
Development of PROTACs targeting estrogen receptor: an emerging technique for combating endocrine resistance
Rouming
Peng
 a,
Xin
Liu
a,
Chun-Chi
Chen
ab,
Rey-Ting
Guo
*ab and
Jian
Min
*a
a,
Xin
Liu
a,
Chun-Chi
Chen
ab,
Rey-Ting
Guo
*ab and
Jian
Min
*a
aState Key Laboratory of Biocatalysis and Enzyme Engineering, National & Local Joint Engineering Research Center of High-throughput Drug Screening Technology, School of Life Sciences, Hubei University, Wuhan, 430062, China. E-mail: guoreyting@hubu.edu.cn; jianmin@hubu.edu.cn
bDepartment of Immunology and Pathogen Biology, School of Basic Medical Sciences, Hangzhou Normal University, Hangzhou, 311121, China
First published on 30th December 2024
Abstract
Despite the success of endocrine therapies in treating ER-positive breast cancer, the development of resistance remains a significant challenge. Estrogen receptor targeting proteolysis-targeting chimeras (ER PROTACs) offer a unique approach by harnessing the ubiquitin–proteasome system to degrade ER, potentially bypassing resistance mechanisms. In this review, we present the drug design, efficacy and early clinical trials of these ER PROTACs. This review underscores the academic and industrial opportunities presented by this emerging technology, as well as the challenges that must be addressed to translate these findings into effective clinical therapies.
1. Introduction
Human estrogen receptor alpha (hERα) functions as a ligand-dependent transcription factor, a member of the type II nuclear receptor family.1 In conjunction with endogenous estradiol, ERα plays major roles in the development and maintenance of female reproductive tissues as well as many other nonreproductive organs, such as bone, brain, vasculature, liver and pancreas.2 Estradiol expression and ERα signaling were identified as the primary driving factors in the occurrence, growth, proliferation and metastasis for more than 70% human breast cancers.3 Therefore, ERα is an effective target against early-stage breast cancer.Currently, ER-positive breast cancer patients are advised to undergo 5–10 years of adjuvant endocrine treatment.4 Endocrine therapies rely on aromatase inhibitors (such as anastrozole, letrozole, and exemestane) that prevent estradiol synthesis, or selective estrogen receptor modulators (such as tamoxifen and raloxifene) and degraders (such as fulvestrant) that inhibit ERα function.5,6
As a standard nuclear receptor, the core architecture of ERα includes a DNA-binding domain C (DBD, amino acids 304–554), a ligand-binding domain E (LBD, amino acids 304–554) and two activation functions (AF1 in the amino terminal A/B domain and AF2 in the LBD),7 as shown in Fig. 1a. ER LBD comprises 12 α helices to form a “sandwich” antiparallel α-helical structure with a substantial pocket for ligand binding, where helix 12 (h12) serves as a “switch” to interact with coregulators.8 In the presence of a bound agonist (such as estradiol), h12 covers the ligand binding pocket (LBP) like a lid.9 This conformation allows the binding of coactivators (CoA) and activates downstream ER signaling pathways (Fig. 1b). In the presence of a bound antiestrogen (such as tamoxifen), h12 switches and blocks effective recognition of coactivators, thereby interrupting the downstream ER signaling pathways (Fig. 1c).10
 | ||
| Fig. 1 a) Domain structure of estrogen receptor α; b) with a bound agonist estradiol, h12 folds back and covers the LBP to bind CoA (PDB ID: 5WGD); c) with a bound antagonist tamoxifen, h12 moves to block the CoA binding (PDB ID: 3ERT). | ||
Although endocrine therapy has been effective in early-stage breast cancer treatment, functional acquired mutations in the ESR1 gene are relatively common in patients with metastatic ER-positive breast cancer.11,12 Crystallography and biophysics studies show that hot ESR1 mutations (such as Y537S, D538G and Y537N) stabilize the active conformation by forming internal hydrogen bonds.13 These conformation changes increase constitutive activity without any ligand and reduce the binding affinity of endocrine therapy drugs.14 Moreover, ESR1 mutations may also affect the interaction between ERα and other signaling pathway proteins, such as those in the PI3K/AKT and MAPK pathways.15 Consequently, these mutations promote tumor growth, metastasis and resistance to treatment. Thus, research on new endocrine therapy drugs is underway, focusing on their effectiveness in preclinical models of ESR1 mutations to improve the prognosis of patients with ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer.16
Early studies indicate that new endocrine therapy drugs, such as oral SERDs (selective estrogen receptor degraders) and PROTACs (proteolysis targeting chimeras), offer effective treatment options for patients with ESR1 mutations.17–19 Currently, Giredestrant by Roche, Camizestrant by AstraZeneca, Imlunestrant by Eli Lilly, Palazestrant by Olema Pharmaceuticals, and ARV-471, a joint effort by Arvinas and Pfizer, are undergoing phase III clinical trials for ER-targeting breast cancer medication.19 Furthermore, compared to traditional SERDs such as fulvestrant, elacestrant and GDC0810, ER PROTAC ARV-471 exhibits a higher rate of target protein degradation, making it more effective in inhibiting ESR1 mutation-positive breast cancer.20
The first ER PROTAC was reported in 2003 by the Crews and Deshaies group.21 Since then, the evolution of ER PROTAC has spanned from its initial conceptualization to clinical translation. In this review, we will explore the ongoing advancements and upcoming development of ER PROTAC degraders.
2. PROTAC technology revolutionized traditional therapies
Taking ER PROTAC as an example, the mechanism of PROTAC is depicted in Fig. 2. The PROTAC molecule is composed of three important parts: a ligand that specifically binds to ER, a ligand that recruits a specific E3 ubiquitin ligase, and a variable linker.22 By simultaneously binding to the target protein and E3 ligase, PROTAC forms a ternary complex and initiates polyubiquitination and subsequent degradation of the POI.23 Once the target proteins are degraded, PROTAC molecules can dissociate and participate in the next catalytic cycle, continuing to induce the degradation of other target proteins.24 The unique catalytic cycle and event-driven pharmacology distinguish PROTAC from conventional ER inhibitors and degraders.25 | ||
| Fig. 2 a) Mechanism of the ERα signaling pathway using SERMs and SERDs. b) Mechanism of action of ER PROTAC degraders (created with https://www.BioRender.com). | ||
