
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper binding alters the core structure of amyloid fibrils formed by Y145Stop human prion protein†
Vidhyalakshmi
Sridharan
a,
Tara
George
a,
Daniel W.
Conroy
a,
Zach
Shaffer
a,
Witold K.
Surewicz
b and
Christopher P.
Jaroniec
 *a
*a
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA. E-mail: jaroniec.1@osu.edu
bDepartment of Physiology and Biophysics, Case Western Reserve University, Cleveland, OH, USA
First published on 8th October 2024
Abstract
Transmissible spongiform encephalopathies (or prion diseases) such as Creutzfeldt-Jacob disease, mad cow disease, and scrapie are characterized by accumulation in the brain of misfolded prion protein aggregates (PrPSc) that have properties of amyloid fibrils. Given that transition metal ions, such as copper and zinc, appear to be important for physiological functions of cellular PrP (PrPC) as well as for prion disease pathogenesis, exploring their role in the protein aggregation process is of considerable interest. Copper(II) in particular is well-known to bind to the four tandem octapeptide repeats (PHGGGWGQ) located in the N-terminal region of PrP (human PrP amino acids 60–91), as well as to additional histidine binding sites outside the octarepeat region with distinct binding modes depending on Cu2+ concentration. Here, using the Y145Stop human prion protein variant (huPrP23-144) as a model and a combination of multidimensional solution and solid-state NMR spectroscopy, atomic force microscopy and thioflavin T fluorescence assays we probed the binding of Cu2+ to monomeric huPrP23-144 and the impact of this binding on fibril assembly kinetics and their structural properties. Remarkably, we found that fibrils formed by huPrP23-144 containing one molar equivalent of bound Cu2+ adopt a core structure that is distinct from that found for huPrP23-144 in the absence of Cu2+ but, instead, corresponds to a conformational strain formed by huPrP23-144 containing the A117V mutation. A similar huPrP23-144 A117V-like amyloid core structure was adopted by a Cu2+-bound Δ51-91 huPrP23-144 deletion variant lacking the entire octarepeat region, suggesting that Cu2+ binding to His residues 96, 111 and 140 located near the C-terminus of huPrP23-144 is primarily responsible for the observed change in fibril conformation, potentially due to partial structuring of the intrinsically disordered huPrP23-144 by the bound Cu2+ during the fibril assembly process. We also found that fibrils formed by Cu2+-bound huPrP23-144 adopt the native huPrP23-144-like rather than A117V-like structure when the fibrillization reaction is seeded with pre-formed huPrP23-144 amyloid.
1. Introduction
Misfolding of proteins from their native states into β-sheet rich amyloids under physiological conditions is associated with a number of neurodegenerative diseases.1 One class of these disorders are transmissible spongiform encephalopathies (or prion diseases) which include Creutzfeldt–Jakob disease in humans, scrapie in sheep, and mad cow disease in cattle. The main pathogenic event in all these disorders is the conformational conversion of the cellular prion protein (PrPC) into infectious “scrapie” isoform (PrPSc) that has properties of amyloid fibrils.2–5It is well-established that, akin to several other proteins involved in neurodegenerative diseases,6,7 prion protein both in vitro8 and in vivo9 binds copper(II) and other transition metal ions. Therefore, there is significant interest in detailed understanding of the mechanism of this interaction and its role in normal function of PrPC as well as in disease pathogenesis. In a series of previous studies, Cu2+ binding to PrP has been investigated in vitro using a variety of biophysical techniques, including electron paramagnetic resonance (EPR) and solution nuclear magnetic resonance (NMR).6,10–14 Collectively, these studies have demonstrated that the four tandem octapeptide repeats PHGGGWGQ within the N-terminal part of the protein [residues 60-91 in human PrP (huPrP)] can bind up to four equivalents of Cu2+, and that histidine residues outside of the octarepeat region (H96, H111 and H140) serve as additional Cu2+ binding sites. Moreover, at sub-stoichiometric concentrations, Cu2+ ions coordinate to multiple histidine side-chains with a sub-nanomolar affinity.
Copper(II) binding to PrPC has been suggested to play a role in PrPC trafficking and endocytosis, synaptic adhesion and signalling, and brain metal homeostasis.13,15,16 Even more important, it was found that Cu2+ ions can modulate PrP amyloid formation in vitro,17–21 and that binding of this metal ion correlates with formation in vivo of structurally distinct PrPSc strains associated with two different subtypes of sporadic Creutzfeldt–Jakob disease.22
To gain molecular level insight into the mechanism by which binding of Cu2+ ions may lead to formation of structurally distinct PrP amyloid strains, we focus here on fibrils formed by the C-terminally truncated Y145Stop human PrP variant (huPrP23-144) associated with a familial prionopathy.23 This PrP variant has been previously extensively used as a model for investigating in vitro formation of distinct prion amyloid strains and the phenomenon of seeding barriers.24 In particular, our previous solid-state NMR and cryo-EM studies of huPrP23-144 amyloid fibrils have established that: (i) fibrils contain a relatively rigid parallel-in-register β-core region spanning residues ∼112–141 and a dynamically disordered ∼90-residue N-terminal domain,25–29 (ii) deletion of most of the N-terminal domain, up to residue ∼98, results in fibrils with a wild-type (WT) core structure and assembly kinetics,30 and (iii) mutations and deletions of certain residues within the amyloid core region can alter the kinetics of amyloid assembly and/or protein conformation within the fibril core.27,31–34
In the present study, we used a combination of multidimensional magic-angle spinning (MAS) solid-state NMR—a technique that has previously been successfully applied toward structural studies of other amyloid-metal ion complexes35–40—with solution NMR, atomic force microscopy (AFM) and thioflavin T (ThT) fluorescence to investigate Cu2+ binding to huPrP23-144 and its impact on the amyloid core conformation and fibril assembly kinetics. Additionally, since, akin to full-length prion protein, huPrP23-144 contains multiple Cu2+ binding sites, we have assessed the relative importance of Cu2+ binding to these different sites by performing similar studies with the Δ51-91 huPrP23-144 deletion variant that, despite lacking the entire octarepeat region, adopts the WT huPrP23-144 conformation in the amyloid state.30
2 Results and discussion
2.1 Cu2+ binding to monomeric huPrP23-144 in solution
First, we investigated Cu2+ binding to monomeric, intrinsically disordered huPrP23-144 by using solution NMR spectroscopy. Fig. 1 shows 2D 15N–1H heteronuclear single quantum coherence (HSQC) NMR spectra of huPrP23-144 recorded in the presence of increasing amounts of Cu2+ in the 0.5–2 molar equivalent range. These spectra clearly demonstrate Cu2+ concentration-dependent decrease in cross-peak intensity for multiple residues, primarily those located in the octarepeat region and in the vicinity of histidines 96, 111 and 140, due to paramagnetic relaxation enhancement.41 This is indicative of Cu2+ binding at all of these sites and consistent with earlier reports regarding high-affinity interactions between Cu2+ ions and multiple histidine residues in PrP.6,10–14 Given that at sub-stoichiometric Cu2+ concentrations the Cu2+ ions coordinate to multiple histidine side-chains and that paramagnetic relaxation enhancements for backbone 1HN nuclei depend strongly on several factors including 1HN–Cu2+ distance and local protein dynamics, we did not attempt to correlate the differences in peak intensities for the individual residues with Cu2+ binding affinities of the different histidine sites.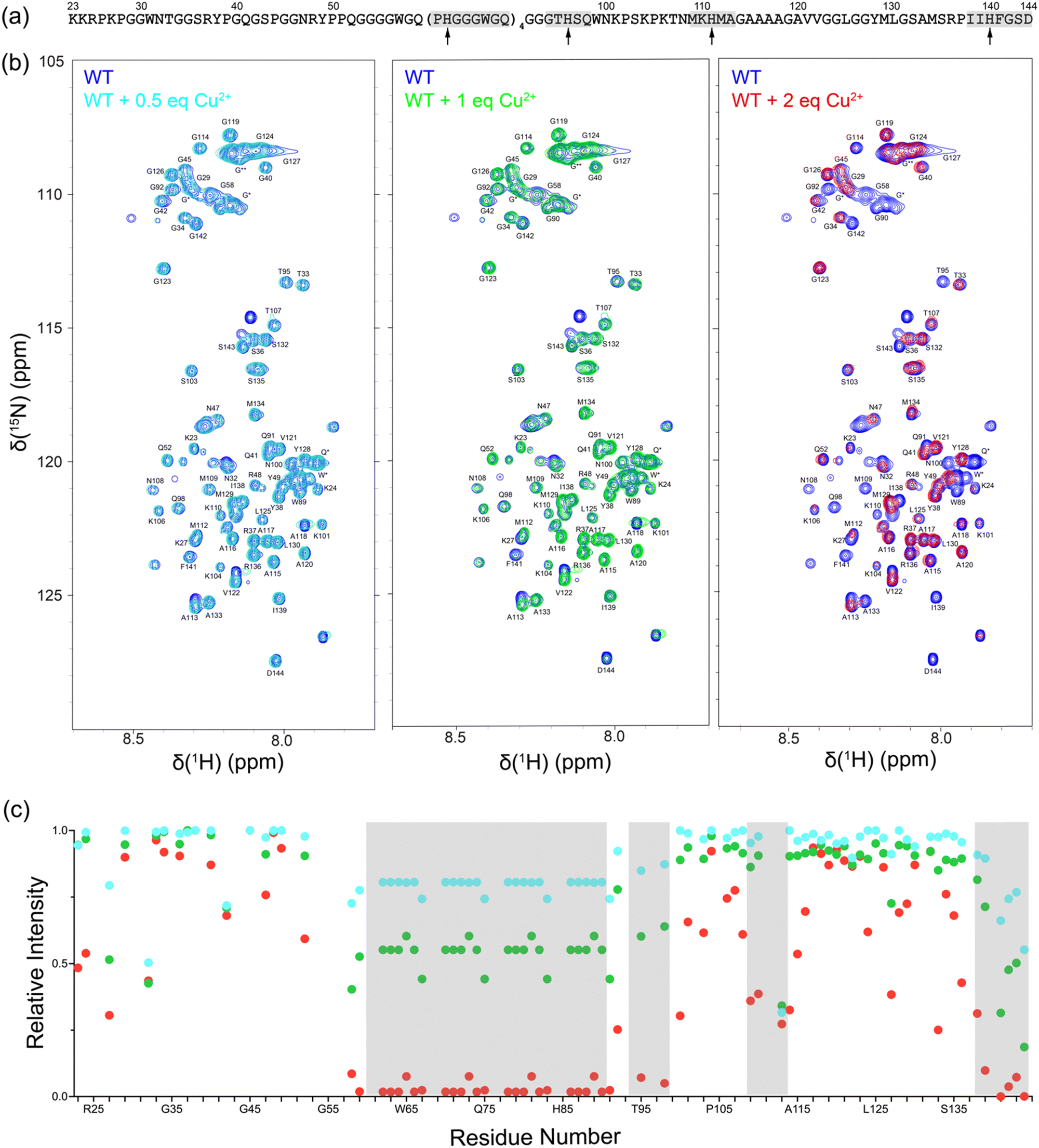 | ||
| Fig. 1 (a) Amino acid sequence of huPrP23-144. The locations of histidine residues are indicated by arrows and regions around these histidines showing the highest Cu2+ binding propensity and most pronounced reduction in resonance intensity due to paramagnetic relaxation enhancement [cf., panel (c)], including the 60–91 segment that contains the tandem octapeptide repeats and regions around H96, H111 and H140 are highlighted in gray rectangles. (b) 850 MHz 2D 15N–1H HSQC solution NMR spectra of monomeric huPrP23-144 in the presence of 0.5, 1 and 2 molar equivalents of Cu2+ as indicated in the insets. Overlapping resonances marked with * and ** correspond to octarepeat region residues, with ** denoting glycine residues preceded by other glycines. (c) Peak intensities for huPrP23-144 residues in the presence of 0.5 (cyan), 1 (green) and 2 (red) molar equivalents of Cu2+ relative to the corresponding intensities in the absence of Cu2+ as a function of residue number. The gray rectangles correspond to regions with the highest Cu2+ binding propensity highlighted in panel (a). | ||
2.2. Cu2+-bound huPrP23-144 fibril formation kinetics and morphology
Next, huPrP23-144 at concentration of 400 μM bound to varying amounts of Cu2+ was converted to amyloid fibrils in autocatalytic manner by addition of potassium phosphate buffer, and the kinetics of fibril formation were monitored by the ThT fluorescence assay (Fig. 2a).42 In the absence of Cu2+, both WT and Δ51-91 huPrP23-144 amyloids formed with a lag phase of ∼3–4 hours, in agreement with earlier studies.30,43 In the presence of one molar equivalent of bound Cu2+, fibril formation by both proteins was characterized by a relatively small increase in the lag phase to ∼4–5 hours. Binding of increasing amounts of Cu2+, up to four molar equivalents, to huPrP23-144 resulted in further modest lengthening of the lag phase for spontaneous (i.e., non-seeded) huPrP23-144 fibrillization reaction. The finding that with one equivalent of bound Cu2+ the lag phase to amyloid formation for Δ51-91 huPrP23-144 is longer than that for WT huPrP23-144 may be associated with somewhat different conformations adopted by these nominally dynamically disordered proteins upon Cu2+ binding, albeit no high-resolution data are available to substantiate this. Furthermore, as expected, in the presence of pre-formed WT huPrP23-144 fibril seeds the lag phase of fibril formation for Cu2+-bound huPrP23-144 was completely eliminated. | ||
| Fig. 2 (a) Kinetics of amyloid formation by WT and Δ51-91 huPrP23-144 bound to varying amounts of Cu2+ (filled circles; see inset) and by WT huPrP23-144 bound to one molar equivalent of Cu2+ and seeded with 2% (w/w) of pre-formed WT huPrP23-144 amyloid (open circles) monitored by ThT fluorescence. (b) and (c) Representative AFM images and height profiles for amyloid fibrils formed by (b) WT and (c) Δ51-91 huPrP23-144 bound to one molar equivalent of Cu2+. | ||
Morphologies of the amyloid fibril samples used for solid-state NMR studies were routinely assessed by using AFM. As shown in Fig. 2b and 2c, respectively, fibrils formed by WT and Δ51-91 huPrP23-144 containing one molar equivalent of bound Cu2+ have a uniform long, thread-like appearance that resembles that of corresponding fibrils obtained in the absence of bound Cu2+. On the other hand, the presence of bound Cu2+ appears to somewhat alter the fibril morphology resulting in smoother, less twisted fibrils as illustrated by the corresponding fibril height profiles.
2.3. Solid-state NMR studies of Cu2+-bound huPrP23-144 amyloids
In order to further verify that amyloid fibrils used for solid-state NMR studies indeed contain bound Cu2+ ions after multiple cycles of washing with 50 mM phosphate buffer at pH 6.4 prepared in absence of CuCl2 using ultrapure bioreagent grade (>99.0%) potassium phosphate salts and corresponding to the fibrillization conditions, we carried out standard inversion-recovery measurements of bulk amide 1H longitudinal relaxation times (T1) (Fig. S1, ESI†). These measurements show a significant ∼3–4-fold reduction in amide 1H T1 for fibrils formed by WT and Δ51-91 huPrP23-144 bound to one molar equivalent of Cu2+ relative to the corresponding fibrils generated in the absence of bound Cu2+ ions, in line with results obtained for microcrystalline proteins containing covalent Cys-EDTA-Cu2+ side-chains.44,45 For fibrils formed by huPrP23-144 containing two molar equivalents of bound Cu2+, an additional ∼3-fold reduction in the amide 1H T1 was observed.To gain residue-specific insight into the effect of bound Cu2+ ions on the molecular conformation and the degree of order within the core region of huPrP23-144 fibrils, we recorded fingerprint 2D 15N–13Cα chemical shift correlation solid-state NMR spectra for these fibrils generated in the absence of Cu2+ or in the presence of one molar equivalent of bound Cu2+. These spectra, shown in Fig. 3a, reveal that fibrils formed by Cu2+-bound huPrP23-144 adopt a core conformation that is highly ordered at the molecular level, but clearly distinct from that of WT huPrP23-144 amyloid. Remarkably, spectra of the fibrils formed by Cu2+-bound huPrP23-144 were found—in a consistent and reproducible manner (Fig. S2, ESI†)—to be very similar to those reported previously by our group for fibrils formed by the A117V mutant of huPrP23-14427,34 as well as several other huPrP23-144 single residue core mutants.33 The huPrP23-144 A117V amyloid core was previously found to map to residues ∼121–138 (vs. ∼112–141 for WT huPrP23-144 fibrils) in a mostly β-strand conformation, and appears to correspond to one particularly stable fold that is accessible to PrP23-144 within its amyloid structural landscape.27,34 Additionally, we found that huPrP23-144 bound to two molar equivalents of Cu2+ also adopts the A117V-like amyloid core structure (Fig. S3, ESI†); based on this finding solid-state NMR studies of amyloids formed by huPrP23-144 containing more than two equivalents of bound Cu2+ were not pursued. As additional controls, we also recorded 2D 15N–13Cα solid-state NMR spectra of huPrP23-144 fibrils with one molar equivalent of Cu2+ added to the sample post fibril formation (Fig. 3b), and fibrils formed by huPrP23-144 containing one equivalent of bound Cu2+ seeded with pre-formed WT huPrP23-144 amyloid (Fig. 3c). In both cases, fibrils clearly exhibit the WT huPrP23-144 amyloid core structure with concomitant reduction of cross-peak intensity for residues A113 and I138-H140 likely caused by backbone 1H, 13C and 15N transverse paramagnetic relaxation enhancements41,46 due to Cu2+ binding at H111 and H140 (as well as H96 and other more distant sites). Collectively, these solid-state NMR data indicate that, in the absence of seeding with pre-formed WT huPrP23-144 amyloid, Cu2+ binding to multiple sites in monomeric huPrP23-144 (including the octapeptide repeats and H96, H111 and H140) leads to huPrP23-144 adopting an alternate stable A117V-like core fold that is distinct from the WT fold. Given that, akin to the Ala to Val mutation at position 117 in huPrP23-144,47 the binding of one molar equivalent of Cu2+ to huPrP23-144 has a relatively minor effect on the fibrillization kinetics (cf., Fig. 2a), adoption of this A117V-like core structure by Cu2+-bound huPrP23-144 may potentially be a consequence of a certain degree of structuring of the dynamically disordered huPrP23-144 by the bound Cu2+ ions.10–14
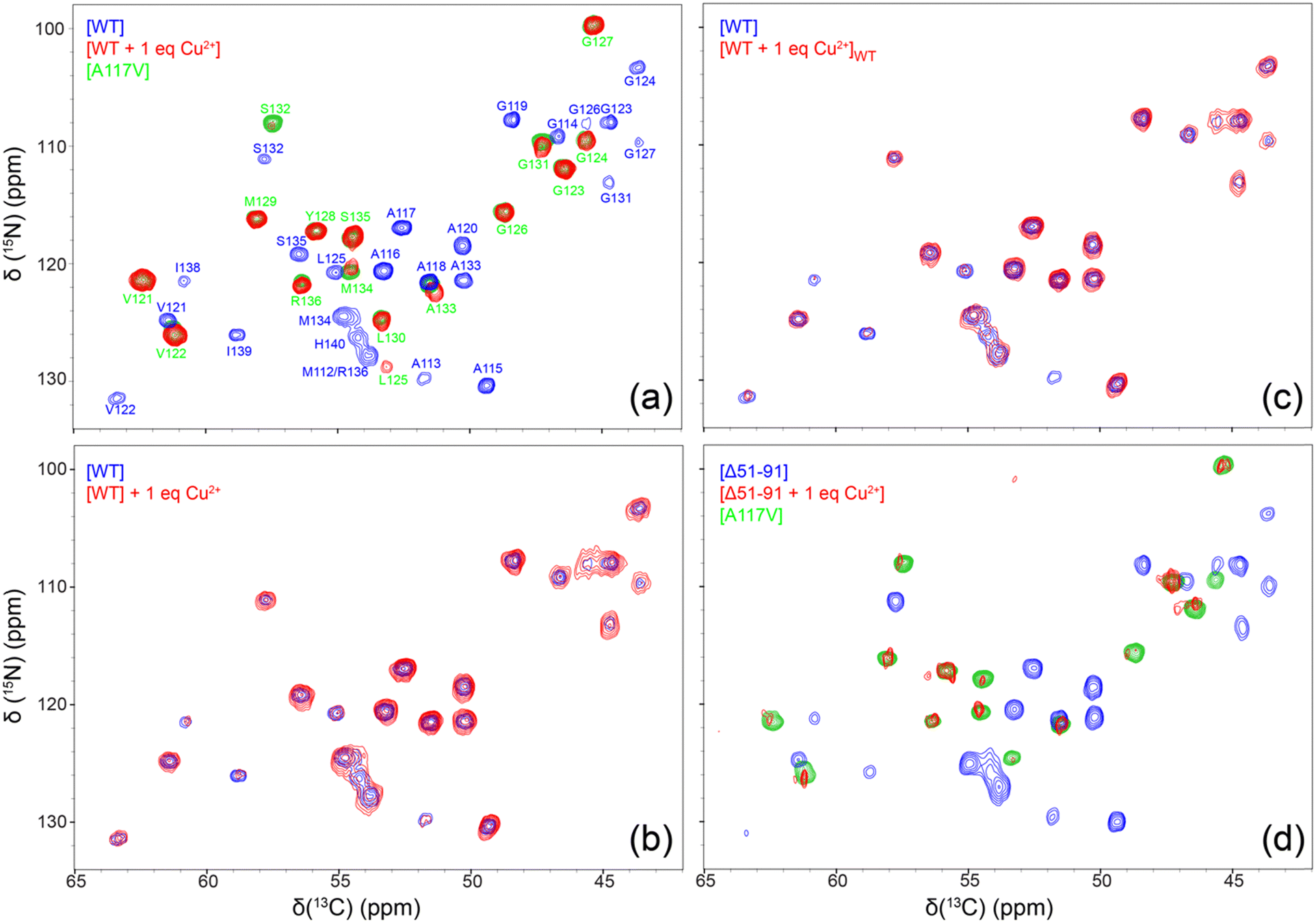 | ||
