
Razing the scaffolding: the elimination of non-catalytic functions of kinases through targeted protein degradation
Sarah
Pogash
a and
Steven
Fletcher
 *ab
*ab
aDepartment of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 N. Pine St., Baltimore, MD 21201, USA. E-mail: steven.fletcher@rx.umaryland.edu
bUniversity of Maryland Marlene & Stewart Greenebaum Cancer Centre, 20 S. Greene St., Baltimore, MD 21201, USA
First published on 15th April 2025
Abstract
Overexpression and activation of kinases often results in cancer initiation and progression through both catalytic and non-catalytic functions that promote rapid proliferation, growth, survival, and metastasis of cells. Catalytic functions are effectively blocked with the use of ATP-competitive inhibitors, however drug-resistant mutations are emerging all the time. Further, single-agent ATP-competitive inhibitors sometimes fail to eliminate oncogenic properties of the targeted kinase, likely due to (non-targeted) non-catalytic functions that are maintained. Non-catalytic functions – such as scaffolding roles, where the kinase may interact with other proteins to coordinate cellular activities or protect them from degradation by the proteasome – may be targeted through the development of protein–protein interaction (PPI) inhibitors, although this is a highly challenging endeavour. To overcome the limitations of classical (ATP-competitive) inhibitors (and circumvent the formidable feat required in the development of PPI inhibitors), which operate through “occupancy-driven” pharmacology, targeted protein degradation, as showcased by proteolysis targeting chimeras (PROTACs), is fast becoming a highly sought-after goal for a large plethora of protein targets, and is governed by “event-driven” pharmacology. Because PROTACs result in the degradation of the protein of interest, these compounds are predicted to both catalytic and non-catalytic functions of a targeted kinase. Herein, we focus on the development of PROTACs that target (i) the scaffolding roles of focal adhesion kinase (FAK) that are associated with the formation of signaling units involved in migration and invasion events and (ii) the scaffolding roles of Aurora-A kinase (AURKA), which play a role in the protection of MYC proteins from proteasomal degradation.
Introduction
Kinases contribute significantly to the maintenance of cellular homeostasis by phosphorylating/activating downstream substrates in response to extra- or intra-cellular stimuli.1–3 Dysregulation in the expression levels and/or the activity of kinases has been linked to a wide range of human cancers,4 as well as other disease states.1–3,5,6 More specifically in cancer, kinases function to promote rapid cell proliferation, survival, growth, and metastasis through both catalytic and non-catalytic functions.7 For example, the V600E B-Raf (BRAF) mutation that occurs in over 50% of malignant melanoma cases, constitutively activates the mitogen-activated protein kinase (MAPK) cascade ultimately driving the proliferation of melanoma cells, highlighting an example of a kinase's catalytic function that leads to cancer progression.2,7,8 FDA-approved, ATP-competitive (catalytic) inhibitors of mutated BRAF include vemurafenib, dabrafenib, and encorafenib.9 Similarly, internal tandem duplication (ITD) mutations to FMS-like tyrosine kinase 3 (FLT3) results in constitutive activation of the FLT3 protein and subsequent activation of downstream signalling pathways related to cell survival and proliferation in patients with acute myeloid leukaemia (AML).10,11Although catalytic functions of kinases have been highly exploited in the pharmaceutical industry for the development of inhibitors/drugs, non-catalytic functions of kinases may also promote cancer progression.1 Such non-catalytic functions may become apparent when comparing the phenotypic output in animals with inactive mutants versus knocked out kinases, which can be as extreme as survival or mortality.1 Non-catalytic functions of kinases can be classified into 3 broad categories including: kinase-DNA interactions, allosteric regulation of other enzymes, and scaffolding roles.1,2,6 This review focuses on targeting both the scaffolding roles of focal adhesion kinase (FAK) that lead to the formation of signalling complexes,12–14 as well as the evasion of proteasomal degradation of MYC proteins through their scaffolding interactions with Aurora-A kinase (AURKA).15–17
In recent years, it has been discovered that the conformation of a kinase's activation loop can influence whether it functions as a scaffold or as an allosteric regulator.1,6 Determining the conformation of the activation loop associated with non-catalytic functions of kinases in cancer is crucial for the development of effective therapeutics. Although non-catalytic functions are independent of kinase activity, many are coupled with active conformations of the kinase.1,2 As of 2022, the FDA has approved 73 small-molecule kinase inhibitors (SMKI), with most of these drugs being either type 1 or 2 ATP-competitive inhibitors that target the ATP binding site in the kinases active or inactive form, respectively.3,7 Because the ATP-binding site is highly conserved, type 1 and 2 inhibitors are often non-selective, targeting more than one member of a kinase family (e.g. tofacitinib pan-JAK inhibitor18), and giving rise to highly efficacious compounds; however, in some cases, multi-kinase inhibitors may be associated with toxicity issues as they may also be promiscuous and target other kinase families.7,19 A majority of SMKIs target tyrosine kinases (TK),19 including: FLT3 (gilteritinib),10,11,20 human epidermal growth factor receptor (EGFR/HER, gefitinib);21 vascular endothelial growth factor receptor (VEGFR, axitinib);22 Janus kinases (JAK, tofacitinib);18 and anaplastic lymphoma kinase (ALK, lorlatinib).23 Other common targets with approved drugs include: BRAF (vemurafenib);9 mitogen-activated protein kinases 1 and 2 (MEK1/2, cobimetinib);24 phosphatidylinositol 3-kinase (PI3K, alpelisib);25 and cyclin-dependent kinases 4 and 6 (CDK4/6, ribociclib).26 Despite the successful deployment of SMKIs, the long-term clinical efficacy may be limited by acquired drug resistance due to gatekeeper mutations (gatekeeper residues control inhibitor access to the ATP-binding site), where patients may initially respond to treatment but after continual exposure the disease becomes resistant to the treatment.2,3,6,7 For instance, gilteritinib, a FLT3 inhibitor, experiences drug resistance due to the gatekeeper mutation F691L.10,11,20 Additionally, it has been demonstrated that single-agent kinase inhibitors are sometimes unable to eliminate oncogenic properties of the targeted kinase likely due to scaffolding (non-catalytic) roles that are not affected by active site inhibition.6
To overcome acquired drug resistance of classical ATP-competitive inhibitors, one strategy is to develop second-generation inhibitors that are (initially) impervious to the resistance-causing mutations.3,27 There are seven FDA-approved kinase inhibitors that have been designed specifically to overcome gatekeeper mutations, however it is predicted that the kinases will mutate again in response to the new drugs,3,7 and so this may result in a perpetual and costly situation of constant drug re-design. Therefore, it is important to devise complementary approaches to overcome all the challenges with classical inhibitors. Some approaches include developing covalent inhibitors, which has been discussed previously,28–31 yielding permanent inactivation of the target protein, as opposed to transient inhibition with a traditional small-molecule inhibitor, or developing degraders,3,7 which afford destruction of the target protein. The design rationale for both the aforementioned approaches often leverages successful small-molecule inhibitors and their conversion into covalent inhibitors or targeted protein degraders by attaching an electrophilic warhead or a ligand that is recognized by an E3 ligase, respectively. The Achilles heel of covalent inhibitors is if the key nucleophilic residue mutates, then the inhibitor may no longer be able to react with the protein target.28–31 While there is a concern that drug-resistant mutations will weaken the binding of targeted protein degraders, they function by a different mechanism of action that can be described as “event-driven” pharmacology, as opposed to small-molecule inhibitors that function by “occupancy-based” pharmacology.3,7 Because of their “event-driven” pharmacology, targeted protein degraders need not bind with exquisite affinities and for a prolonged period of time. Rather, they just need to bind long enough for the protein degradation event to be initiated, and this may lead to overcoming drug resistance (see next section). Indeed, it has been demonstrated that targeted protein degraders can be active against treatment-resistant cancers, such as the dasatinib-derived molecule SIAIS056 that degrades clinically relevant mutated BCR–ABL1 fusion protein that is resistant to treatment with imatinib and dasatinib in chronic myeloid leukemia.32
In addition to the possibility of overcoming resistance mutations, both catalytic and non-catalytic functions may be eliminated with targeted protein degraders, making them highly advantageous over other approaches,2,5,6 which circumvents the challenges of developing protein–protein interaction (PPI) inhibitors to disrupt non-catalytic functions; these obstacles include the lack of a small-molecule tractable (traditional) binding site and an established bioactivity assay.33–36 For instance, apart from its catalytic activity, EGFR acts as a scaffold to bind and activate other receptor tyrosine kinases, a function that also contributes to treatment resistance with EGFR inhibitors, like gefitinib that is used to treat non-small cell lung cancer.32 Therefore, the development of EGFR degraders promises to abolish both its catalytic and non-catalytic functions.32
A highly successful class of degraders comprises those termed PROTACs, an acronymized form of PROteolysis TArgeting Chimeras, and these will be described more fully in the following sub-section. While PROTACs designed to inhibit a protein's catalytic function are “coming of age” having now entered clinical trials,37 the deployment of PROTACs to target non-catalytic scaffolding functions of proteins is a field that is very much in its infancy. Nevertheless, given the exponential publication rate of PROTAC papers since their inception in 2001,38 we expect this niche will blossom rapidly. Herein, we focus on two of the more developed applications: the development of PROTACs to inhibit the scaffolding (non-catalytic) functions of FAK12–14 and AURKA,17,39 in contrast to traditional small-molecule inhibitors that inhibit only the catalytic functions of these enzymes.
