
High entropy alloying strategy for accomplishing quintuple-nanoparticles grafted carbon towards exceptional high-performance overall seawater splitting†
Gokul
Raj‡
 a,
Ravi
Nandan‡
a,
Kanhai
Kumar
a,
Demudu Babu
Gorle
a,
Ambresh B
Mallya
b,
Sameh M.
Osman
c,
Jongbeom
Na
*de,
Yusuke
Yamauchi
cef and
Karuna Kar
Nanda
*ag
a,
Ravi
Nandan‡
a,
Kanhai
Kumar
a,
Demudu Babu
Gorle
a,
Ambresh B
Mallya
b,
Sameh M.
Osman
c,
Jongbeom
Na
*de,
Yusuke
Yamauchi
cef and
Karuna Kar
Nanda
*ag
aMaterials Research Centre, Indian Institute of Science, Bangalore-560012, Karnataka, India. E-mail: nanda@iisc.ac.in
bMicro Nano Characterization Facility, Centre for Nano Science and Engineering, Indian Institute of Science, Bangalore-560012, India
cChemistry Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
dMaterials Architecturing Research Center, Korea Institute of Science and Technology (KIST), 5 Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: jongbeom@kist.re.kr
eAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia
fDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464–8603, Japan
gInstitute of Physics (IOP), Bhubaneshwar-751005, India
First published on 31st August 2023
Abstract
High entropy alloys (HEAs), a novel class of material, have been explored in terms of their excellent mechanical properties. Seawater electrolysis is a step towards sustainable production of carbon-neutral fuels such as H2, O2, and industrially demanding Cl2. Herein, we report a practically viable FeCoNiMnCr HEA nanoparticles system grafted on a conductive carbon matrix for promising seawater electrolysis. The comprehensive kinetics analysis of the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and chlorine evolution reaction (CER) confirms the effectiveness of our system. As an electrocatalyst, HEAs grafted on carbon black show trifunctionality with promising kinetics, selectivity and enduring performance, towards seawater splitting. We optimize high entropy alloy decorated/grafted carbon black (HEACB) catalysts, studying their synthesis temperature to scrutinize the effect of alloy formation variation on the catalysis efficacy. During the catalysis, selectivity between two mutually competing reactions, CER and OER, in the electrochemical catalysis of seawater is controlled by the reaction media pH. We employ Mott–Schottky measurements to probe the band structure of the intrinsically induced metal–semiconductor junction in the HEACB catalyst, where the carrier density and flat band potential are optimized. The HEACB sample provides promising results towards overall seawater electrolysis with a net half-cell potential of about 1.65 V with good stability, which strongly implies its broad practical applicability.
New conceptsOne of the bottlenecks of water splitting is the requirement of high overpotentials to override the oxygen evolution reaction (OER) kinetics. It makes the overall process less economical and highly energy-consuming. Moreover, water splitting generally requires pure water as the starting material, further complicating the overall process. On the other hand, brine is widely available as a source of hydrogen, and it covers 70% of the Earth's entire surface. However, hydrogen production using brine faces a serious challenge with Cl− competing with OH− resulting in a complex and unfavorable situation for the overall reaction to proceed. Herein, a high entropy nanoalloy system that is robust and corrosion resistant, is designed and developed that can ensure the selectivity of the chlorine evolution reaction (CER) over OER by controlling the experimental conditions and ambiance. To understand the science, we have employed Mott–Schottky analysis (MS analysis) to unveil the rationality of fast interfacial electron transfer arising due to the band position and its bending near to the interface. This scientific understanding can be easily extended to develop various electrocatalysts for selective oxygen or chlorine evolution reactions using brine water for the complementary hydrogen evolution reaction (HER) taking place at the cathode. |
Introduction
High entropy alloys (HEAs) are a diverse class of materials containing five or more elements with atomic percentages usually ranging from 5 to 35% with diverse mechanical and chemical properties.1,2 The presence of multi-constituent metals escalates the entropy of the overall system, particularly for a five-component system with metals in equimolar ratio, the mixing entropy accounts for about 1.61R, where R is the gas constant.3–5 This very high entropy accounts for the interesting properties of HEAs.6,7 HEAs are usually procured as FCC, BCC, HCP, or a mixture of these phases but ideally with zero intermetallic phase segregation in the system.8 These novel alloys initially garnered significant attention in the field of materials science and engineering due to their unique properties: many of these alloys exhibit a higher strength-to-weight ratio, along with remarkable corrosion and fracture resistance.9–13 However, this varied class of materials is still not understood in terms of their comprehensive electrochemistry for producing various energy-demanding products.Limited energy resources and ever-increasing power requirements are barriers faced by the human race today. According to up-to-date statistics in 2021, the Covid-19 pandemic significantly increased global energy demand.14–17 To address this issue, constant efforts have been made to develop advanced energy generation and storage systems, like fuel cells and batteries.18 Hydrogen has been deemed as one of the high energy dense and potential future alternatives to fossil fuels. The water splitting process powered by renewable sources like solar, wind, and hydropower could become a reliable and renewable energy production pathway.19 The overall water splitting reaction comprises two important half reactions: the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). However, the current operating cell voltage for commercial electrolyzers (1.8 to 2.2 V) surpasses the theoretical value of 1.23 V by a huge margin, making the overall process less economical.20 To address this issue, a highly active and durable electrocatalyst is required to accelerate the active species adsorption/desorption process efficiently, thereby reducing the energy barrier of such thermodynamically uphill processes. The current state of the art catalysts for HER and OER are based on precious metals like Pt, RuO2, and IrO2, dispersed on suitable supporting matrixes.4,9,21 However, despite their high efficiency in catalytic reactions, their widespread adoption is hampered by their scarcity, high cost, and poor stability. The problem can be addressed by developing affordable, durable, and exceptionally active catalyst materials, possibly by tweaking possible extrinsic factors in transition metals and their alloys.
In light of the theoretical volcano plot of HER, the catalytic activity primarily relies on the strength of the catalyst-H bond.22 The multi-component alloys containing diverse elements like Cu, Zn, Fe, Mo, etc., exhibit complex synergistic interactions inherited from numerous and diverse active sites.23 Interestingly, metals like Ni, Co, and Fe bond with H with a weak interaction, whereas Mo and W strongly bond with H.24 But when we combine all these diverse metals into a single alloy, it is envisaged this will provide excellent catalytic output. This is often explained in terms of their inherent synergistic effect, which is often referred to by the term ‘HEA cocktail effect’.25 Another factor that favors such alloy catalysis is the partially filled d-band orbitals of these metals which can assist in efficiently completing electron exchanges during a reaction cycle.26 However, despite these advantages, such transition metal-based alloys exhibit serious shortcomings like poor corrosion resistance and leaching, giving them low stability, making them poor candidates for practical purposes. Recently some unique HEAs have been developed for various applications – Pt18Ni26Fe15Co14Cu27 for methanol oxidation through a one-pot oil phase method,27 AlFeMnTiM (M = Cr, Co, Ni) for efficient degradation of azo dyes28 – and disordered CoCuGaNiZn, AgAuCuPdPt HEAs and AuAgPtPdCu predicted through DFT, have been developed experimentally for carbon dioxide reduction catalysis with outstanding performance and high turnover frequency.29,30 In addition, FeCoNiCuZn oxide-based multi-metal alloy has been fabricated recently via a urea-based aqueous solution method for nitrobenzene electrocatalytic reduction;31 in a similar vein, a highly efficient HER catalyst based on Ni20Fe20Mo10Co35Cr15 has also been reported recently with excellent corrosion resistance in both acidic and basic reactions.32 These findings indicate the high prospect and potential of such alloy systems for evolving into exemplary catalysts.
Along with typical water electrolysis, seawater electrolysis could alleviate significant demand resulting in practical products:6,33 since 70% of the Earth's entire surface is covered with brine, seawater electrolysis would provide a very attractive and reliable supply system of hydrogen, oxygen, and chlorine if appropriate catalysts can be developed to selectively generate essential and expensive gases by controlling the reaction kinetics. Generally, in the electrolysis of brine solution, water molecules compete with the Cl− ions at the anode side to release chlorine gas through the chlorine evolution reaction (CER), which remains an important hurdle in this method.32,34 As a counter-reaction to the anodic potential, it is beneficial to obtain hydrogen gas via the HER as the cathode product; if this could be managed by the selective oxidation of chloride ions and oxide ions along with other competing species like bromide and sulfates in seawater, this process would be of enormous economic benefit.35 Thermodynamically, the OER is preferred over the CER reaction since the standard potential for the OER and the CER are 1.23 V and 1.36 V, respectively.34 However, the two-electron kinetics of CER seem more active compared to the four-electron kinetics of OER, which makes the chloride oxidation process favorable with slightly lower overpotential. Therefore, one must use these basic distinctions to tweak the reaction with an efficient catalyst to get the desired product via an economically feasible pathway.