| Fig. 3 2D 15N–13Cα solid-state NMR chemical shift correlation spectra (red contours) of amyloid fibrils formed by (a) WT huPrP23-144 containing one molar equivalent of bound Cu2+, (b) WT huPrP23-144 incubated with one molar equivalent of Cu2+ following fibril formation, (c) WT huPrP23-144 containing one molar equivalent of bound Cu2+ seeded with 2% (w/w) pre-formed WT huPrP23-144 fibrils, and (d) Δ51-91 huPrP23-144 containing one molar equivalent of bound Cu2+. As appropriate, the spectra are overlaid with reference spectra corresponding to WT [panels (a)–(c)] or Δ51-91 huPrP23-144 [panel (d)] fibrils (blue contours) and huPrP23-144 A117V fibrils (green contours) with resonance assignments shown in panel (a). All spectra were recorded at 800 MHz, 11.111 kHz MAS rate and effective sample temperature of ca. 5 °C. | ||
To evaluate the relative importance of individual Cu2+ binding sites for the apparent conformational strain switching from WT to A117V-like core fold upon Cu2+ binding to the monomeric huPrP23-144 prior to amyloid formation, we extended our solid-state NMR studies above to include the Δ51-91 huPrP23-144 deletion variant. The latter variant, which was previously found to adopt the WT huPrP23-144 amyloid core fold,30 is missing the entire octarepeat region and, thus, able to bind Cu2+ ions through H96, H111 and H140. As shown In Fig. 3d, the 2D 15N–13Cα solid-state NMR spectra of fibrils formed by Δ51-91 huPrP23-144 with one molar equivalent of bound Cu2+ are also indicative of the A117V-like core structure, in analogy to Cu2+-bound WT huPrP23-144—note that the reduction in overall spectral intensity for fibrils of Δ51-91 huPrP23-144 with one molar equivalent of bound Cu2+ relative to the corresponding fibrils not containing bound Cu2+ ions is related to a limited amount of sample present in the solid-state NMR rotor, and that the variations in peak intensities for individual residues in WT and Δ51-91 huPrP23-144 fibrils are due to μs–ms protein backbone dynamics in the fibril core.26,28 This finding suggests that the binding of Cu2+ ions to three histidine residues, H96, H111, H140 (which are located outside the octarepeat region in WT huPrP23-144), is the primary driver of the observed fibril conformational change.
3. Concluding remarks
Previous studies of amyloids formed by several PrP23-144 variants have revealed that fibrils consist of a ∼20–30 residue C-terminal β-core preceded by an extended dynamically disordered N-terminal domain.27,31–34 Furthermore, these studies showed that relatively conservative mutations or deletions of core residues can lead to the formation of structurally distinct amyloid strains. In the present study, we investigated the binding of Cu2+ to monomeric huPrP23-144 and its impact on amyloid fibril assembly and conformation, using a combination of solution and solid-state NMR spectroscopy as the primary tools. The most remarkable finding of our study is that the binding of a single molar equivalent of Cu2+ to monomeric huPrP23-144 greatly impacts its fibrillization, leading to formation of a structurally distinct amyloid strain that resembles that previously found for fibrils formed by A117V huPrP23-144 and several other huPrP23-144 single residue core mutants.27,33,34 Additionally, we found that upon seeding with pre-formed WT huPrP23-144 amyloid Cu2+-bound huPrP23-144 adopts a fibril structure resembling that of WT huPrP23-144, but not A117V huPrP23-144. The latter observation is suggestive of efficient conformational adaptation and templating of the Cu2+-bound huPrP23-144 monomers onto the WT huPrP23-144 fibril scaffold.Since huPrP23-144 contains multiple Cu2+ binding sites, including the tandem PHGGGWGQ octapeptide repeats spanning residues 60–91 and histidines 96, 111 and 140, analogous studies were performed for the Δ51-91 deletion mutant of huPrP23-144 that, despite lacking the entire octarepeat region, adopts a WT amyloid core fold.30 Akin to WT huPrP23-144, the latter variant in the presence of one molar equivalent of bound Cu2+ was also found to form fibrils with a A117V-like structure, suggesting that Cu2+ binding to histidine residues 96, 111 and 140 is sufficient to induce the observed conformational change, possibly due to the bound Cu2+ resulting in a partial structuring of the intrinsically disordered monomeric huPrP23-14410–14en route to amyloid assembly.
