
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer electrolytes for potassium batteries: incorporating ionic liquids to enhance the room temperature ionic conductivity†
Jinyu Chenab,
Sohelia Ebrahimi-Koodehic,
Boyan Ilievd,
Yuval Steinberge,
Michal Leskes e,
Thomas J. S. Schubertd,
Elizabeth Castillo-Martínezc,
Dominic Bresserab and
Maider Zarrabeitia*ab
e,
Thomas J. S. Schubertd,
Elizabeth Castillo-Martínezc,
Dominic Bresserab and
Maider Zarrabeitia*ab
aHelmholtz Institute Ulm (HIU), Helmholtzstasse 11, 89081 Ulm, Germany. E-mail: maider.ipina@kit.edu
bKarlsruhe Institute of Technology (KIT), P. O. Box 3640, 76021 Karlsruhe, Germany
cDepartment of Inorganic Chemistry, Universidad Complutense de Madrid, 28040 Madrid, Spain
dIolitec Ionic Liquids Technologies GmbH, Im Zukunftspark 9, 74076 Heilbronn, Germany
eDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, 761000 Rehovot, Israel
First published on 7th July 2025
Abstract
Exploring the next generation of batteries based on sustainable materials is crucial for transitioning to a post-lithium-technology era. Potassium-based technology is one of the candidates that could fulfil the sustainability criteria. The electrolyte plays a crucial role in battery performance, being responsible for ionic transport, cycling performance, working temperature, and safety. Polyethylene oxide (PEO)-based solid polymer electrolytes (SPEs) have been extensively studied. However, one of the drawbacks of SPEs is their poor ionic conductivity at room temperature (RT). Quasi-solid and solvent-free polymer electrolytes are a promising solution to this issue, combining the benefits of a liquid phase and SPEs. This work focuses on the development of cross-linked PEO, fluorinated K salt, and ionic liquid (IL)-based solvent-free SPEs for potassium batteries. The designed cross-linked ternary solvent-free SPEs are thoroughly characterized both physicochemically and electrochemically, achieving ionic conductivities of up to 1.6 × 10−3 S cm−1 at 20 °C. The solvent-free SPEs were tested in K cells at 20 °C, using a Prussian white (PW) cathode as a proof-of-concept. The effects that different fluorinated anions, such as bis(fluoromethanesulfonyl) imide (FSI−) and bis(trifluoromethanesulfonyl) imide (TFSI−), have on the electrochemical performance were analysed by investigating the solid electrolyte interphase (SEI) formed on the K metal surface through electrochemical impedance spectroscopy (EIS), X-ray photoelectron spectroscopy (XPS) and magic-angle spinning solid-state nuclear magnetic resonance spectroscopy (MAS-ssNMR).
1. Introduction
The use of conventional fossil energy sources has led to several issues, including global warming, air pollution, and resource depletion.1,2 Fortunately, since 2011, renewable energy sources, such as wind power and solar photovoltaic technology, have experienced a significant increase in market share within the global electricity generation sector. Large-scale energy storage systems can mitigate the intermittency and instability associated with renewable energy sources.2–4 As a result, batteries have naturally become the focus of energy storage systems.5 Over the past few decades, lithium-ion batteries (LIBs) have become essential for portable electronic devices, electric vehicles, and energy storage systems.6–8 However, the limited and uneven global distribution of lithium resources, coupled with concerns regarding certain raw materials (e.g., cobalt, lithium, etc.), has spurred active research into alternative battery technologies that prioritise sustainability and cost-effectiveness.4,9,10Sodium- and potassium-ion batteries (NIBs and KIBs) have garnered particular attention as alternatives to LIBs.4,9,10 The concentrations of Na and K in the Earth's crust are 2.3 wt% and 1.5 wt%, respectively, whereas Li is present at a much lower concentration of 0.0017 wt%.4 This difference in abundance could lead to lower Na and K production costs than Li.11,12 In addition, the standard electrode potential of K is lower than that of Na, with −2.93 V compared to −2.71 V vs. SHE, and closer to Li (−3.04 V vs. SHE).11,12 Moreover, the K+/K redox potential in non-aqueous solvents (i.e., propylene carbonate (PC)) is the most negative among the three alkali metals (−3.002 V vs. SHE).13 This implies that replacing Li and Na with K in conventional battery designs can theoretically result in higher voltages and similar energy densities.13,14 Additionally, Al can serve as the current collector in KIBs, just as it does in NIBs, since no alloying occurs between K and Al.15 This allows for a significant reduction in battery cost and weight compared to LIBs.4 Given these advantages, it is believed that KIBs can serve as future large-scale energy storage systems.4,12,13,15 Indeed, the US company Group 1 announced in 2024 the 1st 18650 KIB based on a PW cathode, delivering a specific energy of 180 Wh kg−1 over 3000 cycles, which is competitive with LiFePO4-based LIBs.16
Commercial batteries are primarily based on flammable liquid electrolytes, which raises safety concerns for all battery applications. Solid-state electrolytes have garnered substantial attention in recent years due to their advantages, including higher stability, non-flammability, high safety, and ease of processing.17–19 Unlike the inherent rigidity and brittleness of inorganic solid electrolytes, SPEs exhibit excellent mechanical flexibility, toughness, and processability, making them a promising solution for addressing interfacial issues.18,20 PEO had been explored for the production of SPEs, even before the term “SPEs” was established, as early as 1971 by M. Armand.20 PEO exhibits remarkable solubility for various salts and excellent structural flexibility, attributed to the presence of flexible –CH2CH2O– units. These unique characteristics enable PEO to form strong chelating complexes with cations, facilitating the dissolution of a diverse range of salts.20,21 However, PEO tends to crystallize at temperatures below 60 °C. In contrast, the motion of polymer chains primarily occurs in the amorphous phase.22,23 Therefore, the ion transport that relies on the motion of polymer chains is hindered below 60 °C; consequently, PEO electrolytes typically exhibit poor ionic conductivity at RT, ranging from 10−7 to 10−6 S cm−1.24,25 Moreover, PEO-based electrolytes lose their mechanical integrity at temperatures exceeding the melting point of PEO (around 56 °C).26
Over the past few decades, considerable efforts have been made to enhance the ionic conductivity of PEO at RT. However, the results have not been entirely satisfactory yet.3 For instance, the addition of nanofillers to the polymer matrix has been reported to enhance ionic conductivity.27 Another promising strategy is the addition of ILs, which appears to enhance the ionic conductivity of PEO-based SPEs at RT effectively.3,28–31 The incorporation of non-flammable ILs not only improves ionic conductivity but also enhances the safety of electrolytes, significantly strengthening the stability at the interface with alkali metals.30 Regarding IL chemistry, pyrrolidinium-based ILs have been extensively studied due to their high electrochemical stability.3 In addition, fluorinated anions, such as FSI− and TFSI−, are also well-studied due to their compatibility with alkali metals and their high thermal and electrochemical stability. For example, Roscher et al.32 have incorporated IL (Pyr14FSI or Pyr14TFSI) into Na-based solvent-free SPE, revealing greater flexibility of the polymer chains, as well as improved ion transport and electrochemical stability. Moreover, Boschin et al.33 demonstrate that both TFSI− and FSI− can inhibit the crystallisation of PEO, especially the FSI−. On the other hand, Appetecchi et al.34 developed a novel poly(diallyldimethylammonium) bis(trifluoromethanesulfonyl) imide-based solvent-free SPE, which was doped with Pyr14TFSI and LiTFSI, achieving an ionic conductivity of 10−4 S cm−1 at 20 °C. Subsequently, Chen et al.3 conducted an investigation using infrared spectrum characterisations and further DFT calculations to elucidate the plasticising effect of FSI−. It was found that the FSI− adheres to the PEO backbone, enhancing its coordination ability and creating more coordination sites. In the case of potassium batteries, despite PEO-based electrolytes having been reported,35 there is a lack of research dedicated to improving the performance of SPEs by employing ILs. To the best of our knowledge, only Kotronia et al.36 reported the preparation of ternary ionic gel electrolytes using K salts, IL, and poly(ionic liquid), but these do not form mechanically stable self-standing films.
In general, the mechanical properties of PEO can be improved by cross-linking, which also reduces its crystallinity.21,23 The well-stacked and entangled chains in the cross-linked PEO phase contribute to the mechanical strength of linear PEO.26 Among the various PEO cross-linking techniques, the ultraviolet (UV) light-induced cross-linking method is commonly used for fabricating cross-linked PEO-based SPEs.23,26,37,38 For instance, Falco et al.38 have demonstrated that UV-induced cross-linking is an effective method for reducing the crystallinity of PEO electrolytes, thereby improving the Li+ transport number and ionic conductivity.
In this work, K-based solvent-free SPEs have been developed by adopting the optimal ratio of PEO, IL and K salt from our previous work.32 The used ILs are Pyr12O1FSI (1-(2-methoxyethyl)-1-methylpyrrolidinium bis (fluoromethylsulfonyl)imide) and Pyr12O1TFSI (1-(2-methoxyethyl)-1-methylpyrrolidinium bis (trifluoromethylsulfonyl)imide), and the K salts are KFSI (potassium bis(fluoromethylsulfonyl)imide) and KTFSI (potassium bis(trifluoromethylsulfonyl)imide). In addition, this work also incorporates methoxy groups into the IL cation. The presence of oxygen atoms in the side-chain of the pyrrolidinium enhances its thermal stability, increases its ionic conductivity, and reduces the viscosity typically associated with alkyl side-chain pyrrolidinium-based ILs. The performance of the solvent-free SPE membranes was tested in coin cells operating at 20 °C, utilising PW (K1.37Mn[Fe(CN)6]1.03) as the cathode active material. All solvent-free SPE membranes have undergone comprehensive characterisation to investigate their thermal and electrochemical stability, thermal phase transitions, and electrochemical properties. The compatibility and surface chemistry of the K∥solvent-free SPEs interface were analysed by EIS, XPS, and MAS-ssNMR. The FSI−-based solvent-free SPE exhibits high ionic conductivity of 1.6 × 10−3 S cm−1 at 20 °C and 6.0 × 10−3 S cm−1 at 60 °C. It also presents remarkable K interface compatibility, as FSI−-based IL can promote the formation of a stable and inorganic-rich SEI against K, thereby reducing interfacial resistance and, in turn, cell polarisation, which enhances the electrochemical performance of K∥solvent-free SPE∥PW cells.
2. Experiment
2.1. Materials suppliers
The following chemicals were used without further purification: PEO (WSR 301, Mw = 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000, Dow Chemical), benzophenone (≥99.0%, Sigma Aldrich), K metal (in mineral, 98%, Sigma Aldrich), Pyr12O1FSI (IOLITEC GmbH, 98%), Pyr12O1TFSI (IOLITEC GmbH, 98%), KFSI (IOLITEC GmbH, 98%), KTFSI (IOLITEC Gmbh, 99%), carbon black (Super C45, Imerys), sodium carboxymethylcellulose (Na-CMC, Sigma Aldrich), and styrene-butadiene rubber (SBR, 40% dispersion, ZEON). The chemical structure of the main salts and ILs of the solvent-free SPEs is shown in Scheme 1A.
000000, Dow Chemical), benzophenone (≥99.0%, Sigma Aldrich), K metal (in mineral, 98%, Sigma Aldrich), Pyr12O1FSI (IOLITEC GmbH, 98%), Pyr12O1TFSI (IOLITEC GmbH, 98%), KFSI (IOLITEC GmbH, 98%), KTFSI (IOLITEC Gmbh, 99%), carbon black (Super C45, Imerys), sodium carboxymethylcellulose (Na-CMC, Sigma Aldrich), and styrene-butadiene rubber (SBR, 40% dispersion, ZEON). The chemical structure of the main salts and ILs of the solvent-free SPEs is shown in Scheme 1A.
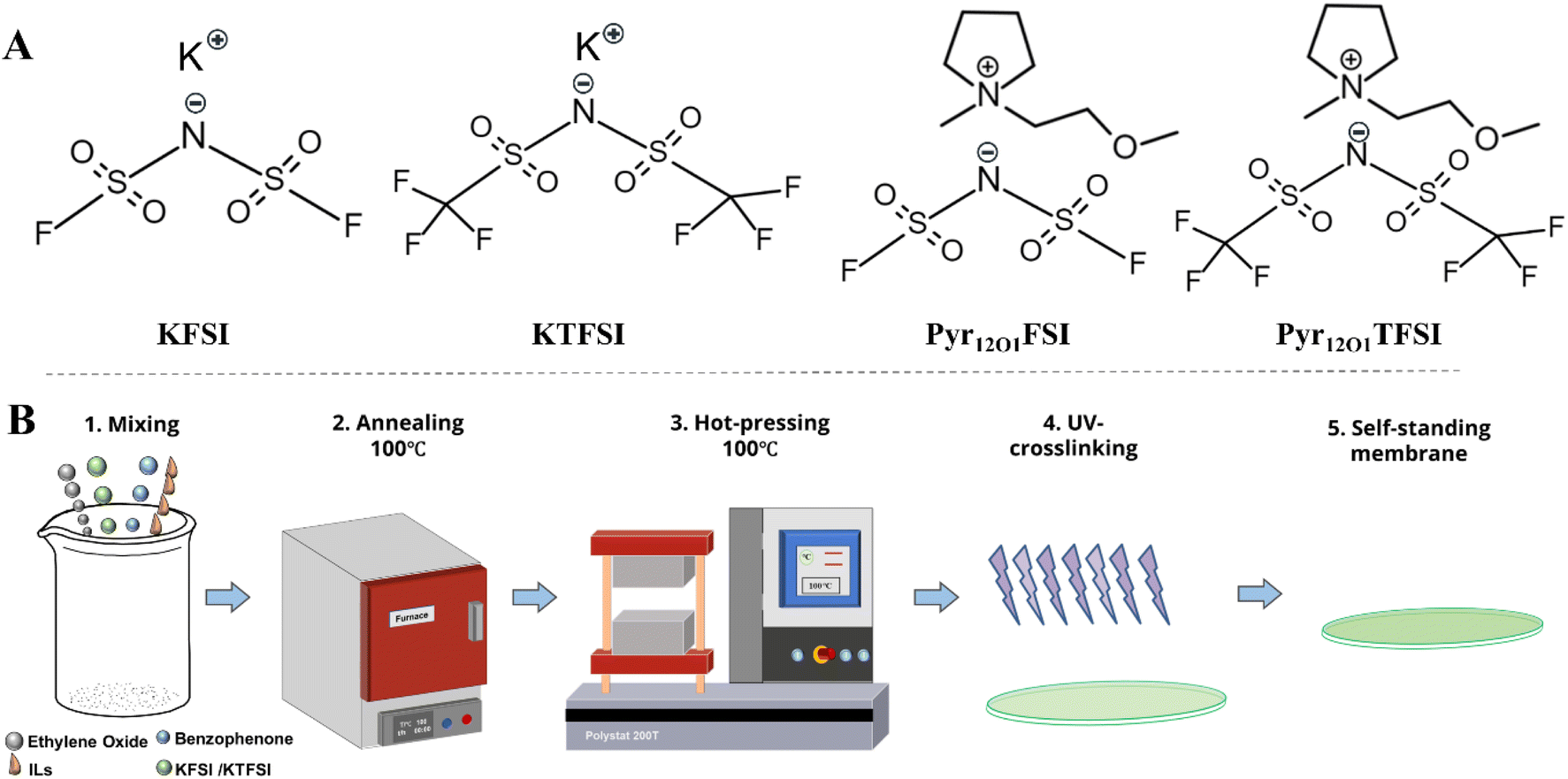 | ||
| Scheme 1 (A) The chemical structure of the K salts and ILs, and (B) schematic for the fabrication procedure of the cross-linked solvent-free SPEs. | ||
2.2. Cathode synthesis and electrode preparation
PW was synthesised by a simple precipitation method at low temperatures.39 The procedure began by preparing two separate aqueous solutions: 0.04 M EDTA-2K·4H2O and 0.04 M (CH3COO)2Mn·4H2O (A), and 0.04 M K4Fe(CN)6·3H2O (B). Then, solution A was added to B dropwise at 0 °C under N2 atmosphere and aged for 2 h without stirring. The resultant precipitants were then washed with water and ethanol several times and dried under vacuum overnight at RT.The PW electrodes were prepared by dispersing the active material (70 wt%), Super C45 conductive carbon (10 wt%), Na-CMC (5 wt%), and SBR (5 wt%) in DI water. The mixture was thoroughly blended using a homogeniser. The obtained slurry was cast onto Al foil (battery-grade, Wellcos) using a doctor blade (wet thickness: 150 μm) and dried overnight in an oven at 80 °C. The resulting PW electrode sheet was punched to a diameter of 12 mm and further dried in a Büchi oven at 80 °C for 24 h under vacuum. Subsequently, the electrodes were pressed at 4 t cm−2 for 30 s and then dried again in a Büchi oven at 80 °C for 2 h under vacuum before cell assembly. The PW electrodes were directly transferred into a sealed vessel and stored in an ultrapure Argon-filled glovebox (MBraun, H2O, and O2 content <0.1 ppm). The final PW electrode active mass loading was ca. 1.2 mg cm−2.
2.3. Preparation of the cross-linked solvent-free solid polymer electrolyte
Scheme 1B shows the fabrication procedure of the cross-linked solvent-free SPEs. The solvent-free SPE membranes were manufactured in a dry room (dew point below −70 °C) in accordance with our previous work.32 All chemicals were subjected to a vacuum drying process using mechanical and turbomolecular pumps. The water content of dried raw materials is shown in Table S1.† PEO, K salt, and benzophenone (cross-linking agent, 50 mg) were mixed. Later, IL was added to obtain a mixture of PEO:K salt:IL with a molar ratio of 10:1:4. The resulting gum-like mixture was then annealed at 100 °C overnight. The transparent and homogeneous mixture was hot pressed at 100 °C with an initial and final pressure of 10 and 15 bar, respectively. The obtained solvent-free SPE membranes (thickness ∼120 μm) were cross-linked by UV light irradiation (Cube photo-irradiator, 350 W Hg lamp) for 3 min on both sides. Finally, transparent and self-standing solvent-free SPEs were skilfully fabricated. SPE without IL, named PEO10−1, is prepared using the same protocol as the reference. Based on the different K salts and IL anions, the samples were labelled as summarised in Table 1.
| Polymer | K salt | IL | Labelled as | FSI− (%) |
|---|---|---|---|---|
| PEO | KFSI | Pyr12O1FSI | FSI: FSI | 100 |
| KTFSI | Pyr12O1FSI | TFSI: FSI | 80 | |
| KFSI | Pyr12O1TFSI | FSI: TFSI | 20 | |
| KTFSI | Pyr12O1TFSI | TFSI: TFSI | 0 |
2.4. Physicochemical characterisation
X-ray diffraction (XRD) patterns were recorded to determine the crystal structure of PW and solvent-free SPEs. The PW structure was also confirmed by Fourier-transform infrared spectroscopy (FTIR) (see Fig. S1†). The thermal stability and phase transitions of solvent-free SPEs were evaluated by thermogravimetric analysis (TGA) (TG 209F1 Libra, Netzsch) and differential scanning calorimeter (DSC) (Discovery DSC, TA Instruments), respectively. Detailed equipment information and experimental parameters were provided in the ESI.†2.5. Electrochemical characterisation
EIS experiments were conducted to determine the temperature-dependent ionic conductivity. In contrast, further EIS and stripping/plating tests were conducted to investigate the compatibility of the solvent-free SPE with K. Linear sweep voltammetry (LSV) was used to assess the electrochemical stability window of all solvent-free SPEs. The cyclic performance, cycling stability, and rate capability of the solvent-free SPEs were evaluated by assembling K∥solvent-free SPE∥PW cells in a CR2032 coin cell configuration. Detailed equipment information, test steps, and test parameters were provided in the ESI.†2.6. Chemical composition of the solid electrolyte interphase
The chemical composition of the formed SEI on K was investigated using XPS and MAS-ssNMR. The equipment details and sample preparation procedure were provided in the ESI.†3. Results and discussion
3.1. Structural and thermal characterisation
The XRD patterns of all solvent-free SPEs and pure PEO are presented in Fig. 1A. Pure PEO displays two distinct crystallisation peaks at 19° and 23° 2θ, which are characteristic of PEO and have been observed in previous reports.40 In the case of solvent-free SPEs, no crystallisation peaks are observed, indicating an amorphous structure already at RT. Khudyshkina et al.40 reported that the crystallinity of solvent-free SPEs exhibits a trend of initially decreasing at low salt contents, being amorphous, and then increasing with increasing K salt content (an optimised PEO:K salt ratio of 1:12). In addition, the plasticising effect of IL further reduces the crystallinity of PEO.41 Therefore, the PEO:K salt:IL with a 10:1:4 molar ratio yields the optimum proportion for obtaining amorphous solvent-free SPEs at RT, as our group previously determined.
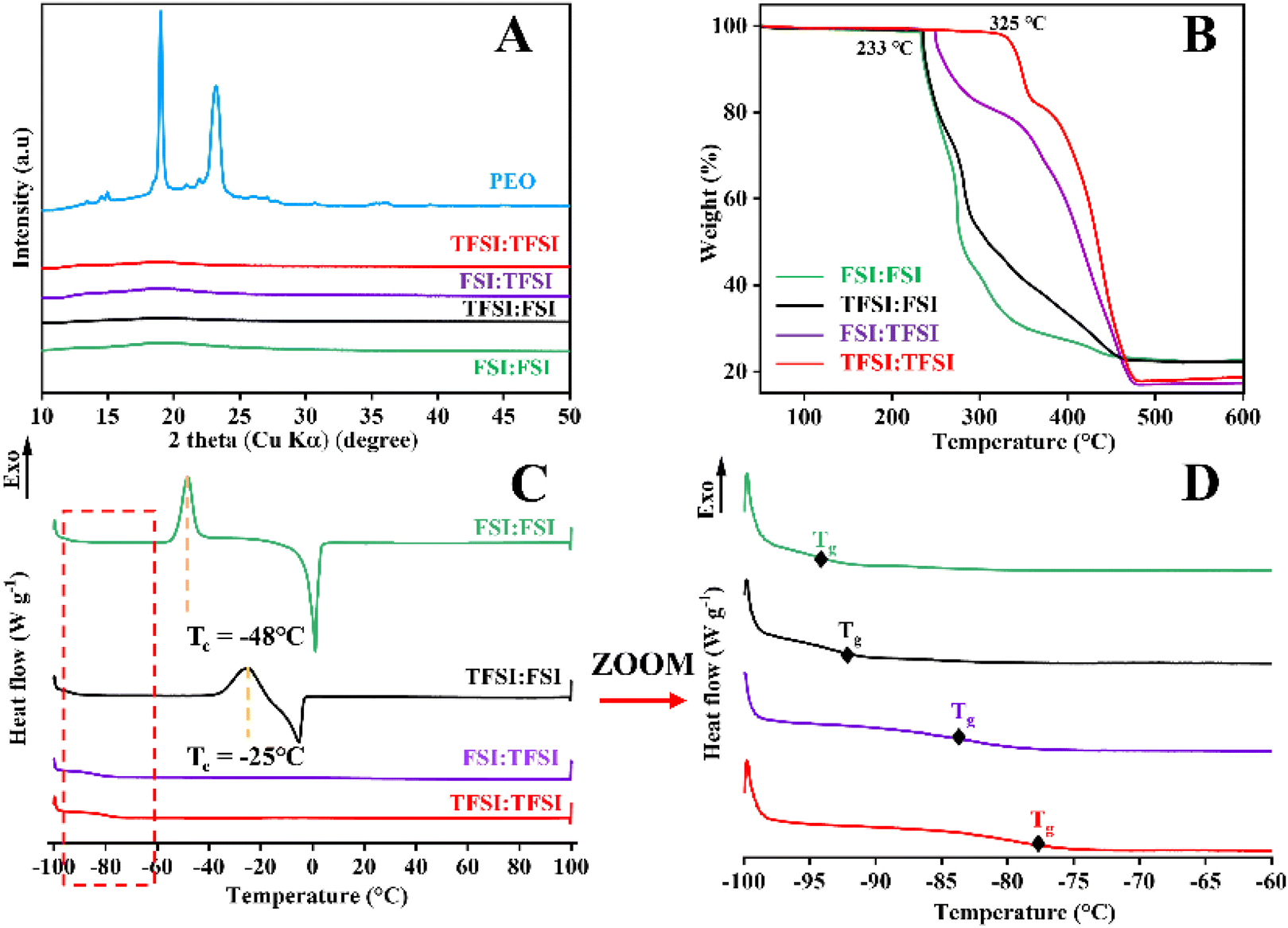 | ||
| Fig. 1 (A) XRD patterns of all solvent-free SPEs and pure PEO, (B) TGA curves (heating rate of 5 K min−1, inert N2 atmosphere), (C) DSC curves (heating cycles between −100 °C and 100 °C, heating rate of 5 K min−1, for all solvent-free SPEs), and (D) enlarged image of the selected area in (C). | ||
Thermal characteristics are important indicators for evaluating the safety performance of solvent-free SPEs, as they are closely related to the applicable temperature range of potassium batteries. The TGA curves of the solvent-free SPEs are illustrated in Fig. 1B, while the TGA curves of the raw K salts and ILs are presented in the ESI (Fig. S2A).† All solvent-free SPEs exhibit rapid thermal decomposition above 233 °C, attributed to the decomposition of the PEO, indicating high thermal stability. The fact that solvent-free SPEs experience mass loss only at such high temperatures suggests that the water content is extremely low, which is a prerequisite for electrolytes. Based on the TGA analysis and in agreement with the results obtained for the pure K salts, the highly concentrated TFSI−-based solvent-free SPEs (FSI:TFSI and TFSI:TFSI) exhibit greater thermal stability. Notably, TFSI:TFSI demonstrates the highest thermal stability up to 325 °C.
Fig. 1C and D reveal the thermal phase behaviour and glass transition temperature (Tg) of the solvent-free SPEs studied by DSC during the heating scan (see Fig. S2B† for the reversible behaviour). All solvent-free SPEs are amorphous above 20 °C, in line with the XRD results. This is primarily due to the incorporation of IL in the solvent-free SPEs, along with the cross-linking treatment, which ensures low crystallinity and improves their mechanical properties. The Tg values of the manufactured solvent-free SPEs are as follows: −94 °C (FSI:FSI), −93 °C (TFSI:FSI), −83 °C (FSI:TFSI) and −80 °C (TFSI:TFSI), showing that the incorporation of TFSI− increases the Tg, i.e., reduces the mobility of the polymer, and probably the ionic conductivity (vide infra). In addition, the DSC of FSI:FSI and TFSI:FSI, i.e., the electrolytes with high FSI− concentration (see Table 1), exhibit peaks close to −48 °C and −25 °C, respectively, corresponding to the cold crystallisation temperature (Tc). This result reveals that the incorporation of Pyr12O1FSI IL exhibits a weak interaction with PEO, K+, and FSI−, resulting in cold crystallisation behaviour. It was previously reported for Li-based SPEs that the use of different IL concentrations and/or different anions would have an impact on the occurrence of cold crystallisation.42,43 In order to understand the influence of the IL on the SPE properties, PEO:K salt (10:1 molar ratio) without IL was prepared. The PEO10−1 exhibits a melting peak at 47 °C, indicating the presence of a semi-crystalline structure, which provides evidence that ILs plasticize the PEO chains (Fig. S2C†). Furthermore, the cross-linking effect was also investigated by fabricating pure cross-linked PEO. As shown, the Tg of pure cross-linked PEO (−53 °C, Fig. S2D†) is lower than that of the non-cross-linked one,40 indicating that the cross-linking treatment also contributes to reducing the Tg and, in turn, increasing the ionic conductivity. Therefore, the observed low Tg values and crystallinity indicate higher segmental mobility of the polymer matrix, which is supported by the ion conductivity measurements (see next section).
3.2. Electrochemical characterization
Fig. 2A displays the ionic conductivity of solvent-free SPEs within the temperature range of 10 to 90 °C, which was determined by EIS. As an example, a fitted Nyquist plot is shown in Fig. S3A,† corresponding to the impedance data collected at 10 °C for the FSI:FSI.As observed in Fig. 2A, the experimental ionic conductivities of all solvent-free SPEs follow the Vogel–Tammann–Fulcher (VTF) behaviour, and their values are aligned with the Tg trend.43,44 FSI:FSI with the highest content of FSI− exhibits the lowest Tg, resulting in the highest segmental mobility of the PEO matrix and, consequently, showing the highest conductivity at all tested temperatures. In fact, FSI:FSI exhibits the highest ionic conductivity of 1.6 × 10−3 S cm−1 at 20 °C among other reported K-based SPEs31,43 (see the comparison of the ionic conductivity of the studied solvent-free SPEs with other reported works in Table S2†). Even though the FSI:FSI is the best candidate in terms of ionic conductivity, all solvent-free SPEs have demonstrated outstanding ionic conductivity at RT (20 °C), exceeding 9.0 × 10−4 S cm−1 in the lowest case. PEO10−1 (reference SPE without IL) shows significantly lower ionic conductivity at all tested temperatures, but the difference is more pronounced at lower temperatures than 60 °C. Indeed, the PEO10−1 exhibits only ionic conductivity of 5.2 × 10−7 S cm−1 at 20 °C (Fig. S3B†). It can be briefly concluded that the addition of IL rapidly enhances the ionic conductivity of solvent-free SPEs, demonstrating a beneficial approach for developing mechanically handleable PEO-based solvent-free SPEs that can operate at RT.
 | ||
| Fig. 2 (A) Temperature-dependent ionic conductivity, and (B) anodic and cathodic LSV curves (Al foil as working electrode, K as counter and reference electrode, scan rate of 0.1 mV s−1). Stripping/plating cycles in symmetrical K∥solvent-free SPE∥K cells at 20 °C and current densities of 0.1 mA cm−2: (C) FSI:FSI, (D) FSI:TFSI, (E) TFSI:FSI, (F) TFSI:FSI. Note the different scales on the x and y axes. | ||
To study the electrochemical stability window (ESW) of the solvent-free SPEs, LSV was performed, including both anodic and cathodic scans, as depicted in Fig. 2B. In line with their thermal properties, the FSI:FSI exhibit the lowest electrochemical stability, displaying the narrowest ESW. By increasing the TFSI− concentration, the ESW can be widened. This phenomenon arises from the easier reduction and oxidation of the FSI− compared to the TFSI−. However, it is reported that KFSI-containing electrolytes exhibit better passivation effects on K metal.45,46 Hence, even though the ESW is narrower, FSI:FSI may perform well within the working potential window up to 4.4 V vs. K+/K.
To corroborate the electrochemical stability of these solvent-free SPEs against K metal, a stripping/plating test was performed in symmetric K∥solvent-free SPE∥K cells, applying current density (j) of 0.1 mA cm−2 at 20 °C (1 h per process). The results are presented in Fig. 2C–F. The stripping/plating curves reveal that only FSI:FSI and TFSI:FSI exhibit stable interfaces against K, as shown in Fig. 2C and D, respectively. Once more, this indicates that the lower electrochemical stability of the FSI− is crucial for being reduced on the K metal surface, forming a more stable SEI that can accommodate the plated K. Meanwhile, the highly TFSI−-concentrated solvent-free SPEs, such as FSI:TFSI and TFSI:TFSI, exhibit poor compatibility. Specifically, the FSI:TFSI demonstrates inadequate interfacial stability, making it impossible to conduct a stripping/plating test (Fig. 2E); while the TFSI:TFSI displays a significant overpotential (close to 3 V, Fig. 2F). In contrast, the FSI:FSI exhibits more than 40 days (1000 h) of operation with remarkably low polarization of around 0.4 V, which is even lower than with organic electrolytes.45 The gradual decrease and eventual stabilization of the voltage polarization in the initial cycles might be attributed to the formation of the SEI. In fact, zooming in on the last 20 cycles of stripping/plating curves (see Fig. S4†), voltage spikes can be observed, indicating the decomposition of FSI− upon cycling. These results reveal the reactivity of the FSI− against K metal, which could facilitate the formation of a stable SEI. Therefore, the FSI:FSI configuration is the most suitable solvent-free SPE among those reported to date for K-cells.
3.3. Proof of concept: potassium-cells
As a proof of concept, all manufactured solvent-free SPEs have been employed as electrolytes, assembling K-cells using PW as the cathode active material. The 1st voltage profiles of the K∥solvent-free SPE∥PW cells are shown in Fig. 3A. From the 1st galvanostatic charge/discharge (GCD) profiles, it is observed that cells with the highest FSI− content exhibit improved charge–discharge performance, similar to that observed in liquid cells. Increasing the TFSI− concentration in the solvent-free SPEs (FSI:TFSI and TFSI:TFSI) leads to greater cell polarization, making it unsuitable as an electrolyte. Considering the FSI:FSI is the most promising solvent-free SPE in terms of acceptable specific capacity and low polarization, its cycling stability is further investigated (Fig. 3B). Even so, the cycling performance of K-metal cells using the other solvent-free SPEs is presented in Fig. S5A and B.† The FSI:FSI containing K-cell delivers a specific capacity of 97 mA h g−1 and a capacity retention of 38% after 100 cycles, with a stable coulombic efficiency (CE) of nearly 95% after several initial cycles. The non-optimal CE is primarily due to the presence of parasitic reactions at low current densities on the surface of the K metal, as indicated by the stripping/plating test, which affect the observed capacity fading. | ||
| Fig. 3 Galvanostatic cycling tests in coin-type half-cells or using the El-cell configuration, employing PW cathodes as working and K metal as counter and reference electrodes between 4.4 and 2.7 V (temperature 20°C ± 2 °C). (A) The first galvanostatic charge/discharge profiles of the PW, using the manufactured solvent-free SPEs as electrolytes in a half-cell, (B) the discharge capacity at 0.1C and corresponding CE of FSI:FSI-containing cell, (C) corresponding GCD test results in the El-cell, (D) illustration of the three-electrode battery assembly (El-cell), and (E) corresponding Nyquist plots. | ||
The K∥PW cells using the other investigated solvent-free SPEs show poorer electrochemical performances. For example, the K-cell with TFSI:FSI exhibits an initial specific capacity comparable to that of the FSI:FSI-based cell. However, it displays even larger capacity decay throughout the entire testing period (Fig. S5A†). Additionally, the CE decreases during cycling, indicating partial decomposition of the solvent-free SPE. On the other hand, the FSI:TFSI-based cell exhibits rapid capacity decay in the initial ten cycles, followed by low-capacity stabilization (only 35 mA h g−1) and a very unstable CE.
To understand the capacity fading of FSI:FSI-containing cells, the stability of the formed cathode-electrolyte interphase (CEI) was investigated using EIS. The impedance has been studied in three-electrode cell configurations (El-cell, Fig. 3D) using PW as the working electrode and K as the counter and reference electrode. The results reveal that the interfacial resistance remains stable at OCV, even after 6 h (see Nyquist plot in Fig. 3E). After the 1st GCD, the overall resistance increases, suggesting CEI formation. However, after the 10th GCD, the interfacial resistance decreased, indicating that the CEI is probably stabilizing after cycling. This suggests that the observed capacity fading in the FSI:FSI-based K cells is not related to the CEI but rather to the K interface. Therefore, the SEI has also been studied and discussed in the next section. A similar test was carried out using TFSI:TFSI as the electrolyte (Fig. S6†) to understand its poor electrochemical properties. However, the significant cell polarization induced by the TFSI:TFSI does not permit investigation of the resistance response upon cycling.
Since the K-cell containing FSI:FSI electrolyte exhibits the best electrochemical performance, further investigation has been conducted, including a rate capability test. The rate capability of the K∥FSI:FSI∥PW cell is illustrated in Fig. 4A and B. The cell delivers a specific discharge capacity of 82, 54, 17, and 5 mA h g−1 at 0.1C, 0.2C, 0.5C, and 1C, respectively. It is observed that as the current density increases, the cell's capacity gradually diminishes, as expected. One of the main reasons could be an increase in polarization resulting from an increase in the C rate, as observed in the voltage profile (Fig. 4A). Therefore, the K+ diffusion is kinetically limited. Fortunately, the PW remains capable of providing a stable discharge capacity at a high discharge rate of 0.5C. However, further optimisation should be carried out to enhance the electrochemical properties of the cell. Notably, the K∥FSI:FSI∥PW cell recovers its capacity after applying a back 0.1C, indicating that the FSI:FSI maintains its structural integrity and functionality across various current densities.
 | ||
| Fig. 4 Electrochemical performance of K∥FSI:FSI∥PW cells between the operation voltage of 4.4–2.7 V vs. K+/K at 20°C ± 2 °C. (A) Charge and discharge curves, and (B) rate capability from 0.1C to 1C and corresponding CE, (C) 1st, 2nd, and from 10th to 100th charge–discharge curves, and (D) cycling performance and corresponding CE at 1C. | ||
Fig. 4C and D show the specific capacity of K∥FSI:FSI∥PW cell at 1C and 20 °C, delivering a capacity of 40 mA h g−1 after 100 cycles and CEs close to ∼99%. In the first 50 cycles, the PW displays a gradual increase in capacity, which can be attributed to the decomposition of the electrolyte to form a SEI on the K surface, as well as some kinetic issues due to the poor wettability that is improved upon cycling.
3.4. Investigation of the interfacial properties of solvent-free SPEs∥K
The reactivity and properties of the formed SEI on the surface of K metal against solvent-free SPE significantly affect the performance of the battery, including the cycling stability, reversibility, and safety. To understand how different anions influence the surface of K, the FSI:FSI and TFSI:TFSI systems have been selected, and the transport and chemical properties of the formed interphase have been investigated. To this end, two solutions were prepared, consisting of KFSI: Pyr12O1FSI (solution 1) and KTFSI: Pyr12O1TFSI (solution 4), in a 1:4 molar ratio (Fig. 5A1). Note that, after cycling, the solvent-free SPE is attached to the K metal and PW cathode, making it impossible to detach and study the surface properly. Therefore, an alternative approach was developed, such as immersing a piece of fresh K metal in those solutions for several weeks (note that the K salt:IL ratio maintains the same as in the solvent-free SPEs).
 | ||
| Fig. 5 (A) Photographs of K in a solution consisting of K salt and IL (1:4 molar ratio); the composition of the solution is described in the box on the left (A1). The photographs of K in a solution over time are illustrated in (A2–A6). (B) EIS experiments performed in three-electrode cell configurations (El-cell, WE, CE and RE are K-metal), and the corresponding Nyquist plots of K∥K cell using as solvent-free SPES B1 FSI:FSI, and B2 TFSI:TFI. | ||
The K metal placed in both solutions (0 days, Fig. 5A2) initially exhibits a metallic colour, suggesting a clean surface. After 7 days (Fig. 5A3), the surface of K in KTFSI:Pyr12O1TFSI solution turned dark purple, while no significant changes were observed in the K immersed in the KFSI:Pyr12O1FSI. After 14 days (Fig. 5A4), KTFSI:Pyr12O1TFSI solution became turbid, suggesting that K pieces and/or surface species had precipitated. In contrast, no obvious changes are still noted in the KFSI:Pyr12O1FSI solution, although the K appears less shiny than before. After 28 days (Fig. 5A5), the KTFSI:Pyr12O1TFSI solution became more cloudy, while the KFSI:Pyr12O1FSI remained stable. To identify the species formed on the surface of the K, small metal pieces from each solution were collected for XPS (Fig. 6) and MAS-ssNMR (Fig. 7) analysis. Finally, after 35 days (Fig. 5A6), the K in the KFSI:Pyr12O1FSI solution is no longer shiny and has turned lavender, indicating the formation of new surface species, potentially SEI species. Meanwhile, the species formed on the surface of the K in the KTFSI: Pyr12O1TFSI appeared to dissolve rather than create a protective layer. The formation of the SEI (FSI:FSI system), rather than its dissolution into the IL solution (for TFSI:TFSI), can dramatically improve the stability against K metal, as indicated by the stripping/plating tests.
 | ||
| Fig. 6 XPS of the K metal surface and bulk (depth of 8 nm, Ar+ etching speed of 0.8 nm min−1) after immersing for 28 days in (A) KFSI:Pyr12O1FSI or (B) KTFSI:Pyr12O1TFSI solutions (the grey open points represent the experimental data, and the solid lines represent the fitting results). | ||
 | ||
| Fig. 7 MAS-ssNMR spectra of pristine and K reacted FSI:FSI and TFSI:TFSI. (A) 1H spectra were acquired with 8 scans and a relaxation delay of 5 s. (B) 13C spectra were acquired with 384 scans and a relaxation delay of 60 s. (C) 19F spectra were acquired using 16 scans and a 10 s relaxation delay for the pristine samples and 128 scans with a 60 s relaxation delay for the reacted samples. Spinning sidebands, marked with an asterisk, are visible in the spectra due to the samples spinning used to increase the spectral resolution. | ||
Fig. 5B shows the Nyquist plots of the K∥K cells, tested with FSI:FSI or TFSI:TFSI solvent free SPEs. The impedance of the K∥FSI:FSI∥K cell is lower than that of the TFSI:TFSI-based K cell. Indeed, the impedance of the FSI:FSI-based K cell (Fig. 5B1) remains stable after 6 h of rest, in agreement with the lack of differences observed on the solution and K surface during the initial 7 days, as shown in the photograph. After the 1st stripping/plating test, the interfacial resistance slightly increased, which might be related to the formation of the SEI due to electrochemically driven electrolyte reduction reactions. Fig. 5B2 displays the Nyquist plot of the K cell containing TFSI:TFSI, which exhibits a higher initial interfacial resistance compared to the K cell with FSI:FSI. The interfacial resistance continues to increase significantly over time, remaining unstable even after 6 h, in agreement with the darkening of the K observed in the photographs. Unfortunately, due to the high overpotential observed in the K symmetric cells using TFSI:TFSI electrolyte (see Fig. 2F), it was not possible to perform stripping/plating after OCV to further investigate the impedance response.
The chemical composition of the surface layer on top of the K metal is analyzed in detail by XPS. Fig. 6 illustrates the XPS spectra of F 1s, K 2p, C 1s, and S 2p photoelectron regions of the formed surface layer and after 10 min of Ar ion sputtering, which corresponds to 8 nm depth profiling on the K immersed in the two solutions, i.e., KFSI:Pyr12O1FSI and KTFSI:Pyr12O1TFSI for 28 days.
The F 1s spectra display a strong peak at ∼688 eV for K in both solutions, whether on the surface or in bulk (8 nm), corresponding to the FSI− or TFSI− species.47,48 However, the K surface is also covered by KF, detected at 684 eV, when immersed in KFSI:Pyr12O1FSI solution, corresponding to the decomposition of FSI−.49–51 Caracciolo et al.52 analysed the SEI composition of the K surface after being immersed in a KFSI-based liquid electrolyte (using EC:DEC as solvent) for 21 days and concluded that the reactivity was driven by the degradation of FSI−, resulting in an inorganic-rich SEI, similar to it is observed in FSI:FSI solvent-free SPE. Nonetheless, no KF signal was detected at the surface of the K metal in contact with KTFSI:Pyr12O1TFSI solution (Fig. 6B), indicating that the TFSI− is chemically more stable, not decomposing and forming a KF-rich SEI. A similar behavior was observed for the Na-based system, in fact.32 Hence, the inorganic-rich SEI promoted by the formation of KF can effectively inhibit the formation of K dendrites,53 in agreement with the better electrochemical properties observed for the PW tested in FSI:FSI cells. Additionally, as the detection depth increases to 8 nm, the KF signal disappears on the surface of K immersed in KFSI:Pyr12O1FSI solution, indicating that the SEI formed is thinner than 8 nm (F 1s, Fig. 6A).
The K 2p spectra of both K metals show that the K surface is mainly covered by fluorinated salts, such as KFSI/KF or KTFSI, in agreement with the F 1s region.54,55 Interestingly, the K immersed in the KTFSI: Pyr12O1TFSI solution exhibits a new peak, which corresponds mostly to the K metal and appears prior to depth profiling, indicating that K in the TFSI:TFSI system does not form a dense SEI. This is consistent with the K dissolving over time, as seen in the photographs of Fig. 5A.
Regarding the carbon-based species (C 1s), two main peaks are observed at ∼285 and 287–286 eV, corresponding to hydrocarbons and C–N bonds from the Pyr12O1+ of the IL. In addition, the K surface immersed in KFSI:Pyr12O1FSI solution exhibits a small peak at 288.5 eV, which may be due to the presence of some O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds resulting from the reactivity of the K metal with moisture or air.49,50
O bonds resulting from the reactivity of the K metal with moisture or air.49,50
The S 2p region displays the strongest peak originating from FSI− and TFSI− (169–170 eV). However, the K metal in contact with KFSI:Pyr12O1FSI solution also displays another peak at 168.4 eV (S 2p3/2), which can be attributed to new S–O bonds, such as those of S2O32− or S2O52−, produced from the decomposition of FSI−, and in agreement with an inorganic-rich SEI.52,56
In short, by comparing the chemistry of the formed SEI in the surface and bulk of K in the two solutions, it can be concluded that FSI− reacts with K to form an inorganic-rich SEI (KF, K2S2O3, and K2S2O5) that could protect the bulk K from a further decomposition reaction, while TFSI− cannot create an optimum SEI, leading to high interfacial resistance and in turn, large polarization.
Moreover, FSI:FSI and TFSI:TFSI have been analyzed using MAS-ssNMR at pristine and after reaction with K. The reaction has been performed by briefly grinding the pristine solvent-free SPEs with K in a mortar and pestle. Fig. 7A displays the 1H spectra of the pristine and K-reacted FSI:FSI and TFSI:TFSI. The spectra have been truncated in the intensity axis to emphasize the low-intensity resonances corresponding to the solvent-free SPE decomposition products. In the FSI:FSI, new resonances between 0.7–1.3 ppm in the spectrum of the reacted solvent-free SPEs are attributed to –CH3− or –CH2− fragments, likely originating from the decomposition of PEO or the IL. A prominent resonance at 2.4 ppm and lower intensity resonance at 2.7 ppm are consistent with CHx groups adjacent to a N atom from the IL.
The 1H spectrum of the reacted TFSI:TFSI exhibits some impurities from a different sample containing Pyr12O1+, reflected by three large peaks between 0.5–2 ppm. Despite these impurities, a comparison of the pristine and reacted TFSI:TFSI reveals decomposition products at approximately 1 and 1.3 ppm, similar to the FSI:FSI. However, the spectrum of the reacted TFSI:TFSI does not contain new resonances of N-bonded species that appeared in the FSI:FSI reacted sample at 2.4 ppm.
The 13C spectra (Fig. 7B) have relatively low sensitivity, which makes it difficult to detect low-population species such as reaction products. In the spectrum of the FSI:FSI, a unique resonance is observed at ∼54 ppm, while the TFSI:TFSI spectrum displays a minor peak at ∼46 ppm. These resonances are attributed to –CH3− and –CH2− groups, corroborating the decomposition assignments made in the 1H spectra.
The 19F spectrum of FSI: FSI (Fig. 7C) reveals the formation of fluorine-containing decomposition products. While the dominant resonance at ∼50 ppm corresponds to the FSI−, additional decomposition products are detected at ∼−70 ppm, attributed to fluoromethyl compounds (CF3-R) formed through the reaction of K with the electrolyte. In the TFSI:TFSI, the primary 19F resonance at ∼−80 ppm corresponds to CF3-S in the TFSI−. At very low intensity, similar decomposition products are detected as those observed in the FSI:FSI, resonating at ∼−70 ppm. Notably, unlike the XPS results, MAS-ssNMR 19F spectra did not detect the formation of KF (expected at ∼−125 ppm), probably due to the very low KF concentration formed, mainly on the surface of the K, as indicated by the XPS. Overall, these findings suggest that grinding the solvent-free SPEs with K metal results in limited decomposition for both FSI:FSI and TFSI:TFSI. Nevertheless, the 1H and 13C spectra indicate that FSI:FSI exhibits slightly more decomposition products than TFSI:TFSI, which is in line with the XPS results.
4. Conclusion
In this work, a simple hot-press and UV cross-linking method has been used to fabricate self-standing solvent-free SPEs for potassium batteries. The ionic conductivity of all solvent-free SPEs is enhanced by incorporating ILs. The influence of anion concentration on solvent-free SPEs, i.e., FSI− – or TFSI−, has been roughly investigated. The PEO:KFSI:Pyr12O1FSI demonstrated excellent ionic conductivity of 1.6 × 10−3 S cm−1 at RT (20 °C) and exhibits the best electrochemical performance in K cells using PW as a cathode. The improved electrochemical properties of the PEO:KFSI:Pyr12O1FSI can be attributed to the reactivity of the FSI− against K, which leads to the formation of an inorganic-rich SEI comprised of KF and S–O species. The results reveal that while the FSI− is a promising candidate for forming a stable SEI, the TFSI− does not exhibit similar capabilities. In addition, the FSI− improves the compatibility and stability with PW, resulting in better cycling stability and CE compared to TFSI−. In summary, this work prepared and evaluated a solvent-free SPE with obvious advantages, positioning it as a potential candidate for potassium batteries. Additionally, understanding the mechanism behind the SEI formation against K metal, and CEI with PW will assist researchers in optimising the SPEs for potassium-based systems in future studies.Data availability
Data for this article, including diffraction, thermal, electrochemical impedance spectroscopy, electrochemical cycling and spectroscopy data, are available at the Zenodo repository at https://doi.org/10.5281/zenodo.15167912.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the Bundersministerium für Forschung, Technologie und Raumfahrt (BMFTR) with her “SPIRIT” project (03XP0186) within the M.Era-net framework and the Helmholtz Association. Grant PCI2022-133005, funded by MCIN/AEI/10.13039/501100011033 and the European Union PRTR funding through projects, is also acknowledged. Project DEAL enabled and organized open-access funding. A. Ares-Dorado and J. Carretero-Gonzalez are acknowledged for the fruitful discussion and supporting on PEO10-1 SPE.References
- International Energy Agency, https://www.iea.org/reports/world-energy-outlook-2024, 2024.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS PubMed.
- G. Chen, Y. Bai, Y. Gao, Z. Wang, K. Zhang, Q. Ni, F. Wu, H. Xu and C. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 43252–43260 CrossRef CAS PubMed.
- X. Min, J. Xiao, M. Fang, W. Wang, Y. Zhao, Y. Liu, A. M. Abdelkader, K. Xi, R. V. Kumar and Z. Huang, Energy Environ. Sci., 2021, 14, 2186–2243 RSC.
- S. Yoda and K. Ishihara, J. Power Sources, 1997, 68, 3–7 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1–24 Search PubMed.
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS PubMed.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- W. Zhang, J. Lu and Z. Guo, Mater. Today, 2021, 50, 400–417 CrossRef CAS.
- Y. Xu, T. Ding, D. Sun, X. Ji and X. Zhou, Adv. Funct. Mater., 2023, 33, 2211290 CrossRef CAS.
- R. Rajagopalan, Y. Tang, X. Ji, C. Jia and H. Wang, Adv. Funct. Mater., 2020, 30, 1–35 CrossRef.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- B. John, V. Anoopkumar and T. D. Mercy, ACS Appl. Energy Mater., 2020, 3, 9478–9492 CrossRef.
- M. Zarrabeitia, J. Carretero-González, M. Leskes, H. Adenusi, B. Iliev, T. J. S. Schubert, S. Passerini and E. Castillo-Martinez, Energy Mater., 2023, 3, 300046 CAS.
- Y. Xu, M. Titirici, J. Chen, F. Cora, P. L. Cullen, J. S. Edge, K. Fan, L. Fan, J. Feng, T. Hosaka, J. Hu, W. Huang, T. I. Hyde, S. Imtiaz, F. Kang, T. Kennedy, E. J. Kim, S. Komaba, L. Lander, P. N. Le Pham, P. Liu, B. Lu, F. Meng, D. Mitlin, L. Monconduit, R. G. Palgrave, L. Qin, K. M. Ryan, G. Sankar, D. O. Scanlon, T. Shi, L. Stievano, H. R. Tinker, C. Wang, H. Wang, H. Wang, Y. Wu, D. Zhai, Q. Zhang, M. Zhou and J. Zou, J. Phys. Energy, 2023, 5, 021502 CrossRef CAS.
- Group1, https://www.prnewswire.com/, 2024.
- R. Verma, P. N. Didwal, J. Y. Hwang and C. J. Park, Batteries Supercaps, 2021, 4, 1428–1450 CrossRef CAS.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, 1–22 Search PubMed.
- H. Yin, C. Han, Q. Liu, F. Wu, F. Zhang and Y. Tang, Small, 2021, 17, 1–23 Search PubMed.
- H. Zhang and M. Armand, Isr. J. Chem., 2021, 61, 94–100 CrossRef CAS.
- M. B. Armand, Rev. Mater. Res., 1986, 16, 245–261 CrossRef CAS.
- H. Zhang, C. Liu, L. Zheng, F. Xu, W. Feng, H. Li, X. Huang, M. Armand, J. Nie and Z. Zhou, Electrochim. Acta, 2014, 133, 529–538 CrossRef CAS.
- C. Xin, K. Wen, S. Guan, C. Xue, X. Wu, L. Li and C. W. Nan, Front. Mater., 2022, 9, 1–9 Search PubMed.
- X. Zhu, R. N. Ali, M. Song, Y. Tang and Z. Fan, Polymers, 2022, 14 Search PubMed.
- M. L. Lehmann, G. Yang, D. Gilmer, K. S. Han, E. C. Self, R. E. Ruther, S. Ge, B. Li, V. Murugesan, A. P. Sokolov, F. M. Delnick, J. Nanda and T. Saito, Energy Storage Mater., 2019, 21, 85–96 CrossRef.
- M. L. Lehmann, G. Yang, D. Gilmer, K. S. Han, E. C. Self, R. E. Ruther, S. Ge, B. Li, V. Murugesan, A. P. Sokolov, F. M. Delnick, J. Nanda and T. Saito, Energy Storage Mater., 2019, 21, 85–96 CrossRef.
- Y. Fu, Z. Gu, Q. Gan and Y. W. Mai, Mater. Sci. Eng., R, 2020, 747, 137335 Search PubMed.
- W. Wang, Z. Fang, M. Zhao, Y. Peng, J. Zhang and S. Guan, Chem. Phys. Lett., 2020, 747, 137335 CrossRef CAS.
- V. K. Singh, S. K. Singh, H. Gupta, Shalu, L. Balo, A. K. Tripathi, Y. L. Verma and R. K. Singh, J. Solid State Electrochem., 2018, 22, 1909–1919 CrossRef CAS.
- Q. Liu, Y. Liu, X. Jiao, Z. Song, M. Sadd, X. Xu, A. Matic, S. Xiong and J. Song, Energy Storage Mater., 2019, 23, 105–111 CrossRef.
- A. Hofmann, M. Schulz and T. Hanemann, Electrochim. Acta, 2013, 89, 823–831 CrossRef CAS.
- D. Roscher, Y. Kim, D. Stepien, M. Zarrabeitia and S. Passerini, Batteries Supercaps, 2023, 6, e20230009 Search PubMed.
- A. Boschin and P. Johansson, Electrochim. Acta, 2016, 211, 1006–1015 CrossRef CAS.
- G. B. Appetecchi, G. T. Kim, M. Montanino, M. Carewska, R. Marcilla, D. Mecerreyes and I. De Meatza, J. Power Sources, 2010, 195, 3668–3675 CrossRef CAS.
- M. Elmanzalawy, E. Sanchez-Ahijón, O. Kisacik, J. Carretero-González and E. Castillo-Martínez, ACS Appl. Energy Mater., 2022, 5, 9009–9019 CrossRef CAS.
- A. Kotronia, K. Edström, D. Brandell and H. D. Asfaw, Adv. Energy Sustainability Res., 2022, 3, 2100122 CrossRef CAS.
- L. Porcarelli, C. Gerbaldi, F. Bella and J. R. Nair, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- M. Falco, C. Simari, C. Ferrara, J. R. Nair, G. Meligrana, F. Bella, I. Nicotera, P. Mustarelli, M. Winter and C. Gerbaldi, Langmuir, 2019, 35, 8210–8219 CrossRef CAS PubMed.
- X. Wu, Z. Jian, Z. Li and X. Ji, Electrochem. Commun., 2017, 77, 54–57 CrossRef CAS.
- A. D. Khudyshkina, P. A. Morozova, A. J. Butzelaar, M. Hoffmann, M. Wilhelm, P. Theato, S. S. Fedotov and F. Jeschull, ACS Appl. Polym. Mater., 2022, 4, 2734–2746 CrossRef CAS.
- I. Osada, H. De Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- E. Simonetti, M. Carewska, G. Maresca, M. De Francesco and G. B. Appetecchi, J. Electrochem. Soc., 2017, 164, A6213–A6219 CrossRef CAS.
- P. Stigliano, C. Ferrara, N. Pianta, A. Gentile, L. Mezzomo, R. Lorenzi, V. Berbenni, R. Ruffo, G. B. Appetecchi and P. Mustarelli, Electrochim. Acta, 2022, 412, 140123 CrossRef CAS.
- K. M. Diederichsen, H. G. Buss and B. D. McCloskey, Macromolecules, 2017, 50, 3831–3840 CrossRef CAS.
- M. Hamada, R. Tatara, K. Kubota, S. Kumakura and S. Komaba, ACS Energy Lett., 2022, 7, 2244–2246 CrossRef CAS.
- T. Hosaka, S. Muratsubaki, K. Kubota, H. Onuma and S. Komaba, J. Phys. Chem. Lett., 2019, 10, 3296–3300 CrossRef CAS PubMed.
- M. Zarrabeitia, L. Gomes Chagas, M. Kuenzel, E. Gonzalo, T. Rojo, S. Passerini and M. Á. Muñoz-Márquez, ACS Appl. Mater. Interfaces, 2019, 11, 28885–28893 CrossRef CAS PubMed.
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Nano Energy, 2019, 55, 327–340 CrossRef CAS.
- Z. Feng, R. Chen, R. Huang, F. Zhang, W. Liu and S. Liu, Metals, 2023, 13 Search PubMed.
- W. Zhang, W. K. Pang, V. Sencadas and Z. Guo, Joule, 2018, 2, 1534–1547 CrossRef CAS.
- Z. Jiang, N. Li, L. Li, F. Tan, J. Huang and S. Huang, Adv. Mater., 2024, 2311127, 1–14 Search PubMed.
- L. Caracciolo, L. Madec, G. Gachot and H. Martinez, ACS Appl. Mater. Interfaces, 2021, 13, 57505–57513 CrossRef CAS PubMed.
- H. Yang, W. Wang, Z. Huang, Z. Wang, L. Hu, M. Wang, S. Yang and S. Jiao, Adv. Mater., 2024, 2401008, 1–13 Search PubMed.
- H. Wang, D. Zhai and F. Kang, Energy Environ. Sci., 2020, 13, 4583–4608 RSC.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- V. Murray, D. S. Hall and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A329–A333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02762d |
| This journal is © The Royal Society of Chemistry 2025 |