Multi-elemental HEAs have emerged as a type of catalyst material with promising physicochemical properties, capable of managing multiple reactions simultaneously because of their numerous active sites.36 Moreover, the catalytic behavior of HEAs can be enhanced by coupling them with suitable substrates such as conductive carbon, doped graphite materials, and MXenes.37,38 Herein, we report a single-step pyrolysis technique to develop a high entropy alloy of Cr, Mn, Fe, Co, and Ni on conductive carbon black (HEACB) for the effective catalysis of HER, OER, and CER reactions, utilizing the synergistic effects of the mixed metal (cocktail effect) high entropy alloy. This work involves an in-depth analysis of kinetics, impedance spectroscopy, and Mott–Schottky analysis for each reaction. This work has the potential to open new avenues towards the development of a near-ideal catalyst that satisfies all the criteria outlined in the catalyst figure of merit. By tweaking and optimizing their inherent structural and chemical characteristics, we can aim for efficient seawater splitting. Additionally, this research aims to gain a deeper understanding of the reaction kinetics and any limitations associated with them.
Results and discussion
Characterization of the composite alloy materials
Fig. 1a shows the X-ray diffraction (XRD) patterns of the samples prepared under different annealing temperatures. After annealing at 500 °C, the diffraction peaks corresponding to the BCC phase appear broad. As the annealing temperature increases to 750 °C, besides the diffraction peaks of the BCC–structured solid solution, diffraction peaks corresponding to the FCC and HCP phases emerge. This indicates that the alloy has a multi-phase structure with mixed BCC, FCC, and HCP phases. In addition, we observe a superlattice reflection peak (indicated by *) at ∼30.24°, corresponding to the ordered B2 phase. This indicates that the alloy either has a single B2-ordered solution structure or a disordered BCC + B2 mixed phase structure. Further, the intensity of the BCC, HCP, and B2 phases decreases and completely disappears with increasing annealing temperature from 750 to 1000 °C. Meanwhile, the intensity of the diffraction peaks of the FCC phase increases with temperature, which indicates the complete formation of a solid alloy structure in the FCC phase. We observe an additional peak at ∼35.56° (indicated by *) with a dramatic decrease in intensity after annealing, probably due to impurities in the structure. In addition, we observe a broad XRD peak at ∼25.18° at all annealing temperatures, confirming the presence of carbon black in the composites. | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of HEACB samples at different synthesis temperatures. (c) HEACB 1000 bright filed TEM image, (d and e) HAADF-STEM images, (f–j) elemental maps: (f) Fe, (g) Co, (h) Ni, (i) Mn, (j) Cr, and (k–p) the line scan maps with the corresponding intensity profiles of individual elements. | ||
The presence of graphitic and defective carbon sites in the material is confirmed by Raman spectroscopy, as shown in Fig. 1b. The typical Raman peaks observed at 1340 (D band), 1569 (G band), and 2685 cm−1 (2D band) correspond to sp3 defects and in-plane vibrations of sp2 hybridized carbon atoms.39 The 2D peak suggests C–C bonds with sp2 domains in the material with little strain between the bonds. The ID/IG ratio provides information on structural defects, and the defect densities (ID/IG ratio, Table S1, ESI†) of the composite alloys are 0.65, 0.76, and 1.10, produced at 500, 750 and 1000 °C, respectively.40 Hence, the defect densities increase with higher annealing temperatures, which can be attributed to the resulting distortions. Furthermore, the presence of defects reduces the intensity of I2D/ID, and this value decreases as the annealing temperature increases due to the declining crystallinity of the sp2 domains.
To obtain a direct visualization of the samples at different annealing temperatures, we employed scanning electron microscopy (SEM) analysis (Fig. S1, ESI†). There is no discernible effect on the morphology of the particles at 750 and 1000 °C. Fig. S1e–k (ESI†) show the elemental mapping from energy-dispersive X-ray spectroscopy (EDS) analysis within a selected area (HEACB 1000). This emphasizes the homogeneous distribution of all elements throughout the structure, hence confirming the successful alloy formation. The percentages of constituent elements in the alloy were determined by EDS spectrum analysis, with the data provided in Table S2 (ESI†). The alloy exhibits an almost uniform distribution of constituent elements, indicating the formation of HEAs. However, the alloy contains a higher amount of carbon compared to other elements, suggesting the presence of carbon black as the primary catalyst substructure in the material.
To obtain more detailed and highly resolved images of our sample, we employed transmission electron microscopy (TEM). The bright field TEM image in Fig. 1c and the high-angle annular dark field (HAADF) scanning mode image in Fig. 1d, show the HEA nanoparticles embedded in the carbon black matrix. A representative particle in the HAADF-scanning transmission electron microscopy (STEM) mode was scanned at high resolution, as shown in Fig. 1e. This image was then subjected to elemental mapping (Fig. 1f–j) to visualize the distribution of elements. The elemental mapping reveals a uniform distribution of all five elements in the nanoparticles, thereby confirming the formation of a homogeneous solid solution in the HEAs. The line scan (Fig. 1k) conducted over two randomly selected nanoparticles (Fig. 1l–p) also yields similar results, confirming the successful stabilization of HEA particles on a highly conductive carbon black support.
To further shed light on the elemental details in HEACBs, we performed X-ray photoelectron spectroscopy (XPS) studies (Fig. 2 and Fig. S2–S5, ESI†). The C 1s peak is maintained at 284.78 eV by applying charge correction for all samples. The deconvoluted HRXPS spectra of Fe, Cr, Mn, Ni, and Co (Fig. 2a–e) indicate the existence of multiple oxidation states, revealing the inherent complex electronic nature of HEAs.25 Such a multielement alloy nature at the atomic level in HEAs is beneficial for offering near-continuous adsorption energy and can be advantageous for tuning the overall physicochemical properties of electrocatalysts for various catalytic activities.25
 | ||
| Fig. 2 XPS spectra of (a) Fe 2p, (b) Cr 2p, (c) Mn 2p, (d) Ni 2p (e) Co 2p and (f) C 1s of the HEACB 1000 catalyst. | ||
The deconvoluted C 1s high-resolution XPS (HRXPS) spectrum in Fig. 2f suggests the presence of carbon moieties such as C–C (284.78 eV), C–N (285.53 eV), C–O (287.09 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.5 eV)25 (Table S3, ESI†). Incorporating the metal core into the carbon matrix allows for the adjustment of adsorption energies for different electroactive species.41 We also found that raising the reaction temperature from 500 to 1000 °C, the overall metal concentration raised and reached a maximum at 750 °C and then came down to a lower concentration at 1000 °C, as shown in Fig. S2 (ESI†). This change in metal concentration is due to the loss of segregated phases of all elements due to evaporation at higher temperatures, which favors alloy formation.31 By analyzing the wide spectra of all three samples, we can see distinct oxide peaks, representing the oxidation of the sample during handling and material synthesis (Fig. S5, ESI†). Correlating the results from the C 1s HRXPS, we can ascertain that these oxygen peaks indicate surface oxygen groups like aldehyde (–CHO) and carboxylic acid (–COOH).42 To gain insight into the presence and involvement of such surface oxygen, we analyze the convoluted XPS spectrum of oxygen in detail in Tables S4, S5 and Fig. S5 (ESI†). As the literature suggests, oxygen is present in different locations in a sample – oxygen atoms in the lattice (OL) (∼530 eV),31 oxygen atoms in the vicinity of an oxygen vacancy (Ov) at ∼531 eV,31 oxygen atoms on the outer face of the lattice structure (Os) with a lower coordination number at ∼532 eV,31 and oxygen species adsorbed (Oads) which may be due to the adsorption of water from the environment, located at ∼533 eV.31 Interestingly, we observe as the temperature increases from 500 °C to 1000 °C, that there is an intensification of Os peaks. Among these, Os peaks always occupy the major contribution which once again signifies the presence of surface functionalities. At higher temperatures of 1000 °C, we find that the concentration of surface oxygen increases, indicating improved order of alloy material formation. This aligns with accelerated alloy formation and supports the notion that higher temperatures facilitate the creation of high entropy alloys. Consequently, the HEACB 1000 system is anticipated to possess a greater abundance of active alloy sites, leading to enhanced catalytic activity, as confirmed by our electrocatalytic investigations. It is evident from numerous reports that such oxygen is beneficial in inducing defects in the system and enhancing the overall catalytic activity by favoring the preferred reaction sites for specific intermediates.31
O (288.5 eV)25 (Table S3, ESI†). Incorporating the metal core into the carbon matrix allows for the adjustment of adsorption energies for different electroactive species.41 We also found that raising the reaction temperature from 500 to 1000 °C, the overall metal concentration raised and reached a maximum at 750 °C and then came down to a lower concentration at 1000 °C, as shown in Fig. S2 (ESI†). This change in metal concentration is due to the loss of segregated phases of all elements due to evaporation at higher temperatures, which favors alloy formation.31 By analyzing the wide spectra of all three samples, we can see distinct oxide peaks, representing the oxidation of the sample during handling and material synthesis (Fig. S5, ESI†). Correlating the results from the C 1s HRXPS, we can ascertain that these oxygen peaks indicate surface oxygen groups like aldehyde (–CHO) and carboxylic acid (–COOH).42 To gain insight into the presence and involvement of such surface oxygen, we analyze the convoluted XPS spectrum of oxygen in detail in Tables S4, S5 and Fig. S5 (ESI†). As the literature suggests, oxygen is present in different locations in a sample – oxygen atoms in the lattice (OL) (∼530 eV),31 oxygen atoms in the vicinity of an oxygen vacancy (Ov) at ∼531 eV,31 oxygen atoms on the outer face of the lattice structure (Os) with a lower coordination number at ∼532 eV,31 and oxygen species adsorbed (Oads) which may be due to the adsorption of water from the environment, located at ∼533 eV.31 Interestingly, we observe as the temperature increases from 500 °C to 1000 °C, that there is an intensification of Os peaks. Among these, Os peaks always occupy the major contribution which once again signifies the presence of surface functionalities. At higher temperatures of 1000 °C, we find that the concentration of surface oxygen increases, indicating improved order of alloy material formation. This aligns with accelerated alloy formation and supports the notion that higher temperatures facilitate the creation of high entropy alloys. Consequently, the HEACB 1000 system is anticipated to possess a greater abundance of active alloy sites, leading to enhanced catalytic activity, as confirmed by our electrocatalytic investigations. It is evident from numerous reports that such oxygen is beneficial in inducing defects in the system and enhancing the overall catalytic activity by favoring the preferred reaction sites for specific intermediates.31
Oxygen evolution reaction
Electrochemical water splitting reaction is deemed to be one of the reliable and renewable means of hydrogen and oxygen production with zero carbon emission to the environment.43 Water splitting is essential in numerous technological applications like fuel cells, metal–air batteries, chemical industries, etc. Statistics show that in contradiction to the manifold advantages of electrochemical synthesis, only around 4% of the world's hydrogen demand can be satisfied by water electrolysis.44 One of the major hindrances to their practical application is the sluggish kinetics of the four-electron-based oxygen evolution reaction (OER). The overall reaction thus runs at a high voltage of 1.8 to 2.0 V, which demands huge electrical input and makes the overall process less feasible. To overcome this hurdle, we require an efficient catalyst to catalyze both the half-cell reactions, i.e., hydrogen evolution reaction (HER) and OER reactions together, which will significantly reduce the production cost of these gases. Among these two half-cell reactions, OER is more challenging.45,46Herein, we investigate the electrochemical performance of our as-synthesized high entropy alloy samples viz HEACB 1000, HEACB 750, and HEACB 500, towards OER in a 1 M KOH medium using a standard three-electrode configured CHI 750E electrochemical workstation. Linear sweep voltammetry (LSV) profiles on a glassy carbon electrode (GCE) were taken for all the samples in the OER polarization window. Fig. 3a shows the LSV curves of HEACB 1000, 750, and 500 samples compared to the current state-of-the-art catalyst RuO2. As illustrated in Fig. 3a and Fig. S6a (ESI†), HEACB 1000 can push current at a rate of 10 mA cm−2 with a lesser onset and an overpotential of 350 mV, followed by HEACB 750 (352 mV) and finally HEACB 500 (389 mV). These values reveal the superiority of the HEACB 1000 over the others in efficiently operating OER kinetics.
 | ||
| Fig. 3 (a) LSV curves of commercial RuO2 and HEACB samples in the OER polarization region in 1 M KOH. (b) Tafel slopes analysis extracted from the kinetic region of LSV curves, (c) electrochemical impedance spectroscopic analysis of HEACB samples in 1 M KOH, and (d) LSVs of the fresh, poisoned, and washed HEACB 1000 in the OER polarization region. | ||
Even though the overpotential was slightly higher than the commercial RuO2, in terms of reaction kinetics and overall reaction stability, HEACB 1000 is competent enough compared with RuO2. The Tafel slope of a catalyst can often be correlated directly to the reaction mechanism and kinetics. A lower Tafel value indicates a less energy-demanding reaction pathway and more active site exposure hence faster kinetics. Fig. 3b shows that HEACB 1000 possesses a shallower Tafel slope of 95 mV dec−1, compared with HEACB 750 (112 mV dec−1) and HEACB 500 (118 mV dec−1), and is also better compared with the commercial RuO2 catalyst (108 mV dec−1) and various recently reported OER catalysts (Table S10, ESI†).
Electrochemical impedance spectroscopy (EIS) is carried out to obtain a perception of the charge transfer resistance at the electrode/electrolyte interface in a 1 M KOH, representing the ease with which the electron transfer can happen at the heterojunction with the catalyst's intrinsic conductivity. Tafel slopes are often expected to have direct implications with charge transfer resistance measured through EIS. Fig. 3c shows the EIS plot of the as-synthesized samples, manifesting the Rct + Rs values (sum of charge transfer and solution resistance) of HEACB 1000 as 12.3 Ω, HEACB 750 as 19.7 Ω, and HEACB 500 as 33.8 Ω. Usually, an EIS spectrum with a smaller circular diameter represents swift and augmented charge transfer capability.38 This shows that our sample HEACB 1000 is also best in terms of charge transfer conductivity. The charge conduction between the electrolyte and multi-alloy interface appears to be more feasible due to the influence of highly conductive carbon on which the high entropy alloy has been dispersed.
The electrochemically active surface area (ECSA) represents the degree of active sites on our catalytic bed, and it can be probed in terms of the double layer capacity (Cdl) of a sample. The cyclic voltammetry response of HEACB 1000, 750, and 500 in 1 M KOH in the non-faradaic region and the corresponding Cdl value derived from the charging current vs. scan rate plot are shown in Fig. S6 and S7 (ESI†). We perceive Cdl values to be in the order: HEACB 1000 (11.3 mF cm−2) > HEACB 500 (10.2 mF cm−2) > HEACB 750 (5.4 mF cm−2). It is worth noting that HEACB 500, even though it has a higher area than HEACB 750, has the lowest activity, and this can be ascribed to the better intrinsic activity of HEACB 750 due to the better alloy formation compared to the lower temperature annealed HEACB 500 sample. Since ECSA has a direct correlation to the Cdl values, HEACB 1000 stands out to be the one with the highest ECSA.44
The direct involvement of the mixed metal cocktail effect of the high entropy alloy and their related active sites on the catalytic activity can be evaluated by exercising a poisoning test using cyanide ligands.47 Being a strong π acceptor, CN− can bind with the metal centers strongly, and will block the approaching electroactive species, thus reducing the catalytic efficiency. Fig. 3d shows the LSV curves of the poisoning experiment, where we can observe that after poisoning the catalyst with 0.1 M NaCN solution, the activity of the sample drastically lowered. This observation reinforces our assumption that the alloy metal centers are the key area behind the promising OER performance. When the poisoned catalyst is washed with DI water to free the metal centers from the ligand hindrance, activity regeneration can be seen which returns near to the level of the fresh sample. The failure to completely recover the activity after poisoning, is expected to be due to the combination of incomplete removal of the cyanide moieties and sample loss incurred during this cleaning process.
For an industrial catalyst to be truly effective, it must show high levels of activity and durability, even when subjected to continuous catalytic processes. So, the best-performing HEACB 1000 catalyst was tested for six hours in 1 M KOH using chronoamperometry analysis, as shown in Fig. S6d (ESI†), to study its stability in the OER polarization region. The current–time response indicated more than ∼75% activity retention post long-term chronoamperometric operation in the harsh electrolytic medium. The activity reduction we observe can likely be attributed to the harsh and corrosive conditions utilized during the OER process. Additionally, we suspect that some sample loss may have occurred on the electrode surface because of prolonged oxygen gas evolution. However, as seen in Fig. S6d (ESI†), after the initial decline in the activity, the catalyst reaches a stable state of action where the stability seems to be retained for a longer time. This observation is quite promising for its practical use in OER applications.
Hydrogen evolution reaction
Electrochemical hydrogen evolution reaction (HER), one of the half-reactions in total water electrolysis, is an economically promising and greener way to deliver hydrogen fuel for the ever-increasing energy demands of humanity. This carbon-free high-energy dense fuel is a major requirement in fuel cells, automobiles, spacecrafts, etc. We analyze the HER performance of our catalyst on a similar 3 electrodes configured setup in 0.5 M H2SO4 electrolytic media. For activity comparison and benchmarking, commercial Pt/C has been employed. Fig. 4a shows the LSV at a 10 mV s−1 scanning rate of all the prepared alloy samples. Fig. S8a (ESI†) shows the onset and overpotential comparison of the HEACB sample synthesized at different pyrolysis temperatures. | ||
| Fig. 4 (a) LSV curves of HEACB samples and commercial Pt–C in the HER polarization region in 0.5 M H2SO4, (b) Tafel plots for the respective polarization curves, and (c) electrochemical impedance spectroscopic analysis of HEACB samples in 1 M H2SO4, and (d) chronoamperometric test for HEACB 1000 in 0.5 M H2SO4 at room temperature in the HER polarization potential (inset: H cell in which water electrolysis is carried out). | ||
It is evident from the comparison plot that the HEACB 1000 with a high degree of single-faced high entropy alloys shows excellent results. In terms of these parameters related to the reaction potential, HEACB 1000 demands a lower onset potential of 72 mV and an overpotential of 190 mV to drive a current of 10 mA cm−2 in the HER polarization region. HER occurs by following two reaction pathways, Volmer–Tafel and Volmer–Heyrovsky pathways.24
Volmer–Heyrovsky pathway:
| H2O + e− → Hads + OH− (Volmer) |
| Hads + H2O + e− → H2 + OH− (Heyrovsky) |
Volmer–Tafel pathway:
| H2O + e− → Hads + OH− (Volmer) |
| Hads + Hads → H2 (Tafel) |
Scrutinizing the Tafel slope in HER gives a lot of insight into the reaction mechanism and nature of reaction kinetics compared to OER, where later the reaction mechanism seems to be much more complex than HER.46,48Fig. 4b shows the Tafel plots for the samples along with the commercial Pt/C catalyst. Among the HEACB catalysts, HEACB 1000 exhibits the most favorable performance, as evidenced by its Tafel slope of 64 mV dec−1. Tafel slope, along with the turnover frequency, exchange current density, and faradaic efficiency values (see Tables S6–S8, ESI†), indicates that HEACB 1000, with a comparatively better and high degree of alloy formation, is capable of promoting HER catalysis efficiently with stability, and at the same time has cheaper production costs compared with precious metal-based catalysts. The value of the Tafel slope 64 mV dec−1 propounds that the rate-determining step is the Heyrovsky step (product desorption step) for HEACB 1000, and hence has faster kinetics. Whereas in the case of HEACB 750 and HEACB 500, we witness higher Tafel values suggesting that the rate-determining step is shifted to the Volmer step, i.e., the active species adsorption step, which makes their catalysis less energy efficient and thermodynamically uphill.
To correlate and corroborate the above kinetics and activity trend, we subject the samples to EIS measurements to study the nature and charge transfer resistance of the catalyst at the heterojunction in an acidic environment. Fig. 4c illustrates the EIS plot for all HEACB samples. We could infer a similar trend to what we observe in basic media in the charge transfer resistance of all three catalysts, which again strengthens our observation of the trend in reaction kinetics of our samples towards HER. The electrochemical active surface area (ECSA) also plays a prominent role in defining the overall activity of the catalyst because it is a measure of the available and accessible active sites on the catalyst surface. Fig. S8b and c (ESI†) show the cyclic voltammetric response and the respective double-layer capacitance measurement plot in a region away from reaction species polarization. As we expect, HEACB 1000 is found to be incorporated with a more active surface area (Cdl = 15.4 mF cm−2) concerning the other two samples (HEACB 750, Cdl = 5.4 mF cm−2 and HEACB 500, Cdl = 8.6 mF cm−2). But in the case of HEACB 750, we find the ECSA is lower than the HEACB 500. This proves the less inherent activity of HEACB 500 samples despite the favorable surface area factor. The comparatively higher alloying nature of NPs present in the HEACB 750 than in HEACB 500 enhances its intrinsic activity, which might have helped it to override the surface area factor and raised it to be a more efficient HER catalyst than HEACB 500.
The direct contribution of the highly active alloy metal centers to the excellent HER activity has been proven through a poisoning test. The poisoning agent used in this test was a 0.1 M cyanide ion solution, similar to the OER activity check. Fig. S8d (ESI†) shows the core observation made in the poisoning experiment. After poisoning, there was a drastic reduction in the performance, indicating the complete or partial blockage of the active metal centers, thus hindering the catalysis. The increased activity observed after removing CN− ions from the electrode surface highlights that the activity of our synthesized catalyst is solely attributed to the presence of active high entropy alloy nanoparticles grafted on the conductive carbon bed. The long-term stability of a catalyst is always of great concern when we look towards practical application of the catalyst materials, such as in the case of bulk water electrolyzers. Fig. 4d shows the stability response curve of our best-performing, HEACB 1000, catalyst in 0.5 M H2SO4 solution in the HER polarization region. It is evident from the current response that the sample shows very promising stability in the HER potential range, where we can see that even after continuous operation for almost 8 hours there is no considerable degradation to HER performance. Moreover, the catalyst activity is enhanced during the stability run, possibly due to the activation of metal species on the catalytic surface and the suitable charge transfer environment.
Chlorine evolution reaction
Chlorine is one of the most demanded elements in the industry, required for the synthesis of many chemical products like plastics, disinfectants, bleaching powders, pesticides, and various inorganic salts, as well as products in the electronic industry.5,34 The current chlorine requirements are satisfied by chloroalkyl electrolysis, a process that demands enormous investment and seriously threatens environmental health by emitting pollutants. To overcome these shortcomings and fulfill chemical demand, an efficient electrochemical oxidation process using a robust and efficient catalyst is needed. A novel strategy is to utilize vastly available seawater as the electrolyte in the water-splitting reaction. This process appears to be a very interesting and reliable source of hydrogen, oxygen, and chlorine production, if the reaction can be selectively steered toward our targeted products. Under normal conditions, in the electrolysis of seawater, water molecules compete with Cl− ions at the anode side to release chlorine; from the counter-reaction, we are able to extract hydrogen gas from the cathode end, which makes the overall process economical and promising in terms of the production of two industrially demanded gases.35 If we can manage the targeted oxidation of chloride and oxide ions, while also dealing with other substances like bromide and sulfates present in seawater, this process would have a substantial impact for the energy and chemical production industries.9,34Herein, we try to explore the chlorine evolution reaction kinetics on the catalytic surface of our novel high entropy alloy grafted conductive carbon-based samples. The goal is to enhance the electrolytic process by promoting selectivity between chloride and oxide ions, resulting in non-parasitic CER and OER reactions. Fig. 5a shows the LSV response of the HEACB samples in a 1 M HCl and 4 M NaCl medium at a scan rate of 10 mV s−1 on a 3-electrode configuration-based electrochemical workstation. Thermodynamically the OER is preferred over the CER reaction. However, the two-electron kinetics of CER compared to the four-electron sluggish kinetics of OER, makes the chloride oxidation process favorable with slightly lower overpotential. Fig. 5a and b compare the relative activity of HEACB 1000, HEACB 750, and HEACB 500 with different degrees of high entropy alloy content, towards CER activity. The HEACB 1000 shows the least onset potential with an overpotential of only 30 mV to deliver a current of 10 mA cm−2, whereas HEACB 750 and HEACB 500 exhibit overpotentials of 79 mV and 163 mV, respectively.
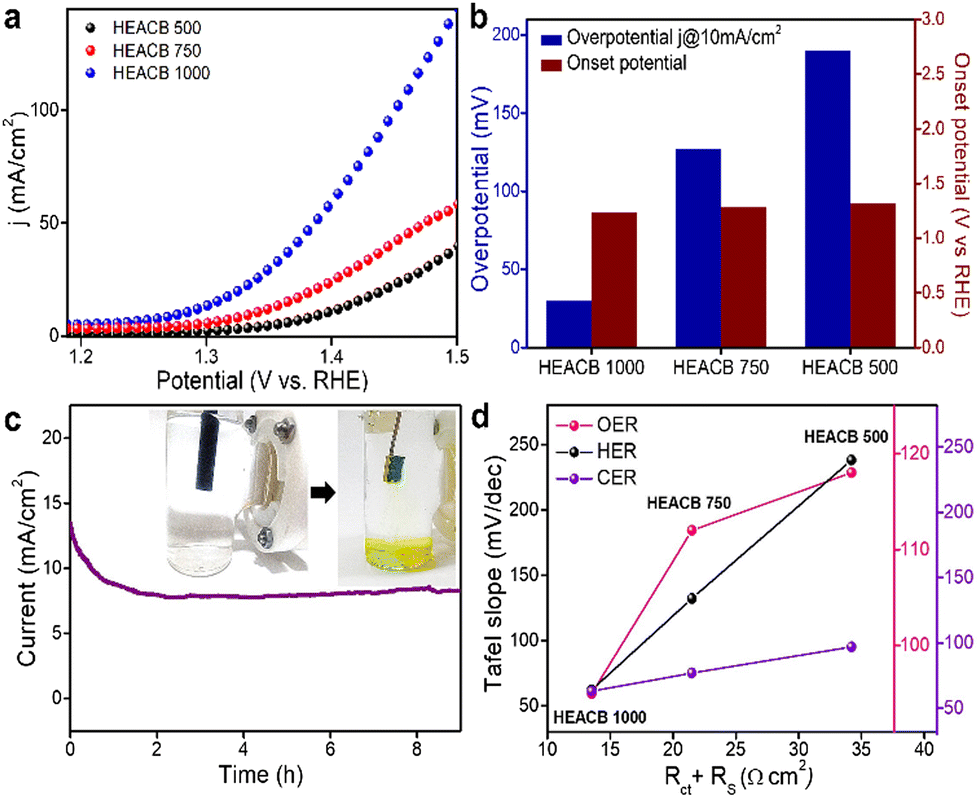 | ||
| Fig. 5 (a) LSV curves of HEACB samples in the CER polarization region in 0.5 M H2SO4 and 4 M NaCl. (b) Comparison plot of onset and overpotential of HEACB samples. (c) Chronoamperometry test for HEACB 1000 at room temperature in the CER polarization potential, and (d) Tafel slope plot comparing HEACB samples with system resistance derived from electrochemical impedance spectroscopic analysis. | ||
The kinetics of CER is well studied on RuO2 surfaces, and their reaction mechanism is explored. The reaction proceeds via the Volmer–Heyrovsky pathway with the desorption step being the rate-determining step,
| 2Cl− → Clad + e− + Cl− (Volmer step) |
| Clad + Cl− → Cl2 + e− (Heyrovsky step) |
To show the involvement of metal core centers in the catalysis, we have staged a poison test with cyanide ions, as shown in Fig. S9b (ESI†). The contrast in the behavior before and after poisoning substantiates the active metal centers in the catalyst responsible for CER activity. To promote any catalyst towards possible practical seawater splitting application of chloride electrolysis, it is required to analyze how suitable the catalyst is in terms of its stability, especially in a corrosive and harsh seawater environment. Chronoamperometric stability analysis was carried out where the HEACB 1000 samples were run in the CER polarisation region in 4 M NaCl and 1 M HCl media for a continuous operation of about 8 h, as shown in Fig. 5c. The stability response shows that the HEACB 1000 retains activity without much degradation, even in a deteriorating and hostile reaction media.
To understand the deteriorative effects on the catalyst material, detailed chemical and structural characterization of our best-performing sample, HEACB 1000, after continuous operation of 15 hours was performed. By conducting this study, we aimed to determine if our catalyst can endure the severe reaction conditions of seawater splitting and if it is a viable option for use as an electrocatalyst in practical applications. Fig. S10–S12 and Table S9 in the ESI,† show the detailed chemical and structural characterization of HEACB 1000 after continuous HER, CER, and OER reactions in a seawater-simulated electrolyte media for about 15 hours of continuous operation. The improved durability of the catalyst can be attributed to the intimate contact between the dispersed HEA nanoparticles and the conductive carbon matrix. During the pyrolysis process, the conductive carbon develops a strong bond with the nanoparticles, preventing their leaching or detachment from the catalytic framework in the harsh electrolytic environment. This intimate contact can ensure the stability and longevity of the catalyst during prolonged operation. Thus, the trifunctional HEACB 1000 seems to be a suitable and promising catalyst for practical adoption in seawater electrolysis.
The EIS results directly show the kinetic parameters which determine the charge transfer efficiency.31,49,50 A plot was made between charge transfer resistance and the Tafel slope as the kinetic parameter, as shown in Fig. 5d. We can see direct correspondence between these intrinsic factors with HEACB 1000 having the most favorable Tafel–Rct plot, reinforcing the experimental findings we discerned in the case of HER, CER, and OER. For HEACB 1000, we see that the Tafel slope and system resistance is lowest for HER, OER and CER, showing its better efficiency towards catalysis. We also subjected the catalysts to BET surface area measurements, as shown in Fig. S13 (ESI†), and found that the surface area of the samples followed a similar trend as in the case of Cdl HEACB 1000 (84 m2 g−1) > HEACB 500 (76 m2 g−1) > HEACB 750 (64 m2 g−1), which once again verified our inference from electrochemistry. BET surface area analysis also matches exactly with the trend in ECSA values we extracted using double-layer capacitance, strengthening the view that the extrinsic factors are favorable for the HEA 1000 alloy sample towards the reaction catalysis. The BET N2 adsorption isotherm resembles a type 4 isotherm indicating the mesoporous nature, where multilayer adsorbate formation occurs initially and is then followed by the capillary condensation of the adsorbate inside the pores, before reaching the saturation pressure of the adsorbate in the liquid form.39 Fig. S13b (ESI†) shows the pore size distribution of the three HEACB samples, indicating that their pore size comes in the mesoporous range of 3 to 30 nm. These factors further explain the enhanced catalyst performance, as the high surface area offers more exposed active sites, improved electrolyte penetration, and faster ion diffusion through the heterojunction. This observation also thus justifies the Cdl measurement made for ECSA estimation as well as the observed activity trends. The better performance of the HEACB 1000 can be inferred as the high degree of formation of single-phase entropy alloy on the carbon matrix concerning the lower temperature annealed HEACB 750 and HEACB 500, which is evident from the XRD analysis. Thus, the cocktail effect of the five metals in the HEACB 1000 alloy, along with the extrinsic factors like surface area/ECSA and better elemental distribution over the carbon, seems to reduce the energy barrier for these reactions by offering suitable and accessible active sites and optimizing the interaction with the reaction intermediates.
One of the main hurdles in seawater hydrolysis is the oxidation of competing Cl− and H2O molecules so that thermodynamically more favorable OER occurs and hinders the CER process. Their selectivity and control are hence, always of prime importance, especially when we plan for seawater electrolysis on a larger scale. Fig. 6a compares the LSV polarization curve at HEACB1000 in pH = 1, with and without chloride ions to study the possible CER and OER responses. Without chloride ions, the process is simply the OER in an acidic medium which seems to be very inferior. With the introduction of NaCl, the chloride ions source, the current response indicates noticeable improvements during the oxidation polarization region suggesting the preferred reaction is CER. Thus, in simulated seawater conditions, the preferred process is CER at HEACB1000 (Fig. 6a and b). Whereas at pH 14 (Fig. 6c), the LSV responses suggest OER emerges as the major reaction (i.e., without chloride ions) while the introduction of chloride ions trims down the OER process owing to the competing nature of chloride and oxide ions. This demonstrates effective control over the selectivity of the two competing reactions. Thus, the catalyst HEACB 1000 was highly efficient in selectively favouring the kinetics of the reaction of interest by controlling the pH of the reaction media. Fig. 6b and d show the comparative abilities of the catalyst in driving current under a specific potential, which indirectly tells us about the exchange current density. The higher the exchange current density the better the reaction kinetics, and HEACB 1000 is the best among the synthesized materials.
 | ||
| Fig. 6 (a) HEACB 1000 activity comparison in 0.5 M H2SO4, with and without NaCl in reaction medium towards CER and OER. (b) Corresponding current density comparison plot at 1.5 V vs. RHE based on (a). (c) HEACB 1000 activity comparison in 1 M KOH with and without NaCl in reaction medium towards CER and OER. (d) Bar diagram showing the current carrying efficiency of HEACB 1000, 750, and 500 concerning their reaction overpotential for CER in 0.5 M H2SO4 + 4 M NaCl reaction medium. | ||
Mott–Schottky (MS) analysis
The excellent catalytic activity of the high entropy metal alloy carbon composite and the need to disperse the high entropy alloy particle on a carbon frame can be explained in terms of their metal–semiconductor heterojunctions formed at their interface. We utilize the concept of Mott–Schottky analysis on our HEACB samples.24 Based on the dispersion of the alloy nanoparticle onto conductive carbon support and the inferred intimate interaction through Raman spectra and XPS (as shown in Fig. 1b and 2), it is likely that a metal alloy/semiconductor interface will form. Electron flows are expected to occur during the formation of such interfaces; where electrons flow from an electron-dense system towards the less electron-dense side until the Fermi level equilibrates.51,52To investigate this phenomenon, we performed detailed Mott–Schottky analysis of HEACB 1000, HEACB 750, and HEACB 500, as indicated in Fig. 7, at a constant current frequency of 1 kHz and amplitude of 20 mV in the potential range −0.2 to 1.2 V vs. RHE. The inverse capacitance square vs. the potential difference between the catalyst–electrolyte interface verifies the existence of a metal–semiconductor junction in our HEACB 1000 system, which is evident from the formation of three distinct potential regions as shown in Fig. 7a. The positive slope of the plot indicates n-type semiconductor behavior, where we expect electron flow from the n-type carbon framework towards the alloy nanoparticles until their Fermi level equilibrates, thus optimizing the HEA particles in terms of their band structure for efficient reaction catalysis.24,53
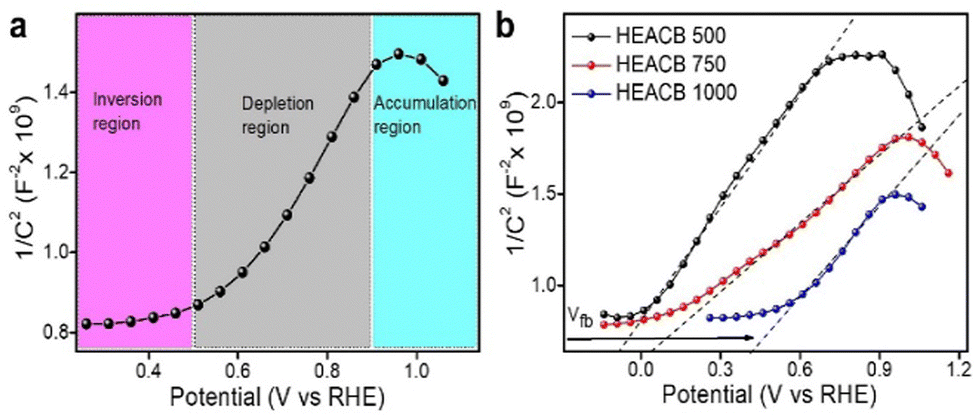 | ||
| Fig. 7 (a) Mott–Schottky plots for HEACB 1000 showing the different potential regions in an electrolytic solution of 1 M KOH. (b) Combined Mott–Schottky plots of HEACB 1000, 750, and 500 samples (X-intercept shows the respective flat band potential (Vfb)). | ||
We can extract two parameters from the graph: the flat band potential (Vfb) and charge carrier density. The X intercept of the MS plot (Fig. 7b) indicates the Vfb of each sample, and the magnitude of the slope provides the charge carrier density.54,55 If we compare the three HEACB samples, we see a considerable shift in the flat band potential towards the positive side, on going from HEACB 500 (−0.078 V) to HEACB 1000 (0.038 V). This positive shift indicates that the Mott–Schottky junction formed helps to reduce the band bending and thus further explains the faster charge transfer kinetics. This optimization was best in the case of HEACB 1000 where the band bending is found to be the minimum. If we analyze the slope of the MS plot, we can see they are in the order HEACB 750 < HEACB 1000 < HEACB 500. Since the slope has an inverse relation with the charge carrier density, we can infer that the HEACB 750 and HEACB 1000 have comparatively much higher charge carrier densities than HEACB 500, which might be a factor that supports the relatively better activity in the catalysis of HER, OER, and CER.
The parameters we obtain from MS analysis have direct information on the kinetics of reactions. Fig. 8a and Fig. S14a and b (ESI†) show the Tafel slope relationship with that of the flat band potential obtained from the MS analysis for the OER, HER and CER, respectively. We can see that for all three processes, the kinetics of the reaction and Vfb followed an exponential relation, where we see HEACB 1000 at the most optimized region of interaction for all three processes, hence verifying our experimental observations. Thus, MS analysis gives us a clear picture of the relative band structure and carrier density of our synthesized alloy carbon composites and therefore supports the catalytic behavior of the samples.51
 | ||
| Fig. 8 (a) Tafel slope–flat band potential relationship plot for the OER. (b) Total water splitting performance of HEACB samples in the OER polarization region in 1 M KOH. (c) Radar plot comparison with respect to multifunctional behavior in terms of their relative activity percentages, along with its respective bar diagram (d). | ||
Total water splitting
The HEACB 1000 catalyst exhibits excellent HER and OER performance, suggesting the potential use of this material as a catalyst in a practical water electrolyzer. To analyze the suitability of the catalyst in a real-life application, we have simulated the environment of an alkaline water electrolyzer setup with the bifunctional catalyst HEACB 1000 as both the cathode and anode material.56,57 1 M KOH is used as the electrolyte medium, and LSVs are recorded at a scan rate of 10 mV s−1. As shown in Fig. 8b, we have tried total water splitting with different combinations of anode and cathode materials, including the current state of the art HER and OER catalysts, Pt/C, and RuO2, respectively. From the plot, the HEACB 1000–HEACB 1000 system performs the best with a potential of 1.65 V against RHE to reach a current density of 10 mA cm−2, which is better than both the RuO2–RuO2 and Pt/C–Pt/C combinations of systems, towards total water splitting. The water-splitting performance of the HEACB 1000 couple is found to be close to that of the RuO2–Pt/C combination. The overall performance of the HEACB 1000–HEACB 1000 system is better than many recently reported catalysts (Table S10, ESI†). This suggests that the HEACB catalyst is a suitable candidate for total water splitting. To estimate how efficiently the catalyst utilizes the electrons to push the reaction of interest, estimation of the faradaic efficiency (F.E.) is important. The evolved gas was quantified during HER and OER reactions, and the concomitant F.E. was deduced accordingly. The HEACB 1000 shows a decent level of F.E. values with 87.3% and 91.5% for OER and HER, respectively. The exchange current density and turnover frequency (TOF) shed light on the intrinsic activity and the density of active sites existing in the catalyst material. HEACB 1000 shows a decent exchange current density of 7.5 mA cm−2, 14 mA cm−2, and 15.2 mA cm−2 for OER, HER, and CER, respectively, indicating the ability of the catalyst to promote efficient charge transfer. The TOF values derived from the active site density estimated from the non-faradaic region of the catalyst in a neutral phosphate buffer show that the intrinsic activity of HEACB 1000 is comparatively better among the HEACB 750 and HEACB 500 (Tables S6–S8, S11–S16 and Fig. S15, ESI†), which again justifies the high performance of HEACB 1000. In conclusion, the HEACB 1000 catalyst provides a reliable platform for complete seawater splitting with impressive catalytic parameters, such as TOF, exchange current density, and faradaic efficiency.Since the catalyst is active for HER-CER in an acidic environment and HER-OER in a basic environment, this material can be used as a potential seawater electrolyzer with two modes of operation which are switchable using the pH of the electrolyte media, thus opening an avenue towards sustainable water splitting for chemical production. The radar and bar diagram in Fig. 8c and d show the multifunctional aspects of the three catalysts, along with their ECSA and impedance data comparison. The HEACB 1000 has the highest relative percentage activity, which is visible in the illustrated radar plot (Fig. 8c and Fig. S16, S17, ESI†). These figures strongly convey the multifunctional aspect of our synthesized HEA catalyst, thus emphasizing its ability to accelerate the kinetics using its multiple reaction sites from the five different transition metals.
