
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional chiral silanol ligands for enantioselective catalysis†
Yun-Pu Changa,
Kevin Blanco-Herreroa,
Turki M. Alturaifib,
James C. Fettinger a,
Peng Liub and
Annaliese K. Franz*a
a,
Peng Liub and
Annaliese K. Franz*a
aDepartment of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA. E-mail: akfranz@ucdavis.edu
bDepartment of Chemistry, University of Pittsburgh, 219 Parkman Avenue, Pittsburgh, PA 15260, USA
First published on 13th June 2025
Abstract
We report transition metal catalysis using novel chiral metal-chelating ligands featuring a silanol coordinating group and peptide-like aminoamide scaffold. The catalytic properties of the silanol ligand are demonstrated through an enantioselective Cu-catalyzed N–H insertion affording unnatural amino acid derivatives in high selectivity. Our investigations into the silanol coordination mode include DFT calculations, ligand structure investigations, and X-ray structure analyses, which support the formation of an H-bond stabilized silanol-chelating copper carbenoid complex. A π–π stacking interaction revealed by DFT calculations is proposed to enable selectivity for aryl diazoacetate substrates, overcoming some of the traditional limitations of using these substrates.
Introduction
Enantioselective catalysis is widely considered as the most efficient method to prepare enantioenriched compounds.1–3 In metal-catalyzed reactions, chiral ligands are utilized to control reactivity and stereoselectivity. Considerable efforts have been made to design and synthesize classes of ligands4,5 where heteroatom coordinating sites are key to chiral ligand design principles6,7 allowing for stereocontrol and tuning of reactivity. Common metal-coordinating groups include phosphines,8–12 phosphites,13–15 amines,8,16 imines,17,18 and alcohols16–21 (Fig. 1A).7 Although silicon has been incorporated in the backbone of a chiral ligand scaffold,22–27 and the hydridosilane introduced as a coordinating group,28–33 there are no examples of silanols in metal-coordinating ligands for transition-metal catalyzed enantioselective synthesis. | ||
| Fig. 1 (A) Common ligands and coordinating groups in asymmetric catalysis. (B) This work: silanol coordinating group in a multifunctional chiral ligand design. | ||
The silanol (Si–OH) group provides an opportunity to design ligands with new coordination modes for enhanced activity and stereoselectivity in asymmetric catalysis. Metal siloxides are more Lewis acidic than metal alkoxides due to back-bonding interactions  34,35 and the angle-dependent donor capacity of metal siloxides may bring balanced reactivity to catalytic cycles.36,37 Furthermore, longer Si–O bonds can contribute to improved steric modulation and selectivity. Achiral silanols have been previously utilized as ancillary ligands for a variety of metal-catalyzed transformations38 such as alkyne metathesis,37,39,40 Pd-catalyzed cross-coupling41–44 and ortho-metalation.45–49 Strohmann has reported chiral zinc(II)-silanoates,50 including a stereogenic-silicon silanoate,51 although no catalytic activity was documented. Bolm reported a notable example of a ferrocene-based chiral organosilanol ligand for an asymmetric phenyl-transfer reaction with stoichiometric zinc reagents.52 In this work, we have developed a novel modular multifunctional chiral ligand scaffold possessing silanol and amide coordination sites (Fig. 1B). This class of ligand overcomes current limitations in substrate scope for the N–H insertion reaction and represents the first example of a silanol in a chelating ligand for transition-metal catalyzed enantioselective synthesis.
34,35 and the angle-dependent donor capacity of metal siloxides may bring balanced reactivity to catalytic cycles.36,37 Furthermore, longer Si–O bonds can contribute to improved steric modulation and selectivity. Achiral silanols have been previously utilized as ancillary ligands for a variety of metal-catalyzed transformations38 such as alkyne metathesis,37,39,40 Pd-catalyzed cross-coupling41–44 and ortho-metalation.45–49 Strohmann has reported chiral zinc(II)-silanoates,50 including a stereogenic-silicon silanoate,51 although no catalytic activity was documented. Bolm reported a notable example of a ferrocene-based chiral organosilanol ligand for an asymmetric phenyl-transfer reaction with stoichiometric zinc reagents.52 In this work, we have developed a novel modular multifunctional chiral ligand scaffold possessing silanol and amide coordination sites (Fig. 1B). This class of ligand overcomes current limitations in substrate scope for the N–H insertion reaction and represents the first example of a silanol in a chelating ligand for transition-metal catalyzed enantioselective synthesis.