Although there are more than 600 E3 ligases in the human body, only a few have been employed in the development of PROTACs to date.26,27 The most commonly used E3 ligases are Von Hippel–Lindau (VHL) and cereblon (CRBN).28 Others like inhibitors of apoptosis protein (IAP), Kelch-like ECH-associated protein (KEAP1) and mouse double minute 2 (MDM2) are also being investigated.29
3. Peptide-based ER PROTAC degraders
In early exploration of ER PROTAC, the peptide sequence was utilized to specifically recruit E3 ligase.30 These early ER PROTAC molecules typically consist of two components: estradiol that binds to ER and a peptide sequence that binds to the E3 ubiquitin ligase. Representative early peptide-based ER PROTAC degraders are shown in Fig. 3.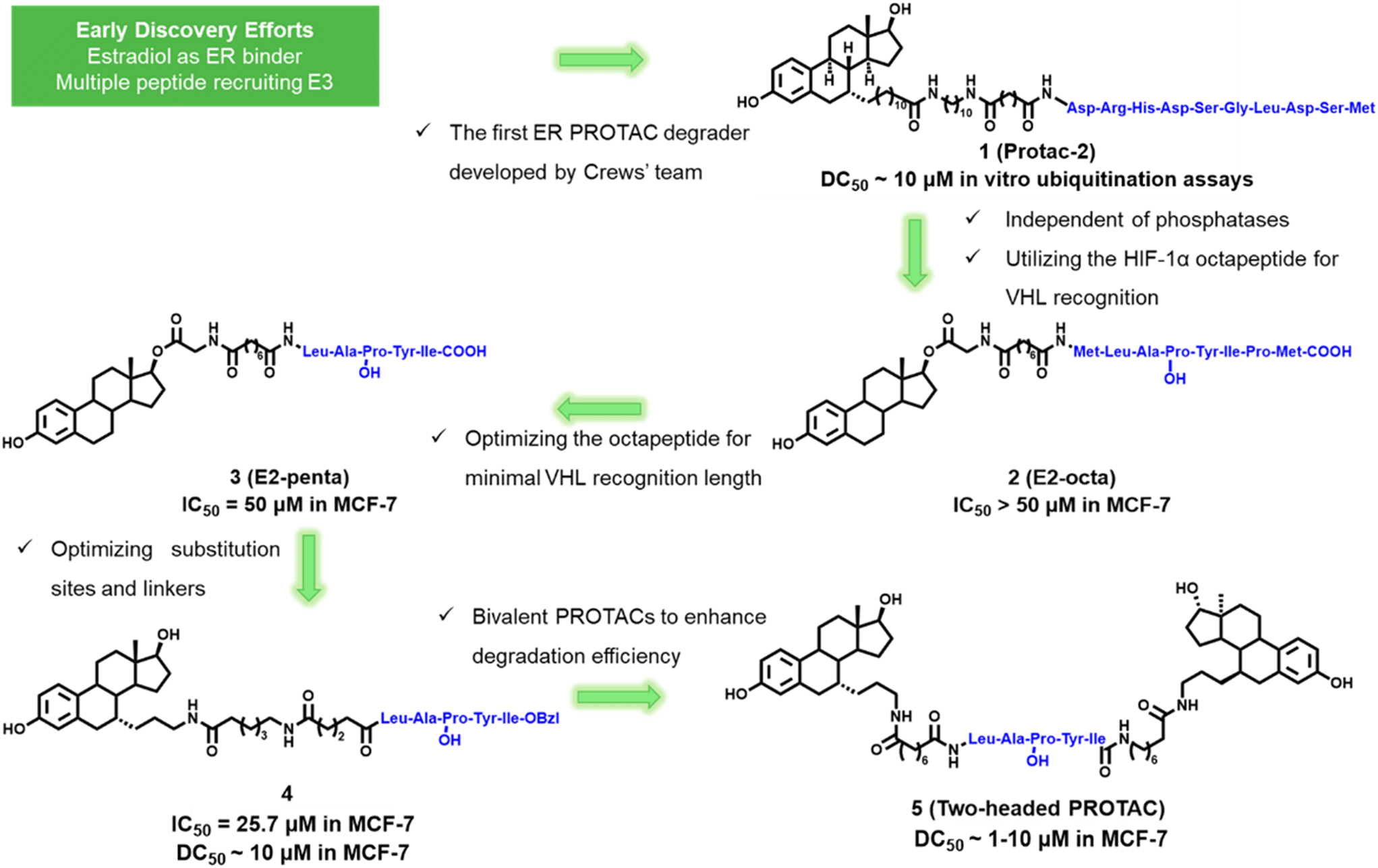 | ||
| Fig. 3 Early peptide-based ER PROTAC degraders 1–5. | ||
In 2003, the Deshaies and Crews group reported the first ER PROTAC degrader named compound 1 (Protac-2). This ER PROTAC comprises an IκBα phosphopeptide segment (DRHD![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) GLDSM) linked to estradiol (E2). Mechanistic studies confirmed that Protac-2 recruited ER to the SCF-TRCP ubiquitin ligase through noncovalent interactions, thereby promoting ubiquitination and degradation of ER in vitro.21 This study lays the foundation for the development of ER PROTAC and demonstrates its potential applications in breast cancer treatment.
GLDSM) linked to estradiol (E2). Mechanistic studies confirmed that Protac-2 recruited ER to the SCF-TRCP ubiquitin ligase through noncovalent interactions, thereby promoting ubiquitination and degradation of ER in vitro.21 This study lays the foundation for the development of ER PROTAC and demonstrates its potential applications in breast cancer treatment.
In 2004, the Kim group from UCLA employed an E2-linked octapeptide from HIF-1α to create compound 2 (E2-octa).31 To identify the minimum length recognized by pVHL, they deleted the amino acids at both ends of the HIF-1α octapeptide, creating a series of shortened peptide E2-based PROTACs. Degradation experiments revealed compound 3 (E2-penta) to be the most efficacious.32 Subsequent studies showed that E2-penta effectively inhibited MCF-7 (IC50 = 50 μM) and T47D (IC50 = 16 μM) cell proliferation, yet ineffective in estrogen-independent cell SKBr3. This implies that E2-penta is specifically targeted for ER. The immunoprecipitation assays indicated that ER degradation was achieved via the proteasome-dependent pathway.33
In 2010, the Kim group optimized E2-penta into compound 4 by changing the attachment point from the O-17 position to the C-7α position to improve stability and binding affinity. Compound 4 showed superior binding affinity to tamoxifen and effectively degrades the endogenous ER in MCF-7 cells.34 They also developed a novel class of two-headed PROTACs by connecting two E2 molecules to this HIF-1α pentapeptide. Two headed PROTAC compound 5 efficiently induces degradation of the ER in a concentration-dependent and proteasome-dependent manner. It also gives superior ER degradation when compared to one-headed PROTACs.35
These first-generation peptide-based ER PROTAC degraders demonstrated the feasibility of the PROTAC concept in treating breast cancer. However, achieving ideal stability and cellular permeability presents significant challenges. Thus, researchers are developing new types of small molecule PROTACs to overcome these hurdles.36
In 2016, the Li group from Peking University developed a facile template for helix formation using an N-terminal aspartic acid cross-linked stabilized peptide (TD) to create peptidomimetic estrogen receptor modulators (PERMs).37 To improve the anti-proliferative activity of TD-PERMs, they attached them to a HIF-1α pentapeptide, resulting in compound 6 (TD-PROTAC, Fig. 4).38 These TD-PROTACs exhibited markedly improved efficacy in ERα transcription and anti-proliferation activities. In vivo experiments indicate that TD-PROTAC induces tumor regression at a dose of 10 mg kg−1 administered intravenously (IP) in the MCF-7 mouse xenograft model.
 | ||
| Fig. 4 Conformation-restricted peptide-based ER PROTACs 6 (TD-PROTAC) and 7 (PROTAC I-6). | ||
In 2019, the Qian group from China Pharmaceutical University developed cell-permeable peptide-based PROTACs by incorporating a γ-methyl-containing Npg side chain.39 The optimized compound 7 (PROTAC I-6, Fig. 4) exhibited increased anti-proliferative activity (IC50 = 9.7 μM) and markedly boosted ERα degradation activity. Furthermore, PROTAC I-6 demonstrated more potent suppression of tumor growth than tamoxifen.
4. Small molecule ER PROTAC degraders
The discovery of small ligands for E3 ligase laid the groundwork for the rational design of small molecule PROTACs.40 Notably, Von Hippel–Lindau (VHL)-based and CRBN-based PROTACs are predominant.4.1 VHL-based ER PROTAC degraders
Encoded by the Von Hippel–Lindau gene, the VHL protein acts as the receptor for substrate recognition in Cullin2-RING ligases (CRL2), a member of the intricate RING-finger E3 ligase family.41 The VHL complex E3 ligase consists of VHL, elognins B and C, cullin 2, and RING box protein 1. It is responsible for the recognition and ubiquitination of specific substrates, such as the hypoxia-inducible factor HIF-1α.42In 2014, GlaxoSmithKline documented a series of ER PROTACs employing estradiol as the ER ligand linked to a VHL ligand, with compound 8 (Fig. 5) representing the general structure (WO2014108452A1).43 The efficacy of these compounds was evaluated, and each demonstrated ERα degradation activity at a 1 μM concentration.
 | ||
| Fig. 5 Chemical structures of VHL-based ER PROTAC degraders 8, 9 and 10. | ||
In 2018, Accutar Biotech's patent disclosed a series of ER PROTACs using 4-OH tamoxifen as the ER ligand to connect the VHL ligand by variable linkers.44 Compound 9 (Fig. 5) displayed below is the patent's representative structure, potentially the actual configuration of AC0682. AC0682 was reported to induce ERα degradation at a nanomolar level in wild-type and ESR1-mutated MCF-7 cells. Recently, phase I clinical trials (NCT05489679 and NCT05080842) of AC0682 were terminated. The next generation AC699 is recruiting patients to evaluate its safety, PK, and efficacy in treating ER+/HER2− advanced or metastatic breast cancer patients.45 The chemical structure of AC699 remains unclosed.
AstraZeneca's 2019 patent revealed their ER PROTACs using AZD9496 as the ERα ligand and VHL ligand as the E3 ligase binder.46 AZD9496 is an oral SERD developed by AstraZeneca.47,48 The representative compound 10 (Fig. 5) achieved 100% degradation of ERα at 0.3 nM (IC50 < 1 nM).
In 2019, the Wang group from University of Michigan, Ann Arbor discovered a highly potent ER PROTAC degrader compound 12 (ERD-308, Fig. 6b).49 The rational design started from the cocrystal structure of raloxifene with ER LBD. Molecular docking studies revealed that substituting the piperidine ring with the N,N-diethylamino group of tamoxifen led to the exposure of an ethyl group to the solvent, making it an appropriate PROTAC linking site (Fig. 6a). By adjusting the length and composition of the linker, and optimizing N-substituent groups, a range of compounds were synthesized and evaluated for the degradation activity. Among them, ERD-308 achieved the best degradation activity with DC50 values of 0.17 nM and 0.43 nM in MCF-7 and T47D ER+ breast cancer cell lines, respectively.
 | ||
| Fig. 6 a) X-ray structure of the crystal structure of raloxifene complexed with ERα (PDB code: 7KBS). b) Chemical structures of compounds 11 (ERD-148) and 12 (ERD-308). | ||
In the subsequent studies, compound 11 (ERD-148, Fig. 6b) was shown to exhibit remarkable ER degrading potency.50,51 The minor difference between ERD-308 and ERD-148 was the linker composition, as shown in Fig. 6b. ERD-148 degrades not only unphosphorylated ERα but also the phosphorylated ERα in the cells. The complete degradation led to greater suppression of estrogen-dependent wildtype and estrogen-independent ESR1-mutated (Y537S and D538G) MCF-7 cells. Compared to the FDA approved SERD fulvestrant, both ERD-308 and ERD-148 exhibit a more potent efficacy to induce ER degradation and suppress cell proliferation.
In 2022, researchers from IRB Barcelona used 4-OHT as an ERα affinity ligand and designed PROTACs with different linkers and E3 ligase ligands.52 Among them, compound 13 (TVHL-1, Fig. 7) recruits the E3 ligase VHL, achieving a DC50 of 4.5 nM in MCF-7 cells.
 | ||
| Fig. 7 Click chemistry-enabled ER PROTACs. VHL-based ER PROTAC degraders compound 13 (TVHL-1) and 14 (ERE-PROTAC). | ||
Current ER therapeutic drugs mainly target the ligand-binding domain, a region prone to mutations that result in drug resistance.53 In 2022, the Tan group from Tsinghua University developed PROTACs that specifically target the DNA-binding domain (DBD) of ERα to overcome endocrine resistance.54 They synthesized a nucleic acid-conjugated compound 14 (ERE-PROTAC, Fig. 7) molecule through click chemistry.55 In this molecule, the natural DNA-binding sequence ERE (estrogen response element) is responsible for binding to ERα, while the small molecule VH032 is responsible for recruiting von Hippel–Lindau (VHL) E3 ubiquitin ligase.56
In 2023, the Zhou group from Wuhan University developed a novel class of ERα PROTACs based on the oxabicycloheptane sulfonamide (OBHSA) scaffold.57 Among them, compound 15 (ZD12, Fig. 8) exhibited superior antitumor efficacy and ERα degradation activity against a broad spectrum of ER+ BC cell lines, including ERα mutant BC cells. It also shows remarkable antitumor efficacy and ERα degradation activity in both tamoxifen sensitive and resistant BC mouse models.
 | ||
| Fig. 8 VHL-based ER PROTACs 15 (ZD-12) and 16 (18c) with the OBHSA scaffold. | ||
Based on compound ZD12, they further designed a new class of dual-targeting PROTAC degraders, aimed at simultaneously targeting ERα and aromatase (ARO).58 Among these, compound 16 (18c, Fig. 8) shows the most potent activity in ERα/ARO degradation and cell proliferation. In the MCF-7 xenograft model, 18c promoted the degradation of ERα/ARO, and inhibited tumor growth.
4.2 CRBN-based ER PROTAC degraders
Up to now, all oral PROTAC degraders that have progressed to clinical trials utilize cereblon ligands.59 CRBN plays a crucial role in the regulation of the human immune system and anti-tumor mechanisms.60 CRBN forms the CRL4–CRBN complex with CUL4–RBX1–DDB1, which is involved in the ubiquitination and degradation of various substrate proteins.61 Over the past few years, CRBN ligands have steadily gained popularity as E3 ligase ligands.62,63The groundbreaking ARV-471 (Vepdegestrant, compound 17, Fig. 9) was developed by Arvinas and Pfizer.64 It became the first ER PROTAC to enter the clinic in 2019 and received FDA fast-track designation in 2024.19 The chemical structure of ARV-471 links the lasofoxifene scaffold and lenalidomide through a piperidine-linked piperazine liner.65–67 At nanomolar concentrations, it effectively induces ERα degradation in MCF-7 cells and inhibits the growth of wild-type (IC50 = 5.3 nM) and multiple mutant MCF-7 cell lines.68 In a preclinical Y537S PDX model, ARV-471 was administrated orally at a dose of 10 mg kg−1 and inhibited tumor growth by 99%, demonstrating better tumor growth inhibition than fulvestrant. When used in combination with CDK4/6 inhibitors, mTOR inhibitors, or PI3K/mTOR pathway inhibitors, ARV-471 has shown stronger tumor regression than monotherapy or fulvestrant combined with the same agent.64 ARV-471 is currently in several clinical trials for the treatment of patients with locally advanced or metastatic ER+/HER2− breast cancer (NCT06125522, NCT05573555, NCT05548127).
 | ||
| Fig. 9 Development of 17 (ARV-471) by Arvinas and Pfizer. | ||
In 2023, the Wang group reported a novel ER PROTAC based on a novel CRBN ligand.69 They initially used lasofoxifene as the ER binder and TX-16 as the new CRBN ligand. The optimized degrader compound 18 (ERD-3111, Fig. 10) attained sub-nanomolar DC50 values in ER degradation (DC50 = 0.5 nM) and showed superior oral bioavailability.70 Given orally in wild-type and two clinically relevant ESR1 mutations (Y537S, D538G) xenograft models, ERD-3111 achieved significant tumor regression and complete tumor growth inhibition, comparable to ARV-471. ERD-3111 exhibited as a promising ERα degrader for further extensive evaluation in the treatment of ER+ breast cancer.
 | ||
| Fig. 10 Development of compounds 18 (ERD-3111), 19 (ERD-1233) and 20 (ERD-12310A) by the Wang group.69–72 | ||
In the subsequent development of more potent and orally efficacious ER PROTACs, the Wang group first focused on the design of new cereblon ligands with high binding affinities and excellent drug-like properties. Recently, they reported two orally efficacious ER PROTACs ERD-1233 (compound 19) and ERD-12310A (compound 20), as shown in Fig. 10.71,72 Both of them utilized the lasofoxifene scaffold as the ER binding moiety. ERD-1233 used a novel CRBN ligand RR-11055, which possesses improved cereblon binding affinity compared with TX-16. Through extensive refinement of the linker, ERD-1233 employed a 6,5-spiro linker with one oxygen atom in the 5-membered ring. ERD-1233 exhibited high efficiency in degrading ERα in MCF-7 and T47D cell lines without significant inhibition of hERG or CYP. Compared with ARV-471, ERD-1233 showed better oral bioavailability and stronger antitumor effects at 20 mg kg−1 PO dose in the MCF-7 tumor xenograft model.