Conclusion
In summary, we have fabricated a novel conductive carbon-supported high entropy alloy (HEACB) based catalyst by employing a facile single-step pyrolysis technique at different synthetic temperatures to optimize the alloy particle formation. The HEACB 1000 sample shows great potential in electrocatalysis as it requires less overpotential and has lower onset and Tafel slopes for HER, OER, and CER reactions. It requires an overpotential of 190 mV, 350 mV, and 30 mV for HER, OER, and CER reactions, respectively to achieve a current density of 10 mA cm−2. The enhanced catalytic performance of the HEACB 1000 compared with other synthesized samples is ascribed to the higher degree of alloy formation, the incorporated multi-metallic active sites, and intimate contact with the carbon catalytic bed, thus inducing a synergistic effect and optimizing the interaction with intermediates formed during the reaction. The catalyst performs well in selectively producing CER and OER reactions, depending on the pH of the reaction media. It exhibits good stability and durability in the complete water-splitting process, with a half-cell potential of 1.65 V at 10 mA cm−2. The catalyst's band structure and charge transfer efficiency were analyzed through Mott–Schottky analysis, impedance spectroscopy, and other structural characterizations. This works points to a novel strategy to design and synthesize high entropy alloy-based catalysts through a simple feasible technique with a detailed study of the possible interaction at the heterojunction. This strategy can be applied to other transition metals, resulting in a cost-effective and dependable catalyst for complete seawater splitting.Experimental section
Materials and reagents
Chromium(III) nitrate hexahydrate (Cr(NO3)3·6H2O, M.W. = 346.10 g mol−1), manganese(II)nitrate hexahydrate (Mn(NO3)2·6H2O, M.W. = 287.04 g mol−1) iron(III)nitrate hexahydrate (Fe(NO3)3·6H2O, M.W. = 349.9 g mol−1), cobalt(II)nitrate hexahydrate (Co(NO3)2·6H2O, M.W. = 291.03 g mol−1), nickel(II)nitrate hexahydrate (Ni(NO3)2·6H2O, M.W. = 290.79 g mol−1), and carbon black (CAS number: 1333-86-4) required for the synthesis, were purchased from Sigma-Aldrich and used as received. Potassium hydroxide (KOH, M.W. = 56.11 g mol−1) pellets, sulfuric acid (H2SO4, 99.9%, M.W. = 98.08 g mol−1), sodium chloride (NaCl, M.W. = 58.44 g mol−1), hydrochloric acid (36%, M.W. = 36.46 g mol−1) from the same manufacturer were used for the electrochemical studies.Synthesis of materials
The equimolar (0.5 M) aqueous solution of chromium(III) nitrate hexahydrate (Cr(NO3)3·6H2O, M.W. = 346.10 g mol−1), manganese(II) nitrate hexahydrate (Mn(NO3)2·6H2O, M.W. = 287.04 g mol−1) iron(III) nitrate hexahydrate (Fe(NO3)3·6H2O, M.W. = 349.9 g mol−1), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, M.W. = 291.03 g mol−1), nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, M.W. = 290.79 g mol−1) were synthesised separately and assorted, under constant and continuous stirring by employing a magnetic stirrer and heated to about 50 °C to enhance the precursor miscibility. After two hours of continuous temperature treatment on a magnetic hot plate, about 2 g of carbon black and 15 mL of ethanol were blended with the above-procured solution. This mixture was stirred at 200 rpm, maintaining the same temperature to ensure homogeneous mixing of metal cations to ensure high reaction probability. The above heat treatment was followed by freeze drying to yield a fluffy darkish brown precursor powder. Using a mortar and pestle, this powder was ground well and then subjected to high-temperature pyrolysis of about 1000 °C with an effective ramping rate of 10 °C min−1, left for about two hours in an inert protected environment on a tube furnace setup under a constant nitrogen gas flow of about 100 sccm. To optimize the synthesis strategy and have a comparative study of samples with different degrees of HEA alloy incorporation, three independent samples were fabricated at different synthesis temperatures of 500, 750, and 1000 °C, labelled as HEACB 500, HEACB 750, and HEACB 1000, respectively.Characterization
In the preliminary phase of the investigation, we subjected the samples to crystal structural assessment using X-ray diffraction studies (XRD) by employing an X-ray diffractometer (PANalytical) with a monochromatized Cu Kα radiation of 1.54 Å wavelength. Direct visualization of the morphology of the sample was obtained by utilizing a scanning electron microscope (SEM, FESEM FEI inspect 50) and transmission electron microscope (TEM, Tecnai T20) with an accelerating voltage of 200 kV. X-ray photoelectron spectrometer AXIS ULTRA DLD Kratos possessing a monochromatic Al Kα radiation source of 1486.6 eV, was used to render high-resolution X-ray photoelectron spectroscopy (HR-XPS) of individual elements, to examine the elemental composition, oxidation states, and the various chemical bonds existing in the catalytic material.Electrochemical measurements
The electrochemical studies were accomplished using a CHI 750E based instrument supporting a three-electrode configuration, where a commercial glassy carbon electrode (GCE) of 0.07 cm2 and rotating ring disk electrode (RRDE) of 0.196 cm2 geometrical surface area, were employed as the major reaction working electrodes, Ag/AgCl electrode was used as the reference electrode, and platinum wire as the counter electrode. The GCE and RRDE were well polished with alumina powder with an average particle size of 0.005 μm to procure an atomically smooth electrode surface. About 7 mg of catalyst material was added to a 1000 μL ethanol–water mixture. On to 100 of the above solution, 20 μL of 5 wt% Nafion 117 solution was added, which was then sonicated for 15 min to obtain a well-dispersed thick catalyst ink. 7 μL of this catalyst ink was used for each electrochemical study. IR compensation is done during each measurement to minimize the uncompensated solution resistance. All LSV measurements were taken at a scan rate of 5 mV s−1 with RDE, unless specified.Author contributions
G. R.: performed the majority of experiments, materials synthesis, methodology, conceptualization, structural and electrochemical data curation, analysis, writing, and editing of the manuscript. R. N: material synthesis, methodology, conceptualization, scientific discussion, review, and editing of the manuscript. K. K.: XPS data acquisition and analysis, SEM imaging, BET, Raman spectroscopy, writing manuscript, and its editing. D. B. G.: CER conceptualization and its electrochemical measurements, manuscript writing, and its editing. A. B. M: TEM imaging, J. N.: image processing, manuscript review, editing, writing. S. M. O.: manuscript review, writing. Y. Y.: scientific discussion, conceptualization, experimental suggestions, review, and editing of the manuscript. K. K. N. conceptualization, funding acquisition, supervision, scientific discussion, project administration, review, and editing of the manuscript. All the authors agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Equations used for the calculation of faradaic efficiency (F.E.)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 58
58
F.E. = Qexperimental/QTheoretical = VnF/(VmA)
V = volume of gas evolved in liters
V m = molar volume of gas at 298 K (24.5 L)
n = number of electrons involved
F = faradaic constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C)
485 C)
A = integral area of current response with time
Equations used for the calculation of exchange current density
J 0 = RT/(nFARct)J 0 = exchange current density
R = gas constant 8.314 J K−1
n = no. of electrons involved
F = faradaic constant (96485 C)
R ct = charge transfer resistance
A = area of the electrode
T = temperature in kelvin
Equations used for the calculation of turn over frequency (TOF)
TOF = lJl × A/(n × F × N)N = S/(2 × F × u)
J = current density
A = area of electrode
n = no. of electrons involved in the reaction
F = faradaic constant
N = active site density
S = integral area of cyclic voltammetry response in the non-faradaic region in a neutral pH phosphate buffer
u = scan rate (V s−1)
Acknowledgements
G. R. would like to thank the Ministry of Human Resource Development (MHRD) for the research scholarship. The authors also acknowledge the Department of Science and Technology (DST), India, for funding the BET adsorption–desorption facility (DST-SERB, EMR/2016/005843 and DST-FIST, SR/FST/PS11-009/2010) for the research. R. N. and Y. Y. acknowledge financial support from the Japan Society for the Promotion of Science (JSPS, No. P22063), the UQ-Yonsei University International Project, the JST-ERATO Yamauchi Materials Space-Tectonics Project (JPMJER2003). J. N. acknowledges financial support from the KIST institutional program (2E32511). This work has been supported by the Researchers Supporting Project (RSP2023R405) from King Saud University. The authors would like to acknowledge the Materials Research Centre (MRC) for providing the XRD facility, the Centre for Nanoscience and Engineering (CeNSE), and Inorganic and Physical Chemistry (IPC) department, IISc for providing the SEM and TEM facilities. Additionally, the authors are grateful to Dr Omeshwari Bisen and Dr Hemam Rachana Devi in the Materials Research Centre, IISc Bangalore, for their help with BET analysis, Raman spectroscopic measurements, and helpful scientific discussions. This work was performed in part at the Queensland Node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers.References
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- D. Sauceda, P. Singh, G. Ouyang, O. Palasyuk, M. J. Kramer and R. Arróyave, Mater. Horiz., 2022, 9, 2644–2663 RSC.
- V. Chaudhary, R. Chaudhary, R. Banerjee and R. V. Ramanujan, Mater. Today, 2021, 49, 231–252 CrossRef CAS.
- S. Akrami, P. Edalati, M. Fuji and K. Edalati, Mater. Sci. Eng., R, 2021, 146, 100644 CrossRef.
- M. W. Glasscott, Curr. Opin. Electrochem., 2022, 34, 100976 CrossRef CAS.
- Y. Wang, Y. Wu, K. Sun and Z. Mi, Mater. Horiz., 2019, 6, 1454–1462 RSC.
- S. S. Jana and T. Maiti, Mater. Horiz., 2023, 10, 1848–1855 RSC.
- Y. Kang, B. Jiang, J. Yang, Z. Wan, J. Na, Q. Li, H. Li, J. Henzie, Y. Sakka, Y. Yamauchi and T. Asahi, ACS Nano, 2020, 14, 17224–17232 CrossRef CAS PubMed.
- P. Li, Y. Yao, W. Ouyang, Z. Liu, H. Yin and D. Wang, J. Mater. Sci. Technol., 2023, 138, 29 CrossRef.
- A. K. Ipadeola, A. K. Lebechi, L. Gaolatlhe, A. B. Haruna, M. Chitt, K. Eid, A. M. Abdullah and K. I. Ozoemena, Electrochem. Commun., 2022, 136, 107207 CrossRef CAS.
- Q. Zeng and Y. Xu, Mater. Today Commun., 2020, 24, 101261 CrossRef CAS.
