unreacted (entries 9 and 10). These results indicate that a stoichiometric amount of Ag salt relative to Cu(OAc)2 is appropriate to achieve optimal catalytic performance for this C–H functionalization.
|
||
|---|---|---|
| Entry | Catalyst (mol%) | Yield (%) |
| a From entry 14 in Table 1 for comparison. | ||
| 1 | Cu(OAc)2·H2O (10), LiSbF6 (10) | 68 |
| 2 | Zn(OAc)2·2H2O (10), AgSbF6 (10) | 0 |
| 3 | Ni(OAc)2·4H2O (10), AgSbF6 (10) | 0 |
| 4 | CuCl2 (5), AgSbF6 (10) | 37 |
| 5 | Cu(SbF6)2 (5) | 40 |
| 6 | Cu(OAc)2·H2O (10), AgSbF6 (3) | 62 |
| 7a | Cu(OAc)2·H2O (10), AgSbF6 (10) | 77 |
| 8 | Cu(OAc)2·H2O (10), AgSbF6 (30) | 35 |
| 9 | Cu(OAc)2·H2O (10), AgSbF6 (100) | Trace |
| 10 | Cu(OAc)2·H2O (10), AgSbF6 (300) | Trace |

We then investigated the reactivity pattern of indoles pre-substituted at the C5 position. Under the established conditions, substrate 1y featuring a simple methyl substituent at C5 underwent C4–H alkylation followed by intramolecular cyclization, giving 3,4-fused indole 5y in 64% yield. Accordingly, the developed copper-catalytic system also demonstrated potential to facilitate C4–H alkylation (Scheme 2).
 | ||
| Scheme 2 Reaction employing C5-substituted indole as a substrate. | ||
To elucidate the origin of the regioselectivity and reaction processes, we next performed quantum chemical computation (Fig. 2).63,64 Calculations on the basis of dispersion-corrected density functional theory (DFT-D3)65 were performed in a spin-unrestricted formalism to account for the Cu(II) species.66,67 First, we investigated elementary reactions between structurally simplified N-methylindole RTSM and Cu-carbene species RTCu, generated from the diazo compound. Computation at the UCAM-B3LYP/6-311+G**/SDD//UB3LYP/6-31G*/Def2-SVPP level of theory in dichloromethane revealed that electrophilic addition of the carbene species to the C5 position of the indole proceeds with a moderate activation barrier of 13.85 kcal mol−1, affording CP1C5 via TS1C5. To elucidate the regioselectivity, we examined the reactivity at other positions of the indole ring. Notably, while the C2, C6, and C7 positions exhibited comparable activation energies (TS1C2: +16.02, TS1C6: +10.31, TS1C7: +13.93 kcal mol−1, respectively), the C4 position demonstrated remarkably enhanced reactivity with a dramatically lower activation barrier of only +2.31 kcal mol−1 (TS1C4). The observed pronounced enhancement in the reaction kinetics and marked stabilization were attributed to the coordination of the enone carbonyl functionality to the copper center (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Cu) in both the transition state (2.01 Å, TS1C4) and the corresponding intermediate state (2.03 Å, CP1C4). Conversely, while carbonyl coordination to Cu was also possible in TS1C2, significant geometric distortion was observed, with the enone and indole planes twisted by 57°, suggesting disruption of π-conjugation.
O⋯Cu) in both the transition state (2.01 Å, TS1C4) and the corresponding intermediate state (2.03 Å, CP1C4). Conversely, while carbonyl coordination to Cu was also possible in TS1C2, significant geometric distortion was observed, with the enone and indole planes twisted by 57°, suggesting disruption of π-conjugation.
 | ||
| Fig. 2 Computational analysis for elucidating the origin of regioselectivity. Calculation was carried out based on UCAM-B3LYP/6-311+G**/SDD//UB3LYP/Def2-SVPP level of theory in dichloromethane. Gibbs free energies are reported in kcal mol−1, and bond lengths in ångströms (Å). | ||
Therefore, further computational analyses were performed starting from CP1C4 (Fig. 3). Following a coordination mode change to the more stable dicarbonyl chelate complex (−6.15 kcal mol−1, CP2), facile three-membered ring formation proceeded, driven by charge neutralization (+4.60 kcal mol−1, TS2).68,69 Indeed, visualization of the frontier orbitals of CP2 revealed that the lowest unoccupied molecular orbital (LUMO) is delocalized over the indole unit. The norcaradiene CP3 possessed a structural motif capable of undergoing ring expansion via electrocyclic reaction. Its divinylcyclopropane rearrangement proved both kinetically and thermodynamically unfavorable, however, presumably due to double dearomatization of the indole core (TS4: ΔΔG‡ = +16.04, CP6: ΔG = −30.09 kcal mol−1). Instead, the ionic cleavage of the C–C bond proceeded more rapidly via TS3 (uphill by only 2.65 kcal mol−1), leading to CP4, driven by the electron push effect of the indole nitrogen and the electron pull effect of the Cu-activated 1,3-dicarbonyl structure on the fused cyclopropane.70,71 The subsequent proton transfer event afforded CP5, accompanied by significant energetic stabilization (ΔG = −27.59 kcal mol−1) due to rearomatization of the benzenoid core.72 These computational findings corroborate the experimentally observed regioselectivity and underscore the pivotal role of copper in this catalysis.
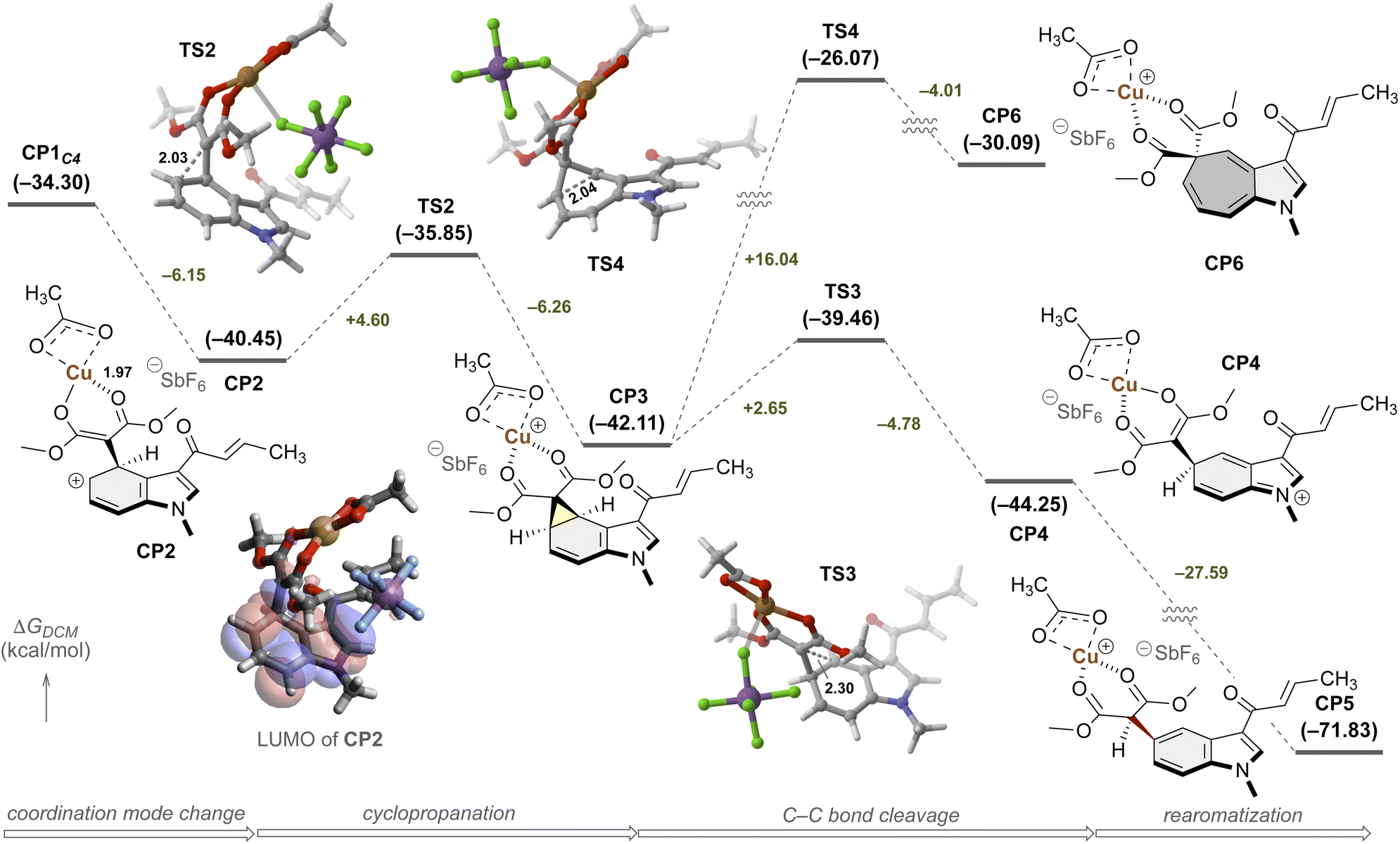 | ||
| Fig. 3 Gibbs free energy profile for reaction coordinates. The computation was implemented at the UCAM-B3LYP/6-311+G**/SDD//UB3LYP/Def2-SVPP level of theory in dichloromethane. Bond lengths are reported in Å. | ||
Conclusions
In summary, we developed a direct, regioselective C5–H functionalization reaction of indoles under copper catalysis. The synthesized products exhibit a structural motif characteristic of indole alkaloids and active pharmaceutical ingredients, underscoring the synthetic utility of this methodology. Experimental mechanistic studies confirmed the crucial role of the combined Cu(OAc)2·H2O/AgSbF6 catalytic system. Computational mechanistic investigations revealed that the initial C4–H alkylation is directed by the carbonyl group at the 3-position of the indoles. Subsequent rapid formation and fragmentation of cyclopropane occur with an activation energy of less than 5 kcal mol−1. Hypothetical competing Buchner reactions were kinetically and thermodynamically unfavorable due to the loss of aromatic stabilization energy. We anticipate that the developed methodology and mechanistic insights will inspire future discoveries for direct and remote C–H functionalization of the benzenoid core in indole scaffolds. Further investigations utilizing metal-carbene species are currently ongoing in our laboratory.Data availability
General information, detailed experimental procedures, characterization data for compounds, and computational details are available in the ESI.†Author contributions
T. I. and M. Y. performed experiments. S. H. and T. N. supported and supervised the research. T. I. and S. H. performed computational studies. S. H. prepared the original draft of the manuscript with feedback from all authors. All authors discussed the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Takeda Science Foundation, and JSPS KAKENHI grant numbers JP22H02741, JP25K01760, and JP25H02003 in Transformative Research Areas (A) JP24A202 Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP). It was also partly supported by a Grant-in-Aid for Transformative Research Areas (A) Digitalization-driven Transformative Organic Synthesis (Digi-TOS, JP24H01059). We thank Dr Shinji Harada (Chiba University) for assistance and advice regarding X-ray crystallographic analysis.Notes and references
- S. Kumar and Ritika, Future J. Pharm. Sci., 2020, 6, 121 CrossRef.
- T. Y. Zheng, J. W. Ma, H. C. Chen, H. Jiang, S. Lu, Z. Shi, F. Liu, K. N. Houk and Y. Liang, J. Am. Chem. Soc., 2024, 146, 25058 CrossRef CAS PubMed.
- T. E. Rose, B. H. Curtin, K. V. Lawson, A. Simon, K. N. Houk and P. G. Harran, Chem. Sci., 2016, 7, 4158 RSC.
- A. J. K. Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef.
- A. Fredenhagen, F. Petersen, M. Tintelnot-Blomley, J. Rosel, H. Mett and P. Hug, J. Antibiot., 1997, 50, 395 CrossRef CAS PubMed.
- W.-L. Wang, Z.-Y. Lu, H.-W. Tao, T.-J. Zhu, Y.-C. Fang, Q.-Q. Gu and W.-M. Zhu, J. Nat. Prod., 2007, 70, 1558 CrossRef CAS.
- A. B. Smith III, N. Kanoh, N. Minakawa, J. D. Rainier, F. R. Blase and R. A. Hartz, Org. Lett., 1999, 1, 1263 CrossRef.
- Y.-C. Xu, J. M. Schaus, C. Walker, J. Krushinski, N. Adham, J. M. Zgombick, S. X. Liang, D. T. Kohlman and J. E. Audia, J. Med. Chem., 1999, 42, 526 CrossRef CAS PubMed.
- P. J. Goadsby and R. Yates, Headache, 2006, 46, 138 CrossRef PubMed.
- Q. P. Dai, X. G. Xie, S. Y. Xu, D. H. Ma, S. B. Tang and X. G. She, Org. Lett., 2011, 13, 2302 CrossRef CAS PubMed.
- J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723 CrossRef CAS PubMed.
- X. Yu, Y. Liu, X. Xie, X.-D. Zheng and S.-M. Li, J. Biol. Chem., 2012, 287, 1371 CrossRef CAS PubMed.
- G. Yoshimura, J. Sakamoto, M. Kitajima and H. Ishikawa, Chem.–Eur. J., 2024, e202401153 CrossRef CAS PubMed.
- S. Liu, X. Zhou, L. Jiao, C. Li and Z. Wang, Chem.–Eur. J., 2023, 23, e202300978 CrossRef PubMed.
- H. You, Z. P. Sercel, S. P. Rezgui, J. Farhi, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2023, 145, 25533 CrossRef.
- Z. Y. Wang, R. L. Guo, X. L. Zhang, M. Y. Wang, G. N. Chen and Y. Q. Wang, Org. Chem. Front., 2021, 8, 1844 RSC.
- L. Cardinale, G. L. Beutner, C. Y. Bemis, D. J. Weix and S. S. Stahl, J. Am. Chem. Soc., 2024, 146, 32249 CrossRef CAS.
- Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966 CrossRef CAS PubMed.
- L.-W. Miao, P. Chen, M.-X. Hu, J. Yin, H. Du and Y.-J. Jiang, Adv. Synth. Catal., 2024, 366, 2226 CrossRef CAS.
- S. Choi, Y. Choi, Y. Kim, J. Lee and S. Y. Lee, J. Am. Chem. Soc., 2023, 145, 24897 CrossRef CAS PubMed.
- P. Bourbon, K. Vitse, A. Martin-Mingot, H. Geindre, F. Guégan, B. Michelet and S. Thibaudeau, Nat. Commun., 2024, 15, 7435 CrossRef CAS.
- N. Tajima, T. Hayashi and S.-i. Nakatsuka, Tetrahedron Lett., 2000, 41, 1059 CrossRef CAS.
- F.-Y. Wang, Y.-X. Li and L. Jiao, J. Am. Chem. Soc., 2023, 145, 4871 CrossRef CAS.
- J. Winkelblech, M. Liebhold, J. Gunera, X. Xie, P. Kolb and S.-M. Li, Adv. Synth. Catal., 2015, 357, 975 CrossRef CAS.
- K. V. Lawson, T. E. Rose and P. G. Harran, Tetrahedron, 2013, 69, 7683 CrossRef CAS PubMed.
- T. Wang, M. A. Bashir, R. Ding, X. Bao, H. J. Zhang, F. Zhong and H. Zhai, ChemistrySelect, 2024, 9, e202304889 CrossRef CAS.
- V. Dočekal, Y. Niderer, A. Kurčina, I. Císařová and J. Veselý, Org. Lett., 2024, 26, 6993 CrossRef PubMed.
- Y. Hussain, C. Empel, R. M. Koenigs and P. Chauhan, Angew. Chem., Int. Ed., 2023, 62, e202309184 CrossRef CAS PubMed.
- A. Gogoi, R. Chouhan and S. K. Das, Org. Lett., 2025, 27, 2461 CrossRef CAS PubMed.
- E. Palomo, A. Krech, Y. J. Hsueh, Z. Li and M. G. Suero, J. Am. Chem. Soc., 2025, 147, 13120 CrossRef CAS.
- M. Peeters, L. Baldinelli, M. Leutzsch, F. Caló, A. A. Auer, G. Bistoni and A. Fürstner, J. Am. Chem. Soc., 2024, 146, 26466 CrossRef CAS PubMed.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC.
- J. Yang, P. R. Ruan, W. Yang, X. M. Feng and X. H. Liu, Chem. Sci., 2019, 10, 10305 RSC.
- Y. Nikolova, B. Fabri, P. M. Lorente, A. Guarnieri-Ibáñez, A. de Aguirre, Y. Soda, G. Pescitelli, F. Zinna, C. Besnard, L. Guénée, D. Moreau, L. D. Bari, E. Bakker, A. I. Poblador-Bahamonde and J. Lacour, Angew. Chem., Int. Ed., 2022, 61, e202210798 CrossRef CAS PubMed.
- X. Wang, W.-Y. Tong, B. Huang, S. Cao, Y. Li, J. Jiao, H. Huang, Q. Yi, S. Qu and X. Wang, J. Am. Chem. Soc., 2022, 144, 4952 CrossRef CAS.
- M. Zhang, T. Zhang, S. Yu, H. Qiu, A. Yusuf, X. Xu, Y. Qian and W. Hu, Chem. Sci., 2023, 14, 11850 RSC.
- Q. Peng, M. Huang, G. Xu, Y. Zhu, Y. Shao, S. Tang, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2023, 62, e202313091 CrossRef CAS PubMed.
- J. Zhang, A. O. Maggiolo, E. Alfonzo, R. Mao, N. J. Porter, N. M. Abney and F. H. Arnold, Nat. Catal., 2023, 6, 152 CrossRef CAS.
- A. H. Bhat, S. Alavi and H. K. Grover, Org. Lett., 2020, 22, 224 CrossRef CAS PubMed.
- W. Zhang, G. Xu, L. Qiu and J. Sun, Org. Biomol. Chem., 2018, 16, 3889 RSC.
- X. Wang, Y. Fu, Z. Guo, A. Lin, Q. Jia and C. Han, Org. Lett., 2025, 27, 493 CrossRef CAS.
- S. Harada, S. Hirose, M. Takamura, M. Furutani, Y. Hayashi and T. Nemoto, J. Am. Chem. Soc., 2024, 146, 733 CrossRef CAS PubMed.
- H. Nakayama, S. Harada, M. Kono and T. Nemoto, J. Am. Chem. Soc., 2017, 139, 10188 CrossRef CAS PubMed.
- S. Harada, M. Kono, T. Nozaki, Y. Menjo, T. Nemoto and Y. Hamada, J. Org. Chem., 2015, 80, 10317 CrossRef CAS PubMed.
- T. Ito, S. Harada, H. Homma, A. Okabe and T. Nemoto, ACS Catal., 2023, 13, 147 CrossRef CAS.
- S. Harada, H. Takenaka, T. Ito, H. Kanda and T. Nemoto, Nat. Commun., 2024, 15, 2309 CrossRef CAS PubMed.
- S. Harada, M. Yanagawa and T. Nemoto, ACS Catal., 2020, 10, 11971 CrossRef CAS.
- X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC.
- Y. Hashimoto, S. Harada, R. Kato, K. Ikeda, J. Nonnho, H. Gröger and T. Nemoto, ACS Catal., 2022, 12, 14990 CrossRef CAS.
- T. C. Maier and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 4594 CrossRef CAS PubMed.
- M.-L. Li, J.-H. Han, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990 CrossRef CAS PubMed.
- M.-Y. Huang, J.-B. Zhao, C.-D. Zhang, Y.-J. Zhou, Z.-S. Lu and S.-F. Zhu, J. Am. Chem. Soc., 2024, 146, 9871 CrossRef CAS PubMed.
- Y.-X. Zheng, L.-G. Liu, T.-Q. Hu, X. Li, Z. Xu, X. Hong, X. Lu, B. Zhou and L.-W. Ye, Nat. Commun., 2024, 15, 9227 CrossRef CAS PubMed.
- K. O. Marichev, K. Wang, K. Dong, N. Greco, L. A. Massey, Y. Deng, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2019, 58, 16188 CrossRef CAS PubMed.
- M. Álvarez, M. Besora, F. Molina, F. Maseras, T. Belderrain and P. J. Pérez, J. Am. Chem. Soc., 2021, 143, 4837 CrossRef PubMed.
- F.-S. Li, X.-Y. Zou, T.-Q. Hu, Q. Sun, Z. Xu, B. Zhou and L.-W. Ye, Sci. Adv., 2024, 10, eadq7767 CrossRef CAS PubMed.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 22017 CrossRef PubMed.
- Please see the ESI† for details.
- Y. Qian, X. Xu, X. Wang, P. J. Zavalij, W. Hu and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 5900 CrossRef CAS PubMed.
- A. Yesilcimen, N.-C. Jiang, F. H. Gottlieb and M. Wasa, J. Am. Chem. Soc., 2022, 144, 6173 CrossRef CAS PubMed.
- J. Zhang, L. G. Hubert-Pfalzgraf and D. Luneau, Polyhedron, 2005, 24, 1185 CrossRef CAS.
- X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC.
- M. Elkin and T. R. Newhouse, Chem. Soc. Rev., 2018, 47, 7830 RSC.
- H. Hayashi, S. Maeda and T. Mita, Chem. Sci., 2023, 14, 11601 RSC.
- M. J. Andrews, A. Carpentier, A. M. Z. Slawin, D. B. Cordes, S. A. Macgregor and A. J. B. Watson, ACS Catal., 2023, 13, 11117 CrossRef CAS.
- F. Kong, S. Chen, J. Chen, C. Liu, W. Zhu, D. A. Dickie, W. L. Schinski, S. Zhang, D. H. Ess and T. B. Gunnoe, Sci. Adv., 2022, 8, eadd1594 CrossRef CAS PubMed.
- I. Morao, J. P. McNamara and I. H. Hillier, J. Am. Chem. Soc., 2003, 125, 628 CrossRef CAS PubMed.
- S. Levin, R. R. Nani and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 774 CrossRef CAS PubMed.
- C. Wang, Z.-H. Liao, R. Wu, K. Chen and S. Zhu, J. Am. Chem. Soc., 2025, 147, 10560 CrossRef CAS.
- T. R. Newhouse, P. S. J. Kaib, A. W. Gross and E. J. Corey, Org. Lett., 2013, 15, 1591 CrossRef CAS PubMed.
- M. Mendel, T. M. Karl, J. Hamm, S. J. Kaldas, T. Sperger, B. Mondal and F. Schoenebeck, Nature, 2024, 631, 80 CrossRef CAS PubMed.
- Rearomatization-assisted cleavage of a cyclopropane ring, yielding a formal C–H insertion product, is documented in the ESI of the following paper, please see: K. L. Smith, C. L. Padgett, W. D. Mackay and J. S. Johnson, J. Am. Chem. Soc., 2020, 142, 6449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2448262. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03417e |
| This journal is © The Royal Society of Chemistry 2025 |