Targeted Protein Degradation (TPD): PROteolysis-TArgeting Chimeras (PROTACs)
PROTACs are a class of targeted protein degraders wherein a ligand for a protein of interest (POI) is chemically linked to a ligand that targets an E3 ligase.5,6,40–42 As shown in Fig. 1, simultaneous binding of the POI and E3 ligase results in ternary complex formation and the polyubiquitination of accessible surface-exposed lysine residues on the POI by an E2 ligase that is closely associated with the E3 ligase. This polyubiquitin tag serves as a protein-destruction signal, and the protein is subsequently recognized and degraded by the proteasome.38,40,41 Hijacking the ubiquitin-proteasome system (UPS), the PROTAC molecule is not consumed by the degradation process and rather has a catalytic mechanism of action, enabling multiple target protein molecules to be degraded by one PROTAC molecule and therefore possibly lower the dose requirement of the parental drug to maintain the desired therapeutic effect.40,43 Furthermore, the mechanism of action of PROTACs may be described as “event-driven pharmacology”, where the PROTAC needs to be bound to each component just long enough for the polyubiquitination process to occur.5,40,43 This is in contrast to the direct inhibition of a protein, such as a kinase by an ATP-competitive inhibitor, wherein the drug's therapeutic effect is realized only when the drug is engaged with the protein, and so high affinity binders are preferred; this latter mode of inhibition has been termed “occupancy-driven pharmacology”.5 It has, thus, been hypothesized that the phenomenon of “event-driven pharmacology” may empower PROTACs to be able to overcome acquired drug resistance.43 Although very high affinity for the POI is not required by PROTACs, mutations to the E3 ligase may impart resistance to the treatment, as may the downregulations of other components of the UPS.43,44 PROTACs also have the potential to convert pan-inhibitors into specific degraders due to differences in the distribution of surface exposed lysine residues or favourable interactions formed between the POI and E3 ligase.42 The advantages of PROTACs renders them extraordinary candidates for the next-generation of therapeutics to overcome limitations of traditional inhibitors in the clinic. | ||
| Fig. 1 Schematic representation of the mechanism of action of PROTACs and molecular glue degraders. In brief, the degrader simultaneously binds the POI and E3 ligase, resulting in polyubiquitination of surface-exposed lysines on the POI ultimately tagging it for destruction by the proteasome. The degrader is recycled and can degrade multiple POI molecules, as it is not consumed by the proteasome. Prepared with https://biorender.com/. | ||
Rational design of PROTACs involves the careful selection of the POI ligand, linker length and flexibility, as well as the E3 ligase to be recruited. It is common practice for the POI ligand to be chosen according to the most clinically advanced inhibitor for the target protein, which may be either a selective- or pan-inhibitor.40 A solvent-exposed portion from the chosen inhibitor functions as a grafting point to attach the linker bearing the E3 ligase ligand, as modifying this functionality should not significantly affect binding affinity. The desired linker length and flexibility are largely based on empirical design; flexible linkers include hydrocarbons and polyethylene glycol (PEG) units,45 while rigid linkers include cyclic structures such as piperidines and piperazines.46 Both the linker length and flexibility are crucial factors in ternary complex formation,45 therefore, the deployment of a library of PROTACs containing linkers with different characteristics may help to determine the optimal composition. Similarly, the decision of which E3 ligase to recruit is not always straightforward, so PROTACs are often designed with different E3 ligase ligands. The majority of PROTACs recruit either cereblon (CRBN), Von Hippel–Lindau (VHL), or inhibitors of apoptosis (IAP) as their E3 ligase by utilizing thalidomide derivatives, VHL ligand 1 derivatives, or bestatin as ligands, respectively.47–50 The application of recruiting different E3 ligases that are expressed in specific tissues are actively being investigated for targeted therapies, as CRBN and VHL are highly expressed in most tissues.44,51 Recently, Adhikari et al. developed a RiPA (rapamycin-induced proximity assay) that can aid in the selection of a proper target protein and E3 ligase pair for the development of PROTACs, by quantifying the level of degradation of the target protein when brought into close proximity to an E3 ligase. They applied this system to the protein WDR5, and their observations were consistent with the literature, where VHL was able to induce the degradation of WDR5 but CRBN was not. They also applied their RiPA system to FBXL12, an E3 ligase that is not typically recruited in PROTACs and observed degradation of a few target proteins. While the authors mentioned that the RiPA system may generate false positives and negatives, it can be used as a starting point while designing PROTACs which may help in reducing the trial-and-error design approach while selecting the E3 ligase.52
Many successful CRBN- and VHL-based PROTACS have been developed for the treatment of cancer,42,43,53 wherein the POI has included AURKA, FAK, BRAF, EGFR, and CDKs, among others.42,53 Other major targets include receptors that are overexpressed in prostate cancer and breast cancer, such as the androgen receptor (AR) and estrogen receptor (ER), respectively.43,53 Recently, vepdegestrant (ARV-471), a PROTAC targeting ER, has entered phase III clinical trials for the treatment of breast cancer.54,55 While most PROTACs are being developed for cancer research, other disease states are also being explored such as neurodegenerative and inflammatory diseases.43 Clinical trials with other PROTACs are underway for the treatment of cancer and auto-immune diseases, such as bavdegalutamide (ARV-110)53,56 and luxdegalutamide (ARV-766)53,57 for prostate cancer and NX-5948 for B-cell malignancies and autoimmune diseases.41,43,53 PROTACs targeting the tau protein showed promising results in the depletion of tau in patient-derived wild-type and mutated neurons, signifying a major advancement in the development of treatments for Alzheimer's disease.58–61 One other example includes Bruton's tyrosine kinase (BTK), which is involved in oncogenic and inflammatory signalling cascades: PROTACs targeting BTK are being developed for the treatment of cancers, and inflammatory and autoimmune diseases.62–65
Although PROTACs have entered clinical trials and show promising results in enhanced therapeutic efficacy, the development of effective PROTACs faces many challenges. The chemical structure of PROTACs comprises three chemical modules (POI ligand, E3 ligase ligand and linker) leading to large molecules with high molecular weights that may compromise the physicochemical properties of the potential drug.42,43,66 Towards predicting if a compound will have good oral bioavailability, Lipinski's rule of 5 was developed to estimate physicochemical properties of compounds, such as cell permeability and absorption, which suggests drug candidates should have less than or equal to the following parameters to be considered a good candidate: 500 Da molecular weight, 5 hydrogen bond donors, 10 hydrogen bond acceptors, and a calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of 5.67 By these rules, PROTACs are not expected to exhibit good oral bioavailabilities as they often significantly deviate from one or more of these parameters. However, it has been suggested that PROTACs permeate the cell membrane via passive diffusion or active transport despite their violation of Lipinski's rule of 5.42 The rational design of PROTACs also poses a significant challenge as mentioned above, although PEG-, piperidine-, and piperazine-based linkers are predicted to improve the solubility of PROTACs and potentially improve cell permeability.45,46 Another challenge encountered in kinase-focused PROTAC research is the potential for degrading kinases in healthy tissues in addition to cancerous tissues, i.e. there is a risk of on-target toxicity effects. Fortunately, many cancers become “addicted” to the kinase that drives them, and so kinase inhibitors are often more lethal to cancer cells than to healthy, non-malignant cells.7 Regardless, it is critical, to investigate this potential risk when converting kinase inhibitors into PROTACs.
P of 5.67 By these rules, PROTACs are not expected to exhibit good oral bioavailabilities as they often significantly deviate from one or more of these parameters. However, it has been suggested that PROTACs permeate the cell membrane via passive diffusion or active transport despite their violation of Lipinski's rule of 5.42 The rational design of PROTACs also poses a significant challenge as mentioned above, although PEG-, piperidine-, and piperazine-based linkers are predicted to improve the solubility of PROTACs and potentially improve cell permeability.45,46 Another challenge encountered in kinase-focused PROTAC research is the potential for degrading kinases in healthy tissues in addition to cancerous tissues, i.e. there is a risk of on-target toxicity effects. Fortunately, many cancers become “addicted” to the kinase that drives them, and so kinase inhibitors are often more lethal to cancer cells than to healthy, non-malignant cells.7 Regardless, it is critical, to investigate this potential risk when converting kinase inhibitors into PROTACs.
Other examples of targeted protein degraders are molecular glues and small molecules bearing hydrophobic tags. In contrast to the bivalent PROTACs, molecular glues are more traditional small-molecule compounds that facilitate and stabilize the interaction between two proteins, such as a POI and an E3 ligase, effectively “glueing” them together.68 Molecular glue degraders function similarly to PROTACs, where the POI is degraded by the proteasome after it is brought into close proximity to an E3 ligase and ubiquitinated.69 One advantage that molecular glues have over PROTACs, is that they typically do not exhibit the hook effect. The hook effect is a phenomenon in which at high concentrations, the PROTAC will saturate the POI or the E3 ligase preventing ternary complex formation (POI–PROTAC–E3 ligase) and subsequent degradation of the POI. On the other hand, molecular glues generally have a high affinity for one of the targets in the complex and then an interaction with the other target is facilitated.70 Molecular glues are estimated to have enhanced physiochemical properties compared to PROTACs, as their structures are smaller and generally comply within Lipinkski's rule of 5.69 Apart from being an E3 ligase ligand for PROTACs, thalidomide is classified as a molecular glue, demonstrating that these molecules can be compact.69,71 The rational design of molecular glues is even more challenging than that of PROTACs because that simple small molecule is required to promote and stabilize the binding of the POI to an E3 ligase, which is no mean feat; indeed, most glues have been discovered by serendipity. Therefore, a current strategy to generate molecular glues is to graft an E3 ligase ligand directly to a small molecule inhibitor with no linker.71 Nomura's group recently published their method of converting small-molecule inhibitors into molecular glue degraders by grafting on EST1060, a scaffold that forms a covalent bond with the E3 ligase RNF126, which may be applied to various targets.71 Like PROTACs, molecular glues are being widely investigated for their application to cancer.
Although PROTACs and molecular glue degraders function via the same mechanism and are the most popular classes of targeted protein degraders, hydrophobic tag-based (HyT) degraders represent a different class of degraders, that pioneered the field of TPD, and, depending on the specific tag enlisted, may or may not be dependent on E3 ligases.72,73 Hydrophobic tags that are commonly used include: adamantane, carborane, Boc3Arginine, norbornene, fluorene, (−)-menthoxyacetyl, and pyrene; all of which have been attached to small-molecule inhibitors targeting specific proteins for cancer research.72,73 Although HyT degraders have not been as widely investigated compared to other classes of degraders, their lack of dependence - in many cases - on E3 ligases may reignite interest in the event that E3 ligase mutations become prevalent, and continued development and success with these types of degraders will be beneficial to the targeted protein degradation approach.
Focal adhesion kinase (FAK)
FAK is a cytoplasmic tyrosine kinase that functions to mediate integrin signalling to regulate cellular growth, survival, and proliferation.12–14 It is overexpressed in a variety of tumour types and is associated with poor prognosis as it regulates many tumorigenic functions including tumour growth, invasion, metastasis, and angiogenesis through both kinase-dependent and kinase-independent functions. Fig. 2 depicts a range of ATP-competitive inhibitors that have been developed to target FAK, such as defactinib,74BI-853520,75PF562271,76 and VS-4718 (PND-1186).77 However, their clinical success is limited, which may be due to the inability of these inhibitors to attenuate scaffolding roles of FAK, such as the formation of signalling complexes at the plasma membrane involved with cancer progression, i.e. kinase-independent functions.14,78,79 Recently, PROTACs targeting FAK have been developed to eliminate both kinase-dependent and kinase-independent functions, shown in Fig. 3.78–81 | ||
| Fig. 2 A selection of ATP-competitive FAK inhibitors. | ||
 | ||
| Fig. 3 Structures of FAK PROTACs: each structure is color-coded, where red is the FAK ligand, black is the linker, and blue is the E3 ligase ligand. | ||
In 2019, the Crews laboratory at Yale University published PROTAC-3 that selectively degrades FAK resulting in the impairment of cell migration and invasion events in human triple-negative breast cancer (TNBC) cells.79 The structure of PROTAC-3 features a very close analogue of defactinib, the most clinically-advanced FAK ATP-competitive inhibitor, conjugated to the VHL ligand 1 through a short PEG linker. They also designed the negative control PROTAC-7, which is a diastereomer (about the hydroxyproline) of PROTAC-3 and thus unable to bind VHL. In TNBC cells, the DC50 of PROTAC-3 against FAK was determined to be 3.0 nM and its Dmax reached 99%, while PROTAC-7 was unable to degrade FAK, as anticipated. In comparison to defactinib, PROTAC-3 has enhanced selectivity for FAK as demonstrated by data they obtained from a DiscoverX KINOMEscan, wherein PROTAC-3 binds to 20 kinases in comparison to the 100 kinases bound by defactinib. They also tested PROTAC-3 in human prostate tumour (PC3) cells and saw degradation of FAK. Western blot analysis of downstream substrates in PC3 cells showed that PROTAC-3 had a more pronounced effect on the activation of paxillin, a protein involved in cell migration, than defactinib. Although both defactinib and PROTAC-3 have no effects on cell viability nor proliferation, PROTAC-3 was demonstrated to significantly impair wound healing by 53% at a concentration of 50 nM in a wound healing assay, while at the same concentration defactinib was unable to impair wound healing. Similarly, in an invasion assay compared to defactinib that is not able to significantly impair cell invasion, PROTAC-3 was able to impair cell invasion by 65%. The authors discussed that AR levels were reduced after treatment with PROTAC-3 but not reduced after treatment with defactinib as evident by western blotting techniques. They concluded that the reduction in AR levels after treatment with their FAK degrader, but not an inhibitor, suggests that AR is involved with cell motility mediated by FAK and its scaffold signalling. Taken together, all these findings highlight the non-catalytic scaffolding functions of FAK in metastatic cancer that are involved with cell migration and invasion can be attenuated with the use of PROTAC-3.79
Boehringer Ingelheim, recently reported two PROTACS, BI-3663 and BI-0319, that selectively degrade FAK in hepatocellular carcinoma and lung adenocarcinoma cell lines in 2019.81 The design of the two PROTACs differ only in the E3 ligase ligand. For their FAK ligand, they selected BI-4464, an analogue of BI-853520 that was previously published as a highly selective FAK inhibitor.75,82BI-3663 and BI-0319 feature BI-4464 coupled to pomalidomide or the VHL ligand 1, respectively, through a linker consisting of 3 PEG units. BI-3663 exhibited a DC50 of 27 nM and a Dmax of 80%, while BI-0319 exhibited a DC50 of 243 nM and a Dmax of 80% in A549, lung adenocarcinoma cells. A diastereomer of BI-0319 completely abolished the degradation activity, through rendering it incapable of binding VHL, as did the co-incubation of the PROTACs with either the parent inhibitor or NEDD8 inhibitors, as expected for CRBN and VHL neddylation-dependent E3 ligases. They observed high selectivity of the PROTACs with multiplexed isobaric tagging mass spectrometry in A549 cells with no significant changes in the abundance levels of other kinases. Both PROTACs did not affect the proliferation of cells more than the parent inhibitor.81 Although the authors did not conduct wound healing or invasion assays, it would not be unexpected that the PROTACs would eliminate non-catalytic functions like PROTAC-3.
FC-11, another FAK PROTAC, was developed by the Rao laboratory from Tsinghua University in 2020.80 The structure of this PROTAC connects pomalidomide to an analogue of PF-562271via a PEG linker. They tested FC-11 initially in PA1, human ovarian cancer, cells and observed a DR10nM (protein degradation relative to DMSO control at 10 nM) of 99% for FAK. To confirm FC-11 degrades FAK via CRBN, they co-treated cells with the PROTAC along with either the parent, CRBN, or proteasome inhibitor and in each case the degradation activity was abolished. FC-11 can degrade FAK with a DC50 value in the picomolar range in five different cell lines for different species. They conducted a washout experiment demonstrating that the levels of FAK can be restored post treatment with FC-11 after about a week. Like the FAK PROTACs previously discussed, FC-11 does not affect the cellular proliferation more than the parent inhibitor.80 They did not investigate cell migration nor invasion in this paper, but it is likely FC-11 can mitigate these functions. In a later publication, the group reported that FC-11 successfully degraded FAK in male mice reproductive tissues and the FAK levels could be restored 2 weeks after treatment was stopped.83 Resulting from degradation of FAK in reproductive tissues, the group reported that there was a significant decrease in the fertility of male mice tested due to reduced sperm motility as well as impairment of embryos fertilized by male mice treated with FC-11 compared to those treated with a vehicle or PF-562271 which did not significantly affect sperm motility nor embryo growth. They also noted that degradation of FAK is not limited to reproductive tissues, and it can degrade the kinase in other organs with different degradation efficiencies excluding the brain.83,84 Overall, this demonstrates that the phenotypic output between mice treated with a FAK inhibitor or a FAK degrader can differ due to the elimination of scaffolding roles with a degrader.
In 2021, the Benowitz laboratory from GlaxoSmithKline reported, a selective FAK PROTAC that connects VS-4718 to a VHL ligand via a short acetamide linker.78 They also developed an enantiomeric control of GSK215 that is unable to bind VHL and abolishes degradation activity. They measured FAK degradation of GSK215 using an ELISA protein quantification assay in A549 cells and determined the DC50 to be 1.3 nM and its Dmax to reach 99%, while the parent inhibitor and enantiomeric control showed no degradation of FAK. To confirm GSK215 mediated FAK degradation, a competition assay was performed with the PROTAC along with the parent inhibitor or the VHL ligand and a reduction of degradation activity of GSK215 was observed. Selectivity of GSK215 was initially measured with a cell lysate kinobead assay confirming high selectivity for binding FAK, however some other proteins could be bound such as RPS6KA1. They further analysed the selectivity of GSK215 using multiplexed proteome dynamics profiling (mPDP) and observed selective degradation of FAK with concentrations up to 10 nM, while at a concentration of 100 nM additional proteins were degraded, such as CDK7, RPS6KA3, MET and GAK. The authors did not report degradation of AR after treatment with GSK215, as the Crews lab did with PROTAC-3,79 to which they suggested that different phenotypic effects may be observed with PROTACs of different structures. They also confirmed that degradation of FAK was dependent on ternary complex formation with a FRET assay, surface plasmon resonance (SPR), and a crystal structure (PDB ID: 7PI4). In A549 and MCF-7 cells, GSK215 repressed 2D cell proliferation but did not affect BT474 cells, while the opposite was observed when observing 3D proliferation. GSK215 was able to suppress cell motility in a wound scratch assay in A549 cells, while the parent inhibitor and enantiomeric control did not affect cell motility demonstrating the ability of the PROTAC to eliminate scaffolding roles of FAK involved in cell migration. A mouse study with GSK215 was conducted and they observed 85% reduction of FAK levels in the liver within 18 hours of treatment with a reduction of 60% of FAK levels remaining for 96 hours post treatment.78 This study demonstrates the ability of GSK215 to degrade FAK and sustain a reduction level of the kinase over a four day period, eliminating both catalytic and non-catalytic functions.
The development of PROTACs targeting FAK show promising results in the prevention of metastatic cancers, as they can significantly impair migration events that are not affected by ATP-competitive inhibitors. Instead, ATP-competitive inhibitors targeting FAK show promising results in the prevention of tumour growth.74,75,85 The advantage of eliminating both catalytic and non-catalytic functions of FAK in cancer progression with PROTACs is promising for the future of cancer therapeutics to aid in the treatment and prevention in patients with poor prognosis. While PROTAC-3 and its parent inhibitor defactinib were unable to affect cell viability,79 FAK PROTACs BI-3663, BI-0319,81 and FC-1180 and their parent inhibitors, BI-4464 and PF-562271, were able to affect cell viability. This effectively highlights the suggestion that the Benowitz laboratory has raised concerning differences in biological observations after treatment with PROTACs of different structures.78
Aurora-A Kinase (AURKA)
Aurora-A Kinase (AURKA) is an isozyme of the aurora kinase family of serine/threonine protein kinases that functions during mitosis.17,39 Further, AURKA functions to ensure centrosome maturation, mitotic timing, microtubule nucleation, and spindle assembly. Like FAK, overexpression and increased activity of AURKA is observed in various cancers and is correlated with resistance to chemotherapeutic treatments and stem-like properties in cells.17,39 Currently, the scaffolding roles of AURKA in cancer are being intensely investigated as therapeutic targets since its protein–protein interaction (PPI) with N-Myc, is well-established, and this PPI effectively protects N-Myc from degradation by the proteasome; c-Myc is also being investigated for its PPI with AURKA.1,5,15–17 Typically, AURKA undergoes targeted proteolysis by anaphase-promoting complex (APC/C) during the cell cycle at mitotic exit, however due to its scaffolding roles with MYC proteins it can be detected in interphase cells.17MYC proteins (c-Myc, N-Myc, and L-Myc) are short lived transcriptional factors that are tightly controlled under normal conditions, but high levels are observed in nearly 50% of all human cancers.15,16,86,87 Although these proteins are validated drug targets, they are devoid of ‘druggable pockets’ due to their intrinsically disordered nature.16 We,88–94 and others,95–97 have experienced limited success in directly targeting the Myc proteins.88,90 Therefore, an emerging strategy to inhibit the MYC proteins involves doing so indirectly by instead targeting AURKA and compromising its stabilization of MYC proteins.39 AURKA/MYC complexes a associated with aggressive tumours and rapid disease progression; accordingly, they represent attractive targets for novel chemotherapies.4 ATP-competitive inhibitors of AURKA have been developed, such as alisertib (MLN8237),98MK-5108,99 and CD532,100 shown in Fig. 4, two of which, alisertib and CD532, are characterized as destabilizing inhibitors since they disrupt AURKA/MYC complexes.16,98,99 Destabilizing AURKA inhibitors function to alter the conformation of the kinase domain, disrupting the ability of AURKA to act as a scaffold.15,16,87,100 Clinical trials with these destabilizing inhibitors have been conducted, however low response rates and toxicities were observed, likely due to the inability to disrupt pre-formed complexes and off-target effects with GABA or closely related Aurora-B, respectively.86,101–103 Furthermore clinical resistance may be observed due to mutations in AURKA making it drug resistant104 or mutations in protein kinase A resulting in its ATP-binding site mimicking that of Aurora-A and demonstrating high affinity for its inhibitors.105 To overcome these limitations, the development of PROTACs, shown in Fig. 5, targeting AURKA are being investigated, a majority of which utilize existing AURKA inhibitors.106–113
 | ||
| Fig. 4 Some ATP-competitive inhibitors of AURKA. | ||
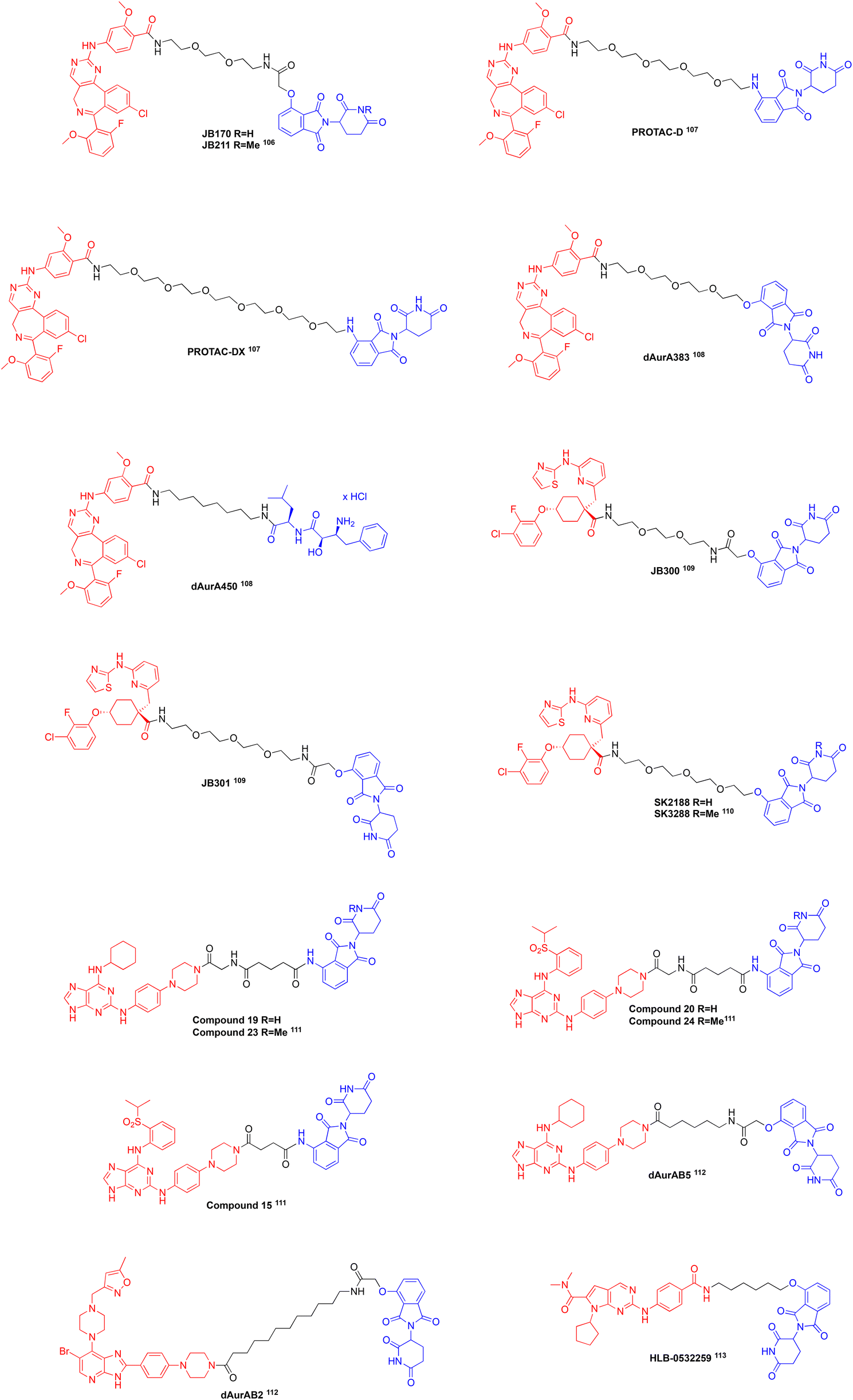 | ||
| Fig. 5 A selection of AURKA PROTACs: each structure above is color-coded, where red is the AURKA ligand, black is the linker, and blue is the E3 ligase ligand. | ||
In 2020, Adhikari et al. developed AURKA degraders connecting alisertib to CRBN and VHL ligands with PEG and aliphatic linkers.106 The most promising PROTACs they reported are JB170 and JB158, both using thalidomide a CRBN binding moiety that induce degradation of AURKA levels by 69% and 62% in MV4-11 leukaemia cells, respectively. Unfortunately, they did not observe a reduction in AURKA levels with VHL-based degraders. As JB170 reduced AURKA to a higher level, they conducted further experiments using this PROTAC. They determined the DC50 of JB170 to be 28 nM using a luciferase detection system and saw a decrease in the half-life of AURKA from 3.8 to 1.3 hours. They prepared the negative control PROTAC, JB211, almost identical to JB170 but crucially presents a methylated glutarimide nitrogen, which prevents the molecule from binding CRBN and they observed no degradation of AURKA after treatment. They further confirmed JB170 acts as a degrader by co-incubation with alisertib or thalidomide and observed the degradation activity was reduced. A quantitative PCR experiment post degrader treatment shows that mRNA levels of AURKA were not reduced even though the protein was degraded. Catalytically inactive versions of AURKA were expressed and degradation was observed demonstrating that depletion is independent of catalytic activity. They conducted a cell viability assay in MV4-11 cells which showed that the number of viable cells was 32% of control levels and there was an increase in the fraction of apoptotic cells 72 hours after treatment with JB170 at a concentration of 1 μM, while JB211 did not significantly induce apoptosis nor reduce cell viability. Using cell lysates, they performed kinobead selectivity profiling observing a higher selectivity for AURKA over AURKB with JB170 compared to alisertib. They also confirmed selectivity with Stable Isotope Labelling by Amino acid in Cell culture (SILAC) mass spectrometry, demonstrating that treatment with JB170 does not deplete the levels of other proteins besides AURKA. JB170 was tested in osteosarcoma (U2OS), hepatocellular carcinoma (HLS) and neuroblastoma (IMR5) cell lines and saw that AURKA was depleted in all cases, but with different potencies and degrees of depletion. Compared with alisertib that arrests cells in G2/M, JB170 arrests S-phase progression which was confirmed by BrdU/PI flow cytometry. The authors hypothesized that the difference of arrest in the cell cycle between the two treatments is due to scaffolding (non-catalytic) roles of AURKA in the S-phase.106 This paper demonstrates the first AURKA PROTAC developed that produces a distinct phenotype compared to its parent inhibitor, alisertib.
In 2021, the Lindon laboratory at the University of Cambridge published their development of AURKA degraders.107 Like Adhikari et al.,106 they designed and synthesized alisertib-based PROTACs that, via PEG linkers of varying lengths, feature a ligand that binds VHL or CRBN. They tested their PROTACs in AURKA-VenusKI and AURKA-VenusTO engineered human retinal pigment epithelial (RPE1) cells, that have endogenously expressed AURKA levels during mitosis and high levels of expression throughout the cell cycle, respectively. In these cell lines, they determined that VHL-based degraders were unable to degrade AURKA while CRBN-based degraders showed depletion. They reported their most efficient degraders with EC50 in the 100 nM range to be PROTAC-D and PROTAC-DX that connect alisertib to pomalidomide through 4 or 6 PEG units, respectively. As the PROTACs were active in both cell lines, they degraded AURKA in both mitotic and interphase cells, however the activity in interphase cells occurred to a lower extent, likely due to scaffolding roles. Additionally, the group expressed an AURKA mutant in RO3306 cells and observed degradation of the mutant. Treatment with alisertib or pomalidomide alone or in combination did not affect AURKA levels, demonstrating that depletion is dependent upon ternary complex formation with AURKA, CRBN, and the PROTAC. Moreover, a competition assay with excess pomalidomide decreased the activity of PROTAC-D. To confirm depletion of AURKA is dependent on PROTAC-D and not APC/C during mitotic exit, they treated cells with both PROTAC-D and an APC/C inhibitor and saw that the depletion of AURKA by PROTAC-D was not affected. They calculated the specificities for AURKA over AURKB to be 8.3, 21.6, and 23.7 with alisertib, PROTAC-D, and PROTAC-DX, respectively. Last, they investigated the phenotypic output of inhibition with alisertib versus treatment of PROTAC-D with immunofluorescent staining compared to a negative control. From this they observed that treatment with PROTAC-D resulted in a loss of AURKA (TPX2-associated pool) within mitotic spindles, but it was conserved at the centrosomes (CEP192-associated pool), while alisertib resulted in loss of all AURKA function including in the centrosome. They hypothesized that this difference could be due to conformation dependent targeting with PROTAC-D, where its preferred target is either free AURKA or TPX2-bound AURKA outside of the centrosome or that CEP192 bound AURKA may block the recruitment of CRBN or other components that successfully result in the ubiquitination of AURKA within the centrosomes.107
Liu et al. developed a library of alisertib-based PROTACs and published synergistic effects observed in acute myeloid leukaemia (AML) cells with a PROTAC cocktail recruiting different E3 ligases.108 Their library of PROTACs contained alisertib linked to ligands for CRBN, cIAP, and VHL E3 ligases through PEG and aliphatic linkers. Initially they observed the degradation activity of the PROTACs by immunoblotting and saw the best degradation with dAurA383 that recruits CRBN, while dAurA425 and dAur450 that recruit VHL and cIAP, respectively, showed moderate activity. They hypothesized that the difference in degradation activities of the PROTACs is attributed to the expression levels of the E3 ligases throughout the cell cycle, where CRBN is highly expressed during mitosis, while VHL and cIAP are highly expressed during interphase. They next determined that the PROTACs had similar dissociation constants as alisertib, using surface plasmon resonance. Ternary complex formation was confirmed with all three PROTACs using a separation of phase-based protein interaction reporter assay in HEK293T cells. They reported degradation activity was observed in KG1A, Kasumi-1, HL60, and U937 cells with immunoblots, where optimal degradation occurred 6 hours after treatment and that degradation was prevented with the use of proteasome inhibitors, bortezomib and MG132, with the PROTACs. Using a CCK8 assay, they determined that the PROTACs inhibited cellular proliferation in KG1A, Kasumi-1, HL60, NB4, U937 and THP1 cells in the low micromolar range and additionally induced apoptosis in KG1A and Kasumi-1 cells.
Proteomic and transcriptomic profiling supported their hypothesis that dAurA383 that recruits CRBN degrades AURKA in mitotic cells, while dAur450 that recruits cIAP degrades AURKA in interphase cells. Furthermore, their transcriptomic analysis showed an increase in CD34, a hematopoietic stem and progenitor cell marker, in response to dAurA383 treatment, but a decrease in the same marker in response to dAurA450 treatment in KG1A cells. This led them to investigate a PROTAC cocktail containing a 1:1 ratio of both PROTACs to enable degradation of AURKA in both mitotic and interphase cells, as well as mitigate stemness activation observed with treatment of dAurA383 alone. They observed synergistic effects with the PROTAC cocktail in KG1A and Kasmi-1 cells that led to inhibition of cell division and a significant induction of apoptosis compared to single PROTAC treatment. The cocktail also decreased stemness markers CD34 and c-Myc in the two cell lines, while also having no effect on AURKB levels. They tested the PROTAC cocktail in KG1A xenograft mice, malignant bone marrow mononuclear cells, and patient derived AML blasts and observed degradation of AURKA, significant inhibition on cell growth, and the induction of apoptosis. Ultimately, this paper conveys that synergistic effects can be observed with cocktails containing PROTACs recruiting different E3 ligases that can lead to a reduction in cancer stemness relating to metastasis.108
In 2022, Bozilovic et al. reported a set of PROTACs that attached MK-5108 to 4-hydroxythalidomide using various PEG and aliphatic linkers.109 They constructed an MV4-11 cell line expressing AURKA tagged with a luciferase protein, and treated the cells with various concentrations of their PROTACs to obtain dose response data. They observed maximal depletion of AURKA after 6 hours of treatment. JB300 and JB301, with linker lengths of 2 and 3 PEG units respectively, were the most potent AURKA degraders. The DC50 values for JB300 and JB301 were 30 nM and 3.2 nM, with maximal depletion of AURKA reaching 78% and 82%, respectively. They conducted immunoblotting analysis after a 6 hour incubation with JB300 that showed depletion of AURKA, but not AURKB. They verified that depletion is due to ternary complex formation by co-incubating MV4-11 cells with JB300 and MK-5108, thalidomide, or MG132 (proteasome inhibitor), and saw decreased depletion of AURKA. They also performed a kinetic degradation assay to observe the effects of JB300 and JB301 single dose treatment over longer treatment periods and verified the results with immunoblotting. After 9 hours of treatment, maximal depletion of AURKA was observed and after 72 hours of treatment there was a slight increase in AURKA levels. They concluded that after a single dose of the PROTACs, depletion of AURKA may be observed over long periods of time. A cell viability assay showed that JB301 induced significant cytotoxicity in comparison to MK-5108. The authors analysed the effects of the parent inhibitor and JB300 on the cell cycle with a BrdU/PI flow cytometry assay. They observed G2/M arrest with MK-5108 and a strong reduction in the S-phase with JB300, like the alisertib-based JB170 developed by the same group,106 suggesting that S-phase arrest observed after treatment of PROTACs is associated with the elimination of scaffolding roles of AURKA.
A proteomic study was conducted to analyse the changes in cellular protein levels after treatment with JB300 and MK-5108 compared to non-treated cells. JB300 showed significant depletion of AURKA and a small amount of PTK2B depletion, while MK-5108 induced an enrichment of AURKA, AURKB, and PTK2B levels.109 The increased levels of AURKA after treatment with MK-5108 demonstrates another advantage of PROTACs over kinase inhibitors, where inhibitors can stabilize and increase the levels of oncogenic proteins as opposed to PROTACs that lead to their destruction. Overall, this paper demonstrates the first MK-5108-based selective AURKA PROTAC with long lasting effects.
The Durinck laboratory at Ghent University published their development of MK5108-based AURKA degraders and identified SK2188 to be their lead compound in 2023.110 The structure of SK2188 features MK5108 attached to 4-hydroxy-thalidomide with a linker consisting of 4 PEG molecules. A negative control, SK3288, was designed and synthesized, differing from the active PROTAC SK2188 by the presentation of an N-methylated glutarimide ring restricting its ability to bind CRBN. They tested their library of PROTACs against NGP neuroblastoma cells that highly express CRBN, respond well to AURKA inhibition, and display an amplification of MYCN and monitored AURKA degradation with immunoblotting. Most of their PROTACs demonstrated a dose-dependent response with SK2188 having the most potent depletion of AURKA reaching a Dmax of 89% 24 hours after treatment and a DC50 value of 3.9 nM, effectively outperforming both MK-5108 and JB170, which resulted in an increase of AURKA levels and a 73% reduction of AURKA, respectively.
The group looked further in the degradation kinetics of SK2188 by treating NGP cells with 500 nM of the PROTAC and monitored AURKA levels at different time points and observed a maximal depletion of AURKA to 93% after 1 hour of treatment with the levels slightly rising overtime. They noted that MYCN levels remained stable upon initial treatment with SK2188, but MYCN levels started to decrease after 24 hours ultimately reaching a Dmax of 73% 72 hours post treatment, which effectively outperforms MK-5108 treatment that results in a Dmax of 40% 48 hours post treatment. SK2188 potently inhibited NGP cell growth and had an IC50 value of 31.9 nM, once again outperforming MK-5108 and JB170 which had IC50 values of 361.7 nM and 876.6 nM, respectively. They investigated downstream effects of SK2188 and MK-5108 treatments in NGP cells and saw a greater depletion of MYCN levels and induction of apoptosis upon treatment with SK2188 compared to MK-5108. To confirm SK2188 functions as an AURKA degrader, they pretreated NGP cells with MG-132 and saw that it effectively prevented SK2188-mediated AURKA degradation. Moreover, treatment with SK3288 failed to decrease AURKA levels and instead led to increasing levels of the kinase, like MK-5108.114 They analysed the selectivity profile of SK2188 with a DiscoverX KINOME scan observing SK2188 binding 15 other kinases to a low extent at a dose of 1 μM and binding only AURKA and TRKA at a dose of 100 nM. Other neuroblastoma cell lines were tested and SK2188 demonstrated 10-fold higher potency than MK-5108 in NGP and IMR-32 cells, while it was unable to outperform MK-5108 in N206 and SK-N-AS cells. Lastly, they tested the effects of SK2188 in patient-derived organoids and observed that it was less potent than MK-5108 in 000GKX and NB059 organoids, but outperformed MK-5108 in NB139 and NB067 organoids with IC50 values determined to be 132.2 and 26.2 nM, respectively. This paper communicates the first published AURKA degrader that validates an indirect degradation of MYCN in response to treatment with SK2188.110 To reiterate our earlier point, one thoroughly investigated non-catalytic role of AURKA is the stabilization of MYCN through a scaffolding interaction, which leads to the accumulation of both proteins in cancer cells and leads to aggressive disease progression.
In 2024, Sflakidou et al. published a series of selective monopolar spindle 1 (TTK) and AURKA degraders from azareversine, MPSI-IN-3, SF1 and SF2, promiscuous ATP-competitive inhibitors.111 They conjugated these inhibitors to either pomalidomide or VHL-1 with various linkers and tested them in MV4-11 HiBit cell lines that have small tags on TTK and AURKA and have slightly enhanced expression levels. They determined that the azareversine-based PROTACs with pomalidomide are more active than those with VHL-1. Compound 19 was determined to be a pan-degrader able to deplete AURKA, AURKB, and TTK, with Dmax values of 78.8%, 43.6%, and 66.5% and DC50 values of 109 nM, 570 nM, and 18 nM, respectively. However, they stated that degradation of the kinases is only partially dependent on E3-ligase recruitment as a negative control 23 also showed degradation activity in both HiBit and native cell lines.
MPSI-IN-3-based degraders showed no significant activity against the kinases.111 Like azareversine-based degraders, SF1-based PROTACs bearing pomalidomide were more active than those bearing VHL-1. Compound 20 and Compound 15 were determined to preferentially degrade AURKA, with compound 20 displaying a DC50 of 68 nM and Dmax of 71% making it 4- and 2-fold more selective for AURKA over AURKB and TTK. Negative control compound 24, a congener of compound 20 with a N-methylated glutarimide ring, minimally degraded all proteins with Dmax values less than 20%, pointing towards the dependence of 20's degradation activity on the CRBN E3 ligase. Moreover, this was corroborated by the co-incubation of 20 with pomalidomide, which resulted in a reduction of AUKRA degradation. Activity of compound 20 is not limited to MV4-11 cells as similar depletion is observed in CALU1 human non-small lung cancer cells. Generally, the parent inhibitors displayed greater cytotoxicity than their respective degraders, however, PROTAC 15 displayed equipotent cytotoxicity with its parent inhibitor SF1. They tested the stability of compound 20 at pH 5.2 and 7.4 and determined it was stable up to 8 hours in both, but the concentrations decreased to 56% at pH 5.2 and 11.2% at pH 7.4 after 24 hours. Finally, they discovered the half-life of compound 20 was 4.4 hours in human plasma. Overall, this report describes PROTACs that actively degrade AURKA, AURKB, and TTK, some of which degrade one of the kinases more selectively than the other two. We believe these should be further investigated to determine their overall selectivities and efficacies in cancer cells because the transformation of promiscuous inhibitors into selective degraders may be especially impactful to the PROTAC field, notwithstanding the potential discovery of an effective preclinical candidate.111
In 2024, Nelson et al. reported AURKA/AURKB dual degraders synthesized from pan-aurora kinase inhibitors CCT137690 and reversine that were also able to degrade MYCN.112 Their rationale for selecting pan-Aurora inhibitors was to create PROTACs capable of reducing both AURKA and AURKB levels, as inhibition of AURKB reduces MYCN-amplified TP53-WT neuroblastoma cell lines in addition to the scaffolding interaction between AURKA and MYCN. They initially observed the degradation of AURKA and AURKB in TP53-WT IMR32 neuroblastoma cells that have a MYCN-amplification in response to treatment with their PROTACs at a concentration of 500 nM using western blotting techniques. Of the synthesized PROTACs, they identified dAurAB2, CCT137690 connected to 4-hydroxythalidomide through an undecyl linker, and dAurAB5, reversine connected to 4-hydroxythalidomide through a pentyl linker, to be their most efficient degraders. From this they determined that dAurAB2 resulted in a 71% reduction in AURKA levels and 72% reduction in AURKB levels, while dAurAB5 resulted in an 84% reduction in AURKA levels and 82% reduction in AURKB levels. They noted that treatment with the parent inhibitors lead to an increase in both aurora kinase levels after treatment, like other groups have observed. After confirming that their PROTACs degraded AURKA and AURKB, they performed the same exact experiment to observe the reduction in MYCN levels, where dAURAB2 reduced the levels by 38% and dAURAB5 reduced the levels by 45%. Next, they performed a dose–response assay testing concentrations between 10–1000 nM and determined dAurAB5 to be more potent than dAurAB2. From this, they reported dAurAB2 to have a DC50 of 59 nM and Dmax of 97% at 1 μM for AURKA and DC50 of 29 nM and Dmax of 89% at 200 nM for AURKB, whereas dAurAB5 had a DC50 of 8.8 nM and Dmax of 91% at 100 nM for AURKA and DC50 of 6.1 nM and Dmax of 96% at 100 nM for AURKB. To confirm that the degradation of these kinases was due to PROTAC treatment, they preincubated cells with the proteasome inhibitor carfilzomib and observed that the depletion of AURKA and AURKB levels was blocked.
The authors subsequently performed a proteomic study, wherein the analysed the changes in 7280 different proteins upon treatment with dAurAB2 and dAurAB5 in Kelly neuroblastoma cells which also have an amplification of MYCN. Of the two they determined that dAurAB2 was more selective as it did not deplete any protein levels that its parent inhibitor (CCT137690) interacts with (FLT3, FGR1 and VEGFR) and only showed decreased levels of SLC25A6 and HSPE1, the former of which they mentioned is a common false positive in the global proteomic assay they conducted. The authors noted that the proteomic data for dAurAB5 showed that it was less selective as it decreased kinases such as AAK1, PTK2, GAK, Q6ZSR9, BANF1 and TTK. As TTK is another risk factor in neuroblastoma in addition to AURKA, AURKB, and MYCN, and it was downregulated in response to treatment with dAurAB5 the authors analysed the cell viability of this PROTAC in IMR32 neuroblastoma cells and HEK293 (control) cells with a MTT assay. From this they reported that dAurAB5 resulted in a 45% reduction in cell viability at a dose of 100 nM and a 55% reduction in cell viability at 1 μM in IMR32 cells, while in HEK293 cells 89% and 86% of cells remained viable after treatment with 100 nM and 1 μM of dAurAB5. This paper demonstrates dual AURKA/AURKB degraders can be beneficial for the exploration in MYCN-amplified cancers such as neuroblastoma.112
In 2025, Tang et al. communicated that PROTAC HLB-0532259 that can promote the degradation of AURKA and indirect degradation of MYCN in neuroblastoma cells.113 Apart from SK2188,110 previously published PROTACs comprising AURKA inhibitors have not reported a subsequent decrease in MYCN levels, thus, the authors suggested that PROTACs containing these ligands may be unable to degrade the AURKA/MYCN complex due to poor selectivity or they are incompatible with the complex conformation. Beginning with the CDK4/6 inhibitor ribociclib, the authors first made some chemical modifications that resulted in a 1000-fold increase in binding affinity to AURKA, and they solved a co-crystal structure (PDB: 9BZG) of their ribociclib analogue bound to AURKA that is compatible to still bind MYCN. Subsequently, they connected their ribociclib variant to thalidomide with a hexyloxy linker arriving at HLB-0532259. They screened this compound against a panel of kinases, and it demonstrated excellent selectivity for AURKA. To confirm they had synthesized an AURKA degrader, they first tested HLB-0532259 in MCF-7 breast cancer cells that do not have an amplification of MYCN to confirm AURKA degradation and observed rapid degradation of AURKA with a DC50 of 20.2 nM and Dmax of 94% after a 4 hour treatment. Mechanistic studies with MG-132 and excess thalidomide demonstrates that depletion of AURKA is dependent upon the ubiquitin–proteasome system (UPS). They then moved on to test HLB-0532259 in SK-N-BE(2) and Kelly neuroblastoma cells with an amplification of MYCN and observed degradation of AURKA with concomitant MYCN degradation. They determined the DC50 of HLB-0532259 for N-Myc to be 179 nM and 229 nM in SK-N-BE(2) and Kelly cells, respectively, but degradation of N-Myc did not occur until about 75% of AURKA is depleted. Furthermore, HLB-0532259 did not induce the degradation of c-Myc. Compared with alisertib and CD532, HLB-0532259 induced a more potent degradation of N-Myc. They confirmed that degradation of AURKA is dependent upon the UPS in a similar manner as mentioned above. They conducted a washout experiment after treatment with HLB-0532259 that demonstrated a small amount of AURKA could stabilize N-Myc back to steady state levels. A proteomic-wide degradation study demonstrated the selectivity of HLB-0532259 for Aurora-A and N-Myc and they validated these results with immunoblotting analysis. They tested HLB-0532259 in various MYCN-amplified neuroblastoma cells and observed that cells underwent apoptosis and potent cytotoxicity occurred with IC50 values ranging from 20.1 to 131 nM. Lastly, a mouse study was conducted and showed substantial tumour regression in mice treated with an injection of HLB-0532259 every 3 days compared to untreated mice. Overall, this publication demonstrates the ability of AURKA PROTACs to promote the indirect degradation of N-Myc, a highly investigated non-catalytic scaffolding function of AURKA.113
The development of highly selective AURKA PROTACs for the treatment of cancers with an overexpression of Aurora-A kinase show promising results in the inhibition of tumour growth and induction of apoptosis. A few of the PROTACs mentioned above demonstrate the ability to indirectly degrade MYC proteins that are also overexpressed in aggressive cancers such as neuroblastoma and liver cancer. It is likely that all the above mentioned PROTACs can indirectly degrade MYC proteins, but more studies to confirm this need to be conducted. The advantages of employing PROTACs over destabilizing inhibitors to prevent the formation of AURKA/MYC complexes include increased selectivity over AURKB, reduction in both AUKRA and MYC protein levels, and the induction of apoptosis. In comparison to alisertib which exhibits IC50 values between 7.6–26.8 nM in neuroblastoma cell lines115 and 8.5–10.8 nM in AML cell lines,116 AURKA PROTACs SK2188 and HLB-0532259 exhibited IC50 values of 31.9 nM and 20.1–131 nM, respectively, demonstrating that these newly developed PROTACs have similar effects on cell viability as alisertib, a clinically advanced AURKA inhibitor. On the other hand, the reported reductions in cell viability, 32% at 1 μM of JB170106 and 45% at 100 nM dAurAB5,112 are less potent than the reduction in cell viability that alisertib induces, about 60% at 100 nM and 70% at 1 μM.116 Despite this, all the forementioned PROTACs in this section are promising for further development and studies. While the preliminary studies to confirm that these compounds effectively function as degraders are encouraging, more side-by-side studies of the PROTACs and their parent inhibitors should be conducted to observe the differences in potency and efficacy in both wild-type and drug-resistant cell lines.
Conclusions
Overexpression and activation of kinases often leads to rapid disease progression, especially in cancer, through both catalytic and non-catalytic functions. ATP-Competitive kinase inhibitors, such as those developed for FAK and AURKA, can significantly inhibit tumour growth by targeting their catalytic functions. However, such inhibitors typically are unable to overcome non-catalytic functions, apart from destabilizing AURKA inhibitors that may disrupt the formation of AURKA/MYC complexes. Clinical application of ATP-competitive inhibitors is limited by off-target effects due to high conservation of the ATP-binding pocket, low response rates, and the risk of developing drug-resistant mutations. PROTACs have the potential to overcome these limitations and reduce dose requirements relating to their catalytic mechanisms of action, while also being able to target non-catalytic functions that often relate to metastatic cancers. In this review, we presented PROTACs developed to target FAK and AURKA that have enhanced efficacies compared to their parental inhibitors in early biological testing in relevant cell lines and in some cases mouse studies. Further biological characterizations with these PROTACs will need to be conducted to further test their efficacy and applicability in various cancers. While the focus of our review was eliminating scaffolding (non-catalytic) roles of kinases, other proteins and enzymes participate in scaffolding roles as well.5,6 For example, PARP1 is a nuclear protein that acts as a scaffold to recruit DNA repair factors in response to cellular stress that results in the breakage of a strand of DNA.117 Cancers harbouring mutations in DNA repair factors are dependent on PARP1, and other pathological conditions are associated with over-activation of PARP1, thus, inhibitors for PARP1 have been identified, however they are highly cytotoxic due to PARP trapping and may bind other PARP proteins.117 A selective PARP1 degrader was developed by Wang et al. and was able to inhibit PARP1 without causing trapping.117 We believe the identification of more scaffolding roles of kinases and other proteins, with subsequent development of PROTACs will offer significant clinical efficacy over classical inhibitors, especially in the cases of aggressive, metastatic cancers.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
SP performed the literature search and wrote the manuscript; SF edited the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank the University of Maryland Schools of Pharmacy and Medicine for their continued support of our work in this area.Notes and references
- J. E. Kung and N. Jura, Structure, 2016, 24, 7–24 CrossRef CAS PubMed
.
- Z. Wang, W. Huang, K. Zhou, X. Ren and K. Ding, J. Med. Chem., 2022, 65, 1735–1748 CrossRef CAS
- Y. Zhou, S. Xiang, F. Yang and X. Lu, J. Med. Chem., 2022, 65, 15540–15558 CrossRef CAS PubMed
- A. Jain, K. B. Lokhande and A. Singh, Med. Hypotheses, 2024, 185, 111320 CrossRef CAS
- C. Kim, X. D. Wang, Z. Liu, S. Zha and Y. Yu, Biochemistry, 2023, 62, 561–563 CrossRef CAS PubMed
- D. Sun, J. Zhang, G. Dong, S. He and C. Sheng, J. Med. Chem., 2022, 65, 14276–14288 CrossRef CAS PubMed
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Mol. Cancer, 2018, 17, 48 CrossRef PubMed
- P. Karki, S. Sensenbach, V. Angardi and M. A. Orman, Metabolites, 2021, 11, 777 CrossRef CAS PubMed
- H. Liu, N. Nazmun, S. Hassan, X. Liu and J. Yang, Cancer Med., 2020, 9, 4881–4896 CrossRef CAS PubMed
- J. C. Zhao, S. Agarwal, H. Ahmad, K. Amin, J. P. Bewersdorf and A. M. Zeidan, Blood Rev., 2022, 52, 100905 CrossRef CAS PubMed
- B. Acharya, D. Saha, D. Armstrong, N. R. Lakkaniga and B. Frett, RSC Med. Chem., 2022, 13, 798–816 RSC
- Y. L. Tai, L. C. Chen and T. L. Shen, BioMed Res. Int., 2015, 2015, 690690 Search PubMed
- Z. Zhang, J. Li, S. Jiao, G. Han, J. Zhu and T. Liu, Front. Cell Dev. Biol., 2022, 10, 1040311 CrossRef PubMed
- X. Zhao and J. L. Guan, Adv. Drug Delivery Rev., 2011, 63, 610–615 CrossRef CAS PubMed
- M. W. Richards, S. G. Burgess, E. Poon, A. Carstensen, M. Eilers, L. Chesler and R. Bayliss, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13726–13731 CrossRef CAS PubMed
- R. Bayliss, S. G. Burgess, E. Leen and M. W. Richards, Biochem. Soc. Trans., 2017, 45, 709–717 CrossRef CAS PubMed
- R. Du, C. Huang, K. Liu, X. Li and Z. Dong, Mol. Cancer, 2021, 20, 15 CrossRef CAS
- H. Kameda, Immunol. Med., 2023, 46, 108–111 CrossRef CAS PubMed
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Nat. Rev. Drug Discovery, 2021, 20, 839–861 CrossRef CAS
- C. C. Goodis, C. Eberly, A. M. Chan, M. J. Kim, B. D. Lowe, C. I. Civin and S. Fletcher, Eur. J. Med. Chem., 2025, 285, 117190 CrossRef CAS
- X. Wang, D. Goldstein, P. J. Crowe and J. L. Yang, Onco Targets Ther, 2016, 9, 5461–5473 CrossRef CAS PubMed
- L. Wang, W. Q. Liu, S. Broussy, B. Han and H. Fang, Front. Pharmacol., 2024, 14, 1307860 CrossRef PubMed
- D. Poei, S. Ali, S. Ye and R. Hsu, Cancer Drug Resist., 2024, 7, 20 CAS
- C. Wang, H. Wang, C. Zheng, Z. Liu, X. Gao, F. Xu, Y. Niu, L. Zhang and P. Xu, Eur. J. Med. Chem., 2021, 218, 113386 CrossRef CAS PubMed
- D. Meng, W. He, Y. Zhang, Z. Liang, J. Zheng, X. Zhang, X. Zheng, P. Zhan, H. Chen, W. Li and L. Cai, Pharmacol. Res., 2021, 173, 105900 CrossRef CAS
- L. M. Spring, S. A. Wander, M. Zangardi and A. Bardia, Curr. Oncol. Rep., 2019, 21, 25 CrossRef
- N. S. Persky, D. Hernandez, M. Do Carmo, L. Brenan, O. Cohen, S. Kitajima, U. Nayar, A. Walker, S. Pantel, Y. Lee, J. Cordova, M. Sathappa, C. Zhu, T. K. Hayes, P. Ram, P. Pancholi, T. S. Mikkelsen, D. A. Barbie, X. Yang, R. Haq, F. Piccioni, D. E. Root and C. M. Johannessen, Nat. Struct. Mol. Biol., 2020, 27, 92–104 CrossRef CAS
- A. M. Chan, C. C. Goodis, E. G. Pommier and S. Fletcher, RSC Med. Chem., 2022, 13, 921–928 RSC
- Faridoon, R. Ng, G. Zhang and J. J. Li, Med. Chem. Res., 2023, 32, 1039–1062 CrossRef CAS PubMed
- D. Patel, Z. E. Huma and D. Duncan, ACS Chem. Biol., 2024, 19, 824–838 CrossRef CAS PubMed
- S. De Cesco, J. Kurian, C. Dufresne, A. K. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2017, 138, 96–114 CrossRef CAS PubMed
- M. R. Burke, A. R. Smith and G. Zheng, Front. Cell Dev. Biol., 2022, 10, 872729 CrossRef PubMed
- D. Wu, Y. Li, L. Zheng, H. Xiao, L. Ouyang, G. Wang and Q. Sun, Acta Pharm. Sin. B, 2023, 13, 4060–4088 CrossRef CAS PubMed
- T. Cierpicki and J. Grembecka, Annu. Rev. Pathol.:Mech. Dis., 2025, 20, 275–301 CrossRef CAS PubMed
- H. Lei, S. Q. Zhang, S. Fan, H. R. Bai, H. Y. Zhao, S. Mao and M. Xin, J. Med. Chem., 2021, 64, 15519–15533 CrossRef CAS PubMed
- U. Jaffry and G. Wells, Biochem. Soc. Trans., 2023, 51, 925–936 CrossRef CAS PubMed
- D. Chirnomas, K. R. Hornberger and C. M. Crews, Nat. Rev. Clin. Oncol., 2023, 20, 265–278 CrossRef CAS PubMed
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed
- V. Bavetsias and S. Linardopoulos, Front. Oncol., 2015, 5, 278 Search PubMed
- D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed
- Z. Liu, M. Hu, Y. Yang, C. Du, H. Zhou, C. Liu, Y. Chen, L. Fan, H. Ma, Y. Gong and Y. Xie, Mol. Biomed., 2022, 3, 46 CrossRef CAS PubMed
- X. Li, W. Pu, Q. Zheng, M. Ai, S. Chen and Y. Peng, Mol. Cancer, 2022, 21, 99 CrossRef CAS PubMed
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed
- Y. Liu, J. Yang, T. Wang, M. Luo, Y. Chen, C. Chen, Z. Ronai, Y. Zhou, E. Ruppin and L. Han, Nat. Commun., 2023, 14, 6509 CrossRef CAS PubMed
- Y. Dong, T. Ma, T. Xu, Z. Feng, Y. Li, L. Song, X. Yao, C. R. Ashby and G. F. Hao, Acta Pharm. Sin. B, 2024, 14, 4266–4295 CrossRef CAS PubMed
- J. Desantis, A. Mammoli, M. Eleuteri, A. Coletti, F. Croci, A. Macchiarulo and L. Goracci, RSC Adv., 2022, 12, 21968–21977 RSC
- Q. Yang, J. Zhao, D. Chen and Y. Wang, Mol. Biomed., 2021, 2, 23 CrossRef PubMed
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed
- T. Ishida and A. Ciulli, SLAS Discovery, 2021, 26, 484–502 CrossRef CAS PubMed
- C. Wang, Y. Zhang, L. Shi, S. Yang, J. Chang, Y. Zhong, Q. Li and D. Xing, J. Enzyme Inhib. Med. Chem., 2022, 37, 1437–1453 CrossRef CAS PubMed
- R. Shah Zaib Saleem, M. P. Schwalm and S. Knapp, Bioorg. Med. Chem., 2024, 105, 117718 CrossRef CAS PubMed
- B. Adhikari, K. Schneider, M. Diebold, C. Sotriffer and E. Wolf, eLife, 2024, 13, RP98450 CrossRef PubMed
- P. Torres-Ayuso and J. Brognard, Mol. Pharmacol., 2022, 101, 191–200 CrossRef CAS PubMed
- E. P. Hamilton, C. Ma, M. De Laurentiis, H. Iwata, S. A. Hurvitz, S. A. Wander, M. Danso, D. R. Lu, J. P. Smith, Y. Liu, L. Tran, S. Anderson and M. Campone, Future Oncol., 2024, 20, 2447–2455 CrossRef PubMed
- S. M. Gough, J. J. Flanagan, J. Teh, M. Andreoli, E. Rousseau, M. Pannone, M. Bookbinder, R. Willard, K. Davenport, E. Bortolon, G. Cadelina, D. Gordon, J. Pizzano, J. Macaluso, L. Soto, J. Corradi, K. Digianantonio, I. Drulyte, A. Morgan, C. Quinn, M. Békés, C. Ferraro, X. Chen, G. Wang, H. Dong, J. Wang, D. R. Langley, J. Houston, R. Gedrich and I. C. Taylor, Clin. Cancer Res., 2024, 30, 3549–3563 CrossRef CAS PubMed
- L. B. Snyder, T. K. Neklesa, R. R. Willard, D. A. Gordon, J. Pizzano, N. Vitale, K. Robling, M. A. Dorso, W. Moghrabi, S. Landrette, R. Gedrich, S. H. Lee, I. C. A. Taylor and J. G. Houston, Mol. Cancer Ther., 2025, 24, 511–522 CrossRef CAS PubMed
- Q. H. Chen, E. Munoz and D. Ashong, Cancers, 2024, 16, 633 CrossRef PubMed
- B. A. I. Thomas, H. L. Lewis, D. H. Jones and S. E. Ward, Biomolecules, 2023, 13, 1164 CrossRef CAS PubMed
- M. C. Silva, G. Nandi, K. A. Donovan, Q. Cai, B. C. Berry, R. P. Nowak, E. S. Fischer, N. S. Gray, F. M. Ferguson and S. J. Haggarty, Front. Cell. Neurosci., 2022, 16, 801179 CrossRef CAS PubMed
- M. C. Silva, F. M. Ferguson, Q. Cai, K. A. Donovan, G. Nandi, D. Patnaik, T. Zhang, H.-T. Huang, D. E. Lucente, B. C. Dickerson, T. J. Mitchison, E. S. Fischer, N. S. Gray and S. J. Haggarty, eLife, 2019, 8, e45457 CrossRef PubMed
- W. Wang, Q. Zhou, T. Jiang, S. Li, J. Ye, J. Zheng, X. Wang, Y. Liu, M. Deng, D. Ke, Q. Wang, Y. Wang and J. Z. Wang, Theranostics, 2021, 11, 5279–5295 CrossRef CAS PubMed
- C. Zhang, X. Sun, P. Song and Y. Rao, Bioorg. Med. Chem., 2024, 115, 117967 CrossRef CAS PubMed
- J. Huang, Z. Ma, Z. Yang, Z. He, J. Bao, X. Peng, Y. Liu, T. Chen, S. Cai, J. Chen and Z. Zeng, Eur. J. Med. Chem., 2023, 259, 115664 CrossRef CAS PubMed
- J. Huang, Z. Ma, X. Peng, Z. Yang, Y. Wu, G. Zhong, T. Ouyang, Z. Chen, Y. Liu, Q. Wang, J. Chen, T. Chen and Z. Zeng, J. Med. Chem., 2024, 67, 2438–2465 CrossRef CAS PubMed
- C. Zhu, Z. Yang, Y. Zhang, Z. Li, G. Li, B. Yang, N. Kang, J. Wang, Y. Sun, N. Ding, Y. Rao and W. Liu, Cell Discovery, 2024, 10, 82 CrossRef CAS PubMed
- A. Hanzl and G. E. Winter, Curr. Opin. Chem. Biol., 2020, 56, 35–41 CrossRef CAS PubMed
- L. Z. Benet, C. M. Hosey, O. Ursu and T. I. Oprea, Adv. Drug Delivery Rev., 2016, 101, 89–98 CrossRef CAS PubMed
- E. G. Weagel, J. M. Foulks, A. Siddiqui and S. L. Warner, Med. Chem. Res., 2022, 31, 1068–1087 CrossRef CAS
- J. M. Sasso, R. Tenchov, D. S. Wang, L. S. Johnson, X. Wang and Q. A. Zhou, Biochemistry, 2023, 62, 601–623 CrossRef CAS PubMed
- M. Konstantinidou and M. R. Arkin, Cell Chem. Biol., 2024, 31, 1064–1088 CrossRef CAS PubMed
- E. S. Toriki, J. W. Papatzimas, K. Nishikawa, D. Dovala, A. O. Frank, M. J. Hesse, D. Dankova, J. G. Song, M. Bruce-Smythe, H. Struble, F. J. Garcia, S. M. Brittain, A. C. Kile, L. M. McGregor, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, ACS Cent. Sci., 2023, 9, 915–926 CrossRef CAS PubMed
- Q. He, X. Zhao, D. Wu, S. Jia, C. Liu, Z. Cheng, F. Huang, Y. Chen, T. Lu and S. Lu, Eur. J. Med. Chem., 2023, 260, 115741 CrossRef CAS PubMed
- S. Xie, J. Zhu, J. Li, F. Zhan, H. Yao, J. Xu and S. Xu, J. Med. Chem., 2023, 66, 10917–10933 CrossRef CAS PubMed
- D. E. Gerber, D. R. Camidge, D. Morgensztern, J. Cetnar, R. J. Kelly, S. S. Ramalingam, D. R. Spigel, W. Jeong, P. P. Scaglioni, S. Zhang, M. Li, D. T. Weaver, L. Vaikus, M. Keegan, J. C. Horobin and T. F. Burns, Lung Cancer, 2020, 139, 60–67 CrossRef PubMed
- S. Tiede, N. Meyer-Schaller, R. K. R. Kalathur, R. Ivanek, E. Fagiani, P. Schmassmann, P. Stillhard, S. Häfliger, N. Kraut, N. Schweifer, I. C. Waizenegger, R. Bill and G. Christofori, Oncogenesis, 2018, 7, 73 CrossRef PubMed
- A. J. Wiemer, S. A. Wernimont, T. D. Cung, D. A. Bennin, H. E. Beggs and A. Huttenlocher, Biochem. Pharmacol., 2013, 86, 770–781 CrossRef CAS PubMed
- I. Tanjoni, C. Walsh, S. Uryu, A. Tomar, J. O. Nam, A. Mielgo, S. T. Lim, C. Liang, M. Koenig, C. Sun, N. Patel, C. Kwok, G. McMahon, D. G. Stupack and D. D. Schlaepfer, Cancer Biol. Ther., 2010, 9, 762–775 Search PubMed
- R. P. Law, J. Nunes, C. W. Chung, M. Bantscheff, K. Buda, H. Dai, J. P. Evans, A. Flinders, D. Klimaszewska, A. J. Lewis, M. Muelbaier, P. Scott-Stevens, P. Stacey, C. J. Tame, G. F. Watt, N. Zinn, M. A. Queisser, J. D. Harling and A. B. Benowitz, Angew. Chem., Int. Ed., 2021, 60, 23327–23334 CrossRef CAS PubMed
- P. M. Cromm, K. T. G. Samarasinghe, J. Hines and C. M. Crews, J. Am. Chem. Soc., 2018, 140, 17019–17026 CrossRef CAS PubMed
- H. Gao, Y. Wu, Y. Sun, Y. Yang, G. Zhou and Y. Rao, ACS Med. Chem. Lett., 2020, 11, 1855–1862 CrossRef CAS PubMed
- J. Popow, H. Arnhof, G. Bader, H. Berger, A. Ciulli, D. Covini, C. Dank, T. Gmaschitz, P. Greb, J. Karolyi-Özguer, M. Koegl, D. B. McConnell, M. Pearson, M. Rieger, J. Rinnenthal, V. Roessler, A. Schrenk, M. Spina, S. Steurer, N. Trainor, E. Traxler, C. Wieshofer, A. Zoephel and P. Ettmayer, J. Med. Chem., 2019, 62, 2508–2520 CrossRef CAS PubMed
- V. Laszlo, Z. Valko, J. Ozsvar, I. Kovacs, T. Garay, M. Hoda, T. Klikovits, P. Stockhammer, C. Aigner, M. Gröger, W. Klepetko, W. Berger, M. Grusch, J. Tovari, I. Waizenegger, B. Dome and B. Hegedus, J. Mol. Med., 2019, 97, 231–242 CrossRef CAS PubMed
- H. Gao, C. Zheng, J. Du, Y. Wu, Y. Sun, C. Han, K. Kee and Y. Rao, Protein Cell, 2020, 11, 534–539 CrossRef CAS PubMed
- M. He, W. Lv and Y. Rao, Front. Cell Dev. Biol., 2021, 9, 685106 CrossRef PubMed
- X. J. Pang, X. J. Liu, Y. Liu, W. B. Liu, Y. R. Li, G. X. Yu, X. Y. Tian, Y. B. Zhang, J. Song, C. Y. Jin and S. Y. Zhang, Molecules, 2021, 26, 4250 CrossRef CAS PubMed
- D. Dauch, R. Rudalska, G. Cossa, J. C. Nault, T. W. Kang, T. Wuestefeld, A. Hohmeyer, S. Imbeaud, T. Yevsa, L. Hoenicke, T. Pantsar, P. Bozko, N. P. Malek, T. Longerich, S. Laufer, A. Poso, J. Zucman-Rossi, M. Eilers and L. Zender, Nat. Med., 2016, 22, 744–753 CrossRef CAS PubMed
- X. Qiu, N. Boufaied, T. Hallal, A. Feit, A. de Polo, A. M. Luoma, W. Alahmadi, J. Larocque, G. Zadra, Y. Xie, S. Gu, Q. Tang, Y. Zhang, S. Syamala, J. H. Seo, C. Bell, E. O'Connor, Y. Liu, E. M. Schaeffer, R. Jeffrey Karnes, S. Weinmann, E. Davicioni, C. Morrissey, P. Cejas, L. Ellis, M. Loda, K. W. Wucherpfennig, M. M. Pomerantz, D. E. Spratt, E. Corey, M. L. Freedman, X. Shirley Liu, M. Brown, H. W. Long and D. P. Labbé, Nat. Commun., 2022, 13, 2559 CrossRef CAS PubMed
- S. Fletcher and E. V. Prochownik, Biochim. Biophys. Acta, Gene Regul. Mech., 2015, 1849, 525–543 CrossRef CAS PubMed
- H. Wang, L. Sharma, J. Lu, P. Finch, S. Fletcher and E. V. Prochownik, Onco Targets Ther, 2015, 6, 15857–15870 Search PubMed
- J. L. Yap, J. Chauhan, K.-Y. Jung, L. Chen, E. V. Prochownik and S. Fletcher, Med. Chem. Commun., 2012, 3, 541–551 RSC
- J. L. Yap, H. Wang, A. Hu, J. Chauhan, K. Y. Jung, R. B. Gharavi, E. V. Prochownik and S. Fletcher, Bioorg. Med. Chem. Lett., 2013, 23, 370–374 CrossRef CAS PubMed
- K. Y. Jung, H. Wang, P. Teriete, J. L. Yap, L. Chen, M. E. Lanning, A. Hu, L. J. Lambert, T. Holien, A. Sundan, N. D. P. Cosford, E. V. Prochownik and S. Fletcher, J. Med. Chem., 2015, 58, 3002–3024 CrossRef CAS PubMed
- S. Shukla, S. Fletcher, J. Chauhan, V. Chalfant, C. Riveros, Y. Mackeyev, P. K. Singh, S. Krishnan, T. Osumi and K. C. Balaji, Cancer Gene Ther., 2022, 29, 1550–1557 CrossRef CAS PubMed
- S. Shukla, C. Riveros, M. Al-Toubat, J. Chardon-Robles, T. Osumi, S. Serrano, A. M. Kase, J. L. Petit, N. Meurice, J. Gleba, J. A. Copland, J. Chauhan, S. Fletcher and K. C. Balaji, Cancers, 2023, 15, 3851 CrossRef CAS PubMed
- N. T. Jacob, P. O. Miranda, R. J. Shirey, R. Gautam, B. Zhou, M. E. de Orbe Izquierdo, M. S. Hixon, J. R. Hart, L. Ueno, P. K. Vogt and K. D. Janda, Bioorg. Med. Chem., 2018, 26, 4234–4239 CrossRef CAS PubMed
- J. Guo, R. A. Parise, E. Joseph, M. J. Egorin, J. S. Lazo, E. V. Prochownik and J. L. Eiseman, Cancer Chemother. Pharmacol., 2009, 63, 615–625 CrossRef CAS PubMed
- D. M. Clausen, J. Guo, R. A. Parise, J. H. Beumer, M. J. Egorin, J. S. Lazo, E. V. Prochownik and J. L. Eiseman, J. Pharmacol. Exp. Ther., 2010, 335, 715–727 CrossRef CAS PubMed
- T. B. Sells, R. Chau, J. A. Ecsedy, R. E. Gershman, K. Hoar, J. Huck, D. A. Janowick, V. J. Kadambi, P. J. Leroy, M. Stirling, S. G. Stroud, T. J. Vos, G. S. Weatherhead, D. R. Wysong, M. Zhang, S. K. Balani, J. B. Bolen, M. G. Manfredi and C. F. Claiborne, ACS Med. Chem. Lett., 2015, 6, 630–634 CrossRef CAS PubMed
- T. Shimomura, S. Hasako, Y. Nakatsuru, T. Mita, K. Ichikawa, T. Kodera, T. Sakai, T. Nambu, M. Miyamoto, I. Takahashi, S. Miki, N. Kawanishi, M. Ohkubo, H. Kotani and Y. Iwasawa, Mol. Cancer Ther., 2010, 9, 157–166 CrossRef CAS PubMed
- W. C. Gustafson, J. G. Meyerowitz, E. A. Nekritz, J. Chen, C. Benes, E. Charron, E. F. Simonds, R. Seeger, K. K. Matthay, N. T. Hertz, M. Eilers, K. M. Shokat and W. A. Weiss, Cancer Cell, 2014, 26, 414–427 CrossRef CAS PubMed
- M. Amin, S. E. Minton, P. M. Lorusso, S. S. Krishnamurthi, C. A. Pickett, J. Lunceford, D. Hille, D. Mauro, M. N. Stein, A. Wang-Gillam, L. Trull and A. C. Lockhart, Invest. New Drugs, 2016, 34, 84–95 CrossRef CAS PubMed
- H. Beltran, C. Oromendia, D. C. Danila, B. Montgomery, C. Hoimes, R. Z. Szmulewitz, U. Vaishampayan, A. J. Armstrong, M. Stein, J. Pinski, J. M. Mosquera, V. Sailer, R. Bareja, A. Romanel, N. Gumpeni, A. Sboner, E. Dardenne, L. Puca, D. Prandi, M. A. Rubin, H. I. Scher, D. S. Rickman, F. Demichelis, D. M. Nanus, K. V. Ballman and S. T. Tagawa, Clin. Cancer Res., 2019, 25, 43–51 CrossRef CAS PubMed
- K. R. Kelly, T. C. Shea, A. Goy, J. G. Berdeja, C. B. Reeder, K. T. McDonagh, X. Zhou, H. Danaee, H. Liu, J. A. Ecsedy, H. Niu, E. Benaim and S. Padmanabhan Iyer, Invest. New Drugs, 2014, 32, 489–499 CrossRef CAS PubMed
- D. A. Sloane, M. Z. Trikic, M. L. H. Chu, M. B. A. C. Lamers, C. S. Mason, I. Mueller, W. J. Savory, D. H. Williams and P. A. Eyers, ACS Chem. Biol., 2010, 5, 563–576 CrossRef CAS PubMed
- A. Pflug, T. Maia de Oliveira, D. Bossemeyer and A. R. Engh, Biochem. J., 2011, 440, 85–93 CrossRef CAS PubMed
- B. Adhikari, J. Bozilovic, M. Diebold, J. D. Schwarz, J. Hofstetter, M. Schröder, M. Wanior, A. Narain, M. Vogt, N. Dudvarski Stankovic, A. Baluapuri, L. Schönemann, L. Eing, P. Bhandare, B. Kuster, A. Schlosser, S. Heinzlmeir, C. Sotriffer, S. Knapp and E. Wolf, Nat. Chem. Biol., 2020, 16, 1179–1188 CrossRef CAS PubMed
- R. Wang, C. Ascanelli, A. Abdelbaki, A. Fung, T. Rasmusson, I. Michaelides, K. Roberts and C. Lindon, Commun. Biol., 2021, 4, 640 CrossRef CAS PubMed
- F. Liu, X. Wang, J. Duan, Z. Hou, Z. Wu, L. Liu, H. Lei, D. Huang, Y. Ren, Y. Wang, X. Li, J. Zhuo, Z. Zhang, B. He, M. Yan, H. Yuan, L. Zhang, J. Yan, S. Wen, Z. Wang and Q. Liu, Adv. Sci., 2022, 9, 2104823 CrossRef CAS PubMed
- J. Bozilovic, L. Eing, B.-T. Berger, B. Adhikari, J. Weckesser, N. B. Berner, S. Wilhelm, B. Kuster, E. Wolf and S. Knapp, Curr. Res. Chem. Biol., 2022, 2, 100032 CrossRef CAS
- M. Rishfi, S. Krols, F. Martens, S.-L. Bekaert, E. Sanders, A. Eggermont, F. De Vloed, J. R. Goulding, M. Risseeuw, J. Molenaar, B. De Wilde, S. Van Calenbergh and K. Durinck, Eur. J. Med. Chem., 2023, 247, 115033 CrossRef CAS PubMed
- E. Sflakidou, B. Adhikari, C. Siokatas, E. Wolf and V. Sarli, ACS Pharmacol. Transl. Sci., 2024, 7, 3488–3501 CrossRef CAS PubMed
- S. E. Nelson, J. R. Tucker, M. G. Prado, L. C. Tierney, S. L. Quigley, A. T. Lumpkin, C. J. Dodd, V. Hasko, S. L. Howie, Aastha, B. K. Ody, J. Yin, J. M. Heemstra and M. Turlington, ChemMedChem, 2025, 20, e202400703 CrossRef CAS
- J. Tang, R. Moorthy, L. E. Hirsch, Ö. Demir, Z. D. Baker, J. A. Naumann, K. F. M. Jones, M. J. Grillo, E. S. Haefner, K. Shi, M. J. Levy, H. B. Gupta, H. Aihara, R. S. Harris, R. E. Amaro, N. M. Levinson and D. A. Harki, Cell Chem. Biol., 2025, 32, 352–362.e10 CrossRef CAS PubMed
- D. Chinn, W. Holland and P. Mack, J. Cancer Res. Clin. Oncol., 2014, 140, 1137–1149 CrossRef CAS PubMed
- M. Michaelis, F. Selt, F. Rothweiler, N. Löschmann, B. Nüsse, W. Dirks, R. Zehner and J. Cinatl, PLoS One, 2014, 9, e108758 CrossRef PubMed
- J. Qi, X. Gao, X. Zhong, N. Zhang, R. Wang, H. Zhang, T. Pan, X. Liu, Y. Yao, Q. Wu, M. Niu and K. Xu, Biomed. Pharmacother., 2019, 117, 109113 CrossRef CAS PubMed
- S. Wang, L. Han, J. Han, P. Li, Q. Ding, Q. J. Zhang, Z. P. Liu, C. Chen and Y. Yu, Nat. Chem. Biol., 2019, 15, 1223–1231 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |