
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigner pseudopeptides: autofluorescent polygonal tubes via Phe-zipper and triple helix†
V. Haridas *ab,
Govind P. Maurya‡
a and
Souvik Dutta‡a
*ab,
Govind P. Maurya‡
a and
Souvik Dutta‡a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India. E-mail: haridasv@chemistry.iitd.ac.in
bDepartment of Chemistry, Indian Institute of Technology Palakkad, Palakkad, Kerala-678623, India. E-mail: haridasv@iitpkd.ac.in
First published on 18th September 2024
Abstract
Chemists are increasingly turning to biology for inspiration to develop novel and superior synthetic materials. Here, we present an innovative peptide design strategy for tubular assembly. In this simple design, a phenylene urea unit is introduced as an aglet at the N-terminus of the peptide. When α-amino isobutyric acid (Aib) is the first residue and phenylalanine (Phe) is the second residue from the phenylene urea entity, it induces an edge-to-face π–π interaction resulting in a turn conformation. The peptides with a unique reverse turn conformation associate to form polygonal peptide tubes via a Phe-zipper arrangement, as evidenced by microscopic and single crystal X-ray studies. Ultra-microscopic imaging revealed that the tubular assembly is hexagonal, square, and triangular in shape. This hierarchical assembly reveals the interplay between π–π interactions and hydrogen bonding. In another design, pseudopeptide 5, wherein a Phe–Phe (FF) unit is linked to phenylene urea, formed polygonal tubes via a triple helical arrangement. Interestingly, the extension of this design to the bis-urea core resulted in vesicular assembly. These supramolecular polygonal tubes and vesicles showed autofluorescence, which allowed confocal imaging. The observed fluorescence is an additional advantage for applications in biological and medical sciences.
Introduction
The functions of biomacromolecules are associated with their uniquely folded three-dimensional structures.1 The folded biomacromolecules can further associate to form functional assemblies. The complex folding and assembly processes are hallmarks of the biomolecular world. The knowledge of molecular association is essential for creating artificial functional systems. Therefore, the design of small self-assembling molecules can provide useful models for an in-depth understanding of the association of biomacromolecules.2 A powerful strategy for designing self-assembling peptides is the scaffold approach.3 In this strategy, a well-designed scaffold places the amino acids/peptides, for facilitating a specific self-assembling pattern.4 The self-assembly of intricately folded biomolecules involves the interplay of various non-covalent forces such as hydrogen bonding, van der Waals interactions, π–π stacking, and hydrophobic effects, resulting in functional assemblies.5 The chirality of self-assembled structures is one of the recently emerging topics in the area of biomolecular assembly.6Self-assembly is a promising approach for fabricating novel functional materials that can be used for various applications, such as, drug delivery, ion transport, biosensing and catalysis.7 The design of functional materials based on molecular self-assembly with supramolecular chirality has received increasing attention in recent years.8 Among them, peptides are recognized as versatile building blocks,9 since such systems open more avenues for biocompatible designs. Additionally, the side chains of amino acids play an important role in supramolecular organization.10
Self-assembly has been used by Ghadiri and co-workers for creating cyclic peptide nanotubes.11 Heterochiral and hybrid peptide nanotubes are excellent candidates for the transmembrane transport of Na+/K+ ions via channel mechanisms.12 Lynn and co-workers used an amyloid-based strategy for peptide nanotube fabrication.13 Gazit and co-workers utilized simple dipeptide nanotubes for the fabrication of silver nanowires.5b,14 Alternatively, carbon nanotubes are extensively explored for several applications.15 Self-assembly is a powerful strategy for the design of functional tubular assemblies; however the designs are still limited.3e,12b–d
Creating versatile molecular inserts to control the conformation regardless of the amino acid sequence is a challenging endeavor.16 Here, we delineate that the introduction of a simple phenylene urea unit at the N-terminus, as an aglet, controls the conformation and thereby facilitates peptide assembly. The simple N-terminal modification not only influences the conformation, but also leads to hierarchical self-assembly. The unique and rare hexagonal, square, and triangular peptide microtubes are a new finding. Additionally, the microtubes have been found to be autofluorescent in nature, further enhancing their value for applications in the fields of biological and medical sciences.
Results and discussion
The introduction of a phenylene urea unit at the N-terminus of the peptide provides additional hydrogen bond donors and acceptors along with a π-bonding unit. We envisioned that the additional donors (NH) and acceptors (CO) could enhance hydrogen bonding (intra and inter), leading to folding and facile self-assembly. In our design, we envisioned that a phenyl unit attached to urea could enhance the potential for π–π interactions.Commercially available phenyl isocyanate upon reaction with the N-deprotected peptide yielded a urea peptide. Simple peptides bearing this phenylene urea unit as an aglet at the N-terminus were synthesized and investigated for their conformation and self-assembly. A series of monourea peptides (1–5) was readily synthesized from the corresponding peptide fragments and phenyl isocyanate.
