
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evolution of bismuth-based metal–organic frameworks for efficient electroreduction of CO2†
Lili
Li
a,
Xinchen
Kang
 ab,
Meng
He
a,
Alena
Sheveleva
ac,
Kui
Hu
a,
Shaojun
Xu
de,
Yiqi
Zhou
fg,
Jin
Chen
a,
Sergei
Sapchenko
a,
George
Whitehead
a,
Iñigo J.
Vitorica-Yrezabal
a,
Laura
Lopez-Odriozola
a,
Louise S.
Natrajan
a,
Eric J. L.
McInnes
ac,
Martin
Schröder
*a,
Sihai
Yang
*a and
Floriana
Tuna
*ac
ab,
Meng
He
a,
Alena
Sheveleva
ac,
Kui
Hu
a,
Shaojun
Xu
de,
Yiqi
Zhou
fg,
Jin
Chen
a,
Sergei
Sapchenko
a,
George
Whitehead
a,
Iñigo J.
Vitorica-Yrezabal
a,
Laura
Lopez-Odriozola
a,
Louise S.
Natrajan
a,
Eric J. L.
McInnes
ac,
Martin
Schröder
*a,
Sihai
Yang
*a and
Floriana
Tuna
*ac
aDepartment of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: M.Schroder@manchester.ac.uk; Sihai.Yang@manchester.ac.uk; Floriana.Tuna@manchester.ac.uk
bBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Science, Beijing, 100190, China
cPhoton Science Institute, University of Manchester, Manchester, M13 9PL, UK
dUK Catalysis Hub, Research Complex at Harwell, Didcot, OX11 0FA, UK
eCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
fDepartment of Materials, University of Manchester, Manchester, M13 9PL, UK
gInstitute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China
First published on 18th August 2022
Abstract
Understanding the structural and chemical changes that reactive metal–organic frameworks (MOFs) undergo is crucial for the development of new efficient catalysts for electrochemical reduction of CO2. Here, we describe three Bi(III) materials, MFM-220, MFM-221 and MFM-222, which are constructed from the same ligand (biphenyl-3,3′,5,5′-tetracarboxylic acid) but which show distinct porosity with solvent-accessible voids of 49.6%, 33.6% and 0%, respectively. We report the first study of the impact of porosity of MOFs on their evolution as electrocatalysts. A Faradaic efficiency of 90.4% at −1.1 V vs. RHE (reversible hydrogen electrode) is observed for formate production over an electrode decorated with MFM-220-p, formed from MFM-220 on application of an external potential in the presence of 0.1 M KHCO3 electrolyte. In situ electron paramagnetic resonance spectroscopy confirms the presence of ·COOH radicals as a reaction intermediate, with an observed stable and consistent Faradaic efficiency and current density for production of formate by electrolysis over 5 h. This study emphasises the significant role of porosity of MOFs as they react and evolve during electroreduction of CO2 to generate value-added chemicals.
Introduction
Electrochemical reduction of CO2 into fuels and chemical feedstocks enables the storage of renewable electrical energy and is a highly desirable process for carbon neutrality.1–5 Formate (or formic acid) has a wide range of applications in industry as a preserving, antibacterial agent as well as liquid fuel, and over one million tonnes of formate is produced annually via carbonylation of methanol.6,7 The electroreduction of CO2 to formate has attracted much attention, and non-precious metals such as Sn, Co, In, Tl, Cd, Hg show catalytic activity for this process.8–12 However, these metals generally exhibit drawbacks, such as high cost, toxicity, and low catalytic selectivity. Bismuth is a relatively benign main group metal environmentally13–15 and tends to show a poor activity for hydrogen evolution reaction (HER),16 which is the main competitive side-reaction during the electrochemical CO2 reduction reaction (CO2RR). Suppressing the HER will greatly increase the Faradaic efficiency (FE) for the formation of carbon-based products.Porous metal–organic frameworks (MOFs) have emerged as efficient catalysts for the CO2RR owing to their atomically dispersed metal sites and porous structure.2 Bismuth-based MOFs are reported to undergo structural evolution to afford active catalysts (primarily Bi nanosheets and particles) during electrochemical reduction of CO2.17–21 Monitoring the evolution of these Bi materials and exploring the catalytic activity of the resultant materials for CO2RR are important to the discovery of efficient new electrocatalysts. Previous studies report various Bi-MOFs constructed from different bridging ligands, but the impact of their porosity on their structural evolution under electrochemical conditions remains largely unexplored.
Herein, we report a comprehensive study of the structural evolution during the CO2RR of three Bi-MOFs, namely MFM-220, MFM-221 and MFM-222, constructed from the same ligand (biphenyl-3,3′,5,5′-tetracarboxylic acid, H4L). The MOFs were synthesised by solvothermal reactions of Bi(NO3)3·5H2O and H4L under different conditions and they show distinct porosity (solvent-accessible void ranging 49.6% to 0%). Powder X-ray diffraction (PXRD), infrared (IR) and Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and scanning electron microscopy (SEM) have been used to characterise the structural evolution of these Bi-MOFs upon reaction with the electrolyte and on application of an external potential; the new materials are denoted as MFM-200/-221/-222-e (e = electrolyte) and MFM-200/-221/-222-p (p = potential), respectively. MFM-220-p, derived from MFM-220 with the highest porosity, shows the best catalytic performance for CO2RR compared with MFM-221-p and MFM-222-p. Thus, in 0.1 M KHCO3 electrolyte solution, a total current density of 23 mA cm−2 at −1.1 V vs. RHE (RHE = reversible hydrogen electrode) and FEformate of 90.4% are observed using an electrode decorated with evolved MFM-220-p. Moreover, MFM-220-p remains active for the CO2RR for at least 5 h. Electron paramagnetic resonance (EPR) spectroscopy using the spin trap, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) reveals the presence of ·COOH radicals as a reaction intermediate and rationalises the observed high catalytic stability. This study demonstrates the important role of porosity of MOFs on their evolution to active electrocatalysts for CO2RR.
Results and discussion
Solvothermal reactions of Bi(NO3)3·5H2O and H4L in CH3CN and dimethylformamide (DMF) with different amounts of HNO3 (5%) and different reaction times (see ESI† for details) afford single crystals of MFM-220 {[Bi2(L)1.5(H2O)2]·3.5DMF·3H2O}, MFM-221 {[Bi(L)]·Me2NH2·1.5DMF} and MFM-222 {[Bi2(HL)·(H2L)·(DMF)·(OH)]} in pure phase (Fig. S1†). MFM-220, MFM-221 and MFM-222 all crystallize in monoclinic systems (Table S1†). We have previously reported22 MFM-220, which shows a non-interpenetrated neutral framework structure constructed from binuclear {Bi2} centres bridged by tetracarboxylate ligands (Fig. 1a and b). MFM-220 co-crystallizes in α- and β-forms with slightly altered coordination environments at the Bi(III) sites (differing only in the coordination of a H2O molecule) but with the same pore size and adsorption properties. In both phases, each Bi(III) ion is coordinated to three carboxylate groups from three different L4− ligands, and the two Bi(III) ions in the binuclear {Bi2} centres share three coordinated oxygen atoms from three bridging carboxylate groups. In α-MFM-220, each Bi(III) ion also coordinates to one terminal H2O to give a coordination number of 8 for Bi1 and 9 for Bi2 (Fig. 1a). MFM-220 displays micropores with a pore size of 6.5 Å, and the accessible solvent voids is 49.6% calculated by Platon (Fig. 1b, S2a and S4†).23 The synthesis and structural characteristics of MFM-221 and MFM-222 have not been reported previously. MFM-221 forms an anionic non-interpenetrated framework where the secondary building units are formed by binuclear {Bi2} moieties. Each Bi(III) ion binds to five ligands with four chelating carboxylates and one in monodentate mode, and adjacent Bi(III) ions are bridged by two carboxylates, giving a coordination number of 9 (Fig. 1c). The pore is occupied by counter-cations Me2NH2+ and uncoordinated DMF molecules (Fig. S2b and S5†), the Me2NH2+ cations being generated by in situ decompositions of the DMF solvent during the reaction. MFM-221 becomes porous after removing free DMF molecules, resulting in a solvent-accessible volume of 33.6% calculated by Platon (Fig. 1d).23 MFM-222 has a neutral, non-interpenetrated and non-porous structure constructed from binuclear {Bi2} centres bridged by tetracarboxylate ligands. The Bi1 and Bi2 centres share one carboxylate oxygen from a ligand and a bridging hydroxyl group (μ2-OH). Bi1 is coordinated to six ligands, two in bidentate and four in monodentate mode. Bi2 is coordinated to five ligands, two in bidentate and three in monodentate mode (Fig. 1e). Bi2 also has a bound DMF molecule giving an overall non-porous structure for MFM-222 (Fig. 1f, S2c and S6†). The purity of bulk materials of MFM-220, MFM-221 and MFM-222 (Fig. S7†) was confirmed by PXRD (Fig. S8†).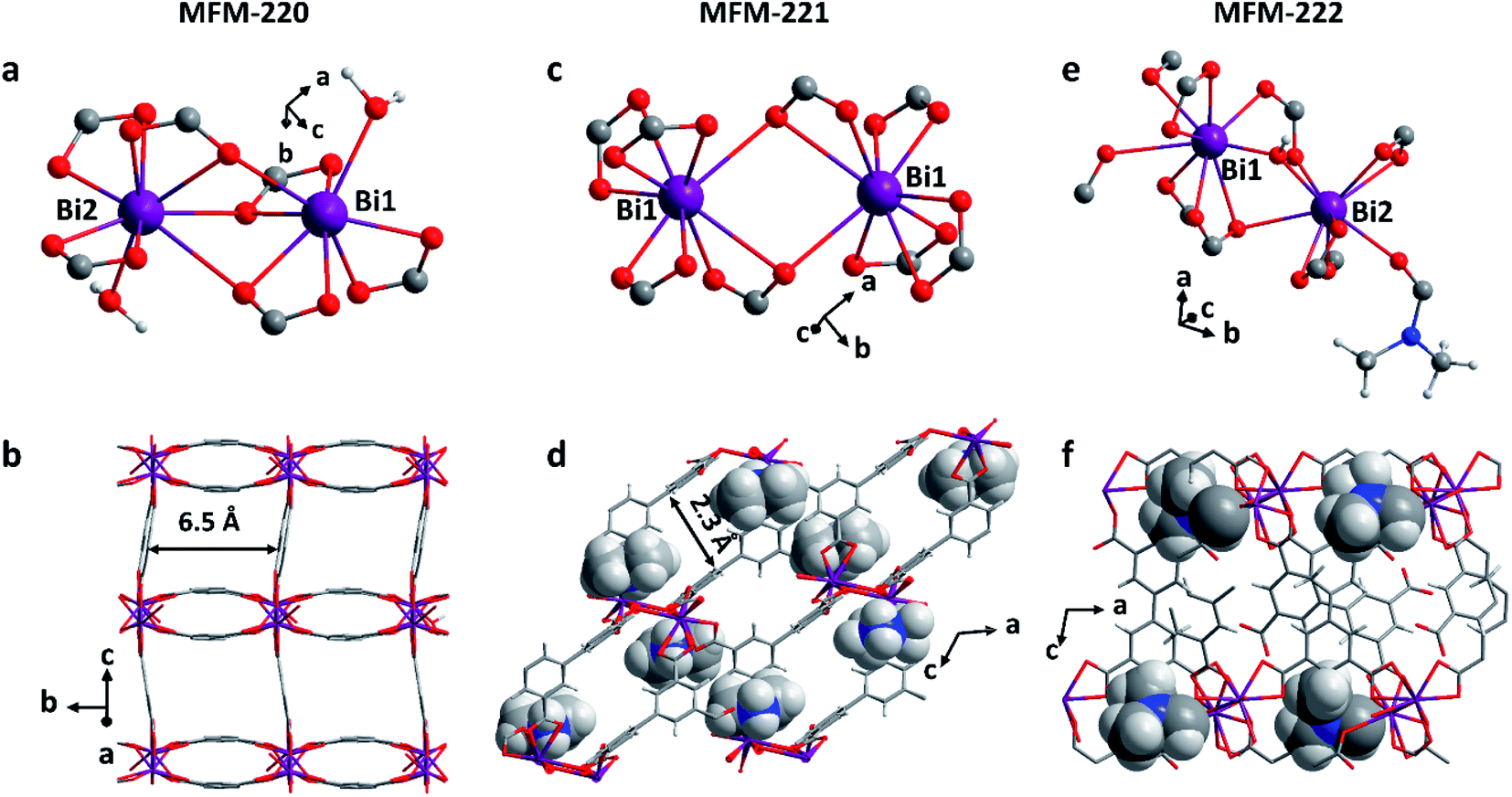 | ||
| Fig. 1 Views of the coordination environments of Bi(III) sites and pore structures of (a and b) MFM-220, (c and d) MFM-221 and (e and f) MFM-222 (Bi, purple; O, red; C, grey; H, light grey; N, blue). The Me2NH2+ cations and coordinated DMF molecules in MFM-221 and MFM-222, respectively, are highlighted in ball-and-stick mode. | ||
MFM-220, MFM-221 and MFM-222 were used as precursors to prepare active catalysts for CO2RR. Carbon paper (CP) was used as the substrate for the preparation of working electrodes. Electrochemical CO2RR was conducted in an H-type cell with a three-electrode configuration (Fig. S9†). Both the catholyte and anolyte were 0.1 M KHCO3 aqueous solutions separated by a Nafion-117 membrane. Before conducting electrocatalysis, we studied the reactivity of these Bi materials in the electrolyte solution only in the absence an external potential. The as-prepared MOF/CP electrodes were placed into the electrolyte for 30 minutes to afford MFM-220-e/CP, MFM-221-e/CP and MFM-222-e/CP. PXRD patterns suggest the disappearance of the Bragg peaks of the pristine MOFs and new peaks assigned to Bi2O2CO3 become evident (Fig. S10†).21 While porous MFM-220 and MFM-221 show complete structural transition to Bi2O2CO3, MFM-222-e exhibits a mixture of MFM-222 and Bi2O2CO3 owing probably to its non-porous nature (Fig. S10c†). IR, Raman and XPS analyses were used to characterise the resultant materials supported on CP. The IR spectrum of MFM-220-e/CP confirms the absence of characteristic bands for the ligands, whilst new bands at 1464, 1387, 1060 and 843 cm−1 are assigned to the carbonate groups in Bi2O2CO3 (Fig. 2a).24,25 Comparison of the Raman spectra of MFM-220 and MFM-220-e/CP revealed (i) the disappearance of bands at 811 and 999 cm−1 (assigned to the C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups in the ligands), (ii) a shift in the Bi–O vibrational band from 150 to 155 cm−1, and (iii) the appearance of a new band at 1061 cm−1 (assigned to the stretching vibration of C–O in Bi2O2CO3), fully consistent with the structural transition to Bi2O2CO3 (Fig. 2b, c and S11†).25–27 The XPS spectrum of MFM-220 shows two peaks of Bi 4f at 165.1 and 159.8 eV, consistent with the expected Bi(III) centers within the material (Fig. 2d). In MFM-200-e/CP, these peaks move to slightly lower binding energy at 164.7 and 159.4 eV, again consistent with a Bi(III) in Bi2O2CO3.28 Similar IR, Raman and XPS results are found for MFM-221-e/CP and MFM-222-e/CP, except that an incomplete structural transition is observed for MFM-222-e (Fig. S11–S14†). Raman spectroscopy as a function of time confirms that MFM-220 undergoes a more rapid structural transition than MFM-221 or MFM-222 upon soaking in electrolyte. MFM-220 and MFM-221 start to evolve after 5 and 10 min, respectively, while MFM-222 shows retention of characteristic Raman features even after 20 min (Fig. S12a–c†).
C groups in the ligands), (ii) a shift in the Bi–O vibrational band from 150 to 155 cm−1, and (iii) the appearance of a new band at 1061 cm−1 (assigned to the stretching vibration of C–O in Bi2O2CO3), fully consistent with the structural transition to Bi2O2CO3 (Fig. 2b, c and S11†).25–27 The XPS spectrum of MFM-220 shows two peaks of Bi 4f at 165.1 and 159.8 eV, consistent with the expected Bi(III) centers within the material (Fig. 2d). In MFM-200-e/CP, these peaks move to slightly lower binding energy at 164.7 and 159.4 eV, again consistent with a Bi(III) in Bi2O2CO3.28 Similar IR, Raman and XPS results are found for MFM-221-e/CP and MFM-222-e/CP, except that an incomplete structural transition is observed for MFM-222-e (Fig. S11–S14†). Raman spectroscopy as a function of time confirms that MFM-220 undergoes a more rapid structural transition than MFM-221 or MFM-222 upon soaking in electrolyte. MFM-220 and MFM-221 start to evolve after 5 and 10 min, respectively, while MFM-222 shows retention of characteristic Raman features even after 20 min (Fig. S12a–c†).
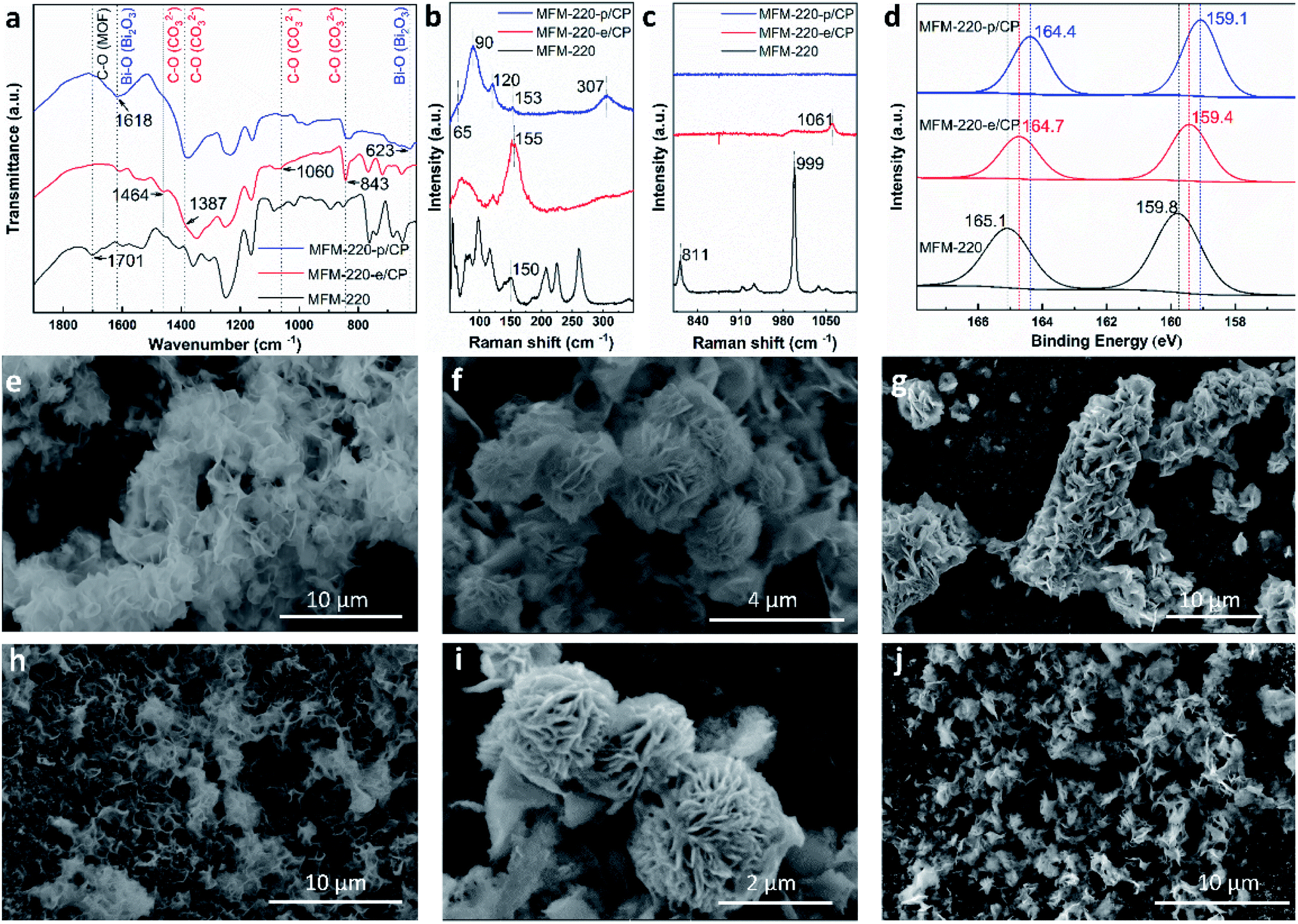 | ||
| Fig. 2 Characterisation of the transformation and evolution of Bi-MOFs to Bi2O2CO3 nanosheets and α-Bi2O3. (a) FT-IR spectra; (b and c) Raman spectra; (d) XPS spectra; SEM images of (e) MFM-220-e/CP, (f) MFM-221-e/CP, (g) MFM-222-e/CP, (h) MFM-220-p/CP, (i) MFM-221-p/CP, and (j) MFM-222-p/CP. | ||
Further structural evolution to MOF-e/CP electrodes occurred upon application of an external potential to give MFM-220-p/CP, MFM-221-p/CP and MFM-222-p/CP. The PXRD patterns of these transformed species show characteristic peaks assigned to α-Bi2O3 with minor amounts of metallic Bi (Fig. S10d–f†). IR, Raman and XPS spectra verified the formation of α-Bi2O3 and Bi (Fig. 2).29 For example, Raman spectra of MFM-220-p/CP show the characteristic bands of α-Bi2O3 at 120, 153 and 307 cm−1 (Fig. 2b).25,30 An additional two new Raman peaks appear at 65 and 90 cm−1 are assigned to the Eg and A1g stretching modes of Bi–Bi bonds, respectively.31–33 The XPS spectra of MFM-220-p/CP show further slight shifts of the 4f peaks for Bi (164.4 and 159.1 eV) to the low energy region compared with MFM-220-e/CP (164.7 and 159.4 eV), indicating the partial reduction of Bi(III) to Bi(0).20 Thus, α-Bi2O3 and Bi co-exist in MFM-220-p/CP. Similar IR, Raman and XPS results are observed for MFM-221-p/CP (Fig. S11–S14†). In contrast, MFM-222-p/CP retains Raman bands at 1052 cm−1 and the IR band at 844 cm−1 (Fig. S11f and S13b†), indicating the presence of a mixture of Bi2O2CO3, α-Bi2O3 and Bi. Analysis of the time-resolved Raman spectra shows that the structural evolution of MFM-220-e and MFM-221-e is completed in 10 and 30 min, respectively, while that for MFM-222-e is not completed even after 50 min (Fig. S12d–f†). The SEM images confirm that pristine MFM-220, MFM-221 and MFM-222 all show well-defined single crystals (Fig. S15†). During the first stage evolution, the morphologies of MFM-220-e/CP, MFM-221-e/CP and MFM-222-e/CP turn into nanosheets (Fig. 2e–g). Little further change to the morphology was observed upon applying the potential (Fig. 2h and i). The SEM-EDX analysis shows the homogeneous distribution of Bi and O in the resulting materials during the two-stage structural evolution process (Fig. S16–S21†). The EPR spectra of MFM-220-p/CP, MFM-221-p/CP and MFM-222-p/CP all show an apparent signal at g = 2.0033, which is assigned to the oxygen vacancies in the resulting α-Bi2O3.34–36 Thus, these results confirm that the MOF-p working electrodes are mainly composed of α-Bi2O3.
The electrocatalytic CO2RR performance of the evolved working electrodes was investigated in an H-cell. A bare CP electrode was included for comparison. Formate was found to be the main carbon-containing product by analysing the products in both gas and liquid phases after the electrolysis by GC, FTIR and 1H NMR spectroscopy. H2 is the only by-product. All three MOF-p/CP electrodes give higher FEformate and current density of formate at all potentials than the bare CP electrode (Fig. 3a and b), with MFM-220-p/CP showing better catalytic performance than MFM-221-p/CP and MFM-222-p/CP. The highest FEformate over MFM-220-p/CP reached 90.4% at −1.1 V vs. RHE with a total current density of 23.0 mA cm−2 after electrolysis for 1 h. The partial current density of formate over MFM-220-p/CP is also higher than that for MFM-221-p/CP and MFM-222-p/CP (20.8, 19.3 and 15.1 mA cm−2, respectively, Fig. 3b). In comparison, the value of FEformate over MFM-221-p/CP and MFM-222-p/CP electrodes are 84.6% and 75.4%, respectively (Fig. 3a). The enhanced catalytic performance of MFM-220-p/CP compared with the cation-blocked MFM-221 and non-porous MFM-222 is attributed to its structural evolution promoted by its highly porous structure. The slow evolution of non-porous MFM-222 and the presence of Bi2O2CO3 in MFM-222-p/CP result in reduced active sites for CO2RR, leading to lower catalytic activity (Fig. S14b†).
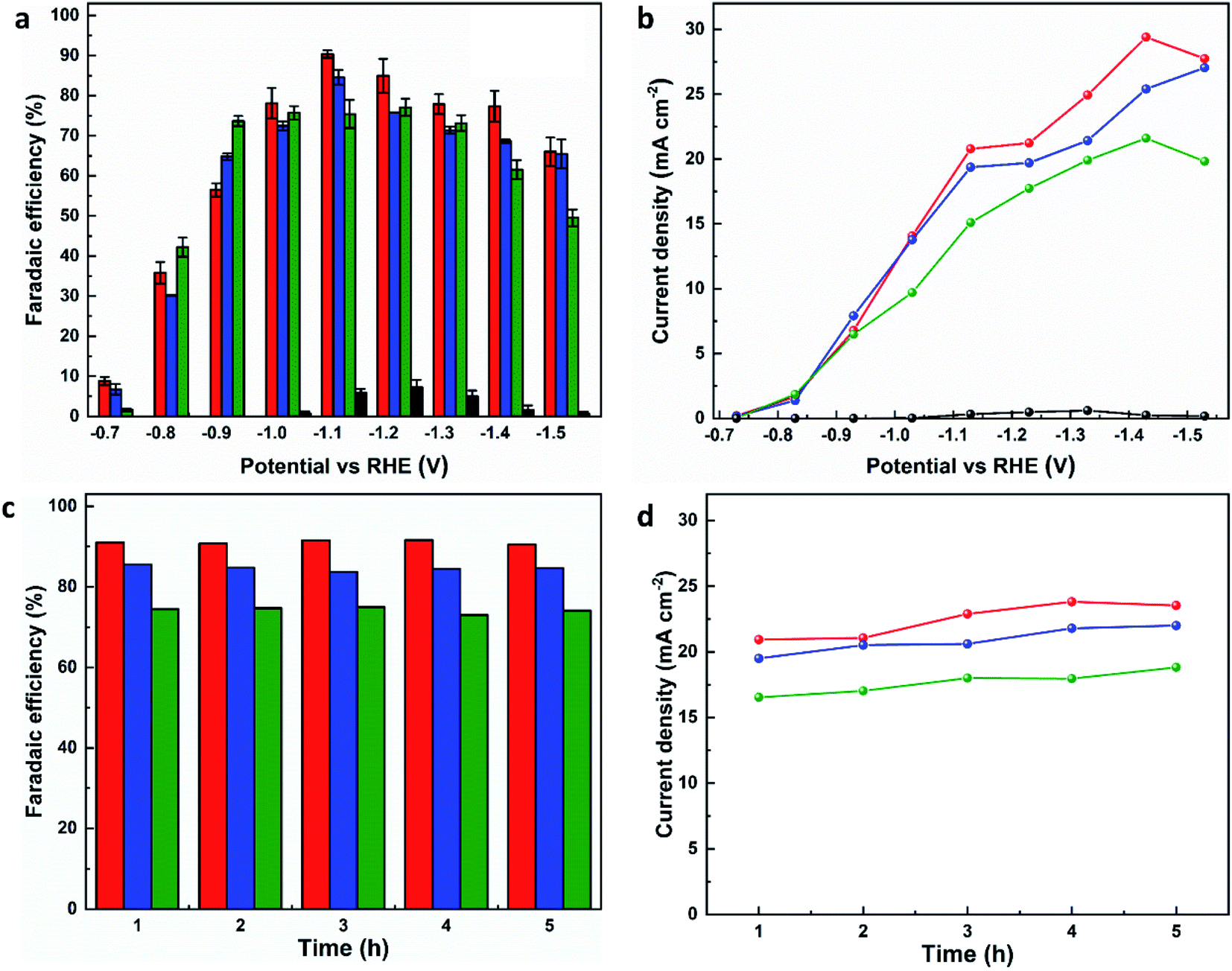 | ||
| Fig. 3 Catalytic performance of MFM-220-p/CP (red lines or column), MFM-221-p/CP (blue lines or column), MFM-222-p/CP (green lines or column), and CP (black lines or column) electrodes for CO2RR in 0.1 M KHCO3. Plots of (a) FEformatevs. potential, (b) current density for formate production vs. potential, (c) FEformatevs. time, and (d) current density of formate vs. time for reduction of CO2 at −1.1 V vs. RHE. | ||
To confirm that CO2 is the sole carbon source for producing formate, a control experiment was conducted in an Ar-saturated electrolyte without CO2, and no formate was detected (Fig. S26†). The change of FEformate and current density with time were recorded at −1.1 V vs. RHE using all three MOF-p/CP electrodes. All three electrodes show stable FEformate and the current density of formate for the electrolysis over at least 5 h (Fig. 3c and d). The overall catalytic performance of MFM-220-p/CP is comparable with the leading Bi-based catalysts studied for CO2RR in an H-cell (Fig. S27 and Table S2†).
Electrochemical impedance spectroscopy (EIS) and the electrochemical active surface area (ECSA) were studied to elucidate the electrochemical activity of these electrodes using the same set-up as above. The charge-transfer resistance of three evolved MOF electrodes was revealed by EIS spectra to characterize the charge exchange between the catalyst and reactant in the electrolyte. As shown in Fig. 4a, MFM-220-p/CP, MFM-221-p/CP and MFM-222-p/CP show a resistance (Rct) to charge transfer of 329.8, 397.6 and 456.8 Ω cm2, respectively. The rapid and thorough transformation of porous MFM-220 results in enhanced conductivity for MFM-220-p/CP, thus promoting its performance for CO2RR. The double-layer capacitance (Cdl) for all three reconstructed MOF electrodes was analyzed by measuring the capacitive current associated with double-layer charging using the scan-rate dependence of cyclic voltammetric stripping.9 MFM-220-p/CP has the highest value for Cdl at 0.2743 mF cm−2, and MFM-221-p/CP, MFM-222-p/CP and CP show values of 0.2511, 0.2119 and 0.1016 mF cm−2, respectively (Fig. 4b). This again is consistent with the observed high catalytic activity of MFM-220-p/CP.
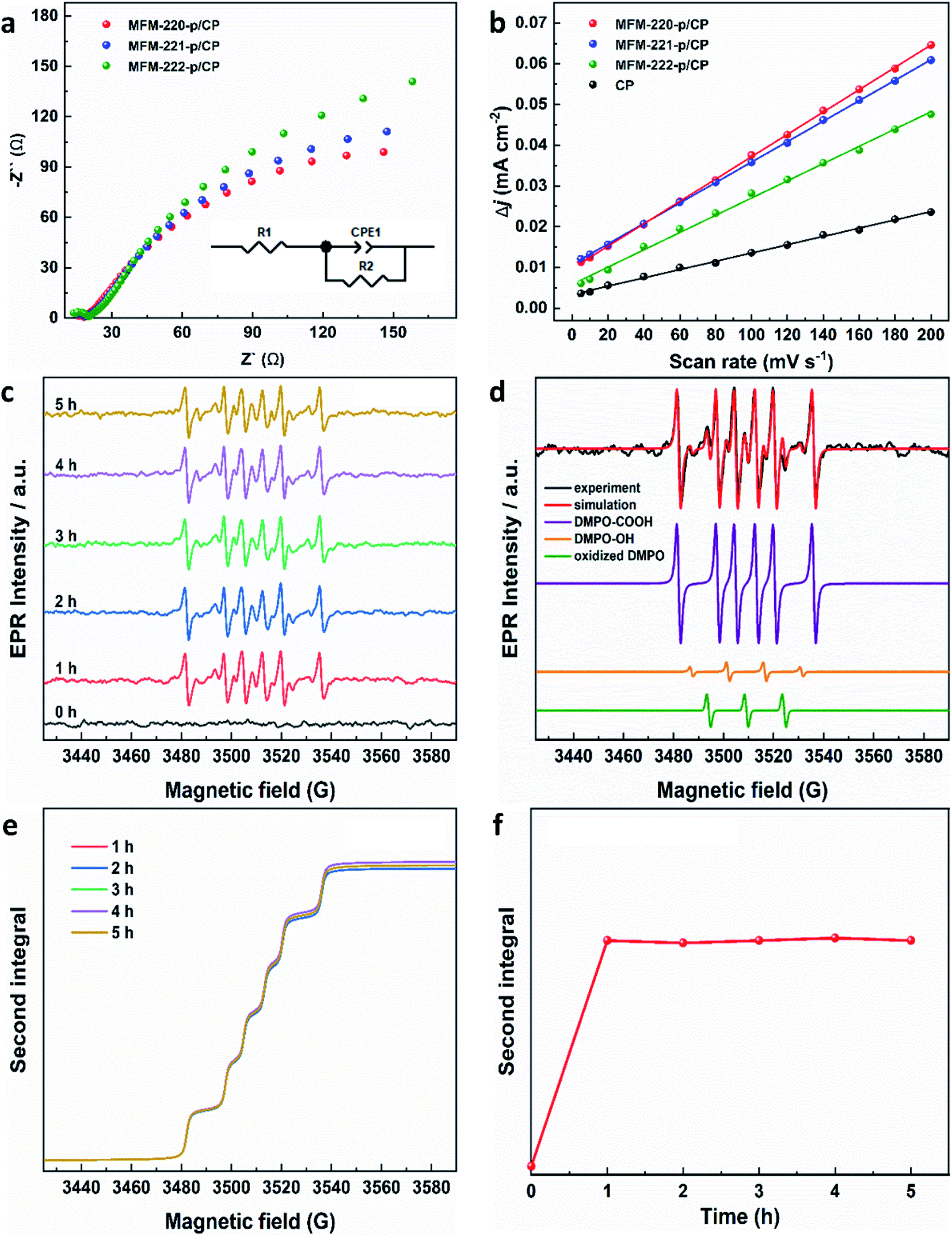 | ||
| Fig. 4 Electrochemical characterization of reconstructed working electrodes: (a) EIS spectra (the inset is the corresponding equivalent circuit); (b) plot of difference in charging current density vs. scan rates. EPR characterization of radicals produced during CO2RR using MFM-220-p/CP at −1.1 V vs. RHE: (c) EPR spectra of aliquots of electrolyte taken at different times; (d) EPR spectra of spin adducts of free radicals observed. The complete set of parameters for simulations are given in Table S3;† (e) second integrals of simulated X-band EPR spectra for DMPO-·COOH adduct vs. time; (f) plot of the second integral of the X-band EPR signals for DMPO-·COOH at room temperature vs. time. | ||
EPR spectroscopy was employed to monitor and characterize as a function of time any intermediate radicals produced during the electroreduction process.37 EPR spectra were measured for aliquots of electrolyte solution taken at time intervals of 1 h during reduction of CO2 at −1.1 V vs. RHE over all three MOF-p/CP electrodes. DMPO was used as a spin trapping agent to identify the short-lived radicals during CO2RR.38 Characteristic spectra of DMPO-adduct radicals were observed for all three electrodes (Fig. 4c, S31 and S32†). The spectra are dominated by a six-line pattern consistent with the DMPO-COOH adduct (hyperfine coupling constants aN = 15.6 G, aH = 22.9 G), with minor quantities of an oxidized DMPO radical (aN = 15.1 G) and the DMPO-OH adduct (aN = 14.7 G, aH = 14.7 G; simulations in Fig. 4d, with parameters in Table S3†).38–40 This is consistent with formate being the only carbon-containing product in this reaction and also established pathways for CO2 reduction via and/or ·COOH radicals.41–43 The hyperfine coupling constants are more consistent with trapped ·COOH radicals rather than trapped
and/or ·COOH radicals.41–43 The hyperfine coupling constants are more consistent with trapped ·COOH radicals rather than trapped 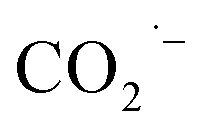 .38 To ensure that the ·COOH radicals come from CO2RR, and not the electrolyte, we also performed the EPR measurements with 0.1 M KHCO3 and 0.1 M KOH as the reference, in the absence of CO2. No EPR signals were detected, confirming that ·COOH is exclusively produced in the presence of CO2 (Fig. S31c†). To monitor the production of ·COOH during electrolysis on three evolved MOF-p/CP electrodes as a function of time, spectra were taken from aliquots at 1 h intervals over a 5 h electrolysis. Analysis of the second integral of the EPR signals (Fig. 4e and f, S32–S34†) shows that the radicals are being formed continuously and at a similar rate (with a continuous supply of CO2) throughout this timescale. This is consistent with the observed stable Faradaic efficiency and current density for formate production over this timescale (Fig. 3c and d).
.38 To ensure that the ·COOH radicals come from CO2RR, and not the electrolyte, we also performed the EPR measurements with 0.1 M KHCO3 and 0.1 M KOH as the reference, in the absence of CO2. No EPR signals were detected, confirming that ·COOH is exclusively produced in the presence of CO2 (Fig. S31c†). To monitor the production of ·COOH during electrolysis on three evolved MOF-p/CP electrodes as a function of time, spectra were taken from aliquots at 1 h intervals over a 5 h electrolysis. Analysis of the second integral of the EPR signals (Fig. 4e and f, S32–S34†) shows that the radicals are being formed continuously and at a similar rate (with a continuous supply of CO2) throughout this timescale. This is consistent with the observed stable Faradaic efficiency and current density for formate production over this timescale (Fig. 3c and d).
In summary, three Bi-MOFs with the same ligand but distinct porosity (ranging from 49.6% to 0%) have been synthesised by tuning the reaction conditions. PXRD, IR, Raman, XPS and SEM-EDX have been used to characterise the structural evolution of these Bi-MOFs upon reacting with electrolyte and applying the external potential. A value for FEformate can reach 90.4% at −1.1 V vs. RHE using evolved MFM-220-p/CP. The high catalytic ability of MFM-220-p/CP is due to the structural evolution promoted by the highly porous structure of MFM-220 (void of 49.6%) compared to the cation-blocked MFM-221 and non-porous MFM-222 (voids of 33.6% and 0%, respectively). EPR spectroscopy identified the formation of ·COOH as a key radical reaction intermediate in this system, and confirmed that the generation of ·COOH radical remained constant during the CO2RR over at least 5 h. This study emphasises the significant impact of the porosity of MOFs on their evolution during the electrochemical CO2RR process.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank EPSRC (EP/P001386, EP/I011870), the Royal Society, the University of Manchester and China Scholarship Council (CSC) for funding, and EPSRC for funding the EPSRC National Facility for Electron Paramagnetic Resonance at Manchester (NS/A000055/1). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 742401, NANOCHEM). X.K. and A.S. are supported by Royal Society Newton International Fellowships. L.L. thanks the University of Manchester for a Dean's Doctoral Scholarship Award.References
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed
.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed
- J. Li, Y. Kuang, Y. Meng, X. Tian, W.-H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou and C.-S. Ku, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed
- S. F. Yuan, R. L. He, X. S. Han, J. Q. Wang, Z. J. Guan and Q. M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14345–14349 CrossRef CAS PubMed
- J. L. Thompson and M. Hinton, Br. Poult. Sci., 1997, 38, 59–65 CrossRef CAS PubMed
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H.-G. Steinrück, K. Lim, Y. Li, M. F. Toney and K. Chan, Nat. Catal., 2019, 2, 55–61 CrossRef CAS
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed
-
Y. i. Hori, in Modern Aspects of Electrochemistry, Springer, 2008, pp. 89–189 Search PubMed
- W. Yang, S. Chen, W. Ren, Y. Zhao, X. Chen, C. Jia, J. Liu and C. Zhao, J. Mater. Chem. A, 2019, 7, 15907–15912 RSC
- M. A. Shahbazi, L. Faghfouri, M. P. A. Ferreira, P. Figueiredo, H. Maleki, F. Sefat, J. Hirvonen and H. A. Santos, Chem. Soc. Rev., 2020, 49, 1253–1321 RSC
- H. Gaspard-Iloughmane and C. Le Roux, Eur. J. Org. Chem., 2004, 2004, 2517–2532 CrossRef
- F. Li, G. H. Gu, C. Choi, P. Kolla, S. Hong, T.-S. Wu, Y.-L. Soo, J. Masa, S. Mukerjee and Y. Jung, Appl. Catal., B, 2020, 277, 119241 CrossRef CAS
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed
- W.-W. Yuan, J.-X. Wu, X.-D. Zhang, S.-Z. Hou, M. Xu and Z.-Y. Gu, J. Mater. Chem. A, 2020, 8, 24486–24492 RSC
- N. Li, P. Yan, Y. Tang, J. Wang, X.-Y. Yu and H. B. Wu, Appl. Catal., B, 2021, 297, 120481 CrossRef CAS
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A.
H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. M. Yang and Y. Wu, Adv. Energy Mater., 2020, 10, 2001709 CrossRef CAS
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS PubMed
- M. Savage, S. H. Yang, M. Suyetin, E. Bichoutskaia, W. Lewis, A. J. Blake, S. A. Barnett and M. Schröder, Chem. - Eur. J., 2014, 20, 8024–8029 CrossRef CAS PubMed
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- P. Taylor, S. Sunder and V. J. Lopata, Can. J. Chem., 1984, 62, 2863–2873 CrossRef CAS
- T. Selvamani, S. Anandan, L. Granone, D. W. Bahnemann and M. Ashokkumar, Mater. Chem. Front., 2018, 2, 1664–1673 RSC
- V. H. Nguyen, T. D. Nguyen and T. Van Nguyen, Top. Catal., 2020, 63, 1109–1120 CrossRef CAS
- W. W. Rudolph, G. Irmer and E. Königsberger, Dalton Trans., 2008, 900–908 RSC
- J. Ma, L. Tian, C. Xu, Y. Zhang, T. Zhang, H. Li, P. Zhao, Y. Liang, J. Wang and X. Fan, Catal. Lett., 2018, 148, 41–50 CrossRef CAS
- E. Kohan and A. Shiralizadeh Dezfuli, J. Mater. Sci.: Mater. Electron., 2019, 30, 17170–17180 CrossRef CAS
- C.-H. Ho, C.-H. Chan, Y.-S. Huang, L.-C. Tien and L.-C. Chao, Opt. Express, 2013, 21, 11965–11972 CrossRef CAS PubMed
- A. Reyes-Contreras, M. Camacho-López, S. Camacho-López, O. Olea-Mejía, A. Esparza-García, J. Bañuelos-Muñetón and M. Camacho-López, Opt. Mater. Express, 2017, 7, 1777–1786 CrossRef CAS
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS
- N. Li, P. Yan, Y. Tang, J. Wang, X.-Y. Yu and H. B. Wu, Appl. Catal., B, 2021, 297, 120481 CrossRef CAS
- D. Wu, G. Huo, W. Chen, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed
- Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei, Z. Jiang, J. Zhong, L. Li and G. Zheng, Small Methods, 2019, 3, 1800449 CrossRef
- S. Neukermans, J. Hereijgers, H. V. Ching, M. Samanipour, S. Van Doorslaer, A. Hubin and T. Breugelmans, Electrochem. Commun., 2018, 97, 42–45 CrossRef CAS
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed
- G. Feng, P. Cheng, W. Yan, M. Boronat, X. Li, J.-H. Su, J. Wang, Y. Li, A. Corma and R. J. S. Xu, Science, 2016, 351, 1188–1191 CrossRef CAS PubMed
- J. Fontmorin, R. B. Castillo, W. Tang and M. J. W. r. Sillanpää, Water Res., 2016, 99, 24–32 CrossRef CAS PubMed
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- H. Cui, Y. Guo, L. Guo, L. Wang, Z. Zhou and Z. Peng, J. Mater. Chem. A, 2018, 6, 18782–18793 RSC
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 899427 (MFM-220), 2126590 (MFM-221) and 2126600 (MFM-222) contains the supplementary crystallographic data for this paper. CCDC 899427, 2126590 and 2126600. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ta04485d |
| This journal is © The Royal Society of Chemistry 2022 |