Within ERD-12310A, there is a [6,6] spiro linker that connects to a novel spiro-piperidine cereblon ligand C-1. ERD-12310A demonstrated a DC50 of 47 pM, making it 10 times more potent than ARV-471. Furthermore, ERD-12310A has shown superior pharmacokinetic properties in mice and rats compared to ARV-471. In the MCF-7 tumor xenograft model, ERD-12310A not only induced tumor regression but also proved to be more effective than ARV-471. Most notably, ERD-12310A exhibited significant inhibition of tumor growth in MCF-7 Y537S mutant xenograft tumors without significant weight loss or toxicity symptoms.
4.3 cIAP1/IAP-based ER PROTACs (SNIPERs)
The IAP (inhibitor of apoptosis proteins) family resists cell apoptosis by inhibiting the function of caspase family proteins to prevent cell death.73 Several IAP family members, such as c-IAP1, c-IAP2, and XIAP, possess E3 ubiquitin ligase activity through their C-terminal RING domain.74,75 This domain can recruit E2 ubiquitin-conjugating enzymes and facilitate the transfer of ubiquitin molecules from E2 to target proteins for degradation.76In 2011, the Hashimoto group from the University of Tokyo attached the cIAP1 ligand bestatin at the C-17 position of E2 through a PEG linker to synthesize ER SNIPERs.77 Western blot analysis showed that compound 21 (Fig. 11) completely degraded ERα at 30 μM in MCF-7 cells.
 | ||
| Fig. 11 Chemical structures of compounds 21 and 22. | ||
In 2016, GlaxoSmithKline employed a raloxifene scaffold as an ER binder to design and synthesize various SNIPERs, with a representative structure of compound 22 (Fig. 11).78 Representative compound 22 induces significant ERα degradation at 1 μM in MCF-7 cells.
In 2012, the Kurihara group linked cIAP1 ligand bestatin to the N atom on the solvent-exposed end of 4-OHT.79 The resulting compounds 23, 24, and 25 (Fig. 12) effectively degraded ERα at 10 μM. Subsequent mechanistic studies indicated that SNIPERs might also affect the activity of SOD (which is an enzyme that scavenges ROS), leading to the production of reactive oxygen species (ROS) and rapid cell death.80
 | ||
| Fig. 12 a) X-ray structure of the crystal structure of 4-hydroxytamoxifen complexed with ERα (PDB code: 3ERT). b) Chemical structures of compounds 23, 24 and 25; c) chemical structure of compound 26 (SNIPER(ER)-87). d) Chemical structure of compound 27 (SNIPER(ER)-110). | ||
In 2017, this group disclosed compound 26 (SNIPER(ER)-87, Fig. 12) by ligating 4-OHT and IAP antagonist LCL161 derivative with a PEG linker.81 SNIPER(ER)-87 was effective in reducing 50% ERα levels at concentrations as low as 3 nM. In the MCF-7 tumor xenograft mouse model, SNIPER(ER)-87 significantly reduced ERα protein levels and thereby inhibited further tumor progression. Mechanistical studies showed that SNIPER(ER)-87 preferentially recruits XIAP rather than IAP1 for the degradation of ERα via the ubiquitin–proteasome pathway.
In 2018, this group replaced the IAP ligand moiety of SNIPER(ER)-87 with several IAP antagonists to achieve higher affinity.82 The improved SNIPERs exhibited higher binding affinity compared to SNIPER(ER)-87 and effectively degraded ERα while simultaneously degrading cIAP1 and XIAP. The most potent compound 27 (SNIPER-110, Fig. 12) showed superior tumor growth inhibition in the MCF-7 tumor xenograft mouse model.
5. Other novel ER PROTAC degraders
In 2020, Genentech developed a HER2-targeting PROTAC conjugate using ADC technology.83 This conjugate releases its degradative payload intracellularly via lysosomal hydrolysis after antibody-mediated delivery. Compounds 28 and 29 (HER2-13 and HER2-14, Fig. 13) showed strong ERα degradation in MCF7-neo/HER2 cells and are stable in vivo. | ||
| Fig. 13 Novel ER PROTAC degraders. | ||
In 2020, the Tang group reported a two-stage strategy for developing ER PROTACs.84 They synthesized novel ERα PROTACs with hydrazone bonds in the first stage and replaced them with amide bonds in the second stage to improve drug-like properties. Compound 30 (A3, Fig. 13) showed effective ERα degradation with DC50 values of 10 nM in MCF-7 cells. This bioconjugation-based approach offers a highly-reactive, high-throughput method for studying the effects of linker combinations on PROTAC activity.
In 2022, the Naito group designed double-stranded nucleotide decoys targeting the transcription factor ERα using the estrogen receptor response element (ERE) sequence.85 They linked these decoys with three E3 ligase ligands (LCL, VH, and POM) to construct chimeric molecules, successfully preparing a new class of transcription factor-PROTACs (TF-PROTACs). The representative compound 31 (LCL-ER(dec), Fig. 13) demonstrated the most potent ERα degradation activity, effectively degrading ERα at 10 μM and inhibiting ERα transcription activity in cells.
In 2023, researchers developed a novel DNA aptamer-based PROTAC. Through SELEX technology, they identified ER(apt)D1, a nucleic acid aptamer with high affinity for ERα.86 By employing copper-catalyzed click chemistry, they successfully conjugated ER(apt)D1 to three E3 ubiquitin ligase ligands (cIAP, VHL, and CRBN) to generate PROTAC molecules. Representative compound 32 (POM-ER(apt)D1, Fig. 13) effectively degraded ERα at 10 μM and significantly inhibited MCF-7 cell proliferation.
In 2023, researchers exploited elevated GSH levels in tumors to design a GSH-responsive ERα PROTAC precursor.87 They linked a para-nitrobenzenesulfonyl group to a VHL ligand's active hydroxyl and used tamoxifen as an ERα ligand, with PEG as a linker to create compound 33 (GSH-ER-P1, Fig. 13). This precursor released more than 70% active product in tumors and efficiently degraded ERα at nanomolar concentrations.
In 2023, the Dong group introduced two hypoxia-activatable groups, nitroimidazole and nitrobenzene, to design hypoxia-responsive ERα-targeting PROTACs.88 The most potent compound 34 (Fig. 13) exhibits good hypoxia reactivity with micromolar-level cell inhibition and ERα degradation activity under hypoxic conditions.
In 2024, the Zhou group developed a series of fluorescent PROTACs by linking high-affinity ER-targeting fluorescent probes to VHL through linkers of varying lengths.89 This approach enables real-time monitoring of protein degradation and has theranostic potential. Compound 35 (A3, Fig. 13), characterized by an emission wavelength of 582 nm and a Stokes shift of 116 nm, showed substantial ERα degradation.
6. Discussion and perspectives
Over the past two decades following Crews and Deshaies group's unveiling of the innovative concept of proteolysis targeting chimeras (PROTACs), significant progress has been made from proof of concept to successful clinical translation.90,91 Given that endocrine therapy resistance is a major challenge in breast cancer treatment, ER PROTACs offer great opportunity to overcome this resistance by degrading ERα. As shown in Fig. 14, most ER ligands used in PROTACs are either estradiol or other highly potent SERM/SERD ligands, featuring an identified solvent exposed site that facilitates attachment to the E3 binding motif. The linkers also underwent extensive optimizations, transitioning from the initial alkyl or PEG linker to cyclic rings with greater conformational constraints. As CRBN-based PROTACs predominate in clinical trials, new CRBN ligands have been developed and utilized in ER PROTACs recently. | ||
| Fig. 14 Summary of the linkers and ligands used in ER PROTAC design. | ||
Currently, ARV-471 has demonstrated exceptional efficacy in clinical trials, being the first oral ER PROTAC degrader with significant potential for success. Although ARV-471 was typically well tolerated in patients, clinical trials noted the occurrence of adverse events. Designing ER PROTACs with greater potency and oral effectiveness for clinical translation presents an urgent need for medicinal chemists.