4. Materials and methods
4.1. Protein expression and purification
Plasmid encoding for huPrP(23-144) with a N-terminal His6-tag and thrombin cleavage site has been described previously.43 As described in previous studies,27,30 uniformly 13C,15N-labeled protein was expressed in E. coli (BL21 DE3) in minimal media containing 13C-glucose and 15NH4Cl as the sole carbon and nitrogen sources, respectively. Protein was purified by Ni-NTA affinity chromatography, and the His6-tag cleaved using biotinylated thrombin. Thrombin and residual His6-tag were removed using streptavidin-agarose beads and dialysis against ultrapure water, respectively. Protein purity was verified by MALDI mass spectrometry and SDS-PAGE.4.2. Preparation of amyloid fibrils for solid-state NMR spectroscopy
Solutions of lyophilized huPrP23-144 at 400 μM concentration in ultrapure water were incubated at 4 °C overnight with varying amounts of Cu2+ added as a 400 mM aqueous solution of CuCl2. Fibril formation was initiated by addition of 1 M potassium phosphate pH 6.4 buffer to a final potassium phosphate concentration of 50 mM at pH 6.4. Amyloid fibrils were formed under quiescent conditions at 25 °C as described previously.32–344.3. Thioflavin T fluorescence and atomic force microscopy
Kinetics of huPrP23-144 fibril formation in the absence and presence of Cu2+ were monitored by using the ThT fluorescence assay.42 The morphologies of fibril samples used for solid-state NMR analysis were evaluated by atomic force microscopy as described in detail in previous studies.30,344.4. NMR spectroscopy
Solution NMR 15N–1H HSQC spectra for huPrP23-144 dissolved at a concentration of 400 μM in aqueous MES pH 6.4 buffer containing varying amounts of Cu2+ (note that MES was used for these experiments instead of phosphate buffer to avoid protein aggregation/fibril formation) were recorded at 25 °C using an 850 MHz Bruker Avance III HD spectrometer equipped with a TCI CryoProbe. Amyloid fibril samples for solid-state NMR studies were incubated for 48 h at 25 °C and centrifuged. Pellets were washed three times with 50 mM potassium phosphate pH 6.4 buffer and packed into Bruker 3.2 mm zirconia rotors. Solid-state NMR 15N–13Cα chemical shift correlation spectra were recorded on an 800 MHz Bruker Avance III HD spectrometer equipped with a 3.2 mm triple-resonance 1H–13C–15N Efree probe. MAS frequency and effective sample temperature were maintained at 11.111 kHz and ca. 5 °C, respectively, and other experimental parameters were similar to those described in previous studies.30,32 Spectra were processed and analyzed using nmrPipe48 and Sparky.49Author contributions
CPJ designed the experiments. VS, TG and ZS prepared samples. VS and DC performed experiments. VS analyzed data and prepared figures. VS, WKS and CPJ wrote the manuscript.Data availability
The data supporting this article have been included as part of the manuscript and ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from NSF (MCB-2303862 to C. P. J.) and NIH (R01GM094357 and S10OD012303 to C. P. J. and R01NS103848 to W. K. S.). The authors thank Christian M. O’Neil for stimulating discussions and acknowledge the contributions of Parvez Alam and Dr Theint Theint to the preliminary ThT fluorescence and AFM assays associated with these studies.References
- F. Chiti and C. M. Dobson, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- S. B. Prusiner, Prions, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13363–13383 CrossRef CAS PubMed.
- N. J. Cobb and W. K. Surewicz, Prion Diseases and Their Biochemical Mechanisms, Biochemistry, 2009, 48, 2574–2585 CrossRef CAS PubMed.
- A. Aguzzi, C. Sigurdson and M. Heikenwaelder, Molecular Mechanisms of Prion Pathogenesis, Annu. Rev. Pathol.: Mech. Dis., 2008, 3, 11–40 CrossRef CAS PubMed.
- A. Kraus, B. R. Groveman and B. Caughey, Prions and the Potential Transmissibility of Protein Misfolding Diseases, Annu. Rev. Microbiol., 2013, 67, 543–564 CrossRef CAS PubMed.
- J. H. Viles, Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion diseases, Coord. Chem. Rev., 2012, 256, 2271–2284 CrossRef CAS.
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensin, Copper, zinc and iron in neurodegenerative diseases (Alzheimer's, Parkinson's and prion diseases), Coord. Chem. Rev., 2012, 256, 2129–2141 CrossRef CAS.
- M. P. Hornshaw, J. R. Mcdermott and J. M. Candy, Copper Binding to the N-Terminal Tandem Repeat Regions of Mammalian and Avian Prion Protein, Biochem. Biophys. Res. Commun., 1995, 207, 621–629 CrossRef CAS PubMed.
- D. R. Brown, K. Qin, J. W. Herms, A. Madlung, J. Manson, R. Strome, P. E. Fraser, T. Kruck, A. von Bohlen, W. Schulz-Schaeffer, A. Giese, D. Westaway and H. Kretzschmar, The cellular prion protein binds copper in vivo, Nature, 1997, 390, 684–687 CrossRef CAS PubMed.
- G. L. Millhauser, Copper and the Prion Protein: Methods, Structures, Function, and Disease, Annu. Rev. Phys. Chem., 2007, 58, 299–320 CrossRef CAS PubMed.
- C. Sánchez-López, G. Rossetti, L. Quintanar and P. Carloni, Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions, Int. J. Mol. Sci., 2018, 20, 18 CrossRef PubMed.
- L. Quintanar and G. L. Millhauser, Methods in Enzymology, Elsevier, 2022, vol. 666, pp. 297–314 Search PubMed.
- G. Salzano, G. Giachin and G. Legname, Structural Consequences of Copper Binding to the Prion Protein, Cells, 2019, 8, 770 CrossRef CAS PubMed.
- E. Walter, D. Stevens, A. Spevacek, M. Visconte, A. Rossi and G. Millhauser, Copper Binding Extrinsic to the Octarepeat Region in the Prion Protein, Curr. Protein Pept. Sci., 2009, 10, 529–535 CrossRef CAS PubMed.
- N. Vassallo and J. Herms, Cellular prion protein function in copper homeostasis and redox signalling at the synapse, J. Neurochem., 2003, 86, 538–544 CrossRef CAS PubMed.
- A. Aguzzi, F. Baumann and J. Bremer, The Prion's Elusive Reason for Being, Annu. Rev. Neurosci., 2008, 31, 439–477 CrossRef CAS PubMed.
- C. E. Jones, S. R. Abdelraheim, D. R. Brown and J. H. Viles, Preferential Cu2+ Coordination by His96 and His111 Induces β-Sheet Formation in the Unstructured Amyloidogenic Region of the Prion Protein, J. Biol. Chem., 2004, 279, 32018–32027 CrossRef CAS PubMed.
- E. Wong, A. M. Thackray and R. Bujdoso, Copper induces increased beta-sheet content in the scrapie-susceptible ovine prion protein PrPVRQ compared with the resistant allelic variant PrPARR, Biochem. J., 2004, 380, 273–282 CrossRef CAS PubMed.
- O. V. Bocharova, L. Breydo, V. V. Salnikov and I. V. Baskakov, Copper(II) Inhibits in Vitro Conversion of Prion Protein into Amyloid Fibrils, Biochemistry, 2005, 44, 6776–6787 CrossRef CAS PubMed.
- M. A. Wells, G. S. Jackson, S. Jones, L. L. P. Hosszu, C. J. Craven, A. R. Clarke, J. Collinge and J. P. Waltho, A reassessment of copper(II) binding in the full-length prion protein, Biochem. J., 2006, 399, 435–444 CrossRef CAS PubMed.
- A. K. Thakur, A. K. Srivastava, V. Srinivas, K. V. R. Chary and C. M. Rao, Copper Alters Aggregation Behavior of Prion Protein and Induces Novel Interactions between Its N- and C-terminal Regions, J. Biol. Chem., 2011, 286, 38533–38545 CrossRef CAS PubMed.
- J. D. F. Wadsworth, A. F. Hill, S. Joiner, G. S. Jackson, A. R. Clarke and J. Collinge, Strain-specific prion-protein conformation determined by metal ions, Nat. Cell Biol., 1999, 1, 55–59 CrossRef CAS PubMed.
- B. Ghetti, P. Piccardo, M. G. Spillantini, Y. Ichimiya, M. Porro, F. Perini, T. Kitamoto, J. Tateishi, C. Seiler, B. Frangione, O. Bugiani, G. Giaccone, F. Prelli, M. Goedert, S. R. Dlouhy and F. Tagliavini, Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 744–748 CrossRef CAS PubMed.
- W. K. Surewicz, E. M. Jones and A. C. Apetri, The Emerging Principles of Mammalian Prion Propagation and Transmissibility Barriers: Insight from Studies in Vitro, Acc. Chem. Res., 2006, 39, 654–662 CrossRef CAS PubMed.
- J. J. Helmus, K. Surewicz, P. S. Nadaud, W. K. Surewicz and C. P. Jaroniec, Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6284–6289 CrossRef CAS PubMed.
- J. J. Helmus, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, Conformational flexibility of Y145Stop human prion protein amyloid fibrils probed by solid-state nuclear magnetic resonance spectroscopy, J. Am. Chem. Soc., 2010, 132, 2393–2403 CrossRef CAS PubMed.
- T. Theint, P. S. Nadaud, D. Aucoin, J. J. Helmus, S. P. Pondaven, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy, Nat. Commun., 2017, 8, 753 CrossRef PubMed.
- M. D. Shannon, T. Theint, D. Mukhopadhyay, K. Surewicz, W. K. Surewicz, D. Marion, P. Schanda and C. P. Jaroniec, Conformational Dynamics in the Core of Human Y145Stop Prion Protein Amyloid Probed by Relaxation Dispersion NMR, ChemPhysChem., 2019, 20, 311–317 CrossRef CAS PubMed.
- Q. Li, C. P. Jaroniec and W. K. Surewicz, Cryo-EM structure of disease-related prion fibrils provides insights into seeding barriers, Nat. Struct. Mol. Biol., 2022, 29, 962–965 CrossRef CAS PubMed.
- Z. Qi, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, Influence of the Dynamically Disordered N-Terminal Tail Domain on the Amyloid Core Structure of Human Y145Stop Prion Protein Fibrils, Front. Mol. Biosci., 2022, 9, 841790 CrossRef CAS PubMed.
- E. M. Jones, B. Wu, K. Surewicz, P. S. Nadaud, J. J. Helmus, S. Chen, C. P. Jaroniec and W. K. Surewicz, Structural polymorphism in amyloids: new insights from studies with Y145Stop prion protein fibrils, J. Biol. Chem., 2011, 286, 42777–42784 CrossRef CAS PubMed.
- T. Theint, P. S. Nadaud, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, 13C and 15N chemical shift assignments of mammalian Y145Stop prion protein amyloid fibrils, Biomol. NMR Assignments., 2017, 11, 75–80 CrossRef CAS PubMed.
- T. Theint, Y. Xia, P. S. Nadaud, D. Mukhopadhyay, C. D. Schwieters, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, Structural Studies of Amyloid Fibrils by Paramagnetic Solid-State Nuclear Magnetic Resonance Spectroscopy, J. Am. Chem. Soc., 2018, 140, 13161–13166 CrossRef CAS PubMed.
- H. H. Dao, M. Z. Hlaing, Y. Ma, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, 13C and 15N chemical shift assignments of A117V and M129V human Y145Stop prion protein amyloid fibrils, Biomol. NMR Assignments., 2021, 15, 45–51 CrossRef CAS PubMed.
- S. Parthasarathy, F. Long, Y. Miller, Y. Xiao, D. McElheny, K. Thurber, B. Ma, R. Nussinov and Y. Ishii, Molecular-Level Examination of Cu 2+ Binding Structure for Amyloid Fibrils of 40-Residue Alzheimer's β by Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 2011, 133, 3390–3400 CrossRef CAS PubMed.
- V. S. Mithu, B. Sarkar, D. Bhowmik, M. Chandrakesan, S. Maiti and P. K. Madhu, Zn++ Binding Disrupts the Asp23-Lys28 Salt Bridge without Altering the Hairpin-Shaped Cross-β Structure of Aβ42 Amyloid Aggregates, Biophys. J., 2011, 101, 2825–2832 CrossRef CAS PubMed.
- S. Parthasarathy, B. Yoo, D. McElheny, W. Tay and Y. Ishii, Capturing a Reactive State of Amyloid Aggregates, J. Biol. Chem., 2014, 289, 9998–10010 CrossRef CAS PubMed.
- A. Sinopoli, A. Magrì, D. Milardi, M. Pappalardo, P. Pucci, A. Flagiello, J. J. Titman, V. G. Nicoletti, G. Caruso, G. Pappalardo and G. Grasso, The role of copper(II) in the aggregation of human amylin, Metallomics, 2014, 6, 1841–1852 CrossRef CAS PubMed.
- L. Vugmeyster, D. F. Au, D. Ostrovsky, B. Kierl, R. Fu, Z. Hu and W. Qiang, Effect of Post-Translational Modifications and Mutations on Amyloid-β Fibrils Dynamics at N Terminus, Biophys. J., 2019, 117, 1524–1535 CrossRef CAS PubMed.
- M. Lee, T. Wang, O. V. Makhlynets, Y. Wu, N. F. Polizzi, H. Wu, P. M. Gosavi, J. Stöhr, I. V. Korendovych, W. F. DeGrado and M. Hong, Zinc-binding structure of a catalytic amyloid from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6191–6196 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat and G. Parigi, Solution NMR of Paramagnetic Molecules, Elsevier Science, 2001 Search PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS PubMed.
- B. Kundu, N. R. Maiti, E. M. Jones, K. A. Surewicz, D. L. Vanik and W. K. Surewicz, Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: Structural clues for prion propagation, Proc. Natl. Acad. Sci., 2003, 100, 12069–12074 CrossRef CAS PubMed.
- P. S. Nadaud, J. J. Helmus, S. L. Kall and C. P. Jaroniec, Paramagnetic Ions Enable Tuning of Nuclear Relaxation Rates and Provide Long-Range Structural Restraints in Solid-State NMR of Proteins, J. Am. Chem. Soc., 2009, 131, 8108–8120 CrossRef CAS PubMed.
- P. S. Nadaud, J. J. Helmus, I. Sengupta and C. P. Jaroniec, Rapid Acquisition of Multidimensional Solid-State NMR Spectra of Proteins Facilitated by Covalently Bound Paramagnetic Tags, J. Am. Chem. Soc., 2010, 132, 9561–9563 CrossRef CAS PubMed.
- C. P. Jaroniec, Structural studies of proteins by paramagnetic solid-state NMR spectroscopy, J. Magn. Reson., 2015, 253, 50–59 CrossRef CAS PubMed.
- E. M. Jones, K. Surewicz and W. K. Surewicz, Role of N-terminal Familial Mutations in Prion Protein Fibrillization and Prion Amyloid Propagation in Vitro, J. Biol. Chem., 2006, 281, 8190–8196 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03593c |
| This journal is © the Owner Societies 2024 |