Results and discussion
At the start of our investigation, we envisioned incorporating a silanol chelating group into tunable chiral oxazoline (L1) and imidazoline (L2) scaffolds which serve as common ligands in various enantioselective transformations (Scheme 1).4,52–55 While L1 showed poor reactivity and selectivity in several test reactions, attempts to synthesize and evaluate imidazoline-silanol L2 (ref. 11) led to the discovery of a novel aminoamide ligand (L3) formed via silanol-facilitated ring-opening of the imidazoline (see ESI†).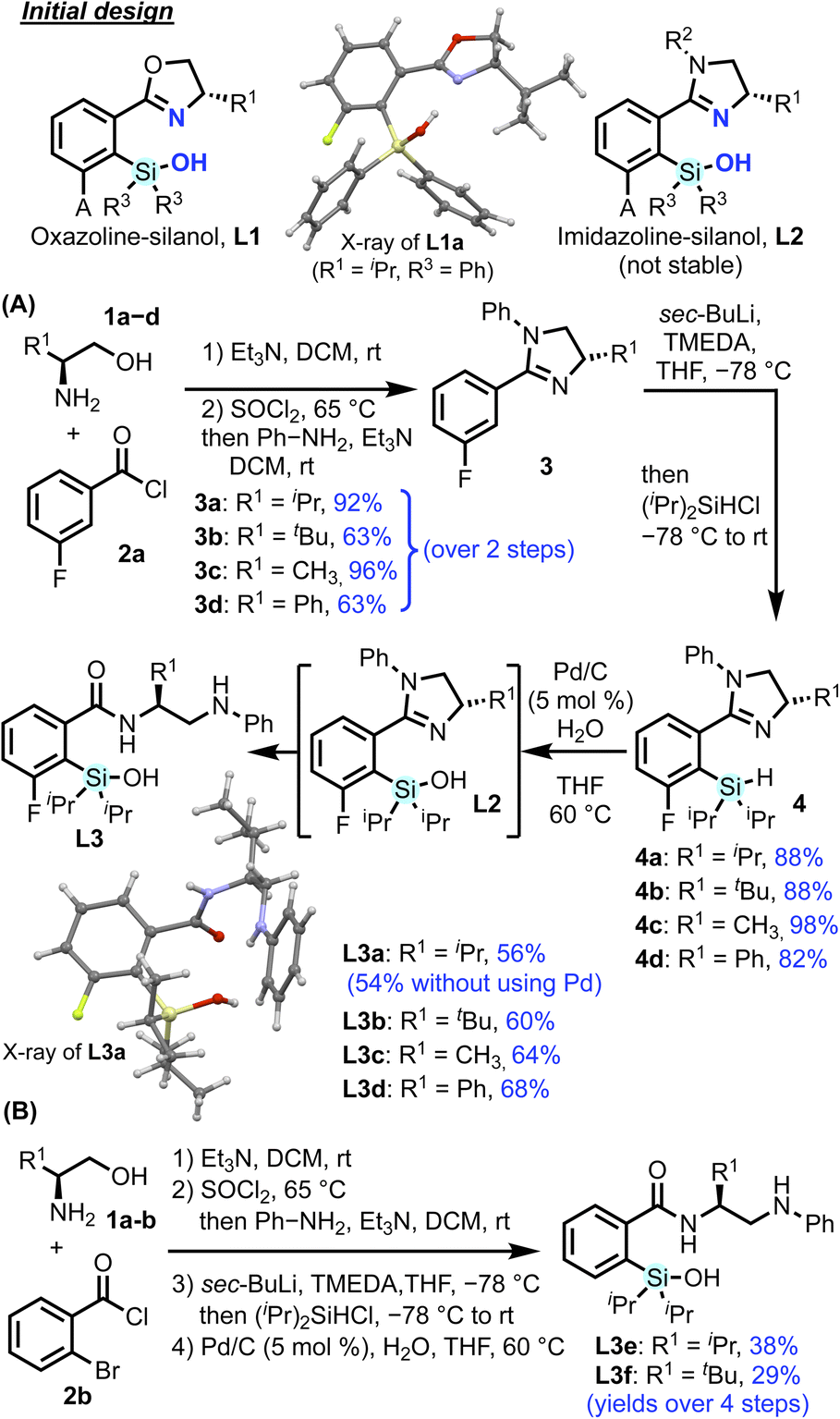 | ||
| Scheme 1 Initial ligand design and synthesis for chiral aminoamide silanol ligands L3a–d (A) and L3e and f (B). | ||
Following this discovery, we prepared a collection of aminoamide silanol ligands (L3) through an efficient modular synthetic sequence (Scheme 1). The general synthetic procedure utilizes amide formation and cyclization to form imidazoline 3, followed by ortho-lithiation56 and addition of a chlorosilane to afford silane 4. Silanols L3 are obtained upon hydrolysis of silane 4 using catalytic Pd/C with H2O.57 We also observed that neighboring group participation of the imidazoline promotes formation of silanol L3a without requiring a Pd/C catalyst (see ESI†). This route from inexpensive starting materials is scalable up to 1.5 grams. All of the aminoamide silanol products are air-stable, crystalline white solids.58 Synthesis via silane 4 provides unique access to L3; hydrolysis of the corresponding silyl chloride and attempts at direct silylation of the amide were not successful (see ESI†).
To demonstrate the silanol as a novel coordinating group for transition metal catalysis, an enantioselective copper-catalyzed N–H insertion reaction serves as a model reaction to highlight our preliminary discoveries and overcome the traditionally low selectivity of aryl diazoacetate in this reaction (Fig. 2).59–69 While initial experiments utilizing oxazoline-silanol L1 resulted in poor enantioselectivity, the remarkable improvement using silanol L3a highlights the importance of the aminoamide backbone. Notably, silanol ligand L3 enables N–H insertion reactions to be performed without a glovebox or Schlenk line, while still maintaining high enantioselectivity. Following initial success with L3a, we optimized the reaction conditions including [Cu] source, solvent, temperature and reaction concentration (see Table S4†). The absence of copper resulted in no reaction, and addition of NaBArF is essential for selectively. While a high background rate is observed without ligand L3, no enantioselectivity is observed. No disiloxane formation is observed with L3 under reaction conditions, and the silanol ligand can be reisolated using column chromatography.70 When the SiOH is replaced with a TMS (L4)71 or COH (L5), a complete loss of enantioselectivity is observed, albeit the background rate still proceeds from unchelated copper catalyst (see Table S5† for more control experiments).
 | ||
Fig. 2 Ligand optimization for enantioselective N–H insertion of α-phenyl-α-diazoester with aniline (see ESI† for raw data). Standard reaction conditions: 5a and 6a (0.2 mmol, 1.0 equiv. each) in DCM (0.05 M) at 23 °C in 1 h; L3c in 2 h. L1b required 16 h for 74% (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 er, opposite enantiomer); however, 1 h timepoint for L1b is graphed for direct comparison. Yield determined using NMR spectroscopy with Ph-TMS as internal standard. Enantiomeric excess (ee%) determined using CSP-HPLC analysis with a Chiralpak OD-H column. :40 er, opposite enantiomer); however, 1 h timepoint for L1b is graphed for direct comparison. Yield determined using NMR spectroscopy with Ph-TMS as internal standard. Enantiomeric excess (ee%) determined using CSP-HPLC analysis with a Chiralpak OD-H column. | ||
To evaluate the modular ligand components we synthesized a series of silanol ligands (L3b–L3g) (Fig. 2). The enantioselectivity of the N–H insertion (L3a–d) is influenced by the steric groups (R1) at the stereogenic center; however, L3a and L3b exhibit similar performance (89:11 and 91:9 er, respectively). The fluoro on the core aryl ring was originally incorporated for synthetic purposes, yet a slight reduction in enantioselectivity was observed for L3e and L3f (80:20 and 86:14 er, respectively). Ligand L3g with a less sterically demanding dimethylsilanol group resulted in a significant decrease in enantioselectivity (89:11 vs. 56:44 er).
Next we evaluated the substrate scope for the enantioselective N–H insertion reaction using silanol ligand L3b. The reaction proceeds in high enantioselectivity (94:6 to 98:2 er) using various alkyl diazoesters 5b–f with aniline (Scheme 2). And the catalytic performance is unaffected by scaling up to 1.0 mmol scale. The silanol ligand also facilitates high yields with substrates prone to β-H elimination (i.e. 7e), proceeding with reduced side products and enhanced yields, effectively addressing issues reported previously.68,72 Additionally, the reaction tolerates an olefin (7f) under the reaction conditions, exhibiting high selectivity; however, moderate yield was observed due to the inevitable competitive β-H elimination.73
 | ||
| Scheme 2 Scope of the enantioselective N–H insertion of α-alkyl-α-diazoesters with aniline.a,b,c aPerformed under standard reaction conditions. bIsolated yields. cDetermined using CSP-HPLC. The absolute configuration was assigned as (R) based on HPLC data for 7d.59 | ||
The silanol ligand overcomes limitations for previous N–H insertion reactions where α-aryl-α-diazoesters traditionally afford low enantioselectivity; employing silanol L3b proceeds with high enantioselectivities for aryl substrates (Scheme 3). The enantioselectivity with phenyldiazoester (91:9 er) is not affected by steric modification on the ester (7a, 7g and 7h). While aryl rings with electron-donating groups afford the highest enantioselectivity, substrates with electron-withdrawing groups also maintain good selectivity. Erosion of enantioselectivity (71:29 er) is observed for substitution at the ortho-position (7l). The electronic profile of the aniline reactant74 was then explored with phenyldiazoacetate (Scheme 3 and 7r–7z). Interestingly, a crossover point is observed in the Hammett plot (Scheme 3A), indicating a change in mechanism where the strong electron-withdrawing anilines (7x and 7y) start acting as achiral proton-shuttle catalysts, thus leading to a decrease in enantioselectivity (Scheme 3B).62,68,69,75
 | ||
| Scheme 3 Scope of the enantioselective N–H insertion of α-aryl-α-diazoesters with anilines providing insight from Hammett plot (A) indicating crossover to proton-shuttle catalysis (B).a,b,c aReactions performed under standard reaction conditions. bIsolated yields. cDetermined using CSP-HPLC. The absolute configuration was assigned as (R) based on the HPLC data for 7h.76 der determined after recrystallization. | ||
Density functional theory (DFT) calculations reveal the binding mode and conformation of ligand L3a in the proposed Cu–carbene complex, supporting the essential role of the silanol as a chelating group in the active complex (Fig. 3A).62,63,68,69 The isopropyl was utilized in all DFT studies since both L3a and L3b exhibit comparable performance in the N–H insertion reaction.77 The analysis demonstrates that the most stable conformation involves both the silanol and amide oxygen atoms coordinating relatively strongly with the copper atom, with bond distances of 2.03 Å and 2.14 Å, respectively. Additionally, intramolecular hydrogen bonding between the silanol and amino group was observed, increasing the Lewis basicity of the silanol oxygen and further stabilizing the silanol–Cu coordination. The ten-membered ring (highlighted in yellow) adopts a chair–chair–half-chair conformation78,79 with no noticeable transannular strain and the isopropyl group placed in the pseudoequatorial position. Moreover, the DFT calculations revealed a π–π interaction between the N-phenyl group on the ligand and the phenyl group on the carbene, evidenced by a distance of 4.20 Å between the centroids of the two benzene rings. Traditionally, aryl diazoacetates have yielded low enantioselectivity in the N–H insertion due to aryl stabilization of the free ylide intermediate,62,68 while alkyl diazoacetates exhibit high selectivity, attributed here to C–H π interactions and lower intermediate stability. Silanol L3 overcomes this traditional limitation through the proposed π–π interaction, thereby enhancing stability in the metal–ligand complex and catalyst-associated ylide.
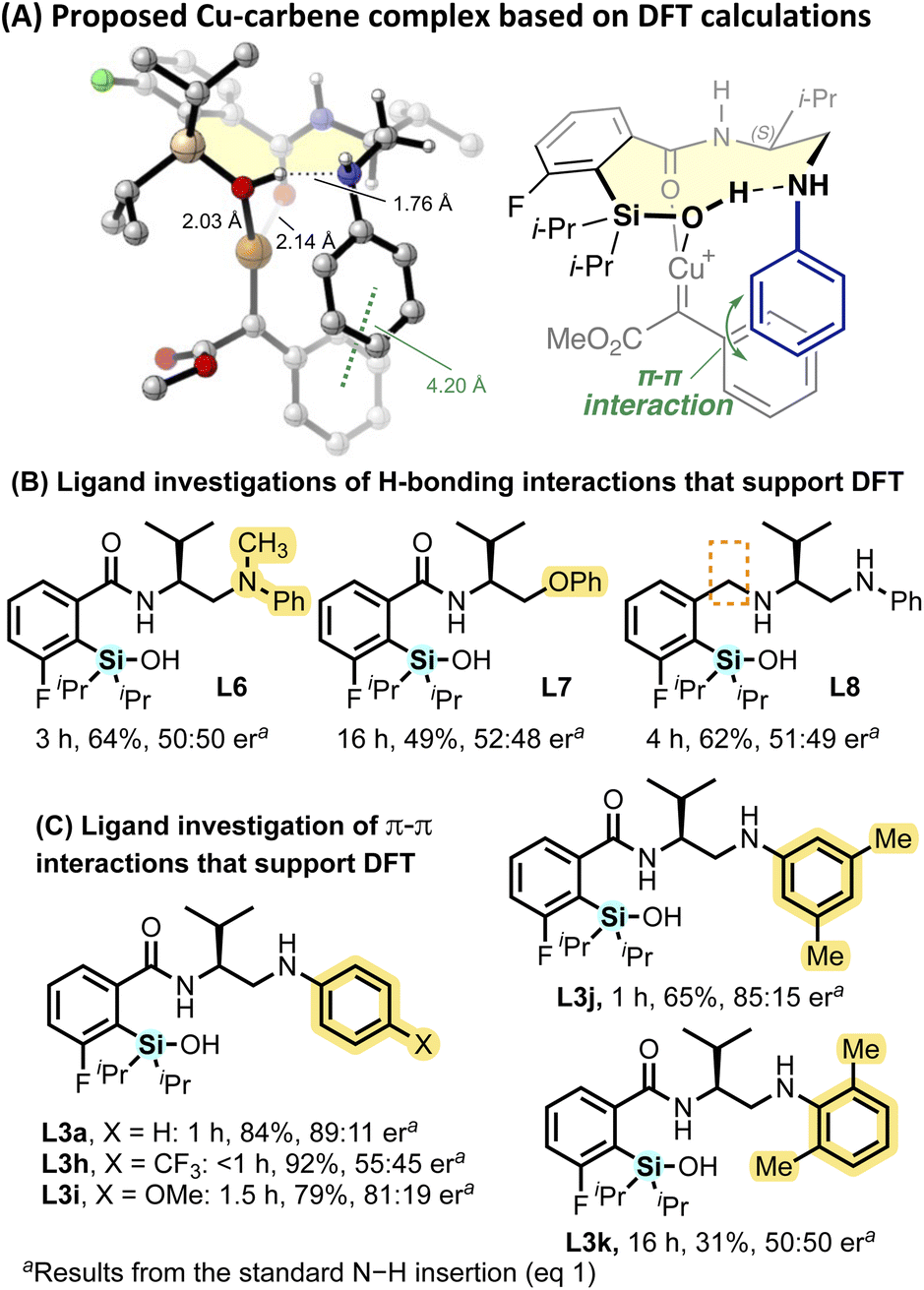 | ||
| Fig. 3 (A) The lowest energy conformer of the proposed Cu–carbene intermediate, featuring ligand L3a and aryl-diazoesters 5a, as determined by DFT calculations at the M06/6-311+G(d,p)–SDD(Cu)/SMD(DCM)//B3LYP-D3/6-31G(d)–SDD(Cu) level of theory. Structures of 10 conformers located within 3 kcal mol−1 of the lowest-energy conformer are provided in the ESI;† (B) ligands investigated with different H-bonding capabilities to support DFT; (C) ligands investigated with capabilities for different π–π interactions to support DFT. | ||
To support results of the DFT calculations, silanols L6–8 were synthesized and evaluated for the enantioselective N–H insertion (eqn (1)) to investigate H-bonding interactions within the metal–ligand complex (Fig. 3B). The N-methylated analog (L6) affords reduced yield (64%) and no selectivity. Similarly, the phenoxy analog (L7) resulted in low enantioselectivity. These observations suggest that the H-bonding interaction between the amine and silanol group is critical to promote a favorable conformation that enhances coordination of the silanol to the metal center and imparts enantioselectivity. Without the carbonyl group, L8 also afforded racemic product, supporting that both the silanol oxygen and the carbonyl oxygen play a key role for coordination in the active complex.
The importance of a π–π interaction is also supported by the distinct influence of the electronic and steric substitution patterns of the N-aryl amine in L3h–k (Fig. 3C). The analog with a p-CF3 group (L3h) shows a loss of enantioselectivity (55:45 er) while the p-OMe group (L3i) retains good enantioselectivity (81:19 er). Comparing ligands L3j and L3k, bearing di-methyl substitution at different positions of the N-aryl ring resulted in a complete loss of selectivity and low reactivity for the ortho-substituted ligand L3k, attributed to steric disruptions of π–π interactions. When conducting the N–H insertion in aromatic solvent, decreased selectivity is observed (78:22 er, Table S4†).
An intermediate [(L3a)2/Na·(H2O)2]+[BArF]− complex was isolated after slow solvent evaporation and single crystal X-ray analysis supports the formation of H-bond stabilized silanol coordination (Fig. 4). In this intermediate, intramolecular H-bonding interactions are observed between the amino and silanol group, with additional intermolecular H-bonding between amino and amide groups. Although not the active metal complex, the silanol coordination observed in this sodium intermediate is consistent with the DFT calculations, highlighting the role of the silanol as a metal chelating group.80,81
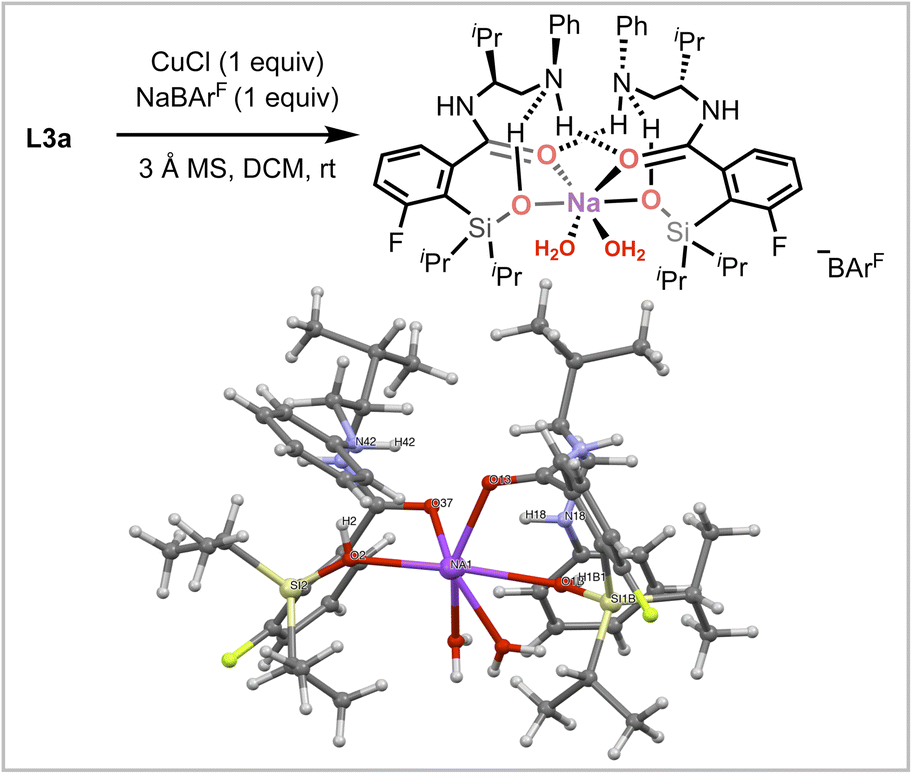 | ||
| Fig. 4 Single-crystal structure of intermediate complex [(L3a)2/Na·(H2O)2]+[BArF]− isolated from slow solvent evaporation over two months. The BArF− anion has been omitted for clarity. Selected bond distances (Å), H-bonding interactions, and angles (deg): Si(2)–O(2): 1.634(4), Na(1)–O(2): 2.284(10), Na(1)–O(37): 2.441(8), O(2)–H(2)…N(42), N(18)–H(18)…O(37), N(42)–H(42)…O(13), O(1B)–H(1B1)…N(18), ∠Si(2)–O(2)–Na(1): 133.7(2). | ||
Conclusions
Our study details the synthesis and application of new chiral aminoamide silanol ligands for enantioselective catalysis. The silanol group facilitates the formation of a H-bond stabilized silanol–copper carbenoid active complex, which is supported by DFT calculations, ligand analog studies, and X-ray structural analysis. Our modular ligand design allows access to multiple ligand variants that demonstrate the influence of steric and electronic substituents on activity and selectivity for an N–H insertion reaction. This pioneering use of silanol ligands in transition-metal catalysis provides valuable insights for ligand design and machine learning training sets to allow a more comprehensive understanding of metal–ligand binding interactions and design features for asymmetric catalysis.Data availability
The data supporting this article have been included as part of the ESI:† experimental procedure, discussion of ligand syntheses characterization data, optimization tables, results of non-linear experiment, copies of spectra, computational details, and Cartesian coordinates of all computed structures are available in the ESI† of this article (PDF). X-ray crystallographic data fare available from the Cambridge Crystallographic Data Centre (CCDC), deposition numbers 2289933 (for L3a), 2289935 (for L3b), 2289936 (for 3a), 2289938 (for S2), 2289939 (for L1a), 2311643 (for [(L3a)2/Na⋅(H2O)2]+[BArF]−).Author contributions
Y. P. C. and K. B. H. performed synthetic, spectroscopic and mechanistic studies. P. L. and T. M. A. coordinated and performed DFT calculations and analyses. J. C. F. performed crystallographic analyses. A. K. F. coordinated the experiments and analyses. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Science Foundation (CHE-1900300 and CHE-2247505), including the dual-source X-ray diffractometer (CHE-1531193) used in this study. The Thermo Q-Exactive High-field Orbitrap used in this study is supported by National Institutes of Health (1S10OD025271-01A1). DFT calculations were carried out at the University of Pittsburgh Center for Research Computing and the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, supported by NSF award numbers OAC-2117681, OAC-1928147, and OAC-1928224. We thank the Shaw group (UC Davis) for providing access to TLC-MS and the Olson group (UC Davis) for the use of IR spectrometer. We also thank the UC Davis NMR facility for their assistance with NMR techniques. YPC thanks the UC Davis graduate studies for 2022–23 Summer Graduate Student Researcher Award. We acknowledge James Vesto (UC Davis) for helpful discussion.Notes and references
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS PubMed.
- A. Fanourakis, P. J. Docherty, P. Chuentragool and R. J. Phipps, ACS Catal., 2020, 10, 10672–10714 CrossRef CAS PubMed.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- J. Margalef, M. Biosca, P. de la Cruz Sánchez, J. Faiges, O. Pàmies and M. Diéguez, Coord. Chem. Rev., 2021, 446, 214120 CrossRef CAS.
- W. Zeng, G.-Y. Chen, Y.-G. Zhou and Y.-X. Li, J. Am. Chem. Soc., 2007, 129, 750–751 CrossRef CAS PubMed.
- A. Pfaltz, I. Drury and J. William, Chem. Inf., 2004, 101, 5723–5726 CAS.
- G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS PubMed.
- F. Menges, M. Neuburger and A. Pfaltz, Org. Lett., 2002, 4, 4713–4716 CrossRef CAS PubMed.
- M. J. Burk, J. Am. Chem. Soc., 1991, 113, 8518–8519 CrossRef CAS.
- H. Park, R. Kumareswaran and T. V. RajanBabu, Tetrahedron, 2005, 61, 6352–6367 CrossRef CAS.
- B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS PubMed.
- B. Bartels and G. Helmchen, Chem. Commun., 1999, 741–742 RSC.
- M. C. Schwarzer, A. Fujioka, T. Ishii, H. Ohmiya, S. Mori and M. Sawamura, Chem. Sci., 2018, 9, 3484–3493 RSC.
- C. A. Krueger, K. W. Kuntz, C. D. Dzierba, W. G. Wirschun, J. D. Gleason, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 4284–4285 CrossRef CAS.
- N. S. Josephsohn, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2001, 123, 11594–11599 CrossRef CAS PubMed.
- A. Matsuzawa, T. Mashiko, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7616–7619 CrossRef CAS PubMed.
- K. Narasaka, N. Iwasawa, M. Inoue, T. Yamada, M. Nakashima and J. Sugimori, J. Am. Chem. Soc., 1989, 111, 5340–5345 CrossRef CAS.
- E. J. Corey and Y. Matsumura, Tetrahedron Lett., 1991, 32, 6289–6292 CrossRef CAS.
- G. T. Notte and J. L. Leighton, J. Am. Chem. Soc., 2008, 130, 6676–6677 CrossRef CAS PubMed.
- Y. Sakaguchi, Y. Iwade, T. Sekikawa, T. Minami and Y. Hatanaka, Chem. Commun., 2013, 49, 11173 RSC.
- K. Maruoka, H. Banno and H. Yamamoto, J. Am. Chem. Soc., 1990, 112, 7791–7793 CrossRef CAS.
- H. Zhang and D. Zhao, ACS Catal., 2021, 11, 10748–10753 CrossRef CAS.
- X. Chang, P. Ma, H. Chen, C. Li and P. Wang, Angew. Chem., Int. Ed., 2020, 59, 8937–8940 CrossRef CAS PubMed.
- H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- B. Yang, J. Gao, X. Tan, Y. Ge and C. He, Angew. Chem., Int. Ed., 2023, e202307812 CAS.
- P. Sangtrirutnugul and T. D. Tilley, Organometallics, 2007, 26, 5557–5568 CrossRef CAS.
- T. Kitano, T. Komuro, R. Ono and H. Tobita, Organometallics, 2017, 36, 2710–2713 CrossRef CAS.
- Z. Mo, A. Kostenko, Y.-P. Zhou, S. Yao and M. Driess, Chem.–Eur. J., 2018, 24, 14608–14612 CrossRef CAS PubMed.
- T. Komuro, D. Mochizuki, H. Hashimoto and H. Tobita, Dalton Trans., 2022, 51, 9983–9987 RSC.
- T. Komuro, Y. Nakajima, J. Takaya and H. Hashimoto, Coord. Chem. Rev., 2022, 473, 214837 CrossRef CAS.
- J. T. Poulton, M. P. Sigalas, K. Folting, W. E. Streib, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1994, 33, 1476–1485 CrossRef CAS.
- M. H. Chisholm, F. A. Cotton, M. W. Extine and R. L. Kelly, J. Am. Chem. Soc., 1978, 100, 3354–3358 CrossRef CAS.
- A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef PubMed.
- J. Heppekausen, R. Stade, A. Kondoh, G. Seidel, R. Goddard and A. Fürstner, Chem.–Eur. J., 2012, 18, 10281–10299 CrossRef CAS PubMed.
- H. Yamagishi, J. Shimokawa and H. Yorimitsu, ACS Catal., 2023, 13, 7472–7487 CrossRef CAS.
- A. Fürstner, J. Am. Chem. Soc., 2021, 143, 15538–15555 CrossRef PubMed.
- J. Hillenbrand, M. Leutzsch, E. Yiannakas, C. P. Gordon, C. Wille, N. Nöthling, C. Copéret and A. Fürstner, J. Am. Chem. Soc., 2020, 142, 11279–11294 CrossRef CAS PubMed.
- S. E. Denmark and R. C. Smith, J. Am. Chem. Soc., 2010, 132, 1243–1245 CrossRef CAS PubMed.
- S. E. Denmark, J. Org. Chem., 2009, 74, 2915–2927 CrossRef CAS PubMed.
- S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed.
- S. A. Tymonko, R. C. Smith, A. Ambrosi, M. H. Ober, H. Wang and S. E. Denmark, J. Am. Chem. Soc., 2015, 137, 6200–6218 CrossRef CAS PubMed.
- C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef CAS PubMed.
- C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef CAS PubMed.
- C. Huang, N. Ghavtadze, B. Godoi and V. Gevorgyan, Chem.–Eur. J., 2012, 18, 9789–9792 CrossRef CAS PubMed.
- M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef CAS PubMed.
- S. M. Sieburth and L. Fensterbank, J. Org. Chem., 1993, 58, 6314–6318 CrossRef CAS.
- C. Golz, P. Steffen and C. Strohmann, Angew. Chem., Int. Ed., 2017, 56, 8295–8298 CrossRef CAS PubMed.
- F. Langenohl, J. Rösler, S. Zühlke, J. Kirchhoff and C. Strohmann, Chem.–Eur. J., 2023, 29, e202202935 CrossRef CAS PubMed.
- S. Özçubukçu, F. Schmidt and C. Bolm, Org. Lett., 2005, 7, 1407–1409 CrossRef PubMed.
- C. C. Bausch and A. Pfaltz, PHOX Ligands, in Privileged Chiral Ligands and Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2011 Search PubMed.
- G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS PubMed.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed.
- S. O. Wilson, N. T. Tran and A. K. Franz, Organometallics, 2012, 31, 6715–6718 CrossRef CAS.
- M. Jeon, J. Han and J. Park, ACS Catal., 2012, 2, 1539–1549 CrossRef CAS.
- L3a was found to be stable to air and atmospheric moisture for >30 months with minimal decomposition and no loss of catalytic activity.
- B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834–5835 CrossRef CAS PubMed.
- S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed.
- L. G. Furniel, R. Echemendía and A. C. B. Burtoloso, Chem. Sci., 2021, 12, 7453–7459 RSC.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC.
- M.-L. Li, J.-H. Yu, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990–994 CrossRef CAS PubMed.
- M.-L. Li, J.-B. Pan and Q.-L. Zhou, Nat. Catal., 2022, 5, 571–577 CrossRef CAS.
- Z. Liu, C. Calvó-Tusell, A. Z. Zhou, K. Chen, M. Garcia-Borràs and F. H. Arnold, Nat. Chem., 2021, 13, 1166–1172 CrossRef CAS PubMed.
- W. Yang, M. Pu, X. Lin, M. Chen, Y. Song, X. Liu, Y.-D. Wu and X. Feng, J. Am. Chem. Soc., 2021, 143, 9648–9656 CrossRef CAS PubMed.
- E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066–12067 CrossRef CAS PubMed.
- S.-F. Zhu, B. Xu, G.-P. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2012, 134, 436–442 CrossRef CAS PubMed.
- Y. Zhu, X. Liu, S. Dong, Y. Zhou, W. Li, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636–1640 CrossRef CAS PubMed.
- The presence of the unaltered silanol ligand L3 was also confirmed after each N–H insertion through 19F NMR spectroscopy.
- The diisopropylmethylsilyl is not readily accessible; however, the trimethylsilyl analog (L4) serves as a direct comparison with the dimethylsilanol (L3g), showing a complete loss of selectivity (50:50 er) compared to L3g.
- W.-S. Huang, Z. Xu, K.-F. Yang, L. Chen, Z.-J. Zheng and L.-W. Xu, RSC Adv., 2015, 5, 46455–46463 RSC.
- P. Guo, Y. Chen, L. Tao, S. Ji, R. Zhang, Z. Zhang, X. Liang, D. Wang, Y. Li and J. Zhao, ACS Catal., 2024, 14, 4690–4698 CrossRef CAS.
- When cyclohexyl amine was used under the optimal reaction conditions, no reaction was observed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J.-B. Pan, X.-G. Zhang, Y.-F. Shi, A.-C. Han, Y.-J. Chen, J. Ouyang, M.-L. Li and Q.-L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202300691 CrossRef CAS PubMed.
- The distinction between L3a and L3b is the steric group at the chiral center, which lacks coordination potential in the metal–ligand complex. We chose L3a for X-ray studies due to its lower cost and higher availability in the lab. Additionally, L3a was used in DFT calculations to ensure consistency with experimental results.
- V. Dragojlovic, ChemTexts, 2015, 1, 14 CrossRef.
- K. B. Wiberg, J. Org. Chem., 2003, 68, 9322–9329 CrossRef CAS PubMed.
- J. C. Slootweg and P. Chen, Organometallics, 2006, 25, 5863–5869 CrossRef CAS.
- E. Iniesta and A. Vidal-Ferran, Chem. Commun., 2020, 56, 6364–6367 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2289933, 2289935, 2289936, 2289938, 2289939 and 2311643. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02528a |
| This journal is © The Royal Society of Chemistry 2025 |