To probe the conformation of the molecules, we initially used NMR spectroscopic methods. The possibility of intramolecular H-bonding in 1 (Fig. 1) was investigated by NMR titration upon addition of DMSO-d6 to the acetonitrile-d3 solution. The addition of DMSO-d6 to the acetonitrile-d3 solution of mono-urea 1 resulted in downfield shifts of NHc, NHa and NHb protons (Fig. 1 and S1a†).
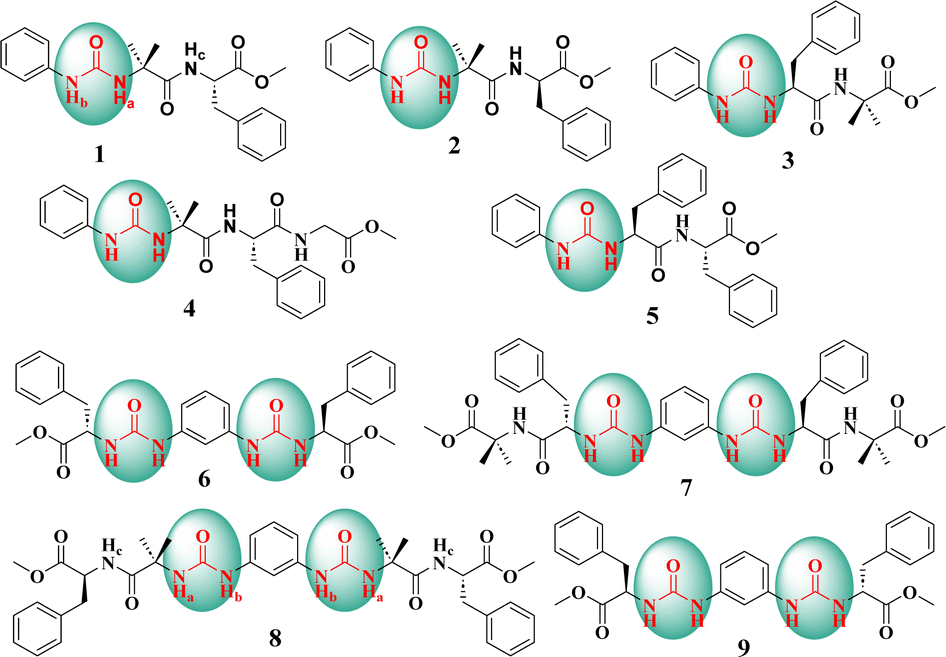 | ||
| Fig. 1 Chemical structures of the amino acid-based mono-urea compounds 1–5 and bis-urea compounds 6–9. | ||
The downfield shifts of Δδ 0.241, Δδ 0.678 and Δδ 0.943 ppm were observed for NHc, NHa and NHb, respectively (Fig. S1b†). The significantly lower downfield shift of NHc, indicates an intramolecular hydrogen bond between NHc and the ester carbonyl group (Fig. S1c†). Upon analyzing all the possibilities of H-bonds (Fig. S2†), we could arrive at the intramolecular C5 hydrogen bonded structure. The J-value of NHc was found to be 5 Hz. The CD data of compound 1, showed two positive bands at 200 and 220 nm, indicating the turn conformation (Fig. 2b).17
 | ||
| Fig. 2 (a) Crystal structure of peptide 1 showing the turn conformation. Intramolecular edge-to-face π–π interaction is shown as green dotted lines, (b) CD spectrum of peptide 1 in acetonitrile, (c) space-filling model of aromatic rings showing the Phe–Phe zipper arrangement. The zoomed-in part shows the orientation of aromatic rings. | ||
The infrared (IR) spectra showed that the stretching frequency of ester carbonyls in 1 and 2 is at 1737 cm−1, while in peptide 5, it is at 1743 cm−1 (Fig. S3†). The lower wavenumber of ester carbonyl stretching in 1 and 2 compared to that in 5, revealed the presence of hydrogen bonding in 1 and 2. Both NMR and IR data together support the intramolecular C5 hydrogen bonding in 1 and 2.
The chemical shifts of CαH of amino acid were used to ascertain the nature of the secondary structure.18 This chemical shift index (CSI) evaluates the folding, if any. The CSI value of Phe in peptides 1 and 2 was found to be “0”, thereby indicating a random coil or a turn conformation (Table S4†).
The crystals of pseudopeptide 1 suitable for single crystal X-ray crystallography were obtained from an acetonitrile solution after slow evaporation. The solid state structure revealed that peptide 1 crystallizes in the P 21 21 21 space group (Fig. 2a). Careful examination of the crystal structure of 1 showed that the aromatic ring of Phe forms an intramolecular edge-to-face π–π interaction with the phenyl ring of the phenylene urea motif, thereby adopting a turn-like conformation (Fig. 2a). The X-ray structure revealed an intramolecular C5 hydrogen bond, involving Phe carbonyl and amide NH. The important backbone torsion angles are listed in Table 1 and the hydrogen bonding interactions are listed in Table S5.† The phenyl ring of Phe is at a distance of 3.5 Å from the aromatic ring of phenylene urea, causing a turn conformation (Fig. 2a). This type of unusual turn has some resemblance to the turns observed in proteins, wherein an intramolecular 10-membered hydrogen bond is observed.
|
||||
|---|---|---|---|---|
| Φ1/° | Ψ1/° | Φ2/° | Ψ2/° | |
| Peptide 1 | 59.41 | 46.31 | −146.41 | 172.60 |
| Peptide 2 | −59.00 | −46.69 | 146.99 | −172.81 |
| Peptide 5 | −61.32 | 138.63 | −59.83 | 135.53 |

The intramolecular CH–π interaction in peptide 1 resembles the 10-membered H-bond found in a classical β-turn (Fig. 3). Considering each aromatic unit as an atom (numbered as 1 to 10 in Fig. 3), the turn structure with CH–π interactions at the terminal is analogous to a β-turn (Fig. 3). In a typical β-turn, the distance between terminal residues (Ciα and Ci+3α) is ∼7 Å,19,20 while in the case of peptide 1, the distance between terminal aromatic units is ∼5.6 Å. The backbone topology and some torsional angles of peptide 1 are similar to those found in the classical type I′ turn.19b,c Therefore, peptide 1 can be considered a topological mimic of the β-turn due to its short Ciα and Ci+3α distance and topology.
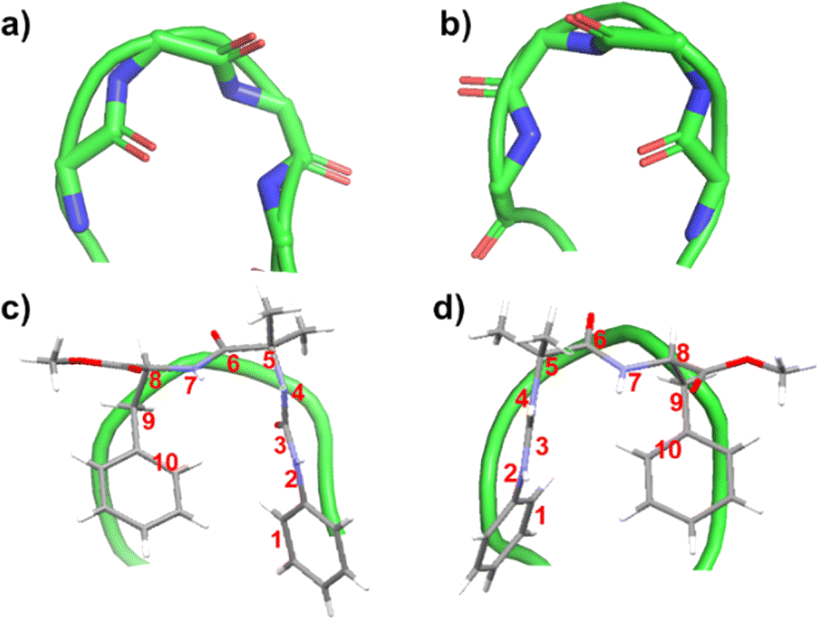 | ||
| Fig. 3 (a) Typical type I′ β-turn (PDB ID: 1KKO) and (b) typical type I β-turn (PDB ID: 2BK9). The green ribbon traces the backbone of protein. Overlayed images of (c) peptide 1 over a typical type I′ β-turn and (d) peptide 2 placed over typical type I β-turn. The numbering has been done considering each aromatic unit as one atom. | ||
Furthermore, peptide 1 assembles through intermolecular H-bonding and π–π interactions. This intermolecular H-bonding is also evident from concentration-dependent NMR studies of peptide 1. Increasing the concentration results in a downfield shift of NHs in CD3CN (Fig. S4†). The broadening of 1H NMR signals is evident in the concentration dependent studies (Fig. S4†), which also indicates association of the molecules at higher concentrations. There are two intermolecular N–H⋯O hydrogen bonds between Aib amide and urea carbonyl involving urea NHs. The distances of these hydrogen bonds are 2.2 and 2.3 Å, respectively (Fig. S5†). The association through intermolecular π–π interactions between aromatic rings and H-bonding completes the tubular structure (Fig. S6a†).
From the crystal structure analysis, the dimensions of the cavity of the nanotube are found to be 5 Å and 14 Å (Fig. S6b†). A careful study of the packing of molecules revealed that the reverse turn further assembles through a Phe-zipper arrangement (Fig. 2c). Ultramicroscopic imaging revealed that the mono urea derivative 1 self-assembled to form a nanotubular structure (Fig. 4a, red arrow). The morphology of 1 was studied by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 4d, red arrow). SEM and TEM imaging show that the width of the tube is in the range of 100–400 nm, which indicates that the tube is formed from the aggregation of several nanotubular structures into a microtube (Fig. 4c, e and g). The tubular structure of 1 was further confirmed by atomic force microscopy (AFM) imaging (Fig. 4f and S7†).
 | ||
| Fig. 4 SEM images of 1 show (a) tubular assembly (indicated by red arrows), (b) intermediate stage of microtube formation (marked by red arrows) and (c) microtube structure. TEM images of 1 show (d) tubular assembly (indicated by red arrows) and (e) microtube structure (indicated by red arrows). AFM image of 1 shows (f) microtubular assembly and (g) schematic representation of self-assembly of 1 to a microtube. | ||
In order to delineate the formation of the microtube, a careful analysis of SEM and TEM images was performed. The microscopic images clearly showed the semi-tubular portion of the microtube (Fig. 4b and d, red arrow). This semi-tubular arrangement is an indication of hierarchical organization of nanotubes to microtubes. The peptide nanotubes further associate through several non-covalent interactions to a microtubular structure. FESEM imaging of 1 at an angle of 10° revealed the open-ended nature of the assembly (Fig. 5a and b). Additionally, we observed hexagonal, square, and triangular tubes with diameters ranging from 500–1600 nm and a wall thickness of 100 nm.
 | ||
| Fig. 5 FESEM images of 1 taken at an angle of 10° from the surface showing the open end of the microtubular assembly. The images reveal that tubular assembly is (a) triangular, square, and (b) hexagonal in shape. Zoomed images are shown in the insets (red arrows showing triangular tubes and yellow arrows showing square-shaped tubes); (c) FESEM images of 5 taken at an angle of 10° from the surface showing the open end of the microtubular assembly. | ||
In another design, we replaced L-Phe of 1 with D-Phe to obtain 2. The single crystal X-ray diffraction study of 2 revealed a similar conformation, but the assembly was right-handed instead of left-handed as in 1. Peptide 2 also showed a Phe-zipper arrangement as in 1. The SEM (Fig. 6a) and FESEM (Fig. S8†) images of 2 also show polygonal tubular assembly.
 | ||
| Fig. 6 SEM images of (a) 2, (b) 4 and (c) 5 showing their tubular morphology, and SEM images of (d) 6, (e) 7 and (f) 8 showing their spherical morphology. The samples were prepared by dissolving 1 mg of respective compounds in 1 mL of ACN. | ||
To investigate the role of π-interactions in the formation of the bioactive turn conformation and the tube assembly, we designed a few mutant versions of peptide 1. The shuffling of the amino acids in 1, resulted in 3. We envisioned that peptide 3 will not be able to make a turn as in 1, and hence would not exhibit nanotube formation. Investigation of the self-assembly of the mono-urea derivative 3 revealed the same. To delineate the scope of the assembly, we designed a series of peptides (Fig. 1 and Table 2), where the first and second residues were systematically changed. We also designed another peptide with Gly as the third residue to peptide 1 to obtain peptide 4, which showed tubular assembly in SEM imaging (Fig. 6b). Hence the strategy is somewhat versatile, as we can add a third residue to design a longer peptide, still making a similar self-assembling pattern.
| Compounds | Morphology of the self-assembled structure |
|---|---|
| Peptide 1 | Tubular |
| Peptide 2 | Tubular |
| Peptide 3 | — |
| Peptide 4 | Tubular |
| Peptide 5 | Tubular |
| Peptide 6 | Vesicle |
| Peptide 7 | Vesicle |
| Peptide 8 | Vesicle |
| Peptide 9 | Vesicle |
In another design, Aib was replaced with Phe to obtain a diphenylalanine peptide with urea at the end. The FF motif is known to assemble to form nanotubes.5b The SEM imaging of 5 showed tubular assembly (Fig. 6c). The FESEM imaging of 5 at an angle of 10° revealed the open-ended nature of the assembly (Fig. 5c).
The X-ray quality crystal of 5 was obtained from an acetonitrile–hexane mixture solution upon slow evaporation. The solid state structure revealed that 5 crystallizes in the P65 space group. Careful analysis reveals that there exist three intermolecular hydrogen bonds in between two consecutive molecules. Both the urea NHs show H-bonding with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the urea group of the next molecule. The amide CO is H-bonded with the amide-NH of the neighboring molecule. The packing showed a left-handed helical arrangement. All the phenyl groups are arranged in a helical fashion, hence forming a triple helical arrangement (Fig. 7c). The X-ray structure revealed no intramolecular CH–π interaction as in 1. However, the phenyl group of the C-terminal Phe (Phe2) shows an intermolecular edge-to-face π–π interaction with the phenyl ring of the phenylene urea motif of the next molecule (distance 2.66 Å) (Fig. 8). Repeating six molecular units with an average twist of ∼60° between each molecule results in a complete turn of 360° (Fig. S9†).
O of the urea group of the next molecule. The amide CO is H-bonded with the amide-NH of the neighboring molecule. The packing showed a left-handed helical arrangement. All the phenyl groups are arranged in a helical fashion, hence forming a triple helical arrangement (Fig. 7c). The X-ray structure revealed no intramolecular CH–π interaction as in 1. However, the phenyl group of the C-terminal Phe (Phe2) shows an intermolecular edge-to-face π–π interaction with the phenyl ring of the phenylene urea motif of the next molecule (distance 2.66 Å) (Fig. 8). Repeating six molecular units with an average twist of ∼60° between each molecule results in a complete turn of 360° (Fig. S9†).
 | ||
| Fig. 7 (a) Single crystal structure of compound 5, (b) left-handed helical arrangement of 5, and (c) the left-handed helical arrangement of (I) phenyl ring of phenylene urea, (II) middle phenyl unit (Phe1), (III) terminal phenyl unit (Phe2), and (IV) overlap of (I), (II) and (III) showing a triple helical arrangement. | ||
 | ||
| Fig. 8 Zoomed-in image of the triple helix formed by peptide 5 showing edge-to-face π–π interactions between the aromatic ring of the urea unit (colored in orange) and the phenyl ring of the second Phe-residue (Phe2) (colored in green). | ||
In peptide 5, the aromatic units make intermolecular contact through π–π interactions. All the three different aromatic units (phenyl of urea, Phe1 and Phe2) are arranged in a helical fashion (Fig. 7) and appear as a left-handed triple helical arrangement. The aromatic ring close to the urea unit is involved in intermolecular edge-to face π–π interaction with the phenyl ring of the C-terminal Phe-residue. This arrangement keeps the phenyl ring of the first Phe-residue (Phe1) also in a helical fashion.
To study the self-assembly by spectroscopic investigation, we monitored the absorption and fluorescence spectra of 1 upon increasing concentration. Careful analysis of the absorption spectra revealed a small absorption at longer wavelengths at higher concentrations (Fig. S10a†). Recently, we have found that autofluorescence is observed in vesicular self-assembly.21 The pseudopeptide 1 at higher concentrations (λex = 365 nm) exhibited fluorescence (Fig. S10b†).
We utilized this autofluorescence to image the assembled pseudopeptide tubes using a confocal microscope. The confocal microscopic images were acquired upon excitation at 405 nm, 488 nm, and 560 nm (Fig. 9a–c). The wider excitation and emission range is a noteworthy observation.
 | ||
| Fig. 9 Confocal images of compound 1 upon excitation at (a) 405 nm, (b) 488 nm, and (c) 560 nm. Confocal images of compound 7 upon excitation at (d) 405 nm, (e) 488 nm, and (f) 560 nm. | ||
We envisaged that two urea moieties could strengthen and extend the association bidirectionally, thereby leading to a new self-assembled system. With this in mind, we synthesized bis-urea peptides 6 and 7 with phenylalanine positioned near the urea unit and another peptide 8 in which phenylalanine was located farthest from the urea unit (Fig. 1). Previously, we reported that Trp-appended bis-urea facilitates vesicle formation through Trp-zipper assembly.22 In the present investigation, the bis-urea peptide appended with Phe exhibited a self-assembly different from that observed with mono-urea peptides. The 1H NMR titration study showed that upon addition of DMSO-d6 to the acetonitrile-d3 solution of 6 resulted in a downfield shift of NHa (Δδ 0.66 ppm) and NHb (Δδ 1.02 ppm) of the urea moiety, indicating the strong intermolecular hydrogen bonding between the molecules (Fig. 1 and S11†).23
The J-values of NHs (8.0 Hz) of compounds 6 and 7 indicate an extended conformation, favorable for self-assembly by stacking.
The CD spectra of 6 and 7, in which the phenylalanine units are close to the urea unit, showed a strong positive cotton effect at 230 nm and a negative band at 210 nm, as a result of exciton coupling (Fig. S12c and d†). The zero-crossover observed at 218 nm, close to the λmax of the absorption spectrum indicates a supramolecular helical arrangement in 6 and 7.24 The positive band (230 nm) and negative band at 210 nm indicate the right-handed helicity of 6 and 7 (Fig. S12c and d†). The chiral helical organization in 6 was further confirmed by synthesizing the D-chiral compound 9 (Fig. 1). The CD spectrum of 9 is opposite in sign to that of 6, inferring left-handed organization (Fig. S13†). To probe the impact of the amino acid sequence on the assembly, we designed peptide 8. The 1H NMR spectra recorded after the addition of DMSO-d6 (15%) to a solution of 8 in CD3CN showed a downfield shift of NHa (Δδ 0.61 ppm) and NHb (Δδ 0.90 ppm) protons of the urea moiety. However, the amide proton NHc displayed a very small (Δδ 0.20 ppm) shift (Fig. S14†), indicating its involvement in intramolecular hydrogen bonding.
The CD spectrum of 8, in which phenylalanine was placed far from the urea moiety, showed two positive bands at 200 and 220 nm, indicative of the turn conformation (Fig. S15b†).17,25 The CSI value of Phe in peptide 8 was found to be “0”, indicating a random coil or a turn conformation (Table S4†). The results underscore that the position of Phe is crucial for the supramolecular helical arrangement. The CD spectrum of 8 (Fig. S15b†) was similar to that of 1 (Fig. 2b), which further supports the presence of a turn conformation in both compounds 1 and 8. In the case of 8, the CD spectrum remains unchanged with varying concentrations, indicating the absence of any chiral assembly (Fig. S15c†).
The urea moiety containing two NH protons and one CO unit can associate through α-type H-bonding. The urea NHs can act as hydrogen bond donors to anionic species. The anion binding ability of bis-urea derivatives 6, 7 and 8 was analyzed by various spectroscopic methods such as UV-vis, fluorescence, CD, and NMR spectroscopy. Various anions have been screened for binding studies. Fluoride (F−), chloride (Cl−), bromide (Br−), iodide (I−) and sulphate (HSO4−) did not show significant binding (Fig. S16†). However, dihydrogen phosphate (H2PO4−) showed considerable binding ability. The CD spectra showed changes upon addition of H2PO4− to 6 and 7. The addition of one equivalent of H2PO4− to 6 and 7 resulted in the inversion of the CD spectra (Fig. 10a and b). The CD spectral changes indicate the chiral switching from a right- to a left-handed helical arrangement upon binding with H2PO4− ions. The observed crossover at 230 nm, corresponds to λmax indicating a left-handed helical arrangement (Fig. 10a and b). The CD spectra of the urea–H2PO4− complex of 6 and 7, changed upon addition of water, indicating the reversible nature of the supramolecular helix (Fig. 10a and b). The addition of H2PO4− to 8, showed a strong negative band at 238 nm and a positive band at 220 nm, indicating a change from a turn conformation to a left-handed helix (Fig. 10c). The reversibility was not observed in the case of 8 upon addition of H2O.
 | ||
| Fig. 10 CD spectra of (a) 6, (b) 7 and (c) 8 alone and with H2PO4−, followed by addition of water. | ||
The UV-vis spectra of urea peptides 6, 7 and 8 showed two bands at 287 and 294 nm (Fig. S17a–c†). On addition of H2PO4− to 6, 7 and 8, a bathochromic shift of about 5 nm was observed along with an isosbestic point at 295 nm, indicating H2PO4− binding (inset Fig. S17a–c†). Anion binding was also investigated by fluorescence spectroscopy. The fluorescence emission spectra of 6, 7 and 8 showed an emission band at 311 nm upon excitation at 280 nm. The emission band showed a bathochromic shift of about 5 nm along with an increase in emission intensity upon phosphate binding (Fig. S17d–f†). The stoichiometry of the complex formed by 6, 7 and 8 with H2PO4− was determined by Job plot using UV-vis spectroscopy. The stoichiometry of the host–guest complex suggested the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (Fig. S18a–c†). The binding constants for H2PO4− were calculated using the Benesi–Hildebrand (BH) plot26 and found to be 2.75 × 103 M−1 (6), 1.1 × 103 M−1 (7) and 1.6 × 103 M−1 (8) (Fig. S18d–f†).
:1 complex (Fig. S18a–c†). The binding constants for H2PO4− were calculated using the Benesi–Hildebrand (BH) plot26 and found to be 2.75 × 103 M−1 (6), 1.1 × 103 M−1 (7) and 1.6 × 103 M−1 (8) (Fig. S18d–f†).
The binding of H2PO4− was further confirmed by the 1H NMR titration experiment. On addition of H2PO4− to the solutions of 6 and 8, the urea NHs (NHa and NHb) were downfield shifted (Fig. S19†), indicating strong binding to H2PO4−. The aromatic protons on the central core showed an upfield shift, indicating electron reorganization in the aromatic ring upon anion binding.22 The aromatic protons of phenylalanine showed a downfield shift upon phosphate binding.
The bis-urea compounds containing urea linkages and amides could facilitate the formation of extended self-assembled structures through non-covalent interactions. The presence of an aromatic amino acid further enhances the association through π–π interactions. We monitored the self-assembly of 6, 7 and 8 using SEM imaging. Vesicular self-assembly (Fig. 6d–f) was observed in all the bis-urea compounds. The vesicles formed by peptides 6 and 7 were found to have diameters in the range of 1100–1400 nm, while the vesicles of 8 were in the range of 500–650 nm in diameter (Fig. S20†). The bis-urea vesicles showed autofluorescence. The confocal images of compound 7 were acquired upon excitation at 405 nm, 488 nm, and 560 nm (Fig. 9d–f).
The bent nature of the 1,3-phenyl unit provides adequate curvature, while the presence of urea and amide bonds favors self-assembly, leading to vesicles. The results from ultramicroscopic imaging revealed that bis-urea-cored molecules have strong tendency to associate to form vesicles. The self-assembly of all Phe-containing compounds to vesicles, irrespective of their conformational preferences, implies the overarching role of π–π interactions in the vesicular assembly.
Conclusions
In conclusion, we presented an innovative strategy for designing a bioactive turn conformation utilizing π–π interactions. Additionally, the turn-forming peptides associate via a Phe-zipper arrangement to form polygonal microtubes. This hierarchical assembly depends on the amino acid sequence and is supported by crystallographic and microscopic studies. Phenylene urea containing the FF peptide assembles to form a left-handed triple helical structure through π–π interactions. The design is further extended to bis-urea peptides that showed vesicular assembly, attributed to the kink in the backbone. Both tubular and vesicular assemblies showed auto-fluorescence, which enabled us to image the self-assembled structure using confocal microscopy.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
VH conceived the idea and designed the molecules. GPM and SD synthesized all the compounds, performed all the experiments, and analysed all the collected data under the supervision of VH. GPM and SD contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding (grant number EMR/2017/003192/OC) from the Science & Engineering Research Board (SERB), Government of India. We thank the Department of Chemistry, Central Research Facility (CRF) and Sophisticated Analytical & Technical Help Institutes (SATHI) of IIT Delhi for all the experimental facilities. We also thank DST: SR/FST/CSII-07/2014 and the Institutions of Eminence (IoE) grant from the University Grants Commission India for funding the single-crystal diffractometer at the Department of Chemistry, IIT Delhi. GPM thanks the University Grants Commission (UGC) for the doctoral fellowship. SD thanks IIT Delhi for the Institute Fellowship. VH thanks IIT Palakkad for the seed grant.References
- (a) J. S. Richardson, Adv. Protein Chem., 1981, 34, 167–339 CrossRef CAS PubMed; (b) C. I. Branden and J. Tooze, Introduction to Protein Structure, Garland Science, 2012 CrossRef.
- (a) G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC; (b) Q. Zhang, Y.-X. Deng, H.-X. Luo, C.-Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D.-H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CrossRef CAS PubMed; (c) I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- (a) W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed; (b) H. Singh, A. Chenna, U. Gangwar, J. Borah, G. Goel and V. Haridas, Chem. Sci., 2021, 12, 15757–15764 RSC; (c) V. Haridas, S. Sadanandan, M. V. S. Gopalakrishna, M. B. Bijesh, R. P. Verma, S. Chinthalapalli and A. Shandilya, Chem. Commun., 2013, 49, 10980–10982 RSC; (d) V. Haridas, S. Sadanandan, Y. K. Sharma, S. Chinthalapalli and A. Shandilya, Tetrahedron Lett., 2012, 53, 623–626 CrossRef CAS; (e) H. Singh, N. Khatoon, S. K. Bhardwaj, P. Kampani, T. K. Nayak and V. Haridas, ChemBioChem, 2023, 24, e202300502 CrossRef CAS PubMed.
- (a) T. Moriuchi and T. Hirao, Chem. Soc. Rev., 2004, 33, 294–301 RSC; (b) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- (a) A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem, 2020, 4, 615–634 CrossRef CAS; (b) M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed; (c) C. H. Görbitz, Chem.–Eur. J., 2001, 7, 5153–5159 CrossRef; (d) I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- (a) P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS PubMed; (b) S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed.
- (a) S. Dhawan, S. Ghosh, R. Ravinder, S. S. Bais, S. Basak, N. M. A. Krishnan, M. Agarwal, M. Banerjee and V. Haridas, Bioconjugate Chem., 2019, 30, 2458–2468 CrossRef CAS PubMed; (b) F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed; (c) B. Shen, Y. Kim and M. Lee, Adv. Mater., 2020, 32, 1905669 CrossRef CAS PubMed.
- (a) G. P. Maurya, D. Verma, A. Sinha, L. Brunsveld and V. Haridas, Angew. Chem., Int. Ed., 2022, 61, e202209806 CrossRef CAS PubMed; (b) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (c) C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, 1904965 CrossRef CAS PubMed.
- (a) S. S. Panda, K. Shmilovich, A. L. Ferguson and J. D. Tovar, Langmuir, 2019, 35, 14060–14073 CrossRef CAS PubMed; (b) R. Garifullin and M. O. Guler, Chem. Commun., 2015, 51, 12470–12473 RSC.
- M. Tanaka, Y. Demizu, M. Doi, M. Kurihara and H. Suemune, Angew. Chem., Int. Ed., 2004, 43, 5360–5363 Search PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- (a) M. J. Strauss, D. Asheghali, A. M. Evans, R. L. Li, A. D. Chavez, C. Sun, M. L. Becker and W. R. Dichtel, Angew. Chem., Int. Ed., 2019, 58, 14708–14714 CrossRef CAS PubMed; (b) D. Ranganathan, V. Haridas, R. Gilardi and I. L. Karle, J. Am. Chem. Soc., 1998, 120, 10793–10800 CrossRef CAS; (c) S. Dhawan, H. Singh, S. Dutta and V. Haridas, Bioorg. Med. Chem. Lett., 2022, 68, 128733 CrossRef CAS PubMed; (d) V. Haridas and S. Jawla, in Supramolecular Nanotechnology, ed. O. Azzaroni and M. Conda-Sheridan, Wiley, 1st edn, 2023, 845–867 Search PubMed.
- (a) K. Lu, L. Guo, A. K. Mehta, W. S. Childers, S. N. Dublin, S. Skanthakumar, V. P. Conticello, P. Thiyagarajan, R. P. Apkarian and D. G. Lynn, Chem. Commun., 2007, 2729–2731 RSC; (b) K. Lu, J. Jacob, P. Thiyagarajan, V. P. Conticello and D. G. Lynn, J. Am. Chem. Soc., 2003, 125, 6391–6393 CrossRef CAS PubMed; (c) A. K. Mehta, K. Lu, W. S. Childers, Y. Liang, S. N. Dublin, J. Dong, J. P. Snyder, S. V. Pingali, P. Thiyagarajan and D. G. Lynn, J. Am. Chem. Soc., 2008, 130, 9829–9835 Search PubMed.
- W. Ji, Y. Tang, P. Makam, Y. Yao, R. Jiao, K. Cai, G. Wei and E. Gazit, J. Am. Chem. Soc., 2021, 143, 17633–17645 CrossRef CAS PubMed.
- (a) I. A. Kinloch, J. Suhr, J. Lou, R. J. Young and P. M. Ajayan, Science, 2018, 362, 547–553 CrossRef CAS PubMed; (b) P. M. Ajayan and S. Lijima, Nature, 1993, 361, 333–334 CrossRef CAS.
- N. L. Truex, Y. Wang and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 13882–13890 CrossRef CAS PubMed.
- (a) T. Muraoka, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 2946–2947 CrossRef CAS PubMed; (b) C. T. Chang, C.-S. C. Wu and J. T. Yang, Anal. Biochem., 1978, 91, 13–31 CrossRef CAS PubMed; (c) A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CrossRef CAS.
- D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS PubMed.
- (a) G. D. Rose, L. M. Glerasch and J. A. Smith, Adv. Protein Chem., 1985, 37, 1–109 CrossRef CAS PubMed; (b) O. Koch and G. Klebe, Proteins, 2009, 74, 353–367 Search PubMed; (c) D. Ranganathan, V. Haridas, S. Kurur, A. Thomas, K. P. Madhusudanan, R. Nagaraj, A. C. Kunwar, A. V. S. Sarma and I. L. Karle, J. Am. Chem. Soc., 1998, 120, 8448–8460 CrossRef CAS.
- C. M. Venkatachalam, Biopolymers, 1968, 6, 1425–1436 CrossRef CAS PubMed.
- R. Sapra, M. Gupta, K. Khare, P. K. Chowdhury and V. Haridas, Analyst, 2023, 148, 973–984 RSC.
- V. Haridas, S. Sadanandan, S. Dhawan, R. Mishra, I. Jain, G. Goel, Y. Hu and S. Patel, Org. Biomol. Chem., 2017, 15, 1661–1669 RSC.
- (a) Y. Zhang, Y. Zhong, A. L. Connor, D. P. Miller, R. Cao, J. Shen, B. Song, E. S. Baker, Q. Tang, S. V. S. R. K. Pulavarti, R. Liu, Q. Wang, Z. Lu, T. Szyperski, H. Zeng, X. Li, R. D. Smith, E. Zurek, J. Zhu and B. Gong, J. Am. Chem. Soc., 2019, 141, 14239–14248 CrossRef CAS PubMed; (b) A. E. Hooper, S. R. Kennedy, C. D. Jones and J. W. Steed, Chem. Commun., 2016, 52, 198–201 RSC.
- (a) K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., 2004, 116, 1249–1253 CrossRef; (b) C. Roche, H.-J. Sun, P. Leowanawat, F. Araoka, B. E. Partridge, M. Peterca, D. A. Wilson, M. E. Prendergast, P. A. Heiney, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and V. Percec, Nat. Chem., 2016, 8, 80–89 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2354491–2354493. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04023f |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |