
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evolution of Mo species and ZSM-5 microstructure with temperature and its impact on methane dehydroaromatisation activity†
Peixi
Cong‡
ab,
Ines
Lezcano-Gonzalez
ab,
Alessandro
Longo‡
c,
Wim
Bras‡
d and
Andrew M.
Beale
 *ab
*ab
aDepartment of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: Andrew.beale@ucl.ac.uk
bResearch Complex at Harwell (RCaH), Rutherford Appleton Laboratory, Harwell, Didcot, Oxfordshire OX11 0FA, UK
cEuropean Synchrotron Radiation Facility (ESRF), 38043 Grenoble Cedex 9, France
dChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge TN 37831, USA
First published on 25th November 2024
Abstract
Mo-ZSM-5 is a well-studied catalyst for its ability to convert methane to aromatics and hydrogen. Since the process is thermodynamically ‘uphill’, high temperatures are necessary to achieve acceptable product yields. However, these temperatures have been shown to negatively impact the stability of the zeolite, with the formation of aluminum molybdates being proposed as particularly responsible for framework collapse. Here we use X-ray absorption and optical Raman spectroscopy as well as X-ray diffraction to characterize the initial Mo species present after introduction to ZSM-5, thermal activation and after structural collapse. The results demonstrate that the formation of mononuclear Mo6+ species coordinated to oxygen is retained despite increasingly high temperature treatment and which is able to perform methane dehydromatisation although with decreasing efficiency as zeolite porosity is lost.
Introduction
Methane dehydroaromatisation (MDA) is a promising reaction to upgrade methane directly into aromatics and hydrogen.1,2 A variety of catalysts based on metal-ion exchanged zeolites have been explored for the MDA reaction. Among the tested zeolite topologies (for example ZSM-5, MCM-22, and MCM-49) and metal cations, the combination of Mo supported on 10-membered ring zeolites (ZSM-5 and MCM-22) remains one of the most active catalysts to date.3 Mo-containing zeolite catalysts are normally prepared by mixing the solid oxides of Mo compounds with zeolite supports followed by calcination at high temperatures, which leads to the decomposition and distribution of the MoOx species into the pores of the zeolite. Extensive research has been carried out to resolve the nature and location of this dispersed MoOx species.4–6 Although there is no consensus, a recent study has shown that after calcination, isolated Mo–oxo species anchored at the channel intersection are the dominant species.7 In general, it is accepted that after migration into the zeolite micropores, the MoOx species anchor at the Brønsted acid sites, releasing water molecules.8 This reaction also distorts or even extracts framework Al and, in some cases, gives rise to the formation of Al2(MoO4)3 species.9Despite all the promise, Mo/H-ZSM-5 suffers from rapid deactivation due to both coke formation and the migration of the active Mo species to the outer parts of the zeolite crystals.10,11 Overcoming this problem has been the focus of many studies, attempting to: (1) modify and optimise the catalyst zeolite support and the active metal cations12,13 and, (2) regenerate the coked catalyst in a reductive or oxidative atmosphere.14 It has been found that at typical MDA reaction temperatures Mo/H-ZSM-5 is not stable enough under the oxidative conditions needed for regeneration, leading to the sublimation of the MoOx species and more importantly, to irreversible alteration to the zeolite framework often resulting in the collapse of the zeolite.8
In the current study, we focus on the thermal stability of the Mo/H-ZSM-5 catalysts with an emphasis on the evolution of the chemical species as the zeolite undergoes structural collapse. The thermal stability of the catalysts was examined firstly by means of differential scanning calorimetry (DSC). The phase and morphological evolution of the catalysts during the course of the collapse was further investigated by powder X-ray diffraction (PXRD) and scanning electron microscopy (SEM), while changes in Mo speciation of Mo cation were characterised by X-ray absorption fine structure (XAFS). It is thought that modality of zeolite framework collapse is intimately related to the microscopic framework Al distribution.
Experimental section
Catalyst preparation
NH4-ZSM-5 was supplied by Zeolyst International (CBV3024E, Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al = 15). SSZ-13 (Si:Al = 15) was synthesized in fluoride media following the reported methods.15 H-form zeolites were obtained by calcination at 550 °C (ZSM-5) and 620 °C (SSZ-13). Mo/H-zeolites were prepared via the established solid-state ion-exchange method (SSIE).16 Mo/H-ZSM-5 samples are designated with the name Mox-Z5, where x denotes the weight percentage of the Mo loading. Samples were calcined to the point of collapse and beyond in static air using a ramp rate of 10 °C min−1.
:Al = 15). SSZ-13 (Si:Al = 15) was synthesized in fluoride media following the reported methods.15 H-form zeolites were obtained by calcination at 550 °C (ZSM-5) and 620 °C (SSZ-13). Mo/H-zeolites were prepared via the established solid-state ion-exchange method (SSIE).16 Mo/H-ZSM-5 samples are designated with the name Mox-Z5, where x denotes the weight percentage of the Mo loading. Samples were calcined to the point of collapse and beyond in static air using a ramp rate of 10 °C min−1.
Catalyst characterization
PXRD patterns were recorded using a Rigaku MiniFlex 600 X-ray diffractometer fitted with Cu Kα radiation. The Kerr-gated Raman spectra were recorded at the ULTRA facility at the Central Laser Facility, using a 400 nm wavelength excitation source. The data were calibrated to the spectra of toluene-impregnated H-ZSM-5. Surface characterization was carried out using N2 physisorption at 77.3 K on a Quadrasorb EVO QDS-30. Samples were outgassed at 350 °C overnight. The Brunauer–Emmett–Teller (BET) method was used for surface area analysis. The t-plot method was used for micropore volume analysis. Solid-state 27Al MAS-NMR were carried out on a standard Bruker 4 mm double-resonance MAS probe at the UCL Department of Chemistry, and samples were fully hydrated prior to each measurement. Differential scanning calorimetry (DSC) was recorded on a NETZSCH STA 449 F3 Jupiter at the UCL Department of Chemistry. Samples were heated up to 1400 °C at 10 °C min−1 under a 30 mL min−1 flow of He. An empty fused alumina sample pan was used as a reference. Scanning electron micrographs (SEM) and energy dispersive X-ray spectroscopy (EDX) analysis were carried out with a JEOL JSM-6610LV at RCaH and JEOL6700F FEG at the UCL Department of Chemistry. XRF elemental distribution maps were recorded at beamline 2-ID-E at the Advanced Photon Source.17X-ray absorption fine structure
Mo K-edge XAFS studies were performed at the European Synchrotron Radiation Facility (ESRF), France, on BM26A Beamline and the Diamond Light Source (DLS), UK, on beamline B18. The measurements were performed in transmission mode using ion chambers with a fast-scanning Si(111) double-crystal monochromator. Each scan took 5 min to acquire at B18 and 10 min at BM26A. Three scans were taken for each sample and averaged. XAFS data reduction and analysis were performed using the Demeter package over a k-range of 2–10 Å and R-range of 0.5–3 Å.18 Linear combination fitting (LCF) of XANES spectra was performed in an energy range of −20 to +70 eV at Mo K-edge. Morlet wavelet transforms are calculated by the Fortran version of the HAMA software developed by Funke et al.19MDA catalytic activity tests
The catalytic testing was carried out by placing 0.6 g of catalyst (150–425 μm sieved fractions) in a tubular quartz reactor with an internal diameter of 0.7 mm. The catalysts were secured in the isothermal zone of the oven by quartz wool. A total gas flow of 30 mL min−1 (GHSV = 1500 h−1) was fed to the reactor by a set of mass flow controllers. The samples were first calcined to 700 °C at 5 °C min−1 under a 20% O2 in He flow. Then still at 700 °C the reactor was flushed by a stream of pure Ar for 20 min, before switching to MDA gas composition (50% CH4 in Ar). The product formation was analyzed by an online OmniStar GSD 320O1 mass spectrometer.Results and discussion
Catalyst characterization
The solid-state Mo-exchanged zeolite materials used in this work are typical for relevant MDA catalyst.2 and 4 wt% Mo/H-ZSM-5 were synthesized from high-temperature calcination of a ground mixture of H-ZSM-5 and MoO3. In Fig. 1(b) is shown the PXRD of the calcined Mo/H-ZSM-5 against the physical mixture. Reflections of crystalline MoO3 in the after high temperature treatment fall below the detection level in sample Mo4-Z5. It is known that during the thermal treatment, Mo species migrate into the pores of zeolite and ion exchange at the Brønsted acid sites. A change in peak position, width, and intensity pertaining to the MFI framework is not observed in the pattern of Mo4-Z5, indicating the zeolite structure is retained with no observable loss in crystallinity. No other crystalline Mo phase such as Al2(MoO4)3 was detected. Mo loading obtained from chemical analysis and texture properties of the Mo/H-ZSM-5 samples are summarized in Table S1 (ESI†). The introduction of Mo leads to a decrease in BET micropore area and volume (up to 16% and 18% respectively) while the severity in reduction directly relates to the Mo loading. This observed decrease is possibly caused by the Mo introduced partially hindering diffusion throughout the micropores and/or a loss of microporous structure due to dealumination,14 which is also evident by the increase in amount of octahedral extra-framework Al (EFAl) and Al2(MoO4)3 species observed by 27Al MAS-NMR (Fig. 2).
 | ||
| Fig. 1 PXRD patterns of zeolite precursors H-ZSM-5 with indexing lines of the references ZSM-5 structures (a) and Mo4-Z5 before (blue line) and after (orange line) calcination with indexing lines of the crystalline MoO3 phase are shown below the patterns (b). | ||
 | ||
| Fig. 2 27Al MAS NMR spectra of H-ZSM-5, Mo2-Z5 and Mo4-Z5 (the * indicates the location of the chemical shift for Al2(MoO4)3 at 14 ppm). | ||
The interaction between Mo and the zeolite framework can lead to extraction of tetrahedral framework Al,20 in some cases leading to an irreversible framework degradation and the formation of mesopores – a process accompanied by the formation of aluminium molybdate.8 To gain insight into Al speciation, Mo-exchanged zeolite catalysts were characterized by 27Al MAS-NMR. The introduction of the Mo ion modifies substantially the NMR spectra of ZSM-5 zeolite (Fig. 2). The extraction of framework Al is undoubtedly evidenced by a significant decrease in the tetrahedral framework Al signals at 54 ppm. This is accompanied by an increase in the peak width, likely caused by an increased quadrupolar broadening effect. This framework dealumination becomes more noticeable with increasing molybdenum loading, while a shift of 2 ppm towards a higher field for the framework Al peak is also observed. Tessonnier et al. reported that peak shift is related to the distortion of framework AlO4 units and alteration of the T–O–T angle.5 Furthermore, an increase in signal intensity around 0 ppm, corresponding to an increase in the amount of octahedral extra-framework Al (EFAl) was observed, along with a new signal at 14 ppm. Kosinov et al. assigned the signals at 14 ppm and −14 ppm to bulk and well-dispersed Al2(MoO4)3 respectively,8 whereas Ma et al. argued that signal at −14 ppm represents the Al2(MoO4)3 species, and it shifts to 14 ppm upon hydration.9 The bulk Al2(MoO4)3 reference measured here shows an intense signal at −13.8 ppm (Fig. S2, ESI†). Given that no bulk crystalline Al2(MoO4)3 phases are detected by PXRD and the Mo samples were hydrated prior to measurement, the signal at 14 ppm likely originates from hydrated Al2(MoO4)3. Signal intensity from both octahedral EFAl and Al2(MoO4)3 surge with increasing Mo loading. Therefore, it can be concluded that Mo ion-exchange leads to framework distortion, significant framework dealumination and formation of octahedral EFAl and Al2(MoO4)3 species. It is interesting to note that these changes have not manifested into long-range structural changes in the zeolite framework as evidenced by the unchanging PXRD patterns (see Fig. 1).
The structure of dispersed Mo oxide species was further investigated by Kerr-gated Raman spectroscopy, as shown in Fig. 3.21 The spectrum of the H-form ZSM-5 zeolite is characterized by two broad bands, one centered around 400 cm−1 with shoulders on either side at 300 and 470 cm−1, and the second at 812 cm−1, due to the vibrations of the MFI framework.22,23 The spectrum of the MoO3 and H-ZSM-5 physical mixture, in contrast, is dominated by strong Raman signals characteristic of MoO3; i.e. 293 cm−1 and 364 cm−1 (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bending), 473 cm−1 and 668 cm−1 edge-sharing (Mo–O(3) stretching), 819 cm−1 (Mo–O–Mo asymmetric stretching), and 995 cm−1 (MoO asymmetric stretching).24
O bending), 473 cm−1 and 668 cm−1 edge-sharing (Mo–O(3) stretching), 819 cm−1 (Mo–O–Mo asymmetric stretching), and 995 cm−1 (MoO asymmetric stretching).24
 | ||
| Fig. 3 Kerr-gated Raman spectra for H-ZSM-5 (blue), Mo2-Z5 (green), Mo4-Z5 (orange), and a physical mixture of MoO3 with H-ZSM-5 (red). | ||
After SSIE and calcination (see Fig. 3), Mo2-Z5 and Mo4-Z5, the Raman bands from MoO3 are clearly diminished, whereas new Raman bands emerge at 858 cm−1 and 944 cm−1. The 944 cm−1 band can be readily attributed to the MoO stretching mode from distorted octahedral and is often attributed to isolated monomeric species,6 while the 858 cm−1 band is due to a Mo–O–Mo antisymmetric stretch, suggestive of the presence of dimeric Mo species. Nevertheless, polymolybdate species also possess symmetric and antisymmetric Mo–O–Mo stretches at 630 cm−1 and 860 cm−1, and MoO terminal stretches at 934 cm−1, so it is difficult to draw definitive conclusions. Further Raman bands at lower frequencies are also observed and their position overlaps with signals from the zeolite framework. The 858 cm−1 band however, presents a slight asymmetry at the low-frequency side, which might be due to a contribution from residual MoO3. These observations indicate after the SSIE and calcination most of the Mo cations migrate into the zeolite channels and become highly dispersed species, although the presence of polymeric molybdate species cannot be completely ruled out.
Thermal stability and zeolite structural evolution
The thermal stability and behavior of Mo/H-ZSM-5 catalysts, including a 6 wt% sample designated by Mo6-Z5 and synthesized using the same method described above, were evaluated with DSC. All four DSC traces (Fig. 4) exhibit an endotherm below 200 which is due to dehydration. Above 1000 °C, an exothermic peak is seen and followed by a clear endothermic step. The exotherms are assigned to the amorphisation and collapse of the zeolite framework and are indicative of heat release upon the collapse. This dynamic has been reported for the collapse of many types of zeolites such as zeolite Y and zeolite A.25,26 | ||
| Fig. 4 DSC traces of H-ZSM-5, Mo2-Z5, Mo4-Z5, and Mo6-Z5. tcs of the Mo exchanged H-ZSM-5 samples and H-form precursor are listed in Table 1. Note the weak shoulders at 900 °C in the Mo4-Z5 and Mo6-Z5 samples are probably due to Mo volatilisation. | ||
The thermal stability of the zeolite studied here is defined by the collapse temperature of the zeolite (tc), which is the maximum of the exotherm. For the H-form zeolite, the measured tc is 1218 °C, which is seen to drop for Mo2-Z5. Interestingly the measured tc rises and plateaus for both Mo4-Z5 and Mo6-Z5, and its magnitude is on par with that of the H-form precursor. This unexpected rise happens against the backdrop of severe framework dealumination and the EFAl formation caused by Mo ions.
To verify the DSC measurement can indeed be correlated to zeolite the structural collapse and to further investigate the phase evolution during framework collapse, Mo2-Z5 and Mo4-Z5 were calcined to temperatures just below (1150 °C), at (1240 °C), and above (1350 °C) the tc. Samples calcined to these temperatures are given the label Mox-Z5-BC, Mox-Z5-@C and Mox-Z5-AC respectively with x denoting the Mo loading. At 1150 °C, below the collapse temperature (tc), all but the most intense low-angle reflections of the MFI structure (Fig. 5a) diminished below the background for both Mo loading. Intensities of the reflections at 2θ angles of 7.92 and 8.87° were integrated for both samples and the result shows that the crystallinity determined for Mo2-Z5-BC and Mo4-Z5-BC are at 8.84 and 8.50% of the initial intensity, respectively. A broad diffuse scattering peak centered at 22° is also observed, and it is believed to originate from amorphous silica/alumina phases. Therefore, at temperatures just below tc, Mo/H-ZSM-5 catalysts are already mostly PXRD amorphous. Interestingly, mullite (Si2Al6O13), an aluminum-rich dense aluminosilicate phase, is detected alongside ZSM-5, suggesting that the recrystallisation of the thermodynamically more stable, denser, aluminous ceramic phase happens before the complete collapse of the zeolite phase.
 | ||
| Fig. 5 PXRD patterns of Mo4-Z5-BC, and Mo2-Z5-BC compared to that of H-ZSM-5 (a), and Mo4-Z5-@C, Mo2-Z5-@C, Mo4-Z5-AC and Mo2AC (b). Indexing lines show position reflections from the mullite (purple) and cristobalite (light green). | ||
Fig. 5b shows the PXRD patterns of Mo/H-ZSM-5 samples calcined to and above the tc. At the onset of tc, the remaining reflections from the MFI zeolite phase completely disappear, and an increase in mullite reflection intensity indicates a greater extent of crystallisation of the dense aluminous ceramic phase. BET specific surface area measurements show that Mo4-Z5-@C and Mo4-Z5-AC possess a total surface area of 8.50 and 2.30 m2 g−1 respectively, confirming the microporous structure of the ZSM-5 is lost completely. Note that recrystallisation of the amorphous silica is not observed at tc. In turn, at temperatures well above tc, recrystallisation of the dense silicate phase in form of α-cristobalite (SiO2) is observed, accompanied by a reduction in intensity of the broad amorphous peak. No molybdenum-containing crystalline phase was detected by the PXRD measurement, suggesting that molybdenum ions remain either highly dispersed in an amorphous phase or trapped in the crystalline phases formed.
The partial collapse of the zeolite framework at temperatures below tc with co-current recrystallisation of the dense ceramic phase evidenced by PXRD point to a possible inhomogeneity in the framework collapse. One possible source for inhomogeneity in collapse behaviour is Al content as numerous studies with isostructural zeolites have concluded that the thermal stability increases with increasing the Si/Al ratio caused by the increasing covalent character associated with more Si–O bonds.27,28 EDX coupled with transmission electron microscopy (Fig. 6) further reveal anisotropy in Al distribution for Mo4-Z5 i.e. Al rich domains appear to be present in the sample. However, with its small(er) particle size (hundreds of nanometers) it is inherently difficult to investigate distribution effects using this particular material. To clearly visualize if Al distribution influences thermal stability, a model SSZ-13 catalyst with a large crystallite size (ca. 30 × 30 × 30 μm) – synthesized using a method described previously.17 Since differentiating between Si and Al is challenging using X-rays, Cu was added to the sample via conventional wet ion-exchange and XRF mapping on the basis that the Cu will provide framework charge compensation mimicking the Al distribution. Fig. S3 (ESI†) contains an XRF map of the Cu exchanged SSZ-13 revealing the Al, Si and Cu distributions from the crystal. Although the signals from the Al and Si appear to overlap, relative to the Si signal most of the Cu intensities are concentrated in a region around 20 μm in length and located at the core of the crystal, and the outer 5–7 μm of the crystal shows almost no Cu intensities at all. This type of distribution is indicative of the so-called “zoning effect”.
 | ||
| Fig. 6 Dark-field TEM image (a) and Si, Al, and Mo elemental EDX maps (b) for Mo4-Z5. | ||
2 and 4 wt% Mo/H-SSZ-13 samples prepared using the SSIE method were calcined to 1300 °C, the tc of 2 wt% Mo/H-SSZ-13 (equivalent to a temperature just below the tc of 4 wt% Mo/H-SSZ-13), and named Mo-S13-@C and Mo-S13-BC, respectively. The zeolite collapse and amorphisation of Mo exchanged model SSZ-13 catalyst are illustrated by SEM (Fig. 7). For Mo-S13-BC, although the calcination temperature is below tc, the Al rich core of the crystal displays severe strain in the form of cavitation and delamination as if the core had imploded under stress. This type of morphology is typical of thermally collapsed zeolites transitioning from a rigid to an amorphous state. Contrary to the core of the crystal, the Si rich shell remains almost completely intact with no sign of fracturing or crumbling while PXRD measurement (Fig. S4, ESI†) shows some of the CHA structure is maintained. When the calcination temperature reaches the tc, the 30 μm sized crystals start to adopt a globular morphology that is reminiscent of a melting solid (Fig. 7d and e). Evidence of flow is clearly demonstrated in SEM micrographs, where the amorphised crystals have merged into a large, single globular unit.
 | ||
| Fig. 7 SEM micrographs of Mo-S13-BC (a)–(c) and Mo-S13-@C (d) and (e). | ||
As summarised by the scheme in Fig. 8, it appears that within a single crystallite of zeolite SSZ-13, the Al rich and Si rich regions are not only amorphised at different temperatures but also with different modalities. The Al rich core is amorphised via framework collapse at a relatively low temperature below tc and forms a rigid amorphous solid first before recrystallisation. The Si rich shell is amorphised in a fashion typical to classical solid melting. This difference in collapse temperature is perhaps not surprising since numerous studies on the effects of varying Si/Al ratio of a series of isostructural zeolites have concluded that the thermal stability increases with increasing the Si/Al ratio. However, previous effort mostly focused on the macroscopic aspect while the microscopic aspect such as the effects of framework Al distribution within a single particle of zeolite has been overlooked.
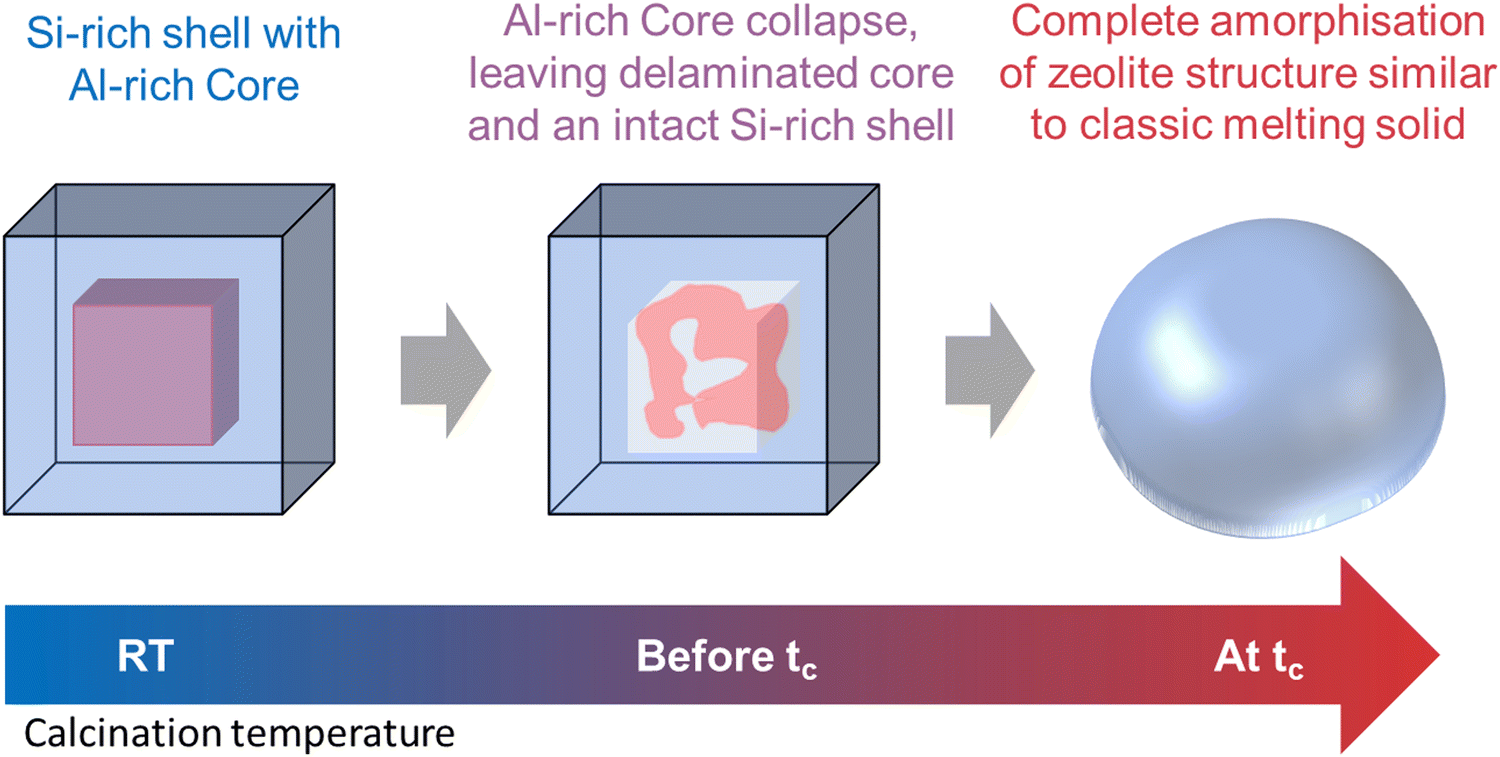 | ||
| Fig. 8 Schematic representation depicting the temperature dependent multi-modal collapse mechanism of a single crystallite of zeolite SSZ-13. | ||
Mo speciation after collapse, a XAFS study
As the state of Mo ions in the collapsed zeolite catalysts was not accounted for by the PXRD, XAFS measurements were performed to obtain insight into the speciation of molybdenum during the collapse. Four reference materials, two calcined 4 wt% Mo/H-ZSM-5, one measured in ambient conditions at RT (MoZ-RT) and the other measured in situ in Ar atmosphere after the 700 °C calcination and cooled to RT (MoZ-700), as well as aluminium molybdate Al2(MoO4)3, and molybdenum trioxide MoO3 are also measured. The collapsed Mo/H-ZSM-5 (Fig. 9a) shows a very similar spectral profile to those of Al2(MoO4)3 and MoZ-700 reference where the pre-edge region is dominated by a pre-edge peak at 20005 eV and it is assigned to the dipole forbidden (quadrupole allowed) 1s → 4d transition, while the white line at 20014 eV is attributed to the 1s → 5p dipole transition. Note the increased intensity for the pre-edge peak for Mo in Al2(MoO4)3 when compared to MoO3 since the former contains monomeric tetrahedral [MoO4]2− species which leads to a relaxing of the dipole selection rule due to the removal of orbital parity in a tetrahedral environment. In the case of Mo/H-ZSM-5, studies have shown evidence for the presence of isolated monomeric Mo–oxo species at low Si/Al ratios,2,6 although the presence of dimeric species cannot be ruled out. More recently, utilizing a combination of in situ and operando XAFS and high-resolution PXRD, Agote-Arán et al. were able to reveal that during calcination the majority of the Mo ions adopt an isolated tetrahedral Mo–oxo arrangement anchored to the straight MFI channels.7 Furthermore, the lack of spectra feature in the post-edge region is in line with the assignment of highly dispersed Mo species that do not possess long-range order. As such, the XANES spectra indicate that similar to the in situ calcined Mo/H-ZSM-5 catalyst, the Mo ions in the collapsed ZSM-5s are also highly dispersed and mostly tetrahedrally coordinated.
 | ||
| Fig. 9 Mo K-edge XANES spectra (a) and k2 weighted FT-EXAFS magnitude (b) of Mo2-Z5-@C, Mo4-Z5-@C, Mo2-Z5-AC, and Mo4- Z5-AC sample, and MoZ-700, MoZ-RT, MoO3, and Al2(MoO4)3 reference (from bottom to top). | ||
Analysis of the energy position of the absorption edge allows the identification of the formal oxidation state (Fig. S5, ESI†) and it shows the Mo ions in all four samples are preserved as +6. Linear combination fitting (LCF) of the XANES spectra (Fig. S5, ESI†) listed in Table S2 (ESI†) demonstrate MoZ-700 is the dominant species in all samples, again confirming the majority of the Mo6+ ions adopting a tetrahedral coordination geometry. The result also suggests that the rest of the Mo ions adopt a distorted octahedral geometry similar to that of hydrated Mo species in calcined Mo/H-ZSM-5 zeolites. It is proposed that tetrahedral coordinated Mo–oxo species formed inside the zeolite during the calcination to 700 °C remain stable and do not undergo thermally induced reduction during further calcination up to 1350 °C and complete collapse of the zeolite framework. As the ZSM-5 framework collapses, the denser new phases trap the Mo–oxo species and seal off their access to moisture, preventing them from reverting back to the octahedral coordination.4,29 The Mo–oxo species on the surface, however, do have access to air/moisture and they undergo rehydration and contribute to the XANES spectra similarly to that in Mo-RT.
The accompanying EXAFS allows independent validation of the XANES results, specifically on local coordination around Mo ions. The k2-weighted Fourier transform (FT)-moduli (Fig. 9b, k-space EXAFS spectra shown in Fig. S7, ESI†) of the collapsed catalysts are dominated by a first-shell contribution and is assigned to near neighbor O ions. First shell fitting of the EXAFS spectra of the thermally collapsed Mo/H-ZSM-5 catalysts (Fig. S8, ESI†) shows the fitted average Mo–O coordination number (CN) of all four collapsed Mo zeolite samples is slightly above 4 (Table 2), consistent with a tetrahedrally coordinated Mo–oxo species. The Mo–O CNs for the two @C samples are higher than those fitted for the AC samples, suggesting a higher fraction of Mo ions present with a coordination number greater than 4. This result is in agreement with the XANES analysis, although it is worth noting the difference in CNs is within the error for determining these values (i.e. 10%). The lack of intensity in FT-moduli beyond the first coordination shell suggests that the structures around the Mo ions lack long-range order. These results from the FT-EXAFS are consistent with isolated [MoO4]2− cations being bonded to double Al sites in the ZSM-5 framework.
| Sample | H-ZSM-5 | Mo2-Z5 | Mo4-Z5 | Mo6-Z5 |
|---|---|---|---|---|
| t c (°C) | 1218 | 1141 | 1198 | 1199 |
| Sample | Scattering path | Coordination number (N) | Bond length (Å) | ΔE (eV) | σ 2 (Å)2 | R-factor |
|---|---|---|---|---|---|---|
| MoZ-700 | MO |
2.0 (6) | 1.69 (4) | −2.7 (2.7) | 0.003 (5) | 0.015 |
| M–O | 2.0 (6) | 1.82 (4) | ||||
| Mo2-Z5-@C | M–O | 4.6 (1.5) | 1.76 (3) | −3.6 (4.8) | 0.012 (5) | 0.02 |
| Mo4-Z5-@C | M–O | 4.7 (1.4) | 1.75 (3) | −2.2 (4.3) | 0.012 (4) | 0.028 |
| Mo2-Z5-AC | M–O | 4.2 (1.2) | 1.77 (2) | 0.9 (4.0) | 0.008 (4) | 0.005 |
| Mo2-Z5-AC | M–O | 4.2 (1.0) | 1.73 (2) | −8.6 (3.4) | 0.004 (3) | 0.039 |
The analysis of second-shell contribution in FT-EXAFS proved to be complicated. The relatively weak scattering intensity and the possible destructive interference among scatterers,30 combined with multiple possible structural models all add complexity to FT-EXAFS-based analysis. To resolve the structure of the Mo species beyond the first coordination shell, we employed wavelet transform (WT) technique for the analysis of EXAFS. WT-EXAFS has the ability to decompose a given EXAFS signal in k-space that allows the resolution of overlapping contributions from different elements. Fig. 10 illustrates the contour plots of the Morlet WT-EXAFS moduli. All the contours contain an intense first-shell peak located at R = 1.21 Å (phase uncorrected) and around 3.7–3.9 Å−1 in χ(k), which is assigned to a first-shell Mo–O contribution. The second-shell contribution (located around R = 3.0 Å) in the contour of MoZ-RT consists of two peaks. For reference, the second shell Mo–Al/Si peak in Mo/H-ZSM-5 is believed to be located around 2.9 Å,30 while the Mo–Mo peaks are in the range of 2.9 to 3.2 Å.20,31 The position of the low χ(k) peak (at 3.7 Å−1) is almost identical to that of oxygen, although the presence of multiple scattering contributions cannot be ruled out. An intense peak centered around 8.3 Å−1 is also observed. The significantly higher χ(k) position designated this peak to contribution from a heavy element, and it is assigned to a Mo–Mo single scattering contribution. The presence Mo–Mo and possible multiple scattering contributions evidence that at RT Mo ions in ZSM-5 are not isolated and are most likely a di/poly-molybdate species.30 This finding echoes the result from Kerr-gated Raman spectra. The contour plot of MoZ-700 shows a single second-shell peak located at χ(k) = 4.7 Å−1. The χ(k) position of this second-shell peak indicates it originates from a backscatterer with Z greater than an O anion but not as heavy as the Mo anion. For Mo/H-ZSM-5, it can only be Al (or Si). As such, the WT-EXAFS analysis indicates that Mo anions calcined to 700 °C exists as an isolated species anchored to the ZSM-5 zeolite framework. These observations are fully consistent with the result reported by Agote-Arán et al.7 The contour plots of Mo4-Z5-@C and Mo4-Z5-AC are almost identical to that of MoZ-700 with a single second-shell peak located at χ(k) = 4.9 Å−1, while the peak at high χ(k) indictive of an Mo–Mo contribution is absent. As such, the WT-EXAFS indicates that the Mo species in the collapsed Mo/H-ZSM-5 catalysts resemble the local structure of the isolated tetrahedral Mo–oxo species attached to the channels of ZSM-5. This result indicates during the collapse of the zeolite framework not only the oxidation state and coordination geometry of the Mo cation are preserved, but also possibly the fragments/building units of the zeolite structure immediately surrounding Mo (i.e., the 10-membered ring where the Mo cation is anchored to) are retained.7
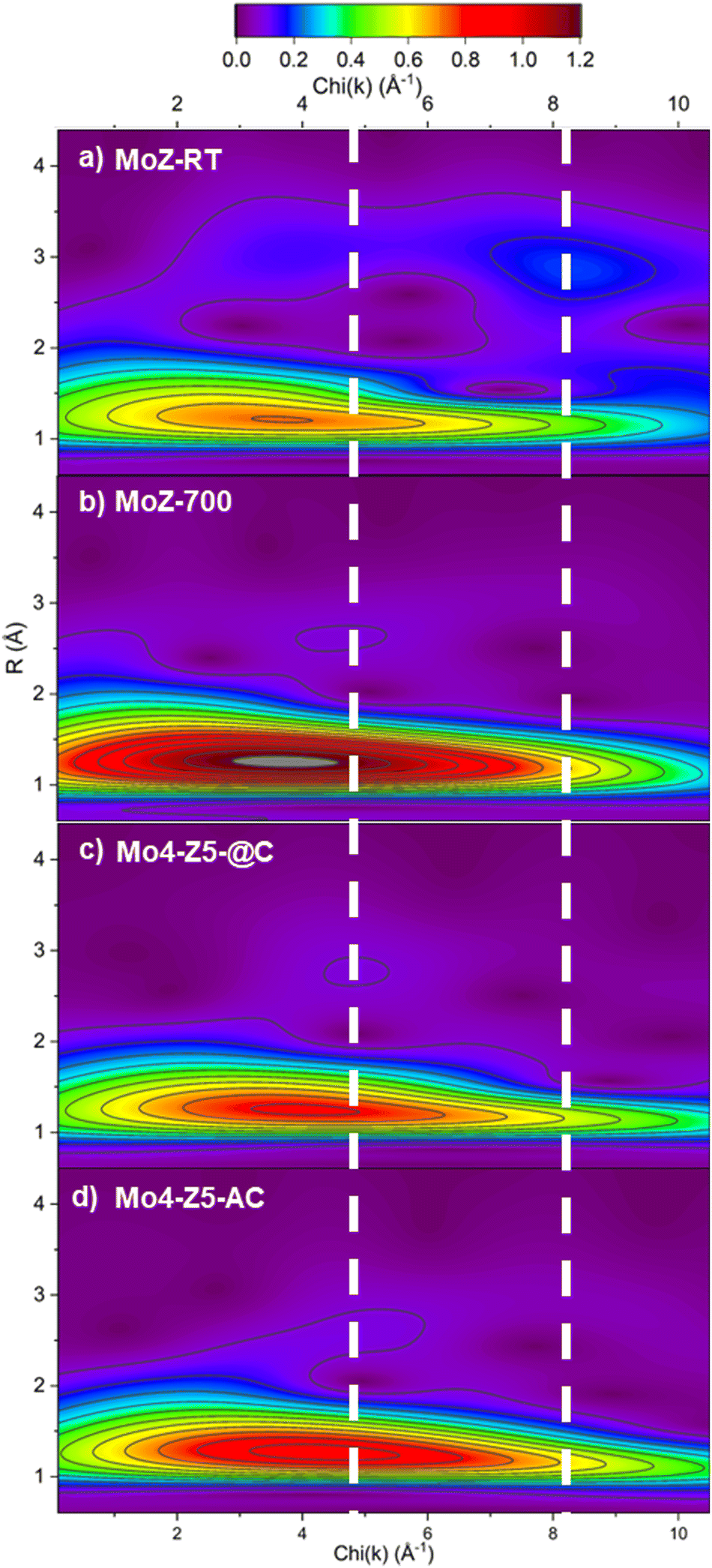 | ||
| Fig. 10 k 2 weighted Morlet WT-EXAFS contour plot of (a) MoZ-RT, (b) MoZ-700, (c) Mo4-Z5-@C, and (d) Mo4-Z5-AC. The white dotted lines mark the k-space position of the Mo–Al/Si and Mo–Mo single scattering path at 4.8 Å−1 and 8.3 Å−1. All four contours are plotted to the same color scale. | ||
Methane dehydroaromatisation activity
Evidence from both XANES and EXAFS analysis converges on the notion that the Mo6+ ions after the high temperature calcination are preserved in a state similar to the Mo6+ ions in the freshly calcined Mo/H-ZSM-5 catalyst before methane activation, while chemical analysis confirmed more than 25% of the original Mo loading is preserved (Table S3, ESI†). This opens up the question of whether these Mo ions are still catalytically active toward the MDA reaction. The resulting benzene response from the MDA reaction are plotted in Fig. S9 (ESI†). Both catalysts were able to activate methane. The observed C6H6 evolution on the collapsed Mo/H-ZSM-5 catalysts is in agreement with studies by Kosinov et al.,32 and Agote-Arán et al.7 that suggest that Brønsted acidity is not required for the aromatisation to occur, since the framework that hosts the Brønsted acid sites has already collapsed. As shown, the benzene response of the two catalysts differs significantly. Relative to Mo/H-ZSM-5, the at collapsed catalyst affords 80% of the original response while the collapsed only managed 15%.Summary and conclusions
This study brings new insights into the thermal stability, the collapse mechanism and nature of the Mo species of the benchmark MDA catalyst Mo/H-ZSM-5. Ex situ Kerr-gated Raman spectra at room temperature suggesting the presence of both monomeric and polymeric molybdate species, the latter species increasing with [Mo]. A consequence of this is the formation of more ‘hydrated’ and non-crystalline Al2(MoO4)3 although interestingly this did not impinge on the thermal stability of the ZSM-5 as the temperature at which crystallinity is lost did not decrease with [Mo] loading remaining on par with the parent H-form.The structural evolution during the collapse of Mo/H-ZSM-5 was then investigated from the perspective of both the zeolite framework and Mo ions. The MFI structure was almost in a state of complete collapse at a temperature just below tc; we note however that previous work has shown that although the XRD pattern indicates that long range crystalline ordering has been lost, many of the structural units (i.e. Secondary Building Units (SBUs) or common fragments (cages)) are often retained immediately after collapse in what has been termed a low density amorphous (LDA) phase.25,33 Further heating leads to complete collapse of the aluminosilicate framework to form a high density amorphous oxide and an aluminium rich dense ceramic phase mullite, while the dense silicate phase α-cristobalite recrystallizes at temperatures above the tc. Further investigation on the mechanism of collapse with a model Mo/H-SSZ-13 crystals demonstrates that (as a result of the Al zoning effect) within a single crystal of zeolite the Al rich and Si rich regions not only amorphized (collapse) at different temperatures, but also with different modalities (illustrated in Fig. 5). The Al rich region amorphised via framework collapse at a temperature below tc and forms a rigid amorphous solid first, while the Si rich shell amorphised in a fashion typical to classical solid melting. These findings can have a profound implication on studies of zeolite in general since zeolites are often characterised by its macroscopic properties, while microscopic properties within a particle of zeolite have often been overlooked. This result offers a possible explanation for the seemingly contradictive tc value observed for Mo/H-ZSM-5s. The observed formation of EFAl and aluminium molybdate species and the resulting framework damage are mostly concentrated in the Al-rich region, while the Si rich region is mostly unaffected. Hence the Si rich region would have thermal stability on par to that of the H-ZSM-5.
On the other hand, the exchanged Mo6+ ions do not undergo autothermal reduction during the high temperature calcination up to 1350 °C. Most of the Mo6+ ions in the collapsed zeolite adopt tetrahedral coordination similar to freshly calcined Mo/H-ZSM-5. These results suggest that the Mo ions were trapped by the collapsed zeolite and do not directly participate in the collapse process and indeed these observations would also be consistent with the retention of ZSM-5 structural units in an LDA which retains many of the key structural features necessary for catalytic activity. WT-EXAFS analysis indicates that the Mo6+ ions can be considered isolated after treatment at reaction temperatures (700 °C) and after the zeolite framework collapse, and more importantly, they form an aluminate species that bear similarities to the freshly calcined tetrahedral Mo6+ species that anchor to the channels of ZSM-5. We note that this suggests an evolution of the Mo6+ species after high temperature treatment from polynuclear to mononuclear and also emphasizes the importance of performing measurements under in situ or ideally under operando conditions.
These results suggest that in Mo-based ZSM-5 catalysts the thermal stability of the zeolite support alone during the reaction and regeneration is important for ensuring good catalytic performance. Interestingly we observe that in already collapsed Mo-zeolites, the newly formed dense phase should not suffer from the stability issue experienced by the zeolite and can act as a kinetically more stable support for the Mo ions. Furthermore, since MDA is an endothermic reaction with aromatisation limited by thermodynamics,8 Mo ions supported on a more stable material may be able to facilitate higher operation temperature, which could shift the equilibrium and achieve higher conversion.
From these observations one can conclude that the macroscopic thermal stability that has been determined from techniques such as PXRD or DSC can be somewhat irrelevant to zeolite catalysis per se, since an Al distribution gradient can exist in zeolite particles in the form of zoning, and the thermal stability of the catalytic relevant framework Al mostly depends on the microscopic Al concentration within the zeolite crystal. In the most extreme cases such as the one demonstrated here in the model SSZ-13, the catalyst may already be deactivated by the irreversible structural change introduced by the collapse of framework Al, as many of the active sites of zeolite catalysts require either (or both) the framework Al or the charge balancing cation anchored to the framework Al to adopt very specific coordination, while the crystallinity of the catalyst is still mostly maintained due to the Si rich part of the zeolite able to survive through the high temperature condition.
Data availability
Processed data (spectra and patterns) are available from the following repository: https://rdr.ucl.ac.uk/.Conflicts of interest
Andrew M. Beale reports a relationship with Finden Ltd. that includes co-ownership of the company in the form of equity and grant funding. All other authors have no known competing financial interest or personal relationship that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank Miren Agote-Arán for performing the catalytic tests, Igor Sazanovich from the STFC-CLF for assistance in performing the Kerr gate Raman measurements, EPSRC for funding (EP/R026939/1 & EP/S016481/1) as well as UCL CDT (EP/L015862/1) and DUBBLE for the funding of a studentship for PC. We would also like to thank the Isis characterisation lab for access to the PXRD instrument.References
- J. J. Spivey and G. Hutchings, Catalytic Aromatization of Methane, Chem. Soc. Rev., 2014, 43(3), 792–803, 10.1039/C3CS60259A.
- I. Lezcano-González, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Molybdenum Speciation and Its Impact on Catalytic Activity during Methane Dehydroaromatization in Zeolite ZSM-5 as Revealed by Operando X-Ray Methods, Angew. Chem., Int. Ed., 2016, 55(17), 5215–5219, DOI:10.1002/anie.201601357.
- P. Schwach, X. Pan and X. Bao, Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects, Chem. Rev., 2017, 117(13), 8497–8520, DOI:10.1021/acs.chemrev.6b00715.
- W. Li, G. D. Meitzner, R. W. Borry and E. Iglesia, Raman and X-Ray Absorption Studies of Mo Species in Mo/H-ZSM5 Catalysts for Non-Oxidative CH4 Reactions, J. Catal., 2000, 191(2), 373–383, DOI:10.1006/jcat.1999.2795.
- J.-P. Tessonnier, B. Louis, S. Rigolet, M. J. Ledoux and C. Pham-Huu, Methane Dehydro-Aromatization on Mo/ZSM-5: About the Hidden Role of Brønsted Acid Sites, Appl. Catal., A, 2008, 336(1–2), 79–88, DOI:10.1016/j.apcata.2007.08.026.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Identification of Molybdenum Oxide Nanostructures on Zeolites for Natural Gas Conversion, Science, 2015, 348(6235), 686–690, DOI:10.1126/science.aaa7048.
- M. Agote-Arán, A. B. Kroner, H. U. Islam, W. A. Sławiński, D. S. Wragg, I. Lezcano-González and A. M. Beale, Determination of Molybdenum Species Evolution during Non-Oxidative Dehydroaromatization of Methane and Its Implications for Catalytic Performance, ChemCatChem, 2019, 11(1), 473–480, DOI:10.1002/cctc.201801299.
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, Stable Mo/HZSM-5 Methane Dehydroaromatization Catalysts Optimized for High-Temperature Calcination-Regeneration, J. Catal., 2017, 346, 125–133, DOI:10.1016/j.jcat.2016.12.006.
- D. Ma, Y. Shu, X. Han, X. Liu, Y. Xu and X. Bao, Mo/HMCM-22 Catalysts for Methane Dehydroaromatization: A Multinuclear MAS NMR Study, J. Phys. Chem. B, 2001, 105(9), 1786–1793, DOI:10.1021/jp002011k.
- N. Kosinov, F. J. A. G. Coumans, E. Uslamin, F. Kapteijn and E. J. M. Hensen, Selective Coke Combustion by Oxygen Pulsing During Mo/ZSM-5-Catalyzed Methane Dehydroaromatization, Angew. Chem., Int. Ed., 2016, 55(48), 15086–15090, DOI:10.1002/anie.201609442.
- M. Agote-Arán, R. E. Fletcher, M. Briceno, A. B. Kroner, I. V. Sazanovich, B. Slater, M. E. Rivas, A. W. J. Smith, P. Collier, I. Lezcano-González and A. M. Beale, Implications of the Molybdenum Coordination Environment in MFI Zeolites on Methane Dehydroaromatisation Performance, ChemCatChem, 2020, 12(1), 294–304, DOI:10.1002/cctc.201901166.
- S. Kikuchi, R. Kojima, H. Ma, J. Bai and M. Ichikawa, Study on Mo/HZSM-5 Catalysts Modified by Bulky Aminoalkyl-Substituted Silyl Compounds for the Selective Methane-to-Benzene (MTB) Reaction, J. Catal., 2006, 242(2), 349–356, DOI:10.1016/j.jcat.2006.06.024.
- H. Wang, L. Su, J. Zhuang, D. Tan, Y. Xu and X. Bao, Post-Steam-Treatment of Mo/HZSM-5 Catalysts: An Alternative and Effective Approach for Enhancing Their Catalytic Performances of Methane Dehydroaromatization, J. Phys. Chem. B, 2003, 107(47), 12964–12972, DOI:10.1021/jp0225931.
- M. T. Portilla, F. J. Llopis and C. Martínez, Non-Oxidative Dehydroaromatization of Methane: An Effective Reaction–Regeneration Cyclic Operation for Catalyst Life Extension, Catal. Sci. Technol., 2015, 5(7), 3806–3821, 10.1039/C5CY00356C.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Determining the Storage, Availability and Reactivity of NH 3 within Cu-Chabazite-Based Ammonia Selective Catalytic Reduction Systems, Phys. Chem. Chem. Phys., 2014, 16(4), 1639–1650, 10.1039/C3CP54132K.
- M. Agote-Arán, A. B. Kroner, D. S. Wragg, W. A. Sławiński, M. Briceno, H. U. Islam, I. V. Sazanovich, M. E. Rivas, A. W. J. Smith, P. Collier, I. Lezcano-González and A. M. Beale, Understanding the Deactivation Phenomena of Small-Pore Mo/H-SSZ-13 during Methane Dehydroaromatisation, Molecules, 2020, 25(21), 5048, DOI:10.3390/molecules25215048.
- N. Omori, A. Candeo, S. Mosca, I. Lezcano-Gonzalez, I. K. Robinson, L. Li, A. G. Greenaway, P. Collier and A. M. Beale, Multimodal Imaging of Autofluorescent Sites Reveals Varied Chemical Speciation in SSZ-13 Crystals, Angew. Chem., Int. Ed., 2021, 60(10), 5125–5131, DOI:10.1002/anie.202015016.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719.
- H. Funke, A. C. Scheinost and M. Chukalina, Wavelet Analysis of Extended X-Ray Absorption Fine Structure Data, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(9), 094110, DOI:10.1103/PhysRevB.71.094110.
- J.-Z. Zhang, M. A. Long and R. F. Howe, Molybdenum ZSM-5 Zeolite Catalysts for the Conversion of Methane to Benzene, Catal. Today, 1998, 44(1–4), 293–300, DOI:10.1016/S0920-5861(98)00202-8.
- I. Lezcano-Gonzalez, E. Campbell, A. E. J. Hoffman, M. Bocus, I. V. Sazanovich, M. Towrie, M. Agote-Aran, E. K. Gibson, A. Greenaway, K. De Wispelaere, V. Van Speybroeck and A. M. Beale, Insight into the Effects of Confined Hydrocarbon Species on the Lifetime of Methanol Conversion Catalysts, Nat. Mater., 2020, 19(10), 1081–1087, DOI:10.1038/s41563-020-0800-y.
- Ö. Attila, H. E. King, F. Meirer and B. M. Weckhuysen, 3D Raman Spectroscopy of Large Zeolite ZSM-5 Crystals, Chem. – A Eur. J., 2019, 25(29), 7158–7167, DOI:10.1002/chem.201805664.
- A. Martinelli, S. Creci, S. Vavra, P.-A. Carlsson and M. Skoglundh, Local Anisotropy in Single Crystals of Zeotypes with the MFI Framework Structure Evidenced by Polarised Raman Spectroscopy, Phys. Chem. Chem. Phys., 2020, 22(3), 1640–1654, 10.1039/C9CP06199A.
- L. Seguin, M. Figlarz, R. Cavagnat and J.-C. Lassègues, Infrared and Raman Spectra of MoO3 Molybdenum Trioxides and MoO3⋯XH2O Molybdenum Trioxide Hydrates, Spectrochim. Acta, Part A, 1995, 51(8), 1323–1344, DOI:10.1016/0584-8539(94)00247-9.
- G. N. Greaves, F. Meneau, A. Sapelkin, L. M. Colyer, I. Gwynn, S. Wade and G. Sankar, The Rheology of Collapsing Zeolites Amorphized by Temperature and Pressure, Nat. Mater., 2003, 2(9), 622–629, DOI:10.1038/nmat963.
- A. M. Beale and G. Sankar, Understanding the Crystallization of Nanosized Cobalt Aluminate Spinel from Ion-Exchanged Zeolites Using Combined in Situ QEXAFS/XRD, Chem. Mater., 2006, 18(2), 263–272, DOI:10.1021/cm050797z.
- T. C. Hoff, R. Thilakaratne, D. W. Gardner, R. C. Brown and J.-P. Tessonnier, Thermal Stability of Aluminum-Rich ZSM-5 Zeolites and Consequences on Aromatization Reactions, J. Phys. Chem. C, 2016, 120(36), 20103–20113, DOI:10.1021/acs.jpcc.6b04671.
- G. Cruciani, Zeolites upon Heating: Factors Governing Their Thermal Stability and Structural Changes, J. Phys. Chem. Solids, 2006, 67(9–10), 1973–1994, DOI:10.1016/j.jpcs.2006.05.057.
- D. Ma, Y. Shu, X. Bao and Y. Xu, Methane Dehydro-Aromatization under Nonoxidative Conditions over Mo/HZSM-5 Catalysts: EPR Study of the Mo Species on/in the HZSM-5 Zeolite, J. Catal., 2000, 189(2), 314–325, DOI:10.1006/jcat.1999.2704.
- R. O. Savinelli and S. L. Scott, Wavelet Transform EXAFS Analysis of Mono- and Dimolybdate Model Compounds and a Mo/HZSM-5 Dehydroaromatization Catalyst, Phys. Chem. Chem. Phys., 2010, 12(21), 5660, 10.1039/b926474d.
- A. Kuzmin and J. Purans, Dehydration of the Molybdenum Trioxide Hydrates MoO3⋯nH2O: In Situ x-Ray Absorption Spectroscopy Study at the Mo K Edge, J. Phys.: Condens. Matter, 2000, 12(9), 1959–1970, DOI:10.1088/0953-8984/12/9/301.
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, Methane Dehydroaromatization by Mo/HZSM-5: Mono- or Bifunctional Catalysis?, ACS Catal., 2017, 7(1), 520–529, DOI:10.1021/acscatal.6b02497.
- F. Meneau and G. N. Greaves, Identifying Low and High Density Amorphous Phases during Zeolite Amorphisation Using Small and Wide Angle X-Ray Scattering, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 238(1–4), 70–74, DOI:10.1016/j.nimb.2005.06.020.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02949f |
| ‡ Previous address: Netherlands Organization for Scientific Research (NWO), ESRF, Grenoble Cedex 9, France. |
| This journal is © the Owner Societies 2024 |