- Y. Shi, B. Yang and P. K. Liaw, Metals, 2017, 7, 43 CrossRef.
- H. Lv, D. Xu, L. Sun, J. Henzie, S. L. Suib, Y. Yamauchi and B. Liu, ACS Nano, 2019, 13, 12052–12061 CrossRef CAS PubMed.
- M. A. Aktar, M. M. Alam and A. Q. Al-Amin, Sustain. Prod. Consum., 2021, 26, 770–781 CrossRef PubMed.
- A. Kumar, P. Singh, P. Raizada and C. M. Hussain, Sci. Total Environ., 2022, 806, 150349 CrossRef CAS PubMed.
- I. Mauleón, Energies, 2021, 14, 4657 CrossRef.
- M. Mohsin, H. W. Kamran, M. Atif Nawaz, M. Sajjad Hussain and A. S. Dahri, J. Environ. Manage., 2021, 284, 111999 CrossRef PubMed.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS PubMed; R. Nandan, G. K. Goswami and K. K. Nanda, Appl. Energy, 2017, 205, 1050–1058 CrossRef; R. Nandan, N. Takamori, K. Higashimine, R. Badam and N. Matsumi, J. Mater. Chem. A, 2022, 10, 15960–15974 RSC; R. Nandan, N. Takamori, K. Higashimine, R. Badam and N. Matsumi, J. Mater. Chem. A, 2022, 10, 5230–5243 RSC; R. Nandan, O. Y. Bisen and K. K. Nanda, J. Phys. Chem. C, 2021, 125, 10378–10385 CrossRef.
- Z.-X. Cai, Y. Xia, Y. Ito, M. Ohtani, H. Sakamoto, A. Ito, Y. Bai, Z.-L. Wang, Y. Yamauchi and T. Fujita, ACS Nano, 2022, 16, 20851–20864 CrossRef CAS PubMed.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- X. Yang, R. Guo, R. Cai, Y. Ouyang, P. Yang and J. Xiao, Int. J. Hydrogen Energy, 2022, 47, 13561–13578 CrossRef CAS.
- C.-L. Huang, K. Sasaki, D. Senthil Raja, C.-T. Hsieh, Y.-J. Wu, J.-T. Su, C.-C. Cheng, P.-Y. Cheng, S.-H. Lin, Y. Choi and S.-Y. Lu, Adv. Energy Mater., 2021, 11, 2101827 CrossRef CAS.
- A. A. Koverga, E. Flórez and J. A. Rodriguez, Int. J. Hydrogen Energy, 2021, 46, 25092–25102 CrossRef CAS.
- G. Raj, D. Das, B. Sarkar, S. Biswas and K. K. Nanda, Sustainable Mater. Technol., 2022, 33, e00451 CrossRef CAS.
- R. Nandan, G. Raj and K. K. Nanda, ACS Appl. Mater. Interfaces, 2022, 14, 16108–16116 CrossRef CAS PubMed; R. Nandan, M. Y. Rekha, H. R. Devi, C. Srivastava and K. K. Nanda, Chem. Commun., 2021, 57, 611–614 RSC.
- J. Liu, C. Tang, Z. Ke, R. Chen, H. Wang, W. Li, C. Jiang, D. He, G. Wang and X. Xiao, Adv. Energy Mater., 2022, 12, 2103301 CrossRef CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- S. Wu, Y. Pan, N. Wang, T. Lu and W. Dai, Int. J. Miner., Metall. Mater., 2019, 26, 124–132 CrossRef CAS.
- J. K. Pedersen, T. A. A. Batchelor, A. Bagger and J. Rossmeisl, ACS Catal., 2020, 10, 2169–2176 CrossRef CAS.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- V. Márquez, M. Fereidooni, J. S. Santos, S. Praserthdam and P. Praserthdam, Electrochim. Acta, 2022, 428, 140975 CrossRef.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- Y. Liu, K. Wang, X. Xu, K. Eid, A. M. Abdullah, L. Pan and Y. Yamauchi, ACS Nano, 2021, 15, 13924–13942 CrossRef CAS PubMed.
- J. Huang, M. Hou, J. Wang, X. Teng, Y. Niu, M. Xu and Z. Chen, Electrochim. Acta, 2020, 339, 135878 CrossRef CAS.
- Y. Gendel, G. Amikam and P. Nativ, in Seawater Electrolysis In Electrochemical Power Sources: Fundamentals, Systems, and Applications, ed. T. Smolinka and J. Garche, Elsevier, 2022, vol. 9, pp. 305–326 Search PubMed.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- Y. Wang, Y. Nian, A. N. Biswas, W. Li, Y. Han and J. G. Chen, Adv. Energy Mater., 2021, 11, 2002967 CrossRef CAS.
- Y. Tang, C. Yang, X. Xu, Y. Kang, J. Henzie, W. Que and Y. Yamauchi, Adv. Energy Mater., 2022, 12, 2103867 CrossRef CAS.
- Y. Yin, J. Wang, T. Li, J. P. Hill, A. Rowan, Y. Sugahara and Y. Yamauchi, ACS Nano, 2021, 15, 13240–13248 CrossRef CAS PubMed.
- H. R. Devi, R. Nandan and K. K. Nanda, ACS Appl. Mater. Interfaces, 2020, 12, 13888–13895 CrossRef CAS PubMed.
- Y. Peng and S. Chen, Green Energy Environ., 2018, 3, 335–351 CrossRef CAS; R. Nandan, P. Pandey, A. Gautam, O. Y. Bisen, K. Chattopadhyay, M. M. Titirici and K. K. Nanda, ACS Appl. Mater. Interfaces, 2021, 13, 3771–3781 CrossRef PubMed; R. Nandan, A. Gautam and K. K. Nanda, J. Mater. Chem. A, 2018, 6, 20411–20420 RSC.
- M. N. Kirikova, A. S. Ivanov, S. V. Savilov and V. V. Lunin, Russ. Chem. Bull., 2008, 57, 298–303 CrossRef CAS.
- C. V. M. Inocêncio, Y. Holade, C. Morais, K. B. Kokoh and T. W. Napporn, Electrochem. Sci. Adv., 2022, e2100206 Search PubMed.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- H. Jing, P. Zhu, X. Zheng, Z. Zhang, D. Wang and Y. Li, Adv. Powder Mater., 2022, 1, 100013 CrossRef.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, Y. Guo, Y. Ide, B. Yuliarto, X. Jiang, Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 3035–3047 RSC.
- M. Liu, B. Niu, H. Guo, S. Ying and Z. Chen, Inorg. Chem. Commun., 2021, 130, 108687 CrossRef CAS; R. Nandan, A. Gautam, S. Tripathi and K. K. Nanda, J. Mater. Chem. A, 2018, 6, 8537–8548 RSC; R. Nandan, A. Gautam and K. K. Nanda, J. Mater. Chem. A, 2017, 5, 20252–20262 RSC.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- T. Chouki, B. Donkova, B. Aktarla, P. Stefanov and S. Emin, Mater. Today Commun., 2021, 26, 101976 CrossRef CAS.
- M. Natalia, Y. N. Sudhakar and M. Selvakumar, Indian J. Chem. Technol., 2013, 20, 392–399 CAS.
- X. Ji, K. Wang, Y. Zhang, H. Sun, Y. Zhang, T. Ma, Z. Ma, P. Hu and Y. Qiu, Sustainable Energy Fuels, 2020, 4, 407–416 RSC.
- B. Sarkar, D. Das and K. K. Nanda, J. Mater. Chem. A, 2021, 9, 13958–13966 RSC.
- Z.-H. Xue, H. Su, Q.-Y. Yu, B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- J. Hou, Y. Sun, Y. Wu, S. Cao and L. Sun, Adv. Funct. Mater., 2018, 28, 1704447 CrossRef.
- H. Zhong, C. Yang, L. Fan, Z. Fu, X. Yang, X. Wang and R. Wang, Energy Environ. Sci., 2019, 12, 418–426 RSC.
- Y. Xu, J. Yang, T. Liao, R. Ge, Y. Liu, J. Zhang, Y. Li, M. Zhu, S. Li and W. Li, Chem. Eng. J., 2022, 431, 134126 CrossRef CAS.
- W. Luo, Y. Wang, L. Luo, S. Gong, Y. Li and X. Gan, Appl. Surf. Sci., 2022, 606, 154808 CrossRef CAS.
- T. ul Haq, S. A. Mansour, A. Munir and Y. Haik, Adv. Funct. Mater., 2020, 30, 1910309 CrossRef CAS; J. Tong, Y. Li, L. Bo, W. Li, T. Li, Q. Zhang, D. Kong, H. Wang and C. Li, ACS Sustainable Chem. Eng., 2019, 7, 17432–17442 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00453h |
| ‡ The authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |