
Comparative performance evaluation of triple quadrupole tandem mass spectrometry and orbitrap high-resolution mass spectrometry for analysis of antibiotics in creek water impacted by CETP discharge†
Arhama T. A. Ansari a,
Ayush Ransingha,
Soumyo Mukherjib,
Andrew Hursthousec,
Fiona L. Henriquezd,
John Connollye and
Suparna Mukherji*a
a,
Ayush Ransingha,
Soumyo Mukherjib,
Andrew Hursthousec,
Fiona L. Henriquezd,
John Connollye and
Suparna Mukherji*a
aEnvironmental Science and Engineering Department, Indian Institute of Technology, Bombay, Mumbai, 400076, India. E-mail: mitras@iitb.ac.in; Tel: +91-(022)-2576-7854
bDepartment of Biosciences and Bioengineering, Indian Institute of Technology, Bombay, Mumbai, India
cSchool of Computing, Engineering & Physical Sciences, University of the West of Scotland, Paisley, UK
dDepartment of Civil and Environmental Engineering, University of Strathclyde, Glasgow, UK
eGlasgow School for Business and Society, Glasgow Caledonian University, Glasgow, UK
First published on 10th July 2025
Abstract
The widespread detection of antibiotics in aquatic environments, particularly in effluent-receiving surface waters, poses significant ecological and public health concerns due to their role in promoting antimicrobial resistance. Accurate trace-level antibiotic measurement is essential for environmental risk assessment and for improving wastewater treatment strategies. This study presents the development, optimization, and validation of two complementary liquid chromatography-mass spectrometry (LC-MS) workflows for the simultaneous quantification of nine antibiotics across five therapeutic classes in creek water impacted by a Common Effluent Treatment Plant (CETP). The performance of a triple quadrupole LC-MS/MS system (LC-QqQ-MS) was compared to that of a high-resolution Orbitrap mass spectrometer (LC-Orbitrap-HRMS). Both instruments demonstrated excellent linearity (R2 > 0.99) and satisfactory recoveries (70–90%) across a wide concentration range. The method detection limits ranged from 0.11 to 0.23 ng L−1 for LC-QqQ-MS and from 0.02 to 0.13 ng L−1 for LC-Orbitrap-HRMS, confirming the superior sensitivity of the high-resolution system approach. Application to real-world creek water samples revealed the ubiquitous presence of multiple antibiotics, with azithromycin and enrofloxacin dominating the detected concentrations, particularly near the CETP discharge point and a nearby waste dumping site. A three-way ANOVA confirmed that antibiotic concentrations were significantly affected by instrument type, sampling site, and antibiotic class along with their interactions. Additionally, non-target screening performed using LC-Orbitrap-HRMS enabled the detection of additional antibiotics belonging to quinolones, sulfonamides and aminoglycosides, further demonstrating the broader analytical scope of high-resolution mass spectrometry. The study highlights the necessity of using advanced analytical tools for the accurate quantification of antibiotics in complex matrices and underscores the environmental risks posed by pharmaceutical pollution in industrial discharge-impacted water bodies.
1. Introduction
The widespread occurrence of emerging contaminants (ECs) in aquatic environments has become a critical concern for both human health and ecosystem integrity. ECs comprise a diverse range of anthropogenic chemicals, including pharmaceuticals, personal care products, pesticides, and industrial additives.1 Among these, antibiotics represent a particularly urgent and complex subclass due to their extensive use in human healthcare, veterinary medicine, and agriculture, coupled with their recognized role in the proliferation of antimicrobial resistance (AMR).2,3 Even at sub-inhibitory concentrations, the continuous discharge of antibiotics into surface waters can exert selective pressure on microbial communities, facilitating the emergence and dissemination of antibiotic-resistant bacteria and resistance genes.4 Ngumba et al. (2016)5 quantified multiple antibiotics and antiretrovirals using SPE-LC-MS/MS in European surface waters, and reported concentrations ranging from 10 to 570 ng L−1. Similarly, LC-MS/MS studies in the US also reported sulfonamide levels up to 11 ng L−1,6 and a broad-spectrum HRMS-based survey in Southern Europe identified 28 pharmaceuticals in river and coastal waters.7 Accurate and reliable quantification of antibiotics at trace levels in environmental matrices is essential not only for comprehensive risk assessments but also for the design and optimization of wastewater treatment strategies aimed at mitigating their release.Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) using triple quadrupole (QqQ) detectors has long been established as the gold standard for targeted analysis of polar and semi-polar contaminants, offering exceptional sensitivity and selectivity.8 Prior to LC-MS/MS analysis, environmental water samples are typically subjected to solid-phase extraction (SPE) for pre-concentration and matrix clean-up, allowing for quantification limits in the nanogram per litre (ng per L) range.9,10 While direct injection approaches have recently gained attention for their faster, simplified workflows particularly in clean or moderately polluted water matrix11,12 traditional offline SPE remains the preferred approach for achieving lower detection limits, superior removal of matrix interference, and reduced measurement uncertainty, especially in complex environmental matrices. Performance of offline SPE, online SPE and direct injection was compared by de la Serena Calleja et al. (2023)13 for the analysis of contaminants of emerging concern in complex environmental matrices, i.e., urban and piggery wastewater. The study found that offline SPE achieved the lowest method quantification limits (MLQs) for over 50% of the analytes. Although offline SPE showed lower precision than online SPE and direct injection, it provided the broadest analyte coverage and captured more information than the other methods.
In recent years, high-resolution mass spectrometry (HRMS) such as quadrupole orbitrap instrument have gained considerable attention for environmental monitoring studies.14 The combination of full-scan data acquisition with accurate mass measurements (≤1 ppm) and high resolving power reaching up to 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, LC-HRMS enables simultaneous targeted quantification and non-targeted screening, as well as retrospective data analysis for unknown or emerging analytes.15,16 The integration of both targeted and non-targeted approaches using LC-MS has proven particularly effective for comprehensively characterizing antibiotic pollution in aquatic environments. Letzel et al. (2015)17 demonstrated how combining target, suspected-target, and non-target LC-MS/MS workflows significantly enhances the detection of both known antibiotics and their transformation products in surface water. Ferrer et al. (2020)18 showed that data-dependent acquisition in Orbitrap HRMS enables robust non-targeted screening and retrospective identification of pharmaceutical residues. In addition, Tlili et al. (2016)19 integrated SPE with LC-MS/MS for efficient simultaneous quantification of 26 drug residues, including 18 antibiotics. These studies collectively highlights the importance of combining targeted and non-targeted methods for a more reliable and holistic assessment of antibiotic contamination in complex water matrices.
000, LC-HRMS enables simultaneous targeted quantification and non-targeted screening, as well as retrospective data analysis for unknown or emerging analytes.15,16 The integration of both targeted and non-targeted approaches using LC-MS has proven particularly effective for comprehensively characterizing antibiotic pollution in aquatic environments. Letzel et al. (2015)17 demonstrated how combining target, suspected-target, and non-target LC-MS/MS workflows significantly enhances the detection of both known antibiotics and their transformation products in surface water. Ferrer et al. (2020)18 showed that data-dependent acquisition in Orbitrap HRMS enables robust non-targeted screening and retrospective identification of pharmaceutical residues. In addition, Tlili et al. (2016)19 integrated SPE with LC-MS/MS for efficient simultaneous quantification of 26 drug residues, including 18 antibiotics. These studies collectively highlights the importance of combining targeted and non-targeted methods for a more reliable and holistic assessment of antibiotic contamination in complex water matrices.
Despite the growing interest in HRMS, as per our knowledge no study has performed a direct comparative evaluations of QqQ-based LC-MS/MS and Orbitrap-HRMS for antibiotic detection in complex environmental samples although some studies have been performed in wastewater. Herrero et al. (2023)20 performed a comparative study of LC-QqQ-MS and Orbitrap-HRMS for the quantification of veterinary drugs (glucocorticoids and polyether ionophores) in sewage samples. They showed a good match for analysis conducted using the two instruments and indicated that LC-QqQ-MS showed more consistent quantification for glucocorticoids. Orbitrap-HRMS offered slightly better sensitivity for the polyether ionophore and also enhanced confirmatory and retrospective analysis capabilities. Additionally, comparisons have been made in the gas chromatography domain. Belarbi et al. (2021)21 compared GC-Orbitrap HRMS with GC-MS/MS for pesticides in wheat and reported that GC-Orbitrap achieved lower limits of detection for 86% of the analytes with acceptable recoveries. However matrix suppression was observed for specific compounds, highlighting the importance of method optimization.
While the majority of environmental monitoring studies have focused on freshwater systems, such as river water, groundwater, drinking water, and wastewater, data on antibiotic contamination in estuarine environments remain relatively scarce.22 Freshwater bodies are particularly vulnerable to antibiotic pollution due to direct inputs from localized anthropogenic sources, implying that most of the data reported reflect only localized contamination profiles. Since all inland water systems ultimately discharge into the marine environment, mapping antibiotic contamination in the estuarine regions is important.23 Tidal creeks, in particular, represent a unique and ecologically important transition zone between freshwater and marine environments. These water bodies are directly influenced by both land-based effluent discharge and tidal flushing. However, estimation of antibiotics in the complex ionic matrix of the creek introduces significant analytical challenges, caused by matrix effects, which can compromise detection sensitivity and quantification accuracy.24,25
In this study, an optimized workflow for antibiotic analysis using integrated SPE–UHPLC–MS was developed and validated for the simultaneous determination of nine antibiotics representing five therapeutic classes including quinolones, macrolides, oxazolidinones, sulfonamides, and β-lactams in creek water impacted by discharges from a Common Effluent Treatment Plant (CETP) which treat wastewater from multiple industries such as pharmaceuticals, petrochemicals, and textile manufacturing industries. In this study a comparative evaluation of two analytical instruments i.e., an LC-QqQ-MS operating in multiple reaction monitoring (MRM) mode (System A), and, an LC-Orbitrap-HRMS employing full-scan ddMS2 mode (System B) was conducted. The comparative assessment focused on key performance metrics including sensitivity, linearity, precision, and matrix effects. Following method validation, both systems were employed to quantify antibiotic concentrations at eight spatially distributed sampling points along a creek impacted by CETP discharge. This approach not only enabled the mapping of antibiotic distribution patterns but also provided insights into the environmental persistence and fate of these contaminants within an industrially influenced aquatic ecosystem. To the best of our knowledge, this study represents the first direct comparison of LC-QqQ-MS and LC-Orbitrap-HRMS methodologies for monitoring of multiple antibiotics in a CETP discharge affected surface water body. Our findings highlight the complementary strengths of both mass spectrometric instruments, highlighting the potential for their use in routine environmental surveillance.
2. Materials and methods
2.1 Chemicals and reagents
This study targeted the analysis of nine antibiotics representing five major therapeutic classes including quinolones, macrolides, oxazolidinones, sulfonamides, and β-lactams. The selected compounds included erythromycin (ERY), azithromycin (AZT), oxacillin (OXC), linezolid (LIN), enrofloxacin (ENF), levofloxacin (LEV), ciprofloxacin (CIP), penicillin G (PEN), and sulfamethazine (SMZ). Analytical-grade standards for each antibiotic were obtained from TCI Chemicals, while corresponding isotope-labelled internal standards were procured from Toronto Research Chemicals (Toronto, Canada). Detailed information on each antibiotic, including surrogate compounds and isotope-labelled internal standards, is summarized in Table S1 of the ESI.† High-purity LC-MS-grade solvents, including methanol and acetone, were supplied by Thermo Fisher Scientific. Reagent-grade sulfuric acid (H2SO4) and formic acid were purchased from Sigma-Aldrich and were used for sample preparation and pH adjustments.2.2 Study area and sample collection
The selected study site was a tidal creek located adjacent to a major industrial zone in Maharashtra, India, which continuously receives treated effluents from a CETP. Sampling was conducted in April 2022 at eight strategically selected locations to capture spatial distribution patterns of antibiotic residues across the creek. The map of sampling locations (Fig. 1) was created using QGIS 3.22. | ||
| Fig. 1 Distribution of sampling points along the creek. *Sites W/S, W/M, and W/R denote locations near the shore, mangrove, and CETP discharge site, respectively; 1U/2U and 1 Da/1Db/2D indicate north and south sampling points at increasing distances from the reference point. The map was generated using QGIS 3.22. | ||
The CETP discharge point was designated as the reference point (W/R). Water samples were collected from both north (W/1U and W/2U) and south (W/1 Da, W/1Db and W/2D) of the reference point at various distances. Samples were also collected along the shoreline (W/S) and mangrove (W/M) zones. At each location, water samples were collected at a standardized depth of approximately 50 cm above the sediment surface using a Niskin Sampler to ensure consistency across sampling sites and transferred to laboratory in an ice box. The samples were immediately stored at 4 °C and processed within 48 hours to minimize degradation and ensure analytical accuracy.
2.3 Sample preparation
All collected water samples were subjected to standardized pre-treatment to ensure consistent and reliable quantification of target analytes. Samples were filtered through a 0.45 μm membrane filter as per the method described by Mohapatra et al. (2016).26 The pH of the filtered samples was then adjusted between 3 and 4 using H2SO4 to optimize extraction efficiency. SPE was employed for sample enrichment and matrix clean-up. Each 500 mL water sample was processed using Oasis HLB cartridges (500 mg) under a controlled flow rate of 5–10 mL min−1. Prior to sample loading, the cartridges were conditioned with 5 mL methanol and 5 mL deionized (DI) water. After sample extraction, the cartridges were washed with DI water and vacuum-dried. Target antibiotics were eluted using 10 mL of methanol. The eluates were evaporated to dryness under a gentle nitrogen stream and reconstituted to a final volume of 1 mL, resulting in a consistent concentration factor of 500 across all samples.2.4 Instrumental analysis
For quantitative and comparative analysis of the selected antibiotics, two high-performance liquid chromatography-mass spectrometry (LC-MS) systems were employed. System A consisted of a TSQ Quantis triple quadrupole mass spectrometer (QqQ-MS, Thermo Fisher Scientific, USA) while System B was a Q Exactive Orbitrap high-resolution mass spectrometer (HRMS, Thermo Fisher Scientific, USA). Both instruments were coupled to an identical Dionex Ultimate 3000 UHPLC system (Thermo Fisher Scientific, USA). Method development was performed using standard antibiotic solutions prepared in LC-MS-grade methanol, enabling the optimization of both chromatographic and mass spectrometric parameters. Chromatographic separation for both systems was carried out on a Hypersil GOLD C18 column (100 × 2.1 mm, 1.9 μm particle size) using a binary gradient of ultrapure water with 0.1% formic acid (solvent A) and acetonitrile (solvent B). The gradient began at 5% B, increased to 30% over 7 minutes, then to 60% at 9 minutes, 90% at 11 minutes, and was held until 12 minutes. Subsequently, the percentage of acetonitrile was reduced to 5% at 13 minutes and held constant until 17 minutes. The column temperature was maintained at 40 °C and the autosampler was set at 10 °C throughout the analysis. Within the framework of this study, the optimized chromatography conditions were kept constant for both the instruments. Mass spectrometry parameters were optimized for each antibiotic via direct infusion of standard solutions in each of the two instruments. Detailed optimization procedures for both the instruments are described in the ESI.† Data acquisition and quantitative analysis were performed using Thermo Xcalibur software.To explore the full scan capabilities of LC-Orbitrap-HRMS (System B), a non-target screening (NTA) workflow was employed to identify additional antibiotic contaminants not included in the target list. Raw MS data acquired in full-scan mode were processed using Thermo Compound Discoverer software (v3.2). The workflow included chromatographic alignment, feature detection, isotopic pattern grouping. Candidate features were filtered through blank subtraction using both solvent and matrix blanks to remove the background signals and laboratory artifacts. Molecular formula prediction was applied to the peaks based on accurate mass and isotopic fidelity. Fragmentation spectra were matched against mzCloud spectral libraries. Only those features with match scores exceeding 70% were retained for further manual evaluation. Compound identification was conducted as suggested by Schymanski et al. (2014)16 Level 2 confidence, defined as a probable structure based on spectral library matching and supported by accurate mass and diagnostic MS/MS fragments. A full description of the NTA workflow, including software parameters and filtering criteria, is provided in the ESI.†
2.5 Method validation, accuracy and precision
The LC-MS/MS method was rigorously validated using antibiotic standards prepared in LC-MS-grade methanol. Calibration curves were constructed for each antibiotic using seven standard concentrations, ranging from 0.1 to 2000 ng L−1, prepared in duplicate. The linearity of the calibration curves was assessed and confirmed by correlation coefficients (R2) exceeding 0.99 for all the analytes, indicative of robust linear responses across the studied concentration range. Limits of detection (LOD) and quantification (LOQ) were calculated using the standard deviation of the response (σ) and the slope of the calibration curve (S), where LOD and LOQ correspond to 3.3*σ/S and 10*σ/S, respectively. The developed method was further validated for its accuracy and precision. Precision was assessed by evaluating both intra-day and inter-day variability, measured as the relative standard deviation (RSD) of replicate analyses. Intra-day precision was determined from five replicate injections of a sample containing a mixture of antibiotics performed on the same day under identical conditions. Inter-day precision was evaluated from five replicate injections conducted on 2 days that were spaced 48 hours apart, providing an assessment of method reproducibility over time.A three–way analysis of variance (ANOVA) was subsequently performed to evaluate the effect of the three factors including instrument (I: System A and System B), Sampling location (S: W/S, W/M, W/R, W/1 Da, W/1Db, W/2D, W/1U,W/2U), and antibiotic type (A: SMZ, AZT, ENF, ERY, LIN, PEN, CIP, LEV) on antibiotic detection and quantification. The model included all the main effects as well as two–way (I*S, I*A, S*A) and three–way interactions effects (I*S*A). The significance of the effects was determined based on the p-values, with p < 0.05 chosen for statistical significance. All analysis were performed using Python 3.11.
2.6 Quality control and quality assurance
A comprehensive QA/QC framework was implemented throughout the study to ensure data reliability. Isotope dilution mass spectrometry was employed to compensate for potential matrix effects and variability in recovery. Isotope-labelled internal standards were spiked in all samples prior to extraction, as well as in all the calibration standards, to ensure consistency across analytical batches. The internal standards were prepared in LC-MS grade methanol. They were added at a concentration that resulted in a final concentration of 100 μg L−1 in the 1 mL reconstituted extract. To evaluate recovery efficiency, recovery experiments were performed by spiking a mixture of antibiotics into creek samples at a concentration of 100 ng L−1, followed by the complete extraction and analysis workflow. Recovery values were calculated using the eqn (1).
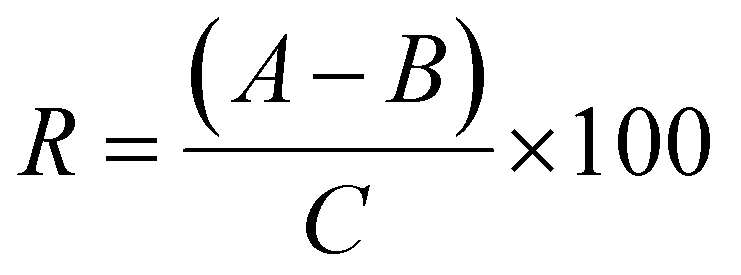 | (1) |
Recovery testing was performed for both LC-QqQ-MS (System A) and LC-Orbitrap-HRMS (System B) to evaluate system-specific recovery values. These recovery values were not used to correct the final antibiotic concentrations, but were assessed to evaluate the robustness of the method and analyte behaviour in the matrix. To monitor contamination and instrument stability, procedural blanks, solvent blanks, and sample duplicates were included in each analytical batch. Milli-Q water, processed identically to the environmental samples, served as a procedural blank to detect laboratory-derived contamination. Solvent blanks were injected after every four-five samples to assess carryover between injections, while sample duplicates ensured the reproducibility of the method across the dataset.
3. Results and discussion
3.1 Optimization of LC-QqQ-MS and LC-Orbitrap-HRMS
The optimization of mass spectrometric parameters was conducted to ensure the sensitive and accurate detection of the target antibiotics across both analytical instruments. System A, employing a triple quadrupole LC-MS/MS configuration (LC-QqQ-MS), was optimized using a univariate, one-factor-at-a-time approach under positive electrospray ionization (ESI) conditions. Data acquisition was performed in multiple reaction monitoring (MRM) mode to achieve maximum selectivity and sensitivity for each antibiotic. Optimal source parameters were established, including a spray voltage of 4.8 kV, a vaporizer temperature of 350 °C, and nitrogen gas flows for sheath and auxiliary gases set at 42 and 10 arbitrary units, respectively. The collision energy (CE) for each analyte was individually fine-tuned within a range of 17–30 eV to ensure the generation of the most intense and characteristic product ion transitions. Argon was used as the collision gas, with a Q2 pressure maintained at 1.5 mTorr, which provided optimal fragmentation efficiency. The optimized retention times, precursor/product ion pairs, and corresponding collision energies for each antibiotic are summarized in Table 1.| Antibiotics | Isotope std | Retention time (min) | A | B | |||
|---|---|---|---|---|---|---|---|
| Precursor ion | Product ion | Collision Energy (eV) | Precursor ion | Product ions | |||
| ENF | ENF-d5 | 5.75 | 360.17 | 316.19 | 19.28 | 360.1705 | 342.16, 316.18, 245.10 |
| LIN | LIN-d3 | 7.13 | 338.15 | 296.12 | 18.02 | 338.1501 | 296.13, 235.12,195.08 |
| AZT | AZT-d3 | 7.64 | 749.51 | 591.44 | 30.91 | 749.5134 | 591.42, 375.26, 158.96 |
| OXC | AMX-d4 | 10.20 | 402.12 | 160.07 | 17.09 | 402.1116 | 243.07, 374.11,160.04 |
| SMZ | SMZ-d4 | 5.14 | 279.09 | 186.07 | 17.21 | 279.0904 | 204.04, 156.01, 186.03 |
| ERY | AZT-d3 | 9.54 | 734.5 | 576.41 | 18.69 | 734.4665 | 576.37, 558.36, 540.35 |
| PEN | AMX-d4 | 6.09 | 335.1 | 160.5 | 28.67 | 335.1059 | 160.57, 217.10, 173.07 |
| CIP | ENF-d5 | 5.82 | 332.14 | 173.08 | 19.02 | 332.1404 | 239.15, 261.13, 173.08 |
| LEV | ENF-d5 | 5.63 | 362.25 | 316.90 | 19.36 | 362.1432 | 316.90, 261.12, 208.03 |
For the high-resolution platform, System B (LC-Orbitrap-HRMS), data acquisition was conducted using a full-scan data-dependent dd-MS2 workflow. Full-scan spectra were acquired across an m/z range of 100–1000 at a resolving power of 70000, while MS/MS spectra were acquired at a resolution of 35000. During the optimization phase, various resolving power settings (17500, 35000, 70000, and 140000) were tested to evaluate the balance between mass accuracy and signal intensity. Although the maximum resolution setting (140000) offered marginal improvements in mass accuracy, it resulted in diminished signal intensities due to longer transient acquisition times and increased susceptibility to ion-ion and ion-neutral gas collisions. Based on this observation, a resolving power of 70000 was selected for full scans, offering an optimal condition between mass accuracy, sensitivity, and acquisition speed. Ionization efficiencies for all antibiotics were evaluated in both positive and negative ESI modes. Consistent with prior reports for structurally similar compounds, the positive ESI mode exhibited superior ionization responses across all nine antibiotics.27 Fragmentation in dd-MS2 mode was performed using stepped normalized collision energies (NCE) of 10, 15, and 30 eV, ensuring comprehensive fragmentation patterns for compound identification and confirmation.
The optimized parameters for precursor ions, product ions, and retention times across both systems are presented in Table 1, while corresponding spectra and fragmentation profiles are shown in Fig. S1 (ESI).† Notably, both LC-MS systems yielded consistent retention times for all antibiotics, highlighting the robustness and reproducibility of the chromatographic method across platforms. Moreover, the comparison of MRM transitions from System A with the dd-MS2 fragmentation spectra from System B confirmed the accurate identification of each target analyte, further validating the analytical reliability of both methods. This dual-instrument optimization not only confirmed the high sensitivity and precision of the LC-QqQ-MS system (System A) for quantitative targeted analysis but also demonstrated the complementary capabilities of the LC-Orbitrap-HRMS system (System B) for both targeted quantification and non-targeted compound discovery. The high resolving power and accurate mass measurements of System B enhance confidence in compound identification, especially when screening complex environmental matrices where unknown or transformation products may co-occur.
3.2 Evaluation of extraction efficiency and matrix effects
Matrix effects are widely recognized as one of the most significant challenges in LC-MS analyses, especially when dealing with complex environmental samples.5 Components intrinsic to these matrices, such as dissolved organic matter, salts, and suspended particulates, often co-elute with target analytes, directly impacting ionization efficiency within the electrospray ionization (ESI) source. This frequently leads to signal suppression and, less commonly, signal enhancement, both of which can compromise the accuracy and sensitivity of quantification.28 Signal suppression in ESI is primarily attributed to three mechanisms: (1) competition for charge between the analyte and co-eluting matrix constituents, which decreases ionization efficiency due to reduced conductivity in the liquid phase; (2) diminished droplet evaporation efficiency resulting from increased surface tension and viscosity caused by matrix components; and (3) gas-phase reactions between analyte ions and interfering molecules, which can lead to a direct reduction in ion yield. The combined effect of these phenomena may cause a notable reduction in method sensitivity, which can be particularly severe in untreated or minimally processed environmental water samples.29To evaluate extraction efficiency and the extent of matrix effects for the selected antibiotics, a recovery experiment was conducted. The recovery values for each antibiotic were assessed on both LC-QqQ-MS (System A) and LC-Orbitrap-HRMS (System B) instruments. As shown in Fig. 2, recovery efficiencies for most antibiotics across both systems ranged from 70% to 90%, which falls within the acceptable limits for environmental sample analysis30 and complies with established validation guidelines including U.S. EPA Method 1694,31 and American Public Health Association (APHA) guidelines (23rd Edition).32 Notably, System B (LC-Orbitrap-HRMS) consistently exhibited slightly higher recovery values than System A (LC-QqQ-MS). This observation likely reflects the reduced susceptibility of System B to ion suppression, which is a known advantage of its high-resolution full-scan acquisition mode, which mitigates matrix interference effects more effectively than targeted MRM acquisition. The employment of stable isotope-labelled internal standards proved advantageous in offsetting residual matrix effects, ensuring reliable quantification despite the complex nature of the samples. These findings are in agreement with earlier reports on other compound classes, such as preservatives and antioxidants33 and steroidal estrogens,34 where the use of isotope-labelled standards significantly improved analytical robustness against matrix-induced variability. Furthermore, the slight differences observed in recovery values between the two LC-MS systems highlight the influence of instrument design on matrix sensitivity. Variations in ESI source architecture and ion optics between instruments often result in different ionization efficiencies, even when analyzing identical samples under comparable conditions.28,33 These results highlight the importance of method-specific recovery evaluations, particularly when translating analytical protocols between different instruments or laboratories.
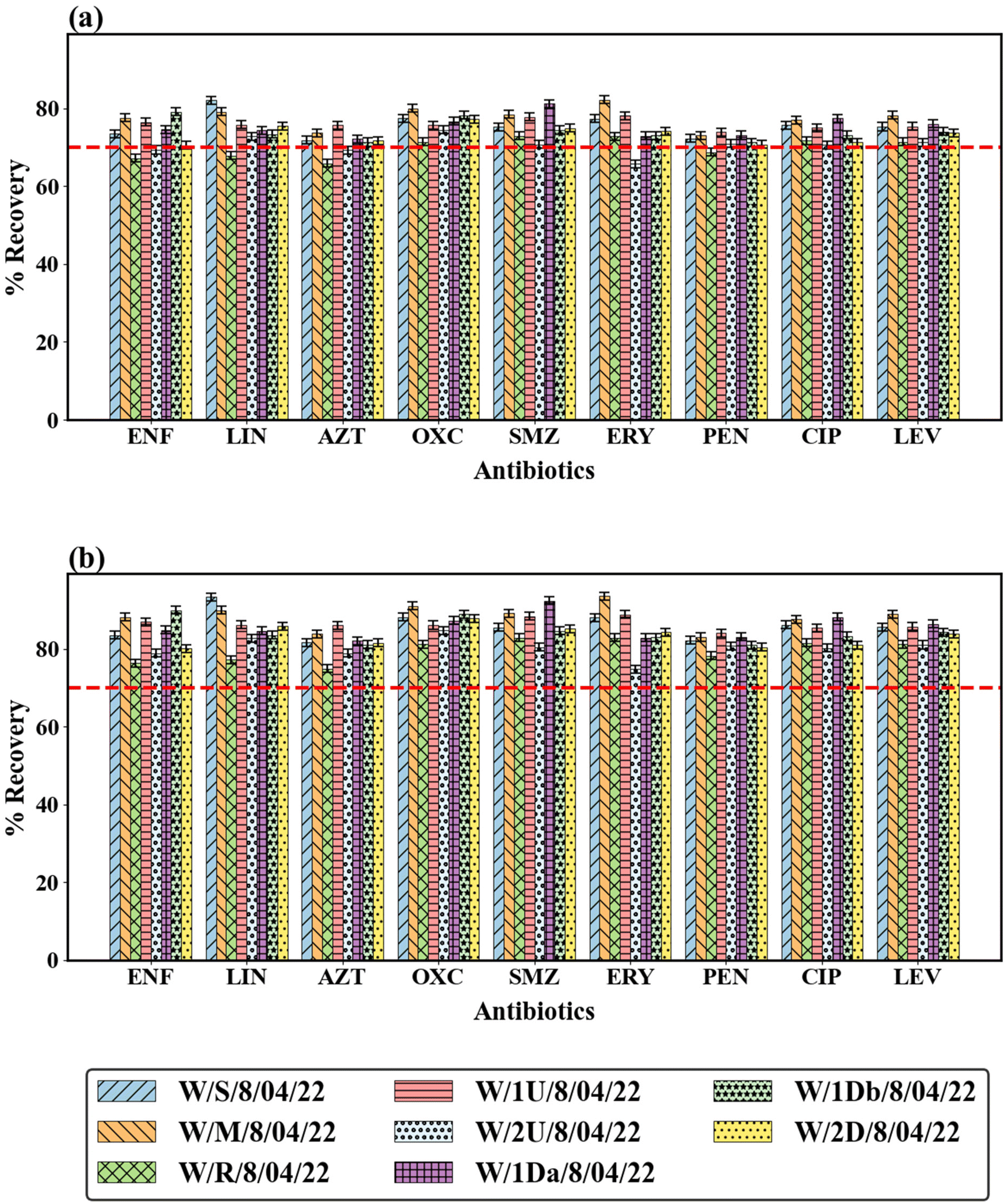 | ||
| Fig. 2 Recovery of selected antibiotics determined by (a) LC-QqQ-MS (System A) and (b) LC-Q-Orbitrap-HRMS (System B). *Error bars represent standard deviation based on duplicate extractions; the line represent 70% recovery, i.e., the acceptable limit recommended by APHA guidelines.32 | ||
3.3 Quantification, performance characteristics, and method validation
Quantification of antibiotics was achieved on both analytical instruments using isotope dilution, where the instrument response was calculated as the ratio of the peak area of the analyte to that of its corresponding isotope-labelled internal standard. Calibration curves were constructed across seven concentration levels each analyzed in duplicate to ensure precision and reproducibility. Both LC-QqQ-MS (System A) and LC-Orbitrap-HRMS (System B) demonstrated excellent linearity, with correlation coefficients (R2) exceeding 0.99 for all nine antibiotics (Table 2). This strong linear response highlights the robustness and reliability of both systems for quantitative analysis across a wide concentration range.| Antibiotics | RSD% within day (RSD% between days) | R2 | LOD, μg L−1 | LOQ μg L−1 | MLOD, ng L−1 | MLOQ, ng L−1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | A | B | A | B | A | B | |
| a Precision is expressed as intra-day RSD% (inter-day RSD %), linearity as R2, and sensitivity as LOD, LOQ, MLOD and MLOQ. | ||||||||||||
| ENF | 2.9 (4.2) | 6.4 (9.7) | 0.99 | 0.99 | 0.08 | 0.05 | 0.26 | 0.17 | 0.17 | 0.11 | 0.51 | 0.33 |
| LIN | 2.4 (3.8) | 3.9 (4.8) | 0.99 | 0.99 | 0.09 | 0.02 | 0.29 | 0.08 | 0.19 | 0.05 | 0.58 | 0.16 |
| AZT | 3.2 (5.5) | 4.5 (9.9) | 0.99 | 0.99 | 0.09 | 0.05 | 0.28 | 0.16 | 0.18 | 0.11 | 0.56 | 0.33 |
| OXC | 3.4 (4.8) | 4.8 (7.7) | 0.99 | 0.99 | 0.05 | 0.03 | 0.17 | 0.10 | 0.11 | 0.07 | 0.34 | 0.21 |
| SMZ | 3.5 (5.4) | 3.6 (8.5) | 0.99 | 0.99 | 0.05 | 0.01 | 0.16 | 0.03 | 0.11 | 0.02 | 0.33 | 0.06 |
| ERY | 2.2 (3.6) | 4.0 (7.9) | 0.99 | 0.99 | 0.11 | 0.05 | 0.34 | 0.14 | 0.23 | 0.1 | 0.69 | 0.28 |
| PEN | 2.7 (4.2) | 3.7 (9.5) | 0.99 | 0.99 | 0.1 | 0.06 | 0.31 | 0.2 | 0.21 | 0.13 | 0.63 | 0.41 |
| CIP | 2.1 (4.2) | 4.5 (9.2) | 0.99 | 0.99 | 0.09 | 0.05 | 0.30 | 0.14 | 0.19 | 0.09 | 0.60 | 0.28 |
| LEV | 2.4 (4.6) | 4.2 (8.4) | 0.99 | 0.99 | 0.09 | 0.05 | 0.30 | 0.14 | 0.19 | 0.09 | 0.60 | 0.28 |
Instrument sensitivity was evaluated through the determination of limits of detection (LOD) and limits of quantification (LOQ), as summarized in Table 2. For the chosen antibiotics, System A exhibited LODs ranging from 0.05 to 0.11 μg L−1 and LOQs ranging from 0.16 to 0.34 μg L−1, while System B achieved slightly superior sensitivity, with LODs between 0.01 and 0.06 μg L−1 and LOQs from 0.03 to 0.20 μg L−1. When adjusted for the concentration factor derived from solid-phase extraction (500-fold), the estimated values of method limit of detection (MLODs) ranged from 0.11 to 0.23 ng L−1 for System A and from 0.02 to 0.13 ng L−1 for System B. These results clearly demonstrate the enhanced sensitivity of the high-resolution Orbitrap platform, particularly for trace-level detection in complex environmental samples. A comparative analysis with previously reported SPE–LC–HRMS methods further underlines the excellent sensitivity of the present approach. Chitescu et al., (2015)35 reported an Orbitrap–HRMS method for the analysis of pharmaceuticals and antifungals in surface waters, achieving MLODs between 0.4 and 5 ng L−1 and MLOQs from 1.5 to 55 ng L−1. Similarly, Kalogeropoulou et al. (2024)36 applied off-line SPE combined with LC–HRMS for seawater analysis, obtaining MLODs ranging from 1.1 to 9.8 ng L−1 and MLOQs between 3 and 26.4 ng L−1. In comparison, System B in the present study demonstrated comparable or superior sensitivity, highlighting its robustness for trace-level quantification in challenging environmental samples.
The precision of both systems was assessed by evaluating intra-day and inter-day variability using standard solutions analyzed in five replicates. For System A, intra-day relative standard deviations (RSDs) ranged from 2.1% to 3.5%, while inter-day RSDs varied between 3.6% and 5.5%. System B exhibited slightly broader variability, with intra-day RSDs ranging from 3.6% to 6.4% and inter-day RSDs between 4.8% and 9.7%. These results affirm the high reproducibility and reliability of both instruments, although the targeted MRM approach of System A provided marginally better precision. The advantage of triple quadrupole systems for quantitative workflows is well established.37 Beyond sensitivity and precision, the fundamental distinction between the two platforms lies in their data acquisition capabilities. System A, which operates in MRM mode, is ideally suited for high-throughput quantitative analysis of predefined compounds due to its superior sensitivity and rapid cycle times. In contrast, full-scan and data-dependent MS2 acquisition mode of System B offers flexibility, enabling not only targeted quantification but also retrospective analysis and non-targeted screening of unknown or emerging contaminants without the need for prior ion transition optimization. This dual-instrument comparison highlights the complementary strengths of both systems. While System A excels in speed and precision for routine environmental monitoring, System B provides a broader analytical scope, making it particularly valuable for advanced environmental investigations where untargeted screening and compound identification are required. The ability of the System B to generate comprehensive full-scan datasets is especially advantageous for post-acquisition data mining, allowing researchers to reprocess samples as new contaminants of concern are identified, a capability beyond the reach of conventional QqQ instruments. Overall, the results reaffirm that the LC-QqQ-MS platform is particularly well-suited for large-scale, targeted environmental surveillance, while the LC-Orbitrap-HRMS offers enhanced versatility and depth for comprehensive environmental monitoring, especially in chemically diverse and data-intensive investigations.
3.4 Performance of the methods on environmental samples
The optimized and validated LC-QqQ-MS (System A) and LC-Orbitrap-HRMS (System B) methods were applied for the quantification of selected antibiotics in water samples collected from multiple locations along the creek impacted by industrial wastewater discharge. Both systems successfully detected a wide range of antibiotic concentrations, spanning from several hundred to several thousand nanograms per litre, highlighting the widespread presence of antibiotic residues in the aquatic environment (Fig. 3). Among the quantified antibiotics, AZT exhibited the highest concentration, 1735 ± 5.7 ng L−1 in System A and 1767 ± 12.4 ng L−1 in System B at the CETP discharge reference site (W/R/8/04/22). These values are significantly higher than those typically reported in other surface water environments. For instance, in the Fenhe River Basin, the average AZT concentration was 74.31 ng L−1, with 100% detection frequency across the 23 sampling sites.38 Similarly, in a comprehensive meta-analysis of 62 river systems across China, the pooled concentration of AZT was just 2.08 ng L−1, confirming that our observed values lie well above typical environmental levels.39 For rivers in Hanoi, AZT was also found at concentration up to 1520 ng L−1.40 Mirzaie et al. (2022) have reported the mean concentration of AZT in seawater and sediment samples as 9 ng L−1 and 6 ng g−1 in the Persian Gulf around Bushehr port.41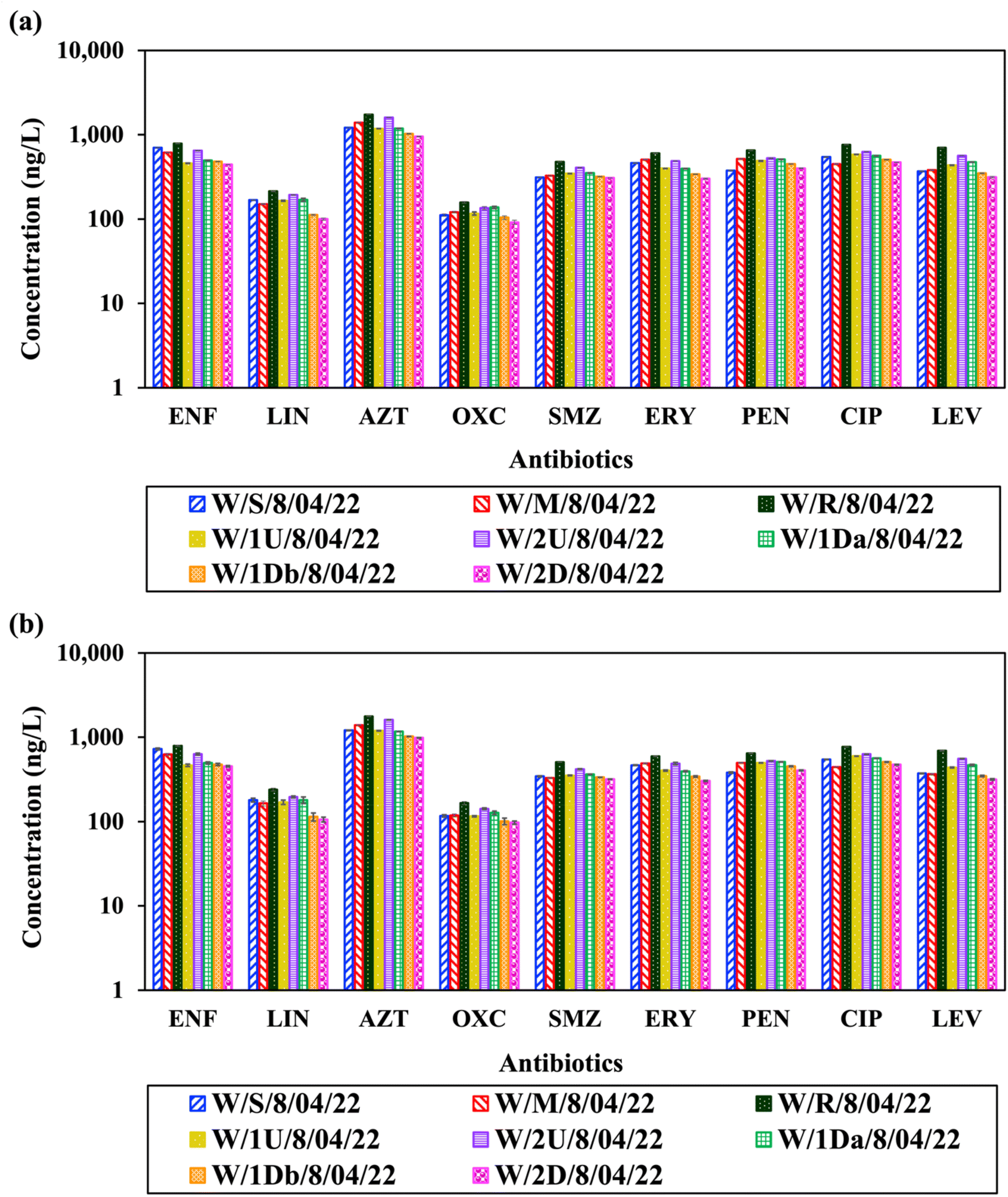 | ||
| Fig. 3 Concentration of selected antibiotics in creek water measured by (a) LC-QqQ-MS (System A) and (b) LC-Orbitrap-HRMS (System B). *Sites W/S, W/M, and W/R denote locations shore, mangrove, and CETP discharge site, respectively; 1U/2U and 1 Da/1Db/2D indicate north and south sampling points at increasing distances from reference point. Error bars represent SD of duplicate analyses. | ||
ENF was the second most abundant antibiotic, with concentrations peaking at 788 ± 5.4 ng L−1 and 793 ± 15.7 ng L−1 on Systems A and B, respectively. ENF was consistently detected across all sampling locations, with concentrations ranging from 443.3 to 787.8 ng L−1 on System A and 453.5 to 792.7 ng L−1 on System B. Other antibiotics were also widely detected, although at lower concentrations. LIN was present in the range of 100–214 ng L−1, and 105–240 ng L−1 based on analysis conducted in System A and System B, respectively. Similarly, OXC and SMZ were detected at moderate concentrations, with OXC ranging from 91.5 to 157.8 ng L−1 and 97.5 to 165.9 ng L−1, and SMZ ranging from 307.3 to 477.8 ng L−1 and 318.6 to 506.8 ng L−1 as per analysis conducted in System A and System B, respectively.
Additional antibiotics including ERY, PEN, CIP, and LEV were consistently detected across sampling points, with concentrations varying from 302 to 650.9 ng L−1 in System A and 305.0 to 643.9 ng L−1 in System B. Specifically, the highest concentrations of ERY and PEN were observed at the CETP discharge site (W/R), measuring 601.2 ± 3.15 ng L−1 and 650.9 ± 3.4 ng L−1, respectively in System A, and 594.7 ± 7.8 ng L−1 and 643.9 ± 5.1 ng L−1, respectively in System B. CIP and LEV showed similar trends, with concentrations reaching 762.5 ± 2.1 ng L−1 and 702.2 ± 1.9 ng L−1, respectively in System A, and 769.9 ± 2.0 ng L−1 and 693.1 ± 7.0 ng L−1, respectively in System B. These elevated quinolone and macrolide concentrations may be attributed to their persistent nature under environmental conditions or continuous external input via untreated or insufficiently treated wastewater. These elevated quinolone residues are consistent with reported seawater concentrations up to hundreds of nanograms per litre.22
The antibiotic concentrations observed in this study notably exceed those reported in Indian surface water systems.42 A study by Mutiyar and Mittal (2014)43 reported ciprofloxacin concentrations ranging from below detection levels to a maximum of 1440 ng L−1 in the Yamuna River downstream of sewage treatment discharges. Kamatham et al. (2024)44 reported antibiotic concentrations ranging between 0.01 and 6.7 ng L−1 for eleven antibiotics including ceftazidime, cefaperazone, sulbactum, piperacillin, tazobactum, ciprofloxacin, amikacin, imipenem, erythromycin, clindamycin and bacitracin across surface water sample collected from multiple locations in South India including Chennai (Tamil Nadu), Nellore (Andhra Pradesh), Hyderabad (Telangana), Kottayam (Kerala), and Bengaluru (Karnataka).44 Another study focusing on urban water bodies in Chennai, encompassing the Buckingham Canal and the Adyar and Cooum rivers, reported erythromycin, sulfamethazine, ciprofloxacin enrofloxacin and azithromycin concentrations up to 372.5 ng L−1, 6.8 ng L−1, 186.1 ng L−1, 4.9 ng L−1, and 29.9 ng L−1, respectively.45
High concentrations of antibiotics have also been reported in surface waters downstream of pharmaceutical manufacturing hubs in India. Fick et al. (2009)46 investigated rivers receiving effluents from a CETP in Patancheru, Hyderabad, processesing wastewater from approximately 90 bulk drug manufacturing facilities. Water samples collected from the receiving rivers including Isakuvagu and Nakkavagu, showed ciprofloxacin levels ranging from 10000 to 2500000 ng L−1, while other fluoroquinolones such as enrofloxacin (below detection to 30000 ng L−1), lomefloxacin (up to 1100 ng L−1), norfloxacin (up to 4700 ng L−1), ofloxacin (up to 4000 ng L−1), and trimethoprim (up to 4000 ng L−1) were also detected in the river water. These values represent some of the highest environmental antibiotic concentrations recorded globally and highlight the severe impact of pharmaceutical discharge on surrounding aquatic ecosystems. To our knowledge, this is the only study that has reported such high concentrations of multiple antibiotics in a surface water body.
Rao et al. (2008)47 also reported the presence of norfloxacin at 48 ng L−1, sulfamethoxazole at 76 ng L−1, and trimethoprim at 87 ng L−1 in the Nakkavagu River. Lower concentrations were also detected in nearby surface water bodies, including Kazipalli Tank and Hussain Sagar Lake, reflecting the dispersion of antibiotics in aquatic systems influenced by industrial and urban discharge. Furthermore, Glorian et al. (2018)48 reported sulfamethoxazole concentrations between 347 and 1260 ng L−1 across the Ganga and Yamuna rivers during five sampling campaigns conducted between 2015 and 2018. Notably, samples from New Delhi and Uttar Pradesh exhibited higher contamination compared to upstream sites in Uttarakhand, reflecting the influence of urban discharge and wastewater loads on antibiotic prevalence in riverine systems.
Distinct spatial distribution patterns were evident across the creek. Lower antibiotic concentrations were typically observed at the location close to shore (W/S) and at the location close to mangroves (W/M; on the opposite side of the shore) compared to the reference point (W/R), where the CETP wastewater was discharged. This trend is possibly caused by the combined effects of dilution, tidal flushing, and possible biotic uptake within the vegetated buffer zones. In contrast, the highest concentrations were recorded at the CETP discharge site (W/R), suggesting the treatment plant as a primary point source for these contaminants. Similar persistence trends have been observed downstream of wastewater treatment plants in Arkansas, USA, where antibiotics were traceable for over 50 km downstream.49
North and south side of W/R sampling points (1U, 1 Da, 1Db, and 2D) showed a gradual decline in concentrations, suggesting progressive dilution and natural attenuation processes such as sorption, photolysis, and microbial degradation, which are commonly observed for antibiotics in surface waters.50,51 Interestingly, the site designated as W/2U exhibited unusually higher antibiotic levels, comparable to or even exceeding those at the CETP discharge point due to proximity to a municipal solid waste dumping area. The elevated concentrations were possibly caused by leachate infiltration or stormwater runoff from the landfill, indicating the role of non-point sources in sustaining antibiotic pollution loads in the creek ecosystem. The quantitative agreement between the two analytical instruments was strong, with differences typically within 10% for most antibiotics. System B often reported slightly higher concentrations, particularly for AZT and LIN. This discrepancy is likely due to the enhanced resolving power of System B, which enables the extraction of narrow mass windows and more effective exclusion of co-eluting matrix interferences, an advantage particularly relevant for complex environmental samples.
The persistent detection of multiple classes of antibiotics at ecologically relevant concentrations raises important environmental and public health concerns. Even sub-inhibitory levels of antibiotics in aquatic systems can exert selective pressure on microbial communities, promoting the proliferation and spread of antimicrobial resistance genes.4 This risk becomes particularly significant when environmental concentration exceed the predicted no-effect concentrations (PNEC) thresholds proposed for the antimicrobials resistance selection.4 Bengtsson-Palme and Larsson (2016)4 proposed PNECs for over 100 antibiotics by extrapolating from minimum inhibitory concentrations (MICs) and applying a safety factor of 10. For fluoroquinolones such as ciprofloxacin, levofloxacin, and enrofloxacin, PNECs ranged from 64 to 250 ng L−1,4 while for macrolides such as azithromycin and erythromycin, the range was higher between 250 and 1000 ng L−1.4 In this study fluoroquinolones were detected at concentrations up to 794 ng L−1 and macrolides were detected at concentrations up to 1700 ng L−1. Several of the measured values exceeded the PNEC for antimicrobial resistance selection in the environment. Additional studies further reinforce this concern. Stanton et al. (2020)52 reported a minimum selective concentration (MSC) for ciprofloxacin at 10.77 μg L−1, corresponding to a PNEC of 1 μg L−152 (after applying a safety factor of 10). Their value was higher than the value for ciprofloxacin proposed by Bengtsson-Palme and Larsson (2016).4 Gullberg et al. (2011)53 reported MSC for two resistant E. coli, mutants as 100 ng L−1 and 2500 ng L−1. Beyond concentration-based thresholds, resistance profiling for antibiotic resistant genes and antibiotic resistant bacteria across aquatic systems in India also reinforce the AMR risk.54,55 These findings emphasize the urgent need for the development of more advanced wastewater treatment processes capable of removing antibiotic and other micropollutants and the implementation of long-term environmental monitoring programs aimed at mitigating the spread of resistance in the environment.
To further evaluate the consistency between the two analytical instruments, concentration data for each antibiotic were directly compared using linear regression (Fig. 4) by plotting the concentration measured by LC-Orbitrap-HRMS (System B) versus concentration measured by LC-QqQ-MS (System A) for each antibiotics. The correlation coefficients (R2) and regression slopes were determined using linear regression by setting the intercept to zero, to determine consistency across the methods. Majority of antibiotics demonstrated excellent linear agreement, with R2 values exceeding 0.9 for all antibiotics, highlighting that both the instrumental methods yield comparable concentration for environmental samples. ENF (Fig. 4a) exhibited near-perfect agreement, with a regression slope of 1.0 confirming the quantitative equivalence of the two instruments across the detected concentration range. Similarly, AZT (Fig. 4c) displayed identical slope of 1.0 illustrating both the accuracy and the reliability of both instruments, even at elevated concentration levels. ERY (Fig. 4f) and PEN (Fig. 4g) each showed a slope of 0.99, further supporting strong inter-instrument consistency. LEV (Fig. 4i) and CIP (Fig. 4h) exhibited slopes of 0.99 and 1.0, respectively, confirming reliable quantification across this antibiotic class. LIN (Fig. 4b) showed a slightly elevated slope of 1.06 suggesting a modest positive bias for System B at higher concentrations, possibly linked to its high mass resolution enabling better isolation of the analyte peaks from matrix interferences. OXC (Fig. 4d) and SMZ (Fig. 4e) showed slopes of 1.01 and 1.04, respectively, indicating comparable concentration with minor variability due to reduced susceptibility to co-eluting matrix ions. Overall, the high degree of agreement across instruments reinforces the reliability of both LC-QqQ-MS and LC-Orbitrap-HRMS for the quantitative analysis of antibiotics in environmental water samples.
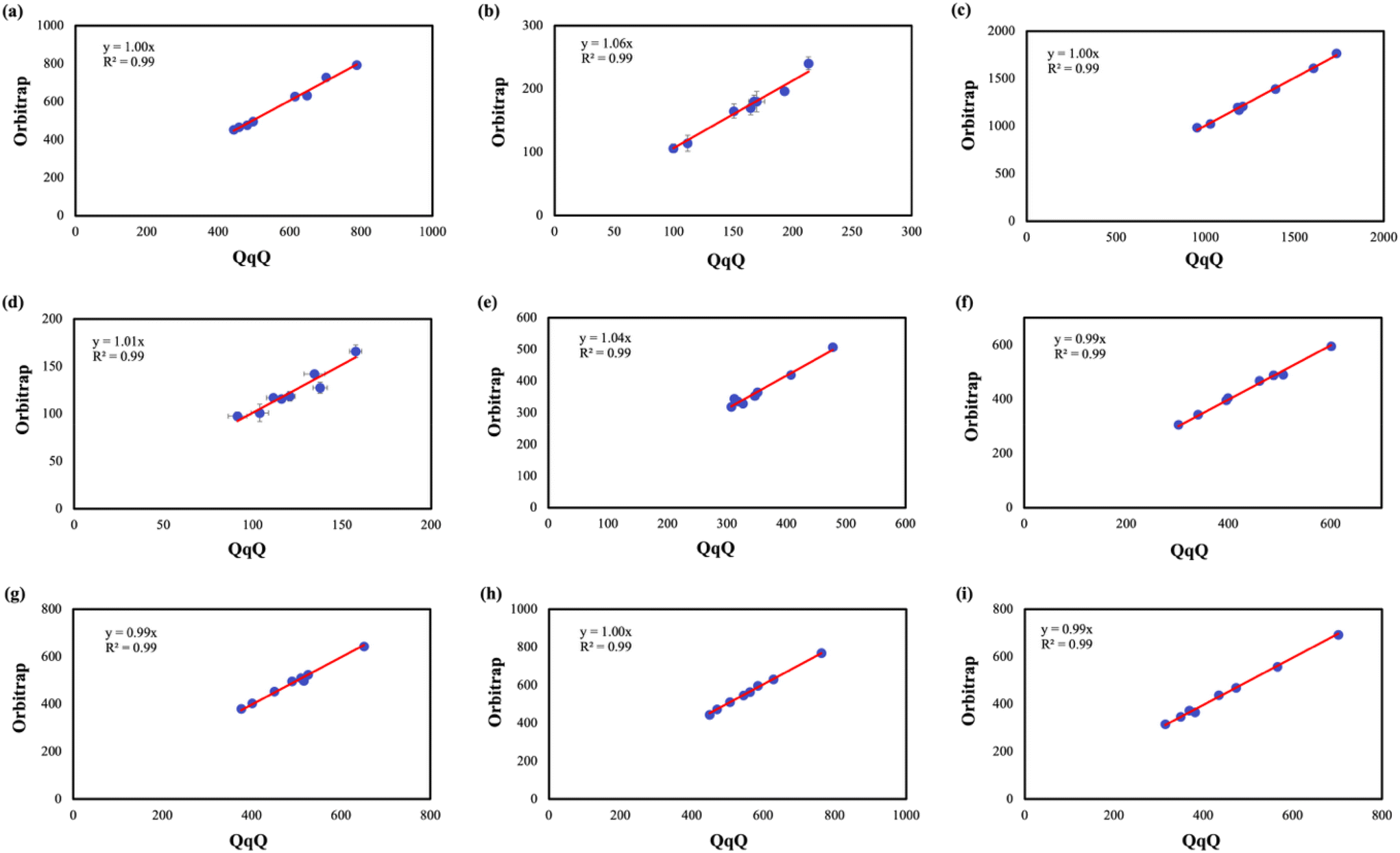 | ||
| Fig. 4 Correlation plots comparing antibiotic concentrations (ng L−1) measured using LC-QqQ-MS and LC-Orbitrap-HRMS (a) ENF, (b) LIN (c) AZT, (d) OXC (e) SMZ (f) ERY (g) PEN (h) CIP and (i) LEV. *Red lines represent the linear regression fits. Slopes and R2 values are displayed on each plot, indicating strong to excellent agreement across most analytes, with minor compound-specific deviations attributed to matrix effects and differences in quantification strategy. Horizontal and vertical error bars indicate the standard deviations of antibiotic concentration measured using LC-QqQ-MS and LC-Orbitrap-HRMS, respectively. | ||
A three–way ANOVA was performed to assess the effects of instrument type (I), sampling location (S), and antibiotic type (A) on measured antibiotic concentrations. The results (Table S2†) indicated that measured antibiotic concentration was significantly affected by each of the individual factors including instrument, sampling location, and antibiotic type. The analysis revealed extremely low p-values for the main effects (p = 4.97 × 10−8 for I, p = 1.9 × 10−182 for S and p = 1.21 × 10−261 for A), suggesting that the type of instrument, sampling location and antibiotic type significantly affected final antibiotic concentration. Additionally, all two-way interaction terms, including, I*S (p = 3.04 × 10−6), I*A (p = 1.39 × 10−7), and S*A (p = 2.98 × 10−152), were also statistically significant. Notably, the three-way interaction (I*S*A, p = 8.85 × 10−4) was significant as well, indicating that the combined influence of instrument, site, and antibiotic type affected the concentration measurements. These results suggest that variations in antibiotic concentrations are influenced not only by individual factors but also by their interactions, reflecting the complex environmental dynamics, instrumental sensitivities, and site- and compound-specific behaviours in the aquatic system.
Despite the excellent linear correlations observed between the two instrumental methods (R2 values exceeding 0.99 across all antibiotics), the three-way ANOVA analysis revealed statistically significant effects of instrument type, sampling location, and antibiotic type on the measured concentrations. In addition, all two-way interactions and the three-way interaction was also found to be significant (p < 0.05). These findings suggest that, although the two instruments track concentration trends very closely, systematic differences in absolute quantification exist across antibiotics and sampling sites. Such variations are likely attributable to differences in instrumental sensitivity, ionization efficiency, and matrix suppression effects that particularly affect quantification of antibiotics in complex environmental samples.
An important finding of this study is that while both LC-QqQ-MS and LC-Orbitrap-HRMS instruments yielded highly comparable spatial concentration trends across all quantified antibiotics, minor yet statistically significant differences in absolute concentration values were observed between the instruments. These systematic biases, although small, highlight the need for caution when comparing quantitative datasets across different mass spectrometry platforms, particularly in regulatory or time-series monitoring contexts. This aspect must be considered when interpreting absolute concentration values in environmental monitoring studies. For environmental surveillance programs, this finding highlights the importance of inter-laboratory calibration and methodological harmonization to ensure appropriate data comparison.
In addition to the nine antibiotics targeted in this study, the LC-Orbitrap-HRMS (System B) was used to perform non-target screening to explore the presence of other antibiotics. Compound identification followed the protocol specified by Schymanski et al. (2014).16 All compounds presented here were identified at Level 2 confidence, based on accurate mass and MS/MS fragmentation pattern matches to spectral libraries. A total of 10 additional antibiotics including fluoroquinolones, sulfonamides, and aminoglycosides were identified across the eight sampling points. Fig. 5 illustrates their relative abundance based on peak area value. Ofloxacin was the most dominant antibiotic, with notably high peak areas observed across the sampling points, suggesting high persistence in the creek. Norfloxacin, sulfadiazine, and sulfamethizole also showed widespread occurrence, although their peak area values were lower.
 | ||
| Fig. 5 Heatmap showing the peak area of antibiotics identified via non-target screening using LC-Orbitrap-HRMS across eight sampling locations. *White spaces indicate antibiotics not detected. | ||
Kanamycin and carbadox were identified only in the three sampling points (W/S, W/M, W/R), suggesting localized pharmaceutical inputs. Sulfisomidine and sulfaquinoxaline, both older-generation sulfonamides, were detected at multiple sampling points however, their peak area were lower compared to other antibiotics. Some antibiotics displayed more site-specific patterns. Sparfloxacin was observed at the sites i.e., W/S, W/M, W/R and W/2U, whereas sedecamycin was present at all sites but at consistently low peak area values. The detection of kanamycin, sparfloxacin, and carbadox only at select locations further highlight the heterogeneous nature of contamination and importance of high-resolution full-scan acquisition for environmental screening. These findings demonstrate the capability of Orbitrap-HRMS to complement traditional targeted analysis by identifying non-prioritized – emerging contaminants that may otherwise go undetected. Incorporating such exploratory data into monitoring programs can enhance the assessment of environmental exposure risks, inform regulatory prioritization, and guide source control strategies. Given the increasing use of HRMS technologies for non-targeted analysis, these results also support their use in future chemical monitoring campaigns, especially in regions affected by complex and dynamic pollutant mixtures.
4. Conclusions
This study provides a comprehensive comparison of two mass spectrometry instruments including LC-QqQ-MS and LC-Orbitrap HRMS, for the quantitative analysis of multiple class of antibiotics in creek water affected by CETP effluent. Both systems demonstrated reliable performance in terms of sensitivity, precision, and recovery, although LC-Orbitrap-HRMS offered superior detection limits and enhanced selectivity due to its high resolving power. The field application confirmed that antibiotics are prevalent in CETP-impacted aquatic environments, with particularly high concentrations at the CETP discharge point highlighting the persistence of antibiotic contaminants even after treatment. The significant effects of instrument type, sampling location, and antibiotic type was revealed through quantitative analysis of antibiotics in environment samples. An important observation from this study is that while both LC-QqQ-MS and LC-Orbitrap-HRMS yielded highly consistent spatial distribution patterns and relative trends, there were minor yet statistically significant differences in the absolute concentrations reported by the two instruments. This is especially crucial in regulatory or monitoring contexts, where absolute concentration thresholds may guide policy decisions or environmental risk assessments. This finding further supports the adoption of Orbitrap-HRMS for environmental monitoring, due to its combined strengths in precise quantification together with the detection of unknown – emerging contaminants in environmental samples. These findings highlight the critical role of advanced mass spectrometry tools in accurate environmental monitoring and the need for integrated surveillance strategies that account for both targeted and broader non-targeted contaminant screening.Author contributions
Arhama T. A. Ansari: conceptualization, methodology, validation, environmental sample collection, investigation, visualization, data curation, resources, writing – original draft, writing – review & editing. Ayush Ransingh: creating Fig. 1 using QGIS 3.22 & environmental sample collection. Soumyo Mukherji: supervision, funding acquisition, & writing – review & editing. John Connolly: funding acquisition, writing – review & editing. Andrew Hursthouse: funding acquisition, writing – review & editing. Fiona L. Henriquez: funding acquisition, writing – review & editing. Suparna Mukherji: supervision, project administration, funding acquisition, conceptualization, methodology, data curation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The work was conducted using the triple quadrupole mass spectrometer and LC-HRMS Orbitrap Central Facility of IIT Bombay, established in the Environmental Science & Engineering Department. The authors gratefully acknowledge the financial support provided by the International Division of the Department of Biotechnology, Government of India, through the Indo-UK AMSPARE project (BT/IN/INDO-UK/AMR-ENV/01/SM/2020-21; December 2020 to December 2024), and by the UK Natural Environment Research Council (NERC) (grant no. NE/T012986/1). A. T. A. A. gratefully acknowledges the support received through the Prime Minister's Research Fellowship (PMRF), provided by the Government of India. Pradnya Vernekar and Harleen Kaur Walia are acknowledged for help with sample collection.References
- R. Wang, H. Tang, R. Yang and J. Zhang, Water Sci. Technol., 2024, 89, 2763–2782 CrossRef PubMed.
- A. K. Singh, R. Kaur, S. Verma and S. Singh, Front. Environ. Sci., 2022, 10, 830861 CrossRef.
- A. Tiwari, P. Kurittu, A. I. Al-Mustapha, V. Heljanko, V. Johansson, O. Thakali, S. K. Mishra, K.-M. Lehto, A. Lipponen, S. Oikarinen, T. Pitkänen, WastPan Study Group, A. Heikinheimo, A. Länsivaara, R. Hyder, E. Janhonen, A.-M. Hokajärvi, A. Sarekoski, A. Kolehmainen, S. Blomqvist, K. Räisänen, C. S. Kopra, T. Möttönen, O. Luomala, A. Juutinen, O. Thakali and S. K. Mishra, Front. Microbiol., 2022, 10, 977106 CrossRef PubMed.
- J. Bengtsson-Palme and D. G. J. Larsson, Environ. Int., 2016, 86, 140–149 CrossRef CAS PubMed.
- E. Ngumba, P. Kosunen, A. Gachanja and T. Tuhkanen, Anal. Methods, 2016, 8, 6720–6729 RSC.
- R. Vaicunas, S. Inamdar, S. Dutta, D. S. Aga, L. Zimmerman and J. Tom Sims, J. Am. Water Resour. Assoc., 2013, 49, 463–474 CrossRef CAS.
- R. Bade, N. I. Rousis, L. Bijlsma, E. Gracia-Lor, S. Castiglioni, J. V. Sancho and F. Hernandez, Anal. Bioanal. Chem., 2015, 407, 8979–8988 CrossRef CAS PubMed.
- S. D. Richardson, Anal. Chem., 2012, 84, 747–778 CrossRef CAS PubMed.
- T. Anumol and S. A. Snyder, Talanta, 2015, 132, 77–86 CrossRef CAS PubMed.
- M. Gorga, M. Petrovic and D. Barceló, J. Chromatogr. A, 2013, 1295, 57–66 CrossRef CAS PubMed.
- R. Malarvili, Y. Y. Low and A. Zaiton, ASEAN J. Sci. Technol. Dev., 2017, 23, 404 Search PubMed.
- V. Albergamo, R. Helmus and P. De Voogt, J. Chromatogr. A, 2018, 1569, 53–61 CrossRef CAS PubMed.
- M. Á. de la Serna Calleja, S. Bolado, J. J. Jiménez and R. López-Serna, Microchem. J., 2023, 187, 108395 CrossRef CAS.
- M. Beccaria and D. Cabooter, Analyst, 2020, 145, 1129–1157 RSC.
- S. Huysman, L. Van Meulebroek, F. Vanryckeghem, H. Van Langenhove, K. Demeestere and L. Vanhaecke, Anal. Chim. Acta, 2017, 984, 140–150 CrossRef CAS PubMed.
- E. L. Schymanski, H. P. Singer, P. Longrée, M. Loos, M. Ruff, M. A. Stravs, C. Ripollés Vidal and J. Hollender, Environ. Sci. Technol., 2014, 48, 1811–1818 CrossRef CAS PubMed.
- T. Letzel, A. Bayer, W. Schulz, A. Heermann, T. Lucke, G. Greco, S. Grosse, W. Schüssler, M. Sengl and M. Letzel, Chemosphere, 2015, 137, 198–206 CrossRef CAS PubMed.
- I. Ferrer, D. L. Sweeney, E. M. Thurman and J. A. Zweigenbaum, J. Am. Soc. Mass Spectrom., 2020, 31, 1189–1204 CrossRef CAS PubMed.
- I. Tlili, G. Caria, B. Ouddane, I. Ghorbel-Abid, R. Ternane, M. Trabelsi-Ayadi and S. Net, Sci. Total Environ., 2016, 563–564, 424–433 CrossRef CAS PubMed.
- P. Herrero, N. Cortés-Francisco, F. Borrull, J. Caixach, E. Pocurull and R. M. Marcé, J. Mass Spectrom., 2014, 49, 585–596 CrossRef CAS PubMed.
- S. Belarbi, M. Vivier, W. Zaghouani, A. D. Sloovere, V. Agasse-Peulon and P. Cardinael, Food Chem., 2021, 359, 129932 CrossRef CAS PubMed.
- Z. Maghsodian, A. M. Sanati, T. Mashifana, M. Sillanpää, S. Feng, T. Nhat and B. Ramavandi, Antibiotics, 2022, 11, 1461 CrossRef CAS PubMed.
- E. Corbett, R. Esiovwa, R. Mooney, K. Rodgers, S. Mukherji, J. Connolly, A. Hursthouse, S. Mukherji and F. L. Henriquez, Environ. Earth Sci., 2025, 84, 88 CrossRef.
- A. Nasiri, R. Jahani, S. Mokhtari, H. Yazdanpanah, B. Daraei, M. Faizi and F. Kobarfard, Analyst, 2021, 146, 6049–6063 RSC.
- G. Siedlewicz, L. Sharma, B. Szymczycha, A. Białk-Bielińska and K. Pazdro, in Pharmaceuticals in Marine and Coastal Environments, ed. J. C. Durán-Álvarez and B. Jiménez-Cisneros, Elsevier, 2021, vol. 1, pp. 275–301 Search PubMed.
- S. Mohapatra, C.-H. Huang, S. Mukherji and L. P. Padhye, Chemosphere, 2016, 159, 526–535 CrossRef CAS PubMed.
- M. J. Nunes, V. Paz, C. M. Cordas, J. P. Noronha and L. C. Branco, Anal. Methods, 2022, 14, 935–948 RSC.
- H. Trufelli, P. Palma, G. Famiglini and A. Cappiello, Mass Spectrom. Rev., 2011, 30, 491–509 CrossRef CAS PubMed.
- J. Liu, F. Jiang, Z. Lu, C. Zhang, P. Liu, M. Huang and G. Zhong, Molecules, 2023, 28, 746 CrossRef CAS PubMed.
- O. I. Lavrukhina, V. G. Amelin, L. K. Kish, A. V. Tretyakov and T. D. Pen'kov, J. Anal. Chem., 2022, 77, 1349–1385 CrossRef CAS.
- United States Environmental Protection Agency (US EPA), Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS, 2007 Search PubMed.
- L. L. Bridgewater, R. B. Baird, A. D. Eaton and E. W. Rice, American Public Health Association, American Water Works Association, and Water Environment Federation, in Standard methods for the examination of water and wastewater, American Public Health Association, Washington, DC, 23rd edn, 2017 Search PubMed.
- I. González-Mariño, J. B. Quintana, I. Rodríguez and R. Cela, Rapid Commun. Mass Spectrom., 2009, 23, 1756–1766 CrossRef PubMed.
- S. Liu, G.-G. Ying, J.-L. Zhao, F. Chen, B. Yang, L.-J. Zhou and H. Lai, J. Chromatogr. A, 2011, 1218, 1367–1378 CrossRef CAS PubMed.
- C. L. Chitescu, G. Kaklamanos, A. I. Nicolau and A. A. M. L. Stolker, Sci. Total Environ., 2015, 532, 501–511 CrossRef CAS PubMed.
- A. Kalogeropoulou, C. Kosma and T. Albanis, Explor. Foods Foodomics, 2024, 2, 767–787 Search PubMed.
- Y. Zhang, X. Ren, Z. Zhou, D. W. Wang, X. Rao, H. Ding and J. Wu, Analyst, 2024, 149, 3444–3455 RSC.
- R. Liu, Q. Long, Y. Liu and L. Wang, Chemosphere, 2025, 370, 143953 CrossRef CAS PubMed.
- C. Li, A. Li, X. Hui, A. Wang, L. Wang and S. Chang, Ecotoxicol. Environ. Saf., 2024, 285, 117022 CrossRef CAS PubMed.
- N. Da Le, A. Q. Hoang, T. T. H. Hoang, T. A. H. Nguyen, T. T. Duong, T. M. H. Pham, T. D. Nguyen, V. C. Hoang, T. X. B. Phung, H. T. Le, C. S. Tran, T. H. Dang, N. T. Vu, T. N. Nguyen and T. P. Q. Le, Environ. Sci. Pollut. Res., 2021, 28, 10622–10632 CrossRef CAS PubMed.
- F. Mirzaie, F. Teymori, S. Shahcheragh, S. Dobaradaran, H. Arfaeinia, R. Kafaei, S. Sahebi, S. Farjadfard and B. Ramavandi, Chemosphere, 2022, 307, 135996 CrossRef CAS PubMed.
- P. K. Mutiyar and A. K. Mittal, Environ. Sci. Pollut. Res., 2014, 21, 7723–7736 CrossRef CAS PubMed.
- P. K. Mutiyar and A. K. Mittal, Environ. Monit. Assess., 2014, 186, 541–557 CrossRef CAS PubMed.
- S. Kamatham, M. Seeralan, U. Sekar and S. Kuppusamy, ACS Omega, 2024, 9, 12801–12809 CAS.
- S. Arun, L. Xin, O. Gaonkar, B. Neppolian, G. Zhang and P. Chakraborty, Sci. Total Environ., 2022, 851, 158195 CrossRef CAS PubMed.
- J. Fick, H. Söderström, R. H. Lindberg, C. Phan, M. Tysklind and D. G. J. Larsson, Environ. Toxicol. Chem., 2009, 28, 2522–2527 CrossRef CAS PubMed.
- R. N. Rao, N. Venkateswarlu and R. Narsimha, J. Chromatogr. A, 2008, 1187, 151–164 CrossRef PubMed.
- H. Glorian, H. Börnick, C. Sandhu and T. Grischek, Water, 2018, 10, 1804 CrossRef CAS.
- B. E. Haggard and L. D. Bartsch, J. Environ. Qual., 2009, 38, 343–352 CrossRef CAS PubMed.
- J. Ji, H. Li and S. Liu, Appl. Sci., 2025, 15, 5182 CrossRef CAS.
- I. Bueno, H. He, A. C. Kinsley, S. J. Ziemann, L. R. Degn, A. J. Nault, A. L. Beaudoin, R. S. Singer, K. H. Wammer and W. A. Arnold, Sci. Total Environ., 2023, 897, 165301 CrossRef CAS PubMed.
- I. C. Stanton, A. K. Murray, L. Zhang, J. Snape and W. H. Gaze, Commun. Biol., 2020, 3, 467 CrossRef CAS PubMed.
- E. Gullberg, S. Cao, O. G. Berg, C. Ilbäck, L. Sandegren, D. Hughes and D. I. Andersson, PLoS Pathog., 2011, 7, e1002158 CrossRef CAS PubMed.
- A. K. Pal, P. Tripathi, J. A. Lal and V. Tripathi, Water, Air, Soil Pollut., 2024, 235, 478 CrossRef CAS.
- B. Ram and M. Kumar, npj Clean Water, 2020, 3, 3 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00482a |
| This journal is © The Royal Society of Chemistry 2025 |