
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,4-Oxazepan-7-one trifluoroacetate: a modular monomer precursor for the synthesis of functional and biodegradable poly(amino esters)†
Tino Mackiola,
Chloé Pascouau a,
Manuel Nagela,
Tamara M. Bizmarka,
Luca Montesela,
Jochen Fischer-Schuchb and
Pol Besenius*a
a,
Manuel Nagela,
Tamara M. Bizmarka,
Luca Montesela,
Jochen Fischer-Schuchb and
Pol Besenius*a
aDepartment of Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55128, Mainz, Germany. E-mail: besenius@uni-mainz.de
bInstitut für Biotechnologie und Wirkstoff-Forschung gGmbH, Hanns-Dieter-Hüsch-Weg 17, 55128 Mainz, Germany
First published on 3rd July 2025
Abstract
N-Acylated poly(amino esters) (PAEs) synthesized via organocatalytic ring-opening polymerization (ROP) offer potential for tailored, functional and degradable polymers. In this study, a universal monomer precursor toward N-acylated-1,4-oxazepan-7-ones (OxP)s was synthesized using a three-step approach, allowing for the introduction of various functional groups. Two novel oxidation sensitive OxP monomers bearing a double bond and a sulfide group were designed, as well as two monomers with alkyl moieties. The organocatalytic ROP of the OxP monomers using 1,8-diazabicyclo[5.4.0]undec-7-en (DBU) and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl thiourea (TU) as catalysts was investigated. Polymerizations were performed under ambient temperature, affording homopolymers with narrow dispersities (Ð = 1.09–1.13). As a proof of concept, a post-polymerization thiol–ene functionalization of the allyl functional PAE was performed via photo-rheology experiments. Finally, the (bio)degradability of the N-acylated poly(amino esters) was evaluated through a series of degradation studies under mild enzymatic catalysis, in neutral phosphate-buffered saline solution and under accelerated conditions.
Introduction
In response to the increasing environmental challenge, degradable polymers, such as aliphatic polyesters or polycarbonates, represent a class of materials that offer a sustainable solution to low-degradable polymers.1,2 These materials can be broken down into non-toxic components over time through hydrolytic and enzymatic degradation mechanisms, making them more eco-friendly and potentially more sustainable.3–10 Poly(amino ester)s (PAEs) represent a class of polymers, with a backbone of repeating units featuring degradable ester bonds along with amine or amide derivatives, enabling the incorporation of various pendant functional side chains.11–16 PEAs are used in biomedical applications as drug or gene delivery vehicles and as materials for wound dressings.17–22 They were found to be non-toxic for cells and their degradability was reported.19,23–25 In addition, the range of applications for these materials can be expanded due to their diverse functionalities and potential for post-modifications, such as crosslinking, coupling, and redox reactions.26–30 PAEs with various pendant groups are widely accessible through step growth polymerization techniques, including polyaddition or polycondenation.11,31–34 Surprisingly, chain-growth polymerization methods such as ring-opening polymerization (ROP) for PAEs are less common.14,35–38 Organocatalytic ROP provides a metal-free and well-established method for the synthesis of well-defined polyesters and polycarbonates under mild reaction conditions.14,35,39–43Wang and Hadjichristidis reported the organocatalytic ROP of the azacaprolactone N-acylated-1,4-oxazepan-7-one (OxP) with a variation of aryl and alkyl substituents.14 The resulting poly(1,4-oxazepan-7-one)s (POxP)s were synthesized with controlled molar masses and narrow dispersities. The water-soluble derivative N-acetyl-1,4-oxazepan-7-one polymer P(OxPMe) showed very slow degradation in phosphate-buffered saline (PBS) solution and was suggested as alternative to non-biodegradable poly(oxazolines). The current literature reported synthetic route and Bayer-Villiger reaction in the final step of the monomer synthesis limits the preparation of a more diverse set of functional OxP monomers other than aryl and alkyl bearing side chains, due to the lability of many functional groups under oxidative conditions. Instead, we hereby report a broadly applicable synthetic approach for the synthesis of a wide range of OxP monomers using 1,4-oxazepan-7-one trifluoroacetate salt (OxPTFA) as a precursor. OxPTFA enables the incorporation of various pendant groups through acylation in the final synthetic step. The synthesis of OxP monomers and their homopolymerization using the DBU/TU organocatalytic system are presented. The reactivity of the different monomers is evaluated by kinetic experiments of the organocatalytic polymerizations, which are analyzed by nuclear magnetic resonance (NMR), size exclusion chromatography (SEC). The (bio)degradability of the water-soluble P(OxPMe) with an acetylated side chain is investigated under mild enzymatic catalysis, in neutral phosphate-buffered saline solution and under accelerated conditions. Finally, a photo-rheology experiment is conducted to study gel formation via thiol–ene cross-linking using the allyl pendant functionalities of PAEs.
Experimental section
Materials
All reagents and solvents were purchased from commercial suppliers and used as received, unless stated otherwise. Dry solvents were obtained from Thermo Fisher Scientific Inc. Deuterated solvents were obtained from Deutero GmbH. 1,8-Diazabicyclo[5.4.0]undec-7-en (DBU) was purified by stirring over CaH2 for 1 hour followed by distillation under reduced pressure and was then stored in the glovebox for use. All operations involving air- and moisture-sensitive chemicals and materials were carried out in flame-dried glassware under argon atmosphere using Schlenk technique or in an argon-filled glovebox (MBraun UNILAB, <0.1 ppm of O2 and <0.1 ppm of H2O).Instrumentation
Synthetic procedures
Synthesis of 1-tert-butoxycarbonyl-4-piperidone (PBoc). 4-Piperidone monohydrate hydrochloride (20.0 g, 0.13 mol, 1.0 eq.) and K2CO3 (21.6 g, 0.16 mol, 1.2 eq.) were dissolved in water (150 mL). Boc2O (31.3 g, 0.14 mmol, 1.1 eq.) was dissolved in THF (50 mL) and added dropwise to the aqueous solution under stirring at ambient temperature. After 16 h, the yellow solution was extracted three times with Et2O. The combined organic layers were dried over Na2SO4, filtered and the solvent removed under reduced pressure to obtain a colorless solid (quantitative yield).
Bayer-Villiger ring-expansion of PBoc (Fig. 1, [2]). mCPBA (43.8 g, 0.20 mol, 1.5 eq.) was dissolved in DCM (200 mL). PBoc (25.9 g, 0.13 mol, 1.0 eq.) dissolved in DCM (50 mL) was added dropwise while cooling. After complete addition, and the reaction stirred at room temperature for 16 h. The formed colorless precipitation was removed and a second portion of mCPBA (9.0 g, 0.04 mol, 0.3 eq.) was added to the reaction solution. After stirring at room temperature for 16 h, the formed colorless precipitate was removed. Saturated sodium thiosulfate solution was added, and the mixture was stirred for 30 min. The organic layer was separated, washed with saturated NaHCO3 solution and water, dried over Na2SO4 and filtered. After removing the solvent under reduced pressure, the crude product was obtained as colorless, slightly yellow solid. Purification by silica gel chromatography (cyclohexane/ethyl acetate = 1/1) yields the desired 4-tert-butoxycarbonyl-1,4-oxazepan-7-one (OxPBoc) (yield = 64%).
 | ||
| Fig. 1 (a) Three-step synthetic route toward 1,4-oxazepan-7-one trifluoroacetate (OxPTFA) starting from 4-piperidone. [1] Boc protection, quantitative yield; [2] Bayer-Villiger ring-expansion, 64%; [2′] oxidation with OXONE®, 80%; [3] Boc-deprotection; 95%. (b) One-step acylation of OxPTFA for the synthesis of four N-acylated-1,4-oxazepan-7-one (OxP) monomers with different side chains and functional groups. | ||
Oxidation of PBoc with OXONE® (Fig. 1, [2′]). PBoc (500 mg, 2.51 mmol, 1.0 eq.) and OXONE® (6066 mg, 17.87 mmol, 7.0 eq.) were dissolved in degassed 2 M phosphate buffer (50 mL, pH = 7). The reaction was stirred for 18 h at room temperature under an argon atmosphere. The solution was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure to obtain OxPBoc as a colorless solid (yield = 80%).
Boc-deprotection of OxPBoc. A solution of TFA in DCM (50% v/v, 25 mL) and OxPBoc (5.0 g, 0.02 mol, 1.0 eq.) were stirred for 45 min at room temperature. The solvent was evaporated under reduced pressure, yielding a yellow oil. Suspending in DCM and precipitating twice in cold Et2O gives a colorless solid. The supernatant was removed, and the solid was dried under reduced pressure to obtain the 1,4-oxazepan-7-one trifluoroacetate salt (OxPTFA) (yield = 95%).
Acylation of OxPTFA. As an example of the acylation of OxPTFA, we provide the experimental details for the synthesis of OxPMe, while all other monomer syntheses and full characterization details are provided in the ESI:† OxPTFA (2.0 g, 8.73 mmol, 1.0 eq.) and K2CO3 (3.6 g, 26.18 mmol, 3.0 eq.) were stirred in DCM (40 mL) at room temperature under an argon atmosphere. Acetyl chloride (1.4 g, 17.46 mmol, 2.0 eq.) was added, and the reaction was stirred for 16 h. The heterogeneous mixture was filtered, and the solvent was removed under reduced pressure to obtain the crude material. The crude material was purified by silica gel chromatography (DCM/methanol = 20/1) (yield = 58%).
Additional methods, experimental conditions and full characterization are provided in the ESI.†
Homopolymer degradation under accelerated conditions. A homopolymer stock solution was prepared by dissolving it in DCM. The stock solution was divided into separate vials (1.5 mL) and the solvent was evaporated under vacuum to form a polymer layer of ≃5 mg in each vial. A solution of sodium hydroxide (NaOH) in methanol (0.005 M) was added to the different vials to reach a final polymer concentration of 10 mg mL−1. The vials were placed in a thermoshaker set at room temperature. After the desired reaction time, the vials were removed from the thermoshaker and the solvent was evaporated under vacuum. The residual product was analyzed by SEC.
Homopolymer degradation under enzymatic conditions and in PBS solution. A similar procedure was employed to prepare a polymer layer (5 mg) in different vials (1.5 mL). A solution of PBS or lipase from Pseudomonas cepacian (LPC, ≥30 U mg−1) in PBS (100 U mL−1) was added to the different vials to reach a final polymer concentration of 10 or 5 mg mL−1. The vials were placed in a thermoshaker set at 37 °C. After the desired reaction time, the vials were removed from the thermoshaker and the solvent was lyophilized. The residual product was analyzed by SEC.
Results and discussion
Synthesis of monomer precursor
OxPTFA was synthesized in three steps starting from 4-piperidone monohydrate hydrochloride with an overall yield of 61% (Fig. 1a). Boc-protection and Bayer-Villiger ring-expansion using meta-chloroperoxybenzoic acid (mCPBA), afforded 4-tert-butoxycarbonyl-1,4-oxazepan-7-one (OxPBoc), which was previously reported as a monomer for ROP by the groups of Hadjichristidis14,35 and Waymouth.36 Using a recently reported greener oxidation procedure by Giraudo et al.,44 we aimed to improve the ring-expansion affording OxPBoc. Using this method, we avoided the use of peroxy acids and carboxylic acid waste products, which are detrimental to the basic ROP methodology if remaining traces are not removed from the OxP building blocks and monomers. Following the OXONE® based procedure, OxPBoc was obtained with a yield of 80% compared to 64% using the mCPBA mediated Bayer-Villiger ring-expansion. Finally, treatment of OxPBoc with a solution of trifluoroacetic acid (TFA) in DCM yielded OxPTFA as a colorless salt after purification. Since ring-opening of 1,4-oxazepan-7-one could potentially occur under acid conditions, we performed kinetic studies to investigate the Boc-deprotection of OxPBoc toward OxPTFA by 1H NMR spectroscopy (Fig. S1†). The complete removal of the Boc-protecting group was confirmed after 30 minutes of reaction time, as observed via the disappearance of the 1H NMR signals of the methyl groups of the Boc-protecting group (δ = 1.41 ppm). No significant side reactions were observed during successful deprotection and a reaction time of up to 1 h, and small amounts (<5%) of impurities were easily removed by precipitation. Only after extended reaction times of 20 h, significant amounts of the ring-open form of OxPTFA, 3-[(2-hydroxyethyl)amino]propanoic acid were observed and confirmed by 1H NMR spectroscopy and ESI-TOF-MS spectrometry. This observation further underlines the robustness of our synthetic route towards a modular OxP monomer platform based on 1,4-oxazepan-7-one trifluoroacetate for the synthesis of functional poly(amino ester) derivatives.Acylation of OxPTFA toward functional monomers
Several OxP monomers were synthesized in a one-step acylation of OxPTFA under alkaline conditions. OxPTFA was reacted with different acyl chlorides to afford the four monomers with yields ranging from 47% to 69% after purification. Purification via silica flush chromatography allowed the isolation of all OxP monomers in high purity. A detailed characterization of the four N-acylated-1,4-oxazepan-7-one monomers via 1H and 13C NMR is provided in Fig. 2, Fig. S2–S3 and S21–S33.† Different moieties, including ethyl methyl sulfide, propylene, butyl, and methyl pendant groups were installed via acylation of the OxPTFA precursor to synthesize a range of OxP monomers (Fig. 1b). 4-(But-3-enoyl)-1,4-oxazepan-7-one (OxPpropylene) bears a double bond in the side chain, which could be further modified post-polymerization, e.g., by addition reactions or crosslinking.45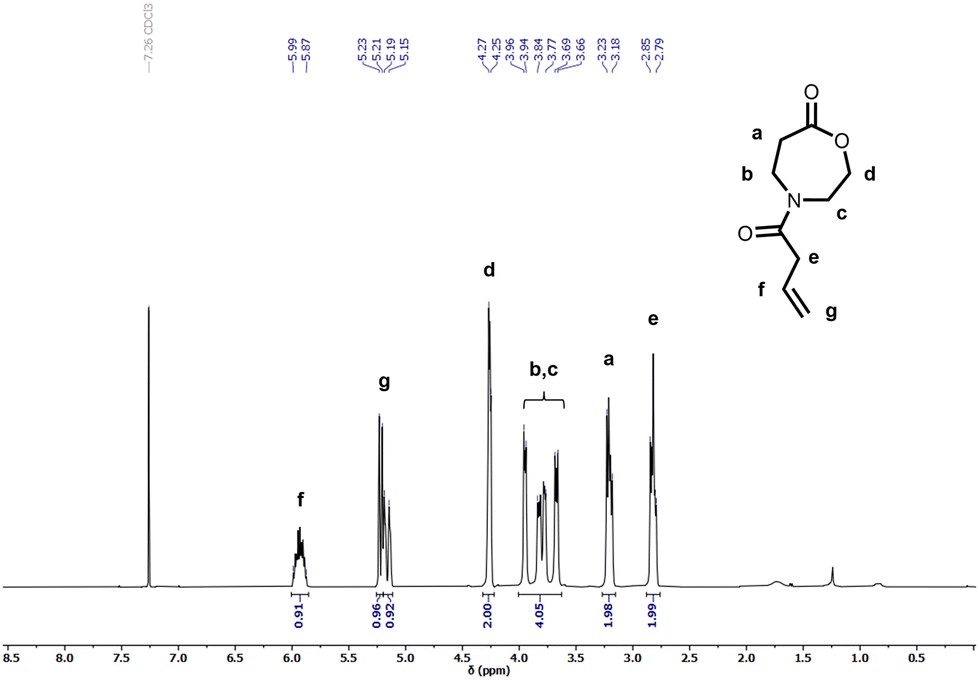 | ||
| Fig. 2 1H NMR spectrum of 4-(But-3-enoyl)-1,4-oxazepan-7-one (OxPpropylene). | ||
4-(3-methylthio)propanoyl-1,4-oxazepan-7-one (OxPEtSMe) contains the analogueous side chain of the amino acid methionine. The thioether group opens the possibility of oxidizing the sulfide to a sulfoxide or sulfone after polymerization, which could impact the hydrophilicity of the polymeric chain.46–54 4-Pentanoyl-1,4-oxazepan-7-one (OxPBut) serves as the aliphatic side chain counterpart to OxPEtSMe. Meanwhile, OxPMe yields a monomer that dissolves readily in water.
Organocatalytic ring-opening homopolymerization of N-acylated-1,4-oxazepan-7-ones
The organocatalytic ROP of the four OxP monomers was carried out at room temperature in DCM using benzyl alcohol (BnOH) as initiator (Scheme 1). The combination of 1,8-diazabicyclo[5.4.0]undec-7-en (DBU) and thiourea catalyst 1-[3,5-bis(trifluoromethyl)phenyl]-3-cyclohexyl thiourea (TU) was used as an organocatalytic system, as it was reported to be efficient for the ROP of polyesters and polycarbonates.14,35,36,41–43,55 Further polymerization parameters were selected as follows: [M]0/[BnOH]0/[DBU]0/[TU]0 = 30/1/3/3; [M]0 = 1 M and t = 80–90 min. The homopolymers were characterized via NMR spectroscopy, SEC and MALDI-TOF MS (Fig. S14–17, Fig. S35–50†). The characterization data for the POxP homopolymers are summarized in Table 1. Polymerizations of the OxPs show high monomer conversions, with values ranging from 89% to complete conversion. The 1H NMR spectrum of P(OxPpropylene) (Table 1, entry 1) is shown as an example for the POxP homopolymers (Fig. 3a). All homopolymers exhibit unimodal distribution, narrow dispersity values determined via SEC (Ð = 1.09–1.13), and good to excellent agreement of the number average molar mass calculated from the 1H NMR spectra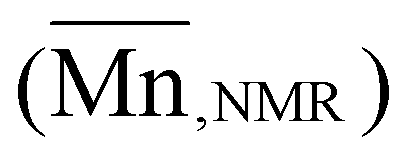 with the theoretical molar masses
with the theoretical molar masses 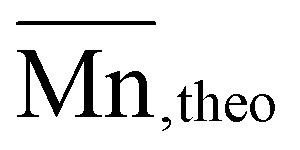 (Table 1). These results are indicative of well controlled chain-growth polymerization. The polymers were further characterized by MALDI-TOF-MS (Fig. S14–17†). The resulting spectra show benzyl alcohol-initiated polymer species for the POxPMe, POxPBut and POxPpropylene homopolymers, with potassium as counter ion and the corresponding Δ m/z of the individual monomer mass.
(Table 1). These results are indicative of well controlled chain-growth polymerization. The polymers were further characterized by MALDI-TOF-MS (Fig. S14–17†). The resulting spectra show benzyl alcohol-initiated polymer species for the POxPMe, POxPBut and POxPpropylene homopolymers, with potassium as counter ion and the corresponding Δ m/z of the individual monomer mass.
 | ||
| Scheme 1 Organocatalytic ROP of N-acylated-1,4-oxazepan-7-ones. | ||
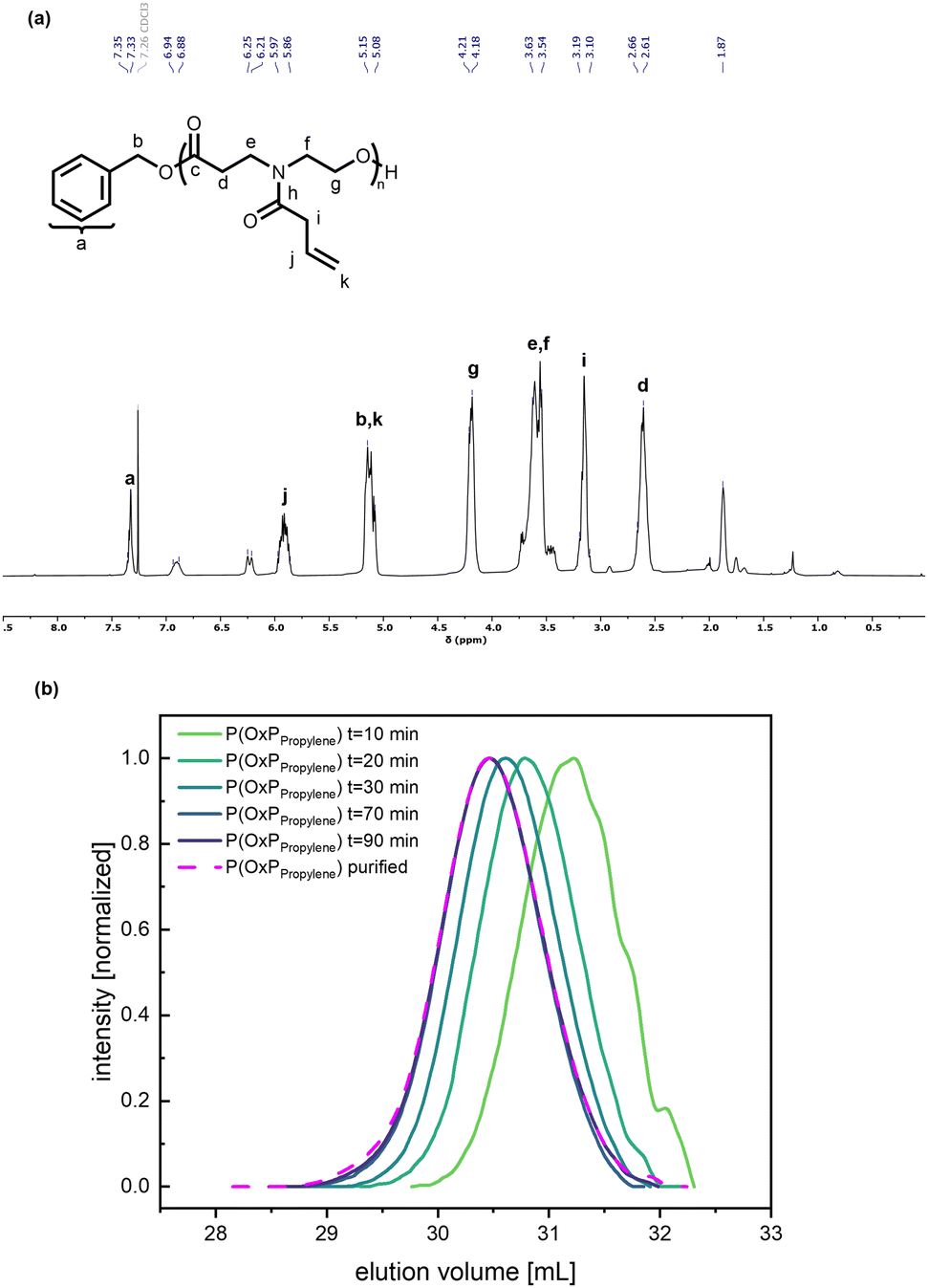 | ||
| Fig. 3 (a) 1H NMR spectrum of P(OxPpropylene) with assigned signals. (b) SEC elution traces of OxPpropylene organocatalytic ROP after given reaction times (green to purple gradient, solid lines). Purified P(OxPpropylene) sample from the t = 80 min sample (pink, dashed line). RI signal; eluent: THF, 25 °C; standard: PMMA. | ||
| Entry | Monomer (M) | Reaction time [min] | Conv.d [%] |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e [g mol−1] e [g mol−1] |
f [g mol−1] |
g [g mol−1] |
Ðg |
|---|---|---|---|---|---|---|---|
| a r.t.; [M]0/[BnOH]0/[DBU]0/[TU]0 = 30/1/3/3; [M]0 = 1 M.b Solvent: dichloromethane.c Solvent: toluene.d Conversion was calculated from 1H NMR spectra of the crude products, using characteristic signals for monomers and polymers, calculated as follows: [M]t/([M]t + [P]t).e Theoretical number-average molar mass, calculated from feed ratio and monomer conversion from crude 1H NMR spectrum as follows: ([M]0/[BnOH]0) × conversion × (M.W. of OxP) + (M.W. BnOH).f Calculated from1H NMR spectrum of isolated product, using signal integrals of the initiator (BnOH) and the polymer.g Determined via SEC analysis (RI signal, eluent: THF, 25 °C, standard: PMMA).h Determined via SEC analysis (RI signal, eluent: DMF, 50 °C, standard: PMMA). | |||||||
| 1 | OxPpropylenea,b |
90 | 100 | 5600 | 4600 | 2400 | 1.10 |
| 2 | OxPButa,b |
90 | 89 | 5400 | 4500 | 3900 | 1.09 |
| 3 | OxPEtSMea,b |
80 | 99 | 6500 | 5200 | 2300 | 1.10 |
| 4 | OxPMea,b |
80 | 100 | 4800 | 4700 | 2500 h | 1.13 h |
| 5 | OxPButa,c |
100 | 94 | 5700 | 5300 | 6000 | 1.09 |

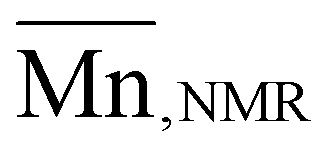

The DSC analysis of the four OxP homopolymers yielded distinct glass transition temperatures (Tg) (Fig. 4). The amorphous character of POxPs is attributed to the irregular orientation of the amide bonds in the polymer backbone.14 The lowest Tg was measured for POxPBut (−13 °C), while the highest Tg was measured for POxPMe (5 °C). This observation is consistent with our expectations, as the Tg decreases with increasing aliphatic side chain length for POxPMe compared to POxPBut.
 | ||
| Fig. 4 DSC thermogram of POxP homopolymers: POxPBut, POxPEtSMe, POxPpropylene, POxPMe. | ||
The well controlled polymerization of OxP monomers allowed the synthesis of a diblock copolymer P(OxPMe)25-b-P(OxPBoc)10 using similar reaction conditions. The synthesis proceeded by polymerization of the OxPMe monomer, followed by addition and polymerization of OxPBoc after complete consumption of the first monomer. The 1H NMR spectrum of the block copolymer (Fig. S51†) showed the presence of both OxPMe and OxPBoc repeating units, which was also confirmed by MALDI-TOF MS (Fig. S52†). The molar mass of the block copolymer determined by NMR  = 5400 g mol−1, is also in good agreement with the theoretical molar mass
= 5400 g mol−1, is also in good agreement with the theoretical molar mass  (Table S2†). Finally, characterization by SEC revealed the synthesis of the diblock copolymer P(OxPMe)25-b-P(OxPBoc)10 with a low dispersity Ð = 1.1 (Fig. S53†).
(Table S2†). Finally, characterization by SEC revealed the synthesis of the diblock copolymer P(OxPMe)25-b-P(OxPBoc)10 with a low dispersity Ð = 1.1 (Fig. S53†).
Kinetic studies of organocatalytic ROP
Kinetic investigations of the organocatalytic ROP for the four OxP monomers with DBU/TU as catalysts were carried out under the previously mentioned reaction conditions. Aliquots of the running polymerization were analyzed via 1H NMR spectroscopy and SEC (Fig. 3, Fig. 5, and Fig. S4–13†). The elugrams of the P(OxPProylene) polymerization (Fig. 3b) at each measured time point of the polymerization reactions are unimodal and give narrow dispersities (Ð < 1.1). As illustrated in Fig. S13,† increases linearly with the monomer conversion.
increases linearly with the monomer conversion.
 | ||
| Fig. 5 Kinetic plot of organocatalytic ROP of OxPs in DCM. Semilogarithmic plots over the reaction time (dots with varying shapes and colors for every OxP homopolymer), fitted by linear regression (line with varying color for every OxP homopolymer). | ||
The semilogarithmic plots of monomer concentration versus reaction time follow a linear trend, indicating first-order kinetics for the polymerizations of the four monomers (Fig. 5). OxPBut has the lowest reactivity, followed by slightly more reactive OxPMe. Polymerization kinetics for OxPBut were also conducted in toluene and were found to be slightly faster than in DCM. Given that not all the monomers are soluble in toluene, DCM was the solvent of choice for all the polymerizations. The reactivities of OxPEtSMe and OxPpropylene were found to be higher than those of OxPMe and OxPBut. The group of Hadjichristidis suggested that aromatic side chains in OxP monomers increase the basicity of the lactone functionality and efficiency of catalyst activation compared to the aliphatic side chains.14 However in our case, electronic effects are less likely to have a similar impact in the case of OxPEtSMe and OxPpropylene compared to OxPMe and OxPBut.
Homopolymer degradation in organic and aqueous media
The degradability of POxPs was evaluated by performing degradation studies under accelerated and enzymatic conditions,56,57 in accordance with previous studies.58–60 The degradation tests were carried out using the water-soluble P(OxPMe) with a molar mass of 3500 g mol−1 and a dispersity of Ð = 1.1. Degradation under accelerated conditions was performed at room temperature (25 °C) using a polymer concentration of 10 mg mL−1 in a low-concentrated solution of NaOH in methanol (0.005 m). As shown in Fig. S18,† the initial homopolymer is almost completely degraded in short reaction times (5–60 min), showing a shift toward lower molar masses (
of 3500 g mol−1 and a dispersity of Ð = 1.1. Degradation under accelerated conditions was performed at room temperature (25 °C) using a polymer concentration of 10 mg mL−1 in a low-concentrated solution of NaOH in methanol (0.005 m). As shown in Fig. S18,† the initial homopolymer is almost completely degraded in short reaction times (5–60 min), showing a shift toward lower molar masses ( = 420 g mol−1, or 88% decrease in
= 420 g mol−1, or 88% decrease in 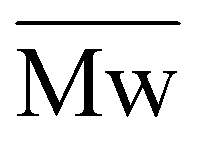 ). Only low molar mass species with a multimodal distribution remain after the test.
). Only low molar mass species with a multimodal distribution remain after the test.
Additionally, degradation studies were performed under enzymatic catalysis conditions at 37 °C using lipase from Pseudomonas cepacian (LPC) in PBS buffer (100 U mL−1). After 15 days and at a polymer concentration of 10 mg mL−1, the formation of low molar mass species is observed, resulting in a decrease in molar mass to  = 2700 g mol−1 (or a 23% decrease in
= 2700 g mol−1 (or a 23% decrease in  ) and a broadening of the dispersity (Fig. S19†). The main polymer species remains present in solution, indicating a slow degradation process. By lowering the polymer concentration to 5 mg mL−1, the molar mass is further decreased to
) and a broadening of the dispersity (Fig. S19†). The main polymer species remains present in solution, indicating a slow degradation process. By lowering the polymer concentration to 5 mg mL−1, the molar mass is further decreased to  = 1400 g mol−1 (or 60% decrease in
= 1400 g mol−1 (or 60% decrease in  ), followed by a broadening of the dispersity (Fig. 6 and Table S1†). Under these conditions, a significant increase in the relative fraction of the lower molar mass species is observed, which confirms faster degradation at a lower polymer concentration and fixed amount of lipase. Control degradation experiments of P(OxPMe) in PBS after 15 days also revealed slight degradation with the formation of lower molar mass species. Further investigation was conducted on a P(OxPMe) precursor (
), followed by a broadening of the dispersity (Fig. 6 and Table S1†). Under these conditions, a significant increase in the relative fraction of the lower molar mass species is observed, which confirms faster degradation at a lower polymer concentration and fixed amount of lipase. Control degradation experiments of P(OxPMe) in PBS after 15 days also revealed slight degradation with the formation of lower molar mass species. Further investigation was conducted on a P(OxPMe) precursor ( of 3900 g mol−1 and Ð = 1.1) to determine the potential degradability of the polymer in PBS at 37 °C, closer to physiological conditions (Fig. S20†). After 50 days of reaction, an increase in the fraction of low molar mass species was observed, as well as an increase in dispersity (
of 3900 g mol−1 and Ð = 1.1) to determine the potential degradability of the polymer in PBS at 37 °C, closer to physiological conditions (Fig. S20†). After 50 days of reaction, an increase in the fraction of low molar mass species was observed, as well as an increase in dispersity ( = 2100 g mol−1, or 46% decrease in
= 2100 g mol−1, or 46% decrease in  ). In summary, the amide linkages in the side chains of the poly(amino ester)s likely increase their hydrolytic stability, but decrease the potential for pH-responsiveness behaviour in PAEs with alkyl side chains.16 Monomer and polymer modification strategies that open opportunities for pH-dependent degradation of PAEs are currently under investigation.
). In summary, the amide linkages in the side chains of the poly(amino ester)s likely increase their hydrolytic stability, but decrease the potential for pH-responsiveness behaviour in PAEs with alkyl side chains.16 Monomer and polymer modification strategies that open opportunities for pH-dependent degradation of PAEs are currently under investigation.
 | ||
| Fig. 6 SEC elution traces of P(OxPMe) degradation under enzymatic conditions: [P(OxPMe)] = 5 mg mL−1, [LPC] = 100 U mL−1, PBS, 37 °C (DMF, standard: PMMA). | ||
Post-polymerization modification of PAE
The diverse functionalities conferred by the PAE pendant chains pave the way for post-modification reactions and tailored materials properties. As a proof of concept, a gelation experiment was performed by thiol–ene coupling using the POxPpropylene homepolymer bearing allyl functional groups in the pendent side chains. Similar to previous gelation studies on poly(oxazoline),61,62 a photo-induced cross-linking experiment was conducted by reacting POxPpropylene with dithiothreitol (DTT) cross-linker using Irgacure-2959 as photoinitiator. The experiment was carried out with an equimolar ratio of thiol to ene in THF at 30 °C to obtain a final 10% (w/v) gel. As shown in Fig. 7, no viscoelastic behavior is visible before irradiation (0–10 min). When UV light is applied (λ = 365 nm, 10 min), G′ increases and crosses G′′ within 30 s, indicating gel formation. The thiol–ene reaction takes place when UV light is applied, thus crosslinking of the polymers occurs with DTT. As a result, a polymer network is formed as observed in the rheological experiment. The POxPpropylene network exhibits a storage modulus G′ of about 3000 Pa, which is a similar gel strength as obtained in the case of the poly(oxazoline)-based network reported in the literature.61,62 These experiments confirm the successful functionalization of POxPpropylene via photo-induced thiol-ene cross-linking, and potential for post-polymerization modification of functional PAEs in general. | ||
| Fig. 7 Storage and loss moduli (G′ and G′′) as a function of time for POxPpropylene photo-induced cross-linking with DTT before and after irradiation (10 min) with 365 nm UV light. | ||
Conclusions
A three-step approach involving Boc-protection, Bayer-Villiger ring-expansion or oxidation with OXONE®, followed by Boc-deprotection was developed to synthesize a universal monomer precursor, OxPTFA, for the synthesis of new functional monomers. A one-step acylation of OxPTFA enabled the synthesis of four monomers with yields ranging from 47 to 69% and various pendant groups, including butyl, propylene, ethyl methyl sulfide, and methyl side chains.The organocatalytic ROP of the four monomers at room temperature using DBU and TU as catalysts, yielded polymers with high monomer conversions (≥ 89%) and low dispersities Ð ≤ 1.16. Kinetic studies of the homopolymerizations exhibited a linear increase in the  with the monomer conversion and first-order kinetics for all four monomers. Furthermore, a photo-rheology experiment involving a thiol–ene cross-linking reaction of POxPpropylene showed that pendant allyl functionalities of PAEs can be used for post-polymerization reactions to open avenues in material functionalization. Finally, degradation studies of POxPMe under accelerated conditions revealed near-complete degradation of the polymer. Conducting the degradation in PBS solution and at body temperature allowed a partial degradation of the polymer, with the formation of species in the low molar mass region. The use of LPC as enzyme promoted the degradation of the polymer, by increasing the fraction of smaller species formed.
with the monomer conversion and first-order kinetics for all four monomers. Furthermore, a photo-rheology experiment involving a thiol–ene cross-linking reaction of POxPpropylene showed that pendant allyl functionalities of PAEs can be used for post-polymerization reactions to open avenues in material functionalization. Finally, degradation studies of POxPMe under accelerated conditions revealed near-complete degradation of the polymer. Conducting the degradation in PBS solution and at body temperature allowed a partial degradation of the polymer, with the formation of species in the low molar mass region. The use of LPC as enzyme promoted the degradation of the polymer, by increasing the fraction of smaller species formed.
The development of this three-step synthetic route toward functional monomers provides new opportunities in the design of functional and degradable poly(amino ester) polymers, including blocked architectures. The ability to introduce diverse functionalities opens the way for post-polymerization reactions and enables fine-tuning of material properties, thereby opening avenues in a range of applications.
Author contributions
Tino Mackiol: conceptualization, data curation, investigation, methodology, validation, visualization, writing – original draft, review & editing; Chloé Pascouau: data curation, investigation, methodology, validation, visualization, review & editing; Manuel Nagel: investigation; Tamara M. Bizmark: investigation; Luca Montesel: investigation; Jochen Fischer-Schuch: conceptualization, methodology, validation, review&editing; Pol Besenius: conceptualization, validation, funding acquisition, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI (Fig. S1–S50†).Acknowledgements
We are grateful for financial support by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (ERC CoG SUPRAVACC 819856, T. M., C. P., P. B.). Funding from the DFG (Deutsche Forschungsgemeinschaft) is acknowledged: T. M. B. and P. B. are members of the SFB 1552 (project no. 465145163). Support by the State of Rhineland-Palatinate with the center SusInnoScience is highly appreciated. We also thank the group of Prof. Holger Frey (Johannes Gutenberg University Mainz) for granting us access to their experimental set-up to perform SEC and DSC experiments.References
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- A. Larrañaga and E. Lizundia, Eur. Polym. J., 2019, 121, 109296 CrossRef.
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS PubMed.
- Y. Xu, F. Zhou, D. Zhou, J. Mo, H. Hu, L. Lin, J. Wu, M. Yu, M. Zhang and H. Chen, J. Biobased Mater. Bioenergy, 2020, 14, 155–168 CrossRef CAS.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- M. Sander, M. Weber, C. Lott, M. Zumstein, A. Künkel and G. Battagliarin, in Synthetic Biodegradable and Biobased Polymers: Industrial Aspects and Technical Products, ed. A. Künkel, G. Battagliarin, M. Winnacker, B. Rieger and G. Coates, Springer International Publishing, Cham, 2024, pp. 65–110, DOI:10.1007/12_2023_163.
- K. O. Siegenthaler, M. Agari, J. Auffermann, M. P. Barth, G. Battagliarin, A. Künkel, F. Lauer, J. Lohmann, A. Nabifar and M. Yamamoto, in Synthetic Biodegradable and Biobased Polymers: Industrial Aspects and Technical Products, ed. A. Künkel, G. Battagliarin, M. Winnacker, B. Rieger and G. Coates, Springer International Publishing, Cham, 2024, pp. 111–175, DOI:10.1007/12_2023_151.
- A. Denk and B. Rieger, in Synthetic Biodegradable and Biobased Polymers: Industrial Aspects and Technical Products, ed. A. Künkel, G. Battagliarin, M. Winnacker, B. Rieger and G. Coates, Springer International Publishing, Cham, 2024, pp. 177–195, DOI:10.1007/12_2022_116.
- S. Kato, T. Ueda, T. Aoshima, N. Kosaka and S. Nitta, in Synthetic Biodegradable and Biobased Polymers: Industrial Aspects and Technical Products, ed. A. Künkel, G. Battagliarin, M. Winnacker, B. Rieger and G. Coates, Springer International Publishing, Cham, 2024, pp. 269–304, DOI:10.1007/12_2023_159.
- S. Gokhale, Y. Xu and A. Joy, Biomacromolecules, 2013, 14, 2489–2493 CrossRef CAS PubMed.
- W. Cheng, D. Wu and Y. Liu, Biomacromolecules, 2016, 17, 3115–3126 CrossRef CAS.
- Y. Liu, Y. Li, D. Keskin and L. Shi, Adv. Healthcare Mater., 2019, 8, 1801359 CrossRef.
- X. Wang and N. Hadjichristidis, ACS Macro Lett., 2020, 9, 464–470 CrossRef CAS PubMed.
- X. Wang, Z. Zhang and N. Hadjichristidis, Prog. Polym. Sci., 2023, 136, 101634 CrossRef CAS.
- M. K. Kuenen, K. S. Reilly and R. A. Letteri, ACS Macro Lett., 2023, 12, 1416–1422 CrossRef CAS.
- D. G. Anderson, D. M. Lynn and R. Langer, Angew. Chem., Int. Ed., 2003, 42, 3153–3158 CrossRef CAS PubMed.
- R. Arote, T.-H. Kim, Y.-K. Kim, S.-K. Hwang, H.-L. Jiang, H.-H. Song, J.-W. Nah, M.-H. Cho and C.-S. Cho, Biomaterials, 2007, 28, 735–744 CrossRef CAS PubMed.
- K. H. Min, J.-H. Kim, S. M. Bae, H. Shin, M. S. Kim, S. Park, H. Lee, R.-W. Park, I.-S. Kim, K. Kim, I. C. Kwon, S. Y. Jeong and D. S. Lee, J. Controlled Release, 2010, 144, 259–266 CrossRef CAS PubMed.
- Y.-J. Gao, Z.-Y. Qiao and H. Wang, Sci. China: Chem., 2016, 59, 991–1002 CrossRef CAS.
- R. A. Cordeiro, A. Serra, J. F. J. Coelho and H. Faneca, J. Controlled Release, 2019, 310, 155–187 CrossRef CAS PubMed.
- H. P. Rahn, J. Sun, Z. Li, R. M. Waymouth, R. Levy and P. A. Wender, Biomacromolecules, 2024, 25, 4305–4316 CrossRef CAS.
- D. M. Lynn, M. M. Amiji and R. Langer, Angew. Chem., Int. Ed., 2001, 40, 1707–1710 CrossRef CAS.
- N. Nun, M. Cruz, T. Jain, Y.-M. Tseng, J. Menefee, S. Jatana, P. S. Patil, N. D. Leipzig, C. McDonald, E. Maytin and A. Joy, Biomacromolecules, 2020, 21, 4030–4042 CrossRef CAS.
- J. Hyun, J. Eom, J. Song, I. Seo, S. H. Um, K. M. Park and S. H. Bhang, Macromol. Biosci., 2021, 21, 2100106 CrossRef CAS.
- I. Taniguchi, W. A. Kuhlman, A. M. Mayes and L. G. Griffith, Polym. Int., 2006, 55, 1385–1397 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- V. A. Ganesh, A. Baji and S. Ramakrishna, RSC Adv., 2014, 4, 53352–53364 RSC.
- G. Becker and F. R. Wurm, Chem. Soc. Rev., 2018, 47, 7739–7782 RSC.
- T. J. Farmer, J. W. Comerford, A. Pellis and T. Robert, Polym. Int., 2018, 67, 775–789 CrossRef CAS.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749–759 CrossRef CAS.
- J. P. Swanson, L. R. Monteleone, F. Haso, P. J. Costanzo, T. Liu and A. Joy, Macromolecules, 2015, 48, 3834–3842 CrossRef CAS.
- J. P. Swanson, M. A. Cruz, L. R. Monteleone, M. R. Martinez, P. J. Costanzo and A. Joy, Polym. Chem., 2017, 8, 7195–7206 RSC.
- X. Wang and N. Hadjichristidis, Macromolecules, 2020, 53, 223–232 CrossRef CAS.
- T. R. Blake, W. C. Ho, C. R. Turlington, X. Zang, M. A. Huttner, P. A. Wender and R. M. Waymouth, Chem. Sci., 2020, 11, 2951–2966 RSC.
- O. A. W. Haabeth, J. J. K. Lohmeyer, A. Sallets, T. R. Blake, I. Sagiv-Barfi, D. K. Czerwinski, B. McCarthy, A. E. Powell, P. A. Wender, R. M. Waymouth and R. Levy, ACS Cent. Sci., 2021, 7, 1191–1204 CrossRef CAS.
- P. Dong, W. Xiong, J. Feng, B. Wang, C. Liang, H. Wang, T. Sun, M. Wei, Q. Shi and X. Xie, Macromol. Rapid Commun., 2025, 2400885 CrossRef CAS , accepted for publication.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- L. Wen, S. Zhang, Y. Xiao, J. He, S. Zhu, J. Zhang, Z. Wu and M. Lang, Macromolecules, 2020, 53, 5096–5104 CrossRef CAS.
- T. F. Burton, J. Pinaud, N. Pétry, F. Lamaty and O. Giani, ACS Macro Lett., 2021, 10, 1454–1459 CrossRef CAS PubMed.
- A. Giraudo, E. Armano, C. Morano, M. Pallavicini and C. Bolchi, J. Org. Chem., 2023, 88, 15461–15465 CrossRef CAS PubMed.
- E. Rossegger, F. Pirolt, S. Hoeppener, U. S. Schubert, O. Glatter and F. Wiesbrock, Eur. Polym. J., 2019, 121, 109305 CrossRef CAS.
- J. Herzberger, K. Fischer, D. Leibig, M. Bros, R. Thiermann and H. Frey, J. Am. Chem. Soc., 2016, 138, 9212–9223 CrossRef CAS PubMed.
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461–15465 CrossRef CAS PubMed.
- R. Otter, C. M. Berac, S. Seiffert and P. Besenius, Eur. Polym. J., 2019, 110, 90–96 CrossRef CAS.
- D. Işık, E. Quaas and D. Klinger, Polym. Chem., 2020, 11, 7662–7676 RSC.
- C. M. Berac, L. Zengerling, D. Straβburger, R. Otter, M. Urschbach and P. Besenius, Macromol. Rapid Commun., 2020, 41, e1900476 CrossRef PubMed.
- F. H. Sobotta, M. T. Kuchenbrod, F. V. Gruschwitz, G. Festag, P. Bellstedt, S. Hoeppener and J. C. Brendel, Angew. Chem., Int. Ed., 2021, 60, 24716–24723 CrossRef CAS PubMed.
- H. Phan, R. Cavanagh, D. Destouches, F. Vacherot, B. Brissault, V. Taresco, J. Penelle and B. Couturaud, ACS Appl. Polym. Mater., 2022, 4, 7778–7789 CrossRef CAS.
- L. V. Arsenie, F. Hausig, C. Kellner, J. C. Brendel, P. Lacroix-Desmazes, V. Ladmiral and S. Catrouillet, Molecules, 2022, 27, 4233 CrossRef CAS.
- Q. Li, K. Wang, L. Fan, Z. Zeng and M. Huo, Polym. Chem., 2023, 14, 1254–1262 RSC.
- O. I. Kazakov, P. P. Datta, M. Isajani, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2014, 47, 7463–7468 CrossRef CAS.
- N. Mohanan, C. H. Wong, N. Budisa and D. B. Levin, Front. Bioeng. Biotechnol., 2022, 10, 854298 CrossRef PubMed.
- F. Freije García and G. García Liñares, J. Polym. Environ., 2024, 32, 2484–2516 CrossRef.
- J. Tran, T. Pesenti, J. Cressonnier, C. Lefay, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2019, 20, 305–317 CrossRef CAS.
- T. Pesenti, E. Gillon, S. Ishii, S. Messaoudi, Y. Guillaneuf, A. Imberty and J. Nicolas, Biomacromolecules, 2023, 24, 991–1002 CrossRef CAS PubMed.
- Y. Deng, S. Schäfer, D. Kronstein, A. Atabay, M. Susewind, E. Krieg, S. Seiffert and J. Gaitzsch, Biomacromolecules, 2024, 25, 303–314 CrossRef CAS PubMed.
- T. R. Dargaville, K. Lava, B. Verbraeken and R. Hoogenboom, Macromolecules, 2016, 49, 4774–4783 CrossRef CAS.
- T. R. Dargaville, D. G. Harkin, J.-R. Park, A. Cavalcanti, E. C. L. Bolle, F. M. Savi, B. L. Farrugia, B. D. Monnery, Y. Bernhard, J. F. R. Van Guyse, A. Podevyn and R. Hoogenboom, Biomacromolecules, 2021, 22, 1590–1599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data (NMR spectroscopy, SEC, and MALDI-TOF-MS analysis) as well as synthetic details and full analytical characterisation. See DOI: https://doi.org/10.1039/d5py00522a |
| This journal is © The Royal Society of Chemistry 2025 |