
Volcano relationships and a new activity descriptor of 2D transition metal–Fe layered double hydroxides for efficient oxygen evolution reaction†
Ziyang
Wu
 a,
Ting
Liao
*ab,
Sen
Wang
c,
Wei
Li
c,
Binodhya
Wijerathne
c,
Wanping
Hu
d,
Anthony P.
O'Mullane
bc,
Yuantong
Gu
ab and
Ziqi
Sun
*bc
a,
Ting
Liao
*ab,
Sen
Wang
c,
Wei
Li
c,
Binodhya
Wijerathne
c,
Wanping
Hu
d,
Anthony P.
O'Mullane
bc,
Yuantong
Gu
ab and
Ziqi
Sun
*bc
aSchool of Mechanical, Medical and Process Engineering, Queensland University of Technology, 2 George Street, Brisbane, QLD 4000, Australia. E-mail: t3.liao@qut.edu.au
bCentre for Materials Science, Queensland University of Technology, 2 George Street, Brisbane, QLD 4000, Australia. E-mail: ziqi.sun@qut.edu.au
cSchool of Chemistry and Physics, Queensland University of Technology, 2 George Street, Brisbane, QLD 4000, Australia
dCentral Analytical Research Facility, Queensland University of Technology, 2 George Street, Brisbane, QLD 4000, Australia
First published on 25th November 2022
Abstract
Iron (Fe) sites play a critical role in boosting the catalytic activity of transition metal layered double hydroxide (LDH) electrocatalysts for the oxygen evolution reaction (OER), but the contribution of the Fe content to the catalysis of Fe-doped LDHs is still not well understood. Herein, a series of two-dimensional (2D) Fe-doped MFe-LDHs (M = Co, Ni, Cu, and Mn) was synthesized via a general molecular self-assembly method to track the role of Fe in their electrocatalytic OER activities. Besides the revelation of the intrinsic activity trend of NiFe > CoFe > MnFe > CuFe, volcano-shaped relationships among the catalytic activity descriptors, i.e., overpotential, Tafel slope, and turnover frequency (TOF), and the Fe-content in MFe-LDHs, were identified. Specifically, a ∼20% Fe content resulted in the highest OER performance for the LDH, while excess Fe compromised its activity. A similar volcano relationship was determined between the intermediate adsorption and Fe content via operando impedance spectroscopy (EIS) measurements, and it was shown that the intermediate adsorption capacitance (CPEad) can be a new activity descriptor for electrocatalysts. In this work, we not only performed a systematic study on the role of Fe in 2D Fe-doped LDHs but also offer some new insights into the activity descriptors for electrocatalysts.
New conceptsMetal (hydr)oxide-based electrocatalysts have been considered as one of the most promising catalysts in the water-electrolyser-based generation of hydrogen energy. In this work, volcano-shaped relationships among the catalytic activity descriptors, i.e., overpotential, Tafel slope, turnover frequency (TOF), and the newly proposed intermediate adsorption capacitance (CPEad), and the Fe contents in bimetallic layered double hydroxides (MFe-LDHs) were identified. Based on operando impedance spectroscopy (EIS) measurements, we revealed that CPEad can be a new activity descriptor to describe the activity of electrocatalysts. According to the volcano relationships, we proposed that a moderate amount of Fe can contribute to the optimal intermediate adsorption behaviours, and hence the maximum catalytic activity of MFe-LDHs, while a higher content Fe will suppress the conversion of the original surface into active sites and the following intermediate adsorption, and thus is not favourable for OER catalysis. Thus, this work provides a new descriptor for describing the activity of electrocatalysts and a general understanding of Fe-doped 2D MFe-LDH electrocatalysts. |
Introduction
Advanced oxygen evolution reaction (OER) electrocatalysts have been considered as the key components in the water-electrolyser-based generation of hydrogen energy.1 However, the multiple-electron transfer process of the OER results in sluggish kinetics and limits the anodic catalyst activity.2–4 Thus, to solve this issue, various electrocatalysts have been developed to overcome the conversion energy barrier in H–O bond cleavage and O–O bond formation involved in the OER, such as benchmark noble metal oxides,5,6 perovskite-type structures,7 and transition metal-based derivatives.8,9 However, some typical electrocatalysts, such noble metal-based electrocatalysts suffer from the challenges of scarcity and durability in alkaline electrolytes, which have dramatically hindered their practical applications.10 Conversely, transition metal-based hydroxides (LDHs), a class of lamellar materials,11,12 demonstrate low cost and high stability in alkaline conditions, enabling this type of materials to be a better choice for industrial hydrogen generation.13–15 Due to these advantages, wide studies have been conducted on the design of LDHs with variable morphologies and compositions to boost the OER in alkaline environments.Notably, the Fe site has been demonstrated to play a vital role in the OER performance of LDH-based electrocatalysts, e.g., NiFe, CoFe and (Ni/Co)FeX (X = N, P or B), where LDHs could achieve over 1000-fold higher activity in electrocatalysis than their Fe-free counterparts.16–19 Although the influence of Fe on the activity of LHDs has gained special attention,20 complex contributions to the OER performance from Fe-containing species have not been clearly understood to date.21–24 For example, the surface reconstruction from metal oxides/hydroxides to oxyhydroxides on oxide-based catalysts25–29 and the phase transitions during electrochemical processes30–33 have been verified to promote the catalytic activity of the oxide-based catalysts. Although comparative studies revealed that the activity of Fe-based LDHs roughly follows the trend of NiFeOxHy > CoFeOxHy > FeOxHy,17,34,35 the mechanisms and real active centres in the Fe-involved OER are still controversial. It has been reported that Fe-doping can not only induce a variation in the valence of M sites but also lead to a transition in the surface phases into active centres in some cases, which can both contribute to an enhancement in activity.36,37 For example, Li et al. reported that Fe3+ doping led to the formation of active Ni4+ sites in NiFe-LDH for an improved performance,36 while Louie et al. indicated that the formation of a new NiOOH phase on the surface played a significant role in enhancing the activity of Ni–Fe oxides.38 However, it is clear that both the surface reconstruction and the active site evolution associated with Fe-doping are related to the intermediate adsorption behaviours on the catalyst surface. Unfortunately, a direct descriptor for the adsorption of intermediates on the catalytic active sites is still under exploration. Although researchers have focused on the adsorption behaviours via DFT simulation,39,40 detection of nucleophile molecules,41 EIS characterization,42–45etc., the quantitative analysis of the detailed evolution of intermediates is still far from satisfactory. Notably, in situ electrochemical impedance spectroscopy (EIS) can provide option for the qualitative analysis of the adsorption capacitance and real active surface area of electrocatalysts, resulting from the adsorption of intermediates at low overpotentials instead of the double layer contribution.46,47 For instance, Ge et al. proved that single-atom ruthenium-doped NiO catalysts could achieve a significant promotion of OH* adsorption during the 5-hydroxymethylfurfural electrooxidation process and the adsorption behaviour was revealed with EIS in detail.48 Recently, Duan et al. also demonstrated that the presence of alkali metal cations could contribute a higher OHad coverage on the surface of platinum via EIS and electrical transport spectroscopy characterization of the HER kinetics.49 Thus, comprehensive characterization and understanding of the mechanism of the Fe-related OER activity in MFe-LDHs are necessary.
Herein, a facile molecular self-assembly strategy was employed to synthesize a series of 2D Fe-doped MFe-LDHs (M = Co, Ni, Cu, and Mn). This wet-chemical approach enabled the synthesized LDH to maintain a similar 2D morphology and a homogeneous distribution of elements in its ultrathin nanosheets. Besides the identification of the activity trend of NiFe > CoFe > MnFe > CuFe for the examined MFe-LDHs, we discovered the volcano relationships among the descriptors of catalytic activity, such as overpotential, Tafel slope, turnover frequency (TOF), and Fe-content in the MFe-LDHs, and around 20 at% Fe content in the MFe-LDHs resulted in the best OER activity for NiFe and CoFe-LDH in 1 M KOH. We further employed the operando electrochemical impedance spectroscopy (EIS) technique to provide qualitative information on the adsorption capacitance resulting from the adsorption of the intermediates on the electrocatalysts, which can be a new descriptor for the activity of the electrocatalysts. A volcano relationship was identified between the intermediate adsorption capacitance CPEad and the Fe content, confirming the suitability of this new descriptor for examining the activity of electrocatalysts by using the operando EIS technique. Furthermore, we revealed that moderate Fe-doping can significantly improve the formation of catalytically active CoOOH and NiOOH oxyhydroxides on the surface of MFe-LDHs, which act as the real active centres for favourable intermediate adsorption, and thus provides the catalysts with superior activity, while an excess Fe content decreases the accessible active sites, and thus compromises the performance. Thus, this work identifies the volcano relationships in 2D MFe-LDHs, proposes a new descriptor to examine the activity via operando EIS measurements, and provides some insights into the origin of the activity of promising LDH electrocatalysts.
Experimental
Materials
Cobalt acetate tetrahydrate (Co(Ac)2·4H2O), nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O), ferric nitrate nonahydrate (Fe(NO3)2·9H2O), manganese triacetate dihydrate (CH3COO)3Mn·2H2O, copper(II) acetate monohydrate (Cu(CO2CH3)2·H2O), ethylene glycol (EG), ethanol (EtOH), hexamethylenetetramine (HMTA), and polyethylene oxide–polypropylene oxide–polyethylene oxide (PEO20-PPO70-PEO20, Pluronic P123) were purchased from Sigma-Aldrich Company.Synthesis of different LDHs
The molecular self-assembly strategy proposed in our previous report was used to synthesize various Fe-doped LDHs.50 In the case of the Co1−xFex and Ni1−xFex LDHs, in which the actual value of x was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis, 13 g EG and 10 g EtOH were employed to dissolve 280 mg P123, followed by the addition of 1 g H2O. Then, 0.5 mmol Co(Ac)2·4H2O or Ni(NO3)2·6H2O, the desired amount of Fe(NO3)2·9H2O, and 0.5 mmol HMTA were added to the mixed solution, which was stirred for 1 h and maintained at 180 °C for 5 h. Specially, the Fe contents were varied from 0.000 mmol (Co1Fe0/Ni1Fe0), 0.005 mmol (the actual formula based on ICP-OES results was Co0.99Fe0.01/Ni0.98Fe0.02), 0.025 mmol (Co0.96Fe0.04/Ni0.94Fe0.06), 0.125 mmol (Co0.82Fe0.18/Ni0.78Fe0.22), and 0.25 mmol (Co0.71Fe0.29/Ni0.65Fe0.35) to 0.35 mmol (Co0.64Fe0.36/Ni0.56Fe0.44) for different Fe-doped Co1−xFex and Ni1−xFex LDHs. Mn0.78Fe0.22 and Cu0.78Fe0.22 LDHs were also synthesized by using 0.5 mmol Mn/Cu and 0.125 mmol Fe in a solvent containing 5 g EG and 10 g ETOH mixture and reacting at 160 °C for 5 h. All the samples were washed with deionized water and EtOH twice, respectively, and collected after drying at 60 °C for 48 h. The synthesis of the 2D Mn and Fe hydroxide nanosheets was carried out under the same conditions as our previous report.50 The Cu-based nanosheets were synthesized using the same formula of Cu0.78Fe0.22 LDH but without the addition of the Fe precursor.Material characterization
The morphology of the synthesized LDHs was characterized by scanning electron microscopy (SEM) using a Zeiss Sigma VP field emission model. For low-resolution image and high-resolution crystal information analysis, a JEOL 2100 transmission electron microscope (TEM) was used for the characterization. Surface chemistry was examined by X-ray photoelectron spectroscopy equipped with a Kratos AXIS Supra photoelectron with Al-Ka radiation (hv = 1486.6 eV). Carbon signal from containment (284.8 eV) was chosen as the calibration benchmark for XPS spectra. To measure the X-ray diffraction (XRD) patterns of the powder samples, a Bruker D8 Advance diffractometer (Co Kα, 35 kV, 40 mA) was employed at a scan speed of 1.5° 2θ/min in the 2θ range of 2° to 90° and a step interval of 0.022°. For the samples loaded on carbon cloth, X-ray diffraction (XRD) with the grazing incidence difference (GID) method was selected to collect the related spectra. In this case, Cu radiation (Cu Kα, 40 kV and 40 mA) was employed for the Rigaku Smartlab diffractometer at a scanning speed of 2°/min and a step interval of 0.02° in the 2θ of 5° to 90°. EVA (V5, Bruker) and the TOPAS package (V6, Bruker) were used for phase analysis. Fourier-transform infrared spectroscopy (FT-IR) was conducted on a Nicolet Nexus 870 FTIR spectrometer. Surface areas were evaluated using a Micromeritics Tristar II 3020 Surface Area and Porosity Analyzer. Atomic force microscopy (AFM, NT-MDT Solver Pro) was used to measure the LDH thickness. Elemental analysis was performed on an inductively coupled plasma optical emission spectrometer (ICP-OES, PerkinElmer Optima 8300 DV). The specimens for elemental analysis were obtained by dissolving the powers in concentrated HNO3 solution, and subsequently concentrated HCl was also employed if the sample was not well dissolved.Electrochemical measurements
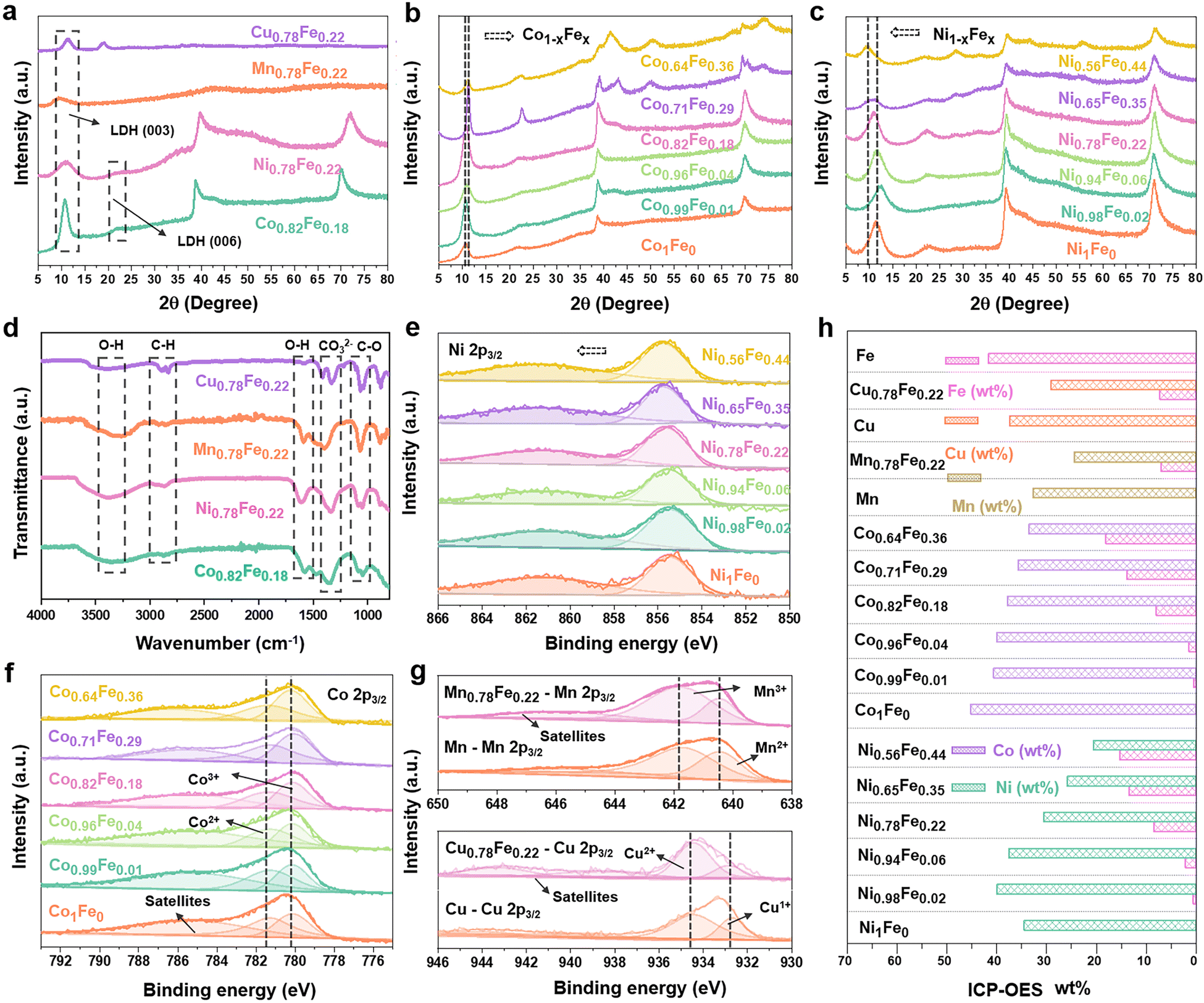
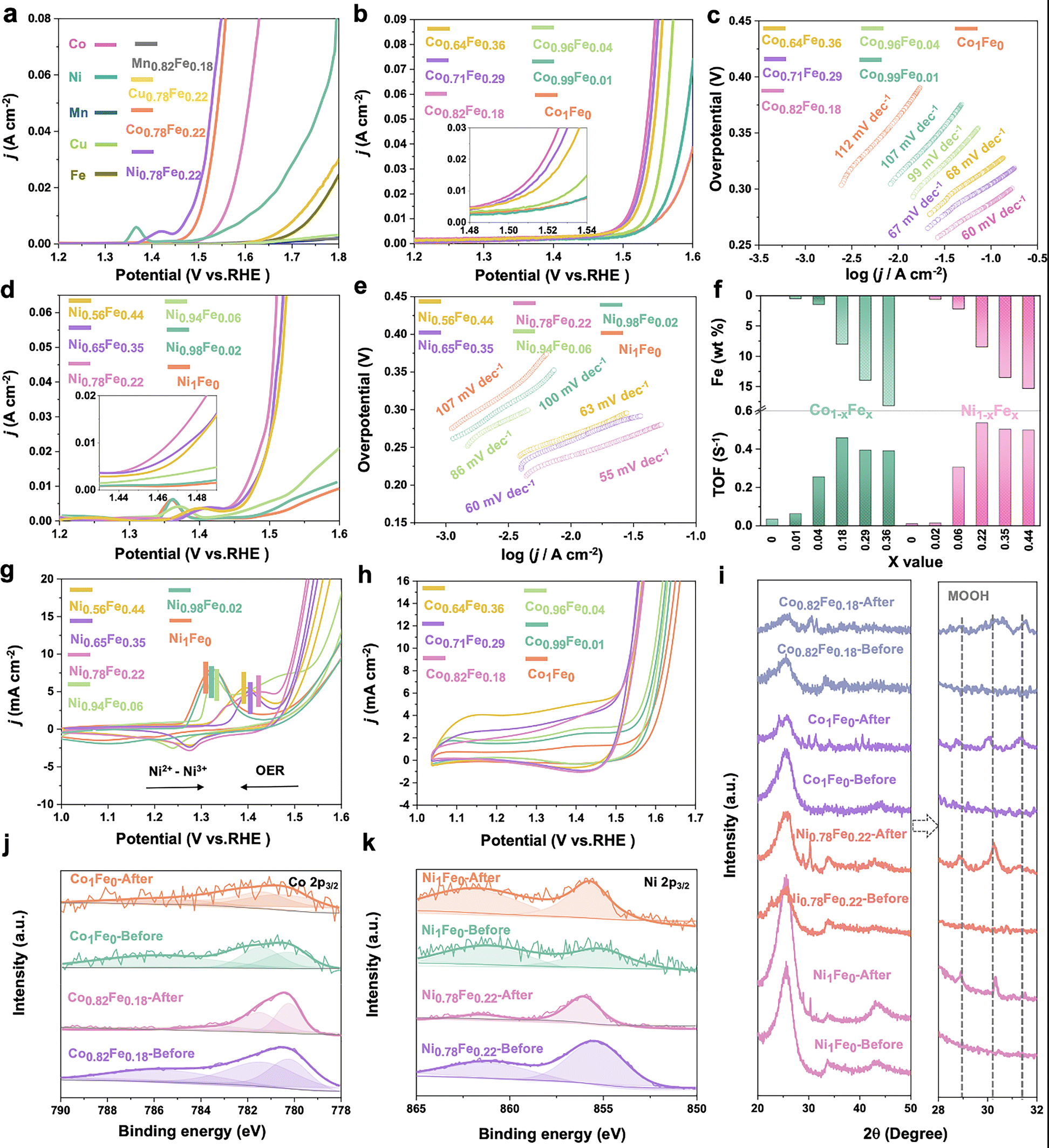
All electrochemical measurements were carried out using a three-electrode system on a CHI 760E electrochemistry workstation, except for the EIS test. The catalyst ink was first prepared by dispersing 4.0 mg the as-synthesized powder in a mixture solution containing 0.8 mL water and 0.2 mL isopropyl alcohol with the addition of 5.0 μL Nafion binder solution. Then, the working electrode was obtained by coating 4.0 μL ink on a polished glass carbon electrode (3 mm in diameter) and completely drying it in an oven (50 °C), which was further coupled with a saturated calomel electrode (SCE) and graphite rod as the reference electrode and counter electrode, respectively. For the long time-stability test, the catalyst ink was loaded on carbon cloth with a rough mass density of 8 mg cm2−. Before the electrochemical characterization, a few Ni-loaded carbon cloth samples were used to purify the Fe impurities in 1 M KOH with cyclic voltammetry (CV) cycles. Then, CV cycles were also conducted over a potential range of 1.0 to 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V (vs. RHE) at a scan rate of 100mVs−1 to achieve stable states in 1 M KOH. Linear sweep voltammetry (LSV) curves of all catalysts for OER were recorded at a scan rate of 5 mV s−1 with 95% iR-compensation in 1 M KOH solution. For the operando EIS characterization, 20 cycles of CV at the sweep rate of 50 mV s−1 were performed before the EIS test for activating the catalysts. Then, EIS was performed in the frequency range of 0.1Hz to 1 MHz with an amplitude of 10mV (BioLogic VSP workstation), and the obtained data were fitted by equivalent circuits with the help of ZSimpWin. The turnover frequency (TOF) values were calculated using the following equations:17
V (vs. RHE) at a scan rate of 100mVs−1 to achieve stable states in 1 M KOH. Linear sweep voltammetry (LSV) curves of all catalysts for OER were recorded at a scan rate of 5 mV s−1 with 95% iR-compensation in 1 M KOH solution. For the operando EIS characterization, 20 cycles of CV at the sweep rate of 50 mV s−1 were performed before the EIS test for activating the catalysts. Then, EIS was performed in the frequency range of 0.1Hz to 1 MHz with an amplitude of 10mV (BioLogic VSP workstation), and the obtained data were fitted by equivalent circuits with the help of ZSimpWin. The turnover frequency (TOF) values were calculated using the following equations:17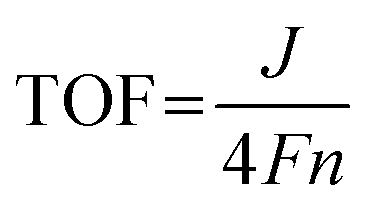 | (1) |
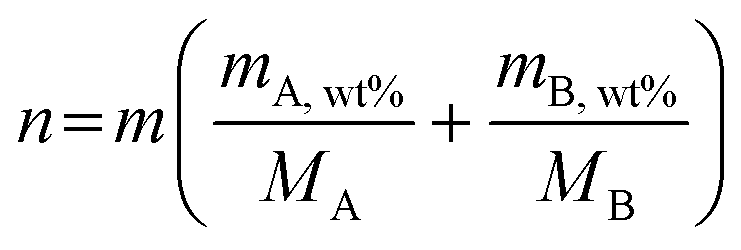 | (2) |
Density functional theory (DFT) calculation
All density functional theory (DFT) calculations were performed using the PWSCF codes as implemented in the Quantum-Espresso package.51 The electron-ion interactions were described by ultrasoft pseudopotentials and exchange–correlation interactions using the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) functional.52,53 The Kohn–Sham (KS) orbitals and the charge density were represented using plane waves (PWs) basis set to a maximum kinetic energy of 50 Ry and 400 Ry, respectively. The MFe-LDH (M = Co, Ni, Cu, and Mn) catalysts were built as supercells to study the OER evolution process. The vacuum spacing in the supercell was 15 Å along the c and b directions to avoid the fake mirror interactions. The long-range dispersion effect was considered using van der Waals correction in Grimme's DFT-D3 scheme.54 Gamma point was used to perform the integration in the Brillouin zone for geometric optimization and 5 × 1 × 5 k-point mesh for electronic structure analysis.55 All structures were optimized at the convergence criteria of 1 × 10−7 eV for the energy and 1 × 10−4 eV Å−1 for the force.The elementary steps of the four-electron oxygen evolution reaction (OER) were studied on the edge of each MFe-LDH catalyst as follows:
| H2O (l) + * → OH* + e− + H+ | (3) |
| OH* → O* + e− + H+ | (4) |
| H2O (l) + O* → OOH* + e− + H+ | (5) |
| OOH* → O2(g) + e− + H+ | (6) |
| ΔG = ΔE + ΔEZPE − TΔS | (7) |
Results and discussion
A series of 2D MFe-LDHs (M = Co, Ni, Cu, and Mn) was synthesized via a generalized molecular self-assembly method, as schematically shown in Fig. 1a, by which the precursor oligomers were confined into the lamellar surfactant micelles, and then crystallized into 2D nanosheets with atomic level thickness.50 In this case, the surfactant/water ratio of the mixed solution was adjusted to balance the hydrolysis rates between the transition metal precursors and the Fe-containing precursor for constructing homogeneous 2D LDH nanosheets. To compare the matrix effect on the Fe-doped LDHs, the LHDs with 0.125 mmol Fe-doping were employed, that is, Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22 and Cu0.78Fe0.22 LDHs. To evaluate the effect of the Fe content on the final electrochemical performance, MFe-LDHs with different Fe/M ratios in the Co1−xFex and Ni1−xFex LDHs were examined, which were denoted as Co1Fe0/Ni1Fe0 (0), Co0.99Fe0.01/Ni0.98Fe0.02 (0.005), Co0.96Fe0.04/Ni0.94Fe0.06 (0.025), Co0.82Fe0.18/Ni0.78Fe0.22 (0.125), Co0.71Fe0.29/Ni0.65Fe0.35 (0.25) and Co0.64Fe0.36/Ni0.56Fe0.44 (0.35). We understand that final stoichiometric ratios of the metallic ions in the synthesized LDHs can deviate from the designed values, and thus the x values in the above-mentioned formulas were the actual values corrected by the ICP-OES technique. As we will discuss later, the obtained Fe/M ratios followed a close stoichiometry, confirming the capability of the molecular self-assembly approach in synthesizing complex LDH materials. After their synthesis, the MFe-LDH samples with 0.125 mmol Fe doping were chosen for the morphology and microstructure investigation. Then, MFe-LDHs with various Fe contents were used to evaluate the surface chemistry and active centres for efficient OER catalysis. | ||
| Fig. 1 (a) Illustration of the fabrication process of LDHs, (b) low-magnification TEM images of Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, Cu0.78Fe0.22-LDHs, and Fe, Co, Ni, and Mn hydroxide nanosheets, and (c) high-resolution TEM images and corresponding elemental mapping of Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, and Cu0.78Fe0.22-LDHs. | ||
The microstructures of the synthesized 2D MFe-LDHs were characterized via the TEM and SEM techniques (Fig. S1, ESI†). Fig. 1b displays the 2D nanosheet morphology of the MFe-LDHs and the corresponding single metal hydroxides observed under TEM. It is clearly demonstrated that a distinct 2D structure was successfully achieved for all the materials, which excludes the influence of the morphology on the properties and performance of the LDHs. The specific surface areas of the obtained nanosheets were examined by the N2 adsorption–desorption method. As shown in Fig. S2 (ESI†), owing to the differences in the density and slight variations in the thickness of the nanosheets, Mn0.78Fe0.22 had the lowest specific surface area of 57.48 m2g−1, and Cu0.78Fe0.22 exhibited a specific surface area of 99.74 m2g−1, followed by 127.78 m2g−1 for Co0.82Fe0.18, and 157.63 m2g−1 for Ni0.78Fe0.22-LDH. The thickness of MFe-LDHs was measured by AFM. The Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, and Cu0.78Fe0.22-LDHs demonstrated a thickness of 2.0, 1.3, 2.2, and 4.2 nm, respectively (Fig. S3, ESI†), which are consistent with their surface areas, except for the heavier Mn0.78Fe0.22-LDH. Fig. 1c presents the corresponding high-resolution TEM images of Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, and Cu0.78Fe0.22-LDHs. The addition of Fe-metal did not significantly alter the crystal structure of the matrix. In Co0.82Fe0.18-LDH, the lattice fringes of 0.23 nm and 0.27 nm can be assigned to the (211) and (111) planes of the Co(OH)2 matrix phase, respectively. Similarly, the spacings of 0.23 nm and 0.27 nm identified in Ni0.78Fe0.22-LDH belong to the (200) and (110) planes of the Ni(OH)2 matrix phase. For Mn0.78Fe0.22 and Cu0.78Fe0.22-LDHs, the (012) and (018) planes with spacing values of 0.23 nm and 0.20 nm, respectively, were confirmed, which also resulted from their hydrotalcite-like phase. The elemental mappings collected via TEM confirmed the uniform distribution of Fe element in the obtained LDHs without segregation (Fig. 1c). It is interesting that the LDHs with different Fe contents displayed different colors (Fig. S4, ESI†), indicating the altered electronic structures and bandgaps with a change in Fe content. Based on the morphology and microstructure characterizations, it is clear that 2D MFe-LDHs can be synthesized with a homogenous graphene-like morphology.
The phase compositions of the various LDHs were examined by XRD. As shown in Fig. 2a, the characteristic peaks related to the interlayer species of the hydrotalcite-like LDHs appeared at around 10° ((003) plane) and 21° ((006) plane) (PDF#00-035-0965) in all the Fe-containing samples, which are direct evidence of the successful synthesis of LDHs rather than oxide nanoparticles.17 Besides the characteristic (003) and (006) peaks for LDHs, the diffractions at around 40° and 70° also suggest the hydrotalcite-like architecture of the Co(OH)2 LDH (PDF#00-071-0089) and Ni(OH)2 LDH (PDF#00-022-0444) matrix phases.58–60 Specially, we evaluated the influence of the Fe content on the crystal structure of both the Co1−xFex (Fig. 2b) and Ni1−xFex LDHs (Fig. 2c), in which the Fe content varied from 0 at% to 44 at%. As a dash line marked for the (003) characteristic peaks, the position shifted to from 10.6° to 11.1° for the Co1−xFex-LDHs with an increase in Fe content, but it shifted to lower degree (11.2° to 9.4°) for Ni1−xFex-LDHs. This could have resulted from different intercalated anions into the interface layers for transition metal hydroxides.61,62 For the Co1−xFex-LDHs with higher Fe contents (Co0.71Fe0.29 and Co0.64Fe0.36), the peaks belonging to the Co(OH)2 LDH at 51.5° (320) and the hydrotalcite-like structure at 40.6° (012) and 73.5° (113) became stronger, indicating that more Fe ions were incorporated in the Co(OH)2 LDH matrix structure and rearranged into a more hydrotalcite-like crystal structure.63 Similarly, the higher Fe-content in Ni0.65Fe0.35 and Ni0.56Fe0.44 also led to an increased intensity for the hydrotalcite-like LDH peak at 54.5° (018). The XRD information suggested that the self-assembled 2D LDH materials are a perfect platform to observe the Fe-associated structural evaluation on the initial Co(OH)2 and Ni(OH)2 LDHs but without damaging the interlayer species.
 | ||
| Fig. 2 (a) XRD patterns of Co0.82Fe0.18, Ni0.78Fe0.22, Cu0.78Fe0.22, and Mn0.78Fe0.22 LDHs. XRD information with the incorporation of different contents of Fe in (b) Co1−xFex-LDHs and (c) Ni1−xFex-LDHs. (d) FT-IR spectra of MFe-LDHs. (e) High-resolution Co 2p XPS spectra of Co1−xFex-LDHs; (f) Ni 2p XPS spectra of Ni1−xFex-LDHs; and (g) high-resolution Mn and Cu XPS spectra of Mn0.78Fe0.22 and Cu0.78Fe0.22 LDHs. (h) Elemental compositions of MFe-LDHs analysed by ICP-OES. | ||
Furthermore, to identify the interlaminar species of the synthesized LDHs, FT-IR spectra were collected. As shown in Fig. 2d, the peaks at ∼3400 and 1600 cm−1 are attributed to the stretching and bending vibrations of the O–H bonds of the interlaminar water molecules, respectively. The existence of carbonate (CO32−) groups was also confirmed by their tensile vibration at around 1390 cm−1. This indicates that the CO32− groups and H2O molecules are interlayer species together with the transition metal ions exist in the positively charged region. Then, the surface chemical environment was revealed by XPS characterization. The oxidate state of Ni should be +2 for the Ni1Fe0-LDH (Ni(OH)2) according to the core level located at 855.4 eV in the Ni 2p3/2 spectra (Fig. 2e). With the addition of Fe, the Ni state kept moving to higher positions and the difference reached 0.4 eV for Ni0.56Fe0.44, indicating strong ion exchange interactions between the Fe and Ni sites.64 For Co1−xFex-LDHs (Fig. 2f), both the states of Co2+ and Co3+ were identified in the crystal structures.65 It is interesting that the higher the Fe content in the LDHs, the higher the Co3+ content. A similar M–Fe ion interaction was also observed in Mn0.78Fe0.22 and Cu0.78Fe0.22-LDHs, where higher amounts of Mn3+ (641.7 eV) and Cu2+ (934.5 eV) compared to Mn2+ (640.5 eV) and Cu1+ (932.6 eV) in the Mn 2p3/2 and Cu 2p3/2 spectra were observed with the addition of Fe ions to their structures.66,67 Thus, the XPS results confirmed that the incorporation of Fe in the LDH structures leads to the higher oxidation state of the matrix metals due to the relatively higher electronegativity of Fe3+.
ICP-OES was conducted to confirm the final compositions of the synthesized MFe-LDHs. Both the weight and molar proportions of the metallic components are listed in Table S1 (ESI†) and summarized in Fig. 2h. Although the actual Fe contents were slightly lower than the designed amounts, the Fe/M ratios were very close to the designed values, confirming that the synthesized LDHs can truly reflect the desired Fe contents.
To evaluate effect of Fe content on the OER activity of the various MFe-LDHs, a three-electrode catalysis system was used to perform electrocatalytic measurements in purified electrolyte. Fig. 3a presents the linear sweep voltammetry (LSV) curves of the MFe-LDHs and the corresponding hydroxide M-OH2 matrix LDHs. It is clear that the addition of Fe to the LDH structures significantly boosted the OER catalytic performance with much lower overpotentials and higher current densities. For the matrix LDHs without Fe-doping, Co-LDH and Ni-LDH exhibited superior activity compared to the other M-LDHs, and specifically, the Co-LDH presented the lowest overpotential (322 mV) to reach 10 mA cm−2 among the examined M-LDHs. Based on these results, subsequently we carefully examined the influence of Fe-doping on the performance of the Co1−xFex (Fig. 3b) and Ni1−xFex-LDHs (Fig. 3d). Fe-doping also showed a similar effect for the Mn and Cu-LDHs. However, as shown in Fig. S5 (ESI†), the enhancement was not as significant as that in Co and Ni LDHs. It is worth noting that ∼20 at% Fe content provided both the Co1−xFex-LDHs and Ni1−xFex-LDHs with the best activity. For instance, the overpotentials of Ni0.78Fe0.22 and Co0.82Fe0.18 were reduced by 34.3% (from 370 to 243 mV) and 16.1% (from 322 to 270 mV) at 10 mA cm−2 compared with the Ni1Fe0 and Co1Fe0-LDHs, respectively (Fig. S6, ESI†). A further increase in the Fe content in the M1−xFex-LDHs (M = Co or Ni) did not further decrease the overpotential but slightly increased at higher current densities, indicating the plateau-like effect of Fe-doping on the electrocatalytic activity. A similar trend was also observed in the Tafel slopes, as shown in Fig. 3c and e. Both Co0.82Fe0.18 (60 mV dev−1) and Ni0.78Fe0.22-LDH (55 mV dev−1) presented the lowest Tafel slopes among the examined catalysts. The turnover frequency (TOF) is regarded as an accurate descriptor of the intrinsic activity of electrocatalysts. The TOF for the MFe-LDHs was calculated to evaluate the normalized activity by dual transition metal sites. Fig. 3f and Table S2 (ESI†) display the calculated TOFs with a variation in the Fe content. A volcano-type trend in the TOF with a variation in the Fe content was also recorded. The TOF value increased with the Fe-content and reached the maximum value at Co0.82Fe0.18 (22.5 at%) and Ni0.78Fe0.22 (18.22 at%), and then decreased with a further increase in the Fe-content. Based on the catalytic descriptors of the overpotential, Tafel slope, and TOF, it is very clear that there is a saturation value for the Fe content in the MFe-LDHs, where an Fe content of around 20 at% provides the best catalytic activity towards the OER on the surface of 2D LDHs.
 | ||
| Fig. 3 Electrocatalytic performance of synthesized MFe-LDHs. (a) Polarization curves at 5 mV s−1 for different MFe-LDHs, (b) polarization curves of Co1−xFex-LDHs, (d) polarization curves of Ni1−xFex-LDHs. (c) Tafel plots of Co1−xFex-LDHs, (e) Tafel plots of Ni1−xFex-LDHs, (f) verified Fe (wt%) contents from ICP-OES results and the measured TOFs (overpotential = 350 mV) of Co1−xFex-LDHs and Ni1−xFex-LDHs; (g) 20th scan of cyclic voltammogram of Ni1−xFex-LDHs, (h) 20th CV of Co1−xFex-LDHs, (i) XRD patterns of Ni, Co, Co0.82Fe0.18, and Ni0.78Fe0.22 before and after the OER process and the enlarged patterns, (j) XPS spectra of Co 2p3/2 of Co and Co0.82Fe0.18-LDHs, and (k) Ni 2p3/2 of Ni and Ni0.78Fe0.22-LDHs. | ||
Given that CV provides reliable information related to the chemical and redox processes during catalytic reactions associated with structural and electronic transformations, we specially compared the 20th cycle of CV scan (Fig. 3g and h) of the Ni1−x1−xFex and Co1−xFex-LDHs. The Ni1−xFex-LDHs demonstrated more obvious surface oxidation behaviours, resulting from Ni2+/Ni3+ transformations rather than Co2+/Co3+ oxidation, where the latter has a broader potential range.17 As marked in Fig. 3g, the Ni2+/Ni3+ redox peaks exhibited evident anodic shifting for the LDHs exhibiting higher TOF values and the gap between Ni2+/Ni3+ and the OER peaks became smaller, indicating a better OER performance. Similarly, a volcano-shape relationship between the redox peak positions and the Fe content was again confirmed.
Stability is another key parameter to evaluate the performance of a catalyst. In this case, the stability of the MFe-LDHs was evaluated via chronoamperometric tests at the potential of around 10 mA cm2, that is, 1.47, 1.5, 1.7, and 1.8 V for Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, and Cu0.78Fe0.22-LDHs, respectively. The Mn0.78Fe0.22 and Cu0.78Fe0.22-LDH showed very poor stability with an obvious drop in performance as soon as the test started and reached only a very low current density (Fig. S7, ESI†), while the Co0.78Fe0.22 and Ni0.78Fe0.22-LDH displayed relatively good stability for up to 15 h at a starting current density of 15 mA cm−2 and 13 mA cm−2, respectively. After 15 h testing, a decrease in the current density of 4% and 35 was recorded for Co0.82Fe0.18 and Ni0.78Fe0.22-LDH, respectively. Subsequently, XPS and XRD characterizations were performed to evaluate the surface evolutions during the OER stability tests. As shown by the XRD patterns in Fig. 3i, phase transformations from Co(OH)2 and Ni(OH)2 to CoOOH and NiOOH, respectively, were identified by the appearance of new peaks with a slight shift at around 28.7°, 30°, and 31.3° in all the LDHs after electrooxidation. The associated changes in chemical states were also reflected in the corresponding Co 2p (Fig. 3j) and Ni 2p (Fig. 3k) XPS spectra. The binding energy of Co 2p3/2 shifted to lower positions and the satellite peak disappeared, revealing a higher content of Co3+ formed on the surface with the formation of CoOOH during the OER process.68 In contrast, Ni 2p3/2 moved to a higher binding energy for Ni0.78Fe0.22-LDH as a result of the electrocatalytic oxidation process and the formation of NiOOH.69 Recently, reports confirmed that CoOOH and NiOOH are the real active centres for OER catalysis after surface reconstruction in the initial stage on the Co/Ni-containing oxide catalysts.70,71 Different to the surface reconstruction observed on the Co0.82Fe0.18 and Ni0.78Fe0.22-LDHs, the Mn and Cu-based LDHs all showed significantly low XPS intensity after the OER, suggesting the dissolution of Mn and Cu from the surfaces during the OER process (Fig. S8, ESI†).22 This reveals that the absence of real oxyhydroxide active centers and the leaching of the metallic elements should account for the low activity of the Mn0.78Fe0.22 and Cu0.78Fe0.22-LDHs, and a proper Fe content could best promote the surface reconstruction into the active oxyhydroxide active centres.
Based on the above-mentioned evaluations, a clear volcano-shape relationship was identified between the OER activity and Fe-content in the Co- and Ni-based LDHs. Although the formation of real oxyhydroxide active centres as a result of surface reconstruction is recognized as one major origins of the activity enhancement, the intermediate adsorption behaviours as another critical activity descriptor is less experimentally understood. In this work, EIS, potential technology to probe the reaction kinetics and the interface properties during the electrocatalysis,72,73 was employed to track the intermediate adsorption evolutions on the Ni1−xFex and Co1−xFex-LDHs during the OER catalysis. The concept is to employ the operando EIS technique to understand the adsorption of the intermediates, i.e., OH*, O*, and OOH*, that closely govern the activity by examining the adsorbed intermediates related charge the relaxation related modes in the spectra. Two capacitance-related elements can be identified from the EIS during OER catalysis, which are the double layer capacitance and the adsorbed intermediate-related charge relaxation.72,74 The adsorption-induced capacitance change was not obvious in the Nyquist plots, but it can be observed in the Bode plots, where the modes appearing in high frequency range correspond to the intrinsic conductivity of the catalysts;75 the middle frequency mode can be attributed to the surface double layer capacitance;42,44 and the mode in the low-frequency region is related to the nonhomogeneous charge distribution at the interface.48 Therefore, analysing the Bode modes in the middle and high frequency areas can contribute to the understanding of the adsorption-associated behaviours. For example, with the earlier formation of low frequency peaks or lower bias needed for middle/low frequency modes, the faster the intermediate-related behaviour happens and a higher OER performance is achieved.43,76,77
To study the intermediate adsorption behaviours of the MFe-LDHs, the Nyquist plots and all the corresponding Bode plots were operando collected with an increased bias, as shown in Fig. S9–S14 and Tables S3–S19 (ESI†). Fig. 4a presents the Bode plots collected on the MFe-LDH catalysts at applied external bias voltages scanned from 1.0 V to 1.7 V. For the Co0.82Fe0.18 and Ni0.78Fe0.22 LDHs, three modes were found. With an increasing bias, the three modes showed a clear decreasing trend and shift in the phase angles compared with Mn0.78Fe0.22 and Cu0.78Fe0.22 LDHs, especially that at the high frequency range. This suggests that Mn0.78Fe0.22 and Cu0.78Fe0.22 LDHs could not be activated by even high bias voltages, and the polarization-dominated responses at the high-frequency areas still existed at a bias higher than the overpotentials in the Bode plots, which well explained the low activity of the Mn0.78Fe0.22 and Cu0.78Fe0.22-LDHs towards the OER. This was also confirmed by the relaxation process of the bare glassy carbon electrode and the Fe, Mn and Cu LDHs, which all demonstrated obvious peaks in a relatively high frequency region with poor OER activity (Fig. S11, ESI†).
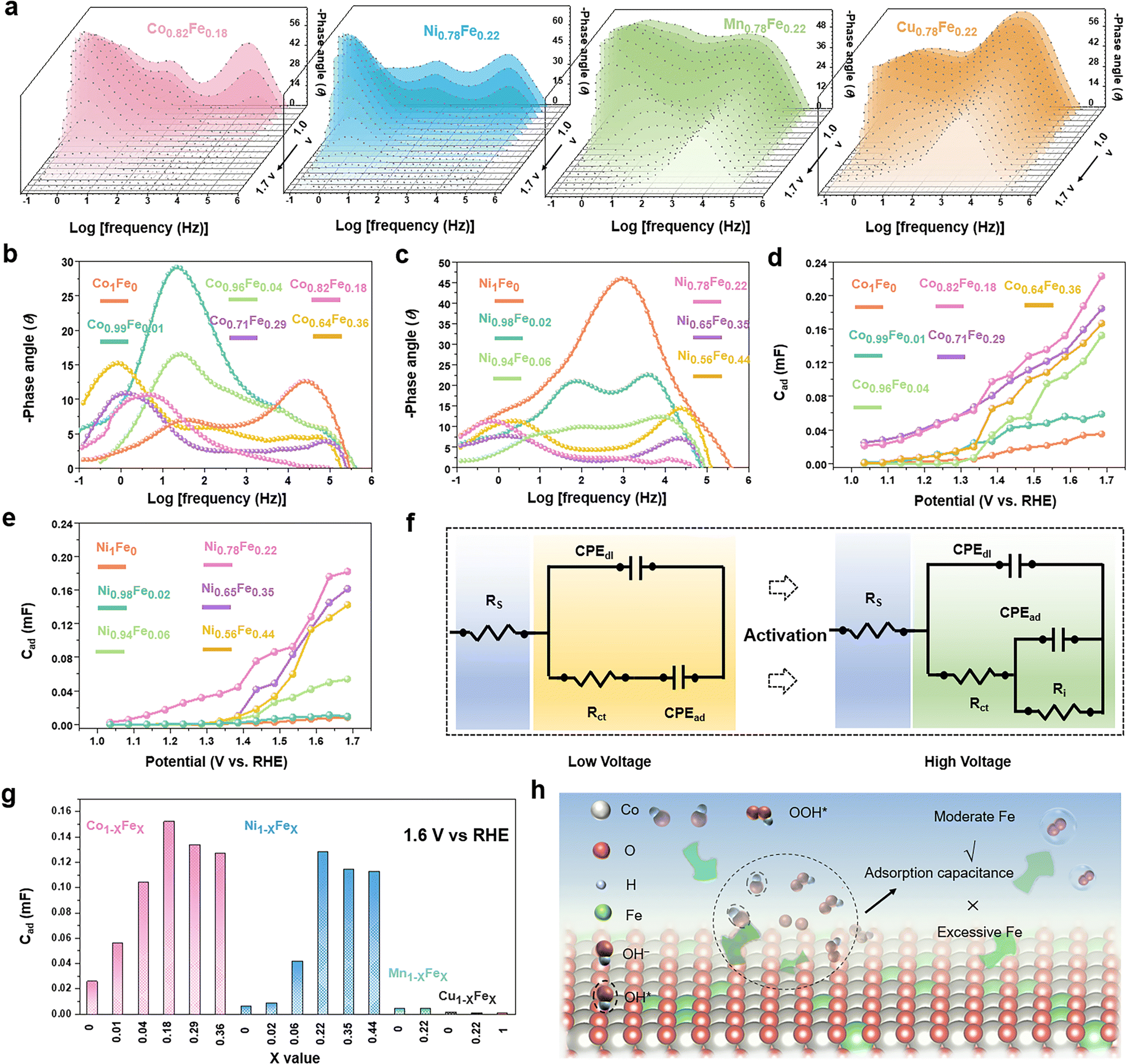 | ||
| Fig. 4 Operando EIS analysis of LDH-based samples. (a) Bode plots of Co0.82Fe0.18, Ni0.78Fe0.22, Mn0.78Fe0.22, and Cu0.78Fe0.22 samples. (b) Bode plots of Ni1−xFex-LDHs, (c) Bode plots of Co1−xFex-LDHs, (d) fitted CPEad values vs. potentials for Co1−xFex-LDHs, (e) fitted CPEad values vs. potentials for Ni1−xFex-LDHs, (f) equivalent circuit models for EIS data fitting, (g) CPEad values for different LDHs at 1.6 V (Vs. RHE) and the corresponding overpotentials at 10 mA cm−2 and (h) proposed mechanism of Fe doping-induced adsorption behaviours of LDHs in the OER process. | ||
For a better understanding of Fe-doping on the adsorption behaviours, operando EIS was performed at a fixed bias of 1.6 V on the Co1−xFex (Fig. 4b) and Ni1−xFex-LDHs (Fig. 4c) with variable Fe contents. For the Co1Fe0-LDHs, the high frequency-dominating mode suggested the low conductivity of the catalysts and the absence of a low-frequency mode indicated the unfavourable adsorption of the OER intermediates on the surface in this stage for efficient OER. The Co0.99Fe0.01 and Co0.96Fe0.04-LDHs mainly demonstrated middle-frequency responses resulting from the nonefficient surface oxidation species. Interestingly, the low-frequency modes were confirmed in the Co0.82Fe0.18, Co0.71Fe0.29, and Co0.64Fe0.36-LDHs, implying the impendence response to the sufficient adsorption of the oxidation species, namely, a better OER performance. A similar trend was also validated in the Ni1−xFex LDHs, where only an obvious peak in the high-middle frequency was observed for Ni1Fe0, but the Ni0.78Fe0.22, Ni0.65Fe0.35, and Ni0.56Fe0.44-LDHs dominated with a low-frequency mode. Thus, proper Fe doping in LDH could contribute to the intermediate adsorption-related behaviours with activation of the catalyst surfaces. With close comparison of the catalysts of Co0.82Fe0.18, Co0.71Fe0.29, and Co0.64Fe0.36-LDHs, the phase angles of the low frequency peaks demonstrated roughly the same values at 1.6 V and this trend was also identified for the Ni0.78Fe0.22, Ni0.65Fe0.35, and Ni0.56Fe0.44 LDHs, suggesting that a higher Fe content did not further significantly improve the OER-associated adsorption and OER processes.
By fitting the Nyquist plots with the equivalent circuits shown in Fig. 4f, the capacitance associated with the relaxation of intermediate adsorption (CPEad) can be quantitatively determined (Fig. S12–14 and Tables S3–S19, ESI†). Fig. 4d and e display the variation in CPEad with a change in the scan voltage for the Co1−xFex and Ni1−xFex-LDHs, respectively. The CPEad increased with an increase in the bias voltage, where the Co0.82Fe0.18 and Ni0.78Fe0.22-LDHs presented the largest values in the corresponding LDHs, which are consistent with the examined catalytic activity. It is clear that Fe-doping significantly improved the intermediate adsorption behaviours and reached a saturation value at around 20 at%. Fig. 3g summarizes the CPEad values for the examined LDHs at a fix voltage of 1.6 V, and a similar volcano relationship exists between the CPEad and the Fe contents. The volcano-shape CPEadvs. Fe content clarified the role of Fe in the faster and higher adsorption of intermediates on the surfaces and the resulting higher OER activity. Based on this understanding, we proposed a possible mechanism, as shown in Fig. 3h, where a moderate Fe content can contribute to favourable intermediate adsorption behaviours, but a higher Fe content exceeding the saturate value can supress the conversion of the original surface into active sites and the following intermediate adsorption, and thus is not favourable for OER catalysis.
According to the above-mentioned examinations, we can conclude that suitable Fe-doping of around 20 at% provides the MFe-LDHs with the most favourable surfaces for the conversion of active sites, the adsorption of intermediates, and superior catalytic activity.
To shed insight into the intrinsic electrocatalytic activity and adsorption behaviours of the MFe-LDHs, DFT calculations were also employed to study the surface configurations, reaction kinetics, and the reaction energy barriers (Fig. 5). Herein, we employed the MFe-LDH crystal structures as DFT models to better compare the role of Fe and its doping content in the simulations. To reveal the effect of the surface reconstruction of MOOH on the OER activity, the catalytic reaction on the NiOOH surface was also examined (Fig. S15 and S16, ESI†). Fig. 5a presents the crystal structures of the MFe-LDHs, where a layered structure can be clearly identified. To determine the electronic density distribution around the metallic sites, the charge density difference of the catalytic site for each catalyst was analysed (Fig. 5b). It can be found that the Mn atom at the edge site of MnFe-LDH featured electron depletion once the coordinated oxygen is removed. The electrons shifted by forming a bond with an inner metal atom, which may reduce the interaction with the surface reactants. In contrast, the exposed active metal sites of CoFe, NiFe, and CuFe LDHs are characterized with the accumulation of extra electrons, which can react with the OER intermediates and boost the OER process. However, the overpopulation of electron concentration at the active metal site may also lead to less available unpaired electrons to interact with the intermediates during the OER process, similar to the CuFe-LDH catalyst. All these electronic features of bimetal MFe-LDH catalysts also shed light on their catalytic behaviour in the OER reaction process. The free energy profile of the OER elementary reaction steps is illustrated in Fig. 5c and d, where the rating-limiting step is the conversion from O* to OOH* intermediates for the NiFe, CoFe, and CuFe-LDHs. However, the MnFe-LDH showed a significantly higher energy barrier for the OOH* to O2 conversion step, indicating a different mechanism for the MnFe-LDH during the OER process. The calculated overpotentials of the different LDHs also suggested that the highest activity is achieved for the CoFe-LDH with a value of 0.48 V, which is lower than 0.6 and 0.82 V for the NiFe and CuFe LDHs, respectively. The MnFe-LDH demonstrated the largest overpotential of 2.76 V. In addition, further simulations were carried out on the Co1−xFex and Ni1−xFex-LDHs with varying Fe contents from 16.7% to 33.3% (Fig. 5e). Interestingly, both the Co1−xFex and Ni1−xFex-LDHs presented the lowest limiting potential for the composition with a 25% Fe doping amount, which is in good agreement with the ∼20% Fe for optimal catalytic activity identified in the experimental results (Fig. 5c). As the result of surface reconstruction, the conversion of the surface into MOOH has been observed. As shown in Fig. S15 and S16 (ESI†), the NiOOH surface presented much lower energy barriers in the formation of HO* and O* intermediates and the conversion of HOO* into O2 at a potential of 0.52 V, demonstrating the vital role of surface reconstruction in oxide-based electrocatalysts. Therefore, the volcano relationship of OER activity as a function of Fe doping content can be summarized in terms of different descriptors of overpotential, TOF, intermediate adsorption, and DFT-calculated limiting potential (Fig. 5f). It is clear that a suitable Fe doping content can contribute to the highest OER activities, while excess Fe doping will compromise the catalytic activity of the MFe-LDHs.
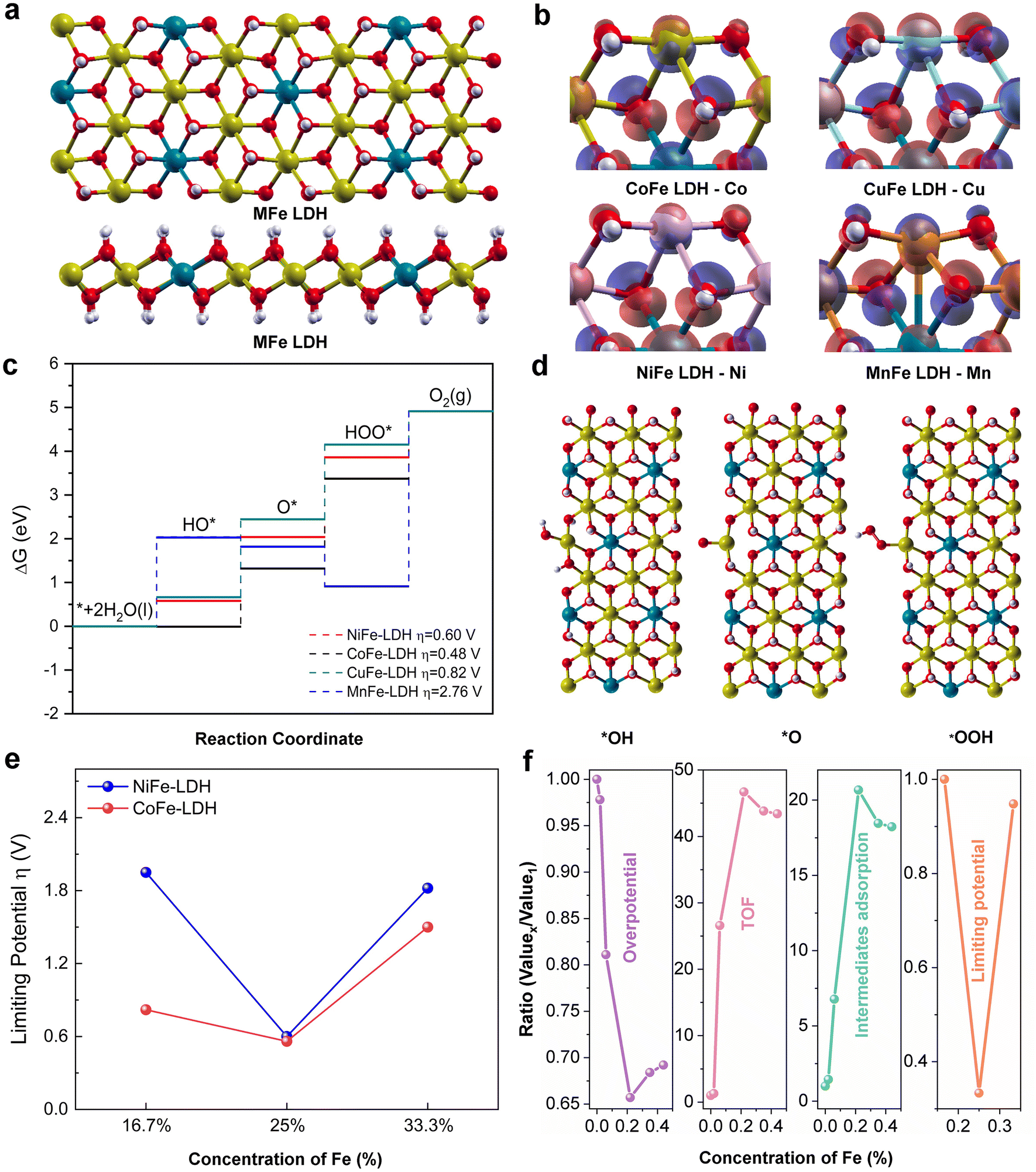 | ||
| Fig. 5 DFT calculations. (a) Top-view and cross-sectional view of the crystal structure of MFe LDHs catalysts. (b) Charge density difference of the catalytic site for each catalyst, where the red and blue colour indicate electron accumulation and depletion, respectively, and the iso-values are 0.005 eV Å−3. (c) Calculated free energy profiles of the OER catalysis steps for each catalyst. (d) Side-view of different oxygen evolution reaction steps. (e) Calculated limiting potentials of NiFe/CoFe LDHs with an increase in Fe concentration. (f) Ratios of the resulting values between the increased Fe-doping sample and the first Fe-doping sample by different descriptors. | ||
Conclusion
We used a facile self-assembly strategy to synthesize a series of Fe-doped 2D MFe-LDHs as efficient OER electrocatalysts. Among the examined MFe-LDHs (M = Co, Ni, Cu, and Mn), the Ni0.78Fe0.22-LDH demonstrated the highest performance with an overpotential of 243 mV at 10 mA cm−2 and great long-term stability. Besides the identification of volcano relationships between the widely accepted activity descriptors of overpotential, Tafel slope, and turnover frequency (TOF) and the Fe contents in the MFe-LDHs, a similar correlation between the intermediate adsorption capacitance (CPEad) measured by operando EIS and the Fe contents was verified. This characterization confirmed that the intermediate adsorption capacitance (CPEad) can be a new activity descriptor for electrocatalysts. All the examinations confirmed that only a moderate amount of Fe could contribute to the optimal intermediate adsorption behaviours, and hence the maximum catalytic activity, while a higher content Fe would supress the conversion of the original surface into active sites and the following intermediate adsorption, and thus is not favourable for OER catalysis. This work not only provides a general understanding of 2D Fe-doped MFe-LDH electrocatalysts, but also provides a new descriptor for evaluating the activity of electrocatalysts.Author contributions
Ziyang Wu designed and conducted this study; Ting Liao and Ziqi Sun supervised the whole project, conceived the concept, and revised the manuscript; Sen Wang, Wei Li, Wanping Hu, Binodhya Wijerathne, Anthony P. O'Mullane, and Yuantong Gu provided their support to the experiments and discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Australian Research Council (ARC) through Future Fellowship grants (FT180100387 and FT160100281) and Discovery Project (DP200103568). The authors acknowledge the support from Central Analytical Research Facility (CARF) in QUT for material characterizations and the generous grants of CPU time from the Australian National Computational Infrastructure Facility and the High-performance Computing Centre of QUT. The authors thank Dr Osama Yousef Ali Ghidan (Sam) and Mr Tal Cooper for their help in ICP data collection and Dr Henry Spratt for XRD measurements and analysis.References
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, Nat. Energy, 2021, 6, 1054–1066 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- H.-S. Kim, B. Kim, H. Park, J. Kim and W.-H. Ryu, Adv. Energy Mater., 2022, 12, 2103527 CrossRef CAS.
- J. Dai, Y. Zhu, Y. Chen, X. Wen, M. Long, X. Wu, Z. Hu, D. Guan, X. Wang, C. Zhou, Q. Lin, Y. Sun, S.-C. Weng, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2022, 13, 1189 CrossRef CAS PubMed.
- Y. He, L. Liu, C. Zhu, S. Guo, P. Golani, B. Koo, P. Tang, Z. Zhao, M. Xu, C. Zhu, P. Yu, X. Zhou, C. Gao, X. Wang, Z. Shi, L. Zheng, J. Yang, B. Shin, J. Arbiol, H. Duan, Y. Du, M. Heggen, R. E. Dunin-Borkowski, W. Guo, Q. J. Wang, Z. Zhang and Z. Liu, Nat. Catal., 2022, 5, 212–221 CrossRef CAS.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- B. Zhang, L. Wang, Z. Cao, S. M. Kozlov, F. P. García de Arquer, C. T. Dinh, J. Li, Z. Wang, X. Zheng, L. Zhang, Y. Wen, O. Voznyy, R. Comin, P. De Luna, T. Regier, W. Bi, E. E. Alp, C.-W. Pao, L. Zheng, Y. Hu, Y. Ji, Y. Li, Y. Zhang, L. Cavallo, H. Peng and E. H. Sargent, Nat. Catal., 2020, 3, 985–992 CrossRef CAS.
- S. Liu, S. Geng, L. Li, Y. Zhang, G. Ren, B. Huang, Z. Hu, J.-F. Lee, Y.-H. Lai, Y.-H. Chu, Y. Xu, Q. Shao and X. Huang, Nat. Commun., 2022, 13, 1187 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- Y. Xu, Z. Wang, L. Tan, Y. Zhao, H. Duan and Y.-F. Song, Ind. Eng. Chem. Res., 2018, 57, 10411–10420 CrossRef CAS.
- L. Qian, Z. Lu, T. Xu, X. Wu, Y. Tian, Y. Li, Z. Huo, X. Sun and X. Duan, Adv. Energy Mater., 2015, 5, 1500245 CrossRef.
- X. Lu, H. Xue, H. Gong, M. Bai, D. Tang, R. Ma and T. Sasaki, Micro Nano Lett., 2020, 12, 86 CrossRef CAS PubMed.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W.-F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- Y. Song, M. Song, P. Liu, W. Liu, L. Yuan, X. Hao, L. Pei, B. Xu, J. Guo and Z. Sun, J. Mater. Chem. A, 2021, 9, 14372–14380 RSC.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W.-X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- Y. Song, B. Xu, T. Liao, J. Guo, Y. Wu and Z. Sun, Small, 2021, 17, 2002240 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- S. Cordoba, R. Carbonio, M. L. Teijelo and V. a. Macagno, Electrochim. Acta, 1987, 32, 749–755 CrossRef CAS.
- M. B. Stevens, L. J. Enman, E. H. Korkus, J. Zaffran, C. D. M. Trang, J. Asbury, M. G. Kast, M. C. Toroker and S. W. Boettcher, Nano Res., 2019, 12, 2288–2295 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- S. Lee, L. Bai and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 8072–8077 CrossRef CAS PubMed.
- D. Liu, H. Ai, J. Li, M. Fang, M. Chen, D. Liu, X. Du, P. Zhou, F. Li, K. H. Lo, Y. Tang, S. Chen, L. Wang, G. Xing and H. Pan, Adv. Energy Mater., 2020, 10, 2002464 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- X. Li, H.-Y. Wang, H. Yang, W. Cai, S. Liu and B. Liu, Small Methods, 2018, 2, 1700395 CrossRef.
- J. Mei, J. Shang, T. He, D. Qi, L. Kou, T. Liao, A. Du and Z. Sun, Adv. Energy Mater., 2022, 12, 2201141 CrossRef CAS.
- J. Bai, J. Mei, T. Liao, Q. Sun, Z.-G. Chen and Z. Sun, Adv. Energy Mater., 2022, 12, 2103247 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. d Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- Z. Qiu, C.-W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581 RSC.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- J. Mei, Q. Zhang, H. Peng, T. Liao and Z. Sun, J. Mater. Sci. Technol., 2022, 111, 181–188 CrossRef.
- Y. Dou, C.-T. He, L. Zhang, M. Al-Mamun, H. Guo, W. Zhang, Q. Xia, J. Xu, L. Jiang, Y. Wang, P. Liu, X.-M. Chen, H. Yin and H. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100077 CrossRef.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- H.-C. Huang, J. Li, Y. Zhao, J. Chen, Y.-X. Bu and S.-B. Cheng, J. Mater. Chem. A, 2021, 9, 6442–6450 RSC.
- D.-Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473–3479 CrossRef CAS PubMed.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- N. Zhang, Y. Hu, L. An, Q. Li, J. Yin, J. Li, R. Yang, M. Lu, S. Zhang, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202207217 CAS.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- M. Kim, K. L. Firestein, J. F. S. Fernando, X. Xu, H. Lim, D. V. Golberg, J. Na, J. Kim, H. Nara, J. Tang and Y. Yamauchi, Chem. Sci., 2022, 13, 10836–10845 RSC.
- S. Watzele, Y. Liang and A. S. Bandarenka, ACS Appl. Energy Mater., 2018, 1, 4196–4202 CrossRef CAS.
- R. Ge, J. Li and H. Duan, Sci. China Mater., 2022, 65, 3273–3301 CrossRef CAS.
- R. Ge, Y. Wang, Z. Li, M. Xu, S.-M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CAS.
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M.-S. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- Q. Wang and D. O’Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- S. Moolayadukkam, S. Thomas, R. C. Sahoo, C. H. Lee, S. U. Lee and H. S. S. R. Matte, ACS Appl. Mater. Interfaces, 2020, 12, 6193–6204 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS PubMed.
- Z. Zheng, D. Wu, G. Chen, N. Zhang, H. Wan, X. Liu and R. Ma, Carbon Energy, 2022, 4, 901–913 CrossRef CAS.
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert, B. R. Cuenya, W. Schuhmann and M. Muhler, ChemSusChem, 2017, 10, 156–165 CrossRef CAS PubMed.
- C. Wu, X. Zhang, Z. Xia, M. Shu, H. Li, X. Xu, R. Si, A. I. Rykov, J. Wang, S. Yu, S. Wang and G. Sun, J. Mater. Chem. A, 2019, 7, 14001–14010 RSC.
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, J. Mater. Chem. A, 2016, 4, 9578–9584 RSC.
- D. Guo, Z. Wu, Y. An, X. Li, X. Guo, X. Chu, C. Sun, M. Lei, L. Li, L. Cao, P. Li and W. Tang, J. Mater. Chem. C, 2015, 3, 1830–1834 RSC.
- W. Cheng, H. Zhang, D. Luan and X. W. Lou, Sci. Adv., 2021, 7, eabg2580 CrossRef CAS PubMed.
- R. Li, B. Hu, T. Yu, H. Chen, Y. Wang and S. Song, Adv. Sci., 2020, 7, 1902830 CrossRef CAS PubMed.
- M. A. Z. G. Sial, S. Baskaran, A. Jalil, S. H. Talib, H. Lin, Y. Yao, Q. Zhang, H. Qian, J. Zou and X. Zeng, Int. J. Hydrogen Energy, 2019, 44, 22991–23001 CrossRef CAS.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- S. Zhang, T. Yu, H. Wen, Z. Ni, Y. He, R. Guo, J. You and X. Liu, Chem. Commun., 2020, 56, 15387–15405 RSC.
- R. L. Doyle and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 5224–5237 RSC.
- R. L. Doyle and M. E. G. Lyons, J. Electrochem. Soc., 2013, 160, H142–H154 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2009, 631, 62–70 CrossRef CAS.
- B. D. Cahan and C. T. Chen, J. Electrochem. Soc., 1982, 129, 700–705 CrossRef CAS.
- F. Kong, W. Zhang, L. Sun, L. Huo and H. Zhao, ChemSusChem, 2019, 12, 3592–3601 CrossRef CAS PubMed.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh01217k |
| This journal is © The Royal Society of Chemistry 2023 |