Despite the progress of PROTAC technology in the field of ER positive breast cancer, it also confronts intricate and demanding challenges. First, a PROTAC molecule is composed of three parts.92 The creation of an effective ER PROTAC molecule requires a precise balance of these three elements to ensure stability, affinity, biological activity and toxicity of the molecule.93 The complexity of PROTAC molecules renders the drug design extremely difficult. Second, achieving oral bioavailability is a significant hurdle in the realm beyond Rule-of-5 (bRo5) for molecular weight exceeding 500 Da.94,95 Current evaluation methods may not accurately assess the pharmacokinetics and pharmacodynamics of PROTACs.96 Predicting the potential toxicity risks is also challenging.97,98 Third, there is a possibility of potential drug resistance of PROTACs.99–101 Most ER PROTACs currently rely on VHL and CRBN E3 ligases. To overcome the growing examples of mutational resistance in VHL and CRBN, discovery and identification of new E3 ligases and ligands is also required.28
Certain elements can be fine-tuned to overcome future constraints of PROTACs. First, choosing the appropriate E3 ubiquitin ligase ligand guarantees that ER PROTAC molecules can successfully attract the E3 ligase and enhance the ER ubiquitination process. It's crucial to take into account the cell permeability and the bioavailability of the E3 ligase ligand, along with its binding affinity to the E3 ligase. For example, in the development of ERD-1233, novel cereblon ligands were developed based on thalidomide to improve the drug-like properties and degradation efficiency. The new CRBN ligand RR-11055 exhibited much more potent binding affinity than lenalidomide and thalidomide. It also demonstrated excellent plasma and microsomal stability in different species.
Second, the dimensions and chemical composition of the linker greatly influence PROTACs' druggability and specificity. Extensive chemical optimization is required to balance the hydrogen bonding donor, lipophilicity, and polar surface area to determine the optimal linker. For example, in the optimization of ERD-12310A, the amide bond in the linker was removed to decrease the topological polar surface area, thus improving oral bioavailability.
Third, with the numerous advantages of target protein degradation (TPD), many derivative technologies have emerged, such as molecular glues, pc-PROTAC, HaloPROTAC, AUTAC, ATTEC, RIBOTACs and more.22,102–105 These innovative alternative pathways could potentially surpass the constraints inherent in PROTACs targeting ER.
Furthermore, understanding the structure of the ternary complex of ER PROTAC will help to further clarify the mechanism of its degradation activity and allow for deeper structure-based optimization and modification.106–108 There are very few high-resolution structures of PROTAC ternary complexes solved using either X-ray crystallography or cryo-EM.109–111 Integrating computational tools with structures will enable more comprehensive structure-based drug design of ER PROTACs.112
Overall, with continuous breakthroughs and improvements in PROTAC technology, there is hope for the development of more safe, efficient, precise, and controllable ER PROTAC degraders in the future. The development of PROTAC technology will rely on interdisciplinary collaboration, including the joint efforts of experts in fields such as chemistry, biology, pharmacology, computational science, and clinical medicine. It is expected that more PROTAC degraders targeting estrogen receptors will soon enter clinical trials, providing new treatment options and hope for breast cancer patients.
Data availability
No primary research results, software or code, have been included and no new data was generated or analyzed as part of this review.Author contributions
R. Peng: writing; X. Liu: editing; C.-C. Chen, R.-T. Guo and J. Min: conceptualization, investigation, writing, review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (82103994) and the China Postdoctoral Science Foundation (2020M672435).References
- T. A. Grese, J. P. Sluka, H. U. Bryant, G. J. Cullinan, A. L. Glasebrook, C. D. Jones, K. Matsumoto, A. D. Palkowitz, M. Sato, J. D. Termine, M. A. Winter, N. N. Yang and J. A. Dodge, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14105–14110 CrossRef CAS PubMed.
- G. Lazennec, T. R. Ediger, L. N. Petz, A. M. Nardulli and B. S. Katzenellenbogen, Mol. Endocrinol., 1997, 11, 1375–1386 CrossRef CAS PubMed.
- J. Burstein Harold, N. Engl. J. Med., 2020, 383, 2557–2570 CrossRef CAS PubMed.
- H. Pan, R. Gray, J. Braybrooke, C. Davies, C. Taylor, P. McGale, R. Peto, I. P. Kathleen, J. Bergh, M. Dowsett and F. Hayes Daniel, N. Engl. J. Med., 2017, 377, 1836–1846 CrossRef PubMed.
- R. Patel, P. Klein, A. Tiersten and J. A. Sparano, npj Breast Cancer, 2023, 9, 20 CrossRef CAS PubMed.
- A. Nardone, C. De Angelis, M. V. Trivedi, C. K. Osborne and R. Schiff, Breast, 2015, 24 Suppl 2, S60–S66 CrossRef PubMed.
- K. De Bosscher, S. J. Desmet, D. Clarisse, E. Estébanez-Perpiña and L. Brunsveld, Nat. Rev. Endocrinol., 2020, 16, 363–377 CrossRef CAS PubMed.
- M. Ruff, M. Gangloff, J. Marie Wurtz and D. Moras, Breast Cancer Res., 2000, 2, 353–359 CrossRef CAS PubMed.
- A. M. Brzozowski, A. C. W. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engstrom, L. Ohman, G. L. Greene, J.-A. Gustafsson and M. Carlquist, Nature, 1997, 389, 753–758 CrossRef CAS PubMed.
- W. Huang, Y. Peng, J. Kiselar, X. Zhao, A. Albaqami, D. Mendez, Y. Chen, S. Chakravarthy, S. Gupta, C. Ralston, H.-Y. Kao, M. R. Chance and S. Yang, Nat. Commun., 2018, 9, 3520 CrossRef PubMed.
- W. Toy, Y. Shen, H. Won, B. Green, R. A. Sakr, M. Will, Z. Li, K. Gala, S. Fanning, T. A. King, C. Hudis, D. Chen, T. Taran, G. Hortobagyi, G. Greene, M. Berger, J. Baselga and S. Chandarlapaty, Nat. Genet., 2013, 45, 1439–1445 CrossRef CAS PubMed.
- D. R. Robinson, Y. M. Wu, P. Vats, F. Su, R. J. Lonigro, X. Cao, S. Kalyana-Sundaram, R. Wang, Y. Ning, L. Hodges, A. Gursky, J. Siddiqui, S. A. Tomlins, S. Roychowdhury, K. J. Pienta, S. Y. Kim, J. S. Roberts, J. M. Rae, C. H. Van Poznak, D. F. Hayes, R. Chugh, L. P. Kunju, M. Talpaz, A. F. Schott and A. M. Chinnaiyan, Nat. Genet., 2013, 45, 1446–1451 CrossRef CAS PubMed.
- S. W. Fanning, R. Jeselsohn, V. Dharmarajan, C. G. Mayne, M. Karimi, G. Buchwalter, R. Houtman, W. Toy, C. E. Fowler, R. Han, M. Lainé, K. E. Carlson, T. A. Martin, J. Nowak, J. C. Nwachukwu, D. J. Hosfield, S. Chandarlapaty, E. Tajkhorshid, K. W. Nettles, P. R. Griffin, Y. Shen, J. A. Katzenellenbogen, M. Brown and G. L. Greene, eLife, 2018, 7, e37161 CrossRef PubMed.
- J. A. Katzenellenbogen, C. G. Mayne, B. S. Katzenellenbogen, G. L. Greene and S. Chandarlapaty, Nat. Rev. Cancer, 2018, 18, 377–388 CrossRef CAS PubMed.
- S. K. Herzog and S. A. W. Fuqua, Br. J. Cancer, 2022, 126, 174–186 CrossRef CAS PubMed.
- J. Min, J. C. Nwachukwu, C. K. Min, J. W. Njeri, S. Srinivasan, E. S. Rangarajan, C. C. Nettles, V. S. Guillen, Y. Ziegler, S. Yan, K. E. Carlson, Y. Hou, S. H. Kim, S. Novick, B. D. Pascal, R. Houtman, P. R. Griffin, T. Izard, B. S. Katzenellenbogen, J. A. Katzenellenbogen and K. W. Nettles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101657118 CrossRef CAS PubMed.
- P. Shao, J. Med. Chem., 2021, 64, 11837–11840 CrossRef CAS PubMed.
- R. K. Rej, J. E. Thomas II, R. K. Acharyya, J. M. Rae and S. Wang, J. Med. Chem., 2023, 66, 8339–8381 CrossRef CAS PubMed.
- M. Jian, L. Xin, P. Rouming, C. Chun-Chi, W. Wei and G. Rey-Ting, Acta Mater. Med., 2024, 3, 57–71 Search PubMed.
- J. M. Tsai, R. P. Nowak, B. L. Ebert and E. S. Fischer, Nat. Rev. Mol. Cell Biol., 2024, 25, 740–757 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, R. Verma, A. Ransick, B. Stein, C. M. Crews and R. J. Deshaies, Mol. Cell. Proteomics, 2003, 2, 1350–1358 CrossRef CAS PubMed.
- K. Li and C. M. Crews, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC.
- F. Liu, J. Chen, K. Li, H. Li, Y. Zhu, Y. Zhai, B. Lu, Y. Fan, Z. Liu, X. Chen, X. Jia, Z. Dong and K. Liu, Mol. Cancer, 2024, 23, 148 CrossRef PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- L. T. Kramer and X. Zhang, Curr. Res. Chem. Biol., 2022, 2, 100020 CrossRef CAS.
- Y. Xiao, Y. Yuan, Y. Liu, Z. Lin, G. Zheng, D. Zhou and D. Lv, J. Med. Chem., 2024, 67, 11580–11596 Search PubMed.
- Y. Liu, J. Yang, T. Wang, M. Luo, Y. Chen, C. Chen, Z. E. Ronai, Y. Zhou, E. Ruppin and L. Han, Nat. Commun., 2023, 14, 6509 CrossRef CAS PubMed.
- T. Sobierajski, J. Małolepsza, M. Pichlak, E. Gendaszewska-Darmach and K. M. Błażewska, Drug Discovery Today, 2024, 29, 104032 CrossRef CAS PubMed.
- Y. Z. Au, T. Wang, L. H. Sigua and J. Qi, Cell Chem. Biol., 2020, 27, 637–639 CrossRef CAS PubMed.
- D. Zhang, S.-H. Baek, A. Ho and K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 645–648 CrossRef CAS PubMed.
- D. Zhang, H. S. Baek, A. Ho, H. Lee, S. Y. Jeong and K. Kim, Comb. Chem. High Throughput Screening, 2004, 7, 689–697 CrossRef CAS PubMed.
- A. Rodriguez-Gonzalez, K. Cyrus, M. Salcius, K. Kim, C. M. Crews, R. J. Deshaies and K. M. Sakamoto, Oncogene, 2008, 27, 7201–7211 CrossRef CAS PubMed.
- K. Cyrus, M. Wehenkel, E.-Y. Choi, H. Lee, H. Swanson and K.-B. Kim, ChemMedChem, 2010, 5, 979–985 CrossRef CAS PubMed.
- K. Cyrus, M. Wehenkel, E.-Y. Choi, H. Swanson and K.-B. Kim, ChemBioChem, 2010, 11, 1531–1534 CrossRef CAS PubMed.
- B. Ma, D. Liu, Z. Wang, D. Zhang, Y. Jian, K. Zhang, T. Zhou, Y. Gao, Y. Fan, J. Ma, Y. Gao, Y. Chen, S. Chen, J. Liu, X. Li and L. Li, J. Med. Chem., 2024, 67, 10336–10349 CrossRef CAS PubMed.
- H. Zhao, Q. Liu, H. Geng, Y. Tian, M. Cheng, Y.-H. Jiang, M. Xie, X.-G. Niu, F. Jiang, Y.-O. Zhang, Y. Lao, Y.-D. Wu, N. Xu and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 12088 CrossRef CAS PubMed.
- Y. Jiang, Q. Deng, H. Zhao, M. Xie, L. Chen, F. Yin, X. Qin, W. Zheng, Y. Zhao and Z. Li, ACS Chem. Biol., 2018, 13, 628–635 CrossRef CAS PubMed.
- Y. Dai, N. Yue, J. Gong, C. Liu, Q. Li, J. Zhou, W. Huang and H. Qian, Eur. J. Med. Chem., 2020, 187, 111967 CrossRef CAS PubMed.
- T. Ishida and A. Ciulli, SLAS Discovery, 2021, 26, 484–502 CrossRef CAS PubMed.
- N. Setia, H. T. A. Almuqdadi and M. Abid, Eur. J. Med. Chem., 2024, 265, 116041 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, J. Wang and D. Xing, Eur. J. Med. Chem., 2022, 227, 113906 CrossRef CAS PubMed.
- S. A. Campos, J. D. Harling, A. H. Miah and I. E. D. Smith, Proteolysis Targeting Chimeras (PROTACS) Directed to the Modulation of the Estrogen Receptor, WO Pat., WO2014108452A1, 2014 Search PubMed.
- J. Fan and K. Liu, Novel Compounds Having Estrogen Receptor Alpha Degradation Activity and Uses Thereof, US Pat., US20180208590A1, 2018 Search PubMed.
- R. M. Layman, M. R. Patel, D. Cosgrove, M. Danso, N. Mota, M. E. Zettler, K. C. Pehlivan and E. Hamilton, Cancer Res., 2024, 84, CT075 Search PubMed.
- R. B. Kargbo, ACS Med. Chem. Lett., 2019, 10, 1367–1369 CrossRef CAS PubMed.
- C. De Savi, R. H. Bradbury, A. A. Rabow, R. A. Norman, C. de Almeida, D. M. Andrews, P. Ballard, D. Buttar, R. J. Callis, G. S. Currie, J. O. Curwen, C. D. Davies, C. S. Donald, L. J. L. Feron, H. Gingell, S. C. Glossop, B. R. Hayter, S. Hussain, G. Karoutchi, S. G. Lamont, P. MacFaul, T. A. Moss, S. E. Pearson, M. Tonge, G. E. Walker, H. M. Weir and Z. Wilson, J. Med. Chem., 2015, 58, 8128–8140 CrossRef CAS PubMed.
- H. M. Weir, R. H. Bradbury, M. Lawson, A. A. Rabow, D. Buttar, R. J. Callis, J. O. Curwen, C. de Almeida, P. Ballard, M. Hulse, C. S. Donald, L. J. Feron, G. Karoutchi, P. MacFaul, T. Moss, R. A. Norman, S. E. Pearson, M. Tonge, G. Davies, G. E. Walker, Z. Wilson, R. Rowlinson, S. Powell, C. Sadler, G. Richmond, B. Ladd, E. Pazolli, A. M. Mazzola, C. D'Cruz and C. De Savi, Cancer Res., 2016, 76, 3307–3318 CrossRef CAS PubMed.
- J. Hu, B. Hu, M. Wang, F. Xu, B. Miao, C.-Y. Yang, M. Wang, Z. Liu, D. F. Hayes, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 1420–1442 CrossRef CAS PubMed.
- T. L. Gonzalez, M. Hancock, S. Sun, C. L. Gersch, J. M. Larios, W. David, J. Hu, D. F. Hayes, S. Wang and J. M. Rae, Breast Cancer Res. Treat., 2020, 180, 611–622 CrossRef CAS PubMed.
- B. Hu and J. Hu, Breast Cancer Res. Treat., 2024, 203, 383–396 CrossRef CAS PubMed.
- G. Loren, I. Espuny, A. Llorente, C. Donoghue, X. Verdaguer, R. R. Gomis and A. Riera, Eur. J. Med. Chem., 2022, 243, 114770 CrossRef CAS PubMed.
- S. Li, D. Shen, J. Shao, R. Crowder, W. Liu, A. Prat, X. He, S. Liu, J. Hoog, C. Lu, L. Ding, O. L. Griffith, C. Miller, D. Larson, R. S. Fulton, M. Harrison, T. Mooney, J. F. McMichael, J. Luo, Y. Tao, R. Goncalves, C. Schlosberg, J. F. Hiken, L. Saied, C. Sanchez, T. Giuntoli, C. Bumb, C. Cooper, R. T. Kitchens, A. Lin, C. Phommaly, S. R. Davies, J. Zhang, M. S. Kavuri, D. McEachern, Y. Y. Dong, C. Ma, T. Pluard, M. Naughton, R. Bose, R. Suresh, R. McDowell, L. Michel, R. Aft, W. Gillanders, K. DeSchryver, R. K. Wilson, S. Wang, G. B. Mills, A. Gonzalez-Angulo, J. R. Edwards, C. Maher, C. M. Perou, E. R. Mardis and M. J. Ellis, Cell Rep., 2013, 4, 1116–1130 CrossRef CAS PubMed.
- X. Zhang, Z. Zhang, X. Xue, T. Fan, C. Tan, F. Liu, Y. Tan and Y. Jiang, ACS Pharmacol. Transl. Sci., 2022, 5, 1109–1118 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- M. D. Driscoll, G. Sathya, M. Muyan, C. M. Klinge, R. Hilf and R. A. Bambara, J. Biol. Chem., 1998, 273, 29321–29330 CrossRef CAS PubMed.
- B. Xie, Z. Yin, Z. Hu, J. Lv, C. Du, X. Deng, Y. Huang, Q. Li, J. Huang, K. Liang, H.-B. Zhou and C. Dong, J. Med. Chem., 2023, 66, 6631–6651 CrossRef CAS PubMed.
- L. Xin, C. Wang, Y. Cheng, H. Wang, X. Guo, X. Deng, X. Deng, B. Xie, H. Hu, C. Min, C. Dong and H.-B. Zhou, J. Med. Chem., 2024, 67, 8913–8931 CrossRef CAS PubMed.
- J. S. Scott, I. N. Michaelides and M. Schade, RSC Med. Chem., 2024 10.1039/D4MD00769G.
- J. Yamamoto, T. Ito, Y. Yamaguchi and H. Handa, Chem. Soc. Rev., 2022, 51, 6234–6250 RSC.
- E. S. Fischer, K. Böhm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. J. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thomä, Nature, 2014, 512, 49–53 CrossRef CAS PubMed.
- D. Mi, Y. Li, H. Gu, Y. Li and Y. Chen, Eur. J. Med. Chem., 2023, 256, 115444 CrossRef CAS PubMed.
- J. An and X. Zhang, Bioorg. Med. Chem., 2024, 104, 117683 Search PubMed.
- S. M. Gough, J. J. Flanagan, J. Teh, M. Andreoli, E. Rousseau, M. Pannone, M. Bookbinder, R. Willard, K. Davenport, E. Bortolon, G. Cadelina, D. Gordon, J. Pizzano, J. Macaluso, L. Soto, J. Corradi, K. Digianantonio, I. Drulyte, A. Morgan, C. Quinn, M. Békés, C. Ferraro, X. Chen, G. Wang, H. Dong, J. Wang, D. R. Langley, J. Houston, R. Gedrich and I. C. Taylor, Clin. Cancer Res., 2024, 30, 3549–3563 CrossRef CAS PubMed.
- X. Han and Y. Sun, Cell Rep. Phys. Sci., 2022, 3, 101062 CrossRef CAS.
- Y. Qian, A. P. Crew, H. Dong, J. Wang and C. M. Crews, Indole Derivatives as Estrogen Receptor Degraders, WO Pat., WO2018053354A1, 2018.
- A. P. Crew, Y. Qian, H. Dong, J. Wang, K. R. Hornberger and C. M. Crews, Tetrahydronaphthalene and Tetrahydroisoquinoline Derivatives as Estrogen Receptor Degraders, WO Pat., WO2018102725A1, 2018.
- L. B. Snyder, J. J. Flanagan, Y. Qian, S. M. Gough, M. Andreoli, M. Bookbinder, G. Cadelina, J. Bradley, E. Rousseau, J. Chandler, R. Willard, J. Pizzano, C. M. Crews, A. P. Crew, J. Houston, M. D. Moore, R. Peck and I. Taylor, Cancer Res., 2021, 81, 44–44 CrossRef.
- Z. Chen, B. Hu, R. K. Rej, D. Wu, R. K. Acharyya, M. Wang, T. Xu, J. Lu, H. Metwally, Y. Wang, D. McEachern, L. Bai, C. L. Gersch, M. Wang, W. Zhang, Q. Li, B. Wen, D. Sun, J. M. Rae and S. Wang, J. Med. Chem., 2023, 66, 12559–12585 CrossRef CAS PubMed.
- R. K. Rej, Z. Chen, R. K. Acharyya, D. Wu, B. Hu, G. Xu, R. Nagilla, H. Metwally, Y. Wang, B. Wen, D. Sun, L. Bai and S. Wang, Cancer Res., 2024, 84, 4510–4510 Search PubMed.
- R. K. Acharyya, R. K. Rej, B. Hu, Z. Chen, D. Wu, J. Lu, H. Metwally, D. McEachern, Y. Wang, W. Jiang, L. Bai, J. Tošović, C. L. Gersch, G. Xu, W. Zhang, W. Wu, E. S. Priestley, Z. Sui, F. Sarkari, B. Wen, D. Sun, J. M. Rae and S. Wang, J. Med. Chem., 2024, 67, 19010–19037 Search PubMed.
- R. K. Rej, B. Hu, Z. Chen, R. K. Acharyya, D. Wu, H. Metwally, D. McEachern, Y. Wang, W. Jiang, L. Bai, L. S. Nishimura, C. L. Gersch, M. Wang, B. Wen, D. Sun, K. Carlson, J. A. Katzenellenbogen, G. Xu, W. Zhang, W. Wu, E. S. Priestley, Z. Sui, J. M. Rae and S. Wang, J. Med. Chem., 2024, 67, 20933–20965 Search PubMed.
- T. K. Oberoi-Khanuja, A. Murali and K. Rajalingam, Cell Death Dis., 2013, 4, e784 CrossRef CAS PubMed.
- P. Vandenabeele and M. J. M. Bertrand, Nat. Rev. Immunol., 2012, 12, 833–844 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, L. Shi, S. Yang, J. Chang, Y. Zhong, Q. Li and D. Xing, J. Enzyme Inhib. Med. Chem., 2022, 37, 1437–1453 CrossRef CAS PubMed.
- L. Kiss, D. Clift, N. Renner, D. Neuhaus and L. C. James, Nat. Commun., 2021, 12, 1220 CrossRef CAS PubMed.
- Y. Itoh, R. Kitaguchi, M. Ishikawa, M. Naito and Y. Hashimoto, Bioorg. Med. Chem., 2011, 19, 6768–6778 CrossRef CAS PubMed.
- J. D. Harling and I. E. D. Smith, IAP E3 Ligase Directed Proteolysis targeting Chimeric Molecules, WO Pat., WO2016169989A1, 2016 Search PubMed.
- Y. Demizu, K. Okuhira, H. Motoi, A. Ohno, T. Shoda, K. Fukuhara, H. Okuda, M. Naito and M. Kurihara, Bioorg. Med. Chem. Lett., 2012, 22, 1793–1796 CrossRef CAS PubMed.
- K. Okuhira, Y. Demizu, T. Hattori, N. Ohoka, N. Shibata, T. Nishimaki-Mogami, H. Okuda, M. Kurihara and M. Naito, Cancer Sci., 2013, 104, 1492–1498 CrossRef CAS PubMed.
- N. Ohoka, K. Okuhira, M. Ito, K. Nagai, N. Shibata, T. Hattori, O. Ujikawa, K. Shimokawa, O. Sano, R. Koyama, H. Fujita, M. Teratani, H. Matsumoto, Y. Imaeda, H. Nara, N. Cho and M. Naito, J. Biol. Chem., 2017, 292, 4556–4570 CrossRef CAS PubMed.
- N. Ohoka, Y. Morita, K. Nagai, K. Shimokawa, O. Ujikawa, I. Fujimori, M. Ito, Y. Hayase, K. Okuhira, N. Shibata, T. Hattori, T. Sameshima, O. Sano, R. Koyama, Y. Imaeda, H. Nara, N. Cho and M. Naito, J. Biol. Chem., 2018, 293, 6776–6790 CrossRef CAS PubMed.
- P. S. Dragovich, P. Adhikari, R. A. Blake, N. Blaquiere, J. Chen, Y.-X. Cheng, W. den Besten, J. Han, S. J. Hartman, J. He, M. He, E. Rei Ingalla, A. V. Kamath, T. Kleinheinz, T. Lai, D. D. Leipold, C. S. Li, Q. Liu, J. Lu, Y. Lu, F. Meng, L. Meng, C. Ng, K. Peng, G. L. Phillips, T. H. Pillow, R. K. Rowntree, J. D. Sadowsky, D. Sampath, L. Staben, S. T. Staben, J. Wai, K. Wan, X. Wang, B. Wei, I. E. Wertz, J. Xin, K. Xu, H. Yao, R. Zang, D. Zhang, H. Zhou and Y. Zhao, Bioorg. Med. Chem. Lett., 2020, 30, 126907 CrossRef CAS PubMed.
- B. L. Roberts, Z.-X. Ma, A. Gao, E. D. Leisten, D. Yin, W. Xu and W. Tang, ACS Chem. Biol., 2020, 15, 1487–1496 CrossRef CAS PubMed.
- M. Naganuma, N. Ohoka, G. Tsuji, H. Tsujimura, K. Matsuno, T. Inoue, M. Naito and Y. Demizu, ACS Med. Chem. Lett., 2022, 13, 134–139 CrossRef CAS PubMed.
- H. Tsujimura, M. Naganuma, N. Ohoka, T. Inoue, M. Naito, G. Tsuji and Y. Demizu, ACS Med. Chem. Lett., 2023, 14, 827–832 CrossRef CAS PubMed.
- Z. Zhou, H. Fan, D. Yu, F. Shi, Q. Li, Z. Zhang, X. Wang, X. Zhang, C. Dong, H. Sun and W. Mi, Bioorg. Med. Chem., 2023, 96, 117526 CrossRef CAS PubMed.
- B. Xie, B. Xu, L. Xin, Y. Wei, X. Guo and C. Dong, Bioorg. Chem., 2023, 137, 106590 CrossRef CAS PubMed.
- X. Wang, L. Xin, X. Deng, C. Dong, G. Hu and H.-B. Zhou, Eur. J. Med. Chem., 2024, 267, 116184 CrossRef CAS PubMed.
- D. Zollman and K. McAulay, Nat. Chem. Biol., 2024, 20, 1559–1561 Search PubMed.
- M. He, C. Cao, Z. Ni, Y. Liu, P. Song, S. Hao, Y. He, X. Sun and Y. Rao, Signal Transduction Targeted Ther., 2022, 7, 181 CrossRef CAS PubMed.
- S.-L. Paiva and C. M. Crews, Curr. Opin. Chem. Biol., 2019, 50, 111–119 CrossRef CAS PubMed.
- M. W. Krone and C. M. Crews, Cell Chem. Biol., 2024 DOI:10.1016/j.chembiol.2024.10.004.
- B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS PubMed.
- M. Schade, J. S. Scott, T. G. Hayhow, A. Pike, I. Terstiege, M. Ahlqvist, J. R. Johansson, C. R. Diene, C. Fallan, A. Y. S. Balazs, E. Chiarparin and D. Wilson, J. Med. Chem., 2024, 67, 13106–13116 CrossRef CAS PubMed.
- D. W. Bartlett and A. M. Gilbert, Chem. Soc. Rev., 2022, 51, 3477–3486 RSC.
- A. Mares, A. H. Miah, I. E. D. Smith, M. Rackham, A. R. Thawani, J. Cryan, P. A. Haile, B. J. Votta, A. M. Beal, C. Capriotti, M. A. Reilly, D. T. Fisher, N. Zinn, M. Bantscheff, T. T. MacDonald, A. Vossenkamper, P. Dace, I. Churcher, A. B. Benowitz, G. Watt, J. Denyer, P. Scott-Stevens and J. D. Harling, Commun. Biol., 2020, 3, 140 CrossRef CAS PubMed.
- Y. Atilaw, V. Poongavanam, C. S. Nilsson, D. Nguyen, A. Giese, D. Meibom, M. Erdelyi and J. Kihlberg, ACS Med. Chem. Lett., 2021, 12, 107–114 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, L. Nian, C. Wang, M. Yu, W. Zhang, Z. Li and C. Chen, Blood, 2023, 142, 6584 CrossRef.
- S. Gooding, N. Ansari-Pour, F. Towfic, M. Ortiz Estévez, P. P. Chamberlain, K.-T. Tsai, E. Flynt, M. Hirst, D. Rozelle, P. Dhiman, P. Neri, K. Ramasamy, N. Bahlis, P. Vyas and A. Thakurta, Blood, 2021, 137, 232–237 CrossRef CAS PubMed.
- L. Zhang, B. Riley-Gillis, P. Vijay and Y. Shen, Mol. Cancer Ther., 2019, 18, 1302–1311 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, Mol. Cell, 2019, 76, 797–810.e710 CrossRef CAS PubMed.
- J. L. Childs-Disney, X. Yang, Q. M. R. Gibaut, Y. Tong, R. T. Batey and M. D. Disney, Nat. Rev. Drug Discovery, 2022, 21, 736–762 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- D. Zaidman, J. Prilusky and N. London, J. Chem. Inf. Model., 2020, 60, 4894–4903 CrossRef CAS PubMed.
- E. Rovers and M. Schapira, J. Chem. Inf. Model., 2024, 64, 6162–6173 CrossRef CAS PubMed.
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- M. E. Matyskiela, T. Clayton, X. Zheng, C. Mayne, E. Tran, A. Carpenter, B. Pagarigan, J. McDonald, M. Rolfe, L. G. Hamann, G. Lu and P. P. Chamberlain, Nat. Struct. Mol. Biol., 2020, 27, 319–322 CrossRef CAS PubMed.
- A. Kroupova, V. A. Spiteri, Z. J. Rutter, H. Furihata, D. Darren, S. Ramachandran, S. Chakraborti, K. Haubrich, J. Pethe, D. Gonzales, A. J. Wijaya, M. Rodriguez-Rios, M. Sturbaut, D. M. Lynch, W. Farnaby, M. A. Nakasone, D. Zollman and A. Ciulli, Nat. Commun., 2024, 15, 8885 CrossRef CAS PubMed.
- D. Zollman and K. McAulay, Nat. Chem. Biol., 2024, 20, 1559–1561 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |